

Abstract
The conserved central region (CR) of PrPC has been hypothesized to serve as a passive linker connecting the protein’s toxic N-terminal and globular C-terminal domains. Yet, deletion of the CR causes neonatal fatality in mice, implying the CR possesses a protective function. The CR encompasses the regulatory α-cleavage locus, and additionally facilitates a regulatory metal ion-promoted interaction between the PrPC N- and C-terminal domains. To elucidate the role of the CR and determine why CR deletion generates toxicity, we designed PrPC constructs wherein either the cis-interaction or α-cleavage are selectively prevented. These constructs were interrogated using nuclear magnetic resonance, electrophysiology, and cell viability assays. Our results demonstrate the CR is not a passive linker and the native sequence is crucial for its protective role over the toxic N-terminus, irrespective of α-cleavage or the cis-interaction. Additionally, we find that the CR facilitates homodimerization of PrPC, attenuating the toxicity of the N-terminus.
Keywords: cellular studies, electrophysiology, neurodeneration, nuclear magnetic resonance, prion
1 |. INTRODUCTION
Transmissible spongiform encephalopathies, also known as prion disorders, are a class of neurodegenerative diseases that occur due to misfolding of the mainly α-helical cellular prion protein (PrPC) into the ß-sheet rich PrP scrapie isoform (PrPSc).1,2 Prion diseases include Creutzfeldt-Jakob disease (CJD) and Gerstmann-Sträussler-Scheinker syndrome (GSS) in humans, Mad Cow disease in bovine. The neurotoxicity found in prion diseases is dependent on the host’s expression of PrPC.3 PrPC is natively found as a glycosylphosphatidylinositol (GPI) anchored glycoprotein located on the extracellular side of the plasma membrane and associated with lipid rafts.4 PrPC is expressed ubiquitously throughout the brain and enriched in neurons.5–7
Mature PrPC is composed of 208 amino acids (mouse numbering: residues 23–230), distributed over two major domains. The globular C-terminal domain (residues 126–230) consists of three α-helices and one short anti-parallel ß-sheet, collectively stabilized by a disulfide bond (Figure 1A).8 The partially structured N-terminal domain (residues 23–104) contains several functional segments. Among these are the octarepeat (OR) segment ((PHGG(G/S)WGQ)4; residues 59–90) that takes up copper and zinc at physiological concentrations9–13 and the polybasic N-terminus (residues 23–31), both asserting regulatory control over certain transmembrane receptors. For example, Zn2+- and Cu2+-binding to the OR modulates AMPA receptor (AMPAR) activity14 and NMDA receptor activity,15,16 respectively. More generally, expression levels of PrPC in mice alters the anatomic distribution of Zn2+ and Cu2+ throughout the brain.17
FIGURE 1.

A, Linear schematic of PrPC. B, Sequences used in this study. CR mutations were made to modulate α-cleavage. In the CR mutation panel, the residues highlighted in red are where α-cleavage occurs between (α1-α3). Residues highlighted in orange are the P3’-P2’ or P3–P2 relative to the cleavage site. In the OR mutation panel, the residues highlighted in red are the histidine residues that Cu2+ bind to (WT PrPC) or what those histidine residues were mutated to in His to Ala PrPC. C, Two regulatory process of PrPC. PrPC can undergo α-cleavage to generate a released N1 fragment and the membrane C1 fragment. Additionally, the binding of Cu2+ to the OR drives a domain-domain cis-interaction between the Cu2+-bound OR and the globular C-terminal domain. The CR is shown in red. The OR is shown in green. The polybasic N-terminus is shown in orange
Recent studies find that the partially structured N-terminus and the globular C-terminal domains of PrPC directly interact (Figure 1C).18,19 This cis-interdomain interaction is mediated by both Zn2+ (20) and Cu2+ (19,21–23) binding to the OR domain as well as electrostatic interactions between the polybasic N-terminus and a negatively charged patch on the globular C-terminal domain.23–25 These results are of interest due to recent literature describing neurotoxicity driven by antibodies, such as POM1, that target the C-terminal domain of PrPC,26 generating cellular responses similar to that elicited by administration of PrPSc such as generation of reactive oxygen species, activation of unfolded protein response, and downregulation of similar genes.27 Specifically, the POM1 epitope is located to the PrPC C-terminal surface that stabilizes the metal ion-promoted, protective cis-interaction.18,21 Interestingly, the toxicity elicited by C-terminal antibodies19 is blocked by the co-administration of N-terminal antibodies,26 which suggests that the N-terminus of PrPC is a toxic effector domain regulated by the globular C-terminal domain. Remarkably, N-terminal antibodies are able to reduce PrPSc-induced toxicity in cerebellar organotypic slices,27 further suggesting the toxic potential of the N-terminus of PrPC.
The intervening linker connecting the two major domains is called the central region (CR) (residues 105–125) and is highly conserved among mammalian species.28,29 Notably, in healthy tissues this region is a locus for proteolysis, termed α-cleavage (Figure 1C)30,31 that generates a N-terminal fragment (N1) and a membrane bound C-terminal domain (C1), both having proposed biological functions.32,33 In addition to cleavage, the CR along with the polybasic N-terminus are high affinity docking sites for amyloid beta (Aß) oligomers.34,35
Developing a mechanistic understanding of the toxicity elicited by the prion protein is of the upmost importance since cell surface expression of this species transmits toxic transmembrane signals in both prion and Alzheimer’s disease. Over the years, insight into the mechanisms come from deletions targeted to the CR. It was initially reported in transgenic mice that Δ32–134 PrPC (ΔF PrPC) generates spontaneous neurodegeneration and death within 2–5 months, which is reversed upon the coexpression of WT PrPC.36 Later it was found that retaining more of the N-terminus of PrPC (Δ94–134) results in myelin degeneration and death within 20–30 days.37 Paradoxically, the shortest studied deletion encompassing just the short CR, Δ105–125 (ΔCR PrPC), leads to the most severe neurodegenerative phenotype, with consequent fatality about one week after birth.38,39
Due to its profound neurotoxicity, ΔCR PrPC has been the focus of numerous recent investigations.19,23,38,40–46 ΔCR PrPC is trafficked and localized to the extracellular membrane similar to WT PrPC.41 In cell culture, whole cell electrophysiological patch clamp experiments find that ΔCR PrPC induces large inward spontaneous currents,42,46 which correlate with a decrease in cellular viability in a drug-based cellular assay (DBCA).47 Both spontaneous currents and drug-induced toxicities can be blocked by the concurrent overexpression of WT PrPC or by deletion of the polybasic N-terminus in ΔCR PrPC.48 Interestingly, these currents are also induced in WT PrPC expressing cells with C-terminal antibodies,19 paralleling the C-terminal antibody experiments previously described. Additionally, C-terminal antibody and ΔCR PrPC-induced currents can be blocked by co-administration of N-terminal antibodies or N-terminal ligands (eg, Cu2+).19,49 These results point toward the CR at playing a central role in regulating the toxic N-terminus––a role that may unify the disparate PrPC dependent modes of neurotoxicity.
Despite the focus on ΔCR PrPC neurodegeneration, the mechanism by which the CR regulates the toxic N-terminus is unknown. It has been thought previously that the CR is simply a passive linker between the metal binding N-terminus and the globular domain. Opposing this view is the observation that the CR sequence is highly conserved and, as noted above, its deletion causes neonatal fatality in transgenic mice and spontaneous currents in cell culture. Two possible explanations can be envisioned to explain the role of the CR: (1) this region is necessary to facilitate the metal-driven cis-interaction; and (2) it is required because it is the locus for α-cleavage. In the absence of either or both of these functions of the CR, PrPC may acquire toxic activities. The aim of this study was to use protein design, NMR, electrophysiology, cleavage assays, cellular crosslinking, and cell survival studies to fully investigate the role of the CR in PrPC structure and toxicity of ΔCR PrPC. Our results demonstrate that the CR is not a passive linker between the N- and C-terminal domains. Instead, we find that the CR facilitates PrPC-PrPC dimerization or in some other fashion provides conformational control over the proteins toxic N-terminal segment, thereby serving as a regulator of PrPC-mediated neurotoxicity.
