

Abstract
Deubiquitinating enzymes (DUBs) regulate the removal of the polyubiquitin chain from proteins targeted for degradation. Current approaches to quantify DUB activity are limited to test tube-based assays that incorporate enzymes or cell lysates, but not intact cells. The goal of this work was to develop a novel peptide-based biosensor of DUB activity that is cell permeable, protease-resilient, fluorescent, and specific to DUBs. The biosensor consists of an N-terminal β-hairpin motif that acts as both a ‘protectide’ to increase intracellular stability and a cell penetrating peptide (CPP) to facilitate the uptake into intact cells. The β-hairpin was conjugated to a C-terminal substrate consisting of the last four amino acids in ubiquitin (LRGG) to facilitate DUB mediated cleavage of a C-terminal fluorophore (AFC). The kinetics of the peptide reporter were characterized in cell lysates by dose response and inhibition enzymology studies. Inhibition studies with an established DUB inhibitor (PR-619) confirmed the specificity of both reporters to DUBs. Fluorometry and fluorescent microscopy experiments followed by mathematical modeling established the capability of the biosensor to measure DUB activity in intact cells while maintaining cellular integrity. The novel reporter introduced here is compatible with high-throughput single cell analysis platforms such as FACS and droplet microfluidics facilitating direct quantification of DUB activity in single intact cells with direct application in point-of-care cancer diagnostics and drug discovery.
Keywords: Deubiquitinating enzymes (DUBs), Peptide-based biosensors, Cell penetrating peptides, Protease-resilient peptides, Single cell analysis, Fluorometry
Graphical abstract
Introduction
Recent research has established the role of the members of the ubiquitin-proteasome-system (UPS) as important pharmacological targets.1–3 The UPS is an intracellular biochemical pathway primarily responsible for identifying misfolded and dysregulated proteins and targeting them to the proteasome for degradation.4 The process starts with a cascade of three enzymes: E1 ubiquitin activating enzymes, E2 ubiquitin conjugating enzymes, and E3 ubiquitin ligases responsible for attaching a monoubiquitin or a polyubiquitin chain to a lysine residue on a target protein. In addition to playing signaling roles, protein polyubiquitination may direct a target protein to the 26S proteasome. Polyubiquitination is a reversible process mediated by a class of enzymes called deubiquitinating enzymes (DUBs). DUBs cleave the isopeptide bond formed between the C-terminal carboxylic acid of ubiquitin and the amine group on the side chain of lysine or ornithine to detach the polyubiquitin chain and rescue the proteins from proteasomal degradation.4 Deubiquitination is believed to correct errors in the polyubiquitination process by ensuring correct ubiquitin chain linkage and chain length.5 DUBs have been shown to be associated with several human pathologies including multiple myeloma (MM),6 neurodegenerative diseases,2 and cardiac disease.7 Regulation of protein degradation makes members of the UPS, including the proteasome and DUBs, attractive pharmacological targets with significant efforts underway to identify new drugs such as small molecule inhibitors to regulate their activity.8 In the past decade, the proteasome inhibitor Bortezomib (Velcade) found remarkable clinical success in the treatment of multiple myeloma, a cancer characterized by malignant plasma cells in the bone marrow that leads to a decreased immune response, weakening of the bones, kidney failure, and congestive heart failure.6 The increased proteolytic activity of the proteasome is associated with a malignant transformation in multiple myeloma which results in Bortezomib-induced apoptosis in myeloma-derived cells by inhibiting the proteasome.9 A significant challenge with proteasome-targeted therapeutics is drug resistance leading to relapse and death in the treated patients.6 Recent research has discovered that increased activity of DUBs resulted in MM patients developing a resistance to Bortezomib, while combinational treatment of DUB inhibitors and proteasome inhibitors led to enhanced Bortezomib efficacy.1, 3, 10, 11 This finding coupled with the cost of molecularly-targeted therapeutics, and the potential for severe side effects like peripheral neuropathy, has created an urgent need for more advanced biochemical screening methods for members of the UPS including the proteasome and DUBs.12 Such biochemical tools also have vast applications in fundamental research on deubiquitination biochemistry.
Several molecular probes have been developed for measuring DUB activity in vitro. One of the most widely used approaches to measure DUB activity consists of a full length ubiquitin (Ub) with a fluorophore conjugated to the C-terminus (e.g, 7-amino-4-methylcoumarin-AMC, λex = 354, λem =442).13,14 The underlying principle for this reporter is that as long as the fluorophore is bound to the substrate (Ub), minimal fluorescence will be observed in the sample. DUB-mediated cleavage of the reporter frees the C-terminal fluorophore and results in an easily detectable fluorescent signal that can be measured using methods like fluorometry. This reporting scheme has been improved by incorporating more sensitive fluorophores,15, 16 different analytical outputs,16, 17 or FRET- (Förster resonance energy transfer)-based pairings.18 However, all of these reporters are limited to enzyme-only and lysate-based experiments and are not compatible with intact cells. Although, lysate-based studies facilitate DUB profiling to reflect the native function and regulation of DUBs, cell lysis can cause a dilution of the cytoplasmic and nuclear proteins leading to the potential dissociation of protein complexes necessary for DUB activity.19 Therefore, more physiologically-relevant screening methods are required to better mimic the conditions in vivo for both clinical diagnostic and fundamental research applications. Additionally, bulk measurments of blended average cell responses are incapable of accoutning for the significant heterogeneity associated with cancer cells which results in the inability to identify distinct subpopulations such as low occurrence, drug resistant cells. More recently, the need for intracellular measurements of DUB activity in intact cells has been identified and attention has been shifted towards the development activity-based probes for intracellular detection and quantification of members of the UPS with minimal to no damage to the cell membrane. Interesting examples include works by An and Statsyuk20 and Gui and colleaguesl.19 An and Statsyuk described the development of a cell-membrane permeable small-molecule probe named ABP1 that covalently labels ubiqutin-like (UBL) proteins in vitro and in cells in the presence of E1 enzymes and ATP. This mechanism-based small-molecule probe can be used to discover and to detect active UBL proteins and to monitor the intracellular activity of E1 enzymes inside intact cells.20 Gui and colleagues employed cell-penetrating peptides (CPPs), particularly cyclic polyarginine (cR10), to deliver an activity-based DUB reporter into cells which facilitated DUB profiling in intact HeLa cells, identifying active DUBs using immunocapture and label-free quantitative spectrometry. They also used this reporter to assess DUB inhibition by small-molecule inhibitors in intact cells.19
In this work, a smiliar approach was undertaken to deliver a peptide-based reporter into the intracellular environment using a cell penetrating peptide. A DUB recognition substrate consisting of the last 4 amino acid residues of ubiquitin (LRGG) was conjugated to a β-hairpin sequence motif (RWVRVpGRWIRQ) recently characterized by Safa et al. as a cell penetrating peptide (CPP) with rapid uptake and enhanced protease-resilience.21 This CPP was shown to penetrate intact cells within 10 minutes and remain stable in the intracellular environment during the course of several hours with a half-life of ~400 minutes in HeLa lysates. The β-hairpin motif of the peptide-based reporter confers enhanced protease-resilience making it ideal for performing long-term, dynamic measurements of DUB activity in intact single cells. First, an in-depth enzymology analysis was performed to demonstrate the sensitivity and specifity of the probe to DUBs in HeLa and OPM2 (a model multiple myeloma cell line) cell lysates with reaction rate kinetics comparable to a commercially available DUB reporter referred to as Peptide 3 [Z-LRGG-AMC]. Dose-response inhibition studies revealed a statistically significant effect on the rates of DUB-mediated hydrolysis of the peptide substantiating its specficity to DUBs. This was followed by microscopic characterization of peptide uptake including cell viability staining and time- and concentration-dependent cell permeability studies. These studies found that unlike the majority of the commercially available DUB reporters, including Peptitde 3, the novel reporter Peptide 1 was capable of penetrating the plasma membrane of intact cells. Finally, the application of the reporter to measure DUB activity in intact HeLa cells was demonstrated by fluorometry studies. A mathematical model was developed for the two-step process of cell penetration and DUB-mediated cleavage of the peptide-based reporter which revealed fundamental results about the enzymology of DUBs and served as a quantitative baseline for future single cell studies using this reporter. These analyses demonstrated that while enzyme-substrate reactions in intact cells fit the Michaelis-Menten equation, this process is more complex when dealing with intact cells. Non-linear regression analysis and mathematical modeling of enzyme-substrate interactions in intact cells facilitated detailed quantification of enzyme-substrate reaction kinetic parameters. Finally, DUB activity was directly visualized in intact cells using fluorescent microscopy. This quality makes this reporter compatible with state-of-the-art single cell technologies such as FACS and novel microfluidic platforms combinations of which make novel bioanalytical platforms for high-throughput quantification and visualization of DUB activity in single intact cells.
