

Abstract
Background
Despite significant endeavor having been applied to identify effective therapies to treat glioblastoma (GBM), survival outcomes remain intractable. The greatest nonsurgical benefit arises from radiotherapy, though tumors typically recur due to robust DNA repair. Patients could therefore benefit from therapies with the potential to prevent DNA repair and synergize with radiotherapy. In this work, we investigated the potential of salinomycin to enhance radiotherapy and further uncover novel dual functions of this ionophore to induce DNA damage and prevent repair.
Methods
In vitro primary GBM models and ex vivo GBM patient explants were used to determine the mechanism of action of salinomycin by immunoblot, flow cytometry, immunofluorescence, immunohistochemistry, and mass spectrometry. In vivo efficacy studies were performed using orthotopic GBM animal xenograft models. Salinomycin derivatives were synthesized to increase drug efficacy and explore structure-activity relationships.
Results
Here we report novel dual functions of salinomycin. Salinomycin induces toxic DNA lesions and prevents subsequent recovery by targeting homologous recombination (HR) repair. Salinomycin appears to target the more radioresistant GBM stem cell–like population and synergizes with radiotherapy to significantly delay tumor formation in vivo. We further developed salinomycin derivatives which display greater efficacy in vivo while retaining the same beneficial mechanisms of action.
Conclusion
Our findings highlight the potential of salinomycin to induce DNA lesions and inhibit HR to greatly enhance the effect of radiotherapy. Importantly, first-generation salinomycin derivatives display greater efficacy and may pave the way for clinical testing of these agents.
Keywords: DNA damage, drug discovery, glioblastoma, homologous recombination
Key Points.
Salinomycin activates autophagy to impair homologous recombination.
Salinomycin induces DNA breaks via replication fork collapse.
Salinomycin derivatives show significantly increased efficacy.
Importance of the Study.
GBM is an aggressive cancer known to resist standard DNA damage–based therapies. Therapeutic strategies with the ability to induce DNA damage and inhibit DNA repair hold merit and could be used to increase the effect of radiotherapy. Here, we identified that salinomycin has the potential to induce DNA damage and disrupt HR-mediated repair. Development of first-generation salinomycin derivatives improves drug efficacy and paves the way for testing of these agents in the clinic.
Glioblastoma (GBM) is a highly aggressive tumor with a poor prognosis. Current treatment involves surgical resection, ionizing radiation (IR), and DNA alkylation with temozolomide (TMZ).1 GBM commonly displays resistance to these therapies and tumors invariably recur. This is in part due to the robust DNA repair mechanisms present within these tumors.2–4 The 2 major repair pathways capable of resolving DNA double-strand breaks (DSBs) are relatively exclusive during cell-cycle progression.5 Non-homologous end joining (NHEJ) is most active during G1-phase.6 To date, no therapeutic benefit has been implicated by targeting NHEJ in GBM.3,4 Homologous recombination (HR), which functions during S- to G2-phase, is the principal pathway used to resolve DNA DSBs arising from replication fork collapse. Evidence shows that upregulation of HR repair components correlates with worse prognosis in GBM.7 Furthermore, GBM is dependent on HR repair to promote resistance against DNA alkylating-based therapies, including TMZ.8,9 To eliminate radioresistance, current alkylating-based strategies require the concomitant use of DNA repair inhibitors.10,11 While effective, this approach poses significant toxicity risk. The present effort has shed light on characterizing a single agent capable of inducing both DNA damage and repair inhibition with favorable synergistic properties.
Salinomycin is an ionophore first isolated from Streptomyces albus. Its potent anticancer function was more recently identified in the treatment of breast cancer, where it showed greater efficacy than paclitaxel and targeted the cancer stem cell population.12 Calzolari and colleagues first discovered that salinomycin synergized with tumor necrosis factor apoptosis inducing ligand, leading to apoptosis in GBM.13 Subsequent studies have outlined the potential of salinomycin to target GBM as well as synergize with TMZ.14–16 Despite these past studies, little is known about the direct function of salinomycin. We therefore sought to characterize in detail the effect of salinomycin and understand its synergistic properties. Obstacles to the clinical translation of salinomycin do exist—these include potential issues of adverse effects from administration at high doses and selective penetrance of the blood–brain barrier.17 However, clinical reports also indicate that salinomcyin is well tolerated at low doses.18 Given the favorable anticancer properties of salinomycin, we sought to develop first-generation derivatives with the hope of increasing efficacy and in turn reducing potential toxicity issues.
Our study confirms the findings of others showing the potent antitumor effect of salinomycin and the ability to target the stem cell–like population in GBM. Further, we identify previously uncharacterized functions of salinomycin, demonstrating its ability to induce DNA DSBs and prevent HR. These properties enable salinomycin to synergize with radiotherapy in vitro and in vivo. We also developed salinomycin derivatives with enhanced pharmacokinetic (PK) properties. A salinomycin-benzene derivative displayed enhanced PK while retaining its synergistic properties. In summary, this study reveals previously unknown functions of salinomycin and defines a first-generation derivative which may be suitable for future clinical testing.
Methods
Antibodies, primers, buffers, synthesis of derivatives, drug toxicity profiling, and proteomics details can be found in the Supplementary Material.
Cell Culture
GBM specimens were collected at the Royal Brisbane and Women’s Hospital (ethical approval, HREC/17/QRBW/577). GBM patient-derived cells (WK1: classical, SB2: mesenchymal, MMK1, and JK2: proneural subtypes) were cultured under glioma neural stem (GNS) conditions. GNS cell lines have been fully characterized. Data are available at Q-Cell19 (https://www.qimrberghofer.edu.au/our-research/commercialisation/q-cell/).
Animal Orthotopic Xenograft
The animal ethical committee of Queensland Institute of Medical Research (QIMR) approved the use of non-obese diabetic/severe combined immunodeficiency (NOD/SCID) and NOD/recombination activating gene 1 (NOD/RAG) mice for orthotopic engraftment and intracranial and intraperitoneal injection. Mice received stereotactic-guided injection of live WK1-luc cells (1 × 104–5 × 105). Dead cells were excluded using trypan blue prior to engraftment. For intratumoral drug administration studies, a guide screw (Plastics One) was used. Dimethyl sulfoxide (DMSO), salinomycin, and its derivative were given followed by mock or IR (2 Gy). QML Pathology provided the drug toxicity service. Tumor progression was monitored by bioluminescent imaging. As per QIMR ethical guidelines, animals were sacrificed upon signs of tumor burden.
Ex Vivo Explant Slice Assay
De-identified GBM specimens were collected immediately postsurgery, specimens were sliced (750 µM thickness) using a vibratome (World Precision Instruments), transferred onto a polytetrafluoroethylene membrane insert (Millipore), and maintained in Dulbecco’s modified Eagle’s medium:Hank’s Balanced Salt Solution media with horse serum.20
DNA Repair Reporter
DNA DSB repair was determined by measuring NHEJ (linearized HindIII pEGFP-N3) and HR (pDRGFP) activity as described.21
Immunoblotting
Whole cell lysate was used. Forty micrograms were separated with 6%, 8%, and 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and transferred to nitrocellulose membranes (BioRad) using a wet transfer system.