2 |. MATERIALS AND METHODS
2.1 |. Plasmids
pcDNA3.1 (+) Hygro plasmids (Invitrogen) encoding WT and ΔCR PrPC used for mammalian cell transfections have been described previously.42,44,50 pJ414 vector (DNA2.0) encoding WT PrPC used for recombinant protein expression has been previously described.21 To generate the vectors for G5, G5α1, G5α23, and His to Ala PrPC (both for the PCDNA 3.1 (+) Hygro plasmid and pj414 vector), Gibson cloning was used.51 Briefly, primers were purchased from Invitrogen to linearize the plasmid while deleting out the selected area to be replaced using Phusion High-Fidelity PCR Master Mix (New England Biolabs). Linearization reactions were run on a 1% of agarose gel and linearized DNA was extracted with GeneJET Gel Extraction Kit (Thermo Fisher Scientific). Gibson reactions were run using Gibson Assembly Master Mix (New England Biolabs) and transformed into E coli (DH5α (DE3) Invitrogen). Colonies were grown and pure DNA was extracted using Qiagen Mini prep kits. Constructs were then verified by DNA sequencing. Plasmids used in mammalian cell culture were further grown and purified using GenElute HP Endotoxin-Free Plasmid Maxiprep Kit (Sigma-Aldrich). Point mutations were introduced using PCR-based site-directed mutagenesis with mutagenic primers (Invitrogen) and Phusion High-Fidelity PCR Master Mix (New England Biolabs). Constructs were verified by DNA sequencing.
2.2 |. Cell lines
HEK 293T cells (ATCC CRL-3216, Lot # 62729596) were maintained in high glucose DMEM supplemented with 10% of fetal bovine serum (Life Technologies) and GlutaMAX (Gibco). PrPC knockout (PrPKO) N2a cells have been described previously.52 PrPKO N2a cells were maintained in low glucose DMEM supplemented with nonessential amino acids (Corning), 10% of heat-inactivated fetal bovine serum (Life Technologies), GlutaMAX, and MycoZap Plus-CL (Lonza). HEK 293T and PrPKO N2a cell lines used in this study were mycoplasma free and were maintained at 37°C in a 5% of carbon dioxide incubator.
HEK 293T cells and PrPKO N2a cells used for western blotting were transiently transfected using LipoD293 In Vitro DNA Transfection Reagent (SignaGEN Laboratories) with PrPC encoding pcDNA3.1(+)Hygro plasmids in 6-well plates. Fifteen to eighteen hours after cells were transfected, the media was changed and cells were allowed to recover for 24 hours. PrPKO N2a cells used for electrophysiology were transiently transfected using Lipofectamine 2000 with pEGFP-N1 (Clontech).
2.3 |. Cell preparation and Western Blotting
Whole cell lysates were prepared by washing cells 2x with PBS. Cells were then lysed with lysis buffer (50 mM of tris(hydroxymethyl)aminomethane (Tris) (pH 8), 150 mM of sodium chloride (NaCl), 1 mM of ethylenediaminetetraacetic acid (EDTA), 1% of Triton X-100, 10% of Glycerol, supplemented with Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific)) and quantified using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). To remove N-linked glycans, cell lysates were treated with recombinant PNGase F (New England Biolabs) under denaturing conditions according to the manufacturers’ protocol. Completed PNGaseF reactions were boiled in SDS-PAGE buffer and run on a 4%−20% of Mini-PROTEAN TGX Precast Protein Gels (Bio-Rad) along with Precision Plus Protein WesternC Blotting Standards (Bio-Rad). SDS-PAGE gels were subsequently washed with water three times totaling 15 minutes and transferred to a nitrocellulose membrane using Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were blocked using 5% of bovine serum albumin in TBS-T. PrPC constructs were probed with PrPC Antibody (M-20) (Santa Cruz Biotechnology, sc-7694, goat origin) who’s epitope matches near the C-terminus of PrPC. The PrPC antibody was then detected with HRP Rabbit Anti-Goat IgG (Abcam: ab6741) and the ladder was detected with Precision Protein StrepTactin-HRP Conjugate (Bio-Rad). Blots were exposed to Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific) and images were taken using ChemiDoc XRS + System (Bio-Rad) and analyzed using Image Lab Software (Bio-Rad).
Cell surface PrPC was analyzed by treating transfected cells with 0.1 units Phosphatidylinositol-Specific Phospholipase C Protein (PIPLC) (Life Technologies) in 200 μL of phosphate-buffered saline (PBS) (+,+) rocking gently for two hours at 4°C. Supernatants containing released PrPC were collected were spun at 300 × g for five minutes at 4°C to pellet any cells that were dislodged from the plate. Supernatants were then transferred to a separate tube and glycerol was added to a final concentration of 5%. SDS-PAGE samples were prepared by adding nonreducing SDS-PAGE sample buffer and boiling for five minutes. SDS-PAGE gels and western blots were run as described above.
2.4 |. Protein expression
Recombinant PrP constructs encoding the various mouse PrPC(23–230) constructs in the pJ414 vector (DNA 2.0) were transformed into and expressed using E coli (BL21 (DE3) Invitrogen).21
Bacteria was grown in M9 minimal media supplemented with 15NH4Cl (1 g/L) (Cambridge Isotopes) for 1H-15N HSQC experiments or in LB media (Research Product International). Cells were grown at 37°C until reaching an optical density (OD) of 1–1.2, at which point expression was induced with 1 mM of isopropyl-1-thio-D-galactopyranoside (IPTG). PrPC constructs were purified as previously described.20 Briefly, proteins were extracted from inclusion bodies with extraction buffer (8 M of guanidinium chloride (GdnHCl), 100 mM of Tris, 100 mM of Sodium Acetate (pH 8)) at room temperature and were purified by Ni2+-immobilized metal-ion chromatography (IMAC). Proteins were eluted from the IMAC column using elution butter (5 M of GdnHCl, 100 mM of Tris, 100 mM of Sodium Acetate (ph 4.5)) and were brought to pH 8 with 6 M of potassium hydroxide (KOH) and left at 4°C for 2 days to oxidize the native disulfide bond. Proteins were then desalted into 50 mM of potassium acetate buffer (pH 4.5) and purified by reverse-phase HPLC on a C4 column (Grace). Pure protein was lyophilized and stored at −20°C until needed. The purity and identity of all constructs were verified by analytical HPLC and mass spectrometry (ESI-MS). Disulfide oxidation was confirmed by reaction with N-ethylmaleimide and subsequent ESI-MS analysis.
2.5 |. NMR
Lyophilized uniformly 15N-labeled PrPC constructs were first suspended in water until fully solubilized and concentrations were checked using the absorbance at 280 nm (A280) with the proper extinction coefficient. NMR samples were made to 300 μM in 10 mM of 2-(N-morpholino)ethanesulfonic acid (MES) buffer with 10% of D2O and the pH was adjusted to 6.1 with 600 mM of hydrochloric acid. Apo samples were loaded into a Shigemi NMR tube (Wilmad Glass, BMS-005B) and a 1H- 15N HSQC spectrum as collected at 37°C on an 800-MHz spectrometer (Bruker, Billerica, MA) at the University of California, Santa Cruz NMR Facility (Santa Cruz, CA). The sample was then removed from the tube and one equivalent of Cu2+ from a 10 mM of copper chloride solution in water (determined accurately by flame atomic absorption) was added and the pH was adjusted to 6.1 if necessary. The sample was loaded back into the Shigemi NMR tube and the sample height was adjusted to match the sample height of the apo sample and another 1H-15N HSQC spectrum was collected. NMR spectra were analyzed with NMRPipe53 and Sparky. Structural analysis was performed with Chimera.54 Protein assignments were achieved using previously determined values.21
2.6 |. ADAM8 cleavage assay
ADAM8, ADAM10, and ADAM17 were purchased from R&D systems. ADAM8 was activated according to the manufactures protocol. Activated ADAM8 was then diluted into ADAM8 dilution buffer (20 mM of Tris, 5 mM of calcium chloride (CaCl2), and 25 mM of potassium chloride (KCl) (pH 7.4), aliquoted, flash frozen in liquid nitrogen, and stored at −80°C until needed. ADAM10 and ADAM17 were diluted into ADAM10 assay buffer (25 mM of Tris, 2.5 μM of zinc chloride, and 0.005% of Brij-35 (pH 7.4)), aliquoted, flash frozen in liquid nitrogen, and stored at −80°C until needed.
Working stocks of purified PrPC were prepared to 40 μM in ADAM8 dilution buffer (for ADAM8 assay) or ADAM10 assay buffer (ADAM10 or ADAM17 assay). Cleavage assays were previously described by McDonald et al Briefly, assays were set up by mixing 15 μL of PrPC and 15 μL of ADAM protease and reacted overnight at 37°C. Reactions were quenched by the addition of 5 μL of 1% formic acid and stored on ice or at 4°C until needed. About 30 μL of the supernatant were pipetted into autosampler vials and loaded into an LTQ LC/MS autosampler (Thermo Fisher Scientific). About 20 μL of cleavage products was drawn from the vial and separated with a C4 HPLC column (Higgins Analytical) using a 60-min gradient of water/acetonitrile mobile phases. The A280 was continuously recorded by a photodiode array, whereas mass spectra were continuously taken using an LTQ mass spectrometer (Thermo Fisher Scientific). The C4 column was flushed with 95% of acetonitrile to remove any residually bound protein, and then, re-equilibrated with 95% water between each sample run.