Materials and Methods
Chemicals
Safety-catch resin (aliphatic), Fmoc-protected amino acids, 2-(6-Chloro-1H-benzotriazole-1-yl)-1,1,3,3 - tetramethylaminium hexafluorophosphate (HCTU), trifluoroacetic acid (TFA) and rink amide SS resin were purchased from Advanced ChemTech (Louisville, KY). Boc-Gly-AFC was purchased from AnaSpec (Fremont, CA). Ethyl cyano(hydroxyimino)acetato)-tri-(1-pyrrolidinyl)-phosphonium (PyBOP) was purchased from Novabiochem (Billerica, MA). Dimethylformamide (DMF) was purchased from Protein Technologies, Tucson, AZ. Diisopropylethylamine (DIEA), triisopropylsilane, and N-methylmorpholine (NMM) tetrakis(triphenylphosphine) palladium (0) (palladium), 5(6)-carboxyfluorescein (FAM), chloroform (CHCl3), and methanol (MeOH) were purchased from Sigma Aldrich, St. Louis, MO. Dichloromethane (DCM) and acetonitrile were purchased from VWR, Atlanta, GA. Nα-Fmoc-Nδ-allyloxycarbonyl-L-ornithine (Fmoc-Orn(Aloc)-OH) was purchased from Chem-Impex International, Inc, Wood Dale, IL. 1-Hydroxy-6-(trifluoromethyl) benzotriazole (HOBt) and (Ethyl cyano(hydroxyimino)acetato)-tri-(1-pyrrolidinyl)-phosphonium (PyBOP) were purchased from Novabiochem, Billerica, MA. Glacial acetic acid was purchased from Alfa Aesar, Ward Hill, MA. Acetic anhydride was purchased from Fisher Scientific, Fair Lawn, NJ. All chemicals were used without further purification. Peptide 3 [Z-Leu-Arg-Gly-Gly-AMC] was purchased from Boston Biochem, AMC [7-Amino-4-methylcoumarin], AFC [7-Amino-4-trifluoromethylcoumarin], and dimethyl sulfoxide (DMSO) were purchased from Sigma Aldrich, St. Louis, MO. M-PER (mammalian protein extraction reagent) was purchased from ThermoFisher Scientific, Carlsbad, CA.
Peptide Synthesis and Purification
The synthesis of Peptide 1 [Ac-RWVRVpGO(FAM)WIRQ-LRGG-AFC, Figure 1B] and Peptide 2 [Ac-RWVRVpGO(FAM)WIRQ-LVLRLRGG-AFC, Figure 1B] were based on the procedure outlined by Backes and Ellman (1999).22 The first residue, Fmoc-Gly-OH, was manually coupled to the safety-catch resin in the following manner. Safety-catch resin (637 mg, 0.7 mmol/g) was added to a 50 mL syringe fitted with a teflon frit. The resin was swollen by adding DMF (6 mL) and gently shaking for 30 minutes. The resin was drained and then Fmoc-Gly-OH (528 mg, 4 eq.) and DIEA (464 μL, 6 eq) dissolved in 6 mL NMP was added and the resin slurry was shaken gently for 10 minutes. PyBOP (900 mg, 3.9 eq) was added and the syringe was sealed and shaken overnight.
Figure 1. Sequence and structure of peptides used in this study.

(A) The structure of the peptide-based DUB reporter (herein referred to as Peptide 1) which contains a β-hairpin motif (black), a DUB substrate derived from the C-terminus of ubiquitin (blue), and a fluorophore (AFC: 7-Amino-4-trifluoromethylcoumarin) for analytical detection (green). The β-hairpin sequence acts as both a cell penetrating peptide (CPP) and protectide to increase intracellular stability. (B) The names and sequences of the peptides used in this study with the color scheme matching the structure in (A) for the β-hairpin (black), substrate (blue), and fluorophore (green). The two experimental β-hairpin-conjugated reporters (Peptides 1 and 2) vary in the length of the DUB recognition sequence (blue). A commercially available DUB substrate (Peptide 3) was used as positive control and contained a different fluorophore (AMC: 7-Amino-4-methylcoumarin). Peptide 4 is a variant of Peptide 1 that is constitutively fluorescent (FAM: 5,6-carboxyfluorescein) to visualize peptide uptake.
The resin was drained and the coupling procedure was repeated once more. The loading level was determined to be 0.26 mmol/g by the procedure outlined in Chan and White.23 With the preloaded Fmoc-Gly-safety-catch resin in hand, the remaining sequences of Peptides 1 and 2 were synthesized on a Tribute peptide synthesizer (Protein Technologies, Tucson, AZ) utilizing a standard Fmoc peptide chemistry protocol on a 100 μmol scale. Five-fold excess of Fmoc-amino acids (Fmoc-Arg(Pbf)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Val-OH, Fmoc-d-Pro-OH, Fmoc-Gly-OH, Fmoc-Ile-OH, Fmoc-Gln(Trt)-OH, Fmoc-Leu-OH, and HCTU, in the presence of 10 equivalents of NMM, were used for each of the amino acid coupling steps (10 min) with NMP as the solvent. Once the peptide synthesis was complete, the resin was manually washed with DMF (3×30 sec) and drained. The peptidylsulfonamide resin was activated by suspending the resin in DMF (3 mL) then adding DIEA (87 μL, 5 eq.) and iodoacetonitrile (170 μL, 23.5 eq.). The resin suspension was gently shaken in the dark for 24 hours. The resin was then drained and washed with 3 mLs each of DMF (3 × 30 sec) followed by THF (3 × 30 sec). Just prior to the completion of the resin activation, Boc-Gly-AFC (193 mg, 5 eq.) was stirred in the dark in a round bottom flask containing a solution of TFA:DCM (3 mL, 1:1) for 10 minutes. The solvent mixture was removed by a rotary evaporator and the residue was dissolved in THF (3 mL). DIEA (348 μL, 20 eq.) was added to the H-Gly-AFC solution and transferred to the now activated resin. The resin slurry was gently shaken in the dark for 24 hours after which the resin was filtered and washed several times with small amounts of THF. The combined THF solutions were evaporated and the residue was dissolved in a solution of TFA:water (3 mL, 1:1). The solution containing the peptide was shaken in the dark for 4 hours to fully deprotect the peptide. Cold diethyl ether was then added to the peptide solution to precipitate the crude peptide. The peptide was centrifuged for 10 minutes (4,000 rpm) and the ether layer decanted. Fresh cold diethyl ether was added, and the pelleted peptide was resuspended. The peptide was centrifuged again, and the procedure was repeated 5 times in total. After the final ether wash, the peptide pellet was dissolved in 5 mL acetonitrile:water (1:1) containing 0.1% TFA, frozen and lyophilized. Peptide 4 [Ac-RWVRVpGO(FAM)WIRQLRGG-NH2] was synthesized on a Tribute peptide synthesizer (Protein Technologies, Tucson, AZ) utilizing a standard Fmoc peptide chemistry protocol on a 100 μmol scale using rink amide resin (189 mg, 0.53 mmol/g). Five-fold excess of Fmoc-amino acids (Fmoc-Arg(Pbf)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Val-OH, Fmoc-d-Pro-OH, Fmoc-Gly-OH, Fmoc-Ile-OH, Fmoc-Gln(Trt)-OH, Fmoc-Orn(Aloc)-OH), Fmoc-Leu-OH and HCTU, in the presence of 10 equivalents of NMM were used for each of the amino acid coupling steps (10 min) with DMF as the solvent. A solution of acetic anhydride, NMM and NMP (1:1:3) was added to the Fmoc-deprotected resin and shaken for 30 minutes to acetylate the N-terminus of the peptide. Once the peptide synthesis was complete, the resin was washed with DMF (3×30 sec) then DCM (3×30 sec). The Aloc group was removed with three-fold excess of palladium in 4 ml of CHCl3-HOAc-NMM (37:2:1) under nitrogen for 2 hrs. The resin was then washed with DCM (3×30 sec) followed by DMF (3×30 sec). In the dark, FAM was coupled to the delta nitrogen of the ornithine side chain with four-fold excess of FAM, HOBt, PyBOP and DIEA in 3mL DMF for 24 hrs and then repeated for 8 hours. The peptide resin was washed with DMF (3×30 sec) and DCM (3×30 sec). The peptide was cleaved from the resin and side-chain deprotected using TFA/water/TIPS (4 mL, 95:2.5:2.5) for 3 hours and collected in a 50 mL centrifuge tube. The cleavage reaction was repeated for 10 minutes. The cleavage solutions for the peptide were combined and concentrated in vacuo. Cold diethyl ether was then added to the peptide solution to precipitate the crude peptide. The peptide was centrifuged for 10 minutes (4,000 rpm) and the ether layer decanted. Fresh cold diethyl ether was added, and the pelleted peptide was resuspended. The peptide was centrifuged again, and the procedure was repeated 5 times in total. After the final ether wash, the peptide pellet was dissolved in 5 mL water containing 0.1% TFA, frozen and lyophilized. HPLC analysis was performed on all peptides with a Waters 616 pump, Waters 2707 Autosampler, and 996 Photodiode Assay Detector which are controlled by Waters Empower 2 software. The separation was performed on an Agilent Zorbax 300 SB-C18 (5 um, 4.6 × 250 mm) with an Agilent guard column Zorbax 300 SB-C18 (5 um, 4.6 × 12.5 mm). Elution was done with a linear 5% to 55% gradient of solvent B (0.1% TFA in acetonitrile) into A (0.1% TFA in water) over 50 min at a 1 mL/min flow rate with UV detection at 442 nm. Preparative HPLC runs were performed with a Waters prep LC Controller, Waters Sample Injector, and 2489 UV/Visible Detector which are controlled by Waters Empower 2 software. The separation was performed on a Agilent Zorbax 300SB-C18 PrepHT column (7 um, 21.2× 250 mm) with Zorbax 300SB-C18 PrepHT guard column (7 um 21.2 × 10 mm) using a linear 5% to 55% gradient of solvent B (0.1% TFA in acetonitrile) into A (0.1% TFA in water) over 50 min at a 20 mL/min flow rate with UV detection at 215 nm. Fractions of high (>95%) HPLC purity of each peptide and with the expected mass were combined and lyophilized.
