

Abstract
Cholesterol 25-hydroxylase (CH25H) is an interferon-stimulated gene that converts cholesterol to the oxysterol 25-hydroxycholesterol (25HC). Circulating 25HC modulates essential immunological processes including antiviral immunity, inflammasome activation and antibody class switching; and dysregulation of CH25H may contribute to chronic inflammatory disease and cancer. Although 25HC is a potent regulator of cholesterol storage, uptake, efflux and biosynthesis, how these metabolic activities reprogram the immunological state of target cells remains poorly understood. Here, we used recently designed toxin-based biosensors that discriminate between distinct pools of plasma membrane cholesterol to elucidate how 25HC prevents Listeria monocytogenes from traversing the plasma membrane of infected host cells. The 25HC-mediated activation of acyl-CoA:cholesterol acyltransferase (ACAT) triggered rapid internalization of a biochemically defined fraction of cholesterol, termed ‘accessible’ cholesterol, from the plasma membrane while having little effect on cholesterol in complexes with sphingomyelin. We show that evolutionarily distinct bacterial species, L. monocytogenes and Shigella flexneri, exploit the accessible pool of cholesterol for infection and that acute mobilization of this pool by oxysterols confers immunity to these pathogens. The significance of this signal-mediated membrane remodelling pathway probably extends beyond host defence systems, as several other biologically active oxysterols also mobilize accessible cholesterol through an ACAT-dependent mechanism.
The mucosal epithelium functions as a primary physical barrier that separates microbial communities from deep tissues. If this barrier is breached, a dedicated array of innate immune cells stimulate inflammatory responses to clear local infections. Of particular importance to the mucosal immune system is the multipotent cytokine type II interferon (IFN-γ). Genetic deficiencies in either IFN-γ or its receptor, IFNGR, increase host susceptibility to infection by Listeria monocytogenes, Mycobacterium tuberculosis and Salmonella enterica1–3. IFN-γ-activated macrophages induce a complex transcriptional profile consisting of hundreds of IFN-stimulated genes (γ-ISGs), many of which promote bactericidal functions1. Due to the proximity of macrophages to epithelial barrier tissues in the mucosa, it is plausible that some γ-ISGs produce molecules that enhance epithelial-barrier immunity to invading pathogens when secreted. However, the pathways of direct communication between IFN-γ-activated macrophages and epithelial cells remain poorly characterized.
L. monocytogenes is an opportunistic pathogen that frequently contaminates food products. Following ingestion, L. monocytogenes penetrates the mucosal membranes of the gut and disseminates to distal organs such as the liver or spleen. Tissue transmission results in listeriosis, a disease associated with high mortality rates in immunocompromised and susceptible individuals. In addition to being a significant health burden, L. monocytogenes has been used as a model pathogen to study the innate and adaptive immune responses to bacterial infection4,5. Here, we used L. monocytogenes as a model enteric pathogen to determine whether a subset of γ-ISGs produce soluble molecules that enhance the cell-intrinsic defence mechanisms of the barrier epithelium when secreted by macrophages. High-throughput complementary DNA screening revealed that the expression of Cholesterol 25-hydroxylase (CH25H) and secretion of its product, 25-hydroxycholesterol (25HC), protected target epithelial cells from L. monocytogenes infection. Unexpectedly, secreted 25HC acutely remodelled cholesterol accessibility at the epithelial cell surface, thereby preventing cell-to-cell transmission of L. monocytogenes. We further show that a subset of structurally related oxysterols mobilize plasma membrane (PM) ‘accessible’ cholesterol, revealing a signalling pathway that can rapidly alter the lipid profile at the surface of target cells. These studies uncover an unexpected mechanism that links type II IFN signalling and cholesterol metabolism. Our findings may have broad implications for the role of circulating oxysterols on the mammalian immune system.
Results
Identification of CH25H as a potent antibacterial component of the type II IFN response pathway.
We sought to determine whether IFN-γ-activated macrophages express extrinsic factors that, when secreted, protect barrier tissues from bacterial infection in trans. To test this idea, primary bone marrow-derived macrophages (BMDMs) were treated with murine IFN-γ (mIFN-γ) and the conditioned medium from these cells was transferred to a monolayer of naive HEK293A epithelial cells (Fig. 1a). The HEK293A cells were then infected with a low dose of green fluorescent protein (GFP)-expressing L. monocytogenes, which models the single-cell infection conditions observed in physiological settings (Extended Data Fig. 1a–c). Remarkably, HEK293A cells exposed to mIFN-γ-conditioned media were highly resistant to L. monocytogenes infection compared with cells exposed to vehicle-conditioned media (Fig. 1b,c). We concluded that activated macrophages secrete an antibacterial molecule, as direct application of mIFN-γ to HEK293A cells had no effect on their permissiveness to infection (Fig. 1d).
Fig. 1 |. IFN-γ-activated BMDMs secrete an antibacterial factor.
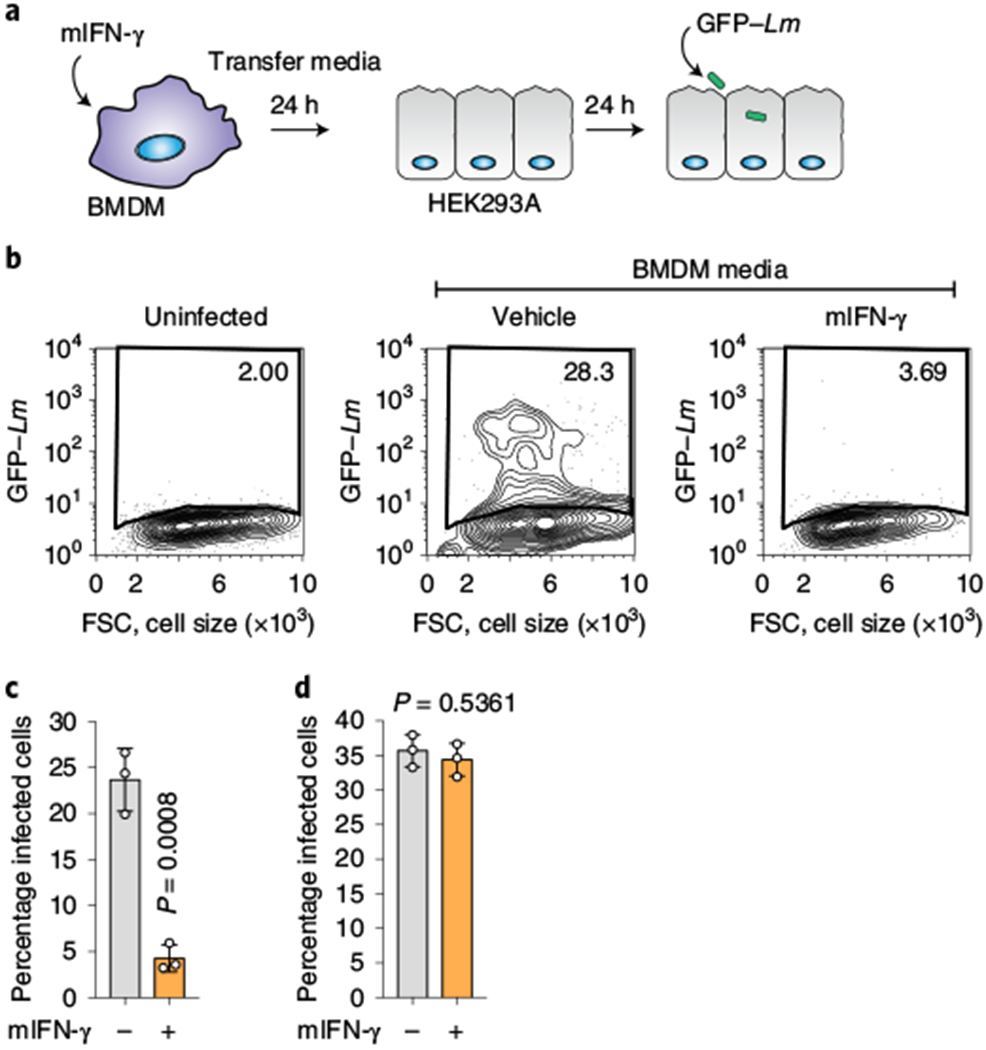
a, Schematic of the medium-transfer assay designed to investigate antibacterial products produced and secreted by mIFN-γ-stimulated BMDMs. b, HEK293A cells cultured in conditioned medium from mIFN-γ-stimulated BMDMs were infected with GFP-expressing L. monocytogenes (multiplicity of infection (m.o.i.) = 2; 22 h) as indicated. The flow cytometry plots show the percentage of GFP-positive HEK293A cells. FSC, forward scatter. c, Quantification of the assay described in b (BMDM-conditioned medium). d, HEK293A cells were not affected by residual mIFN-γ present in BMDM-conditioned media. HEK293A cells were treated with 500 U ml−1 mIFN-γ for 24 h and infected with GFP-expressing L. monocytogenes the next day (m.o.i. = 2; 22 h). Infection was quantified by flow cytometry as in c. c,d, The bars represent the mean values. The error bars show the s.d. from three independent experiments and statistical significance was determined using a Student’s unpaired t-test (two-tailed). Lm, L. monocytogenes.
A gain-of-function screening platform was designed to identify γ-ISGs that produce soluble antibacterial molecules (Fig. 2). After evaluating the transcriptional response of cells and tissues stimulated with IFN-γ (see Methods), we cloned 414 highly representative γ-ISGs into a lentiviral reporter vector (Fig. 2a). STAT1-deficient human skin fibroblasts (SDFs; a cell type that does not respond to IFN, thereby eliminating spurious activation of ISGs) were transduced with lentivirus in a one-gene to one-well format. The SDF cells were then infected with GFP-expressing L. monocytogenes and subsequently evaluated using multi-dimensional flow cytometry (Fig. 2a). Eight γ-ISGs exhibited potent antibacterial activity, the strongest effect being due to the expression of CH25H, which suppressed L. monocytogenes infection by 78.5% (Fig. 2b,c). In a secondary analysis, we found that media collected from CH25H-expressing SDF cells, and to a lesser extent from MYD88-expressing cells, protected naive SDF cells from L. monocytogenes infection (Fig. 2d). We chose to focus on the cell-extrinsic mechanism of CH25H given that this gene is induced by both IFNs and TLR-dependent signalling pathways6–8 and because CH25H-conditioned medium protected cells from L. monocytogenes infection (Fig. 2d) to a similar extent as conditioned medium from mIFN-γ-stimulated macrophages (Fig. 1c).
Fig. 2 |. Functional cDNA screen of γ-ISGs identifies CH25H as an inhibitor of L. monocytogenes infection.
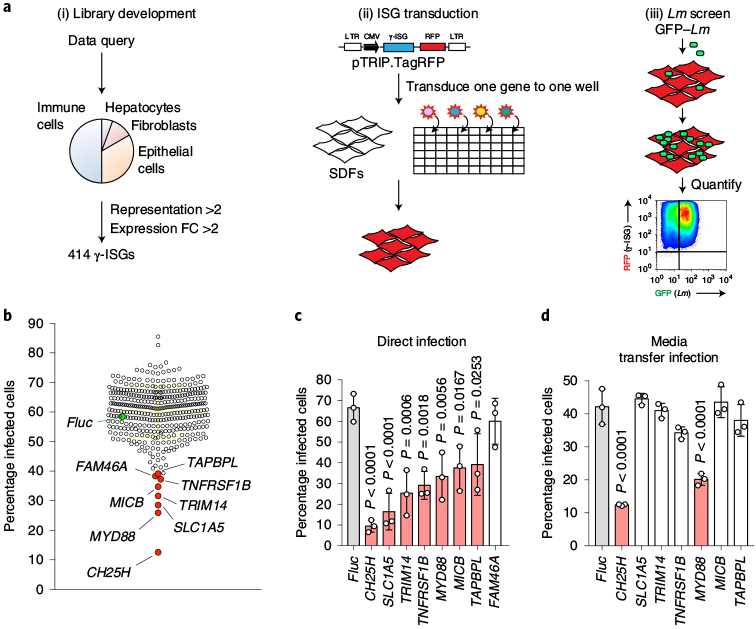
a, Schematic of the γ-ISG lentiviral library development and screening strategy for antibacterial γ-ISGs. SDFs were transduced for 48 h and then infected with GFP-tagged L. monocytogenes (m.o.i. = 5; 8 h). FC, fold change; LTR, long terminal repeat; CMV, cytomegalovirus; RFP, red fluorescent protein. b, Level of L. monocytogenes infection in the presence of overexpressed γ-ISG. Two replicate screens were performed and the average infection level for each ISG is shown. The horizontal yellow line represents the mean and the error bars show the s.d. Inhibitory γ-ISGs are indicated in red, control firefly luciferase (Fluc) is indicated in green. c, Lentiviral stocks of the genes identified in b were reproduced and the antibacterial activity was assessed. SDFs were transduced with each lentivirus and then infected with GFP-expressing L. monocytogenes (m.o.i. = 10; 6 h) after 48 h. d, To determine whether the confirmed inhibitors of L. monocytogenes infection from c possessed secreted antibacterial activity, SDFs were transduced with each ISG for 48 h, and naive SDFs were bathed in conditioned media and subsequently infected with GFP-expressing L. monocytogenes (m.o.i. = 5; 6 h). c,d, Infection was quantified by flow cytometry. The bars represent the mean values. The error bars show the s.d. from three independent experiments and statistical significance in comparison to Fluc was determined using a one-way analysis of variance (ANOVA) with Dunnett’s correction.
25HC potently inhibits Listeria infection of epithelial cells.
CH25H is an endoplasmic reticulum (ER)-localized enzyme that hydroxylates the iso-octyl side chain of cholesterol, producing 25HC (Fig. 3a)9. This gene is expressed at low levels in naive macrophages and is potently induced by IFN-γ8. We observed a substantial increase in the level of 25HC in the media collected from mIFN-γ-stimulated BMDMs (Fig. 3b). In contrast, 25HC was not secreted from activated BMDMs generated from Ch25h−/− mice and no other oxysterols were differentially expressed in these samples.
Fig. 3 |. 25HC inhibits L. monocytogenes infection in local tissue environments.
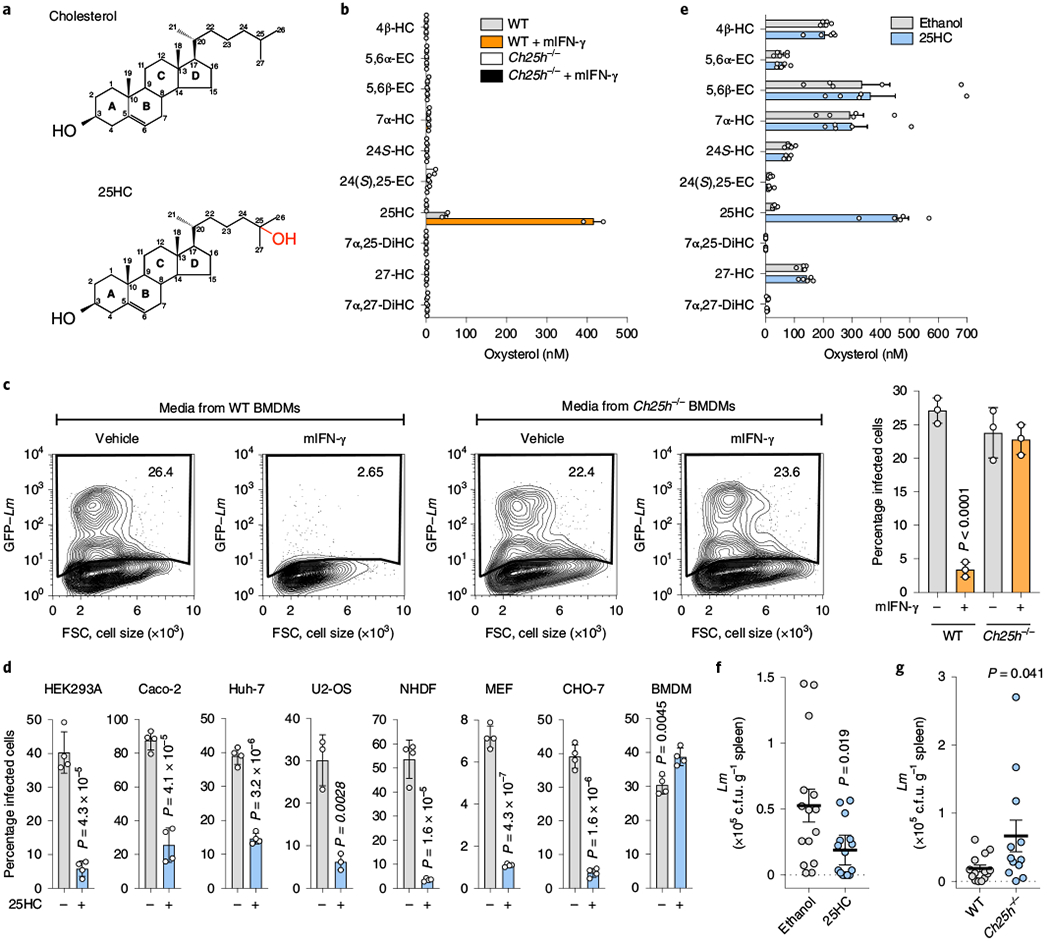
a, Chemical structures of cholesterol (top) and 25HC (bottom). b, Concentration of the indicated oxysterols in media collected from WT or Ch25h−/− BMDMs treated with either mIFN-γ or vehicle for 24 h. The bars represent the mean values. The error bars show the s.d. from two independent experiments. See Methods for the oxysterol nomenclature. c, HEK293A cells were cultured in media collected from WT or Ch25h−/− BMDMs treated with mIFN-γ or vehicle as described in Fig. 1a and infected as in Fig. 1b. Left, representative flow cytometry plots showing the percentage of infected cells. Flow cytometry plots are representative of three independent experiments. Right, percentage of infected cells determined from three independent flow cytometry experiments. The bars represent the mean values. The error bars show the s.d. and statistical significance was determined using a Student’s unpaired t-test (two-tailed). d, Quantification of L. monocytogenes infection of immortalized cell lines, primary normal human dermal fibroblasts (NHDFs) and BMDMs treated with 25HC. See Methods for the infection conditions. The bars represent the mean values. The error bars show the s.d. from four independent experiments for all except U2-OS (n = 3) and statistical significance was determined using a Student’s unpaired t-test (two-tailed). e, Concentration of the indicated oxysterols in the serum collected from ethanol- (vehicle, n = 5) or 25HC-treated (n = 5) mice as described in f. The bars represent the mean values. The error bars show the s.e.m. f, Mice injected with either ethanol (vehicle, n = 15) or 25HC (n = 15) in the peritoneal cavity and infected with L. monocytogenes by oral gavage as described in Methods. Bacterial tissue transmission was determined 72 h post-infection by enumerating the bacterial colony-forming units (c.f.u.) recovered from whole-spleen homogenates. g, Ch25h−/− (n = 12) and WT (n = 14) mice were orally infected with L. monocytogenes as described in Methods. Bacterial tissue transmission was determined as in f. f,g, The horizontal lines represents the mean. The error bars are the s.e.m. and statistical significance was determined using a Student’s unpaired t-test (two-tailed).
