

Abstract
The outcomes for atypical EGFR and HER2 alterations remain underexplored. We performed a single-center retrospective study to determine unique clinical characteristics, treatment outcomes, and resistance mutations among this subgroup. Patients with atypical EGFR and HER2 alterations have a unique metastatic phenotype with higher rate of synchronous lung and bone metastases, different proportion of resistance mutations relative to typical EGFR mutations, and poorer responses to targeted and immunotherapies. Novel approaches are urgently needed.
Background:
The clinicopathologic characteristics, acquired resistance patterns, and outcomes among patients with atypical EGFR mutations and HER2 alterations remain underexplored.
Patients and Methods:
A single-center retrospective review was conducted. Oncogenes assessed include typical EGFR (t-EGFR; exon 19 del and L858R), atypical EGFR (a-EGFR; G719X, exon 20, L861Q), HER2 (exon 19, exon 20, amplifications), gene fusions (ALK, ROS1, RET), RAS (KRAS, NRAS), and RAF (BRAF V600E). Progression-free survival (PFS), overall survival (OS), disease control rate, and objective response rate (Response Evaluation Criteria in Solid Tumors 1.1) were collected.
Results:
Among 570 patients, we found 55 a-EGFR mutations (13 G719X, 38 exon 20, 4 L861Q) and 31 HER2 alterations (2 exon 19 mutations, 27 exon 20 insertions, 2 amplifications). Patients with EGFR and HER2 alterations had increased lung and bone metastases relative to patients with gene fusions, RAS/RAF mutations, and no identified driver oncogenes (P < .001). Patients with EGFR exon 20 insertions had a median PFS to EGFR tyrosine kinase inhibitors (TKIs) of 5 months and an OS of 16 months–significantly worse than exon 19 del and L858R (Bonferroni correction; P < .001), but not G719X or L861Q. Relative to t-EGFR mutations, T790M and MET amplification occurred less frequently as acquired resistance mechanisms among a-EGFR samples (P < .001). Ten patients with a-EGFR mutations and HER2 alterations received single-agent immune checkpoint inhibitors (ICIs) with no radiographic responses and a median PFS of 2 months.
Conclusion:
EGFR and HER2-mutated NSCLC have a high rate of synchronous lung and bone metastases. Patients with a-EGFR mutations have inferior responses to EGFR-directed therapies with lower rates of acquired T790M and MET amplification. Responses to ICIs are uniformly poor. Novel therapeutic approaches are needed.
Keywords: Bone metastases, ERBB2, Exon 20 insertion, Immune checkpoint inhibitor, Tyrosine kinase inhibitor
Introduction
The identification of activating mutations in the epidermal growth factor receptor (EGFR) gene and the development of tyrosine kinase inhibitors (TKIs) that selectively target these mutations has revolutionized the management of patients with non–small-cell lung cancer (NSCLC).1–5 Most clinical trials evaluating EGFR TKIs have only included patients harboring typical drug-sensitive EGFR mutations, specifically exon 19 deletions and the exon 21 L858R substitution.6–16 Collectively, these mutations (hereafter referred to as typical EGFR mutations; t-EGFR) represent approximately 80% to 85% of all EGFR mutations.1–4,17,18 The remaining 15% to 20% of EGFR mutations (hereafter referred to as atypical; a-EGFR) are largely composed of point mutations (G719X and L861Q) or exon 20 insertion mutations.17–23
The ERBB2 gene (encoding the HER2 protein) is also a proto-oncogene that can activate downstream signaling through PI3K-AKT and MEK-ERK pathways.24 In contrast to other members of the ErbB family, no ligand has been described for HER2, which is activated by either homodimerization or heterodimerization with other members within the ErbB family. HER2 alterations have been identified as oncogenic drivers in 2% to 3% of NSCLC.25–27 The most common HER2 alteration is a 12 bp in-frame insertion (encoding YVMA) in exon 20. Preclinical studies have shown that mutations in the kinase domain result in constitutive phosphorylation and activation of the HER2 receptor.26–30 Additionally, increased signalling through ERBB2/HER2 gene amplification is another important HER2 alteration, though importantly, gene amplification may be defined in different ways (eg, absolute mean copy number per cell or the ratio of gene to the centromere on the same chromosome). As a continuous variable, the threshold for defining positivity has not yet been established in NSCLC.31
Patients with t-EGFR mutations have high objective response rates (ORR) and meaningful progression-free survival (PFS) to first-generation reversible EGFR TKIs (eg, erlotinib, gefitinib),9,10,12,14 second-generation irreversible TKIs (eg, afatinib, dacomitinib),6,11,15,16 and third-generation irreversible TKIs (eg, osimertinib).8,13 In contrast, patients with a-EGFR and HER2 alterations have experienced variable efficacy with EGFR- and HER2-directed therapies.21,23,25,26,32–34 The role of immune checkpoint inhibitors (ICIs) alone or with chemotherapy in patients with EGFR and HER2-mutated NSCLC continues to evolve.29,30,35–38
In this retrospective study, we assessed the clinicopathologic characteristics of patients with a-EGFR mutations and HER2 alterations, treatment outcomes of patients with a-EGFR mutations treated with TKIs, acquired resistance patterns among a-EGFR mutations, outcomes of patients with HER2 alterations treated with HER2-directed therapy, and outcomes of patients with a-EGFR mutations and HER2 alterations treated with ICIs (either as single agent or in combination with platinum doublet chemotherapy).
Patients and Methods
Patients and Clinical Characteristics
All patients with NSCLC (according to the American Joint Committee on Cancer (AJCC) 8th edition of the tumor, node, metastasis classification system) evaluated at University of Colorado from June 2009 to March 2019 were eligible for assessment. Patients were identified through a database that included all patients treated through the University of Colorado health system through an institutional review board–approved protocol. The patient population contained a mix of patients who were diagnosed and treated within the University of Colorado health system and patients who were treated elsewhere and subsequently referred to our institution.
Outcomes data and imaging results were collected by retrospective chart review. We captured age, sex, smoking status, histology, tumor grade, clinical stage, and metastatic sites. EGFR and HER2 subtypes were recorded and included t-EGFR mutations (exon 19 deletion, L858R), a-EGFR mutations (G719X, exon 20 insertions, L861Q), and HER2 alterations (exon 19 mutations, exon 20 insertions, and HER2 gene amplifications defined as HER2/CEP17 ≥ 3). Where available, we identified comutations and programmed death ligand 1 (PD-L1) expression at diagnosis.
We performed a formal analysis to look for statistically significant differences in the patterns of metastatic spread between oncogene groups including mutations within the ErbB family (EGFR and HER2), gene fusions (ALK, ROS1, and RET), mutations within the Ras/Raf pathway (K-RAS, N-RAS, and BRAF V600E), and other mutations (no identifiable oncogene driver). Metastatic sites considered for this analysis included the lung (contralateral lung metastases), pleura (either pleural plaques or malignant effusions), brain, bone, liver, and the adrenal glands.