Cell Culture and Lysate Generation
HeLa cells (LSU AgCenter Tissue Culture Facility) were maintained in Dulbecco’s modified eagle medium (DMEM) with 10% v/v fetal bovine serum (FBS, VWR Life Sciences Seradigm). OPM2 cells (a kind gift from Nancy Allbritton, UNC) were maintained in RPMI 1640 media supplemented with 12% FBS, 21.8 mM glucose, 8.6 mM HEPES (pH 7.4) and 1.0 mM sodium pyruvate. All media components were from Corning (Atlanta, GA) unless otherwise noted. Cell lysates (both HeLa and OPM2) were generated by harvesting 1×106 cells/mL, followed by washing 2X and pelleting in phosphate buffered saline (PBS; 137 mM NaCl, 10 mM Na2HPO4, 27 mM KCL, and 1.75 mM KH2PO4 at pH 7.4). The cell pellet was re-suspended in an approximately equivalent volume of mammalian protein extraction reagent (M-PER) to the volume of the cell pellet (~1000–2000 μL) then vortexed for 10 min at room temperature. Following this, the mixture was centrifuged at 14,000 × g for 15 min at 4°C and the supernatant transferred to a centrifuge tube and stored on ice until use. Total protein concentration was determined using a NanoDrop (Thermo Scientific, Madison, WI).
Analysis of Enzyme-Substrate Kinetics in Cell Lysates
Peptides 1–3 were reconstituted in dimethyl sulfoxide (DMSO) and final peptide concentration was determined by diluting the peptide in 5 M Guanidium Hydrochloride and using a NanoDrop spectrophotometer (Thermo Scientific) and Beer’s Law. The stock peptide was diluted as needed in assay buffer (50 mM HEPEPS, pH 7.4, 150 mM NaCl, 2 mM DTT, and 0.02% Tween-20) supplemented with 4 mg/mL cell lysates to reach the desired peptide concentration with a final volume of 100 μL in a 96-well plate. To interrogate the role of DUBs on peptide hydrolysis, the reaction mixtures were supplemented with PR-619 at two concentrations (20 or 50 μM) or a vehicle control of DMSO. Peptide only (noise/background) samples were included in the 96-well plate to confirm that the rates observed were due to DUB-mediated hydrolysis initiated by the cell lysates. The 96-well plate was maintained at a temperature of 30°C in the dark for the duration of the experiment. The fluorescent signals emitted as a result of DUB-mediated cleavage of AFC (λex= 405 nm and λem= 490 nm) or AMC (λex= 355 nm and λem= 460 nm) were quantified using a plate reader (Perkin Elmer Wallac 1420 Victor2 multilabel HTS counter). Readings were collected every 30 min for 6 h. A calibration curve was generated for known concentrations of AFC or AMC to correlate the fluorescent signal (AU) to concentration (μM). The approximate concentration of free fluorophore was used to calculate reaction rates (μM/min) for each substrate concentration using linear regression analysis.
Analysis of Enzyme-Substrate Kinetics in Intact Cells
HeLa cells cultured in T-75 flasks were washed with PBS and trypsinized to facilitate detachment. Cells were diluted in extracellular buffer (ECB; 5.036 mM HEPES pH 7.4, 136.89 mM NaCl, 2.68 mM KCl, 2.066 mM MgCl2•6H2O, 1.8 mM CaCl2•2H2O, and 5.55 mM glucose) to a final concentration of 104 cells/ml and loaded into the 96-well plate. For this experiment, the Peptides 1 and 3 was reconstituted in sodium phosphate buffer (2.26 mM NaH2PO4•H2O and 8.43 mM Na2HPO4•7H2O) to avoid uptake issues associated with DMSO. The stock peptide was added to the 96-well plate to achieve the desired final concentration and a total operating volume of 100 μL. Intact cells were incubated with peptide solutions at varying concentrations in the 96-well plate at 30°C in the dark. Fluorescent signals for AFC and AMC were quantified using a plate reader as describe above. Data was collected every 60 min for 6 h. A peptide-only control experiment, in absence of intact cells, was performed to confirm that the observed rates were due to DUB activity in the cells. Additionally, a negative control experiment incubating cells with only AFC was also performed with the intact cells.
Quantification and Visualization of Peptide Uptake in Intact Cells
HeLa cells were seeded on Falcon culture slides (Corning) 24 h prior to experimentation. On the day of experiment, the culture media was removed, and the cells washed 2X with ECB. Cells were incubated with a 30 μM peptide solution of Peptide 4 for varying durations (10, 30, 60, and 90 min) to analyze time-dependent peptide internalization. To assess cell viability and intracellular localization, the 90-minute sample was co-incubated with 4 μM ethidium homodimer-1 (Life Technologies, Carlsbad, CA) and 8 μM Hoechst (Thermo Fisher Scientific, Rockford, IL) stains for an additional 30 minutes and immediately imaged. Concentration-dependent experiments were performed with cells incubated with varying concentrations of peptide for 90 min. Cellular fluorescence was visualized using a Leica DMi8 inverted microscope outfitted with a FITC filter cube, 20X objective (Leica HC PL FL L, 0.4X correction), and phase contrast and brightfield applications. Digital images were acquired using the Flash 4.0 high speed camera (Hamamatsu) with a fixed exposure time of 500 ms for FITC, DAPI, and Rhodamine filters, and 10 ms for brightfield. Image acquisition was controlled using the Leica Application Suite software. All images were recorded by using the same parameters. Fluorescent images (gray scale) were processed and quantified in gray values using the particle analysis and thresholding tabs in Image J. The data were fitted to Hill model using non-linear regression analysis with Levenberg-Marquardt algorithm using Origin Pro.
Direct Visualization of DUB Activity in Single Intact Cells
HeLa cells were seeded on culture slides as previously described above. On the day of experiment, the culture media was removed, and the cells washed 2X with ECB. Concentration-dependent studies were performed by incubating varying concentrations (80, 100, 120, and 160 μM) of Peptide 1 with the cells for 90 minutes at 37°C. Samples were immediately imaged using brightfield and DAPI channels. To visualize the increase in DUB-mediated cleavage of Peptide 1 over time, the 160 μM sample was further incubated with ECB supplied with 5% Fetal Bovine Serum (FBS) and imaged after 60, 90, and 120 minutes using brightfield and DAPI filters.