Inhibitors and Small Interfering RNA Knockdown
To examine protein degradation, MG132 (20 µM), MLN4924 (1 µM), CDC34 (50 µM), or PYR-41 (50 µM) was used to inhibit 26S proteasome and E1, E2, and E3 ubiquitin, respectively. 3-Methyladenine (5 mM) and leptomycin B (LMB (20 nM) were employed to inhibit autophagosome formation and nuclear protein export. Nucleosides (40 µM) were used as a supplement. Small interfering (si)RNA control, extracellular signal-regulated kinase 1 and 2 (ERK1/2), and autophagy related 7 (ATG7) were performed per manufacturing protocol. Aphidicolin (APH) (10 µg/mL) or nocodazole (NOCO) (1 µM) was used for cell synchronization.
Quantitative Real-Time PCR
RNA extraction was performed according to the manufacturer’s instructions (Sigma-Aldrich). All reactions were performed in triplicate by using a SYBR green master mix at quarter reaction and carried out in a ViiA 7 Real-time PCR Cycler (Applied Biosystems). Average cycle thresholds were determined by ABI software. Transcript level of mRNA was determined by the relative expression of β-actin.
Salinomycin-Biotin Pulldowns
Whole cell lysate was pre-cleared with dynabeads (streptavidin, Invitrogen). Separately, salinomycin-biotin and biotin (100 µg) were prepared by incubating with dynabeads. Coated beads were rinsed with NaCl (400 mM) solution. Whole cell lysate (1 mg) was incubated with salinomycin-biotin or biotin-bounded dynabeads at 4°C overnight. Samples underwent 0.05% NP-40 washes to remove nonspecific binding and protein eluted with urea (8 M) for label-free quantification (see Supplementary Material).
Immunofluorescence
Cells were permeabilized with cytoskeletal buffer, fixed with 2% paraformaldehyde, and blocked with 5% bovine serum albumin. Antibodies were performed at 1:500 anti-γH2AX and 1:100 anti-RAD51, followed by the use of fluorophore-conjugated antibody 1:400 (Invitrogen) and Hoechst stain. Images were acquired by AxioScop2 (Zeiss), 780 nonlinear optical confocal (Zeiss), or IN Cell (GE Healthcare).
Nucleotide Pool Extraction
Cell pellets were resuspended in ice-cold methanol (60%), vortexed, and heated at 95°C. Supernatant was collected and nucleotides level was determined as described.22
Single-Strand Fiber and DNA Pulse Labeling
Native single-strand (ss)DNA fiber (non-denatured) analysis was performed as described.23 Cells were pulse-labeled with iododeoxyuridine (IdU) (75 µM) for 16 hours. The reaction was stopped by ice-cold versene and treated accordingly. Approximately 100 ssDNA fibers were scored per experiment. To determine native single-strand bromodeoxyuridine (BrdU) foci, cells were pulse-labeled with IdU (10 µM) for 16 hours prior to treatment. For double-stranded (ds)BrdU labeling, G1/S synchronized cells were treated and then replaced with fresh culture containing 10 µM of IdU. Cells were incubated for 6 hours prior to fixation, permeabilization, and DNA denaturation by HCl (1 M).
Cell Cycle, Cell Death, Cell Division Proliferation and Flow Cytometry
Cells were fixed with 70% ethanol (−20°C) overnight and subsequently incubated with propidium iodide. Cell cycle was measured using flow cytometry. Cell death was performed by using an annexin V‒fluorescein isothiocyanate apoptosis detection kit (BD Pharmingen) or NucGreen viable dye (Invitrogen). We employed the IncuCyte system (Essen Instruments) to determine proliferation. For surface marker analysis, cells were immunolabeled with CD133–Alexa Fluor 488 (Miltenyi) and performed as described.24 In cell division analysis, cells were pre-labeled with carboxyfluorescein succinimidyl ester (CFSE) (10 µM) before quenching. Data were analyzed by ModFit.
Neurosphere Formation
Orthotopic xenograft tumor was dissociated, treated, and grown as a single cell suspension. Neurospheres were captured 7 days later with a digital microscope (Evos Cell Imaging).
Immunohistochemistry
Samples were fixed in 10% neutral buffered formalin and embedded in paraffin. Sections were stained with hematoxylin and eosin (H&E). Antigen retrieval was performed using a pH 6.0 citrate buffer (125°C) for 15 min. Sections were probed with the respective antibodies and stained as described.24
Synthesis of Salinomycin Derivatives
The salinomycin-biotin, 20-O-acetyl (Sal-Ac), 20-O-benzoyl (Sal-Bz), and the truncated F1 and F2 derivatives were prepared as described (see Supplementary Material).25–27
Data Analysis
Unless otherwise stated, all data represent the means ± SEM of 3 independent experiments. Where appropriate, comparisons were conducted using a two-tailed Student’s t-test or ANOVA. Asterisks indicate statistical significance (*P < 0.05; **P < 0.01; ***P < 0.001).
Results
Salinomycin Prevents the Resolution of DNA Damage
Radioresistant GBM relies on the DNA damage response (DDR) cascade to survive IR and TMZ treatment.28 However, a growth inhibitory effect was observed when GNS cells received salinomycin alone and in combination with IR (2 Gy) (Fig. 1A). Unresolved DNA DSBs were noted in the majority of salinomycin-treated cells, as indicated by the presence of γH2AX foci in the nucleus (Fig. 1B, Supplementary Fig. 1A). We postulated that salinomycin could promote a defect in cell-cycle arrest, DNA repair inhibition or both. In the presence of salinomycin, cell-cycle arrest appears normal (Supplementary Fig. 1B). We next investigated the more predominant of the 2 major DNA DSB repairs, NHEJ. Salinomycin-treated cells indicated functional pS2056DNA-PKcs, which is required for NHEJ (Supplementary Fig. 1C). This was further confirmed using an NHEJ green fluorescent protein (GFP) reporter essentially showing no difference (Fig. 1C). Investigation of the HR pathway using a similar reporter demonstrated salinomycin-treated cells had complete HR inhibition (Fig. 1D).21 RAD51, a major component for HR, was absent in the nucleus of salinomycin-treated cells following IR (Fig. 1E, Supplementary Fig. 1D).
Fig. 1.

Salinomycin prevents the resolution of DNA DSBs. (A) Salinomycin (Sal) dose response 72 hours post-treatment ± IR (2 Gy). (B) 24 hours post-Sal (0.5 µM–10 µM) treatment. DNA DSBs induction was assessed by γH2AX foci (green) and nuclear staining (blue). Each dot represents the foci number of individual cells. (C, D) DNA DSB (NHEJ and HR) repair was determined by quantifying GFP positive cells (above diagonal line) at 48 hours post-DMSO or post-Sal treatment (middle). NHEJ and HR was depicted as fold change (bottom). (E) RAD51 positive cells (>5 foci per nucleus) were assessed following DMSO (blue) or Sal (red) treatment (10 µM) followed by IR (2 Gy). Statistical significance: **P < 0.01, ***P < 0.001.
