

Abstract
Background
Glioblastoma-initiating cells (GICs) comprise a tumorigenic subpopulation of cells that are resistant to radio- and chemotherapies and are responsible for cancer recurrence. The aim of this study was to identify novel compounds that specifically eradicate GICs using a high throughput drug screening approach.
Methods
We performed a cell proliferation/death-based drug screening using 10 560 independent compounds. We identified dihydroorotate dehydrogenase (DHODH) as a target protein of hit compound 10580 using ligand-fishing and mass spectrometry analysis. The medical efficacy of 10580 was investigated by in vitro cell proliferation/death and differentiation and in vivo tumorigenic assays.
Results
Among the effective compounds, we identified 10580, which induced cell cycle arrest, decreased the expression of stem cell factors in GICs, and prevented tumorigenesis upon oral administration without any visible side effects. Mechanistic studies revealed that 10580 decreased pyrimidine nucleotide levels and enhanced sex determining region Y–box 2 nuclear export by antagonizing the enzyme activity of DHODH, an essential enzyme for the de novo pyrimidine synthesis.
Conclusion
In this study, we identified 10580 as a promising new drug against GICs. Given that normal tissue cells, in particular brain cells, tend to use the alternative salvage pathway for pyrimidine synthesis, our findings suggest that 10580 can be used for glioblastoma therapy without side effects.
Key Points
1. Chemical screening identified 10580 as a novel GIC-eliminating drug that targets DHODH, an essential enzyme for the de novo pyrimidine synthesis pathway.
2. Compound 10580 induced cell cycle arrest, apoptosis, and differentiation in GICs.
Keywords: chemical screening; DHODH; glioblastoma-initiating cells, GICs; Sox2; O-GlcNAc
Importance of the Study.
Despite the tremendous efforts for developing new therapeutic methods for glioblastoma (GBM), the prognosis of GBM remains poor. One of the reasons is the lack of methods that can eradicate GICs, which show invasive ability, tumorigenicity, and resistance to chemoradiotherapies. We established human temozolomide-resistant GICs (GICRs), which are thought to contribute recurrence. We performed a large-scale drug screening using GICRs and identified a potential lead compound, 10580. We show here that 10580 specifically induced cell cycle arrest, apoptosis, and differentiation in GICs by directly inhibiting DHODH, a key enzyme in the de novo pyrimidine synthesis pathway. Given that normal tissue cells, in particular brain cells, tend to use the alternative salvage pathway for pyrimidine synthesis, our findings suggest that 10580 can be used for GBM therapy without side effects.
Glioblastoma (GBM) is a malignant glioma with a median survival of approximately 15 months.1,2 Despite the development of multimodal treatments involving surgery, chemotherapy, and radiotherapy, the overall survival rate of patients with GBM has not improved over the past few decades. The discovery of GBM-initiating cells (GICs) has strongly affected the direction of GBM research, as these cells have been shown to possess strong tumorigenic ability and to be resistant to irradiation and anticancer drugs such as temozolomide (TMZ).3–7 It is therefore crucial to characterize GICs and to find new drugs that specifically target and kill them. However, the identification of new compounds through drug screening is problematic because bona fide GICs have not been successfully maintained as a homogeneous population in culture.8,9 We previously established TMZ-resistant GIC lines (GICRs), in which bona fide GICs are likely to be enriched, and performed a small-scale drug screen using these cell lines; in this screen, we identified multiple drugs that killed both GICs and GICRs but not normal neural stem cells (NSCs).10 Unfortunately, the identified drugs were toxic to mice upon long-term administration. Herein, we screened a large number of drugs, including uncharacterized compounds, and elucidated their pharmacological mechanisms with the goal of identifying compounds that eradicate cancer but are not toxic to mice.
Materials and Methods
Animals and Chemical Reagents
All experiments with mice were performed according to protocols approved by the Animal Care and Use Committees of Hokkaido University. The mice were obtained from Hokudo, and chemicals and growth factors were purchased from Invitrogen and PeproTech, respectively, except as otherwise indicated.
Cell Culture
Human GIC lines, E3, E6, and E16, and GICR lines, E3R, E6R, and E16R, were cultured as described previously.10,11 To examine cell proliferation, the cells were labeled with bromodeoxyuridine (BrdU; 10 μM) for 6 h, fixed, and stained for BrdU as previously described.12 To prevent 10580-dependent cell death, E6 and E16 GICs were cultured in the presence of the pan-caspase inhibitor benzyloxycarbonyl-L-valyl-L-alanyl-[(2S)-2-amino-3-(methoxycarbonyl)propionyl] fluoromethane (Z-VAD-FMK) (50 μM, Peptide Institute) for 2 days and then used for MTT assay and sex determining region Y–box 2 (SOX2) immunostaining.