The LC/MS spectra from each sample run were first analyzed by MS Bioworks. The mass spectrum ladder for each peak separated by the C4 column was deconvoluted using Bioworks to reveal the parent mass of the cleavage product (data not shown). The masses of the observed peaks were cross-referenced against the predicted masses of hydrolysis of all possible peptide bonds of the particular PrPC construct being assayed to determine, which cleavage product was produced. For all cleavage fragments enzymatically produced, observed masses were within 1 atomic mass unit of the mass of a predicted cleavage fragment.
2.7 |. Cell viability assay using WST-1
The cell viability assay conducted was described previously43 and adapted for this study. A 96-well plates for were seeded with 1 × 104 cells in 100 μL of high glucose DMEM and grown overnight. Wells were transfected with 50 ng of PrPC DNA using LipoD293 In Vitro DNA Transfection Reagent (SignaGEN Laboratories). Media was changed 18 hours later and replaced with either DMEM or DMEM supplemented with G418 (Life Technologies) and cells were allowed to grow for 48 hours. After this time 10 μL of Cell Proliferation Reagent WST-1 (Roche) was added to each well and allowed to incubate at 37°C for 1–2 hours. After this time, the A450 was measured for each well using a Perkin-Elmer EnVision plate reader. Background subtractions were made by subtracting the A450 of well with just DMEM and 10 μL of WST-1 reagent from the A450 of the sample wells. Viability was measured by dividing the A450 value of the G418 treated cells by the A450 of the untreated cells.
2.8 |. Electrophysiology
Recordings were made from PrPKO N2a cells 24–48 hours after transfection. Transfected cells were recognized by green fluorescence resulting from co-transfection with pEGFP-N1. Whole-cell patch clamp recordings were collected using standard techniques. Pipettes were pulled from borosilicate glass and polished to an open resistance of 2–5 megaohms. Experiments were conducted at room temperature with the following solutions: internal, 140 mM of cesium-glucuronate, 5 mM of cesium chloride, 4 mM of magnesium-ATP, 1 mM of disodium-GTP, 10 mM of ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), and 10 mM of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.4 with cesium hydroxide); external, 150 mM of NaCl, 4 mM of KCl, 2 mM of CaCl2, 2 mM of magnesium chloride (MgCl2), 10 mM of glucose, and 10 mM of HEPES (pH 7.4 with NaOH). Current signals were collected from a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA), digitized with a Digidata 1440 interface (Molecular Devices), and saved to disc for analysis with pCLAMP 10 software.
3 |. RESULTS
3.1 |. Blocking cis-interaction does not elicit toxicity
Deletion of the CR leads to a weakening of the Cu2+-driven cis-interaction19 as well as elimination of α-cleavage.31 Either of these two major changes could lead to the spontaneous neurotoxicity. To investigate whether neurotoxicity is due to a weakened Cu2+-driven cis-interaction, His to Ala PrPC was designed, in which all of the histidine’s within the OR were mutated to alanine’s (Figure 1B). By deleting out the OR histidine’s, the OR cannot bind Cu2+, in turn eliminating the primary driving force for the cis-interaction.18
To ensure His to Ala PrPC undergoes α-cleavage to a similar extent as WT PrPC, both constructs were transfected into either HEK293T cells or PrP knock out (PrPKO) N2a cells (Figure 2A,B). Whole cell lysates were treated with PNGaseF to remove N-linked glycans and analyzed by western blot. Both WT and His to Ala PrPC displayed two main bands, one corresponding to full-length protein (FL) and the other corresponding to the C-terminal fragment generated by α-cleavage (C1). Quantitating the intensity of the C1 band revealed that His to Ala PrPC (48 ± 6.7% and 27 ± 8.8% C1 band in HEK293T and PrPKO N2a cells, respectively) was cleaved to a similar extent as WT PrPC (37 ± 8.1% and 32 ± 0.6% C1 band in HEK293T and PrPKO N2a cells, respectively).
FIGURE 2.

Mutating the OR histidine’s to alanine’s (His to Ala PrPC) retains cleavage in cells and does not drive spontaneous currents. Western blots of PNGaseF treated lysates of HEK293T cells (A) or PrPKO N2a cells (B) transfected with either WT or His to Ala PrPC. Uncleaved full length band is denoted by FL and the C-terminal side of α-cleavage is denoted by C1. The bar graph below each blot show the quantitation by densitometric analysis of %C1 band relative to the sum of FL and C1 bands. Error bars represent ± SD from at least three independent experiments. In both cell lines, α-cleavage of His to Ala PrPC is comparable to WT PrPC. C, Representative current recordings of either His to Ala or ΔCR PrPC transfected PrPKO N2a cells show that His to Ala PrPC does not have spontaneous currents
Whole cell patch clamp experiments were conducted on PrPKO N2a cells held at −70 mV to test if inward currents were generated by blocking the metal-driven cis-interaction (Figure 2C). ΔCR PrPC transfected cells displayed strong inward spontaneous currents, which is in agreement with a previous study.42 In contrast, His to Ala PrPC transfected cells did not show large inward currents. This demonstrates that eliminating Cu2+ binding to the OR is not sufficient enough to produce ΔCR PrPC-like currents. It can then be inferred that there must be other factors leading to the currents generated by ΔCR PrPC than loss of the cis-interaction facilitated by histidine residues in the OR. This is in agreement with previous studies showing deletion of the OR (ΔCR/Δ59–90) has no effect on the magnitude of the currents generated relative to ΔCR PrPC.19,44
3.2 |. Blocking cleavage with flexible linker generates currents
ΔCR PrPC lacks the stretch of 21 highly conserved amino acids that encompass the α-cleavage sites (Figure 1). A previous in vitro study showed that there are multiple α-cleavage sites within the CR (α1-α3, Figure 1B). Proteolysis within this segment separates the N-terminal domain, resulting in a putative loss of intrinsic PrPC function.31,55 To address the possibility that the toxicity of ΔCR PrPC is due to misregulation from loss of α-cleavage, the WT PrPC CR was replaced with a flexible glycine-serine-rich linker (G5 PrPC, Figure 1). This glycine-serine rich linker was chosen for its solubility and high degree of flexibility to allow for the Cu2+-driven cis-interaction in addition to its resistance to proteolysis.56
To investigate if G5 PrPC retains the Cu2+-driven cis-interaction, paramagnetic relaxation enhancement NMR was employed.21 With the intrinsic paramagnetism of Cu2+, resulting from its d9 electron configuration, there is an increase in the relaxation rate of the NMR active nuclei when Cu2+ is in close proximity. This causes a broadening and a decrease in the intensity of the resonance lines, which is inversely proportional to the distance of Cu2+ to the backbone amide bond. Uniformly 15N-labeled protein was prepared and 1H-15N HSQC spectra were collected in the absence and presence of 1 eq. of Cu2+. The intensity of the resonance crosspeaks with 1 eq. of Cu2+ (I) was divided by the intensity of the resonances in the absence of Cu2+ (Io) to generate intensity ratios (I/Io) for each backbone NH. I/Io vs residue plots were made for both WT PrPC and G5 PrPC comprising amino acids 126–230 (Figure 3A). Residues considered strongly affected by the addition of Cu2+ (those that are affected by greater than 1σ) are shown in blue on the surface representations of the PrPC C-terminal domain (PDB: 1XYX) below each intensity ratio plot. The residues affected localize to three main patches: the C-terminal end of the loop going into helix 1 (Patch 1), the N-terminal end of helix 2 (Patch 2), and the N-terminal half of helix 3 (Patch 3). Both WT and G5 PrPC show similar residue-specific patterns demonstrating that G5 PrPC retains the Cu2+-driven cis-interaction.
FIGURE 3.