Salinomycin Promotes Autophagy to Target Homologous Recombination
Nuclear RAD51 foci were analyzed to determine HR induction. GNS cells were pretreated with LMB, a nuclear protein export inhibitor, followed by combined salinomycin and IR. RAD51 recruitment to the nucleus was absent, indicating salinomycin could be inhibiting this process (Fig. 2A). Salinomycin did not affect RAD51 mRNA levels (Supplementary Fig. 2A). However, we noted loss of RAD51 protein following salinomycin treatment. Furthermore, treatment with the proteasome inhibitor MG132 did not recover RAD51, indicating protein degradation was not occurring via this pathway (Fig. 2B). To further explore salinomycin function, a biotin conjugate was generated. Proteins that bound to the salinomycin-biotin were subjected to label-free quantification by liquid chromatography–tandem mass spectrometry. Analysis using MaxQuant identified 95 interacting proteins (Fig. 2C). Gene enrichment analysis (Gene Ontology, Kyoto Encyclopedia of Genes and Genomes, and Reactome) revealed 3 functional clusters (Fig. 2D).29,30 Two of these were closely associated: mitogen-activated protein kinase (MAPK) signaling and proteolysis. Validation of MAPK (pT202/T204ERK1/2) signaling confirmed salinomycin-induced autophagy, as shown by the conversion of light chain (LC)3I into LC3II (Fig. 2E, Supplementary Fig. 2B and 2C).31,32 Inhibiting autophagosome (A) formation confirmed that the loss of RAD51 was autophagy mediated. Since the ubiquitin-conjugating enzymes (E3, E2, and E1) are different protein degradation pathways, inhibition of these cascades was unlikely to recover RAD51 (Fig. 2F). To demonstrate autophagy-mediated loss of RAD51, we targeted ATG7, which is essential for autophagosome maturation. ATG7 knockdown using 2 independent siRNAs rescued RAD51 protein levels (Fig. 2G, Supplementary Fig. 2D). Furthermore, ATG7 siRNA treated cells (autophagy deficient) showed HR recovery and displayed partial resistance to salinomycin (Fig. 2H, Supplementary Fig. 2E). These data combined indicate that salinomycin exerts its role not in NHEJ but through autophagy to inhibit HR.
Fig. 2.

Salinomycin induces autophagy to target HR. (A) GNS cells were given LMB (20 nM) to inhibit nuclear protein export followed by DMSO or Sal (10 µM) with IR (2 Gy). Six hours posttreatment RAD51 foci were assessed (white arrows). (B) One hour prior to treatment, GNS cells were given the proteasome inhibitor (MG132, 20 µM) following DMSO or Sal treatment (10 µM). RAD51 protein was determined by immunoblot. (C) Schematic of the label-free quantification approach used for drug–protein interaction study. A Venn diagram depicted the number of interacting proteins with Sal-biotin or biotin (false discovery rate <0.05). (D) The functionally grouped network of proteins that interacted with Sal-biotin. Three proteins per term as nodes linked with ≥0.4 kappa score level. Overrepresented term per group was shown. (E) Immunoblot to validate MAPK and autophagy in Sal-treated cells (10 µM). (F) GNS cells received ubiquitin E3 (PYR-41, 50 µM), E2 (CDC34, 50 µM), E1 (MLN4924, 1 µM) ligases and autophagosome (A) (3-Methyladenine, 5 mM) inhibitors as indicated (+) for one hour prior to Sal treatment (10 µM). The vertical line represents a splice mark between samples from the same gel. (G) RAD51 and LC3 levels were assessed by immunoblot in siControl versus siATG7 knockdown cells. Following siRNA knockdown, cell proliferation was assessed 48 hours post-Sal treatment (2.5 µM–10 µM). Statistical significance: **P < 0.01, ***P < 0.001.
Salinomycin Induces Replication Fork Breakage
Nucleotide metabolism was the third cluster identified in our mass spectrometry analysis. This indicated that salinomycin could target nucleotide production and affect DNA replication.33 Ribonucleotide reductase is responsible for deoxyribonucleotide triphosphate (dNTP) synthesis, and investigation showed its catalytic subunit ribonucleotide reductase regulatory subunit M2 (RRM2) was downregulated following salinomycin treatment (Fig. 3A).34 In the event of an exhausted nucleotide pool, replication fork stall can occur and give rise to ssDNA. To prevent DNA deterioration, replication protein A (RPA) is recruited.35 Indeed, salinomycin depleted dNTPs (Fig. 3B) and caused ssDNA formation, which correlated with increased RPA foci (Supplementary Fig. 3A and 3B). Oxidative damage was ruled out as a source for ssDNA induction (Supplementary Fig. 3C). Using an independent approach to assess replication fork stall, we performed ssDNA fiber analysis and confirmed salinomycin gave rise to ssDNA fibers (Fig. 3C). Without HR, replication fork stall leads to DNA DSBs and is detrimental to cell survival.36,37 To investigate this phenomenon, salinomycin-treated cells were assessed for the colocalization of ssDNA and γH2AX foci. We observed a >2-fold increase for both markers (Fig. 3D, Supplementary Fig. 3D). When GNS cells undergo replication fork stall, cell-cycle arrest typically occurs at S-phase. Aphidicolin was employed to synchronize GNS cells in G1/S phase. Upon release, salinomycin-treated cells showed S-phase blockage (Fig. 3E, Supplementary Fig. 3E). Subsequent analysis of dsBrdU uptake confirmed that replication was absent (Supplementary Fig. 3F). To strengthen the notion that salinomycin causes replication fork breakage, we examined the accumulation of γH2AX foci during cell-cycle progression. In an asynchronous condition, moderate accumulation of γH2AX foci was observed following salinomycin treatment (Fig. 3F, Supplementary Fig. 3G). However, this effect was exacerbated in APH-synchronized cells when progressing from G1/S to S phase to initiate replication. Salinomycin caused rapid γH2AX foci induction as a result of replication fork breakage. γH2AX foci were absent following APH synchronization in DMSO-treated cells (Fig. 3G, Supplementary Fig. 3H). To further assess replication fork breakage, we also synchronized GNS cells in G2/M phase with NOCO. During the progression from G2/M to G1 phase, we did not detect DNA DSBs following salinomycin treatment (Fig. 3H, Supplementary Fig. 3I). Additionally, we attempted to rescue the effect of DNA DSBs by supplementing nucleosides, which showed reduced γH2AX foci and partial recovery in cell survival (Fig. 3I and 3J). Taken together, these data indicate salinomycin induces replication fork breakage in rapidly dividing GBM cells.
Fig. 3.