Several lines of evidence indicated that 25HC was the sought-after antibacterial molecule. First, unlike the case of wild-type (WT) BMDMs, media collected from mIFN-γ-stimulated Ch25h−/− BMDMs failed to suppress L. monocytogenes infection of HEK293A cells in trans (Fig. 3c). Second, 25HC was secreted from cells ectopically expressing CH25H but not the catalytically inactive H242Q/H243Q mutant9, and consequently the expression of CH25HH242Q/H243Q failed to inhibit L. monocytogenes infection (Extended Data Fig. 2a,b). Third, exogenous 25HC potently suppressed intracellular bacterial colonization of cells originating from diverse tissues and mammalian species, including those used to model L. monocytogenes infection of barrier tissues in humans (for example, Caco-2 intestinal epithelial cells and Huh-7 hepatocytes; Fig. 3d and Extended Data Fig. 2c). Unlike its protective effect on epithelial cells, 25HC rendered BMDMs more susceptible to infection when compared with vehicle-treated cells (Fig. 3d and Extended Data Fig. 2c), a finding that is in line with previous reports10–11. Thus, the autocrine (for example, macrophage) and paracrine (for example, epithelial cells) signalling functions of 25HC have diverse effects on the host defence systems. Finally, to determine whether 25HC indeed suppresses L. monocytogenes infection of mucosal tissues in vivo, 25HC was injected into mice, resulting in a transient increase of circulating 25HC from 31.7 ± 3.41 nM to 457 ± 39.0 nM, with no quantifiable effect on other oxysterols (Fig. 3e). After the vehicle- and 25HC-treated mice were infected with L. monocytogenes by oral administration, the bacteria readily disseminated from the gut mucosa to systemic sites in vehicle-treated control mice (Fig. 3f); however, the bacterial burden was reduced in mice treated with 25HC (Fig. 3f). Moreover, tissue dissemination of L. monocytogenes in orally infected Ch25h−/− mice was enhanced compared with the WT controls (Fig. 3g). Combined, these studies support the role of 25HC in conferring local antibacterial immunity.
25HC suppresses contact-dependent Listeria cell-to-cell spread.
The antibacterial activity of 25HC could not be explained by direct regulation of bacterial growth, inhibition of L. monocytogenes virulence factor expression or induction of mammalian cell death (Extended Data Fig. 3a–d). Furthermore, 25HC had little effect on the initial stages of L. monocytogenes infection, including cellular adhesion/invasion, vacuole escape, assembly of the actin polymerization machinery required for cytosolic motility and intracellular bacterial replication (Extended Data Fig. 4a–d). However, we noted that the incidence of membrane protrusions required for intracellular L. monocytogenes to cross cell-to-cell contact junctions was substantially reduced by 25HC (Fig. 4a,b and Supplementary Video 1). A foci-forming assay revealed that 25HC suppressed bacterial dissemination across a monolayer of host cells by 77.8 ± 5.1% compared with the vehicle control (Extended Data Fig. 4e). These findings were corroborated in donor-to-recipient infection assays, in which a small number of pre-infected HEK293A cells (donors) were seeded onto a monolayer of uninfected HEK293AΔMETΔCH1 cells (recipients), which lack the receptors c-Met and E-cadherin required for L. monocytogenes extracellular invasion12 (Fig. 4c). Notably, 25HC treatment shielded recipient cells from L. monocytogenes infection (Fig. 4d), resulting in a marked reduction in the total bacterial burden (Fig. 4e,f). Collectively, these studies indicate that 25HC protects uninfected epithelial cells from contact-dependent transmission of L. monocytogenes. This led us to explore the effects of 25HC on the PM of epithelial cells.
Fig. 4 |. 25HC restricts L. monocytogenes cell-to-cell dissemination.
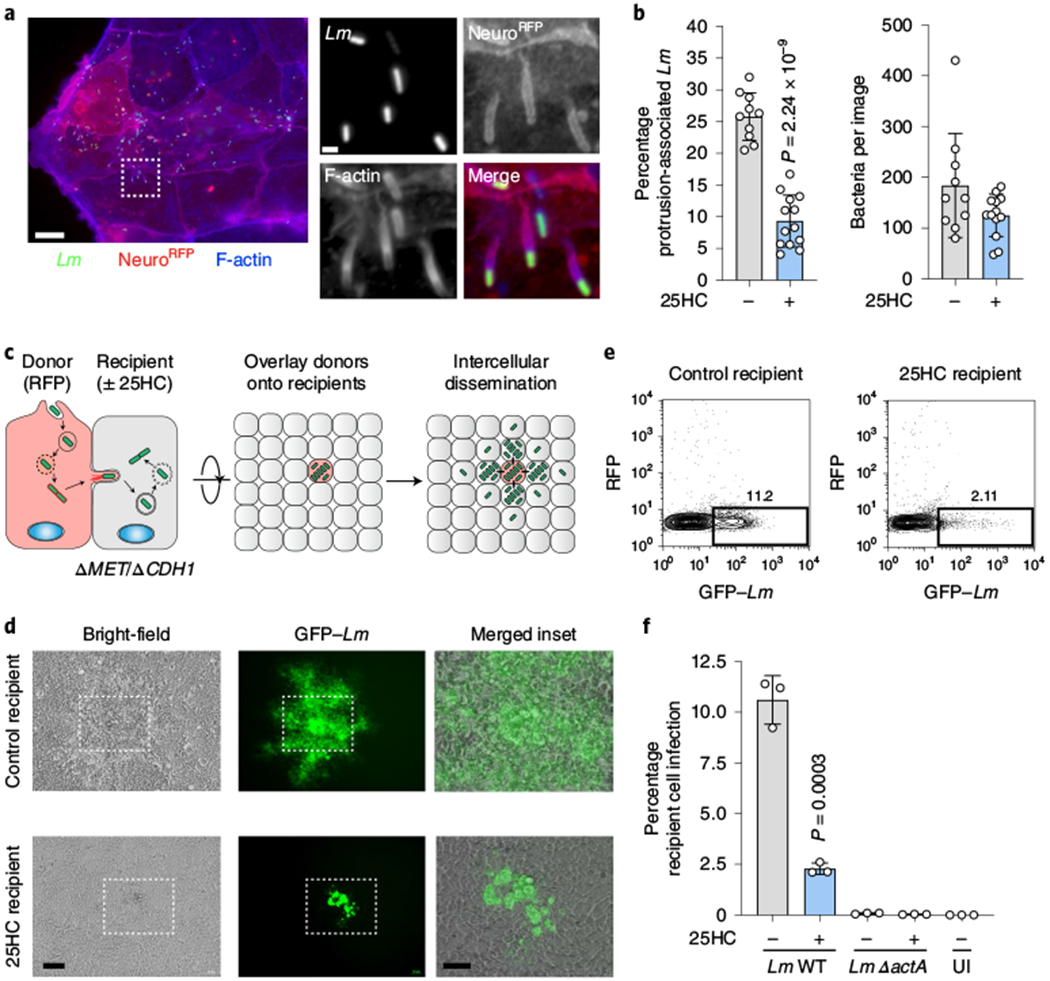
a,b, Representative images (a) and quantification (b) of membrane protrusions induced by L. monocytogenes infection of Caco-2 cells expressing NeuroRFP. Alexa Fluor 647 phalloidin staining was used to visualize F-actin. a, Scale bars, 20 μm (left) and 1 μm (right; magnifications of the region in the white box on the left). b, Left, frequency of host membrane protrusions per field of view collected across three independent experiments (vehicle treatment (−), n = 10 and 25HC, n = 13). Right, the total number of bacteria were similar between the two indicated treatments. The bars represent the mean values. The error bars show the s.d. and statistical significance was determined using a Student’s unpaired t-test (two-tailed). c, Schematic depicting the donor-to-recipient experiment. d, Representative images of recipient HEK293AΔMETΔCDH1 cells seeded with donor cells (WT HEK293A) infected with GFP-expressing L. monocytogenes as in c. The white boxes show the region of the merged inset (right). Scale bars, 100 μm (left and middle) and 50 μm (right). The cells were seeded at a donor:recipient ratio of 1:200. Images were acquired 24 h after donor-cell seeding. Images are representative of three independent experiments. e, Representative flow cytometry plots (n = 3) of donor (RFP-positive) and recipient (RFP-negative) cells infected with L. monocytogenes (GFP-positive). The percentage of L. monocytogenes infection in recipient cells (rectangular gate) is shown. f, Percentage of recipient cells infected, determined using the gating strategy in e. The bars represent the mean values. For the WT L. monocytogenes comparison (bars 1 and 2), the error bars show the s.d. from three independent experiments and statistical significance was determined using a Student’s unpaired t-test (two-tailed). Spread-deficient L. monocytogenes ΔactA served as a negative control and uninfected (UI) recipient cells are shown.
25HC rapidly mobilizes accessible cholesterol from the PM.
Oxysterols such as 25HC have long been known to regulate the cholesterol levels of the cell13–18. However, no significant change in total cellular cholesterol was detected in 25HC-treated cells over the time frame required to elicit an antibacterial response (Extended Data Fig. 5a). This observation prompted us to investigate whether 25HC regulates the organization of PM cholesterol, independent of new cholesterol synthesis, uptake or efflux. Although cholesterol is the most abundant PM lipid, the majority of PM cholesterol is sequestered by proteins and lipids such as sphingomyelin19–21. When the levels of PM cholesterol rise above a saturation point for lipid and protein sequestration (approximately 35 mol% (molar percentage) of total PM lipids), an accessible pool of cholesterol accumulates19–21. The abundance of accessible cholesterol and sphingomyelin-sequestered cholesterol in the PM can be monitored using domain 4 of anthrolysin O (ALOD4) encoded by Bacillus anthracis and ostreolysin A (OlyA) encoded by Pleurotus ostreatus—non-lytic toxins that exhibit all-or-none binding specificity for accessible cholesterol and sphingomyelin–cholesterol complexes, respectively21–23 (Fig. 5a). As expected, both the accessible cholesterol and sphingomyelin–cholesterol complexes were readily detected on the PMs of CHO-7 cells as well as several cell types used throughout this study (Fig. 5b, lane 1, and Extended Data Fig. 5b, lane 1 of each panel). Strikingly, although 25HC had only a minor effect on the sphingomyelin–cholesterol complexes, accessible cholesterol was severely depleted from the PM within 1 h of 25HC treatment and completely eliminated by 4 h (Fig. 5b, lanes 1–4). In contrast, 7α-HC—an oxysterol that inhibits cholesterol synthesis in a manner similar to 25HC13,15—had no effect on either cholesterol pool (Fig. 5b, lanes 5–8). Furthermore, 7α-HC was not detected in the media of mIFN-γ-activated BMDMs (Fig. 3b) and did not suppress L. monocytogenes infection (Extended Data Fig. 5c).
Fig. 5 |. 25HC reorganizes PM cholesterol.
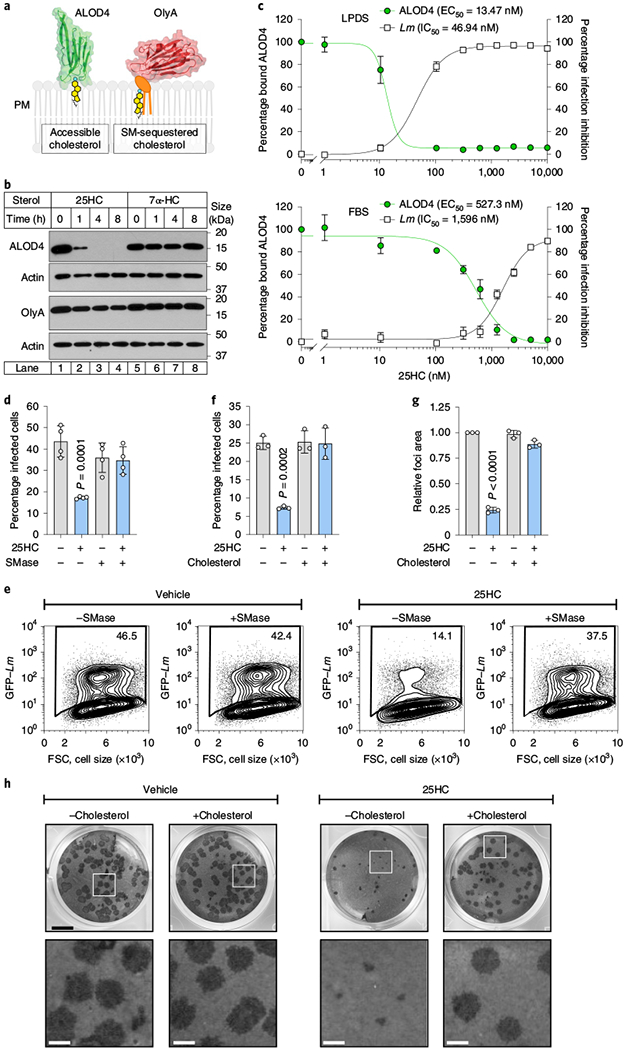
a, Structure and membrane-binding model for ALOD4 (left; residues 404–512 of ALO; Protein Data Bank: 3CQF61) and OlyA (right; Protein Data Bank: 6MYJ21), which bind to accessible cholesterol and sphingomyelin (SM)-sequestered cholesterol, respectively. b, Immunoblot showing ALOD4 and OlyA bound to the membranes of CHO-7 cells treated with 5μM of the indicated oxysterol. The blots are representative of three independent experiments. c, Percentage of ALOD4 bound to the membranes and percentage of L. monocytogenes inhibition in CHO-7 cells treated with the indicated concentrations of 25HC for 16 h in media supplemented with 5% LPDS (low lipoprotein content; upper graph) or 10% FBS (high lipoprotein content; lower graph). The mean values from three independent experiments are plotted and the error bars show the s.e.m. EC50, half-maximum effective concentration; IC50, half-maximum inhibitory concentration. d,e, Graph (d) and representative flow cytometry plots (e) showing the percentage of CHO-K1 cells infected with L. monocytogenes after treatment with vehicle (ethanol) or 25HC (5μM; 16 h) and SMase, as described in Methods. The bars represent the mean values. The error bars show the s.d. from four independent experiments and statistical significance in comparison to the vehicle was determined using a one-way ANOVA with Dunnett’s correction. f, Percentage of HEK293A cells infected with L. monocytogenes after treatment with vehicle (ethanol) or 25HC (5μM; 16 h). The treated cells were incubated with cholesterol-methyl-β-cyclodextrin (MCD) complexes (40 μM) for 1 h before infection with GFP-expressing L. monocytogenes (m.o.i. = 10; 6 h) and then analysed by flow cytometry. The bars represent the mean values. The error bars show the s.d. from three independent experiments and statistical significance in comparison to the vehicle was determined using a one-way ANOVA with Dunnett’s correction. g,h, Quantification (g) and representative images (h) of foci-forming assays showing the effect of cholesterol repletion on bacterial dissemination in HEK293A cells treated with 25HC (5μM). The foci areas for the experiment presented in g, h were quantified as in Extended Data Fig. 4e. The bars represent the mean values. The error bars show the s.d. from three independent experiments and statistical significance in comparison to the vehicle was determined before normalization using a one-way ANOVA with Dunnett’s correction. h, Scale bars, 5 mm (top) and 1mm (bottom; magnification of the white squares in the top images).