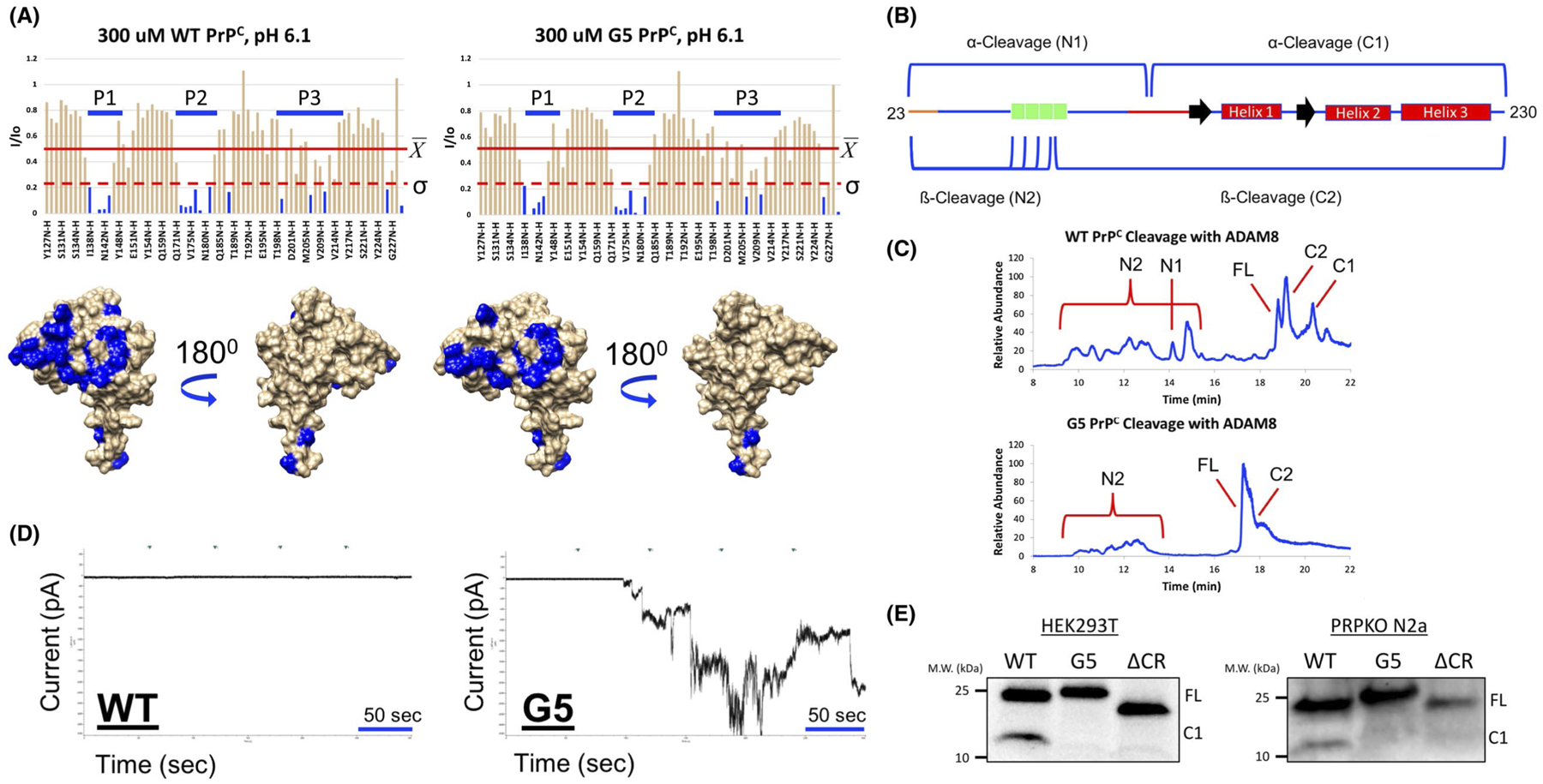
Mutating the CR to a flexible GS linker retains the copper-driven
cis-interaction, blocks alpha cleavage, and generates spontaneous currents. A,
I/Io vs residues plot of WT or G5 PrPC constructs
titrated with 1eq. of Cu2+ at pH 6.1 and 37°C. The average
 and standard deviation
(σ) of only similar residues of each construct were
taken. Below each plot is the surface representation of the C-terminus of
PrPC (PDB: 1XYX). Residues in blue are the residues where the
I/Io values are affected by
greater than one standard deviation. Both WT and G5 PrPC constructs
have similar residues affected to a similar degree, thus, the
cis-interaction is retained. B, Schematic diagram of
PrPC showing the possible cleavage products that can be produced
from in vitro cleavage assays. C, LC/MS traces of in vitro cleavage assays using
recombinant WT or G5 PrPC constructs reacted with ADAM8 overnight at
37°C. Reactions were loaded onto a C8 column and were eluted using a
gradient of water and acetonitrile. When analyzed using the LC/MS, WT
PrPC produces α- and ß-cleavage where G5
PrPC produces only ß-cleavage. D, Representative current
recordings of either WT or G5 PrPC transfected PrPKO N2A cells. The
data show that G5 PrPC has spontaneous currents, which are
characteristic of toxicity. E, Western blots of PNGaseF treated lysates of
HEK293T or PrPKO N2a cells transfected with DNA coding for either WT,
ΔCR, or G5 PrPC. Uncleaved full length protein is denoted by
FL and the C-terminal side of α-cleavage is denoted by C1. In HEK293T
cells, WT PrPC produces only α-cleavage, where ΔCR and
G5 PrPC did not undergo any cleavage events. In PrPKO N2a cells, WT
PrPC underwent α-cleavage, where ΔCR and G5
PrPC were not cleaved at all
and standard deviation
(σ) of only similar residues of each construct were
taken. Below each plot is the surface representation of the C-terminus of
PrPC (PDB: 1XYX). Residues in blue are the residues where the
I/Io values are affected by
greater than one standard deviation. Both WT and G5 PrPC constructs
have similar residues affected to a similar degree, thus, the
cis-interaction is retained. B, Schematic diagram of
PrPC showing the possible cleavage products that can be produced
from in vitro cleavage assays. C, LC/MS traces of in vitro cleavage assays using
recombinant WT or G5 PrPC constructs reacted with ADAM8 overnight at
37°C. Reactions were loaded onto a C8 column and were eluted using a
gradient of water and acetonitrile. When analyzed using the LC/MS, WT
PrPC produces α- and ß-cleavage where G5
PrPC produces only ß-cleavage. D, Representative current
recordings of either WT or G5 PrPC transfected PrPKO N2A cells. The
data show that G5 PrPC has spontaneous currents, which are
characteristic of toxicity. E, Western blots of PNGaseF treated lysates of
HEK293T or PrPKO N2a cells transfected with DNA coding for either WT,
ΔCR, or G5 PrPC. Uncleaved full length protein is denoted by
FL and the C-terminal side of α-cleavage is denoted by C1. In HEK293T
cells, WT PrPC produces only α-cleavage, where ΔCR and
G5 PrPC did not undergo any cleavage events. In PrPKO N2a cells, WT
PrPC underwent α-cleavage, where ΔCR and G5
PrPC were not cleaved at all
To investigate if G5 PrPC can undergo α-cleavage, HEK293T or PrPKO N2a cells were transfected with WT, G5, or ΔCR PrPC. PNGaseF treated lysates were analyzed by western blot (Figure 3E). Results show that G5 and ΔCR PrPC did not undergo α-cleavage. Cleavage assays using ADAM8 protease, the protease responsible for α-cleavage in skeletal muscle,31,57 were undertaken with recombinant PrPC to test if G5 PrPC can be cleaved in vitro (Figure 3B,C). Results showed that G5 PrPC underwent ß-cleavage in the OR region, which agrees with a previous study.31 In contrast, G5 PrPC is not susceptible to α-cleavage in ADAM8 assays, supporting the primary design goal for this construct.
Electrophysiology measurements were then performed to test if blocking α-cleavage induces currents as seen with ΔCR PrPC (Figure 2D). PrPKO N2a cells were transfected with WT PrPC did not display inward currents (Figure 3D). Conversely, G5 PrPC transfected cells produced large inward currents (Figure 3D). These results suggest that blocking α-cleavage by introduction of the G5 linker may be responsible for inducing inward currents similar to ΔCR PrPC.
3.3 |. Reintroduction of α-cleavage in G5 PrPC generates currents
If the currents generated by G5 PrPC are due to blockage of α-cleavage, then, inward currents should be reduced or eliminated upon reintroduction of cleavage into the G5 linker. G5α1 PrPC was designed by reintroducing α1-cleavage in the G5 linker (Figure 1B). ADAM8 cleavage assays (Figure 4D) and PNGaseF treated cell lysates (Figure 4A,B) show that G5α1 PrPC regains α-cleavage (38 ± 5.6% and 31 ± 6.6% C1 band in HEK293T and PrPKO N2a cells, respectively). Due to the close proximity of α2 and α3 in the linear sequence, G5α23 PrPC was designed which simultaneously adds back both cleavage sites (Figure 1B). In contrast to G5α1 PrPC, G5α23 PrPC exhibited no α-cleavage in vitro and in cells (Figure 4A,B,D). These results show that with the cell lines used, PrPC undergoes α-cleavage at the α1-cleavage sites.
FIGURE 4.