Statistical Analysis and Numerical Modeling
All data visualization, curve-fittings, and statistical analyses were performed using Origin Pro (OriginLab, Northampton, MA) unless otherwise specified. The analyses of enzyme-substrate reactions in cell lysates started with scatter plotting the fluorometry signals (μM) measured for each substrate concentration against time (min). Fluorescent signals measured for each substrate concentration remained stable during the first 60 minutes, suggesting there was a 60-minute lag period before reaction initiation. Beyond 60 minutes, the signals demonstrated a linear increase over time as assessed by linear regression and ANOVA statistics. The slopes of each line corresponded to the reaction rate for the given substrate concentrations. In all cases, r2 values of above 0.95 were reached for the linear fits. Rates were then plotted against substrate concentrations as shown in Figure 2. Next, non-linear regression analysis was performed to fit the classic Michaelis-Menten enzymology model (equation 1) to the rate data using Levenberg Marquardt iteration algorithm to calculate the kinetic constants Km and vmax.1
| (Eq. 1) |
where V denotes reaction rate and S denotes substrate/peptide concentration. Standard Chi-squared tests and ANOVA statistics were used to confirm the goodness of the fit in each case.
Figure 2. Kinetics of DUB-mediated hydrolysis of peptides in HeLa and OPM2 lysates.

Concentration-dependent DUB-mediated cleavage rates of Peptide 1 (A), Peptide 2 (B), and Peptide 3 (C) in HeLa lysates (4 mg/mL) and Peptide 1 (D) Peptide 2 (E), and Peptide 3 (F) in OPM2 lysates (4 mg/mL). Lysates were pre-treated with 20 μM PR-619, 50 μM PR-619, or DMSO. Legends are the same in all plots. Rate data demonstrate Michaelis-Menten kinetics (r2>0.99) in all cases. (G) Michaelis-Menten kinetics parameters calculated for the DMSO curves. All peptides efficiently distinguish samples treated with varying concentrations of the inhibitor ([I]=0, 20, 50 μM) corresponding to the specificity of the peptides to DUBs. ‘****’ denotes p<10−10, ‘***’ denotes p<10−5, ‘**’ denotes p<10−4, ‘*’ denotes p<0.005.
The analysis of enzyme-substrate interactions in intact cells encompassing cell-entry and DUB-mediated cleavage was carried out using a combination of theoretical modeling and non-linear regression analysis using the Hill model. The theoretical steps of enzyme-substrate interactions in intact cells are as follows:
| (Eq. 2) |
where T represents cell surface proteins involved in peptide internalization and S represents the cell-permeable reporter (Peptide 1).
S0: Substrate in solution
T: Cell membrane proteins facilitating substrate permeation into intact cell
Si: Internalized substrate
[S0]: Substrate concentration in the solution (known)
[Si]: Intracellular substrate concentration (unknown)
f: An unknown function governed by uptake dynamics, relating the intracellular and extracellular substrate concentrations
In theory, the enzyme-substrate reaction rates in intact cells must reach their levels in cell lysates upon peptide delivery to the intracellular environment. Therefore;
| (Eq. 3) |
Due to the added complexity of peptide uptake and enzyme-substrate interactions in intact cells, the Michaelis-Menten model was found to be a poor fit for the experimental data. Therefore, curve-fitting with the Hill model (equation 4) using the Levenberg-Marquardt algorithm was carried out.
| (Eq. 4) |
where Y denotes the dynamic or static physiochemical property being modeled [enzyme-substrate reaction rate (V) or internalized peptide content (Si)] and n denotes the sigmoidal Hill coefficient. The Hill model was first introduced by A.V. Hill to describe the equilibrium relationship between oxygen tension and the saturation of hemoglobin in 1910.2 It is widely applicable in modeling biological systems in which the relationship between the enzyme and substrate or the drug and the target is non-linear and saturable.3 Therefore, reaction rates in intact cells were modeled as follows;
| (Eq. 5) |
For brevity, vmax,inact and Km,intact are hereafter referred to as ‘a’ and ‘b’ respectively. Similarly, vmax,lysates and Km,lysates are referred to as ‘c’ and ‘d’ respectively. Equating equations 5 and 3, and solving for [Si] yielded:
| (Eq. 6) |
The numerical values of a and b were later determined by non-linear regression analysis of experimental data in the results section. Equation 6 describes the mathematical relationship between [Si] and [S0] the physiochemistry of which is governed by peptide uptake. This equation follows the general form of the Hill model (equation 4) suggesting that peptide uptake similar to enzyme-substrate reaction rates in intact cells (equation 5) is saturable and can be described by the Hill model. Comparing Eq. 6 to Eq. 4 to define:
| (Eq. 7) |
| (Eq. 8) |
where Si,max is the intracellular concentration (in μM) when the solution concentration approaches infinity (S0→∞), the theoretical maximum reachable intracellular concentration or saturation concentration, while S0,50 is the solution concentration corresponding to [Si] = Si,max/2 (the half-saturation level), and α is the Hill coefficient of sigmoidicity. This translates equation 6 into equation 9.
| (Eq. 9) |
The numerical values of the parameters were calculated and reported in the results section.
Results and Discussion
Design and synthesis of the peptide-based DUB reporter
The hallmark of the majority of available DUB reporters is the conjugation of a C-terminal fluorophore on either full length ubiquitin (e.g., ubiquitin-AFC, ubiquitin-TAMRA)24 or a shortened DUB recognition substrate consisting of the final four C-terminal amino acids from ubiquitin (e.g., Z-LRGG-AMC, herein referred to as Peptide 3).25 Here, two experimental peptide reporters were synthesized consisting of a β-hairpin sequence RWRWR (RWVRVpGRWIRQ, Figure 1A black) on the N-terminus and the DUB recognition sequence derived from the C- terminus of ubiquitin on the C- terminus (e.g., LRGG for Peptide 1, Figure 1A blue). Previous work by Houston et al26 and Safa et al27 confirmed that RWRWR behaves as both a stabilizing protectide and a cell penetrating peptide (CPP). The β-hairpin secondary structure of this sequence confers superior protease resilience, and therefore intracellular stability, by preventing its entry into the catalytic cleft of intracellular proteases and peptidases. Thus, it was hypothesized that conjugating this sequence to a DUB-specific substrate would both increase its intracellular lifetime and facilitate its uptake into intact cells. Prior work by Safa et al confirmed the secondary structure and the extended lifetime (half-life of 420 min in HeLa lysates) of the RWRWR sequence.27 The DUB recognition substrate for Peptide 1 consisted of a four amino acid long recognition sequence from the C-terminus of ubiquitin (LRGG) (Figure 1A, blue), while Peptide 2 consisted of an eight amino acid long recognition sequence from the C-terminus of ubiquitin (LVLRLRGG: Figure 1B, blue). Similar to existing reporting schemes, a C-terminal fluorophore AFC (7-amino-4-trifluoromethylcoumarin, Figure 1A green) was incorporated into the reporter to allow for direct visualization of reporter hydrolysis using fluorometry and fluorescent microscopy. In routine solid phase peptide synthesis (SPPS), the C-terminal amino acid is anchored to the linker through the main chain carboxyl group, thus blocking any further modification to the C-terminus of the peptide. The most widely used resins yield either C-terminal acid or C-terminal amide peptides. When a different modification to the C-terminus of a peptide is desired, other solid phase synthesis strategies must be employed. In the case where a C-terminal fluorophore is desired, the most common synthesis strategy utilizes a linker attached to the resin that will provide wide synthetic flexibility in the choice of fluorophore. There are several linkers that will accomplish this goal;28 however, for this work an approach was employed that was previously demonstrated by Backs and Ellman22, 29 due to its ease of use and the commercial availability of the alkyl sulfonamide resin (“safety catch” resin). AFC-labeled Peptides 1 and 2 were synthesized in good yields and were easily purified (Figure S1). A fourth peptide was synthesized, called Peptide 4, which was a variant of Peptide 1 without the C-terminal AFC (Figure 1B). Instead, 5,6-carboxyfluorescein was conjugated to position eight to produce a peptide that was constitutively fluorescent and allowed for peptide internalization studies.