Salinomycin causes replication-associated DNA DSBs. (A) RRM2 protein expression was assessed by immunoblot following Sal treatment (10 µM). (B) dNTP level was measured 24 hours post-Sal treatment (10 µM). (C) ssDNA combing strategy used (top) and representative ssDNA fiber (middle). GNS cells were pre-labeled with BrdU for 24 hours prior to DMSO or Sal treatment (10 µM) for 8 hours to determine native ssDNA fibers (bottom). (D) GNS cells were pre-labeled with BrdU prior to DMSO or Sal treatment (10 µM) for 8 hours. Positive cells (%) showing dual ssDNA and γH2AX foci (>5 foci per nucleus, bottom). (E) GNS cells were synchronized in G1/S phase using APH. Cell-cycle analysis was conducted following the release and subsequent DMSO or Sal (10 µM) treatment. (F) Analysis of γH2AX positive cells (>5 foci per nucleus) following Sal treatment (10 µM) in asynchronous cells. (G, H) GNS cells were synchronized using APH (G1/S phase) or NOCO (G2 phase). DNA DSBs were determined by quantifying γH2AX positive cells (>5 foci per nucleus) following Sal treatment (10 µM). (I) GNS cells received PBS or nucleoside supplement (40 µM) prior to Sal treatment (10 µM). γH2AX positive cells (>5 foci per nucleus) were assessed 12 hours post-treatment. (J) Cell proliferation was assessed 48 hours post-Sal treatment (1 µM–10 µM) in PBS versus nucleoside supplemented cells (40 µM). Statistical significance: *P < 0.05, **P < 0.01.
Salinomycin Targets the Radioresistant Stem Cell–Like Population
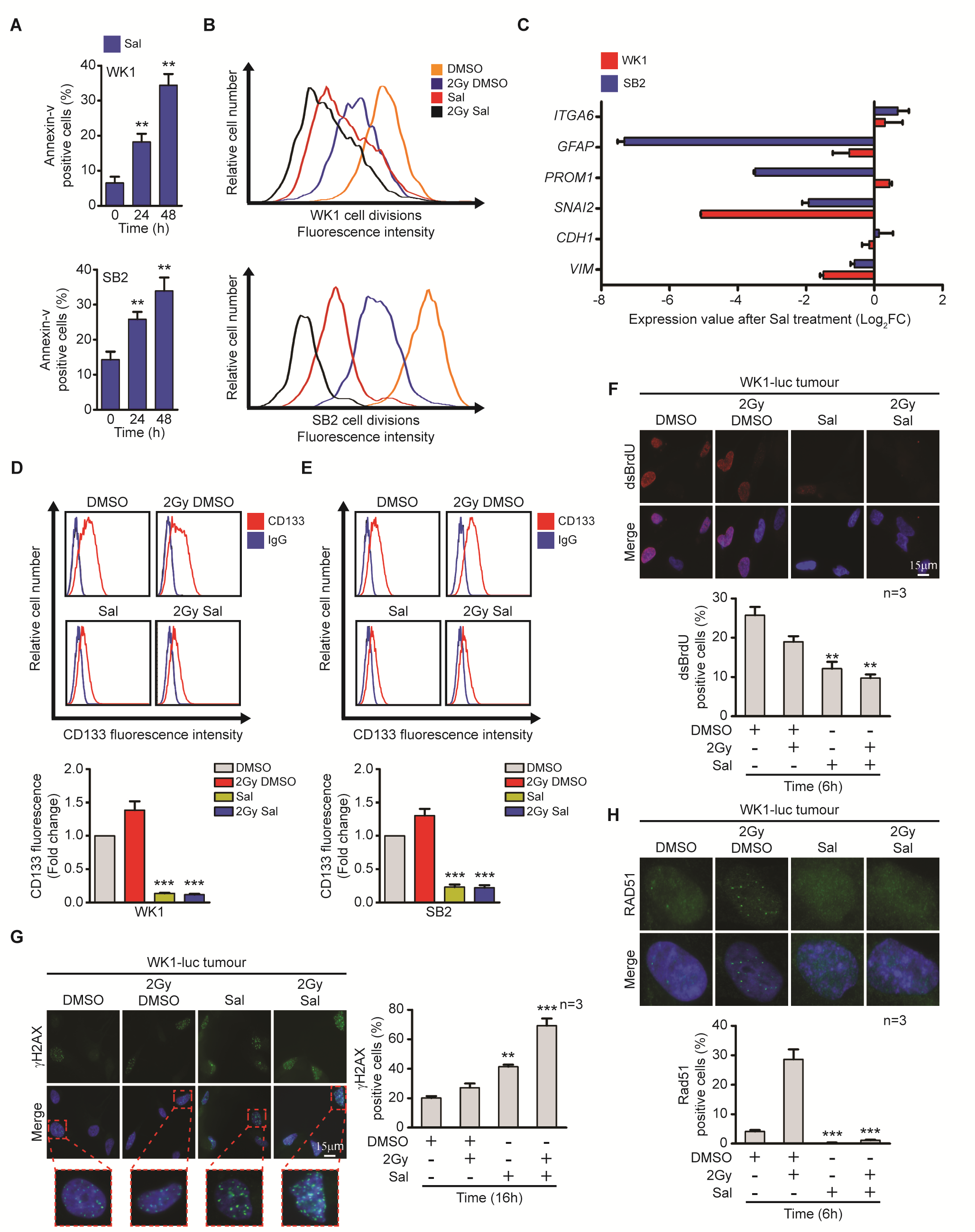
Salinomycin-treated cells showed chromatin condensation, DNA fragmentation, and subsequent cell death (Fig. 4A). Our previous studies indicate that GBM cell survival is heavily reliant on elevated myeloid cell leukemia 1 (MCL-1) levels and that other anti-apoptotic protein members are lowly expressed.38 Following salinomycin treatment, we observed downregulation of MCL-1 followed by apoptosis, which was exacerbated when combined with IR (Fig. 4B and 4C, Supplementary Fig. 4A).38 Previous findings have highlighted the ability of salinomycin to target the more stem cell–like population.14 To confirm these findings, we first tracked cell division via CFSE staining, following treatment and subsequent drug withdrawal. Results showed reduced cell division even after withdrawal (Fig. 4D, Supplementary Fig. 4B). Despite not being exhaustive, CD133 has been proposed as a glioma stem cell marker.2,39 In our analysis, salinomycin eliminated the CD133+ cell population (Supplementary Fig. 4C–E). We also adopted an ex vivo approach where orthotopically engrafted GBMs were dissociated and subsequently treated with salinomycin. Using this assay, salinomycin also eliminated the CD133+ cell population. Even with the removal of salinomycin for 4 days, tumor cells did not recover CD133 expression (Fig. 4E). We also employed a neurosphere assay as a surrogate readout for stemness.24 Following salinomycin treatment and subsequent 7 days recovery, a significant reduction in primary and secondary neurospheres were observed (Fig. 4F). Furthermore, salinomycin prevented ex vivo tumor cells from undergoing replication (Supplementary Fig. 4F). This included DNA DSB formation and HR inhibition (Supplementary Fig. 4G and 4H).