Addition of α-cleavage (α1 or α23) site back into G5 PrPC still generates spontaneous currents. Western blots of PNGaseF treated lysates of HEK293T cells (A) or PrPKO N2a cells (B) transfected with either G5α1, G5α23, or WT PrPC constructs. The bar graph below each blot show the quantitation by densitometric analysis of %C1 band relative to the sum of FL and C1 bands. Error bars represent ± SD from at least three independent experiments. In HEK293T cells, G5α1 PrPC regains α-cleavage where G5α23 PrPC does not. However in PrPKO N2a cells, both constructs have α- and ß- cleavage. C, I/Io vs residues plot of recombinant G5α1 PrPC with 1eq. of Cu2+ at pH 6.1 and 37°C. The average (X) and standard (σ) deviation were taken. Below the plot is the surface representation of the C-terminus of PrPC (PDB: 1XYX). Residues in blue are the residues where the I/Io values are affected by greater than one standard deviation. G5α1 PrPC has similar residues affected to a similar degree as WT PrPC, thus, the cis-interaction is retained. D, LC/MS traces of in vitro cleavage assays using recombinant G5, G5α1, or G5α23 PrPC constructs were reacted with ADAM8 overnight at 37°C. Reactions were quenched with formic acid to a final concentration of 3%, and then, 10 μg of PrPC was loaded onto a C8 column. Cleavage products were eluted using a gradient of water and acetonitrile. When analyzed using LC/MS. G5α1 PrPC produces α- and ß-cleavage where G5 and G5α23 produces only ß-cleavage. E, Representative current recordings of either G5, G5α1, or G5α23 PrPC transfected PrPKO N2a cells. The bar graph next to the current recording is the quantitation of the time the currents are greater than 200 pA. Error bars represent ± SEM from at least three different independent experiments. The data show that even though α-cleavage is regained in G5α1 PrPC, spontaneous currents are still generated
Due to the finding that G5α1 PrPC undergoes α-cleavage, NMR was used to determine if the Cu2+-driven cis-interaction is also retained (Figure 4C). As seen with G5 PrPC, G5α1 PrPC shows similar residue-specific patterns (Figure 4C) when compared to WT, indicating the Cu2+-driven cis-interaction is retained.
Electrophysiology experiments were then conducted to test if adding back susceptibility to α-cleavage reduces the generated inward currents (Figure 4E). Both G5α1 and G5α23 PrPC transfected PrPKO N2a cells still exhibit large inward current. Quantification of the data (measured by the percentage of time the currents exceeded 200 pA) revealed that G5, G5α1, and G5α23 PrPC generated the same time-averaged currents, which is consistent with previous measured values for ΔCR.19,23 This result demonstrates that the large inward currents, which are highly correlated with toxicity in animals, still persists even when PrPC retains susceptibility to α-cleavage.
Cleavage of the N-terminus, without replenishment of cell surface PrPC, must decrease the magnitude of the currents generated since it removes the polybasic extreme N-terminus responsible for current activity.44 If cell surface G5α1 PrPC was cleaved with an efficiency of 20%−40%, then, the currents should show a concomitant decrease of approximately the same amount, which is not observed in our analysis. However, when quantitating the percentage of time the currents are significant, a threshold current value was chosen. Therefore, it is possible that G5α1 PrPC exhibits a weaker current magnitude than G5 PrPC, but it spends the same amount of time over the threshold current value as G5 and G5α23 PrPC. Our results are nevertheless significant since they demonstrate that the full length G5α1 PrPC remaining on the cell surface still generates potent currents similar to ΔCR PrPC. Additionally, WT PrPC on the cell surface is mostly full length, but it contains the CR. Thus, there is something intrinsically encoded into the WT CR sequence that is able to attenuate the toxicity of the N-terminus.
3.4 |. The CR sequence facilitates dimerization
Results thus far show that the inward currents measured in the electrophysiology experiments are not a result of a weakened Cu2+-promoted cis-interaction or from blocking α-cleavage. This suggests that there is something inherent to the sequence of the CR that regulates the toxic N-terminus thereby muting inherent PrPC toxicity. In addition to conservation of this segment, the CR is notable for its high content of hydrophobic and small aliphatic amino acids. It was previously demonstrated that a peptide consisting of residues 105–125 readily forms fibrils that are toxic to cells if they express membrane anchored WT PrPC.58 Given that PrP(105–125) binds to PrPC suggests that the CR of proximal PrPC monomers may facilitate inter-PrPC interactions. In support of this, a previous study using cross-linkable unnatural amino acids in recombinant protein demonstrated that dimerization is facilitated by the CR.59 Dimerization has also been studied in cell culture using bioluminescent complementation,60 native PAGE and cysteine crosslinking.61
It was previously reported that ΔHD (Δ114–133) PrPC, a deletion similar to that in ΔCR PrPC, has a reduced dimerization propensity in N2a cells using native PAGE.61 This group was also able to trap the dimer by adding a cysteine flanking the CR (S131C). We, therefore, used this latter strategy to study if ΔCR or G5 PrPC would exhibit reduced dimerization. If the CR facilitates dimerization, then, deleting it away (ΔCR PrPC) or mutating it (G5 PrPC) should result with reduced dimerization. This hypothesis was tested using S131C mutants of WT, ΔCR, G5, G5α1, and His to Ala PrPC constructs.
The experimental protocol is schematized in Figure 5A. Briefly, if a cysteine on one PrPC monomer is in close proximity and in the correct orientation to another cysteine, a disulfide can form. HEK293T cells were transfected with WT S131C PrPC, and then, treated with phosphoinositide phospholipase C (PIPLC) to release PrPC from the cell surface. Samples were then boiled in SDS buffer free of reducing agents and analyzed by western blot (Figure 5B). Blots were quantified by the percentage dimer band relative to total PrPC in each lane (Figure 5C). In HEK293T cells, WT S131C PrPC displayed an intense band corresponding to approximately 67 ± 4.6% dimer. However, the measured dimer band could be due to membrane crowding coupled to nonspecific collisions between two PrPC monomer’s N-termini. To control for this possibility, a cysteine was added to the N-terminus (S36C) (Figure 5A). PIPLC-treated WT S36C PrPC transfected cells analyzed by western blotting (Figure 5B) showed a very small band migrating as a dimer, and an extremely prominent monomer band. Therefore, the dimerization seen is specific for the CR.
FIGURE 5.

ΔCR and G5 PrPC have a reduced dimer band in cell surface cysteine crosslinking experiments at position S131C. A, Schematic of the reaction occurring in the experiment. If two PrPC molecules come close enough together in the right orientation, then, disulfide formation will occur. Two separate positions were mutated (S36C and S131C). S36C was used to test if crosslinking only occurs due to cell surface crowding and S131C was used to test the specific interaction surface between two cell surface PrPC molecules. B, Western blot of HEK293T cells transfected with one of the constructs either labeled at S36C or S131C. Samples were prepared by incubating cells with 0.1 U of PIPLC in PBS (+,+) for 2 hours rocking at 4°C to remove only cell surface PrPC. Samples were then boiled with SDS-PAGE gel loading buffer that did not contain reducing agent and loaded on to a SDS-PAGE gel. This allowed for the separation of a monomer and dimer band. When the constructs were labeled at S36C, there was little to no dimer band detectable. C, Quantitation of western blots for % Dimer band of samples labeled at S131C relative to the sum of monomer and dimer bands using densitometric analysis. Error bars represent ± SD for at least two different independent experiments. Asterisks denote significant differences when compared to WT S131C PrPC (**P < .05). Results show that ΔCR, G5, and G5α1 PrPC constructs have a reduced dimer band in HEK293T cells, where only ΔCR and G5 PrPC have reduced dimer bands in PrPKO N2a cells
Next, ΔCR, G5, G5α1, and His to Ala PrPC (both S36C and S131C) constructs were tested (Figure 5B). For all constructs, similar to WT PrPC, there was essentially no measurable dimer band for the S36C PrPC mutants, however, dimer bands of varying intensity were observed for the S131C mutants. Compared to WT PrPC, ΔCR S131C and G5 S131C PrPC had a significantly reduced dimer bands of 39% ± 7.8% and 18% ± 6.6%, respectively. G5α1 S131C PrPC resulted in a slightly reduced, but still significant, dimer band of 56 ± 3.7% when compared to WT PrPC. Conversely, His to Ala S131C PrPC had a slightly increased, but still significant, dimer band of 79% ± 6.4% percent relative to WT S131C PrPC. These experiments were then repeated in PrPKO N2a cells (Figure 5C). Results were consistent with HEK293T cells, except that G5α1 S131C and His to Ala S131C PrPC both had a comparable dimer band with WT PrPC. Overall, the mutations to the CR led to the largest decrease in the measured dimer band. These suggest that the CR facilitates dimerization, an interaction that may play a role in regulation of the otherwise toxic N-terminal domain.
3.5 |. Addition of a cysteine in CR partially rescues toxicity
The addition of a cysteine just outside the CR (S131C) forces two PrPC molecules to crosslink, as measured by western blot analysis (shown above). WT S131C PrPC has a significantly larger dimer band when compared to ΔCR S131C and G5 S131C PrPC constructs. However, there is still a dimer band for both ΔCR S131C and G5 S131C. This shows that the two constructs may still interact with an orientation that allows for disulfide bond to formation, thereby forcing an irreversible dimer, albeit at a reduced level with respect to WT PrPC.