Kinetic characterization of the peptide reporter in cell lysates
Standard enzymology studies were carried out to characterize the performance of Peptides 1–3 and demonstrate their specificity to DUBs using lysates derived from HeLa cells and a model multiple myeloma cell line, OPM2 cells (Figure 2). Data presented for each plot in Figure 2 are representative of a minimum of triplicate experiments. Each datapoint represents the slope of the fluorescence (AU) vs. peptide concentration (μM) line calculated using linear regression. In all cases, r2 values of above 0.95 were reached for the linear fits allowing for calculation of the rate data with minimal error. Detailed information on data analysis can be found in the supplementary document. Each experiment was performed at three different conditions in which lysates were pre-treated (1h) with increasing concentrations ([I]=0, 20, and 50 μM) of a commercially available DUB inhibitor (PR-619). The zero concentration corresponds to lysates pre-treated with DMSO as vehicle control. In all cases, non-linear regression analysis was used to fit the rate data to the Michaelis-Menten model and calculate reaction rate constants (Km and vmax) in the absence of PR-619 ([I]=0). The curve-fittings were repeated for samples treated with varying concentrations of PR-619 ([I]=20 and 50 μM) to facilitate a comparison between different conditions and assess the significance of DUB inhibition on rate data translating to the specificity of each reporter to DUBs. As expected, all three peptides were found to exhibit increased reaction rates with increasing peptide concentrations confirming their activity in both HeLa and OPM2 lysates. The results of the inhibition studies revealed that peptides 1 and 3 successfully distinguished between the three cohorts ([I]=0, 20, 50 μM). A statistically significant decrease in reaction rates was observed by increasing the inhibitor concentration from 0 to 20 μM and then again from 20 to 50 μM for peptides 1 and 3 in HeLa and OPM2 lysates and for Peptide 2 in OPM2 lysates. The residual activity observed in lysates treated with 50 μM PR-619 is an indication of incomplete inhibition of DUB activity by the inhibitor, suggesting that higher concentrations of the inhibitor would be needed to further decrease the level of DUB activity until a complete inhibition be potentially reached. To ensure the significance of the findings in this section, the fitted curves were compared against each other using ANOVA statistics resulting in the p-values reported for each comparison in Figure 2. Results confirmed that Peptides 1 and 3 were sensitive to DUB-mediated cleavage and that the observed increase in fluorescent signals were due to DUB activity and not protease-mediated degradation of the peptides. This was expected for Peptide 1 due to the stability of the RWRWR peptide and its resistance to protease and peptidase activity27. Conversely, Peptide 2, which contained the longer DUB recognition sequence, showed a decreased sensitivity to DUB inhibition. Specifically, DUB inhibition did not result in a significant effect on rate data for Peptide 2 in HeLa lysates. This suggested that Peptide 2 has a low specificity to DUBs and that increasing the length of the DUB recognition sequence from 4 amino acid residues did not improve DUB specificity. The numerical values of Michaelis-Menten parameters Vmax and Km were calculated using non-linear regression analysis (Figure 2G).
In order to facilitate a comparison between the performance of the different sequences as DUB reporters, Vmax/Km values, hereafter referred to as V/K values, were calculated and compared among the three different peptides with a higher performing peptide exhibiting the higher V/K value. Rate data for Peptides 1 and 3 demonstrated a perfect fit for the Michaelis-Menten model in both HeLa and OPM2 lysates as confirmed by ANOVA statistics (Figure 2A, C, D, and F), while the rate data for Peptide 2 only exhibited Michaelis-Menten kinetics in OPM2 lysates (Figure 2E) but not in HeLa lysates (Figure 2B). In Figure 2B, since iterations with the Michaelis-Menten model did not converge, reliable values for Vmax and Km could not be calculated. Therefore, these values were not calculated. In order to facilitate an approximation of the V/K value in infinite dilution of the substrate in which case the Michaelis-Menten equation simplifies to a line, the data in Figure 2B were fitted to lines, and the V/K value was approximated with the slope of the DMSO line (Figures 2B and 2G). In HeLa lysates, Peptide 1 demonstrated a higher Vmax value, corresponding to a higher saturation rate, and a higher Km value, corresponding to lower binding affinity, compared to Peptide 3. A 10-fold increase in the V/K value of Peptide 3 (2.1×10−4 min−1) compared to the value calculated for Peptide 1 (2.12×10−3 min−1) suggested the better performance of Peptide 3 as a DUB substrate in HeLa lysates. The V/K value of Peptide 2 (2.99×10−4 min−1) was found to be comparable to Peptide 1; however, due to its poor fit for the Michaelis-Menten model and most importantly, its low specificity to DUBs, this sequence was determined to be a low performing DUB reporter. It was concluded that increasing the length of the DUB recognition sequence did not improve reporter performance in terms of specificity or in terms of kinetics. Similar results had been previously reported by Melvin et al. where longer substrate sequences sometimes resulted in poorer reaction kinetics.30 Thus, the analysis in intact cells was limited to Peptide 1 in the following sections. The performance of the sequences was compared across the two cell lines (HeLa and OPM2). Peptide 1 demonstrated a significantly lower Km in OPM2 lysates (Figure 2D) compared to HeLa lysates (Figure 2 A) leading to higher V/K values corresponding to its better performance in OPM2 lysates; however, Peptide 3 did not follow the same trend, and its parameters remained consistent across the two cell lines (Figures 2 C and F). Peptide 2 also resulted in similar V/K values across the two cell lines (Figures 2 B and E). Therefore, although Peptide 3 was the top performer in terms of V/K in HeLa lysates, the V/K values of the three peptides were close in value in OPM2 lysates. These findings suggested that the difference in reaction rates between Peptide 1 and Peptide 3 was cell-type dependent. To provider a further comparison among the performance of Peptide 1 and Peptide 3, their uninhibited reaction rates in OPM2 lysates were provided in a single plot (Figure S2). The OPM2 cell line was specifically selected for the purpose of this comparison due to its biological relevance. As illustrated in Figure S1, the reaction rates of Peptide 1 in OPM2 lysates were significantly higher than Peptide 3 (p<10−4).
Quantification of peptide uptake dynamics in intact cells
To effectively model peptide performance in intact cells, it was first necessary to characterize peptide uptake. Previous work by Safa et al.27 demonstrated that the RWRWR sequence behaved as a CPP; however, prior studies have found that the attachment of cargoes to CPPs can potentially alter uptake kinetics.31 A constitutively fluorescent variant of Peptide 1 containing 5,6-caroxyfluorescen (FAM) instead of AFC was synthesized (Peptide 4) to visualize uptake in intact cells using a FITC filter set. A 30 μM solution of Peptide 4 was incubated with HeLa cells for 60 minutes and counterstained with the dead stain ethidium homodimer-1 (Eth-D1, visualized using a Rhodamine filter set) (Figure 3). Intracellular peptide distribution was simultaneously visualized by co-staining with Hoechst to denote the nucleus (Figure 3) which demonstrated a homogeneous peptide distribution across the entire cell area. The absence of any signal in the Rhodamine channel confirmed that peptide uptake did not decrease cellular viability and that a 90 min incubation time with 30 μM peptide solution was sufficient to deliver the peptide to cells. Similar uptake dynamics were observed in OPM2 cells (data not shown).
Figure 3. Cell viability and intracellular distribution of the DUB reporter in intact HeLa cells.

Cells incubated with 30 μM Peptide 4 were washed and counterstained with 4 μM ethidium homodimer (EthD-1) and 8 μM Hoechst. Channels were immediately imaged using Brightfield, FITC (peptide), DAPI (nucleus), and Rhodamine (viability assessed by Ethidium Homodimer) filters. Scale bar is 40 μm.