Fig. 4.

Salinomycin targets the radioresistant stem cell–like population. (A) Intact, condensed, and fragmented nuclei (4′,6′-diamidino-2-phenylindole blue) were assessed 24 hours post-DMSO versus post-Sal (10 µM) treatment, quantitation (right). (B) Flow cytometry was performed to determine cell death following Sal treatment (10 µM) with ± IR (2 Gy). (C) Immunoblot was performed to determine MCL-1 protein expression following Sal (10 µM) with ± IR (2 Gy) treatment. (D) CFSE was used to track cell division, GNS cells were treated with Sal (10 µM) ± IR (2 Gy) and allowed to recover for 72 hours prior to labeling. Cell division was assessed 96 hours later. (E, F) WK1-luc tumor cells were isolated from orthotopic xenografts at day 60 and received Sal (10 µM) with ± IR (2 Gy) treatment for 72 hours prior to CD133 flow cytometric expression analysis as indicated; CD133 expression was assessed during treatment (treated) and after Sal had been removed for 96 hours (recovery). Neurosphere formation was assessed 7 days post-treatment withdrawal as indicated. Statistical significance: **P < 0.01, ***P < 0.001.
Salinomycin Synergizes with Ionizing Radiation In Vivo
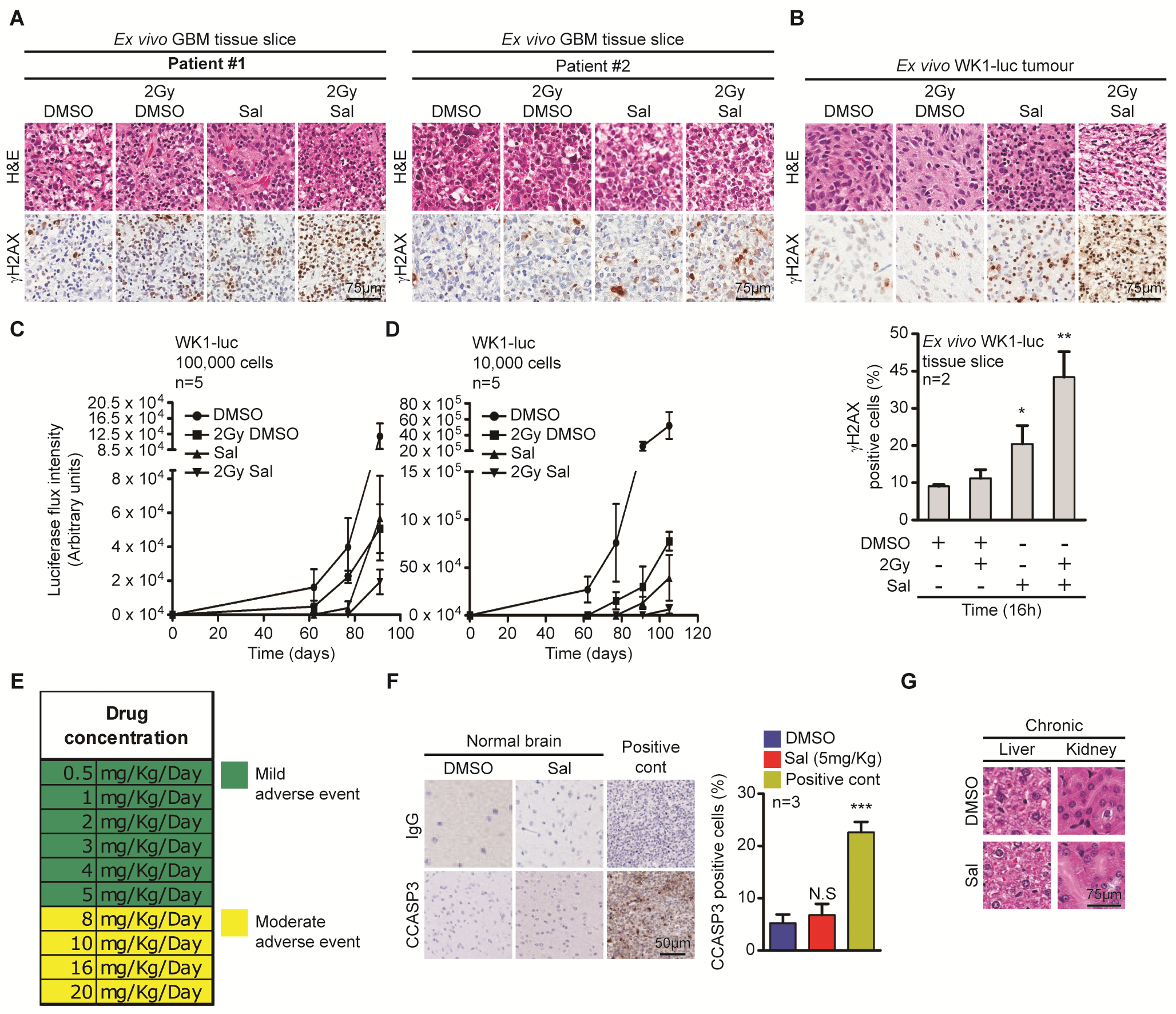
Firstly, to examine the effect of salinomycin in vivo we adopted an organotypic GBM slice culture technique.40 GBM patient specimens were obtained at time of surgery and cut into 750 µM slices and cultured (Fig. 5A). Two GBM patient specimens (#1 and #2) were treated with salinomycin, IR, or the combination. Immunohistochemistry (IHC) analysis showed presence of γH2AX positive cells following salinomycin treatment, which was exacerbated by IR (Fig. 5B, Supplementary Fig. 5A). Orthotopic GBM xenograft tissue was also assessed using the same approach and demonstrated a similar response (Supplementary Fig. 5B). We investigated an additional 2 GBM patient specimens (#3 and #4), using immunoblot. Results also showed elevated γH2AX and loss of RAD51 following salinomycin treatment (Fig. 5C). As an initial in vivo study, WK1-luc cells were pretreated with salinomycin for 72 hours. Prior to engraftment, dead cells were excluded by using trypan blue (Fig. 5D, Supplementary Fig. 5C and 5D). Animals engrafted with 1 × 104 WK1-luc cells showed a median survival of 120 days in DMSO-treated versus IR = 140 days, salinomycin = 152 days, and the combination = 164 days. Animals engrafted with 1 × 105 WK1-luc cells showed a median survival of 112 days for DMSO-treated versus IR = 120 days, salinomycin = 136 days, and combination = 149 days. In both experiments, salinomycin and combination therapy led to an increase in survival. As previously reported, salinomycin may have adverse effects at high doses, especially from systemic injection.17 We therefore adopted an approach of direct intratumoral administration via a guide screw. Our toxicity studies showed salinomycin 5 mg/kg (based on brain weight) was the maximum tolerated dose with no observable signs of neurotoxicity (Supplementary Fig. 5E and 5F). To assess drug efficacy, WK1-luc engrafted animals were randomized to 4 treatment arms: DMSO, IR, salinomycin, and combined treatment. A single IR (2 Gy) dose was administered 48 hours post-engraftment (Fig. 5E, top). Mice received 8 doses of salinomycin (total dose = 40 mg/kg) followed by a control cull 5 days after the last treatment to examine acute systemic toxicity. Salinomycin was well tolerated with no elevation of liver and kidney enzymes to indicate tissue injury (Fig. 5F). Overall survival analysis showed DMSO-treated mice had a median survival of 114 days. When combined, salinomycin (5 mg/kg) was able to synergize with IR to increase overall survival to 134 days (Fig. 5E, right). At endpoint, liver and kidneys were collected, and H&E examination did not indicate observable long-term tissue damage (Supplementary Fig. 5G).