Only responses and clinical outcomes from the first EGFR- or HER2-directed therapy that each patient received were considered. EGFR agents considered for this study included first-generation TKIs (erlotinib, gefitinib), second-generation TKIs (afatinib, dacomitinib), and third-generation TKIs (osimertinib). Patients with a-EGFR mutations enrolled onto clinical trials were excluded from this retrospective review. Outcomes from ICIs (pembrolizumab, nivolumab, or atezolizumab) as single agents or in combination with chemotherapy (using KEYNOTE-189 regimen) were captured. PFS was defined as time from initiation of therapy to time of radiographic progression (as defined by Response Evaluation Criteria in Solid Tumors [RECIST] 1.1) or death. Overall survival (OS) was defined as time from initiation of therapy to time of death.
Given variability in follow-up, we captured RECIST responses on the basis of first scan after starting EGFR- or HER2-directed therapy. ORR represents the sum of partial response (PR) and complete response (CR). Disease control rate (DCR) represents the sum of stable disease (SD), PR, and CR. In defining acquired resistance to EGFR TKI, we utilized criteria proposed by Jackman et al.39 When available, resistance mutations at the time of progression were captured utilizing combination of circulating tumor DNA (Guardant Health, Redwood City, CA) or tissue biopsy samples for only those patients with an objective ORR or a PFS greater than 6 months. Patients with a secondary cancer receiving active cytotoxic treatment, nonmetastatic NSCLC, or incomplete molecular or clinical documentation were excluded.
Molecular Methods
Molecular testing was conducted via Clinical Laboratory Improvement Amendments–certified laboratories and was performed using laboratory assays that were independently validated. Over the course of time of this study, the testing approach for standard-of-care laboratory testing evolved, and therefore testing was performed using a variety of assay platforms. These approaches included the following: Sanger sequencing of relevant targeted regions, single nucleotide base extension assay (SNaPshot), real-time polymerase chain reaction, targeted next-generation sequencing (NGS) using a 26-gene panel (TruSight; Illumina, San Diego, CA), or Archer VariantPlex/FusionPlex Solid Tumor library preparation kit (ArcherDx, Boulder, CO) with raw sequence data analyzed by Archer Analysis 4.1.1.7 software (ArcherDx).
For internally tested cases, fluorescence in-situ hybridization (FISH) for MET and HER2 (ERBB2) was performed on 4 ± 1 μm thick formalin-fixed, paraffin-embedded tumor sections. FISH was performed by manual slide processing technique on neutral formalin-fixed, paraffin-embedded tissue that was pretreated with proteinase K, then hybridized with the PathVysion HER2 DNA Probe kit (Vysis HER2/neu (17q11.2) SpectrumOrange and Vysis 17 centromere SpectrumGreen; Abbott Molecular, Des Plaines, IL). The MET FISH assays were performed with laboratory-developed reagents or commercial reagents encompassing the genomic sequences of MET. CEP7 was used as a control probe to define the relative CNG. This sample was considered positive for MET amplification if MET/CEP7 ≥ 3 and HER2 amplification if the HER2/CEP17 ≥ 3.
For liquid biopsies, we used the Guardant360 circulating tumor DNA assay (Guardant Health, Redwood City, CA). Approximately 50 to 100 ng of DNA is extracted from plasma, and 73 genes are used to identify somatic genomic alterations utilizing massively parallel sequencing of amplified target genes via Illumina HiSeq 2500 or Illumina NextSeq 500 platforms using hg19 as the reference genome. The limit of detection for single-nucleotide variants is 0.1%.40 PD-L1 expression was assessed using a combination of the PD-L1 IHC 22C3 pharmDx and the Ventana PD-L1 (SP263) assays with expression categorized according to the tumor proportion score (percentage of tumor cells with membranous PD-L1 staining).41
Statistical Analysis
Descriptive statistics summarizing patient characteristics included the median, frequency, and percentage for categorical variables. Group comparisons of patient characteristics were performed using the Fisher exact test for categorical variables. Student t test or analysis of variance were used for continuous variables. Survival analysis was performed by Kaplan-Meier estimates using a log-rank test to assess for differences between subgroups. We addressed multiple statistical testing by adjusting the familywise error rate using the Bonferroni method. P ≤ .05 was defined as statistically significant. All statistical analyses were performed by GraphPad Prism 6.00 for Mac (GraphPad Software, La Jolla, CA) and JMP Pro 14.3.0 for Mac (SAS Institute, Cary, NC).
Results
Clinicopathologic Characteristics of Patients With Atypical EGFR Mutations and HER2 Alterations
A total of 603 patients seen at the University of Colorado Cancer Center with stage IV NSCLC from 2009 to 2019 were considered for this study. Of these, 33 were excluded and 570 patients were included for further analysis, as shown in the CONSORT diagram (Supplemental Figure 1 in the online version). We identified 55 a-EGFR mutations (13 G719X, 38 exon 20 insertions, 4 L861Q) and 31 HER2 alterations (2 exon 19 mutations, 27 exon 20 insertions, 2 gene amplifications). No patient with a HER2 exon 20 insertion had synchronous HER2 amplification (HER2/CEP17 ≥ 3). Baseline clinical characteristics are listed in Table 1. The median age of patients with a-EGFR and HER2 alterations was 62 years for both groups (range, 38–91 years). There was a predominance of female patients with a-EGFR mutations (38/55, 69%) and HER2 alterations (21/31, 68%). The majority of patients with a-EGFR mutations (32/55, 58%) and HER2 alterations (23/31, 75%) were never smokers.
Table 1.
Clinicopathologic Characteristics of Patients With Atypical EGFR Mutations and HER2 Alterations
| Characteristic | a-EGFR (N = 55) | HER2 (N = 31) |
| Sex | ||
| Male | 17 (31) | 10 (32) |
| Female | 38 (69) | 21 (68) |
| Race | ||
| White | 39 (71) | 20 (65) |
| Black | 3(5) | 0 |
| Asian | 7(13) | 1 (3) |
| Hispanic | 0 | 2(6) |
| Other | 6(11) | 8 (26) |
| Age (y), median (range) | 62 (38–91) | 62 (47–80) |
| Smoking Status (pack-years) | ||
| Never | 32 (58) | 23 (76) |
| Light (≤10) | 7(13) | 3(8) |
| Heavy (>10) | 16 (29) | 5 (16) |
| Histology | ||
| Adenocarcinoma | 53 (96) | 30 (97) |
| Adenosquamous | 1 (2) | 1 (3) |
| Squamous | 1 (2) | 0 |
| Stage at Diagnosis | ||
| I | 4(8) | 0 |
| II | 2(4) | 1 (3) |
| III | 2(4) | 3(10) |
| IV | 47 (85) | 27 (87) |
| Brain metastasesa | 16 (27) | 9 (29) |
Data are presented as n (%) unless otherwise indicated. All disease was staged by magnetic resonance imaging.