Listeria and Shigella infections are suppressed by the mobilization of accessible cholesterol.
A detailed dose-curve analysis revealed that 25HC induced a switch-like drop in the levels of accessible cholesterol on the PMs of CHO-7 cells grown in lipoprotein-deficient serum (LPDS) as well as in lipoprotein-rich serum (fetal bovine serum, FBS; Fig. 5c and Extended Data Fig. 5d). Furthermore, 25HC induced an equally sharp transition in cellular resistance to L. monocytogenes infection at the precise concentrations required to mobilize accessible cholesterol in either serum condition (Fig. 5c). The effective concentration of 25HC required to mobilize accessible cholesterol was approximately 40-fold higher in cells grown in FBS when compared with those grown in LPDS (Fig. 5c and Extended Data Fig. 5d). This strong correlation between PM cholesterol accessibility and host defence against bacterial infection suggested that elimination of accessible cholesterol is the rate-limiting step in cellular immunity driven by 25HC.
If accessible cholesterol is required for L. monocytogenes to traverse across the PM of infected cells and elimination of this pool by 25HC provides innate protection against intercellular dissemination, then restoring the accessible cholesterol on the PM of 25HC-treated cells should re-establish host permissiveness to bacterial infection. Incubation of cells with sphingomyelinase (SMase), an enzyme that cleaves sphingomyelin and disrupts sphingomyelin-cholesterol complexes19,21, generated new accessible cholesterol on the surface of 25HC-treated cells (Extended Data Fig. 5b). Although SMase had no effect on infection of untreated cells, it potently enhanced the susceptibility of 25HC-treated cells to L. monocytogenes infection (Fig. 5d,e). In agreement with these findings that bacteria exploited the new accessible cholesterol generated by SMase, delivery of exogenous cholesterol in cyclodextrin complexes to 25HC-treated cells also replenished the accessible cholesterol pool (Extended Data Fig. 5e) and restored L. monocytogenes dissemination (Fig. 5f–h).
In addition to regulating L. monocytogenes, an acute reduction in PM accessible cholesterol by 25HC inhibited the intercellular dissemination of Shigella flexneri, a Gram-negative bacterial pathogen that is the causative agent of shigellosis (Extended Data Fig. 6a,b). S. flexneri has a intracellular lifecycle that is analogous to L. monocytogenes, including escape from the phagocytic vacuole, actin-based motility and cell-to-cell spread via membrane protrusions24. Despite this similar pathogenic strategy, S. flexneri employs a distinct repertoire of virulence factors and escapes the phagocytic vacuole through a cytolysin-independent mechanism24. Cholesterol repletion restored S. flexneri dissemination of 25HC-treated cells, further providing evidence that the regulation of PM accessible cholesterol is broadly antimicrobial (Extended Data Fig. 6a,b).
Oxysterols mobilize accessible cholesterol and inhibit bacterial infection via acyl-CoA:cholesterol acyltransferase (ACAT) activation.
While we were led to the effect of 25HC on PM cholesterol remodelling through its immunological effects on the target epithelium, we reasoned that a survey of other biologically active oxysterols could reveal insights into the mechanism of cholesterol mobilization and pathogen suppression (Fig. 6a). Oxysterols harbouring hydroxyl groups on carbons 20, 25 or 27 of the iso-octyl side chain mobilized accessible cholesterol, whereas those with hydroxyl groups on the A-ring (4β-HC) or B-ring (7α-HC) did not affect the levels of accessible cholesterol in the PM (Fig. 6b). Notably, side-chain hydroxylation was not the sole determinant, as 22(R)-HC did not mobilize the accessible cholesterol (Fig. 6b). The differential activity of these oxysterols provided key insights into the specific signalling pathway driving accessible cholesterol mobilization. For example, 20α-HC, 25HC and 27HC potently inhibited proteolytic activation of SREBP2, the master transcriptional regulator of cholesterol uptake and biosynthesis25 (Fig. 6c). However, acute inhibition of SREBP2 was not the initiating signal for rapid PM cholesterol remodelling as two of the oxysterols that had no effect on accessible cholesterol abundance on PMs, 7α-HC and 22(R)-HC potently inhibited SREBP2 activation (Fig. 6c). In addition, 7α-HC and 22(R)-HC activate liver-X-receptor (LXRα/β)26, making it unlikely that the cholesterol efflux pathways controlled by this transcription factor initiate cholesterol remodelling under these conditions.
Fig. 6 |. Side-chain oxysterols mobilize accessible cholesterol through a concerted mechanism.
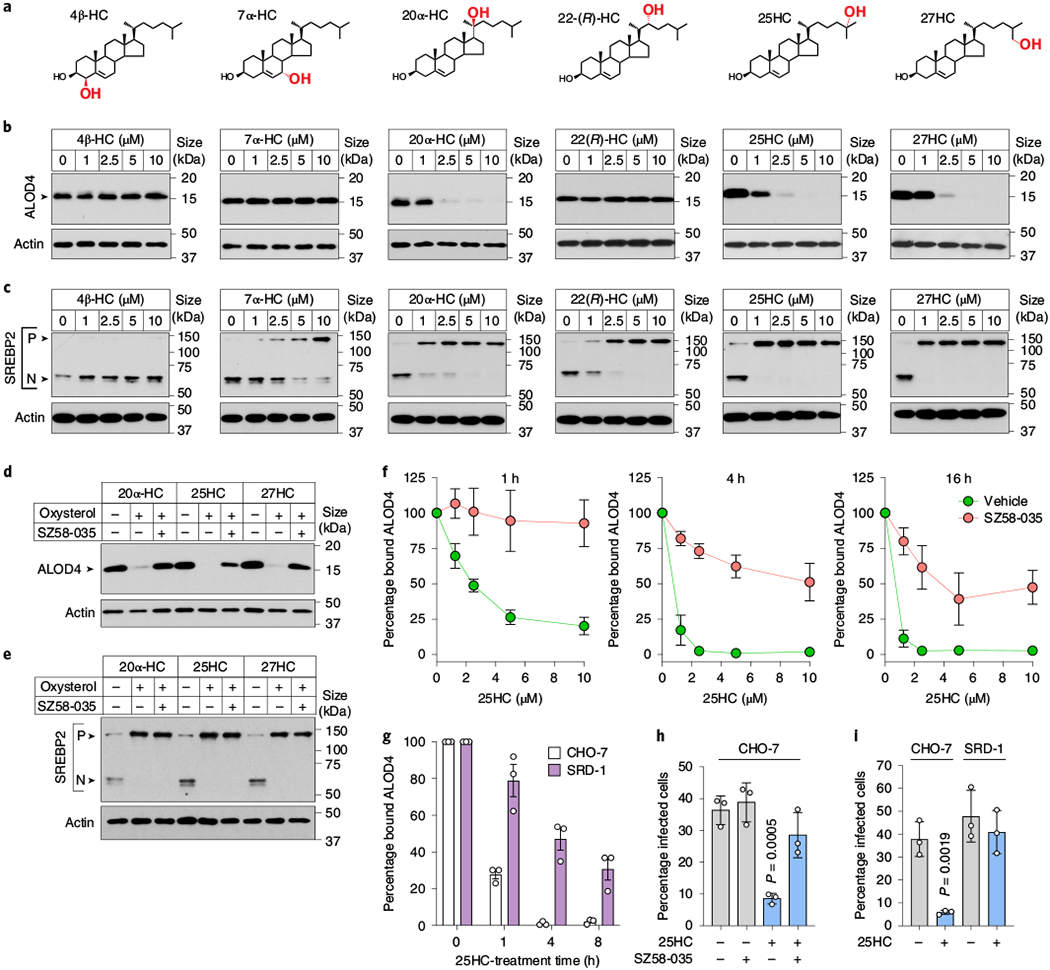
a, Oxysterol structures. b, Immunoblot showing ALOD4 bound to membranes of CHO-K1 cells treated with different concentrations of the indicated oxysterol. Immunoblots are representative of three independent experiments. c, Immunoblot showing SREBP2 processing in sterol-depleted CHO-K1 cells, which were incubated with increasing concentrations of the indicated oxysterols for 4 h. P, precursor form of SREBP2; N, nuclear form of SREBP2. Immunoblots are representative of three independent experiments. d,e, Immunoblots showing ALOD4 bound to the membrane (d) or SREBP2 processing (e) in CHO-K1 cells treated with 10 μM SZ58-035 or vehicle (dimethylsulfoxide) before the addition of the indicated oxysterols as described in b,c. See Methods for additional experimental details. Blots are representative of three independent experiments. f, Quantification of ALOD4 binding to membranes in CHO-7 cells pretreated with 10 μM SZ58-035 or vehicle (dimethylsulfoxide) and treated with the specified concentration of 25HC for the indicated times (1-h 25HC treatment, n = 4; 4-h treatment, n = 3; 16-h treatment, n = 3 except the 10 μM data point where n = 2) the following day. The mean values are plotted and the error bars show the s.e.m. g, Percentage of ALOD4 bound to the membranes of CHO-7 and SRD-1 cells treated with 25HC (5μM) for the indicated times. The bars represent the mean values. The error bars show the s.d. from three independent experiments. h, Percentage of L. monocytogenes-infected CHO-7 cells pretreated with SZ58-035 (10μM) and 25HC (5μM) as described in Methods. The bars represent the mean values. The error bars show the s.d. from three independent experiments and statistical significance in comparison to the vehicle was determined using a one-way ANOVA with Dunnett’s correction. i, Percentage of L. monocytogenes-infected CHO-7 and SRD-1 cells pretreated with 25HC (5μM) as described in Methods. The bars represent the mean values. The error bars show the s.d. from three independent experiments and statistical significance was determined using a Student’s unpaired t-test (two-tailed). h,i, The infection levels were assessed by flow cytometry.
Having eliminated the SREBP2 and LXR pathways as the initiating signal for PM cholesterol remodelling by oxysterols, we focused on the cholesterol storage pathway mediated by ACAT—a membrane-bound enzyme that converts cholesterol in the ER to cholesteryl esters16,27,28. The selective ACAT inhibitor Sandoz 58-035 (SZ58-035; ref. 29) prevented the elimination of accessible cholesterol by 20α-HC, 25HC and 27HC (Fig. 6d). SZ58-035 also inhibited 25HC from inducing cholesterol-rich lipid droplets, suggesting that esterification of cholesterol in the ER triggers rapid cholesterol remodelling on the cell surface (Extended Data Fig. 7a,b). Importantly, SZ58-035 had no effect on the ability of 20α-HC, 25HC and 27HC to suppress SREBP2 activation (Fig. 6e) and alone it did not affect SREBP2 activity in cholesterol-replete or cholesterol-depleted cells, which is consistent with the specificity of this inhibitor (Extended Data Fig. 7c).
Although ACAT inhibition with SZ58–035 prevented rapid internalization of accessible cholesterol even by the highest concentration of 25HC (10 μM; 1-h treatment; Fig. 6f), we noted that some accessible cholesterol was lost from the PM under ACAT-inhibitory conditions over longer periods of 25HC incubation (for example, 16 h; Fig. 6f). We speculated that the loss of accessible cholesterol over longer time periods may be due to 25HC-mediated suppression of SREBP2 (ref. 25), a slower-acting transcriptional response that may prevent the replenishment of PM accessible cholesterol with newly synthesized or lipoprotein-derived cholesterol. In support of this idea, mobilization of PM accessible cholesterol by 25HC was much slower in SRD-1 cells that constitutively express the transcriptionally active fragment of SREBP2 (note that SRD-1 cells are refractory to 25HC-mediated inhibition of the cholesterol uptake and biosynthesis machinery30; Fig. 6g and Extended Data Fig. 7d). Together, these studies reveal a concerted mechanism for oxysterol-mediated remodelling of the PM lipid composition through rapid activation of ACAT and a sustained response through suppression of SREBP2. Importantly, this mechanism is supported by studies on the epithelial cell host defence system, as both ACAT inhibition by SZ58-035 and constitutive expression of nuclear SREBP2 in SRD-1 cells suppressed the antibacterial activity of 25HC (Fig. 6h,i). In contrast, clustered regularly interspaced short palindromic repeats (CRISPR)-mediated deletion of LXRα and LXRβ had no effect on the dissemination of L. monocytogenes in 25HC-producing cells, further highlighting rapid stimulation of ACAT as the primary mechanism for elimination of accessible PM cholesterol (Extended Data Fig. 8a–c).
Discussion
We have identified a previously unknown immuno-metabolic signalling pathway that enhances epithelial cell barrier resistance to infection by two evolutionarily distinct bacterial pathogens (Extended Data Fig. 9). Specifically, 25HC produced by IFN-γ-activated macrophages functions through a paracrine signalling mechanism to inhibit the dissemination of L. monocytogenes across epithelial cell junctions. Among its many targets, the primary mode of action of 25HC in this particular immune context is through the allosteric stimulation of ACAT, an enzyme in the ER that converts cholesterol to cholesteryl esters for storage in lipid droplets. Activation of ACAT triggers the rapid internalization of accessible cholesterol from the cell surface by transport mechanisms that are still not well understood. Nevertheless, we show that cell surface accessible cholesterol is essential for both L. monocytogenes and S. flexneri to penetrate adjacent cells and that acute reduction of this cholesterol pool from the PM underlies the antibacterial activity of 25HC. These mechanistic insights reveal how IFN-γ-activated macrophages may protect mucosal barrier tissues from local microbial infection. More generally, acute remodelling of accessible cholesterol in PMs by oxysterols may impact a broad range of cellular signalling activities, immunological processes and human disease conditions31–35.
How the loss of accessible cholesterol protects against bacterial infection is an open question. We suspect that changes in accessible cholesterol may alter the function of integral membrane proteins required for the formation or resolution of PM protrusions induced by L. monocytogenes and S. flexneri. Alternatively, a loss of accessible cholesterol from the PM may affect cellular signalling pathways that control membrane dynamics or rigidity. It is worth noting that CH25H has been implicated in numerous immunological processes including antiviral immunity8,36, inflammasome activation10,11 and antibody class switching6; and dysregulation of 25HC production may contribute to chronic inflammatory disease37 and cancer38. It is tempting to speculate that 25HC may be controlling these diverse immunological responses at least in part through the mobilization of accessible cholesterol from the cell surface.
It has not escaped our attention that 25HC could inhibit the pore-forming activity of cholesterol-dependent cytolysins, which require accessible cholesterol for membrane interactions22,39. It is therefore noteworthy that 25HC had no effect on L. monocytogenes escape from the phagocytic vacuole, a process that requires the well-characterized cholesterol-dependent cytolysin listeriolysin O (Extended Data Fig. 4b). These studies suggest that 25HC does not directly affect the cholesterol content of the phago-lysosomal system. Consistent with this idea, an acute reduction in PM accessible cholesterol by 25HC also inhibited the intercellular dissemination of S. flexneri, which utilizes effector proteins other than cholesterol-dependent cytolysins to escape the vacuole. Our current data suggest that changes in accessible cholesterol induced by 25HC may be restricted to the PM.
Previous studies showed that the expression of CH25H enhances the permissiveness of macrophages to L. monocytogenes infection10,11, a result that was independently confirmed here (Fig. 3d and Extended Data Fig. 2c). Our finding that the secretion of 25HC from IFN-γ-activated macrophages potently suppresses the transmission of L. monocytogenes across epithelial cell junctions therefore expands the immunological function of CH25H beyond its established role in immune cells. However, it is important to note that although the ability of 25HC to inhibit intracellular transmission of L. monocytogenes was quite robust in vitro, Ch25h−/− mice exhibited only a modest increase in bacterial burden after oral inoculation (Fig. 3g). Because IFN-γ induces hundreds of γ-ISGs, it is unlikely that the loss of a single gene would completely ablate the host type II IFN response to pathogens. Further studies will be needed to clarify the specific role of CH25H in IFN-γ-mediated mucosal immunity to enteric pathogens.
Methods
Mammalian cell culture.