Fig. 5.

Salinomycin synergizes with IR to prolong survival in vivo. (A) GBM patient organotypic slice cultures were harvested 16 hours post-Sal (10 µM) with ± IR (2 Gy) treatment. (B) IHC was conducted to assess γH2AX positive cells post-treatment (patient specimens #1 and #2) and (C) γH2AX and RAD51 were determined by immunoblot (patient specimens #3 and #4). (D) WK1-luc cells received 72 hours of Sal treatment (10 µM) with ± IR (2 Gy). Dead cells were excluded by trypan blue, 1 × 104 and 1 × 105 WK1-luc cells were engrafted orthotopically into NOD/SCID mice and survival determined by Kaplan–Meier plot. (E) Intracranial studies were conducted using a guide screw approach, a total dose of 40 mg/kg (brain weight) of DMSO versus Sal in 8 fractions of 5 mg/kg was given. (F) Control cull (n = 4) was performed 5 days post-treatment. Serum was collected for the analysis of liver (alanine aminotransferase, alkaline phosphatase, and creatinine) and kidney (urea) injuries. Statistical significance: *P < 0.05, **P < 0.01.
Salinomycin-Benzene Derivative Shows Increased Efficacy In Vivo
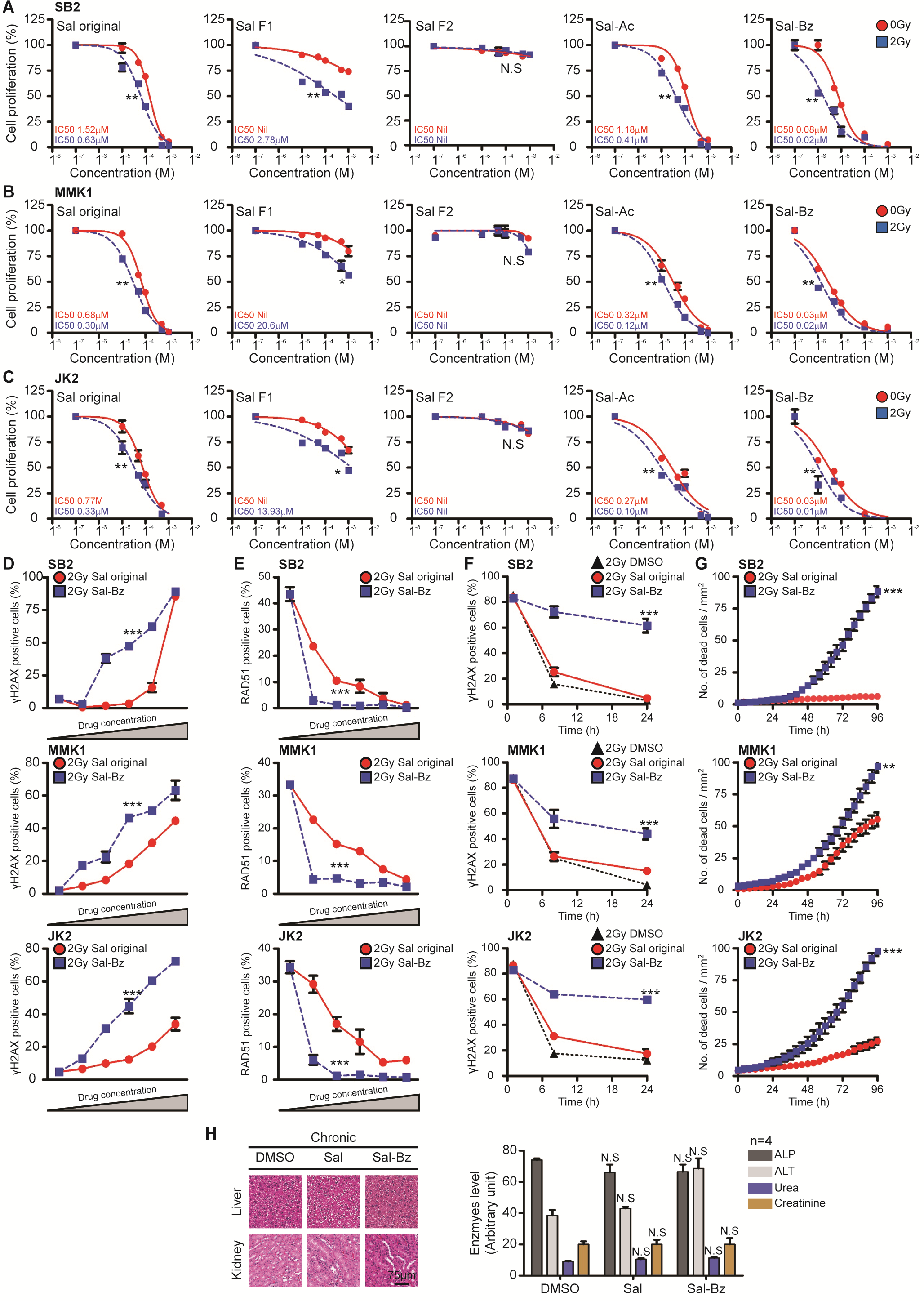
Given the favorable properties of salinomycin, we sought to develop first-generation derivatives with increased efficacy. Two derivatives with alterations at the carbon 20 position, an acetate (Sal-Ac) and a benzene (Sal-Bz), were generated (Fig. 6A). We also generated a salinomycin truncation, introduced at position C9 (Sal-F1 and Sal-F2), to determined which portion of the molecule was responsible for function. Of the 2 derivatives tested, Sal-Bz displayed the highest potency across all GNS cell lines (Fig. 6B, Supplementary Fig. 6A–6C). The half-maximal inhibitory concentration (IC50) of Sal-Bz ranged from 80 nM to 30 nM and synergized with IR (IC50 of 10 nM to 20 nM). Sal-Bz was ~20-fold more potent than the original salinomycin. Conversely, Sal-Ac, Sal-F1, and Sal-F2 were less effective. Similar to salinomycin, Sal-Bz showed identical DNA damage and repair inhibition functions 24 hours posttreatment. Sal-Bz at 500 nM and 1 µM resulted in 27% and 43% of γH2AX positive cells, respectively (Fig. 6C, Supplementary Fig. 6D). In comparison, salinomycin at the same concentration showed reduced DNA damage and repair inhibition (Fig. 6D, Supplementary Fig. 6E). When combined with IR, Sal-Bz showed significant synergy compared with salinomycin at the same concentration (Fig. 6E, Supplementary Fig. 6F), and resulted in greater cell death (Fig. 6F, Supplementary Fig. 6G).