Chemical Screening
In order to identify drugs that induced cell cycle arrest or cell death in GICRs, we conducted chemical screening using a Fujifilm library containing 10 560 compounds dissolved in dimethyl sulfoxide (DMSO) at 5 mM, as described previously.10 This chemical library was constructed with compounds, which were synthesized by a chemist at Fujifilm, and consist of several classes of molecules such as reducing agents and pigment compounds synthesized in the construction of photography and printing technology.
Ligand-Fishing and Sample Preparation for Mass Spectrometry
In order to identify the binding proteins of the hit compound, we designed and synthesized bait compounds tagged with linker and FLAG epitope (positive probe) as described previously.13 We also synthesized linker and FLAG tagged inactive bait compound (negative probe), which has a similar structure with the hit compound, for identifying nonspecific interactors. The detailed method is described in the Supplementary Methods.
Liquid Chromatography–MS/MS Analysis
Digested peptide samples were analyzed using a nanoscale liquid chromatography–tandem mass spectrometry (LC-MS/MS) system as previously described.14 The detailed method is described in the Supplementary Methods.
DHODH Enzyme Assay
The enzymatic assay couples activity of dihydroorotate dehydrogenase (DHODH) with bleaching of the dye 2,6-dichlorophenolindophenol. The detailed method is described in the Supplementary Methods.
Immunochemistry
Tumors were sectioned at 10 µm thickness and immunostained as described previously.11,12 Detailed information of antibodies is described in the Supplementary Methods.
Metabolite Quantification
E6 and E16 cells were treated in culture with 1 µM and 10 µM, respectively, of 10580 for 24 h. Cells were washed with 5% D-mannitol (Wako Pure Chemical), and metabolites were extracted with 70% ice-cold methanol and analyzed. The detailed method is described in the Supplementary Methods.
Vector Construction
Vectors have been constructed as previously described.11,15 The detailed method is described in the Supplementary Methods.
Tumorigenesis
A tumor-bearing mouse test was performed as previously described.11,15 The detailed method is described in the Supplementary Methods.10
Immunoprecipitation and Western Blotting
Immunoprecipitation and western blotting were performed as previously described.16 Detailed information of antibodies is described in the Supplementary Methods. Protein quantification was performed using ImageJ software (National Institutes of Health).17
Statistical Analysis
Student's t-test was used to compare 2 paired groups. Kaplan–Meier curves were used to estimate unadjusted time-to-event variables. P-values less than 0.05 (two-sided) were considered significant.
Results
Drug Screening Identified a Novel Chemotype that Inhibits GICs/GICRs
We performed a drug screen to identify new compounds that kill GICRs or inhibit their proliferation, as shown previously.10 In the first screen, we cultured E3R cells in the presence of various compounds at 10 µM (10 560 independent compounds) for 3 days and then examined cell viability by MTT assay (Fig. 1A, B). We selected 319 compounds that decreased E3R viability to less than 20% and examined their cytotoxicity against GICs (E3 and E6) and GICRs (E3R and E6R) in the second screen (Fig. 1C). We further selected 302 compounds that decreased the viability of GICs and E6R cells to less than 50% and examined their cytotoxicity against normal NSCs and astrocytes in the third screen (Fig. 1D). We identified 12 compounds, in which viability of both NSCs and astrocytes in response to treatment was over 75%. We further performed cell viability assays using the compounds to calculate 50% growth inhibition (GI50) values and selected 7 compounds whose GI50 against E6R was less than 1 µM. Notably, the selected chemicals have a similar structure with a carboxylic acid skeleton. Among them, 2 compounds, 9700 and 10607, are structurally highly similar and strongly prevented proliferation of GICs (E6 and E16) as well as GICRs (Supplementary Table 1). Thus, we determined 9700 and 10607 as hit compounds.
Fig. 1.

Identification of 10580 as a candidate compound for GIC/GICR eradication.
(A) Summary of chemical screening. (B) E3R cell viability plots for 10 560 compounds; 319 compounds that cell viability to less than 20% were entered into the second screen. (C) The cytotoxicity of 319 compounds against E3 (gray column, upper panel), E3R (black column, upper panel), E6 (gray column, lower panel), and E6R (black column, lower panel). Then, 302 compounds that decreased both GIC and GICR viability to less than 50% were included in the third screen. (D) The cytotoxicity of 302 compounds against NSCs (black column) and astrocytes (gray column); 9700 (dashed arrow) and 10607 (solid arrow) were selected as candidate compounds by their GI50 effects against GICs/GICRs and structural analysis.