Time-dependent internalization was observed for Peptide 4 with a low degree of peptide uptake occurring within 10 min of incubation (Figure S3A) compared to the majority of cells demonstrating peptide uptake after 90 min (Figure S3D). Intermediate uptake was observed after incubation times of 30 min (Figure S3B) and 60 min (Figure S3C) with a greater degree of peptide uptake correlating to a longer incubation time. The high background fluorescence observed in Figures S3A and S3B corresponds to the presence of residual free peptide on the coverslip. The observed time-dependent decrease in background fluorescence was attributed to an increase in peptide uptake over time. Additionally, a small number of cells exhibited minimal to no peptide uptake which was denoted by cells with fluorescence level below the background. This finding is consistent with prior work by Safa et al. which found that peptide uptake did not occur in all of the cells.27 Based on these findings, it was decided that a 90-minute incubation time at the given experimental conditions was sufficient to efficiently deliver the peptide to cells. Previous studies with RWRWR27 reported that a 10-minute incubation time was sufficient to deliver the CPP into intact cells. This suggests that conjugating the CPP to the DUB-recognizing sequence decreased uptake dynamics which could be expected due to the increased length of Peptide 4 compared to RWRWR. A similar approach was performed to characterize the concentration-dependence of peptide uptake and provide a baseline for microscopy studies measuring DUB activity in intact cells in the following sections (Figure S4). Prior work by Safa et al. with RWRWR found that peptide uptake was concentration dependent, which has been demonstrated by several others investigating CPPs.27, 31 This was accomplished by incubating HeLa cells with four different concentrations (80, 100, 120, and 160 μM) of Peptide 4 for 90 min at 37°C (Figure S4A–D). Peptide uptake was monitored using fluorescence microscopy to illustrate an overall positive trend between intracellular fluorescence and peptide concentration. HeLa cells incubated with the lowest concentrations of Peptide 4 (Figure S4A) were found to exhibit an accumulation of peptide near the nucleus while increasing concentrations (Figure S4B–C) resulted in a more homogeneous distribution across the cytoplasm until the intracellular distribution was homogeneous in cells incubated with a 160 μM peptide solution (Figure S4D). This concentration-dependent effect of intracellular peptide distribution can be attributed to peptide internalization pathways. Prior reports found that external peptide concentration can alter the internalization mechanism and intracellular peptide distribution.31 Interestingly, a decreased level of cell-to-cell variability was also observed in cells incubated with the higher peptide concentrations (Figure S5A). Based on these findings of a homogeneous intracellular peptide distribution coupled with a decreased level of cell-to-cell variability in terms of peptide uptake, both of which are favorable conditions contributing to consistency in fluorometric quantification of DUB activity, it was decided to use an external peptide concentration of 160 μM in microscopy studies in the following sections. To further mathematize the concentration dependence of peptide, the concentration-dependent fluorescent signals were simulated with the Hill model of peptide uptake (equations 4 and 9). It was confirmed that peptide uptake was saturable demonstrating a good fit for the Hill model (Figure S5B). Hill parameters for this dataset were calculated to be Si, max= (3.38±4.15)*104 gray values for the maximum internal concentration S0, 50=68.8±15.5 μM for external peptide solution concentration corresponding to the half-saturation level, and α=6.26 Hill coefficient of sigmoidicity. This simulation clarifies the concentration dependence of peptide uptake and provides a mathematical baseline for the future microscopy studies with the DUB reporter.
Kinetic characterization of the peptide-based DUB reporter in intact cells
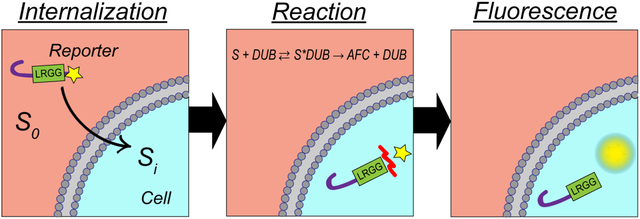
The results reported in the previous sections demonstrated the application of Peptide 1 as a reporter of DUB activity in cell lysates (Figure 2) which was also shown to be capable of cell penetration (Figures 3 and S3–S4). Both DUB mediated cleavage in cell lysates and peptide uptake in intact cells were mathematically characterized using non-linear regression analysis. Kinetics parameters of DUB-mediated cleavage in cell lysates were found to demonstrate Michaelis-Menten kinetics (Figure 2G) while the concentration dependent peptide uptake fitted the Hill model (Figure S5). Here, a mathematical model is developed for measuring DUB activity in intact cells accounting for both peptide uptake and DUB-mediated cleavage. The analysis in this section is restricted to intact HeLa cells due to the convenience of experimentation with an adherent cell line; however, as the activity of the DUB reporter (Peptide 1) was previously evidenced in OPM2 lysates (Figures 2 D–G), similar results are expected in intact OPM2 cells. The mechanism of action of Peptide 1 consisted of two steps: cell entry and DUB-mediated hydrolysis. Figure 4A schematically illustrates the mechanism of action of Peptide 1 starting with peptide internalization and continuing with cleavage reaction (denoted by the red mark) resulting in a fluorescent output.
Figure 4. Enzyme-substrate kinetics of DUB reporters in intact HeLa cells.

(A) Schematic illustration of peptide interaction with intracellular DUBs encompassing peptide uptake and DUB-mediated cleavage resulting in fluorescent output. The red mark in the reaction section schematically illustrates the site of DUB-mediated cleavage. (B) Intact HeLa cells were incubated with varying concentrations of Peptide 1 and Peptide 3 for 6 h. Control experiment (background) was performed by incubating Peptide 1 in a cell-free buffer. Each datapoint represents the slope (μM/min) of the fluorescent signal (μM) vs. time (min) obtained foreach peptide concentration (μM) by linear regression (r2>0.99). Fluorescent signals measured in arbitrary units were converted to μM units by generating calibration curves. Each peptide concentration was assayed in triplicate. (C) Compiled effect of peptide uptake and DUB-mediate hydrolysis of Peptide 1 in intact cells demonstrating a Hill model correlation with substrate concentration (Vintact=vmax, intact.[S]n/(Km, intactn+[S]n)), r2=0.96, vmax, intact=(7.6±1.01)*10−4 μM/min, Km, intact=55.18±5.38 μM, nintact=6.8, and V/K=1.38*10−5).
Similar to results obtained from the lysate studies (Figure 2), the bulk fluorescent signals observed for HeLa cells incubated with varying concentrations of Peptide 1 demonstrated a linear, time-dependent increase as assessed by linear regression. As a result, the slopes of the lines were calculated (μM/min) to obtain the reaction rates in intact cells (Figure 4B, black squares). As such, each data point in Figures 4B and 4C represents the slope (μM/min) of the fluorescent signal (μM) versus time (min) obtained for each peptide concentration (μM) by linear regression (r2>0.99). Each peptide concentration was assayed in three independent experiments. The reaction rates obtained for Peptide 3 (Figure 4B, red circles) and peptide-only background (Figure 4B, blue triangles) were not significantly different from zero and did not increase by increasing peptide concentrations. This confirmed that Peptide 1 was capable of being internalized and cleaved by DUBs to measure the intracellular activity of DUBs. Furthermore, the absence of the concentration-dependent increase in rates quantified for the peptide-only samples confirmed that the observed rates were due to the presence of the cells and the peptide by itself did not produce an increase in fluorescent signals. Next, the Hill model was used to numerically simulate the data for Peptide 1 providing a mathematical description for the process (Figure 4C). Iterations were performed using Levenberg-Marquardt algorithm resulting in a chi-squared value of 6*10−9 and r2=0.96. The goodness of the fit was assessed by ANOVA statistics contrasting the Hill curve to the function y=constant (null hypothesis) which resulted in p<0.005, confirming the goodness of the fit. This allowed for the calculation of the kinetic rate constants in intact cells (Vmax, intact, Km, intact, and n). These values referred to as a, b, and nintact were calculated to be a= vmax, intact = (7.6±1.01) *10−4 μM/min, b = Km, intact=55.18±5.38 μM, and nintact=6.8 corresponding to (V/K) intact=1.38*10−5 (Figure 4C). These results numerically characterize the combined effect of uptake and cleavage in intact cells, while cleavage kinetics for Peptide 1 in HeLa lysates were previously described and parametrized in Figures 2A and 2G. Therefore, the numerical values of c= vmax, lysates = 0.104 μM/min and d = Km, lysates=499.4 are also known up to this point. In order to provide insight into uptake dynamics from a mathematical standpoint, the numerical values of a, b, c, d, and nintact were substituted to solve for internalized peptide concentration Si as a function of extracellular/solution concentration S0. This allowed for the calculation of uptake parameters intracellular saturation concentration Si,max=1.28 μM and external solution concentration corresponding to the half-saturation level S0,50= 55.1 μM. It was shown that the intracellular fluorescence corresponding to peptide uptake reached a saturation level (Figure S5) and therefore could be modeled as a saturable physiochemical equilibrium. Non-linear regression analysis on microscopy fitted the Hill model (Figure S5B) resulting in an Si,max value of 33800 (gray values), a S0,50 of 68.83 μM, and a Hill coefficient of sigmoidicity (α) of 6.26. Although the experimental Si,max value is not directly comparable to the numerically simulated Si,max value due to the incompatibility of the units (gray values vs. μM), the S0,50 and α coefficients obtained from microscopy experiments verify the mathematically simulated values with minimal error. These results identified that like reaction kinetics in intact cells (Figure 4C), uptake dynamics were saturable demonstrating the Hill model. The theoretical intracellular saturation concentration of Si,max = 1.28 μM was sufficient for measuring enzyme activity in single cells considering the small size of the cells leading to significant intracellular fluorescent densities (concentration/cell cross-sectional area) resulting in detectable signals in Figure 4. Interestingly, the value of the term in S0,50 is found to be close to 1. Therefore, S0,50~Km,intact suggested that the Km value in intact cells is mostly governed by uptake characteristics. This motivates future studies to enhance reporter performance by increasing CPP uptake efficiency. The findings in this section first illustrate the application of Peptide 1 to measure intracellular DUB activity, and next provide a mathematical baseline for future applications of this reporter in intact cells encompassing both uptake characteristics and enzyme-substrate reaction kinetics.