Abbreviation: a-EGFR = atypical EGFR.
Brain metastases at time of stage IV disease.
The distribution of metastatic sites in stage IV disease by oncogene is shown in Supplemental Table 1 in the online version. Within the ErbB family (ie, EGFR and HER2), there was an increased incidence of lung metastases (112/220, 76%, vs. 116/322, 35%; P < .002) and bone metastases (117/220, 53%, vs. 124/322, 40%; P < .001) relative to other metastatic sites. We then compared metastatic sites across oncogene families. The incidence of lung and bone metastases within the ErbB family was significantly higher than gene fusions, mutations within the Ras/Raf pathway, and mutations without an identified oncogene driver (P < .001; Figure 1). A decreased incidence of adrenal metastases was noted within the ErbB family when compared to mutations within the Ras/Raf pathway (P = .03) and other mutations (P = .001), but not when compared to gene fusions (P = .842). To assess whether these patterns of metastatic spread varied within the ErbB family, we compared EGFR mutations to HER2 alterations and found no significant differences in the incidence of lung or bone metastases (Supplemental Table 2 in the online version).
Figure 1.
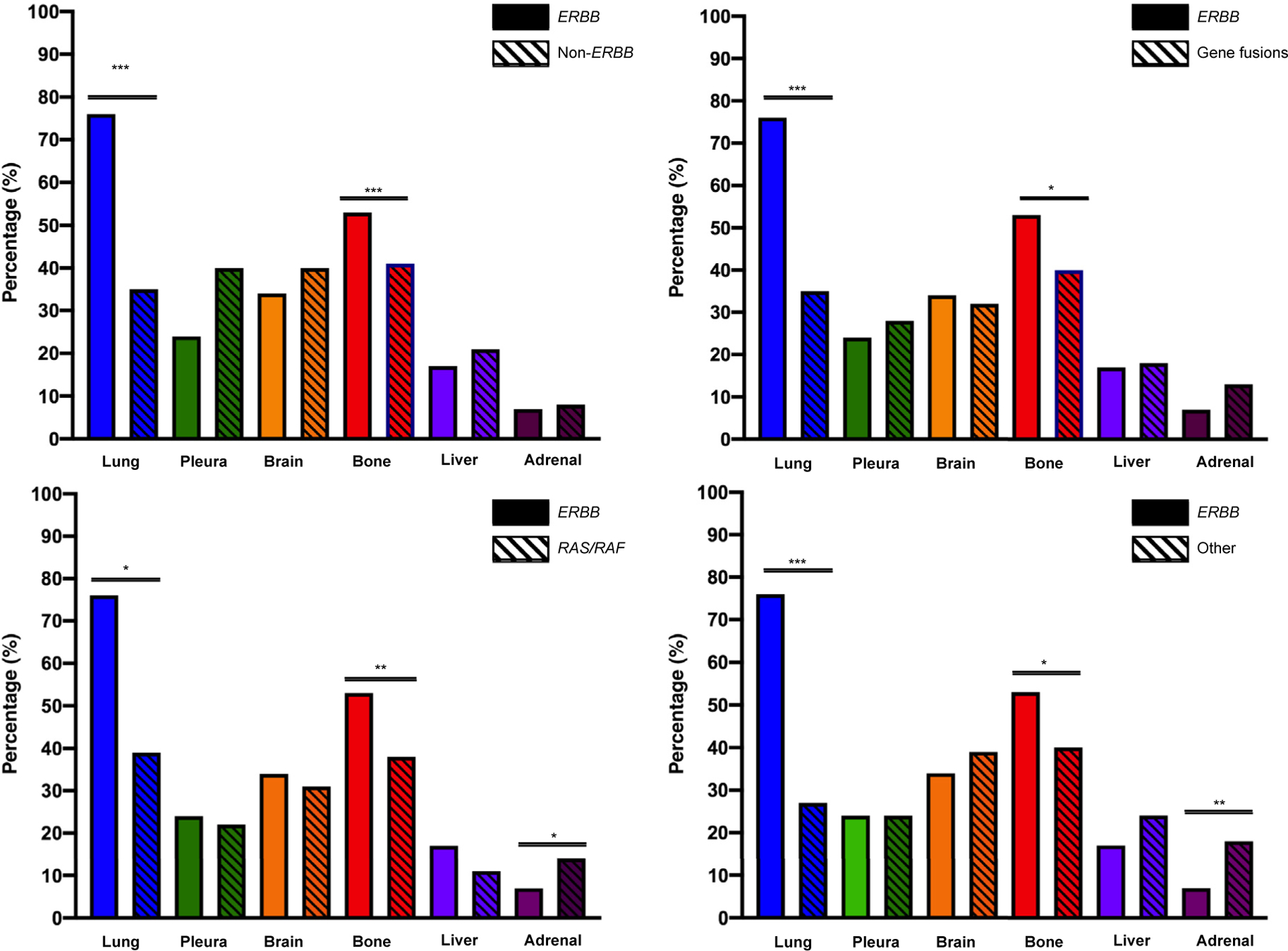
Distribution of Metastatic Sites Based on Driver Oncogene. Lung and Bone Metastases Were Significantly More Common Within the ErbB Family (ie, EGFR and HER2) Relative to Gene Fusions, Mutations in the Ras/Raf Pathway, and Mutations Without an Identified Oncogene Driver
Treatment Outcomes in Patients With a-EGFR Mutations Receiving EGFR-Targeted Therapies
The distribution of EGFR TKIs evaluated in this study is shown in Supplemental Table 3 in the online version. These reflect the first EGFR TKI that each patient received. Both a-EGFR and t-EGFR cohorts received a median of one prior systemic therapy before initiating therapy with an EGFR TKI. The median PFS was significantly worse among patients with a-EGFR mutations compared to t-EGFR mutations (6 vs. 17.5 months; P < .0001; hazard ratio [HR] = 0.33; 95% confidence interval [CI], 0.22–0.50; Figure 2A). This difference in PFS was maintained even when we restricted our analysis to patients who first received afatinib or osimertinib (respectively, 7 vs. 15.5 months; P < .002; HR = 0.81; 95% CI, 0.08–8.23; Figure 2B). Median PFS across EGFR subtypes was as follows: exon 19 del (17 months), L858R (18 months), G719X/L861Q (7 months), and exon 20 insertion (5 months). Median OS across EGFR subtypes was as follows: exon 19 del (63 months), L858R (73 months), G719X/L861Q (30 months), and exon 20 insertion (16 months). After Bonferroni correction, patients with EGFR exon 20 insertions had a significantly worse PFS and OS to first EGFR TKI relative to patients with exon 19 del and L858R (adjusted P < .001), but not G719X/L861Q (adjusted P = 1.00; adjusted P = .89; Figure 2C and D).