The question then arises whether or not forcing dimerization in ΔCR S131C or G5 S131C PrPC constructs rescues the cellular toxicity of these variants. To test this, a quantitative drug-based cellular assay (DBCA)47 was utilized in the place of measuring spontaneous currents. DBCA offers a convenient and rapid throughput means for testing for the presence of compromised cellular membranes, similar to what is measured using electrophysiological spontaneous currents. Previously it was shown that ΔCR PrPC expressing HEK293 cells, which exhibit spontaneous currents, also have a lower cell viability in the DBCA assay when challenged by the addition of G418 for 48 hours. This is proposed to occur due to ΔCR PrPC increasing drug influx by biasing cationic-selective membrane channels or by PrPC directly forming cationic permeable pores through its N-terminus.62
Therefore, transient transfections of PrPC constructs in to HEK293T cells were performed and cell viability was assessed using WST-1. First, the noncysteine constructs were tested (Figure 6A). Consistent with previous results, ΔCR PrPC had a significant decrease in cell viability when compared to WT PrPC. Interestingly, mutation of the OR His residues to Ala also exhibit a reduction in cell viability that is on the threshold of significance. As expected, the other constructs with mutations in the CR (G5, G5α1, and G5α23 PrPC) also showed decreased cell viability when compared to WT PrPC. This is significant for G5α1 PrPC because it is still undergoing cleavage in the cells, yet, has a similar level of reduced cell viability as seen with ΔCR and G5 PrPC. Additionally, two pathological mutations in the CR, G113V, and G130V, both cause a decrease in cell viability, which agrees well with previous studies.44
FIGURE 6.
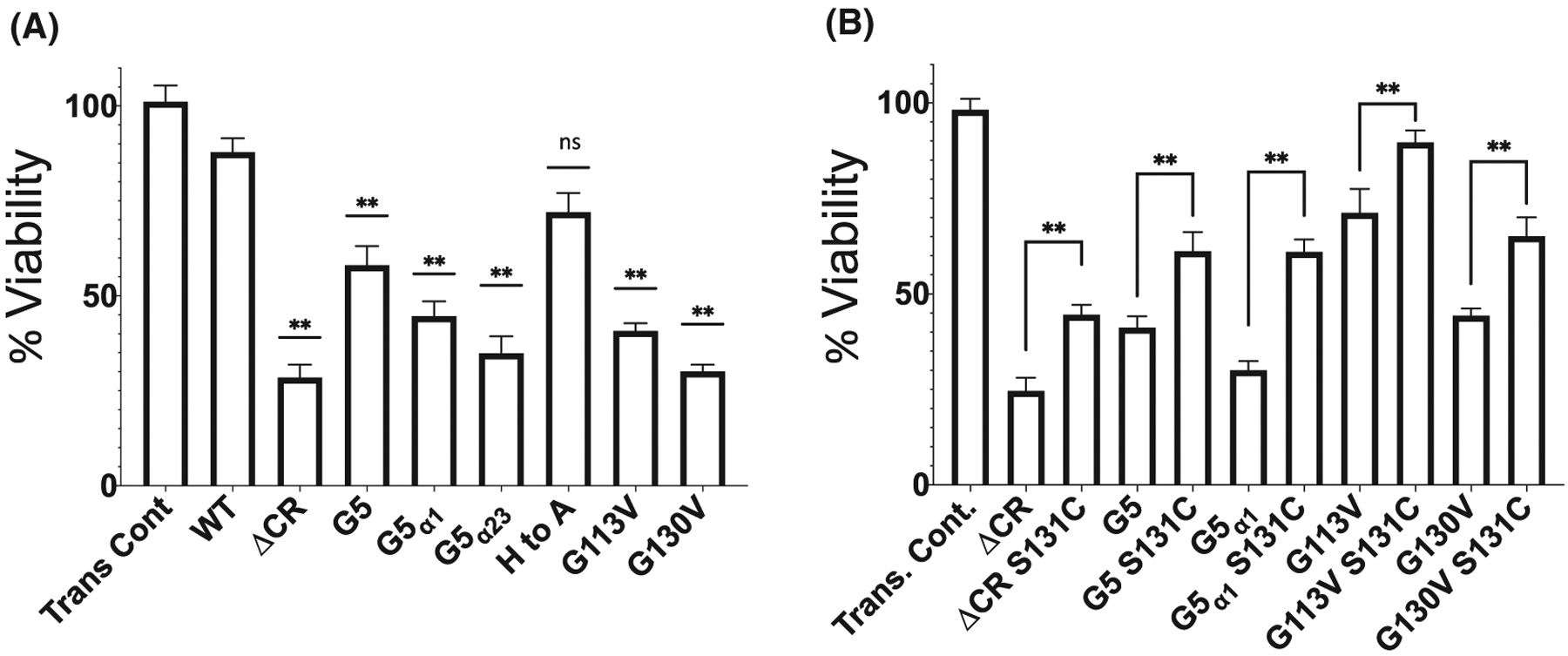
Addition of a cysteine at position 131 partially regains cell viability when challenged with G418. A, HEK293T cells transfected with the indicated PrPC construct were treated with 400 μg/mL of G418 for 48 hours. Cell Viability was assessed by WST-1 reduction as described in the method section. Error bars represent ± SEM for at least three independent experiments. Asterisks denote significantly different when compared to WT (**P < .05). All mutants, except His to Ala PrPC have a reduced cell viability relative to WT. B, Cysteine (S131C) constructs were tested with the noncysteine construct to test if the reduction in cell viability could be increased. Asterisks denotes significantly different when compared to the particular PrPC mutant without the added cysteine (**P < .05). ΔCR, G5, G5α1 PrPC, G113V, and G130V constructs were able to be partially rescued by the addition of a cysteine. The small differences in measured cell viability resulting from batch to batch cell variations fall within standard error
To determine if the addition of a cysteine at position 131 can rescue the reduction in cell viability of PrPC constructs with mutated CR, HEK293T cells were transfected with the cysteine-containing PrPC constructs along with the noncysteine containing constructs of ΔCR, G5, G5α1 PrPC, and two pathological mutations, G113V and G130V (Figure 6B). For each cysteine-containing PrPC construct, there was a partial, but still significant, increase in cell viability when compared to the noncysteine PrPC construct. This result demonstrates that forcing two PrPC molecules together by the CR can decrease the toxicity elicited by the N-terminus.
4 |. DISCUSSION
The molecular basis for the toxicity produced by elimination of the CR of PrPC has remained unclear. Indeed, findings over the last two decades show, paradoxically, that the shorter the deletion, the more toxic the response.63,64 Here, we used protein design, NMR, electrophysiology, and a DBCA to address this fundamental issue. Replacement of the CR region with a flexible linker of equivalent length recapitulates ΔCR PrPC toxicity, as measured by spontaneous electrophysiological currents and toxicity induced by a DBCA. Reintroduction of consensus α-cleavage sites fails to dampen the observed currents. In addition, while it is now established that the WT CR segment supports the protective, metal ion-promoted cis N-terminal—C-terminal interaction, elimination of this interaction by replacement of OR His residues alone does not generate spontaneous currents. However, we do observe a mild increase in toxicity in the DBCA. Together, these results demonstrate that the spontaneous toxicity induced by deletion of the CR is not due exclusively to altering the length of the CR, blocking α-cleavage, or preventing the metal-driven cis-interaction. Rather, our results suggest that specific features of the CR sequence restrain the toxic activity of the PrPC molecule, either by affecting the protein’s interaction with itself, with other cell-surface molecules, or by altering the orientation of the N-terminal domain. Based on our recent structural and physiological studies of PrPC, we propose three hypotheses, to explain how this might occur (Figure 7).
FIGURE 7.

N-terminal toxicity model: WT PrPC’s CR sequence regulates the toxic effects of the extreme N-terminus. We hypothesize this can occur by three different mechanisms. First, the CR facilitates dimerization, which would move the extreme N-terminus away from causing toxicity. Second, Cu2+ binding allows the CR to reposition and hold the N-terminus away from generating toxicity. Third, the CR binds to a coreceptor which helps regulate the toxic potential of the N-terminus. When the CR is substituted with the G5 linker, or deleted in ΔCR PrPC, the regulatory sequence of the CR is deleted and the N-terminus goes unregulated to cause toxic signaling. N-terminal toxicity can also possibly occur with C-terminal antibodies and CR pathological mutations causing GSS, such as G130V
One hypothesis for the role of the CR is that it facilitates dimerization of PrPC (Figure 7). Cellular cysteine crosslinking experiments show that WT PrPC exhibits significantly greater dimerization than ΔCR and G5 PrPC. This result demonstrates that a reduction in dimerization correlates with spontaneous currents and cell viability. Furthermore, enhancement of dimerization by introduction of the cysteine residues partially suppresses G418-induced toxicity relative to the noncysteine versions of G5, ΔCR, and G5α1 PrPC. A full restoration was likely not observed possibly due to a proportion of the constructs remaining in the monomeric form. Due to the incorporation of the non-native cysteine, we cannot be certain that the disulfide-stabilized dimer represents a true physiological state of PrPC; however, our data suggest that the WT CR sequence enhances the efficiency of two PrPC molecules orienting correctly to allow for this disulfide bond to form. This agrees with a previous study that showed dimerization occurs at a specific interface.59
How can dimerization regulate the toxic N-terminus? It is possible that homodimerization orients the otherwise toxic effector N-terminus in a way that prevents its misregulation. A previous study showed that C-terminal antibodies block dimerization as measured by bioluminescent complementation.60 If the N-terminus is regulated by a PrPC homodimer, then, an antibody blocking dimerization would thus dislodge the N-terminus to elicit toxicity. This may explain why binding of C-terminal antibodies to WT PrPC generates spontaneous currents.19 Dimerization may also provide a mechanism by which overexpression of WT PrPC rescues the toxicity of ΔCR PrPC expressing cells. Specifically, an overabundance of WT PrPC would force ΔCR PrPC into a dimer, with cellular surface expression either in cis (same cell)47 or in trans (adjacent cells),45 thus, regulating ΔCR PrPC’s toxic N-terminus.