Fig. 6.

Benzene derivative analysis in vitro and in vivo. (A) Diagram showing the generated structural analogues of salinomycin. (B) A dose response curve of cell proliferation was used to compare derivatives versus Sal 96 hours posttreatment. (C, D) γH2AX or RAD51 positive cells (>5 foci per cell) were assessed by immunofluorescence to compare the response between Sal and Sal-Bz (1 µM–10 µM). (E, F) GNS cells were pretreated for one hour with 1 µM of DMSO, Sal, or Sal-Bz in combination with IR (2 Gy). γH2AX positive cells and cell death were quantified using IncuCyte analysis. (G) WK1-luc cells were intracranially engrafted in NOD/RAG mice; animals received a total dose of 10 mg/kg (brain weight) of DMSO, Sal, or Sal-BZ in 8 fractions of 1.25 mg/kg and survival determined by Kaplan–Meier plot. Statistical significance: **P < 0.01, ***P < 0.001. (H) Simplified model of the mechanism of action of salinomycin.
We next assessed the ability of Sal-Bz to synergize with IR in vivo. Toxicity testing (Sal-Bz 1.25 mg/kg × 8 schedules, total dose 10 mg/kg) showed good drug tolerance with no observable organ damage (Supplementary Fig. 6H). To assess overall survival, mice were engrafted with WK1-luc cells and randomized to 3 treatment arms: DMSO, Sal + IR, and Sal-Bz + IR. A single intratumoral injection of salinomycin or Sal-Bz (1.25 mg/kg) was given prior to IR (2 Gy). This was followed by 7 salinomycin or Sal-Bz treatments (total dose 10 mg/kg). Median survival for DMSO-treated animals was 118 days versus 131 days in the Sal + IR arm. The Sal-Bz + IR arm showed a significantly better response (P < 0.001); by day 200 only a single animal had succumbed to tumor burden when the experiment was terminated (Fig. 6G). Taken together, these data show that the beneficial properties of salinomycin were retained in the Sal-Bz derivative with greater efficacy. Furthermore, Sal-Bz showed significant synergy in vivo when administered with IR compared with salinomycin at the same concentration.
Discussion
Here we show that salinomycin has dual functions of inducing replication fork collapse and HR inhibition, and displays significant synergy when administered with IR. Our findings are consistent with the findings of others demonstrating the synergistic properties of salinomycin when combined with DNA alkylating-based chemotherapies.14,16 Current evidence suggests that autophagy can mediate HR and is likely cancer-type dependent.41,42 We found that salinomycin induces an autophagy-mediated response leading to RAD51 downregulation. The depletion of HR components renders DNA repair unresponsive and leaves the replication machinery prone to DNA damage (see model in Fig. 6H).35 Even when autophagy was inhibited by targeting ATG7, HR-mediated repair could be partially restored following salinomycin treatment. This is due in part to the absence of RRM2, which is critical for nucleotide production. Low dNTP levels support the notion of nucleotide exhaustion in obstructing fork progression, resulting in DNA DSBs. A critical matter with active DNA repair is the issue of restoring tumorigenesis. Alkylating-based drugs such as TMZ and radiotherapy can promote DNA replication fork stall; this DNA damage is resolved by HR, leading to tumor recurrence.8,9 In our study, salinomycin showed its greatest benefit when combined with IR. Significant synergy was observed in our animal studies despite only a single dose of radiotherapy being given, suggesting that the dual properties of DNA damage and repair inhibition increase IR effectiveness.
Our GBM organotypic model is clinically relevant as it preserves tumor heterogeneity and in part recapitulates the native state of radioresistance.40 These GBM patient specimens showed that RAD51 was upregulated in response to radiotherapy, which was subsequently abrogated by salinomycin treatment. Unresolved DNA DSBs were noted in multiple patient specimens. The clinical relevance of salinomycin, in its native form, is uncertain, with potential side effects being observed from systemic administration at high doses.17 Despite this limitation, the field has recognized the benefits of salinomycin and is developing next-generation derivatives with significant potential.18,43 We developed and tested a benzene derivative, which maintained the beneficial characteristics of the original drug while significantly improving efficacy. The addition of a benzene group typically aids cell permeability, allowing greater drug transfer and stability. This enabled us to administer less drug, reducing toxicity while increasing the synergistic effects when combined with radiotherapy.
In summary, we have performed an in-depth analysis of salinomycin, further elucidating its mechanism of action, and validated the efficacy of a first-generation salinomycin derivative using preclinical models of GBM. The translational significance of this study is relevant as salinomycin and its derivatives hold the potential to greatly improve radiotherapy, a modality which has long held the greatest benefit for GBM patients.
Funding
This work was supported by Cancer Council QLD (1061216 and 1098841), Rio Tinto Ride to Conquer Cancer, Brain Cancer Discovery Collaborative, Novo Nordisk Foundation (NNF16OC0023146 and NNF17OC0026056), and the National Institute of Health (AI136581 and AI150451 to B.K).
Conflict of interest statement. All authors declare no conflicts.