Immortalized human SDFs (J.-L. Casanova, Rockefeller University) were cultured in RPMI 1640 medium (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS and 1×non-essential amino acids (NEAA; Gibco, Thermo Fisher Scientific). Caco-2 (American Type Culture Collection, ATCC), U2-OS (ATCC), HEK293T (P. Bieniasz, Aaron Diamond AIDS Research Center), HEK293A (J. Dixon, UC San Diego), immortalized murine embryonic fibroblasts (C. Rice, Rockefeller University) and Huh-7 (J. Schoggins, UTSW Medical Center) cells were cultured in DMEM medium (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS and 1×NEAA. CHO-K1 (ATCC) cells were maintained in DMEM-F12 medium (Gibco, Thermo Fisher Scientific) supplemented with 10% FBS and 1×NEAA. CHO-7 (ref. 40) and SRD-1 (provided by J. Goldstein and M. Brown, UTSW Medical Center) cells were maintained in DMEM-F12 medium supplemented with either 10% FBS or 5% LPDS and 1×NEAA. Normal human dermal fibroblasts were from Lonza and cultured in ATCC fibroblast basal media. All of the cell lines were routinely tested for mycoplasma contamination. HEK293T, Huh-7, U2-OS and immortalized human STAT1-deficient fibroblasts were authenticated by the ATCC human short-tandem repeat profiling cell authentication service.
Reagents.
Recombinant mIFN-γ (Prospec) was reconstituted in sterile MilliQ-filtered water supplemented with a final concentration of 0.1% BSA. The tetrazolium dye 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma and freshly prepared for each experiment by diluting in PBS immediately before use. Cholesterol (Sigma), 7α-hydroxycholesterol (Avanti Polar Lipids), 25-hydroxycholesterol (Steraloids Inc.), epicholesterol (Steraloids Inc.), 4β-hydroxycholesterol (Avanti Polar Lipids, 700036P), 20α-hydroxycholesterol (Avanti Polar Lipids, 700156P), 22(R)-hydroxycholesterol (Avanti Polar Lipids, 700058P) and 27-hydroxycholesterol (Avanti Polar Lipids, 700021P) were obtained from the indicated sources and either dissolved in ethanol (final concentration of 1–2 mg ml−1) or complexed to MCD (Cyclodextrin Technologies Development, Inc.) at a 1:10 molar ratio (final sterol concentration of 2.5 mM) as described previously41. Lipoprotein-deficient serum was prepared as described previously19. Hydroxypropyl-β-cyclodextrin was from Cyclodextrin Technologies Development, Inc. and diluted in complete tissue culture media before use. CellTiter-Glo was purchased from Promega and used according to the manufacturer’s instructions. Monoclonal anti-His was purchased from Millipore. Monoclonal anti-actin, SZ58–035 and SMase were from Sigma. To detect SREBP2, IgG-7D4 (10 μg ml−1) was used. Super signal substrate was obtained from Pierce, anti-rabbit and anti-mouse IgG were from Jackson ImmunoResearch, and Phenix blue X-ray film product F-BX810 was from Phenix Research Products. Anti-β-actin (13E5) rabbit monoclonal antibody was purchased from Cell Signaling Technologies (4970). Lipidspot Alexa Fluor 488 was purchased from Biotium. Alexa Fluor 647 phalloidin was purchased from Thermo Fisher.
BMDM medium-transfer assays.
Six- to nine-week-old male WT and Ch25h−/− (B6.129S6-Ch25htm1Rus/J) C57BL/6J mice (The Jackson Laboratory)6 were euthanized according to the UTSW Animal Research policy and Institutional Animal Care and Use Committee. The tibiae and femurs were isolated, the bone marrow was released by pestle, and red blood cells were lysed using RBC lysis buffer. The cells were centrifuged and the resulting cell pellet was resuspended in DMEM medium (Corning) supplemented with 1×NEAA, 10% FBS and 10% L929-conditioned media, and plated onto 10-cm tissue-coated plates. The following day, non-adherent precursor cells were collected, re-plated onto petri dishes and allowed to differentiate for 5 d in the medium described above before use in experiments.
To generate conditioned media for transfer assays, BMDM (day 6 post culture in L929-conditioned medium) were plated onto multi-well plates at a density of 4.0 × 105 cells cm−2 in DMEM supplemented with 10% FBS, 1×NEAA and 10% L929 supernatant. The medium was removed the next day and replaced with DMEM (with 10% FBS) for 6 h. The cells were washed once in PBS and DMEM (containing 5% LPDS and 1×NEAA) containing either 500 U ml−1 mIFN-γ or vehicle was added. The medium from the BMDMs was collected approximately 24 h post IFN treatment, centrifuged at 500g for 5 min and the supernatants were immediately used for transfer assays or frozen at −80 °C for later use. For each biological replicate, conditioned media were utilized from BMDMs differentiated from distinct mice.
γ-ISG lentiviral library generation.
To screen for γ-ISGs that regulate bacterial infection in human cells, a list of genes was developed using datasets queried from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/), Interferome (http://www.interferome.org) and data from published transcriptomic studies42–54. The datasets assessed the gene transcription of human cells in response to a wide concentration of IFN-γ as well as various treatment durations. The inclusion criterion for the library consisted of upregulation of transcription by twofold or more (P < 0.05) in at least two unique datasets, which narrowed the gene list to 414 genes. The majority of cDNA constructs (pENTR) were obtained from the Invitrogen hORF lite collection and 146 constructs were obtained from a previously published Type I ISG library55. A small subset of genes was acquired from the plasmid repository DNASU56. The genes were recombined into a pTRIP. CMV.IVSb.ires.TagRFP destination vector using a LR clonase II enzyme kit according to the manufacturer’s instructions. Each reaction was transformed into competent DH5α E. coli; the plasmid DNA was extracted using a Qiagen miniprep kit and sequence verified. For the ISG screen, genes with a Z-score ≥ 2.5 were considered inhibitors of L. monocytogenes infection.
Lentiviral production and transduction.
Lentivirus was produced as described previously12. Briefly, HEK293T cells were co-transfected with HIV-1 gag-pol, VSV-G and pTRIP.CMV.IVSb.ISG.ires.TagRFP. After 48–72 h, pseudoparticle-containing supernatants were collected and stored at −80 °C for further use in DMEM medium supplemented with 3% FBS, 1×NEAA, 20 mM HEPES and 4 μg ml−1 polybrene. For the γ-ISG library, lentivirus for each gene was aliquoted into 96-well plates.
For lentiviral transduction, multi-well plates were seeded with 3.5–5.0× 104 cells cm−2, depending on the cell type and assay length. The following day, the medium was replaced with Pseudoparticle medium (3% FBS, cell-type-specific medium, 1×NEAA, 20 mM HEPES and 4 μg ml−1 polybrene). Lentivirus was added to each well and spinoculated at 1,000g for 60 min. After 6 h, the medium was removed and complete medium (with 10% FBS) was added. The cells were allowed to express the transduced gene for 48–72 h before the assays were performed unless otherwise stated in the figure legend. For the γ-ISG screen, 96-well plates seeded with 12,000 SDFs per well and transduced for 48 h before infection with GFP-expressing L. monocytogenes.
Site-directed mutagenesis.
CH25H-HHQQ was generated using a QuikChange lightning site-directed mutagenesis kit (Agilent) following the manufacturer’s instructions. Primers to introduce mutations T726G and C729G were designed using the QuikChange primer design program (http://www.agilent.com/genomics/qcpd). The mutant strand was then synthesized using Pfu-based DNA polymerase blend with pENTR-CH25H as a template. The original template DNA was eliminated by Dpn I treatment and the resulting reaction was transformed into chemically competent DH5α cells. The mutations were further verified by sequencing. Finally, CH25H (H242H243→Q242Q243) was subcloned into the pTRIP–TagRFP vector for expression.
The following primers were used to generate the desired mutation: (F) 5′-GTGCACCACGACCTGCAGCAGTCTCACTTTAACTGC-3′ and (R) 5′GCAGTTAAAGTGAGACTGCTGCAGGTCGTGGTGCAC-3′
L. monocytogenes inoculum preparation and mammalian cell infection.
L. monocytogenes strain 10403S expressing GFP (provided by D. Portnoy, UC Berkeley) was scraped from a glycerol stock into 3 ml brain heart infusion (BHI) medium and incubated overnight at 30 °C without shaking. A 1-ml volume of the overnight culture was washed in PBS and resuspended in a final volume of 1 ml PBS. The optical density at 600 nm (OD600) was measured and adjusted to 0.8. The cells were then infected at the m.o.i. specified for an initial period of 1.5 h, unless otherwise stated. The cells were then washed three times, fresh medium supplemented with 25–50 μg ml−1 gentamicin was added and the infection was allowed to proceed for the time indicated.
For the quantification of bacterial infection by flow cytometry, cells were collected and analysed as described previously12. Briefly, the cells were dissociated using Accumax for 5 min, pelleted at 800g for 3 min and then fixed with 1% paraformaldehyde for 30 min at 4 °C. The cells were then pelleted and resuspended in FACS buffer (3% FBS in PBS). The samples were analysed by flow cytometry using a Stratedigm S1000 flow cytometer and FlowJo Software (Treestar). For RFP-expressing cells, the infection levels were quantified as the percentage of GFP-positive cells in the RFP-positive population. For the γ-ISG screen, 1 × 104 singlet cells were collected. Samples that had fewer than 2,000 singlets or less than 25% RFP-positive cells were excluded from further analysis.
For the quantification of bacterial infection by c.f.u., the cells were first washed three times with PBS at the time of collection. The cells were then incubated with 0.5% Triton X-100 for 5–10 min at 37 °C. The cell lysates were mixed by pipetting and vortexed, and serial dilutions were prepared in PBS, plated onto BHI agar plates and incubated overnight at 30 or 37 °C.
For Fig. 3d and Extended Data Fig. 2c, the cells were seeded onto 24-well plates and treated with 25HC (5 μM) for 16–20 h the following day. The cells were then infected with GFP-expressing L. monocytogenes as described above using the following m.o.i. and incubation times: HEK293A cells were set up at 1.0 × 105 cells per well and infected (m.o.i. = 1) for 18 h, murine embryonic fibroblasts were set up at 4.0 × 104 cells per well and infected (m.o.i. = 8) for 5 h, Caco-2 cells were set up at 5.0 × 104 cells per well and infected (m.o.i. = 0.5) for 18 h, Huh-7 cells were set up at 1.0 × 105 cells per well and infected (m.o.i. = 0.1) for 18 h, U2-OS cells were set up at 1.0 × 105 cells per well and infected (m.o.i. = 0.5) for 20 h, CHO-7 cells were set up at 5.0 × 104 cells per well of a 48-well plate and infected (m.o.i. = 1) for 22 h, and normal human dermal fibroblasts were set up at 2.0 × 104 cells per well of a 24-well plate, treated with 25HC after 2 d as described above and then infected with GFP-expressing L. monocytogenes (m.o.i. = 1) for 22 h. Infection was quantified by flow cytometry for each cell type.
For the BMDM medium-transfer infection assays (Figs. 1b,c and 3c), 15,000 HEK293A cells were seeded per well of a 96-well plate in DMEM medium (containing 5% LPDS, 1×NEAA). The following day, the medium was removed and 0.15 ml of conditioned media collected from macrophages was applied. After 24-h of treatment, the cells were infected with GFP-expressing L. monocytogenes (m.o.i. = 2) as described earlier. Following the initial 90 min of infection, the cells were washed and fresh medium supplemented with 25 μg ml−1 gentamicin was added. After 22 h, infection was quantified by flow cytometry analysis.
For L. monocytogenes infection of BMDMs (Fig. 3d and Extended Data Fig. 2c), macrophages were generated as described earlier and set up at 3.0 × 105 cells per well of a 24-well plate. The following day, the BMDMs were treated overnight (16–20 h) with 25HC (5 μM) and then infected with GFP-expressing L. monocytogenes (m.o.i. = 5) for 45 min. The cells were washed three times with PBS and then fresh medium supplemented with 25 μg ml−1 gentamicin was added. After 24 h, the cells were collected for flow cytometry analysis or lysed with 0.5% Triton X-100 and serial dilutions were plated to enumerate the c.f.u.
A donor-recipient infection assay was carried out to assess the effects of 25HC on L. monocytogenes cell-to-cell spread via flow cytometry. On day 0, WT HEK293A cells were seeded at 5.0 × 104 cells cm−2 and transduced with TagRFP coexpressing Fluc on day 1. In parallel, c-Met- and E-cadherin-deficient HEK293A cells12 were seeded at a density of 7.0 × 104 cells cm−2 (day 0) and treated with 25HC (5 μM) or vehicle (ethanol) on day 1. On day 2, the WT HEK293A cells were infected with GFP-expressing L. monocytogenes at a m.o.i. of one for 1.5 h, washed and then fresh medium (supplemented with 50 μg ml−1 gentamicin) was added. After 2 h, the infected WT HEK293A cells were washed three times with PBS and detached using Accumax. The infected HEK293A cells were overlaid onto monolayers of c-Met and E-cadherin-deficient cells treated with 25HC or vehicle, at a ratio of one infected cell (donor) to 200 uninfected recipient cells, in medium supplemented with 25 μg ml−1 gentamicin. The infection was allowed to proceed for 24 h in the presence of gentamicin before collection for flow cytometry or fluorescent microscopy imaging. Infection was quantified via flow cytometry as the percentage of RFP-negative recipient cells that were GFP-positive (Listeria infected). A control experiment was carried out in parallel, in which donor cells were infected with L. monocytogenes 10403S ΔactA pactA::GFP, a spread-deficient mutant, which was included as a control. The 10403S ΔactA strain (DP-L3078) was provided by D. Portnoy (UC Berkeley). The generation of the DP-L3078 pactA::GFP strain was described previously12.
For Fig. 5c, CHO-7 cells were treated with 5 μM 25HC or vehicle for 16 h in medium supplemented with 5% LPDS or 10% FBS. The cells were infected with GFP-expressing L. monocytogenes (m.o.i. = 1; 22 h) and analysed by flow cytometry. The per cent infection inhibition was calculated as the ratio of the percentage of GFP-positive cells treated with 25HC compared with vehicle-treated cells.
For Fig. 5d,e, CHO-K1 cells were set up at 5.0 × 104 cells per well of a 48-well plate and treated with 5 μM 25HC or vehicle for 16 h the following day. The cells were next treated with SMase as described for Extended Data Fig. 5b and subsequently infected with GFP-expressing L. monocytogenes (m.o.i. = 1; 22 h) in medium supplemented with 25 μg ml−1 gentamicin and SMase or vehicle. The cells were then collected for flow cytometry analysis.
For Fig. 6h, CHO-7 cells were set up at 5.0 × 104 cells per well of a 48-well plate, treated with 10 μM SZ58–035 or vehicle (dimethylsulfoxide) for 2 h the next day and then incubated with 5 μM 25HC (or vehicle) along with 10 μM SZ58–035 or vehicle (dimethylsulfoxide) for 20 h. Subsequently, the cells were infected with GFP-expressing L. monocytogenes (m.o.i. = 1) for 22 h (in medium supplemented with 25 μg ml−1 gentamicin and 10 μM SZ58–035 or vehicle). Infection was quantified by flow cytometry analysis.
For Fig. 6i, CHO-7 or SRD-1 cells were treated with 25HC (5 μM; 16 h) and subsequently infected with GFP-expressing L. monocytogenes (m.o.i. = 25; 6 h). Infection was quantified by flow cytometry analysis.
L. monocytogenes adhesion/invasion assay.
HEK293A cells were seeded at a density of 6.0 × 104 cells cm−2. The cells were treated with 25HC (5 μM) or vehicle for 20 h and then infected with L. monocytogenes (m.o.i. = 10) for 20 min. The cells were washed three times with PBS and 0.5% Triton X-100 was added for 5 min to lyse the cells. Serial dilutions were plated and the recovered c.f.u. were enumerated. The per cent adhesion/invasion was calculated using the following equation: (total c.f.u. recovered) ÷ (initial bacterial inoculum)× 100%.
L. monocytogenes vacuole escape assay.
To measure vacuole escape, U2-OS cells were seeded onto 25-mm coverslips deposited in six-well plates at a density of 7.0 × 104 cells cm−2. The cells were treated with 25HC (5 μM) or vehicle for 16–20 h the following day. Next, the cells were infected with GFP-expressing L. monocytogenes or the DP-L2161 (ΔHly) strain expressing GFP. The cells were infected for an initial period of 1.5 h (m.o.i. = 6) and then washed three times with PBS. Fresh medium containing 50 μg ml−1 gentamicin was added for 2 h, and the cells were then fixed with formaldehyde (3.7%; 10 min), permeabilized with Triton X-100 and stained with Alexa Fluor 647 phalloidin and 4,6-diamidino-2-phenylindole (DAPI). The cells were mounted onto slides with Prolong Gold. The association of L. monocytogenes with actin was observed using a Zeiss Observer Z1 fluorescent microscope with a ×63 objective lens. Images were analysed using Zen 2 Pro software.