Our data are also consistent with a model in which the CR of PrPC pulls the N-terminus away from the membrane when engaged in the metal-driven cis-interaction (Figure 7). The DBCA does show a reduction in cell viability when the metal binding His residues are eliminated in the OR segment. In this context, the CR could serve to reorient the otherwise toxic, polybasic N-terminus, away from the membrane but only when the protein is stabilized by metal ion driven cis-interaction. This proposal agrees with a molecular model generated using restraints from NMR and mass spectrometry crosslinking experiments.23 We have shown previously that, without the CR segment, that the stabilizing cis-interaction is significantly compromised, thus, promoting a toxic fold with the polybasic N-terminus extending away from the protein.19 It is also possible that removal of the CR segment leads to a substantial refolding of PrPC to a toxic state. However, given that CR is outside of the helical, globular domain, which is stabilized by a disulfide bond, we believe that at least this C-terminal segment of the protein retains its fold.
A third plausible model posits that the CR docks to an unknown co-membrane receptor (Figure 7). This model is consistent with data describing PrPC as a regulator of both ionotropic14,65 and metabotropic66,67 glutamatergic receptors. Additionally, ΔCR PrPC expressing cells are sensitive to glutamate-induced excitotoxicity.46 However, ΔCR PrPCinduced currents occur in both mammalian and insect cells,42 which possess very different complements of membrane receptors, suggesting that spontaneous currents originate from intrinsic PrPC properties, and not by misregulation of a membrane co-receptor.
Our findings may provide insight into the distinct phenotypes of inherited prion disease. Familial prion diseases are caused by mutations in two main regions. The first region is near the conserved negatively charged patch on the C-terminus (eg, D177N and E199K),20 mutations in which typically cause CJD or fatal familial insomnia.68 The second region encompasses the CR (eg, P101L, A116V, and G130V), mutations in which typically cause GSS. Both CJD mutations (D177N and E199K), as well as GSS mutations (P101L and A116V) have a near 100% penetrance.69 Several lines of evidence show, however, that there are substantial phenotypic differences between the two sets of disease-causing mutations. These include differences in plaque conformation,28,70–72 generation of spontaneous currents and G418-induced toxicity,44 and metal-driven cis-interaction.20,23 These observations suggest that mutations in the two regions act via different pathological mechanisms. Given that only CR mutations cause spontaneous currents, G418-induced toxicity, and a WT PrPC-like cis-interaction, it is possible that the toxic mechanisms of CR GSS causing mutations originate from an N-terminal toxicity model, similar to ΔCR PrPC, G5 PrPC, and C-terminal antibodies (Figure 7). This can be due to reduction in dimerization or modulation of the conformational landscape of the CR. Conversely, C-terminal CJD causing mutations may produce toxicity by other mechanisms, for example by enhancing aggregation of the protein or otherwise altering its biochemical properties.
In summary, our results demonstrate that the CR is not a passive linker connecting the N- and C-terminal domains. Instead, specific features of the sequence are absolutely crucial for blocking toxicity generated by the otherwise unregulated N-terminus. We propose that the CR either facilitates homodimerization of PrPC or serves to conformationally restrict the N-terminus from driving toxicity. Further elucidation of these regulatory contacts will be important for advancing concepts of prion toxicity.
ACKNOWLEDGMENTS
This work was supported by NIH grants R35 GM131781, S10 OD024980 and S10 OD018455 (to GLM) and R01 NS065244 (to DAH). We thank Dr Gerold Schmitt-Ulms for supplying PrPKO N2a cells.
Abbreviations:
- C1
C-terminal fragment
- CR
central region
- CJD
Creutzfeldt-Jakob disease
- DBCA
drug-based cellular assay
- FL
full length protein
- GSS
Gerstmann-Sträussler-Scheinker syndrome
- N1
N-terminal fragment
- NMR
nuclear magnetic resonance
- OR
octarepeat
- PIPLC
phosphoinositide phospholipase C
- PrPC
cellular prion protein
- PrPSc
prion protein scrapie isoform
- WT
wild type
- ΔCR PrPC
central region deletion mutant
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- 1.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–144. [DOI] [PubMed] [Google Scholar]
- 2.Collinge J Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–550. [DOI] [PubMed] [Google Scholar]
- 3.Brandner S, Isenmann S, Raeber A, et al. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379(6563):339–343. [DOI] [PubMed] [Google Scholar]
- 4.Watt NT, Griffiths HH, Hooper NM. Lipid rafts: linking prion protein to zinc transport and amyloid-beta toxicity in Alzheimer’s disease. Front Cell Dev Biol. 2014;2:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barmada S, Piccardo P, Yamaguchi K, Ghetti B, Harris DA. GFPtagged prion protein is correctly localized and functionally active in the brains of transgenic mice. Neurobiol Dis. 2004;16(3):527–537. [DOI] [PubMed] [Google Scholar]
- 6.Peralta OA, Eyestone WH. Quantitative and qualitative analysis of cellular prion protein (PrP(C)) expression in bovine somatic tissues. Prion. 2009;3(3):161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sales N, Rodolfo K, Hassig R, et al. Cellular prion protein localization in rodent and primate brain. Eur J Neurosci. 1998;10(7):2464–2471. [DOI] [PubMed] [Google Scholar]
- 8.Hornemann S, Korth C, Oesch B, et al. Recombinant full-length murine prion protein, mPrP(23–231): purification and spectroscopic characterization. FEBS Lett. 1997;413(2):277–281. [DOI] [PubMed] [Google Scholar]
- 9.Aronoff-Spencer E, Burns CS, Avdievich NI, et al. Identification of the Cu2+ binding sites in the N-terminal domain of the prion protein by EPR and CD spectroscopy. Biochemistry. 2000;39(45):13760–13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burns CS, Aronoff-Spencer E, Legname G, et al. Copper coordination in the full-length, recombinant prion protein. Biochemistry. 2003;42(22):6794–6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chattopadhyay M, Walter ED, Newell DJ, et al. The octarepeat domain of the prion protein binds Cu(II) with three distinct coordination modes at pH 7.4. J Am Chem Soc. 2005;127(36):12647–12656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Millhauser GL. Copper and the prion protein: methods, structures, function, and disease. Annu Rev Phys Chem. 2007;58:299–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walter ED, Stevens DJ, Visconte MP, Millhauser GL. The prion protein is a combined zinc and copper binding protein: Zn2+ alters the distribution of Cu2+ coordination modes. J Am Chem Soc. 2007;129(50):15440–15441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watt NT, Taylor DR, Kerrigan TL, et al. Prion protein facilitates uptake of zinc into neuronal cells. Nat Commun. 2012;3:1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.You H, Tsutsui S, Hameed S, et al. Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-Daspartate receptors. Proc Natl Acad Sci USA. 2012;109(5):1737–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasperini L, Meneghetti E, Pastore B, et al. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal. 2015;22(9):772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pushie MJ, Pickering IJ, Martin GR, et al. Prion protein expression level alters regional copper, iron and zinc content in the mouse brain. Metallomics. 2011;3(2):206–214. [DOI] [PubMed] [Google Scholar]
- 18.Evans EGB, Millhauser GL. Copper- and zinc-promoted interdomain structure in the prion protein: a mechanism for autoinhibition of the neurotoxic N-terminus. Prog Mol Biol Transl Sci. 2017;150:35–56. [DOI] [PubMed] [Google Scholar]
- 19.Wu B, McDonald AJ, Markham K, et al. The N-terminus of the prion protein is a toxic effector regulated by the C-terminus. Elife. 2017;6 10.7554/eLife.23473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spevacek A, Evans E, Miller J, et al. Zinc drives a tertiary fold in the prion protein with familial disease mutation sites at the interface. Structure. 2013;21(2):236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans EG, Pushie MJ, Markham KA, Lee HW, Millhauser GL. Interaction between prion protein’s copper-bound octarepeat domain and a charged C-terminal pocket suggests a mechanism for N-terminal regulation. Structure. 2016;24(7):1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thakur AK, Srivastava AK, Srinivas V, Chary KV, Rao CM. Copper alters aggregation behavior of prion protein and induces novel interactions between its N- and C-terminal regions. J Biol Chem. 2011;286(44):38533–38545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonald AJ, Leon DR, Markham KA, et al. Altered domain structure of the prion protein caused by Cu(2+) binding and functionally relevant mutations: analysis by cross-linking, MS/MS, and NMR. Structure. 2019;27(6):907–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez J, Sanchez R, Castellanos M, et al. PrP charge structure encodes interdomain interactions. Sci Rep. 2015;5:13623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Markham KA, Roseman GP, Linsley RB, Lee HW, Millhauser GL. Molecular features of the Zn(2+) binding site in the prion protein probed by (113)Cd NMR. Biophys J. 2019;116(4):610–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sonati T, Reimann RR, Falsig J, et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature. 2013;501(7465):102–106. [DOI] [PubMed] [Google Scholar]