DHODH Is the Target of the Hit Compounds
We focused on 10607, as its cytotoxic activity was greater than that of 9700 (Supplementary Table 1). To identify the binding proteins of 10607, we synthesized bait compounds tagged with a linker, polyethylene glycol or C12, and a FLAG epitope (positive control probe) and a control compound (negative control probe) with a structure similar to that of 10607 to exclude nonspecific interactions. We incubated cell lysate with the positive and negative control probes, purified the probe–protein complex using affinity purification with the anti-FLAG antibody, digested the purified complexes with Lys-C endoproteinase and trypsin, and directly analyzed the peptide mixture using nanoflow LC-MS/MS.13,14 Differential analysis of the proteins that interacted with the positive and negative control probes led to the selection of candidate proteins (Supplementary Table 2). To evaluate the binding specificity of candidate proteins with 10607, we performed a competition assay, in which cell lysate was incubated with the positive control probe and FLAG-linker-free 10607 (at a 100-fold higher concentration than the positive control probe), and the proteins that interacted with the positive control probe were analyzed. Among the candidates, DHODH was determined to be the target because its binding with the positive control probes was abolished in the presence of FLAG-linker-free 10607 (Supplementary Table 2).
We confirmed that 10607 and 9700 inhibited DHODH activity using an in vitro enzyme inhibition assay. The half maximal inhibitory concentration (IC50) values of 10607 and 9700 were 28.6 and 120.3 nM, respectively, which were similar to that of a traditional DHODH inhibitor, Brequinar (BRQ) (IC50 11 nM). In addition, ChEMBL data (https://www.ebi.ac.uk/chembl/) revealed that 10607 has 74.7% similarity to DHODH inhibitors, which have a carboxylic acid skeleton.
Pharmacokinetics-Based Selection of a DHODH Inhibitor from a Compound Library
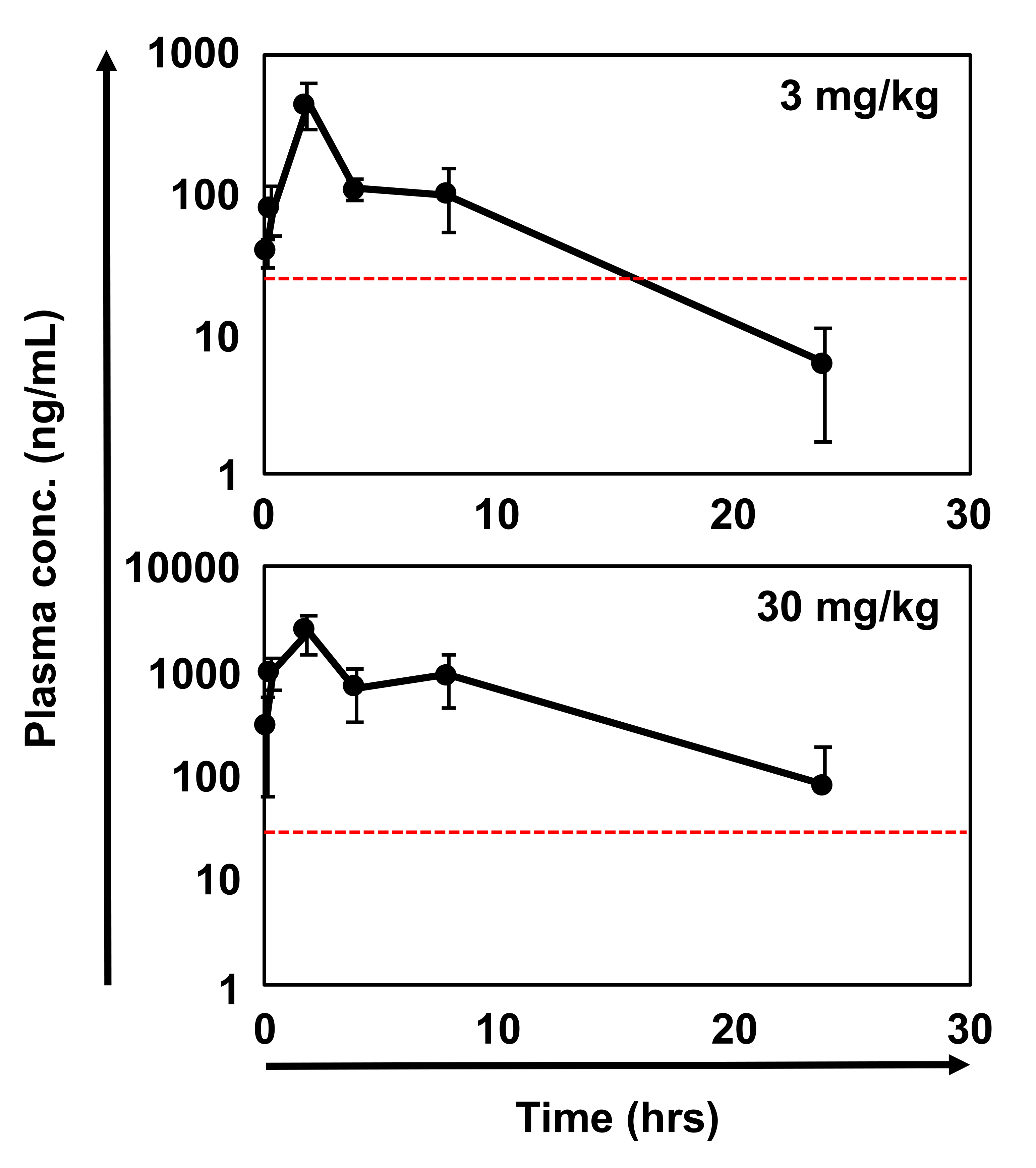
Since 10607 showed low stability in mouse liver microsomes, we screened a focused library of compounds with the same chemotype as the DHODH inhibitor from the Fujifilm library and identified 10580 showing high stability in liver microsome. Both 10607 and 10580 have a 2-amino-5-cyclopropyl nicotinic acid moiety and indole moiety, which are important structures for potent inhibition of GIC growth but do not exist in traditional DHODH inhibitors (Fig. 2A). The pharmacokinetic profile of 10580 (30 mg/kg) showed a sufficient area under the concentration curve of 12230 (ng•h/mL) and a longer blood half-life (T1/2 = 6.25 h), indicating that the administration of 30 mg/kg keeps a sufficient