Figure 2.

Treatment Outcomes for Patients With Atypical EGFR Mutations and Typical EGFR Mutations Treated With EGFR TKIs. Only Responses to First EGFR TKI Received Were Considered. Log-rank Test Was Used to Compare PFS and OS Between 2 Groups. (A) PFS Among Patients With a-EGFR Mutations Compared to t-EGFR Mutations. (B) PFS Among Patients With a-EGFR Mutations Compared to t-EGFR Mutations When Only Afatinib and Osimertinib Were Included. (C) PFS to EGFR TKIs Separated out by EGFR Subtype: Exon 19 Deletion, L858R Mutations, G719X/L861Q, and Exon 20 Insertions. Median PFS Across EGFR Subtypes Was as Follows: Exon 19 Deletion (17 Months), L858R (18 Months), G719X/L861Q (7 Months), Exon 20 Insertion (5 Months). After Bonferroni Correction, Exon 20 Insertions Had Significantly Worse PFS Relative to Exon 19 Del and L858R (P < .001), but Not G719X/L861Q (P = 1.00). (D) Median OS Across EGFR Subtypes Was as Follows: Exon 19 Del (63 Months), L858R (73 Months), G719X/L861Q (30 Months), and Exon 20 Insertion (16 Months). After Bonferroni Correction, Exon 20 Insertions Had Significantly Worse OS Relative to Exon 19 Del and L858R (P < .001), but Not G719X/L861Q (P = .89)
Abbreviations: a-EGFR = atypical EGFR; OS = overall survival; PFS = progression-free survival; t-EGFR = typical EGFR; TKI = tyrosine kinase inhibitor.
First posttreatment imaging after treatment was available for 16 patients with a-EGFR mutations. RECIST responses are shown as a waterfall plot in Figure 3A. The ORR and DCR based on the first EGFR TKI received is shown in Supplemental Table 4 in the online version. Among the 10 patients who received erlotinib, the ORR was 10% and the DCR was 50%. The DCR by a-EGFR mutation was as follows: G719X (2/4, 50%), L861Q (2/3, 66%), and exon 20 insertion (1/3, 33%). Only 3 patients (19%) had PR to EGFR-directed therapies, 2 of whom were patients with exon 20 insertion mutations receiving afatinib. The PFS of a-EGFR mutations to different generations of TKIs is shown in a swimmer plot in Figure 3B. No patient with an EGFR exon 20 insertion who received erlotinib had a PFS greater than 6 months (the median PFS for a-EGFR mutations in our series). In contrast, 5 patients with EGFR exon 20 insertion mutations who received afatinib or osimertinib had a median PFS greater than 6 months. All 4 patients with EGFR L861Q insertions had a PFS ranging from 5 to 7 months. There was notable heterogeneity in the PFS benefit among the EGFR G719X group independent of amino acid pair. Forty percent (4/10) of patients with an EGFR G719X mutation had a PFS of greater than 12 months while an equal percentage of patients had a PFS of 3 months or less. Twenty-one patients underwent comutation testing at diagnosis, and the percentage of TP53 gene mutations was as follows: G719X (3/7, 48%), exon 20 insertion (7/10, 70%), and L861Q (3/4, 75%) with no significant differences between each group.
Figure 3.

Waterfall and Swimmer Plots for Patients With Atypical EGFR Mutations Receiving EGFR TKI Therapy. (A) RECIST Responses to First Posttreatment Imaging After Initiating Therapy With EGFR TKI. (B) Swimmer Plot Demonstrating PFS of a-EGFR Mutations Separated out by Different Generations of TKI. * Indicates That Patient Had New Lesions Detected on First on Treatment Scan, Consistent With Progressive Disease
Abbreviations: PFS = progression-free survival; RECIST = Response Evaluation Criteria in Solid Tumors; TKI = tyrosine kinase inhibitor.
Acquired Resistance Mechanisms Among Patients With Atypical EGFR Mutations Receiving EGFR-Targeted Therapies
One hundred twenty-nine patients with EGFR mutations who had serial molecular testing and clinical follow-up available for review were considered for this analysis. Of the 129 patients, 65 patients with EGFR mutations underwent testing for acquired resistance having met the criteria of prior objective response (n = 9) or PFS ≥ 6 months (n = 56). Acquired resistance was assessed using utilizing circulating tumor DNA (via Guardant) or tissue testing (via Archer VariantPlex or FusionPlex). Patients with a-EGFR mutations underwent testing for resistance mutations at a significantly lower rate than patients with t-EGFR mutations (respectively, 9/55, 16%, vs. 56/74, 76%; P < .001). The distribution of acquired resistance mutations across EGFR mutations and method for detecting acquired resistance is shown in Supplemental Table 5 in the online version. Among the 65 patients tested for acquired resistance, 59 patients received erlotinib, gefitinib, or afatinib-TKIs where the T790M acquired resistance mutation has been observed. The T790M acquired resistance mutation occurred at a significantly lower rate among patients with a-EGFR mutations relative to t-EGFR mutations (respectively, 2/7, 29%, vs. 39/52, 75%; P < .001). The percentage of T790M by EGFR subtype was exon 19 del (26/34, 76%), L858R (13/18, 72%), and G719X (2/7, 33%). T790M was only seen in patients with G719X mutations and did not occur in 2 patients with L861Q and exon 20 mutations.
Among the 65 patients tested for acquired resistance, HER2 and MET amplification assessed using FISH occurred in 66% (6/9) of a-EGFR and 55% (31/56) of t-EGFR samples. MET amplification as an acquired resistance mechanism was significantly higher among patients in the t-EGFR group (respectively, 5/31, 16%, vs. 0/9; P < .001). Only one patient (G719S mutation) with response to EGFR-directed therapy developed HER2 amplification as an acquired resistance mechanism, though this was detected through circulating tumor DNA (Guardant) and not using FISH. No identifiable resistance mechanism was found in 66% (6/9) a-EGFR and 20% (11/56) t-EGFR tested samples, though 50% (3/6) a-EGFR and 54% (6/11) t-EGFR samples were not assessed for HER2 or MET amplification via FISH. Only one patient with a HER2 exon 20 insertion was tested for acquired resistance (via NGS from a tissue biopsy) at time of disease progression while receiving afatinib, with no identifiable resistance mechanism found.
Treatment Outcomes in Patients With HER2 Alterations Receiving Targeted Therapies
Nineteen patients with HER2 alterations had clinical follow-up data. Thirty-seven percent (7/19) of these patients received HER2-directed therapy (which includes TKIs, HER2 monoclonal antibodies, and HER2 antibody–drug conjugates) during their treatment. Patients received a median of 2 lines of systemic therapy (eg, cytotoxic chemotherapy or chemoimmunotherapy combinations) before receiving their first HER2-directed treatment. A swimmer plot demonstrating the PFS of 7 patients with HER2 alterations who received initial HER2-directed therapies is provided in Figure 4A. Four patients received TKIs, 2 patients received trastuzumab with pertuzumab, and 1 patient received trastuzumab emtansine (T-DM1). Five patients had measurable lesions by RECIST, and all had SD based on the first posttreatment scan while receiving HER2-directed therapies (Figure 4B). Patients who received HER2-directed therapies had a higher OS relative to those who received cytotoxic chemotherapy (respectively, 65 vs. 29 months), although this was not statistically significant (P = .1574; HR = 0.30; 95% CI, 0.05–1.73).