L. monocytogenes replication assay.
To measure the L. monocytogenes replication, multi-well plates were seeded with 6.0 × 104 HEK293A cells cm−2. The following day, the cells were treated with 25HC (5 μM) or vehicle overnight. The cell monolayers were then infected with L. monocytogenes at an m.o.i. of 10 for 1.5 h. Subsequently, the cells were washed three times and fresh medium supplemented with 25 μg ml−1 gentamicin was added for 1.5 h. The cells were lysed at the indicated time points by washing with PBS and adding 0.5% Triton X-100 for min. Serial dilutions of the lysates were plated on BHI agar plates and incubated at 30 °C overnight. The c.f.u. were enumerated the following day. Fold replication was calculated as the number of c.f.u. recovered at time point T2 (5.5 h) divided by the c.f.u. recovered at T1 (3 h) or the number of c.f.u. recovered at time point T3 (9.5 h) divided by the c.f.u. recovered at T1 (3 h).
L. monocytogenes membrane protrusion assay.
Membrane-localized RFP was generated by fusing the membrane localization sequence (N-terminal 20 amino acids) of neuromodulin to TagRFP using PCR. The resulting fusion (NeuroRFP) was cloned into pENTR using BP clonase and then recombined into the pLenti CMV Puro DEST (w118–1) destination vector using LR clonase II. Lentivirus was produced in HEK293T cells as described earlier, and Caco-2 cells were transduced with NeuroRFP and then selected under puromycin for 10 d.
On day 0, 9.0 × 104 Caco-2 cells stably expressing NeuroRFP were seeded onto 25-mm coverslips deposited in six-well plates. On day 4, the cells were treated with 25HC (5 μM) or vehicle for 16–20 h. The Caco-2 cells were then infected with GFP-expressing L. monocytogenes (m.o.i. = 0.5) for 90 min, washed three times with PBS and fresh medium supplemented with 50 μg ml−1 gentamicin was added. After 6 h, the cells were washed with PBS, fixed with 3.7% formaldehyde, permeabilized with Triton X-100 and stained with Alexa Fluor 647 phalloidin. The cells were then mounted onto slides using Prolong Gold. Images were taken using a Zeiss Observer Z1 fluorescent microscope with a ×63 objective lens and analysed using the Zen 2 Pro software. The frequency of the total population of GFP-expressing L. monocytogenes associated with a membrane protrusion was quantified per field of view. A similar number of bacteria were evaluated for vehicle- (n = 1,839) or 25HC-treated (n = 1,628) cells in fields of view with comparable numbers of total bacteria. The pLenti CMV Puro DEST (w118–1)57 was a gift from E. Campeau and P. Kaufman (Addgene plasmid no. 17452; http://n2t.net/addgene: 17452; RRID: Addgene_17452). For live-cell imaging (Supplementary Video 1), Caco-2-expressing NeuroRFP cells were set up as above, and 85 images were recorded on an Olympus Flourview FV10i every 8.8 s.
L. monocytogenes foci-forming assay.
Twelve-well plates were seeded with 3.0 × 105 HEK293A cells per well and treated with 25HC (5 μM) or vehicle the following day. After 16 h of treatment, the cells were infected with L. monocytogenes for 1 h (m.o.i. = 0.1) and subsequently washed three times with medium supplemented with 50 μg ml−1 gentamicin. The cell monolayers were then overlaid with 0.75 ml DMEM supplemented with 10% FBS, 0.4% agarose and 50 μg ml−1 gentamicin, and further incubated at 37 °C in a tissue culture incubator. After 72 h, the infection foci were visualized by the addition of 150 μl MTT (5 mg ml−1; Sigma). Images were recorded 16 h after MTT staining and analysed using ImageJ software. For the cholesterol-addition experiments, before infection, the media were removed from the cell monolayers and the cells were incubated with DMEM (with 10% FBS) supplemented with 40 μM cholesterol–MCD complexes. After an incubation of 1 h, the cells were infected and stained with MTT as above. No foci were formed in the uninfected host cells.
In vivo L. monocytogenes infection.
On days 1–3, 8- to 11-week-old male WT C57BL/6J mice were injected intraperitoneally with 25HC (5 mg kg−1) or vehicle (ethanol) once daily. On day 4, the mice were orally infected with 1 × 109 L. monocytogenes by intragastric gavage. The bacterial inoculum was prepared in PBS and mixed at a 2:3 ratio with 50 mg ml−1 CaCO3 before being administered to the mice. The number of bacteria in the inoculum was confirmed by plating serial dilutions onto agar plates before and after infection. Following gavage, the mice were intraperitoneally injected with 25HC (5 mg kg−1) or vehicle. On days 5–7, the mice were injected with 25HC (5 mg kg−1) or vehicle once daily. The spleens of the mice were collected 4 h after the last dosing and homogenized in PBS. Serial dilutions of the spleen homogenates were plated onto BHI agar plates to enumerate the bacterial burden.
Eight- to 11-week-old male WT C57BL/6J and Ch25h−/− (B6.129S6-Ch25htm1Rus/J) mice (The Jackson Laboratory)6 were orally infected with 1 × 109 L. monocytogenes by gavage. The bacterial inoculum was prepared in PBS and mixed at a 2:3 ratio with 50 mg ml−1 CaCO3 before being administered to the mice. The number of bacteria in the inoculum was confirmed by plating serial dilutions onto agar plates before and after infection. The spleens of the mice were collected and homogenized in PBS 3 d post-infection and serial dilutions of the spleen homogenates were plated onto BHI agar plates to enumerate the bacterial burden.
S. flexneri infection and plaque assays.
S. flexneri strain M90T (serotype 5) harbouring the pBBRMCS1-GFP plasmid was inoculated into 5 ml BHI media and incubated at 30 °C with shaking at 225 r.p.m. for 16 h. The following day, 400 μl of the overnight culture was added to 20 ml BHI and incubated at 37 °C with shaking (225 r.p.m.) for 2 h or until the OD600 reached 0.5. The bacteria were then washed with PBS and resuspended in 1 ml Congo red (0.03%) for 15 min at 37°C. For the plaque-forming assays, 4.0 × 105 HEK293A cells were seeded onto poly-d-lysine-coated 12-well plates and treated with medium supplemented with 10% FBS, 1×NEAA and 25HC (5 μM) or ethanol the following day. Following overnight treatment, cholesterol–cyclodextrin complexes (40 μM) or vehicle were added to the cells 1 h before infection. The cells were infected with S. flexneri (m.o.i. = 0.0015) for 1 h, washed extensively and fresh medium supplemented with 50 μg ml−1 gentamicin was added. After 1 h, the cells were washed again, 0.75 ml Avicel solution (2×DMEM, 1.2% Avicel, 10% FBS and 50 μg ml−1 gentamicin) was overlaid and further incubated at 37 °C for 72 h post overlay. The samples were fixed with 3.7% formaldehyde for 15 min. After fixation, the overlay and formaldehyde were aspirated, the monolayers were washed twice with PBS and the cells were stained with crystal violet. Plaque images were scanned and then analysed using the ImageJ software.
Oxysterol measurements.
STAT1-deficient skin fibroblasts were plated onto 12-well plates at a density of 1.0 × 105 cells cm−2. The following day, the cells were transduced with pTRIP–RFP coexpressing Flue, CH25H or CH25H-HHQQ. The cell medium was collected after 48 h. Conditioned media from WT and Ch25h−/− C57BL/6J BMDMs were collected as described earlier for the medium-transfer assays. To measure the serum oxysterol concentration, the mice were injected intraperitoneally with 25HC or vehicle (ethanol) and infected with L. monocytogenes as described above. On day 7, blood was collected and centrifuged. The serum was isolated and submitted for oxysterol analysis. The oxysterol concentration was measured by mass spectrometry as described previously58.
The oxysterol nomenclature used in this study was as follows:
4β-HC, 4β-hydroxycholesterol; 5,6α-EC, 5,6α-epoxycholesterol;
5,6β-EC, 5,6β-epoxycholesterol; 6α-HC, 6α-hydroxycholestanol;
7α-HC, 7α-hydroxycholesterol; 24-OC, 24-oxocholesterol; 24S-HC,
24S-hydroxycholesterol; 24(S),25-EC, 24(S),25-epoxycholesterol; 25HC,
25-hydroxycholesterol; 7β,25-DiHC, 7β,25-dihydroxycholesterol; 27HC,
27-hydroxycholesterol; and 7β,27-DiHC, 7β,27-dihydroxycholesterol.
Buffers and media for toxin-based biosensor assays, total cholesterol measurements and lipid droplet staining.
Medium A: 1:1 mixture of DMEM and Ham’s F12 supplemented with 100 U ml−1 penicillin and 100 μg ml−1 streptomycin sulfate.
Medium B: medium A supplemented with 5% (vol/vol) FBS.
Medium C: medium A supplemented with 10% (vol/vol) FBS.
Medium D: medium A supplemented with 5% (vol/vol) LPDS.
Medium E: medium D supplemented with 50 μM compactin and 50 μM mevalonate.
Medium F: high-glucose DMEM supplemented with 5% (vol/vol) FBS, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin sulfate.
Medium G: high-glucose DMEM supplemented with 10% (vol/vol) FBS, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin sulfate.
Medium H: low-glucose DMEM supplemented with 10% (vol/vol) FBS, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin sulfate.
Medium I: high-glucose DMEM supplemented with 5% (vol/vol) LPDS, 50 μM compactin, 50 μM mevalonate, 100 U ml−1 penicillin and 100 μg ml−1 streptomycin sulfate.
Medium J: low-glucose DMEM supplemented with 5% (vol/vol) LPDS, 50 μM compactin, 50μM mevalonate, 100 U ml penicillin and 100 μg ml−1 streptomycin sulfate.
Medium K: medium E supplemented with 1% (wt/vol) hydroxypropyl-β-cyclodextrin (HPCD).
Buffer A: 50 mM Tris–HCl (pH7.5), 150 mM NaCl and 1 mM TCEP.
Buffer B: 10mM Tris–HCl (pH 6.8), 100 mM NaCl, 1% (wt/vol) SDS, 1 mM EDTA, 1 mM EGTA, 20 μg ml−1 phenylmethylsulfonyl fluoride and protease inhibitors (1 tablet per 20 ml; Roche).
Toxin biosensor protein purification.
His6- and FLAG-tagged ALOD4 (ALOD4) was expressed and purified as previously described59 and O1yA–His6 (OlyA) was expressed and purified as previously described21. Purified protein was stored at 4°C in buffer A and used within 4 (ALOD4) or 2 weeks (OlyA).
Assay for measuring ALOD4 and OlyA binding after time course of 25HC and 7α-HC treatment.
On day 0, CHO-7 or SRD-1 cells were set up in medium D at a density of 3.0 × 104 cells per well of a 48-well plate. On the morning of day 1, the cells were washed twice with PBS, followed by the addition of 500 μl medium B. This procedure was repeated in the evening of day 1. On day 2, the cells were washed twice with PBS, followed by the addition of 200 μl medium B containing 5 μM 25HC or 7α-HC. After incubation for the indicated times at 37°C, the cells were switched to 200 μl medium B containing 3 μM ALOD4 or OlyA. After further incubation for 30 min at 37°C, the cells were washed twice with PBS, harvested and subjected to immunoblot analysis.
Assay for measuring ALOD4 binding after dose curve of 25HC treatment.
On day 0, CHO-7 cells were set up in medium D at a density of 3.0 × 104 cells per well of a 48-well plate. In the evening of day 0, the cells were washed twice with PBS, followed by the addition of 500 μl medium C or D. This procedure was repeated on day 1. On day 2, the cells were washed twice with PBS, followed by the addition of 200 μl medium C or D containing the indicated concentrations of 25HC. After incubation for 16 h at 37°C, the cells were switched to either medium C or D containing 3 μM ALOD4. After further incubation for 30 min at 37°C, the cells were washed twice with PBS, harvested and subjected to immunoblot analysis.
Assay for measuring ALOD4 binding after treatment with 25HC in the absence or presence of SZ58-035.
On day 0, CHO-7 cells were set up in medium D at a density of 3.0 × 104 cells per well of a 48-well plate. On the morning of day 1, the cells were washed twice with PBS, followed by the addition of 500 μl medium B. In the evening of day 1, the cells were washed twice with PBS, followed by the addition of 500 μl medium B containing either dimethylsulfoxide vehicle (1:1,000) or 10 μM SZ58-035. After incubation for 16 h at 37°C, the cells were washed twice with PBS, followed by the addition of 200 μl medium B containing 5 μM 25HC along with either dimethylsulfoxide vehicle or 10 μM SZ58-035. After a further incubation for the indicated times at 37°C, the cells were switched to 200 μl medium B containing 3 μM ALOD4 along with either dimethylsulfoxide vehicle or 10 μM SZ58-035. After an additional incubation of 30 min at 37°C, the cells were washed twice with PBS, harvested and subjected to immunoblot analysis.
Assay for measuring ALOD4 and OlyA binding after SMase treatment.
On day 0, the indicated cell lines were set up at 6.0 × 104 cells per well of 48-well plates in the following media (HEK293A, medium F; Caco-2, medium G; U2-OS, medium H; Huh-7, medium F; CHO-K1, medium B; and CHOU, medium D). On day 1, the cells were washed twice with PBS, followed by the addition of 200 μl of the cell-line-specific lipoprotein-rich medium (see above) with or without 5μM 25HC. After incubation for 4 h at 37 °C, the cells were washed twice with PBS and treated with either 200 μl of the cell-line-specific lipoprotein-rich media (see above) with (lanes 2 and 4) or without (lane 1 and 3) 100 mU ml−1 SMase. After a further incubation for 30 min at 37 °C, the cells were washed twice with PBS, followed by the addition of 200 μl of the cell-line-specific lipoprotein-poor media (medium E, I or J) containing 3 μM ALOD4 or OlyA. After an additional incubation of 30 min at 37 °C, the cells were washed twice with PBS, harvested and subjected to immunoblot analysis.
Assay for measuring ALOD4 binding after treatment with sterol–MCD complexes.
On day 0, the indicated cell lines were set up either at 7 × 105 cells per 10-cm dish (HEK293A) or 6.0 × 104 cells per well of 48-well plates (all other cell lines) in the following media (HEK293A, medium F; Caco-2, medium G; U2-OS, medium H; Huh-7, medium F; CHO-K1, medium B; and CHO-7, medium D). On day 1, the cells were washed twice with PBS, followed by the addition of 200 μl (5 ml for HEK293A) of the cell-line-specific lipoprotein-rich media (see above) with or without 5 μM 25HC. After incubation for 4 h at 37 °C, the cells were washed twice with PBS and treated with 200 μl (5 ml for HEK293A) of either cell-line-specific lipoprotein-rich media (lane 1) or cell-line-specific lipoprotein-poor media (medium E, I or J) containing the indicated concentrations of epicholesterol or cholesterol complexed to MCD (lanes 2–9). After a further incubation for 2 h at 37 °C, the cells were washed twice with PBS, followed by the addition of 200 μl (2 ml for HEK293A) of the cell-line specific lipoprotein-poor media containing 3 μM ALOD4. After an additional incubation of 30 min at 37 °C, the cells were washed twice with PBS, harvested and subjected to immunoblot analysis.
Assay for measuring ALOD4 binding after 4 h incubation with various oxysterols.
On day 0, CHO-K1 cells were set up in medium B at a density of 6.0 × 104 cells per well of a 48-well plate. On day 1, cells were washed twice with PBS and then treated with 200 μl medium B containing the indicated concentration of 4β-HC, 7α-HC, 20α-HC, 22-HC, 25HC, 27HC or vehicle (ethanol). After incubation for 4 h at 37 °C, the medium was removed and the cells were treated with 200 μl medium B containing 3 μM ALOD4. After incubation for 30 min at 37 °C, the cells were washed twice with PBS, harvested and subjected to immunoblot analysis.
Assay to determine oxysterol-mediated regulation of SREBP2.
On day 0, CHO-K1 cells were set up in medium B at a density of 6.0 × 104 cells per well of a 48-well plate. On day 1, the cells were washed twice with PBS and then treated with 200 μl medium K to deplete cholesterol. After 1 h at 37 °C, the cells were washed twice with PBS and then treated with 200 μl medium E supplemented with the indicated concentrations of 4β-HC, 7α-HC, 20α-HC, 22-HC, 25HC, 27HC or vehicle (ethanol). After incubation for 4 h at 37 °C, the cells were washed twice with PBS, harvested and subjected to immunoblot analysis.
Assay to determine oxysterol-mediated regulation of SREBP2 in the absence and presence of SZ58-035.
On day 0, CHO-K1 cells were set up in medium B at a density of 3.0 × 104 cells per well of a 48-well plate. On day 1, the cells were washed twice with PBS and then treated with 200 μl medium B containing dimethylsulfoxide or 10 μM SZ58-035. On day 2, the cells were washed twice with PBS and then treated with 200 μl medium K to deplete cholesterol. After 1 h at 37 °C, the cells were washed twice with PBS and then treated with 200 μl medium E supplemented with the indicated concentrations of 20α-HC, 25HC or 27HC. After incubation for 4 h at 37 °C, the cells were washed twice with PBS, harvested and subjected to immunoblot analysis.
Assay to determine oxysterol sensitivity to SZ58-035.