concentration, over the predicted maximum GI50 concentration of 25.8 ng/mL, in blood for 24 h, which is needed for an in vivo efficacy study (Supplementary Figure 1). Notably, 10580 could be administered orally to test for in vivo efficacy. We then compared the cytotoxicity of 9700, 10607, and 10580 in GICs and GICRs and found that 10580 had the highest cytotoxicity among these compounds (more than 56- and 47-fold higher than 9700 and 10607, respectively) (Fig. 2B, Supplementary Table 1). We further found that 10580-induced cytotoxicity against GICs was higher than that of well-known DHODH inhibitors, including teriflunomide, leflunomide, vidofludimus, and BRQ (Fig. 2C). Thus, we identified 10580, a powerful DHODH inhibitor, as a candidate drug for the eradication of GICs/GICRs.
Fig. 2.

Identification of the DHODH inhibitor 10580 as a new anti-GIC compound.
(A) Chemical structures of 10607, 10580, and traditional DHODH inhibitors. (B) Dose-dependent effects of the candidate compounds, 10607 (open circle), 9700 (open triangle), and 10580 (closed circle) against GICs (E6 and E16 cells) and GICRs (E6R and E16R cells). (C) Dose-dependent effects of the DHODH inhibitors teriflunomide (open circle), leflunomide (open square), vidofludimus (cross), BRQ (open diamond), and 10580 (closed triangle) against GICs (E6 and E16 cells). (D) Pyrimidine biosynthesis pathways. (E) Representative data of metabolites in control (DMSO, white column) and 10580-treated (black column) GICs (E6 and E16 cells). All experiments were repeated at least 3 times with similar results. Error bar: ±SD. Statistical significance was determined by the t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
10580. Inhibits the Cellular Metabolism of Pyrimidine
To further clarify that 10580 inhibits DHODH activity, we performed metabolome analysis and determined the quantities of dihydroorotate (DHO), orotate, and its downstream metabolites, which are used for pyrimidine synthesis (de novo pathway), in 10580-treated GICs (Fig. 2D). DHO levels greatly increased in 10580-treated GICs compared with DMSO-treated control cells (596- and 125-fold change in E6 and E16 cells, respectively), while orotate levels decreased in 10580-treated E16 cells but slightly increased in 10580-treated E6 cells (Fig. 2E, Supplementary Figure 2). The levels of all pyrimidine nucleotides (UMP, UDP, UTP, CMP, CDP, CTP, dCTP, and dTTP), which are downstream metabolites of orotate, decreased in 10580-treated cells, whereas levels of purine nucleotide (ATP, GTP, dATP, and dGTP) did not change or increase in these cells (Fig. 2D, Supplementary Figure 2). These data clearly indicated that 10580 prevents de novo pyrimidine synthesis by inhibiting DHODH activity.