Figure 4.

Swimmer and Waterfall Plots for Patients With HER2 Alterations. (A) Swimmer Plot of 7 Patients With HER2 Alterations Separated by Initial HER2-directed Therapy. (B) Waterfall Plot of First Posttreatment Responses Among 5 Patients With Measurable Lesions Who Received HER2-directed Therapies
Treatment Outcomes in Patients With Atypical EGFR and HER2 Alterations Receiving Immune Checkpoint Inhibitors
Sixteen patients with a-EGFR mutations (5/16, 31%) and HER2 alterations (11/16, 69%) received ICIs during their treatment course. PD-L1 expression was available in 8 patients prior to receiving ICIs. PD-L1 ≥ 50% was seen in 66% (2/3) of a-EGFR samples and 60% (3/5) of HER2 samples. The majority of patients were never smokers (13/16, 81%), with a median pack-year history of zero pack-years. Thirty-one percent (5/16) of patients received ICIs with platinum chemotherapy using the KEYNOTE-189 regimen,42 pathway (K-RAS, N-RAS, and BRAF V600E), and other mutations (no identifiable oncogene driver).Ten patients had posttreatment imaging available for review. No objective responses to single-agent ICI were seen in either the a-EGFR (0/4) or the HER2 (0/6) group. This poor response to single-agent ICI was seen even in 3 patients with PD-L1 ≥ 50%. Partial responses were seen in 2 patients with EGFR exon 20 and HER2 exon 20 insertion mutations who received ICI with platinum chemotherapy using the KEYNOTE-189 regimen.42 Patients treated with ICIs had received a median of 2 prior systemic therapies. Patients with a-EGFR and HER2 alterations who received ICIs with platinum doublet chemotherapy had significantly prolonged PFS relative to those who received ICIs as monotherapy (respectively, 7 vs. 2 months; P < .001; HR = 0.06; 95% CI 0.01–0.53; Figure 5).
Figure 5.

Outcomes of Patients With Atypical EGFR and HER2 Alterations Treated With ICIs. (A) Patients Who Received ICI With Platinum Doublet Chemotherapy (Using KEYNOTE-189 Regimen) Had Significantly Prolonged PFS Relative to Those Who Received Single-agent ICI. (B) Swimmer Plot of Patients With a-EGFR Mutations and HER2 Alterations Receiving ICI Therapy
Abbreviations: a-EGFR = atypical EGFR; ICI = immune checkpoint inhibitor; PFS = progression-free survival.
Discussion
Atypical EGFR and HER2 alterations are activating oncogenes in NSCLC with unique clinical features, varied responsiveness to current TKI therapy, and poor response to single-agent ICI therapy. There was a predominance of female patients with a-EGFR mutations and HER2 alterations, similar to what is seen among patients with t-EGFR mutations. While the majority of patients in the a-EGFR and HER2 groups were never smokers, a notable percentage of these patients (29% and 16%, respectively) were heavy smokers (≥ 10 pack-years). There has been a growing appreciation that different oncogenes can have different patterns of metastatic spread,43 and it appears that NSCLC with mutations in the ErbB family (ie, EGFR and HER2) have a tropism for lung and bone metastases. This metastatic phenotype was statistically significant when compared to patients with gene fusions, mutations in the Ras/Raf pathway, and with no identifiable oncogene drivers. There were no differences in the incidence of lung or bone metastases when EGFR and HER2 mutations (both within the ErBB family) were compared to each other. Our findings are consistent with other studies that report an increased incidence of lung metastases in patients with t-EGFR and HER2 activating mutations.44–46 The increased incidence of bone metastases within the ErbB family deserves mention. Given the propensity of bone metastases among patients with mutations in the ErbB family, the role of osteoclast inhibitors in conjunction with targeted therapy in reducing skeletal fracture risk remains unexplored and is being currently evaluated in the context of an ongoing clinical trial (ClinicalTrials.gov, NCT03958565).
Consistent with multiple other studies, patients with a-EGFR had significantly worse PFS to EGFR TKIs relative to patients with t-EGFR mutations.19,23,33,34,47 This inferior response was seen even among patients who received only afatinib and osimertinib (respectively, 7 vs. 15.5 months; P < .002; HR = 0.81; 95% CI, 0.08–8.23). Patients with EGFR exon 20 insertions and G719X/L861Q mutations had a median PFS of 5 months and 7 months to EGFR TKIs, though only exon 20 insertions had a statistically inferior PFS to exon 19 del and L858R after using Bonferroni adjustment (P < .001).
Single-nucleotide variants in exon 18 (G719X) and exon 21 (L861Q) have been reported to confer a moderate degree of sensitivity to EGFR TKIs.23,33,48,49 A post hoc analysis of NEJ002 study data, which compared first-line gefitinib to carboplatin/paclitaxel for advanced NSCLC with activating EGFR mutations, identified 10 patients with a-EGFR mutations (7 G719X, 3 L861Q). The median PFS for patients with EGFR G719X and L861Q mutations was significantly shorter than patients with t-EGFR mutations who received gefitinib (2.2 vs. 11.4 months; P < .0001), but this did not differ among patients who received platinum doublet chemotherapy (5.9 vs. 5.4 months; P = .847).50 A limitation of the NEJ002 analysis was the imbalance in the number of G719X/L861Q mutations (n = 10) compared to t-EGFR mutations (n = 215). In contrast to the post hoc analysis of the NEJ002 study, a retrospective analysis by Passaro et al23 included patients who received both first-generation EGFR TKI (erlotinib, gefitinib) and second-generation TKI (afatinib). Patients in this study with G719X/L861Q mutations had a numerically lower (but not statistically significant) median PFS of 12.3 months and an OS of 31 months compared to patients with t-EGFR mutations, consistent with data in our study. We found that there was notable heterogeneity among patients with G719X mutations, with 40% (4/10) deriving a PFS benefit to erlotinib that was greater than 12 months, while another 40% demonstrating progression within 3 months of initiating erlotinib therapy. One possibility for this observation could be that acquired T790M occurs at lower rate among G719X mutations relative to sensitizing mutations such as exon 19 del and L858R, and that alternative patterns of acquired resistance may emerge in this subgroup. Another possibility could be related to the presence of a TP53 gene mutation (a known negative predictive factor),3,5 which was detected in 48% of patients with G719X mutations in our series.