Direct quantification of DUB activity in intact cells using fluorescent microscopy
While the fluorometry experiments demonstrated the ability of Peptide 1 to measure DUB activity in intact cells, they were not capable of providing insight into the single cell response. To expand upon the utility of the novel DUB reporter and examine single cell DUB activity, both concentration-and time-dependent studies were performed using Peptide 1 in intact HeLa cells (Figure 5). A concentration-dependent study was first performed by incubating the cells with four increasing peptide concentrations and immediately imaging the cells after peptide removal (Figure 5A). It was determined that an external peptide concentration of 120 μM or higher was required to immediately yield a detectable fluorescent signal in the cells. Cells incubated with lower external peptide concentrations exhibited a diminished fluorescence signal or no signal at all. This confirmed the findings from Figures S4–S5 that indicated that higher concentrations of peptide were needed to achieve reporter uptake in intact cells. Based on this, the time-dependent microscopy studies were performed using an external peptide concentration of 160 μM Peptide 1 to visualize DUB activity in intact cells. For this experiment, the cells were pre-incubated with peptides for 90 min followed by time-dependent visualization of intracellular fluorescence corresponding to DUB activity. After 90 min, the peptide solution was removed and replaced with ECB to halt the influx of fresh peptide and determine how long the reporter could produce a measurable signal in intact cells. The t = 0 min time point corresponds to 90 min of peptide incubation and was shown to yield a measurable fluorescent signal. The intensity of fluorescent signal correlating to DUB-mediated cleavage of the reporter continued to increase, in addition to the evolution of its intracellular distribution in subcellular regions, until 100 min (Figure 5B). Interestingly, regions of DUB activity were initially more concentrated near the nucleus immediately after peptide removal (Figure 5B, t=0 min). Significantly higher intracellular fluorescent intensities were observed in cells after a 60 min period with increased fluorescence around the nuclear envelope (Figure 5B, t=60 min). Continual imaging after 60 minutes illustrated a more uniform distribution of fluorescence across the entire cell cross-sectional area until a stable and more homogeneous intracellular intensity was reached at the 100 min time point. No significant difference was observed in fluorescence intensities beyond this time point (Figure 5B, t=100 min and t=120 min). These findings confirm the ability of Peptide 1 to enter intact single cells and visually assess DUB activity. Finally, to confirm the ability of Peptide 1 to be internalized and reacted upon by DUBs was not cell type specific, a similar experiment was performed by incubating Peptide 1 with OPM2 cells (Figure 6A). OPM2 cells incubated with Peptide 1 showed an increase in intracellular fluorescence due to DUB-mediated hydrolysis of the reporter. Expectedly, OPM2 cells incubated with Peptide 3 did not produce a detectable signal (Figure 6B). These findings further confirm the capabilities of Peptide 1 to enter intact cells and assess DUB activity.
Figure 5. Direct visualization of DUB activity in intact HeLa cells.

(A) HeLa cells seeded on glass coverslips were incubated with varying concentrations of Peptide 1 for 90 minutes at 37 °C. Peptides were removed, cells were washed with ECB and imaged immediately using a DAPI filter. (B) HeLa cells seeded on glass coverslips were pre-incubated with a 160 μM solution of Peptide 1 for 90 minutes at 37 °C. The peptide solution was removed, cells were washed, and further incubated with ECB to discontinue the internalization process and dynamically measure DUB-mediated cleavage of the fluorophore. All time points were visualized using a DAPI filter AFC fluorophore) at the indicated time points and concentrations to demonstrate concentration- and time-dependent increases in intracellular fluorescence due to DUB-mediated hydrolysis of the peptide. Scale bars are 40 μm.
Figure 6. Direct visualization of DUB activity in intact OPM2 cells.

OPM2 cells were incubated with 50 μM of Peptide 1 (A) or Peptide 3 (B) for 120 minutes in microcentrifuge tubes at 37 °C. Peptides were removed and cells were washed with ECB and immediately visualized using a DAPI filter. Coverslips were pre-treated with CellTak to facilitate cellular adhesion. Scale bars are 20 μm.
Conclusions
In this work, a novel reporter of DUB activity (Peptide 1) was synthesized and characterized. Peptide 1 was confirmed to be sensitive and specific to DUBs as assessed by fluorescence-based enzymology studies in HeLa and OPM2 lysates in the presence of varying doses of an established DUB inhibitor (PR-619). Non-linear regression analysis with the Michaelis-Menten model characterized the enzyme-substrate reactions kinetics in cell lysates and provided a comparison with a commercially available counterpart (Peptide 3) used as a positive control. The DUB-mediated hydrolysis reaction rates of Peptide in OPM2 (multiple myeloma-derived cell line) lysates were found to be significantly higher than the positive control. All findings were assessed by ANOVA statistics confirming the significance of the results. Furthermore, microscopy experiments performed with Peptide 4 (a constitutively fluorescent version of Peptide 1) demonstrated the efficient uptake of the CPP-based reporter in intact cells and identified both time and concentration-dependent thresholds for efficient reporter performance. The application of Peptide 1 to measure DUB activity in intact cells was demonstrated using fluorometry. Non-linear regression analysis was performed to model the cell-substrate interactions and a mathematical model was provided to describe the biochemistry of the entire system and serve as a descriptive baseline for future applications of this reporter. These findings led to the conclusion that peptide uptake in cells was saturable following the Hill model which allowed for the estimation of the maximum reachable internalized peptide content and the peptide solution concentration corresponding to the half saturation level. All these findings were experimentally confirmed by fluorescence microscopy. Finally, live cell fluorescence microscopy experiments visualized DUB activity in single intact HeLa and OPM2 cells and identified intracellular regions with increased DUB activity within the cells. The findings in this paper confirmed that unlike many existing, commercially available DUB reporters that are limited to enzyme-only or lysate-based studies, the novel peptide-based reporter introduced here is capable of detecting DUB activity in intact cells. This new reporting scheme has vast potential to be incorporated into high-throughput single cell screening methods such as flow cytometry or droplet microfluidics. Such single cell characterizations can provide invaluable information about cell population distributions in terms of DUB activity and intracellular regions with upregulated DUB activity.
Supplementary Material
Highlights.
New approach to measure deubiquitinating enzyme (DUB) activity in intact cells
Peptide-based reporter incorporates a β-hairpin sequence motif that is a CPP and protectide
Developed a kinetic model to account for both peptide uptake and reaction kinetics
Demonstrated similar reaction kinetics to commercially available peptides in cell lysates
Observed time- and concentration-dependent increase in intracellular fluorescence due to DUBs
ACKNOWLEDGMENT
This work was supported by grants from the National Institute of Biomedical Imaging and Bioengineering, R03EB02935 (ATM), the National Science Foundation, CBET1509713 (ATM), the LSU College of Engineering FIER - Round VII (ATM), and the LSU Council on Research (ATM). The authors would like to thank Dong Liu (LSU Ag Center) for assistance with peptide synthesis, Gavin A. Pappas (LSU Chemical Engineering) for some assistance with preliminary experiments, and Karen McDonough (LSU AgCenter) for assistance with cell culture.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information
This article comes with a supplementary document comprising of five figures. They include analysis on the purity and identify of Peptide 1 (Figure S1), a comparison between reaction kinetics of Peptides 1 and 3 in OPM2 lysates (Figure S2), fluorescent microscopy data showing the uptake of Peptide 4 (Figures S3–S4), and single cell analysis and statistical analysis of Peptide 4 uptake (Figure S5).
The authors declare no competing financial interests.