On day 0, CHO-K1 cells were set up in medium B at a density of 3.0 × 104 cells per well of a 48-well plate. On day 1, the cells were washed twice with PBS and then treated with 200 μl medium B containing dimethylsulfoxide or 10 μM SZ58-035. On day 2, the cells were washed twice with PBS and then treated 200 μl medium B containing either vehicle (dimethylsulfoxide) or 10 μM SZ58-035 and 5 μM 20α-HC, 25HC or 27HC. After 4 h at 37 °C, the medium was removed and the cells were treated with medium B containing 3 μM ALOD4. After incubation for 30 min at 37 °C, the cells were washed twice with PBS, harvested and subjected to immunoblot analysis.
Immunoblot analysis of ALOD4 and OlyA binding.
After the indicated treatments, the cells were washed twice with PBS, followed by the addition of 200 μl buffer B. After incubation for 20 min at room temperature on a shaker, the cell lysates were collected, mixed with 5×loading dye and heated at 95 °C for 10 min. Equal aliquots of cell lysates (10% of the total) were then subjected to either 15% SDS-PAGE for the detection of ALOD4, OlyA and actin or 10% SDS-PAGE for the detection of SREBP2 and actin. For assays measuring ALOD4 binding to HEK293A cells after treatment with sterol–MCD complexes, the cells were harvested by scraping from 10-cm dishes, the protein content of the cell lysates was measured using a bicinchoninic acid assay and equal protein amounts (20 μg per lane) were subjected to 15% SDS-PAGE. The electrophoresed proteins were then transferred to nitrocellulose membranes using the Bio-Rad trans blot turbo system and subjected to immunoblot analysis with the following primary antibodies: anti-actin (1:1,000 dilution), anti-His (1:1,000 dilution) to detect ALOD4 (calculated molecular weight of 16.02 kDa) and OlyA (calculated molecular weight of 17.35 kDa), or IgG-7D4 (10 μg ml−1) to detect SREBP2. The bound antibodies were visualized by chemiluminescence after incubation with either anti-rabbit or anti-mouse IgG, each at a dilution of 1:5,000. The membranes were exposed to Phoenix blue X-ray film at room temperature for 1–30 s for anti-His and 1–120 s for anti-actin and IgG-7D4. Quantification of ALOD4 binding and actin for each sample was performed using the gel analyser feature of FIJI60. The percentage of ALOD4 bound was calculated as the actin-normalized ALOD4 signal relative to vehicle treatment.
Cholesterol measurements.
On day 0, CHO-K1 cells were set up at a density of 2.45 × 105 cells per well of a six-well plate in medium C. On day 1, the cells were treated with either 25HC (5 μM) or vehicle in medium C. On day 2, the cells were treated with either 25HC (5 μM) for 4 h or 1% HPCD in medium C for 1 h. The cells were then washed twice with PBS and lysates were collected by scraping cells into 150 μl 1×Reaction buffer (Amplex red cholesterol assay kit) supplemented with protease inhibitors. Total cellular cholesterol was determined using the Amplex red cholesterol assay kit according to the manufacturer’s instructions. The cholesterol content was normalized to protein content, as determined using the bicinchoninic acid assay.
Lipid droplet staining.
On day 0, CHO-7 cells were set up in medium D at a density of 3.0 × 104 cells per well of a Lab-TEK II eight-well chamber or a 24-well plate. On day 1, the cells were treated with 10 μM SZ58-035 for 2 h and then 25HC (5 μM) in the absence or presence of 10 μM SZ58-035 in medium D. After 16 h, the cells plated on eight-well chambers were washed with PBS, fixed with 1% paraformaldehyde and then stained with 1×LipidSpot for 10 min and DAPI for 2 min. The cells were then observed using a Zeiss Observer Z1 fluorescent microscope with a ×63 objective lens. Images were analysed using the Zen 2 Pro software. For flow cytometry analysis, cells plated on 24-well plates were detached using Accumax, fixed with 1% paraformaldehyde and then stained with 1×LipidSpot for 10 min. The cells were then washed twice with PBS, resuspended in PBS and analysed by flow cytometry.
CRISPR-Cas9-mediated gene editing.
All guides were designed using the Optimized CRISPR design tool (http://crispr.mit.edu/). To generate insertions/deletions in NR1H3 (LXRα) and NR1H2 (LXRβ), paired guides were designed to target exon 3 of either gene. The guides were cloned into the pX335-U6-Chimeric_ BB-CBh-hSpCas9n(D10A) vector. HEK293A cells were transfected with EGFP-N3 and pX335 encoding paired guides targeting NR1H3 or NR1H2. After 48 h of transfection, GFP-positive cells were isolated using FACS to establish clonal cell lines. Following clonal expansion, individual colonies were screened for the loss of LXR-dependent gene transcription and further confirmed by genomic sequencing of the targeted locus.
The following guides were used to target NR1H3:
5′-TCGGCTTCGCAAATGCCGTC-3′ and 5′-CTGGCACTTGCGACGCATGT-3′.
The following guides were used to target NR1H2:
5′-TTCCGGCGCAGTGTGGTCCG-3′ and 5′-CGCAGCTGAGCACGTTGTAG-3′.
The following primers were used to amplify genomic DNA of NR1H3 spanning exon 3: (F) 5′-CCTAGAACATAATGGCACTTGGC-3′ and (R) 5′-GCAAAGCGCTGCTGACG-3′.
The following primers were used to amplify genomic DNA of NR1H2 spanning exon 3: (F) 5′-CTGTAAAGGAGGAGGGTCCG-3′ and (R) 5′-TACAACGGCAAGCACTGGTC-3′.
Quantitative real-time PCR.
Total RNA was isolated from mammalian cells using an RNAeasy kit (Qiagen) according to the manufacturer’s instructions. The RNA quality and concentration were assessed using a NanoDrop-1000 spectrophotometer. Real-time quantitative PCR was carried out using the QuantiFast SYBR Green PCR kit (Qiagen). Relative messenger RNA values were normalized to the housekeeping gene RPS11 (QuantiTech Primer Assay, QT00061516).
The following primers were used for ABCA1:
(F) 5′-AGGTTGCTGCTGTGGAAG-3′ and (R)
5′-TCATGTTGTTCATAGGGTGGG-3′.
Animal experiments.
Preliminary assays were conducted to estimate variance and determine sufficient sample size. The number of mice used per group is reported in the figure legends. For the in vivo infection assays, three independent experiments were carried out. For the in vivo experiments, sex-matched mice of similar age were randomly assigned to each experimental group. The order of mouse dosing and sample collection was randomized between each group for each independent experiment. Investigators were not blinded to the mouse treatment group or genotype during experiments as the cages needed to be identified for multiple dosings over the experiment. Furthermore, the determination of the organ c.f.u. burden and oxysterol serum analysis were quantitative not subjective assays.
Statistical analysis.
Experiments were conducted as at least three biologically distinct independent replicates, unless otherwise stated. Statistical significance was determined using the GraphPad Prism software. For bar graphs, line graphs and scatterplots, data are shown as the mean and the error bars depict the s.d. unless otherwise stated. For figures where normalized data is shown, statistical analysis was performed on raw non-normalized data and compared with the controls. Unpaired two-tailed Student’s t-tests were used to calculate the P values for experiments with only variable. For experiments with multiple variables, significance was calculated using one-way ANOVA with Dunnett’s correction for multiple comparisons. Dose-response curves were fitted to a sigmoidal model using the GraphPad Prism software, and IC50 and EC50 values were also calculated using the GraphPad Prism software. All statistical significance was calculated using GraphPad Prism.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Comparison between flow cytometry and CFUs for L. monocytogenes infection.
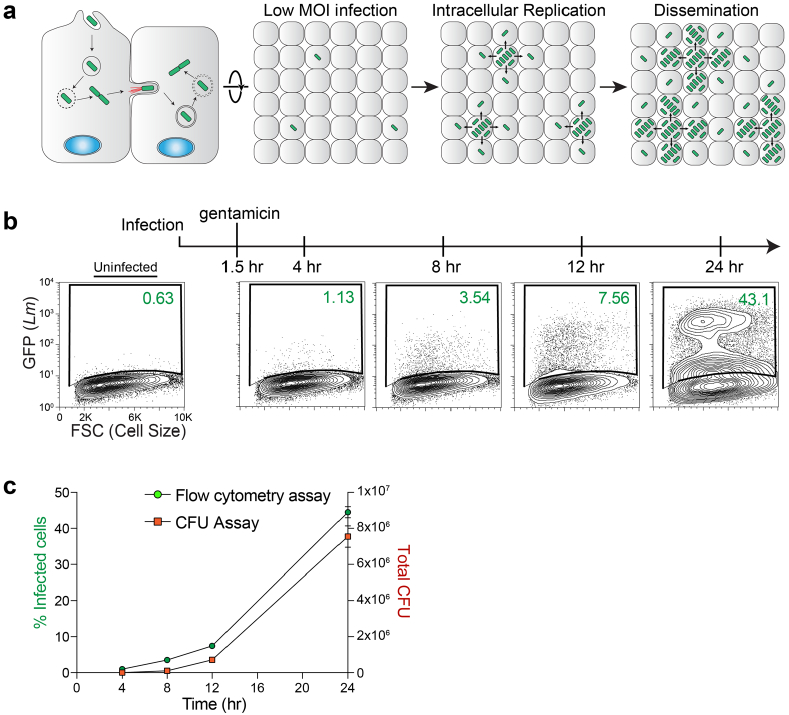
a, Schematic of the L. monocytogenes life-cycle (left) and its replication and intercellular dissemination initiated from a low dose of bacterial infection. HEK293A cells were infected with GFP-expressing L. monocytogenes (MOI=1) so that only a small percentage of the host cell monolayer (<1%) are initially infected. Cell-to-cell spread of L. monocytogenes results in robust infection of the monolayer over time. b, Representative flow cytometry plots of L. monocytogenes (GFP) infection of HEK293A cells at the indicated time points. After 90 minutes of infection, the host cell monolayers were washed and incubated with gentamicin to remove and kill extracellular bacteria. These studies were repeated independently four times with similar results. c, Direct comparison between gentamicin protection assays assessed by flow cytometry (as above) or Colony forming Units (CFUs). Samples were harvested for analysis at the indicated time points after infection. Graph showing the percent of infected cells determined by flow cytometry (y-axis, left) were directly compared to CFUs recovered (y-axis, right). Mean values from 4 independent experiments are plotted, and error bars show s.d. We concluded that flow cytometry is an accurate method of enumerating bacterial burden in host cells.
Extended Data Fig. 2 |. CH25H inhibits L. monocytogenes through 25HC production.
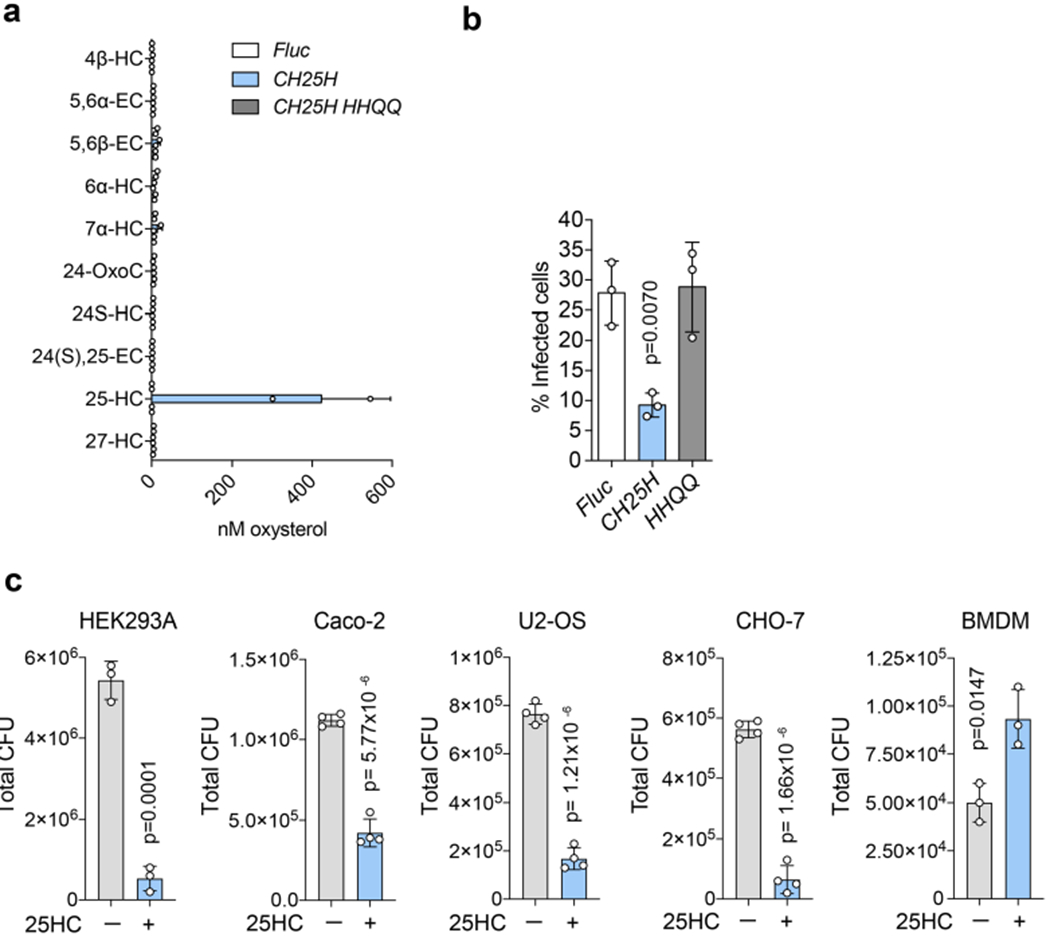
a, Oxysterol measurements in media collected from SDFs transduced with lentivirus encoding Fluc, CH25H, or a catalytically inactive CH25H mutant with the following mutations: H242H243/Q242Q243. Following 48 hours transduction, the concentration of oxysterols secreted into the media was measured by mass spectrometry. Bars represent mean values. Error bars show s.d. from two independent experiments. See Methods for oxysterol nomenclature. b, To determine if CH25H catalytic activity is necessary for its antibacterial function, HEK293A cells were transduced with lentivirus as in (a), infected with GFP-expressing L. monocytogenes (MOI=10) for 6 hours, and analysed by flow cytometry. Bars represent mean values. Error bars show s.d. from three independent experiments and statistical significance was determined by one-way ANOVA compared to Fluc with Dunnet’s correction. c, Bar graph showing the total L. monocytogenes CFUs recovered from gentamicin protection assays performed on the indicated cell lines treated with vehicle (EtOH) or 25HC (5 μM). The specified cell lines were set up, treated, and infected as in Fig. 3d, and CFU enumerated by lysing cells and plating serial dilutions. Bars represent mean values. Error bars show s.d. from three or four independent experiments as indicated and statistical significance was determined by student’s unpaired t-test (two-tailed).
Extended Data Fig. 3 |. 25HC does not directly affect bacterial infectivity or host viability.