Exon 20 insertions account for approximately 5% of EGFR mutations and are associated with poor response to first-generation EGFR TKIs.22,34,47 In a combined post hoc analysis of uncommon EGFR mutations in the LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6 trials, patients with exon 20 insertions had a PFS of 2.7 months and an ORR of 8.7% to afatinib.50 Our study showed that afatinib may result in a higher ORR in patients with EGFR exon 20 insertions (2/4, 50%), though our sample sizes are small and thus limit the strength of our conclusions. In our series, no patient with an EGFR exon 20 insertion had a PFS of greater than 3 months when treated with erlotinib, but all 5 patients who received either afatinib or osimertinib had a PFS greater than 6 months (the median PFS for a-EGFR mutated NSCLC treated with an EGFR TKI in our series). There are ongoing clinical trials assessing the efficacy of different TKIs in patients with EGFR exon 20 insertion mutations such as osimertinib (ClinicalTrials.gov, NCT03414814), poziotinib (NCT03066206), tarloxotinib (NCT03805841), TAK-788 (NCT02716116), and JNJ-372 (NCT02609776).
Acquired resistance remains a major clinical challenge in the management of oncogene-driven NSCLC. Patients with a-EGFR mutations underwent resistance testing at a significantly lower rate than patients with t-EGFR mutations. Among 59 patients who received erlotinib, gefitinib, or afatinib, the percentage of T790M by EGFR subtype was as follows: exon 19 del (76%), L858R (72%), and G719X (40%). T790M was only seen in patients with G719X and did not occur in 2 patients with L861Q and exon 20 insertions. MET amplification (defined as MET/CEP7 ≥ 3) was seen as an acquired resistance mechanism in 16% (5/31) of t-EGFR samples, but was not seen in the a-EGFR group. No identifiable resistance was seen in 66% of patients with a-EGFR mutations. One possibility is that current EGFR-directed therapies ineffectively suppress cancer growth among patients with a-EGFR mutations. Therefore, the selective pressure towards developing acquired resistance mutations is lower than that for t-EGFR mutations. It is worth noting that among a-EGFR mutations with acquired resistance, only half of these cases were tested for HER2 or MET amplification via FISH, and therefore the incidence of gene amplification as a potential resistance mechanism remains underexplored.
There is growing appreciation of HER2 alterations in NSCLC and increasing interest in the development of HER2-targeted therapies. In our series, 37% of patients with HER2 alterations received HER2-directed therapy (which included TKIs, HER2 monoclonal antibodies, and HER2 antibody–drug conjugates) during their treatment. All patients with measurable lesions had SD on their first on-treatment scans. Patients who received HER2-directed therapies had numerically longer median survival relative to patients who received cytotoxic chemotherapy (65 vs. 29 months), although this did not meet threshold for statistical significance. Given our small sample size, we were unable to differentiate responses to HER2-directed therapies based on specific insertion mutations, though this is an area of importance. Afatinib has been explored in the management of patients with HER2 exon 20 insertions, with mixed results.51 In one retrospective series, the ORR to afatinib was 100% (n = 4),25 though this was not replicated in a prospective phase 2 Niche trial of 13 patients, with only one patient experiencing a PR.52 Recent data suggest that pyrotinib, an irreversible pan-HER receptor TKI, demonstrated a superior antitumor effect within a PDX model relative to afatinib (P = .047) and T-DM1 (P = .013). Among 15 patients in the phase 2 cohort, pyrotinib had an ORR of 53.3% (8/15) and a PFS of 6.4 months.53
The efficacy of ICIs in patients with a-EGFR mutations and HER2 alterations also remains underexplored. A subgroup analysis of the IMpower150 trial reported the efficacy of ICIs in combination with platinum doublet chemotherapy and bevacizumab for patients with EGFR mutations and ALK gene fusions,37 though this finding was only significant among patients with t-EGFR mutations. In our series, no patient with a-EGFR or HER2 alterations had a RECIST response to single-agent ICI. Patients with a-EGFR and HER2 alterations who received ICI with platinum doublet chemotherapy (using the KEYNOTE-189 regimen) had significantly improved PFS relative to single-agent ICI (7 vs. 2 months; P < .001; HR = 0.06; 95% CI 0.01–0.53). These poor single-agent ICI responses are similar to those reported in a large cohort study by Mazieres et al29 that found that the PFS to single-agent ICI ranged from 1.4 to 2.8 months depending on EGFR mutation subtype, and 2 to 3.4 months among those with HER2 alterations.29 Increased smoking status positively correlated with improved PFS to single-agent ICI among patients with HER2 alterations (P = .04) and trended toward significance among those with EGFR mutations (P = .06).29 It also remains an open question as to whether there are subtle differences in the efficacy between the KEYNOTE-189 and IMpower150 regimens among patients with a-EGFR mutations and HER2 alterations.
There are several limitations to our study. This was a retrospective single-institution study and was therefore prone to selection bias. Given variability in follow-up among patients in this study, we performed RECIST measurements according to the first on-treatment scan, not best treatment response. It is possible that our response rates were skewed toward measuring SD. Nonetheless, patients with sensitizing mutations will typically demonstrate radiographic responses by the first on-treatment scan,41,54 and we thought that this was a close surrogate for best response to targeted therapy. We were unable to control for multiple confounders like age, performance status, prior lines of therapy, and brain metastases when making PFS and OS comparisons across EGFR subtypes because of our small sample size. Given that our objective was to assess clinical outcomes of patients with a-EGFR and HER2 alterations with currently available targeted agents, we excluded patients who were first treated with agents in clinical trials, and we acknowledge that this may potentially bias our selection of patients. A limited number of patients underwent testing for acquired resistance, tempering our ability to draw strong conclusions regarding mechanisms of resistance, though this is an area ripe for future exploration. Finally, our sample size limited our ability to assess the impact of smoking status and PD-L1 expression on ICI outcomes in patients with a-EGFR and HER2 alterations, though this is an important area of investigation.
Conclusion
Our study has several important findings of clinical relevance. Alterations within the ErbB family (ie, EGFR and HER2) appear to have a unique metastatic phenotype characterized by a higher rate of synchronous lung and bone metastases compared to gene fusions, mutations within the Ras/Raf pathway, or mutations without an identifiable driver. Patients with a-EGFR mutations receiving afatinib and osimertinib have an inferior PFS relative to patients with t-EGFR mutations, with exon 20 insertions having the worst PFS and OS to current EGFR directed therapies. In our series, patients with a-EGFR mutations were much less likely to undergo testing for acquired resistance. A large percentage of patients with no identified resistance mutations did not undergo subsequent testing for HER2 or MET amplification. Elucidating differences in acquired resistance mechanisms between a-EGFR and t-EGFR mutations is an area ripe for further investigation. Finally, patients with a-EGFR mutations and HER2 alterations have uniformly poor responses to single-agent ICI, even with high PD-L1 expression. Novel therapies are urgently needed for patients with these mutations, as current outcomes remain poor.