REFERENCES
- 1.Lim K; Baek K, Deubiquitinating Enzymes as Therapeutic Targets in Cancer. Current Pharmaceutical Design 2013, 19 (22), 4039–4052. [DOI] [PubMed] [Google Scholar]
- 2.Edelmann M; Nicholson B; Kessler B, Pharmacological targets in the ubiquitin system offer new ways of treating cancer, neurodegenerative disorders and infectious diseases. Expert Reviews in Molecular Medicine 2011, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tian Z; D’Arcy P; Wang X; Ray A; Tai Y; Hu Y; Carrasco R; Richardson P; Linder S; Chauhan D; Anderson K, A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014, 123 (5), 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kessler B; Edelmann M, PTMs in Conversation: Activity and Function of Deubiquitinating Enzymes Regulated via Post-Translational Modifications. Cell Biochemistry and Biophysics 2011, 60 (1–2), 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai JJ; Safadi SS; Mercier P; Barber KR; Shaw GS, Ataxin-3 is a multivalent ligand for the parkin Ubl domain. Biochemistry 2013, 52 (42), 7369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hideshima T; Qi J; Paranal R; Tang W; Greenberg E; West N; Colling M; Estiu G; Mazitschek R; Perry J; Ohguchi H; Cottini F; Mimura N; Gorgun G; Tai Y; Richardson P; Carrasco R; Wiest O; Schreiber S; Anderson K; Bradner J, Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America 2016, 113 (46), 13162–13167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gupta N; Li W; McIntyre T, Deubiquitinases Modulate Platelet Proteome Ubiquitination, Aggregation, and Thrombosis. Arteriosclerosis Thrombosis and Vascular Biology 2015, 35 (12), 2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chauhan D; Tian Z; Nicholson B; Kumar K; Zhou B; Carrasco R; McDermott J; Leach C; Fulcinniti M; Kodrasov M; Weinstock J; Kingsbury W; Hideshima T; Shah P; Minvielle S; Altun M; Kessler B; Orlowski R; Richardson P; Munshi N; Anderson K, A Small Molecule Inhibitor of Ubiquitin-Specific Protease-7 Induces Apoptosis in Multiple Myeloma Cells and Overcomes Bortezomib Resistance. Cancer Cell 2012, 22 (3), 345–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tobinai K, Proteasome inhibitor, bortezomib, for myeloma and lymphoma. International Journal of Clinical Oncology 2007, 12 (5), 318–326. [DOI] [PubMed] [Google Scholar]
- 10.D’Arcy P; Brnjic S; Olofsson M; Fryknas M; Lindsten K; De Cesare M; Perego P; Sadeghi B; Hassan M; Larsson R; Linder S, Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nature Medicine 2011, 17 (12), 1636–U150. [DOI] [PubMed] [Google Scholar]
- 11.Hussain S; Zhang Y; Galardy P, DUBs and cancer The role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors. Cell Cycle 2009, 8 (11), 1688–1697. [DOI] [PubMed] [Google Scholar]
- 12.Melvin A; Woss G; Park J; Waters M; Allbritton N, Measuring Activity in the Ubiquitin-Proteasome System: From Large Scale Discoveries to Single Cells Analysis. Cell Biochemistry and Biophysics 2013, 67 (1), 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dang L; Melandri F; Stein R, Kinetic and mechanistic studies on the hydrolysis of ubiquitin C-terminal 7-amido-4-methylcoumarin by deubiquitinating enzymes. Biochemistry 1998, 37 (7), 1868–1879. [DOI] [PubMed] [Google Scholar]
- 14.Bozza W; Zhuang Z, Biochemical Characterization of a Multidomain Deubiquitinating Enzyme Ubp15 and the Regulatory Role of Its Terminal Domains. Biochemistry 2011, 50 (29), 6423–6432. [DOI] [PubMed] [Google Scholar]
- 15.Hassiepen U; Eidhoff U; Meder G; Bulber J; Hein A; Bodendorf U; Lorthiols E; Martoglio B, A sensitive fluorescence intensity assay for deubiquitinating proteases using ubiquitin-rhodamine 110-glycine as substrate. Analytical Biochemistry 2007, 371 (2), 201–207. [DOI] [PubMed] [Google Scholar]
- 16.Tirat A; Schilb A; Riou V; Leder L; Gerhartz B; Zimmermann J; Worpenberg S; Eldhoff U; Freuler F; Stettler T; Mayr L; Ottl J; Leuenberger B; Filipuzzi I, Synthesis and characterization of fluorescent ubiquitin derivatives as highly sensitive substrates for the deubiquitinating enzymes UCH-L3 and USP-2. Analytical Biochemistry 2005, 343 (2), 244–255. [DOI] [PubMed] [Google Scholar]
- 17.Nicholson B; Leach C; Goldenberg S; Francis D; Kodrasov M; Tian X; Shanks J; Sterner D; Bernal A; Mattern M; Wilkinson K; Butt T, Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Science 2008, 17 (6), 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohayon S; Spasser L; Aharoni A; Brik A, Targeting deubiquitinases enabled by chemical synthesis of proteins. Journal of Peptide Science 2012, 18, S75–S75. [DOI] [PubMed] [Google Scholar]
- 19.Gui W; Ott CA; Yang K; Chung JS; Shen S; Zhuang Z, Cell-Permeable Activity-Based Ubiquitin Probes Enable Intracellular Profiling of Human Deubiquitinases. J Am Chem Soc 2018, 140 (39), 12424–12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.An H; Statsyuk AV, Development of activity-based probes for ubiquitin and ubiquitin-like protein signaling pathways. J Am Chem Soc 2013, 135 (45), 16948–62. [DOI] [PubMed] [Google Scholar]
- 21.Safa N; Anderson JC; Vaithiyanathan M; Pettigrew JH; Pappas GA; Liu D; Gauthier T; Melvin AT, CPProtectides: Rapid uptake of well-folded β-hairpin peptides with enhanced resistance to intracellular degradation. Peptide Science 2018, e24092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Backes B; Ellman J, An alkanesulfonamide “safety-catch” linker for solid-phase synthesis. Journal of Organic Chemistry 1999, 64 (7), 2322–2330. [Google Scholar]
- 23.Chan WC; White PD, Fmoc Solid Phase Peptide Synthesis: A Practical Approach. 1 ed.; Oxford University Press: New York, 2000. [Google Scholar]
- 24.Dang LC; Melandri FD; Stein RL, Kinetic and mechanistic studies on the hydrolysis of ubiquitin C-terminal 7-amido-4-methylcoumarin by deubiquitinating enzymes. Biochemistry 1998, 37 (7), 1868–79. [DOI] [PubMed] [Google Scholar]
- 25.Stein RL; Chen Z; Melandri F, Kinetic studies of isopeptidase T: modulation of peptidase activity by ubiquitin. Biochemistry 1995, 34 (39), 12616–23. [DOI] [PubMed] [Google Scholar]
- 26.Houston KM; Melvin AT; Woss GS; Fayer EL; Waters ML; Allbritton NL, Development of β-Hairpin Peptides for the Measurement of SCF-Family E3 Ligase Activity in Vitro via Ornithine Ubiquitination. ACS OMEGA 2017, 2 (3), 1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Safa N; Anderson JC; Vaithiyanathan M; Pettigrew JH; Pappas GA; Liu D; Gauthier TJ; Melvin AT, CPProtectides: Rapid uptake of well-folded β-hairpin peptides with enhanced resistance to intracellular degradation. Peptide Science 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alsina J; Yokum T; Albericio F; Barany G, Backbone amide linker (BAL) strategy for N-alpha-9-fluorenylmethoxycarbonyl (Fmoc) solid-phase synthesis of unprotected peptide p-nitroanilides and thioesters. Journal of Organic Chemistry 1999, 64 (24), 8761–8769. [DOI] [PubMed] [Google Scholar]
- 29.Backes B; Virgilio A; Ellman J, Activation method to prepare a highly reactive acylsulfonamide “safety-catch” linker for solid-phase synthesis. Journal of the American Chemical Society 1996, 118 (12), 3055–3056. [Google Scholar]
- 30.Melvin A; Woss G; Park J; Dumberger L; Waters M; Allbritton N, A Comparative Analysis of the Ubiquitination Kinetics of Multiple Degrons to Identify an Ideal Targeting Sequence for a Proteasome Reporter. Plos One 2013, 8 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brock R, The Uptake of Arginine-Rich Cell-Penetrating Peptides: Putting the Puzzle Together. Bioconjugate Chemistry 2014, 25 (5), 863–868. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.