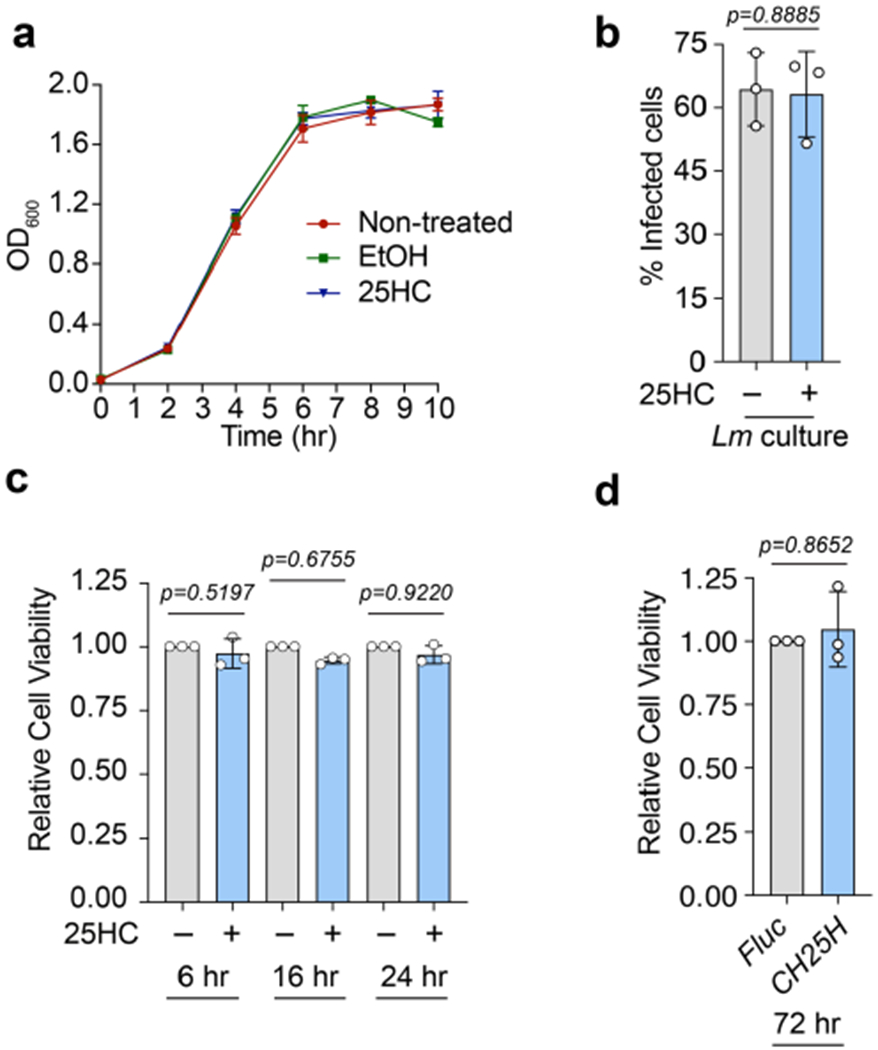
25HC could inhibit L. monocytogenes infection through different mechanisms. For example, it may (1) directly reduce bacterial viability, (2) inhibit the expression or function of bacterial virulence factors, (3) induce host cell death, or (4) regulate host cellular processes that limit bacterial infection. a, To determine whether 25HC directly reduced bacterial viability, a starting bacterial culture was back-diluted in DMEM (10% FBS) supplemented with vehicle (EtOH) or 25HC (5 μM). Bacterial cultures were incubated at 37°C while shaking at 200 rpm, and OD600 was measured for each sample at the indicated time points. Mean values from 3 independent experiments are plotted, and error bars show s.d. b, To determine whether 25HC directly modifies bacterial virulence, GFP-expressing L. monocytogenes were cultured overnight in BHI supplemented with vehicle (EtOH) or 25HC (5 μM) and HEK293A cells were then infected with bacteria from either culture (MOI = 20, 6 hours). Infection was analysed by flow cytometry. Bars represent mean values. Error bars show s.d. from three independent experiments and statistical significance was determined by student’s unpaired t-test (two-tailed). c-d, Host cell viability was assessed in cells treated with 25HC (c) or in cells virally transduced with CH25H (d). HEK293A cells were treated with 25HC (5 μM) or vehicle (EtOH) for 6, 16, or 24 hours (c) or transduced with Fluc or CH25H for 72 hours (d). Cell viability was evaluated by measuring ATP production using CellTiter-Glo assays. Data are normalized to vehicle (EtOH) in (c), and Fluc in (d). Bars represent mean values. Error bars show s.d. from three independent experiments and statistical significance was determined before normalization by student’s unpaired t-test (two-tailed).
Extended Data Fig. 4 |. 25HC has little effect on the early life-cycle stages of L. monocytogenes.
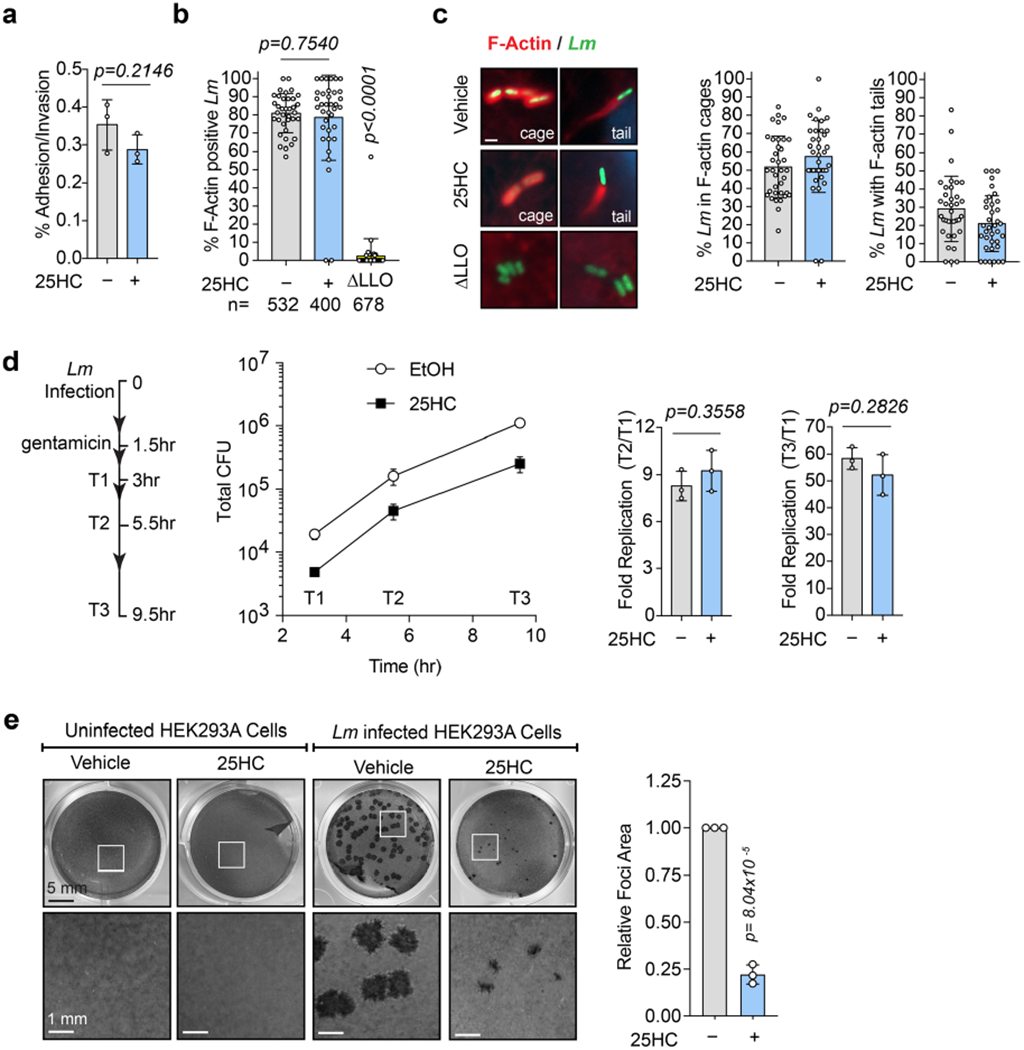
a, 25HC does not inhibit L. monocytogenes adhesion/invasion. The percent adhesion/invasion is shown. Bars represent mean values. Error bars show s.d. from three independent experiments and statistical significance was determined by student’s unpaired t-test (two-tailed). b, 25HC does not inhibit L. monocytogenes vacuole escape. The number of internalized bacteria that escape the phagocytic vacuole was determined by quantifying percent cytosolic L. monocytogenes that polymerize actin (F-actin cages or tails). L. monocytogenes lacking LLO was used as an escape-deficient control. Each data point represents the percent of bacteria that nucleate F-actin per field of view. The total number of individual bacteria assessed for F-actin nucleation is also indicated. Bars represent mean values. Error bars show s.d. and statistical significance was determined by student’s unpaired t-test (two-tailed). c, 25HC does not inhibit L. monocytogenes actin polymerization. Images (left) of GFP-expressing L. monocytogenes (green) and F-actin structures associated with cytosolic bacteria (phalloidin, red). Nuclei were labelled with DAPI (blue). Scale bar, 1 μm. Graph shows the frequency of F-actin structures nucleated by bacteria in host cells treated with vehicle or 25HC (5 μM). These data were collected from the experiments performed in (b). Bars represent mean values, and error bars are s.d. d, 25HC does not inhibit L. monocytogenes replication in host cells. Schematic indicating the time points of sample collection after bacterial infection. The total CFU recovered at each time point is shown (line graph). Bacterial replication was determined by calculating the ratio of CFU recovered at the indicated time points (T2 or T3) relative to the CFUs recovered after 3 hours of infection (T1). Mean values were plotted (left), and bars (right) represent mean values. Error bars show s.d. from three independent experiments and statistical significance was determined using student’s unpaired t-test (two-tailed). e, Left, representative images of L. monocytogenes cell-to-cell dissemination foci in HEK293A monolayers. Scale bar, 1 mm. Right, graph showing mean dissemination foci area formed by L. monocytogenes. Bars represent mean values. Error bars show s.d. of foci area normalized to vehicle-treated cells from three independent experiments. Statistical significance was determined prior to normalization by student’s unpaired t-test (two-tailed).
Extended Data Fig. 5 |. 25HC has no effect on the total cholesterol content of target cells and mobilizes accessible cholesterol in diverse cell types.

a, Total cellular cholesterol measurements of CHO-K1 cells treated for the indicated times with vehicle (EtOH), 25HC (5 μM), or HPCD (1% w/v), normalized to cellular protein content. Bars represent mean values. Error bars show s.d. from six independent experiments and statistical significance was determined by one-way ANOVA compared to vehicle with Dunnet’s correction. b, Immunoblots showing the effects of SMase treatment on cell surface binding of ALOD4 and OlyA in the indicated cell lines. Cells were treated without or with 25HC (5 μM) for 4 hours, followed by treatment with SMase (100 mU/ml) as indicated. Equal aliquots of cell lysates (10% of total) were subjected to immunoblot analysis. Data are representative of three independent experiments. c, CHO-7 cells were treated with 7α-HC (5μM) or vehicle (EtOH) for 16 hours, infected with GFP-expressing L. monocytogenes (MOI=1, 22 hours), and then subjected to flow cytometry analysis. Bars represent mean values. Error bars show s.d. from four independent experiments and statistical significance was determined by student’s unpaired t-test (two-tailed). d, Representative immunoblots from three independent experiments, measured by ALOD4 binding, quantification of which is shown in Fig. 5c. e, Immunoblots showing the effects of exogenously added epicholesterol or cholesterol on cell surface binding of ALOD4 to the indicated 25HC-treated cell lines. Cells were treated with 5 μM 25HC for 4 hours and then incubated with the indicated concentrations of epicholesterol or cholesterol (complexed to MCD) as described in Methods. Equal aliquots of cell lysates (10% of total; or 20 μg/lane for HEK293A) were subjected to ALOD4 immunoblot analysis. Data are representative of three independent experiments.
Extended Data Fig. 6 |. 25HC suppresses spread of S. flexneri through mobilization of accessible cholesterol.
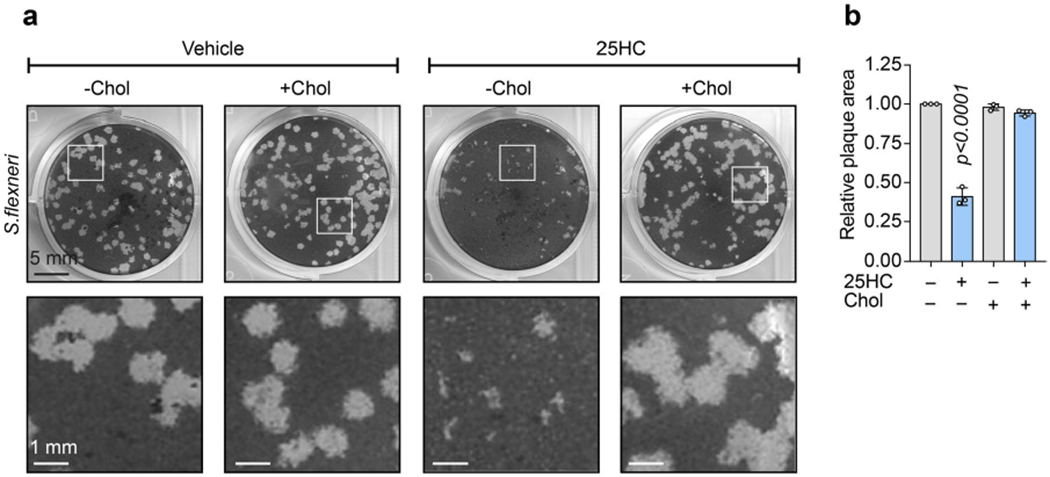
a, S. flexneri invades epithelial cells and disseminates from cell-to-cell. However, compared to L. monocytogenes, S. flexneri uses different molecular mechanisms and virulence factors. To test whether 25HC can inhibit S. flexneri by modulating accessible cholesterol, we carried out plaque-forming assays coupled with cholesterol repletion. HEK293A were treated with 25HC (5 μM) or vehicle for 16 hours, then 1 hour prior to S. flexneri infection, cells were treated with Chol/MCD complexes diluted in media (40 μM), or vehicle. Plaques were analysed 72 hours after avicel overlay. Representative images of three independent experiments are shown. Scale bar, 1 mm. b, Plaque area was quantified for assay described in (a), and normalized to vehicle-treated cells. Bars represent mean values. Error bars show s.d. from three independent experiments and statistical significance was determined before normalization by one-way ANOVA compared to vehicle with Dunnet’s correction.
Extended Data Fig. 7 |. 25HC regulation of cholesterol esterification and ALOD4 binding.
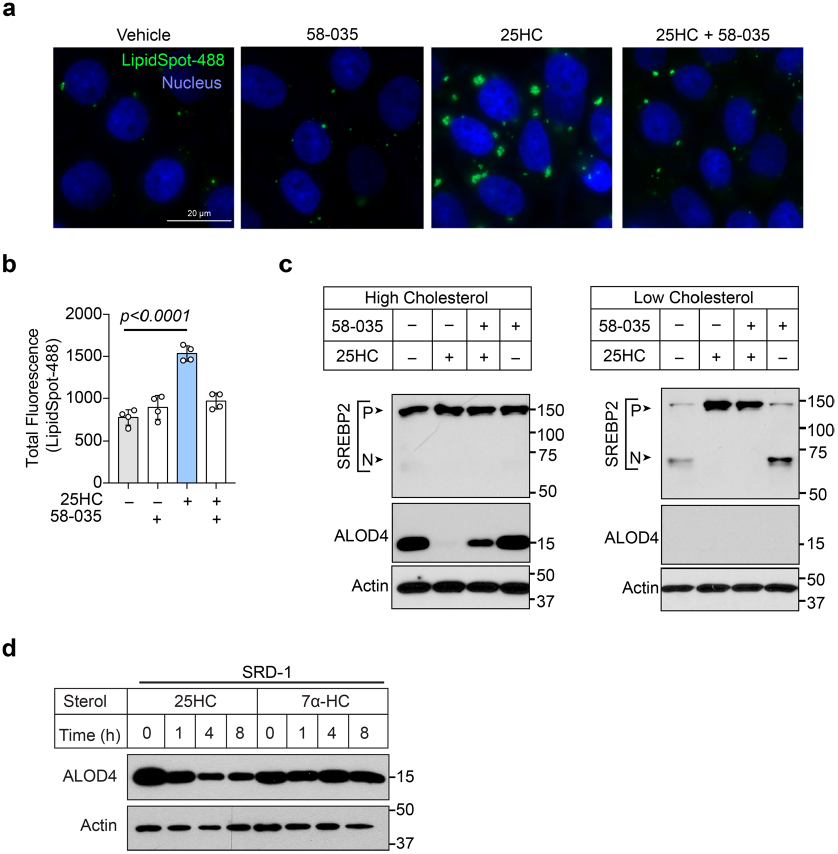
a, b, Examination of lipid droplet formation by microscopy (a; images) or flow cytometry (b; bar graph). CHO-7 cells were treated for 2 hours with SZ58-035 (10 μM) or vehicle (DMSO), and then treated with 25HC (5 μM) along with SZ58-035 (10 μM) or vehicle. For microscopy, CHO-7 cells were plated onto glass culture slides prior to treatments and fixed cells were incubated with DAPI to visualize nuclei (blue) and LipidSpot (green) to detect lipid droplet formation. Representative microscopy images are shown. Scale bar, 20 μm. For flow cytometry measurement of lipid droplets in treated CHO-7 cells, the total fluorescence (LipidSpot, Ex-488 nm) was calculated as the %LipidSpot+ cells multiplied by the geometric mean fluorescence intensity. Bars represent mean values. Error bars show s.d. from four independent experiments and statistical significance was determined by one-way ANOVA compared to vehicle-treated cells, with Dunnet’s correction. c, Immunoblot showing SREBP2 processing in cholesterol-replete (left blot) or cholesterol-depleted (right blot) CHO-K1 cells after treatment without or with 25HC in the presence or absence of 58-035, as described in Methods. P, precursor form of SREBP2; N, nuclear form of SREBP2. Blots are representative of three independent experiments. d, Cholesterol accessibility on PMs of CHO-7 mutant cells (SRD-1) constitutively expressing nuclear SREBP-2 treated with 25HC or 7α-HC was assessed by immunoblot analysis of ALOD4 binding, as described in Methods. A representative immunoblot from three independent experiments quantified in Fig. 6g is shown.
Extended Data Fig. 8 |. 25HC does not suppress L. monocytogenes infection via LXR stimulation.