Supplementary Material
Clinical Practice Points.
Most clinical trials exploring EGFR-directed therapies have only included patients with typical drug-sensitive EGFR mutations. Atypical EGFR mutations, representing only 15% to 20% of EGFR mutations, are less well described. Likewise, HER2 alterations have been identified as oncogenic drivers in 2% to 3% of NSCLC.
We found that mutations within ErbB family (ie, EGFR and HER2) appear to have a unique metastatic phenotype characterized by higher rate of synchronous lung and bone metastases compared to gene fusions, mutations within the Ras/Raf pathway, or those without an identifiable oncogene driver.
Patients with a-EGFR mutations have a worse PFS and OS relative to patients with t-EGFR mutations, even when afatinib and osimertinib are used in the first-line setting.
Patients with a-EGFR mutations were much less likely to undergo resistance testing. Acquired T790M occurred at a lower rate among patients with a-EGFR mutations treated with first and second generation EGFR TKIs. MET amplification was not seen among patients with a-EGFR mutations, though only half of samples were tested for MET and HER2 amplification.
Survival was numerically (though not statistically) improved for patients with HER2 alterations who received HER2-directed therapies over cytotoxic chemotherapy.
Patients with a-EGFR mutations and HER2 alterations have uniformly poor ORRs and PFS to single-agent immunotherapy.
Acknowledgments
D.R.C. P.A.B., and R.C.D. were supported in part by the University of Colorado Lung Cancer Specialized Program of Research Excellence (P50CA058187). E.L.S., J.M.P., and R.C.D. were supported by funds from the University of Colorado Cancer Center Thoracic Oncology Research Initiative. Data collection was made possible in part by a NIH/NCATS Colorado CTSA grant (UL1 TR002535).
Footnotes
Disclosure
T.P. has received honoraria from PRIME Oncology, Physicians Education Resource (PER) LLC, and Genentech/Roche. K.D. has received consulting fees from ArcherDx. D.L.A. received personal frees from AbbVie, Bristol Myers Squibb, and Inivata. E.L.S. has received honoraria or consulting fees from Takeda, Roche/Genentech, and AbbVie. J.M.P. has received honoraria or consulting fees from Roche/Genentech, Takeda, AstraZeneca, GLG, Novartis, and Pfizer. P.A.B. received personal fees from AstraZeneca and Guardant Health. D.R.C. has received honoraria or consulting fees from Ariad, Takeda, Ignyta, and Roche/Genentech, and a sponsored clinical trial research agreement with Takeda. R.C.D. has received consulting fees from Rain Therapeutics, Roche/Genentech, Takeda, AstraZeneca; a sponsored research agreement from Ignyta; licensing fees for patents from Abbott Molecular and Rain Therapeutics; licensing fees for biologic materials from Black Diamond, Pearl River, Ariad, Foundation Medicine, and Genentech; and stock ownership in Rain Therapeutics. The other authors have stated that they have no conflict of interest.
Supplemental Data
Supplemental figure and tables accompanying this article can be found in the online version at https://doi.org/10.1016/j.cllc.2019.11.008.
References
- 1.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304:1497–500. [DOI] [PubMed] [Google Scholar]
- 2.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004; 101:13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sequist LV, Bell DW, Lynch TJ, et al. Molecular predictors of response to epidermal growth factor receptor antagonists in non–small-cell lung cancer. J Clin Oncol 2007; 25:587–95. [DOI] [PubMed] [Google Scholar]
- 4.Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005; 97:339–46. [DOI] [PubMed] [Google Scholar]
- 5.Tokumo M, Toyooka S, Kiura K, et al. The relationship between epidermal growth factor receptor mutations and clinicopathologic features in nonesmall cell lung cancers. Clin Cancer Res 2005; 11:1167–73. [PubMed] [Google Scholar]
- 6.Janne PA, Ou SI, Kim DW, et al. Dacomitinib as first-line treatment in patients with clinically or molecularly selected advanced non–small-cell lung cancer: a multicentre, open-label, phase 2 trial. Lancet Oncol 2014; 15:1433–41. [DOI] [PubMed] [Google Scholar]
- 7.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non–small-cell lung cancer with mutated. EGFR N Engl J Med 2010; 362:2380–8. [DOI] [PubMed] [Google Scholar]
- 8.Mok TS, Wu YL, Papadimitrakopoulou VA. Osimertinib in EGFR T790M-positive lung cancer. N Engl J Med 2017; 376:1993–4. [DOI] [PubMed] [Google Scholar]
- 9.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009; 361:947–57. [DOI] [PubMed] [Google Scholar]
- 10.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non–small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012; 13:239–46. [DOI] [PubMed] [Google Scholar]
- 11.Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013; 31:3327–34. [DOI] [PubMed] [Google Scholar]
- 12.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non–small-cell lung cancer. N Engl J Med 2005; 353:123–32. [DOI] [PubMed] [Google Scholar]
- 13.Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer. N Engl J Med 2018; 378:113–25. [DOI] [PubMed] [Google Scholar]
- 14.Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer—molecular and clinical predictors of outcome. N Engl J Med 2005; 353:133–44. [DOI] [PubMed] [Google Scholar]
- 15.Wu YL, Cheng Y, Zhou X, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation–positive non–small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol 2017; 18: 1454–66. [DOI] [PubMed] [Google Scholar]
- 16.Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non–small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014; 15:213–22. [DOI] [PubMed] [Google Scholar]
- 17.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N Engl J Med 2004; 350:2129–39. [DOI] [PubMed] [Google Scholar]
- 18.Yasuda H, Kobayashi S, Costa DB. EGFR exon 20 insertion mutations in non–small-cell lung cancer: preclinical data and clinical implications. Lancet Oncol 2012; 13:e23–31. [DOI] [PubMed] [Google Scholar]
- 19.Arcila ME, Nafa K, Chaft JE, et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther 2013; 12:220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kosaka T, Tanizaki J, Paranal RM, et al. Response heterogeneity of EGFR and HER2 exon 20 insertions to covalent EGFR and HER2 inhibitors. Cancer Res 2017; 77:2712–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Massarelli E, Johnson FM, Erickson HS, et al. Uncommon epidermal growth factor receptor mutations in non–small cell lung cancer and their mechanisms of EGFR tyrosine kinase inhibitors sensitivity and resistance. Lung Cancer 2013; 80: 235–41. [DOI] [PubMed] [Google Scholar]
- 22.Oxnard GR, Lo PC, Nishino M, et al. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J Thorac Oncol 2013; 8: 179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Passaro A, Prelaj A, Bonanno L, et al. Activity of EGFR TKIs in Caucasian patients with NSCLC harboring potentially sensitive uncommon EGFR mutations. Clin Lung Cancer 2019; 20:e186–94. [DOI] [PubMed] [Google Scholar]