- 27.Herrmann US, Sonati T, Falsig J, et al. Prion infections and anti-PrP antibodies trigger converging neurotoxic pathways. PLoS Pathog. 2015;11(2):e1004662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coleman BM, Harrison CF, Guo B, et al. Pathogenic mutations within the hydrophobic domain of the prion protein lead to the formation of protease-sensitive prion species with increased lethality. J Virol. 2014;88(5):2690–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schatzl HM, Da Costa M, Taylor L, Cohen FE, Prusiner SB. Prion protein gene variation among primates. J Mol Biol. 1995;245(4):362–374. [DOI] [PubMed] [Google Scholar]
- 30.Liang J, Kong Q. alpha-Cleavage of cellular prion protein. Prion. 2012;6(5):453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDonald AJ, Dibble JP, Evans EG, Millhauser GL. A new paradigm for enzymatic control of alpha-cleavage and beta-cleavage of the prion protein. J Biol Chem. 2014;289(2):803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Küffer A, Lakkaraju AK, Mogha A, et al. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature. 2016;536(7617):464–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westergard L, Turnbaugh JA, Harris DA. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J Biol Chem. 2011;286(51):44234–44242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J Biol Chem. 2010;285(34):26377–26383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457(7233):1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shmerling D, Hegyi I, Fischer M, et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998;93(2):203–214. [DOI] [PubMed] [Google Scholar]
- 37.Baumann F, Tolnay M, Brabeck C, et al. Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J. 2007;26(2):538–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christensen HM, Dikranian K, Li A, et al. A highly toxic cellular prion protein induces a novel, nonapoptotic form of neuronal death. Am J Pathol. 2010;176(6):2695–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li A, Christensen HM, Stewart LR, et al. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007;26(2):548–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Westergard L, Christensen HM, Harris DA. The cellular prion protein (PrP(C)): its physiological function and role in disease. Biochim Biophys Acta. 2007;1772(6):629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christensen HM, Harris DA. A deleted prion protein that is neurotoxic in vivo is localized normally in cultured cells. J Neurochem. 2009;108(1):44–56. [DOI] [PubMed] [Google Scholar]
- 42.Solomon IH, Huettner JE, Harris DA. Neurotoxic mutants of the prion protein induce spontaneous ionic currents in cultured cells. J Biol Chem. 2010;285(34):26719–26726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Massignan T, Biasini E, Harris DA. A drug-based cellular assay (DBCA) for studying cytotoxic and cytoprotective activities of the prion protein: a practical guide. Methods. 2011;53(3):214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solomon IH, Khatri N, Biasini E, et al. An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J Biol Chem. 2011;286(16):14724–14736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biasini E, Turnbaugh JA, Massignan T, et al. The toxicity of a mutant prion protein is cell-autonomous, and can be suppressed by wild-type prion protein on adjacent cells. PLoS ONE. 2012;7(3):e33472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biasini E, Unterberger U, Solomon IH, et al. A mutant prion protein sensitizes neurons to glutamate-induced excitotoxicity. J Neurosci. 2013;33(6):2408–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Massignan T, Stewart RS, Biasini E, et al. A novel, drug-based, cellular assay for the activity of neurotoxic mutants of the prion protein. J Biol Chem. 2010;285(10):7752–7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Westergard L, Turnbaugh JA, Harris DA. A nine amino acid domain is essential for mutant prion protein toxicity. J Neurosci. 2011;31(39):14005–14017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDonald AJ, Wu B, Harris DA. An inter-domain regulatory mechanism controls toxic activities of PrP(C). Prion. 2017;11(6):388–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turnbaugh JA, Westergard L, Unterberger U, Biasini E, Harris DA. The N-terminal, polybasic region is critical for prion protein neuroprotective activity. PLoS ONE. 2011;6(9):e25675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gibson DG, Young L, Chuang R-Y, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6(5):343–345. [DOI] [PubMed] [Google Scholar]
- 52.Mehrabian M, Brethour D, MacIsaac S, et al. CRISPR-Cas9-based knockout of the prion protein and its effect on the proteome. PLoS ONE. 2014;9(12):e114594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delaglio F, Grzesiek S, Vuister GeertenW, et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–293. [DOI] [PubMed] [Google Scholar]
- 54.Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera–avisualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. [DOI] [PubMed] [Google Scholar]
- 55.Oliveira-Martins JB, Yusa SI, Calella AM, et al. Unexpected tolerance of alpha-cleavage of the prion protein to sequence variations. PLoS ONE. 2010;5(2):e9107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65(10):1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liang J, Wang W, Sorensen D, et al. Cellular prion protein regulates its own alpha-cleavage through ADAM8 in skeletal muscle. J Biol Chem. 2012;287(20):16510–16520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fioriti L, Quaglio E, Massignan T, et al. The neurotoxicity of prion protein (PrP) peptide 106–126 is independent of the expression level of PrP and is not mediated by abnormal PrP species. Mol Cell Neurosci. 2005;28(1):165–176. [DOI] [PubMed] [Google Scholar]
- 59.Sangeetham SB, Huszár K, Bencsura P, et al. Interrogating the dimerization interface of the prion protein via site-specific mutations to p-benzoyl-L-phenylalanine. J Mol Biol. 2018;430(17):2784–2801. [DOI] [PubMed] [Google Scholar]
- 60.Wüsten KA, Reddy PP, Smiyakin A, et al. A bioluminescent cell assay to quantify prion protein dimerization. Sci Rep. 2018;8(1):14178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rambold AS, Müller V, Ron U, et al. Stress-protective signal-ling of prion protein is corrupted by scrapie prions. EMBO J. 2008;27(14):1974–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Solomon IH, Biasini E, Harris DA. Ion channels induced by the prion protein: mediators of neurotoxicity. Prion. 2012;6(1):40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McDonald AJ, Millhauser GL. PrP overdrive: does inhibition of alpha-cleavage contribute to PrP(C) toxicity and prion disease? Prion. 2014; 8(2):183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yusa S-I, Oliveira-Martins JB, Sugita-Konishi Y, et al. Cellular prion protein: from physiology to pathology. Viruses. 2012;4(11):3109–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stys PK, You H, Zamponi GW. Copper-dependent regulation of NMDA receptors by cellular prion protein: implications for neurodegenerative disorders. J Physiol. 2012;590(6):1357–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goniotaki D, Lakkaraju AKK, Shrivastava AN, et al. Inhibition of group-I metabotropic glutamate receptors protects against prion toxicity. PLoS Pathog. 2017;13(11):e1006733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Um JW, Kaufman AC, Kostylev M, et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron. 2013;79(5):887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aguzzi A, Baumann F, Bremer J. The prion’s elusive reason for being. Annu Rev Neurosci. 2008;31:439–477. [DOI] [PubMed] [Google Scholar]
- 69.Minikel EV, Vallabh SM, Lek M, et al. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. 2016;8(322):322ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Collins S, McLean CA, Masters CL. Gerstmann-Straussler-Scheinker syndrome, fatal familial insomnia, and kuru: a review of these less common human transmissible spongiform encephalopathies. J Clin Neurosci. 2001;8(5):387–397. [DOI] [PubMed] [Google Scholar]
- 71.Kitamoto T, Tateishi J, Tashima T, et al. Amyloid plaques in Creutzfeldt-Jakob disease stain with prion protein antibodies. Ann Neurol. 1986;20(2):204–208. [DOI] [PubMed] [Google Scholar]
- 72.Masters CL, Gajdusek DC, Gibbs CJ Jr. Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Straussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus-induced spongiform encephalopathies. Brain. 1981;104(3):559–588. [DOI] [PubMed] [Google Scholar]