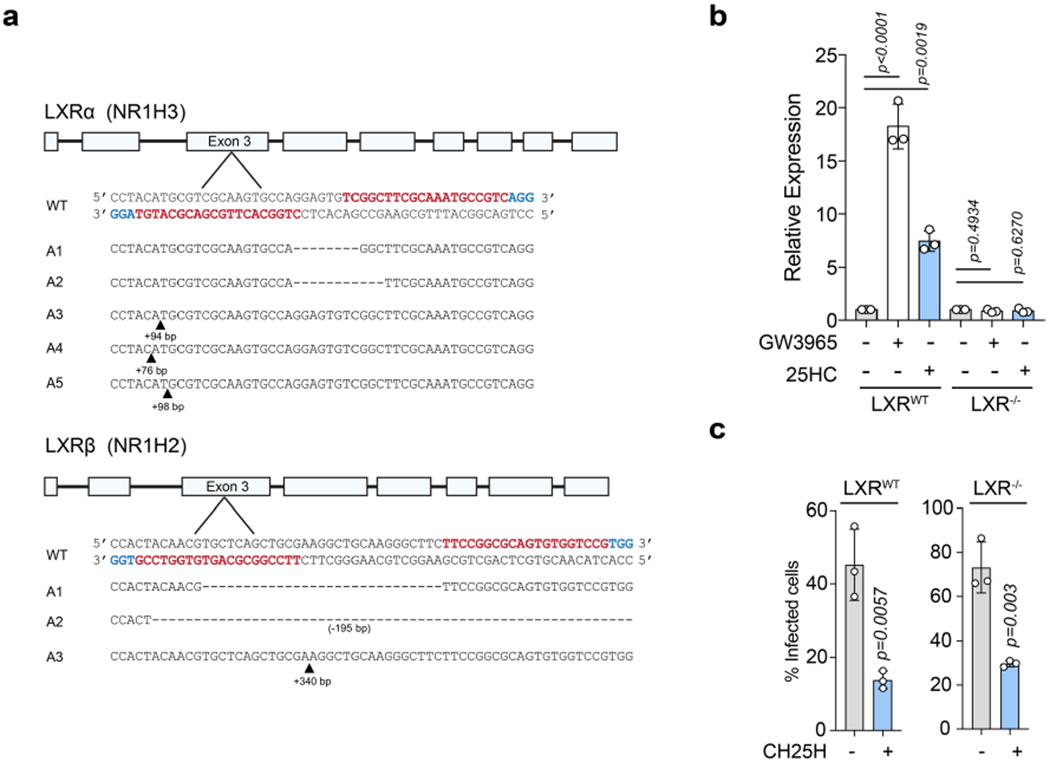
a, Strategy to generate LXRα/β-deficient cells via CRISPR/Cas9 editing. Genomic sequencing of LXRα/β-deficient HEK293A demonstrating indel formation in each targeted exon is shown. The 20 bp guides are highlighted in red, while the 3 bp PAM sequence is highlighted in blue. Alignments to WT reference DNA using Needle software is shown for each allele. b, To confirm loss of LXR signalling, Wild-type and LXRα/β-deficient HEK293A were stimulated with LXR agonists (2.5 μM GW3965 or 5 μM 25-HC) or vehicle for 24 hours. Total RNA was extracted and mRNA levels of the LXR target gene ABCA1 was determined by qPCR. Expression levels were normalized to non-treated samples, for LXR WT and LXRα/β-deficient cells, respectively. Bars represent mean values. Error bars show s.d. from three independent experiments and statistical significance was determined by one-way ANOVA compared to vehicle-treated cells, with Dunnet’s correction. c, Wild-type and LXRα/β-deficient HEK293A cells were transduced with lentivirus co-expressing Tag-RFP and Fluc or CH25H. After 48 hour transduction, cells were infected with GFP-L monocytogenes (MOI =10) for 6 hours and analysed by flow cytometry. Bars represent mean values. Error bars show s.d. from three independent experiments and statistical significance was determined by student’s unpaired t-test (two-tailed).
Extended Data Fig. 9 |. Model of plasma membrane cholesterol remodelling by circulating oxysterols.
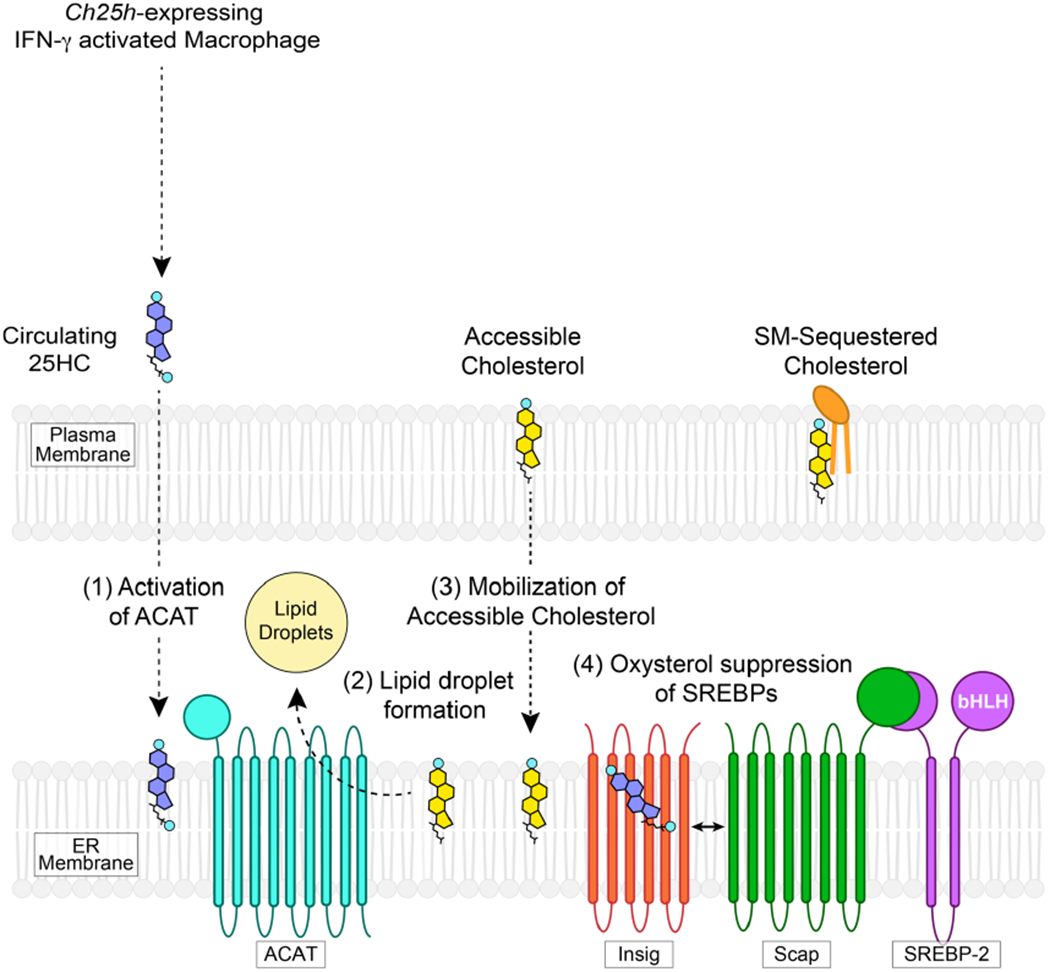
Model of 25HC-mediated regulation of accessible cholesterol. (1) 25HC secreted from IFN-γ activated macrophage enters the target cell and stimulates the enzymatic activity of ACAT (Step 1). ACAT activation results in production of cholesteryl esters that are incorporated into lipid droplets (Step 2). Cholesterol esterification lowers the free cholesterol levels in the ER, triggering internalization of accessible cholesterol from the plasma membrane (Step 3). Long-term suppression of accessible cholesterol is achieved through 25HC-mediated inhibition of the SREBP2 pathway which leads to lower cholesterol synthesis and uptake (Step 4).
Supplementary Material
Acknowledgements
We thank D. Gammon and E. Rex for assistance with the live-cell imaging, D. Vazquez for assistance with protein purification, and R. Debose-Boyd, D. W. Russell, J. Goldstein and M. Brown for reagents and helpful discussions. We also thank the members of the Alto, Schoggins and Radhakrishnan laboratories for their helpful discussions. This research was supported by grants from the National Institutes of Health (grant nos. AI083359 to N.M.A., AI117922 to J.W.S. and HL20948 to A.R.), Welch Foundation (grant nos. I-1731 to N.M.A. and I-1793 to A.R.), and grants to N.M.A. from the Burroughs Welcome (grant no. 1011019) and the Howard Hughes Medical Institute and Simons Foundation Faculty Scholars Program (grant no. 55108499).
Footnotes
Data availability
Data collected during this study is included in the Source Data and Supplementary Information. The data that support the findings of this study are available from the corresponding author on request.
Code availability
No new code was used to analyse the findings in this study.
Competing interests
The authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41564-020-0701-5.
Supplementary information is available for this paper at https://doi.org/10.1038/s41564-020-0701-5.
References
- 1.Schroder K, Hertzog PJ, Ravasi T & Hume DA Interferon-γ: an overview of signals, mechanisms and functions. J. Leukoc. Biol 75, 163–189 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Jouanguy E et al. A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat. Genet 21, 370–378 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Jouanguy E et al. IL-12 and IFN-γ in host defense against mycobacteria and salmonella in mice and men. Curr. Opin. Immunol 11, 346–351 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Radoshevich L & Cossart P Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol 16, 32–46 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Portnoy DA, Auerbuch V & Glomski IJ The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. J. Cell Biol 158, 409–414 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bauman DR et al. 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl Acad. Sci. USA 106, 16764–16769 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park K & Scott AL Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons. J. Leukoc. Biol 88, 1081–1087 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanc M et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 38, 106–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lund EG, Kerr TA, Sakai J, Li WP & Russell DW cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J. Biol. Chem 273, 34316–34327 (1998). [DOI] [PubMed] [Google Scholar]
- 10.Dang EV, McDonald JG, Russell DW & Cyster JG Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell 171, 1057–1071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reboldi A et al. Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon. Science 345, 679–684 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perelman SS et al. Cell-based screen identifies human interferon-stimulated regulators of Listeria monocytogenes infection. PLoS Pathog. 12, e1006102 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kandutsch AA & Chen HW Inhibition of sterol synthesis in cultured mouse cells by 7α-hydroxycholesterol, 7β-hydroxycholesterol, and 7-ketocholesterol. J. Biol. Chem 248, 8408–8417 (1973). [PubMed] [Google Scholar]
- 14.Brown MS & Goldstein JL Suppression of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity and inhibition of growth of human fibroblasts by 7-ketocholesterol. J. Biol. Chem 249, 7306–7314 (1974). [PubMed] [Google Scholar]
- 15.Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS & Goldstein JL Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc. Natl Acad. Sci. USA 104, 6511–6518 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown MS, Dana SE & Goldstein JL Cholesterol ester formation in cultured human fibroblasts. Stimulation by oxygenated sterols. J. Biol. Chem 250, 4025–4027 (1975). [PubMed] [Google Scholar]
- 17.Chang TY, Chang CC, Ohgami N & Yamauchi Y Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Dev. Biol 22, 129–157 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Repa JJ & Mangelsdorf DJ The liver X receptor gene team: potential new players in atherosclerosis. Nat. Med 8, 1243–1248 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Das A, Brown MS, Anderson DD, Goldstein JL & Radhakrishnan A Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. eLife 3, e02882 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Infante RE & Radhakrishnan A Continuous transport of a small fraction of plasma membrane cholesterol to endoplasmic reticulum regulates total cellular cholesterol. eLife 6, e25466 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Endapally S et al. Molecular Discrimination between two conformations of sphingomyelin in plasma membranes. Cell 176, 1040–1053 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gay A, Rye D & Radhakrishnan A Switch-like responses of two cholesterol sensors do not require protein oligomerization in membranes. Biophys. J 108, 1459–1469 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson KA, Endapally S, Vazquez DC, Infante RE & Radhakrishnan A Ostreolysin A and anthrolysin O use different mechanisms to control movement of cholesterol from the plasma membrane to the endoplasmic reticulum. J. Biol. Chem 294, 17289–17300 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ray K, Marteyn B, Sansonetti PJ & Tang CM Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat. Rev. Microbiol 7, 333–340 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Brown MS, Radhakrishnan A & Goldstein JL Retrospective on cholesterol homeostasis: the central role of scap. Annu. Rev. Biochem 87, 783–807 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janowski BA, Willy PJ, Devi TR, Falck JR & Mangelsdorf DJ An oxysterol signalling pathway mediated by the nuclear receptor LXRa. Nature 383, 728–731 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Chang TY, Chang CC & Cheng D Acyl-coenzyme A:cholesterol acyltransferase. Annu. Rev. Biochem 66, 613–638 (1997). [DOI] [PubMed] [Google Scholar]
- 28.Cheng D, Chang CC, Qu X & Chang TY Activation of acyl-coenzyme A:cholesterol acyltransferase by cholesterol or by oxysterol in a cell-free system. J. Biol. Chem 270, 685–695 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Ross AC, Go KJ, Heider JG & Rothblat GH Selective inhibition of acyl coenzyme A:cholesterol acyltransferase by compound 58–035. J. Biol. Chem 259, 815–819 (1984). [PubMed] [Google Scholar]
- 30.Yang J, Sato R, Goldstein JL & Brown MS Sterol-resistant transcription in CHO cells caused by gene rearrangement that truncates SREBP-2. Genes Dev. 8, 1910–1919 (1994). [DOI] [PubMed] [Google Scholar]
- 31.Hannedouche S et al. Oxysterols direct immune cell migration via EBI2. Nature 475, 524–527 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nelson ER et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 342, 1094–1098 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corcoran RB & Scott MP Oxysterols stimulate Sonic hedgehog signal transduction and proliferation of medulloblastoma cells. Proc. Natl Acad. Sci. USA 103, 8408–8413 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dwyer JR et al. Oxysterols are novel activators of the hedgehog signaling pathway in pluripotent mesenchymal cells. J. Biol. Chem 282, 8959–8968 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Kinnebrew M et al. Cholesterol accessibility at the ciliary membrane controls hedgehog signaling. eLife 8, e50051 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu SY et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38, 92–105 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi WS et al. The CH25H–CYP7B1–RORα axis of cholesterol metabolism regulates osteoarthritis. Nature 566, 254–258 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Ortiz A et al. An interferon-driven oxysterol-based defense against tumor-derived extracellular vesicles. Cancer Cell 35, 33–45 e36 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flanagan JJ, Tweten RK, Johnson AE & Heuck AP Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 48, 3977–3987 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Metherall JE, Goldstein JL, Luskey KL & Brown MS Loss of transcriptional repression of three sterol-regulated genes in mutant hamster cells. J. Biol. Chem 264, 15634–15641 (1989). [PubMed] [Google Scholar]
- 41.Brown AJ, Sun L, Feramisco JD, Brown MS & Goldstein JL Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol. Cell 10, 237–245 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Sanda C et al. Differential gene induction by type I and type II interferons and their combination. J. Interferon Cytokine Res 26, 462–472 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Sa SM et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J. Immunol 178, 2229–2240 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Xue J et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang SH et al. Microarray analysis of cytokine activation of apoptosis pathways in the thyroid. Endocrinology 148, 4844–4852 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Waddell SJ et al. Dissecting interferon-induced transcriptional programs in human peripheral blood cells. PLoS ONE 5, e9753 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pawliczak R et al. Influence of IFN-γ on gene expression in normal human bronchial epithelial cells: modulation of IFN-γ effects by dexamethasone. Physiol. Genomics 23, 28–45 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Rock RB et al. Transcriptional response of human microglial cells to interferon-γ. Genes Immun. 6, 712–719 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Johnson-Huang LM et al. A single intradermal injection of IFN-γ induces an inflammatory state in both non-lesional psoriatic and healthy skin. J. Invest. Dermatol 132, 1177–1187 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Indraccolo S et al. Identification of genes selectively regulated by IFNs in endothelial cells. J. Immunol 178, 1122–1135 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Hu X, Park-Min KH, Ho HH & Ivashkiv LB IFN-γ-primed macrophages exhibit increased CCR2-dependent migration and altered IFN-γ responses mediated by Stat1. J. Immunol 175, 3637–3647 (2005). [DOI] [PubMed] [Google Scholar]
- 52.He XS et al. Differential transcriptional responses to interferon-α and interferon-γ in primary human hepatocytes. J. Interferon Cytokine Res 30, 311–320 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fairfax BP et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343, 1246949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheon H et al. IFNβ-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 32, 2751–2763 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schoggins JW et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Seiler CY et al. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res. 42, D1253–D1260 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Campeau E et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE 4, e6529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McDonald JG, Smith DD, Stiles AR & Russell DW A comprehensive method for extraction and quantitative analysis of sterols and secosteroids from human plasma. J. Lipid Res 53, 1399–1409 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Endapally S, Infante RE & Radhakrishnan A Monitoring and modulating intracellular cholesterol trafficking using ALOD4, a cholesterol-binding protein. Methods Mol. Biol 1949, 153–163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bourdeau RW et al. Cellular functions and X-ray structure of anthrolysin O, a cholesterol-dependent cytolysin secreted by Bacillus anthracis. J. Biol. Chem 284, 14645–14656 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.