Authorship statement. Y.C.L., B.S., A.B., J.C., T.J., P.H., and B.D. conceptualized the manuscript. Y.C.L., B.S., C.O., R.D., V.C., A.W., A.B., and B.D. investigated the findings. Y.C.L., K.E., H.Q., Z.B., A.K., A.B., B.S., C.O., J.C., F.L., T.R., K.K., M.L., A.W., T.J., A.B., M.C., F.S., B.M., and B.D. performed the analysis. L.W., R.J., F.L., J.C., P.H., K.K., A.B., V.C., T.J., B.K., and M.C. provided the resource. Y.C.L., A.B., R.J., L.W., T.R., A.B., C.O., R.D., P.H., B.K., V.C., and B.D. performed the writing. A.B., K.K., P.H., T.R., M.L., and B.D. supervised the findings.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1. Hegi ME, Diserens AC, Gorlia T, et al. . MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. [DOI] [PubMed] [Google Scholar]
- 2. Bao S, Wu Q, McLendon RE, et al. . Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. [DOI] [PubMed] [Google Scholar]
- 3. Lim YC, Roberts TL, Day BW, et al. . Increased sensitivity to ionizing radiation by targeting the homologous recombination pathway in glioma initiating cells. Mol Oncol. 2014;8(8):1603–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Short SC, Martindale C, Bourne S, Brand G, Woodcock M, Johnston P. DNA repair after irradiation in glioma cells and normal human astrocytes. Neuro Oncol. 2007;9(4):404–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008;7(18):2902–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwartz EI, Smilenov LB, Price MA, et al. . Cell cycle activation in postmitotic neurons is essential for DNA repair. Cell Cycle. 2007;6(3):318–329. [DOI] [PubMed] [Google Scholar]
- 7. Knijnenburg TA, Wang L, Zimmermann MT, et al. ; Cancer Genome Atlas Research Network Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018;23(1):239–254.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gil Del Alcazar CR, Todorova PK, Habib AA, Mukherjee B, Burma S. Augmented HR repair mediates acquired temozolomide resistance in glioblastoma. Mol Cancer Res. 2016;14(10):928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ohba S, Mukherjee J, See WL, Pieper RO. Mutant IDH1-driven cellular transformation increases RAD51-mediated homologous recombination and temozolomide resistance. Cancer Res. 2014;74(17):4836–4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carruthers RD, Ahmed SU, Ramachandran S, et al. . Replication stress drives constitutive activation of the DNA damage response and radioresistance in glioblastoma stem-like cells. Cancer Res. 2018;78(17):5060–5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sun C, Yin J, Fang Y, et al. . BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33(3):401–416.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gupta PB, Onder TT, Jiang G, et al. . Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138(138):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Calzolari A, Saulle E, De Angelis ML, et al. . Salinomycin potentiates the cytotoxic effects of TRAIL on glioblastoma cell lines. PLoS One. 2014;9(4):e94438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen T, Yi L, Li F, et al. . Salinomycin inhibits the tumor growth of glioma stem cells by selectively suppressing glioma-initiating cells. Mol Med Rep. 2015;11(4):2407–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang F, Zheng Z, Guan J, et al. . Identification of a panel of genes as a prognostic biomarker for glioblastoma. EBioMedicine. 2018;37:68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xipell E, Aragón T, Martínez-Velez N, et al. . Endoplasmic reticulum stress-inducing drugs sensitize glioma cells to temozolomide through downregulation of MGMT, MPG, and Rad51. Neuro Oncol. 2016;18(8):1109–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lagas JS, Sparidans RW, van Waterschoot RA, Wagenaar E, Beijnen JH, Schinkel AH. P-glycoprotein limits oral availability, brain penetration, and toxicity of an anionic drug, the antibiotic salinomycin. Antimicrob Agents Chemother. 2008;52(3):1034–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Naujokat C, Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. J Biomed Biotechnol. 2012;2012:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stringer BW, Day BW, D’Souza RCJ, et al. . A reference collection of patient-derived cell line and xenograft models of proneural, classical and mesenchymal glioblastoma. Sci Rep. 2019;9(1):4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gogolla N, Galimberti I, DePaola V, Caroni P. Preparation of organotypic hippocampal slice cultures for long-term live imaging. Nat Protoc. 2006;1(3):1165–1171. [DOI] [PubMed] [Google Scholar]
- 21. Kijas AW, Lim YC, Bolderson E, et al. . ATM-dependent phosphorylation of MRE11 controls extent of resection during homology directed repair by signalling through Exonuclease 1. Nucleic Acids Res. 2015;43(17):8352–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Diamond TL, Roshal M, Jamburuthugoda VK, et al. . Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem. 2004;279(49):51545–51553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cruz-García A, López-Saavedra A, Huertas P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014;9(2):451–459. [DOI] [PubMed] [Google Scholar]
- 24. Day BW, Stringer BW, Al-Ejeh F, et al. . EphA3 maintains tumorigenicity and is a therapeutic target in glioblastoma multiforme. Cancer Cell. 2013;23(2):238–248. [DOI] [PubMed] [Google Scholar]
- 25. Borgström B, Huang X, Pošta M, Hegardt C, Oredsson S, Strand D. Synthetic modification of salinomycin: selective O-acylation and biological evaluation. Chem Commun (Camb). 2013;49(85):9944–9946. [DOI] [PubMed] [Google Scholar]
- 26. Horita K, Nagato S, Oikawa Y, Yonemitsu O. Highly stereocontrolled total synthesis of the polyether antibiotic salinomycin. IV. Chemical degradation of salinomycin for the structure confirmation of synthesic key intermediates. Chem Pharm Bull. 1989;37(7):1726–1730. [Google Scholar]
- 27. Zhang W, Wu J, Li B, et al. . Structure–activity & structure–toxicity relationship study of salinomycin diastereoisomers and their benzoylated derivatives. Org Biomol Chem. 2016;14(10):2840–2845. [DOI] [PubMed] [Google Scholar]
- 28. Erasimus H, Gobin M, Niclou S, Van Dyck E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat Res Rev Mutat Res. 2016;769:19–35. [DOI] [PubMed] [Google Scholar]
- 29. Bindea G, Mlecnik B, Hackl H, et al. . ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morris JH, Knudsen GM, Verschueren E, et al. . Affinity purification-mass spectrometry and network analysis to understand protein-protein interactions. Nat Protoc. 2014;9(11):2539–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen H, Ma Z, Vanderwaal RP, et al. . The mTOR inhibitor rapamycin suppresses DNA double-strand break repair. Radiat Res. 2011;175(2):214–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J, Whiteman MW, Lian H, et al. . A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J Biol Chem. 2009;284(32):21412–21424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pfister SX, Markkanen E, Jiang Y, et al. . Inhibiting WEE1 selectively kills histone H3K36me3-deficient cancers by dNTP starvation. Cancer Cell. 2015;28(5):557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. D’Angiolella V, Donato V, Forrester FM, et al. . Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell. 2012;149(5):1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chu WK, Payne MJ, Beli P, Hanada K, Choudhary C, Hickson ID. FBH1 influences DNA replication fork stability and homologous recombination through ubiquitylation of RAD51. Nat Commun. 2015;6:5931. [DOI] [PubMed] [Google Scholar]
- 36. Zimmer J, Tacconi EMC, Folio C, et al. . Targeting BRCA1 and BRCA2 deficiencies with G-quadruplex-interacting compounds. Mol Cell. 2016;61(3):449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Toledo LI, Altmeyer M, Rask MB, et al. . ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155(5):1088–1103. [DOI] [PubMed] [Google Scholar]
- 38. Day BW, Stringer BW, Spanevello MD, et al. . ELK4 neutralization sensitizes glioblastoma to apoptosis through downregulation of the anti-apoptotic protein Mcl-1. Neuro Oncol. 2011;13(11):1202–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Singh SK, Hawkins C, Clarke ID, et al. . Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. [DOI] [PubMed] [Google Scholar]
- 40. Bayin NS, Ma L, Thomas C, et al. . Patient-specific screening using high-grade glioma explants to determine potential radiosensitization by a TGF-β small molecule inhibitor. Neoplasia. 2016;18(12):795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Y, Liu F, Wang Y, et al. . Rapamycin-induced autophagy sensitizes A549 cells to radiation associated with DNA damage repair inhibition. Thorac Cancer. 2016;7(4):379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Y, Zhang N, Zhang L, et al. . Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol Cell. 2016;63(1):34–48. [DOI] [PubMed] [Google Scholar]
- 43. Mai TT, Hamaï A, Hienzsch A, et al. . Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat Chem. 2017;9(10):1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.