- 24.Spector NL, Blackwell KL. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2–positive breast cancer. J Clin Oncol 2009; 27:5838–47. [DOI] [PubMed] [Google Scholar]
- 25.Mazieres J, Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: results from the European EUHER2 cohort. Ann Oncol 2016; 27:281–6. [DOI] [PubMed] [Google Scholar]
- 26.Mazieres J, Peters S, Lepage B, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol 2013; 31: 1997–2003. [DOI] [PubMed] [Google Scholar]
- 27.Pillai RN, Behera M, Berry LD, et al. HER2 mutations in lung adenocarcinomas: a report from the Lung Cancer Mutation Consortium. Cancer 2017; 123:4099–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li BT, Ross DS, Aisner DL, et al. HER2 amplification and HER2 mutation are distinct molecular targets in lung cancers. J Thorac Oncol 2016; 11:414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mazieres J, Drilon A, Lusque A, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry [e-pub ahead of print]. Ann Oncol. 10.1093/annonc/mdz167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takezawa K, Pirazzoli V, Arcila ME, et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFR T790M mutation. Cancer Discov 2012; 2: 922–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noonan SA, Berry L, Lu X, et al. Identifying the appropriate FISH criteria for defining MET copy number–driven lung adenocarcinoma through oncogene overlap analysis. J Thorac Oncol 2016; 11:1293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leduc C, Merlio JP, Besse B, et al. Clinical and molecular characteristics of non–small-cell lung cancer (NSCLC) harboring EGFR mutation: results of the nationwide French Cooperative Thoracic Intergroup (IFCT) program. Ann Oncol 2017; 28:2715–24. [DOI] [PubMed] [Google Scholar]
- 33.Shen YC, Tseng GC, Tu CY, et al. Comparing the effects of afatinib with gefitinib or erlotinib in patients with advanced-stage lung adenocarcinoma harboring non-classical epidermal growth factor receptor mutations. Lung Cancer 2017; 110:56–62. [DOI] [PubMed] [Google Scholar]
- 34.Takeda M, Sakai K, Hayashi H, et al. Clinical characteristics of non–small cell lung cancer harboring mutations in exon 20 of EGFR or. HER2. Oncotarget 2018; 9:1132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hastings K, Yu H, Wei W, et al. EGFR mutation subtypes and response to immune checkpoint blockade treatment in non–small cell lung cancer. Ann Oncol 2019; 30:1311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ng TL, Liu Y, Dimou A, et al. Predictive value of oncogenic driver subtype, programmed death-1 ligand (PD-L1) score, and smoking status on the efficacy of PD-1/PD-L1 inhibitors in patients with oncogene-driven non–small cell lung cancer. Cancer 2019; 125:1038–49. [DOI] [PubMed] [Google Scholar]
- 37.Reck M, Mok TSK, Nishio M, et al. Atezolizumab plus bevacizumab and chemotherapy in nonesmall-cell lung cancer (IMpower150): key subgroup analyses of patients with EGFR mutations or baseline liver metastases in a randomised, open-label phase 3 trial. Lancet Respir Med 2019; 7:387–401. [DOI] [PubMed] [Google Scholar]
- 38.Socinski MA, Jotte RM, Cappuzzo F, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med 2018; 378:2288–301. [DOI] [PubMed] [Google Scholar]
- 39.Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non–small-cell lung cancer. J Clin Oncol 2010; 28:357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013; 31:1023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roach C, Zhang N, Corigliano E, et al. Development of a companion diagnostic PD-L1 immunohistochemistry assay for pembrolizumab therapy in non–small-cell lung cancer. Appl Immunohistochem Mol Morphol 2016; 24:392–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gandhi L, Rodríguez-Abreu D, Gadgeel S, et al. Pembrolizumab plus chemotherapy in metastatic nonesmall-cell lung cancer. N Engl J Med 2018; 378:2078–92. [DOI] [PubMed] [Google Scholar]
- 43.Doebele RC, Lu X, Sumey C, et al. Oncogene status predicts patterns of metastatic spread in treatment-naive nonsmall cell lung cancer. Cancer 2012; 118:4502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujimoto D, Ueda H, Shimizu R, et al. Features and prognostic impact of distant metastasis in patients with stage IV lung adenocarcinoma harboring EGFR mutations: importance of bone metastasis. Clin Exp Metastasis 2014; 31: 543–51. [DOI] [PubMed] [Google Scholar]
- 45.Hsu F, Nichol A, Toriumi T, et al. Miliary metastases are associated with epidermal growth factor receptor mutations in non–small cell lung cancer: a population-based study. Acta Oncol 2017; 56:1175–80. [DOI] [PubMed] [Google Scholar]
- 46.Poonia S, Berge EM, Aisner DL, et al. EGFR exon 19 deletion mutations and systemic/central nervous system miliary metastasis: clinical correlations and response to therapy. Clin Lung Cancer 2014; 15:387–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuiper JL, Hashemi SM, Thunnissen E, et al. Non-classic EGFR mutations in a cohort of Dutch EGFR-mutated NSCLC patients and outcomes following EGFR-TKI treatment. Br J Cancer 2016; 115:1504–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiu CH, Yang CT, Shih JY, et al. Epidermal growth factor receptor tyrosine kinase inhibitor treatment response in advanced lung adenocarcinomas with G719X/L861Q/S768I mutations. J Thorac Oncol 2015; 10:793–9. [DOI] [PubMed] [Google Scholar]
- 49.Galli G, Corrao G, Imbimbo M, et al. Uncommon mutations in epidermal growth factor receptor and response to first and second generation tyrosine kinase inhibitors: a case series and literature review. Lung Cancer 2018; 115:135–42. [DOI] [PubMed] [Google Scholar]
- 50.Yang JC, Sequist LV, Geater SL, et al. Clinical activity of afatinib in patients with advanced non–small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol 2015; 16:830–8. [DOI] [PubMed] [Google Scholar]
- 51.Lai WV, Lebas L, Barnes TA, et al. Afatinib in patients with metastatic or recurrent HER2-mutant lung cancers: a retrospective international multicentre study. Eur J Cancer 2019; 109:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dziadziuszko R, Smit EF, Dafni U, et al. Afatinib in NSCLC with HER2 mutations: results of the prospective, open-label phase II NICHE trial of European Thoracic Oncology Platform (ETOP). J Thorac Oncol 2019; 14: 1086–94. [DOI] [PubMed] [Google Scholar]
- 53.Wang Y, Jiang T, Qin Z, et al. HER2 exon 20 insertions in non–small-cell lung cancer are sensitive to the irreversible pan-HER receptor tyrosine kinase inhibitor pyrotinib. Ann Oncol 2019; 30:447–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Costa DB, Kobayashi S, Tenen DG, et al. Pooled analysis of the prospective trials of gefitinib monotherapy for EGFR-mutant non–small cell lung cancers. Lung Cancer 2007; 58:95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.