GICs/GICRs Depend on the De Novo Pyrimidine Synthesis Pathway
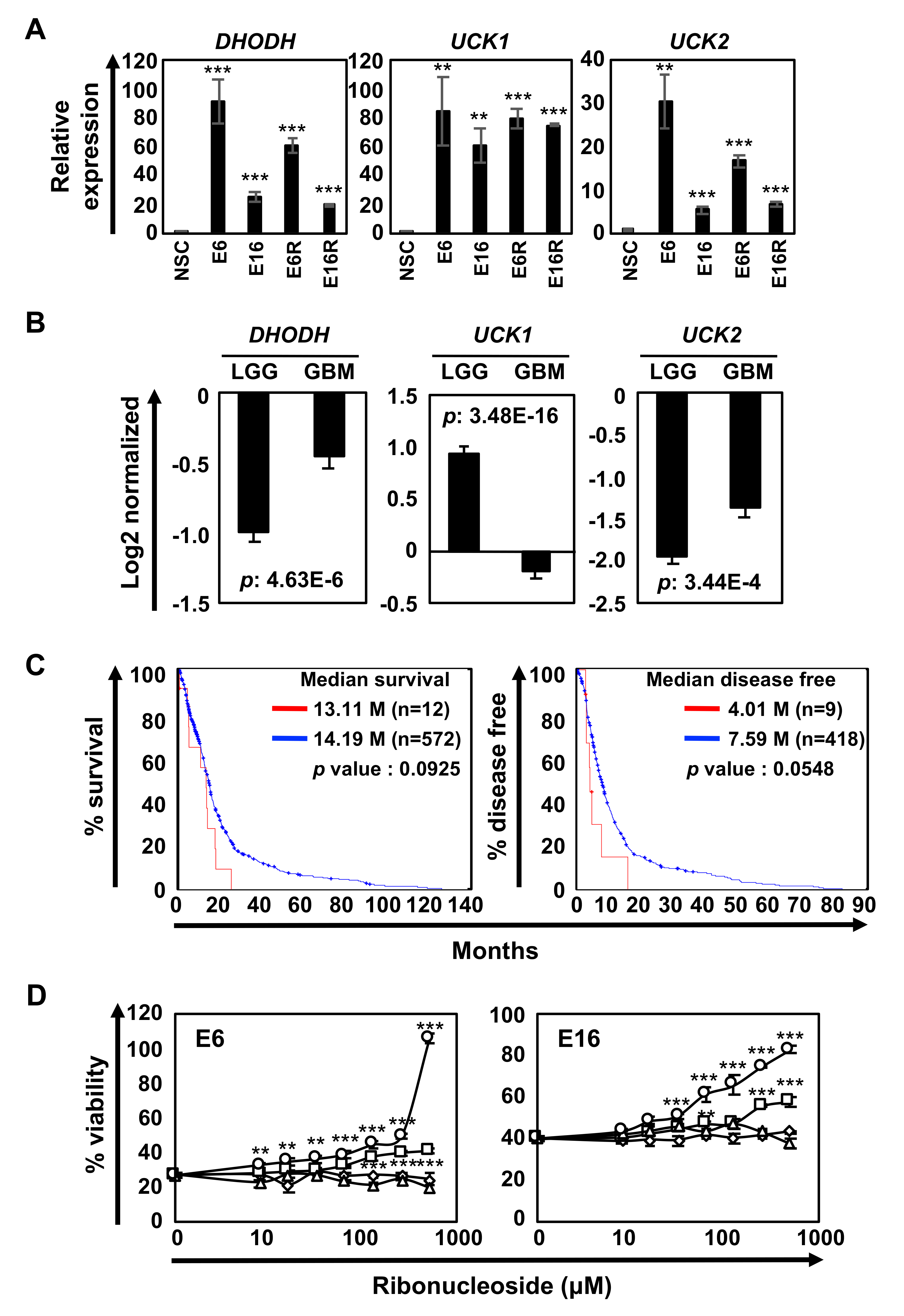
Pyrimidine synthesis is regulated by both the de novo and salvage pathways (Fig. 2D), raising a question of why DHODH inhibitors effectively prevent GIC/GICR proliferation/survival. We examined the expression of DHODH and uridine-cytidine kinases (UCKs), key regulators of the salvage pathway, in NSCs, GICs, and GICRs and found that both DHODH and UCKs were highly expressed in GICs and GICRs compared with NSCs (Supplementary Figure 3A). The Cancer Genome Atlas revealed that DHODH is more expressed in GBM than in lower-grade glioma, while uck1 expression is lower in GBM (Supplementary Figure 3B). Data from cBioPortal (http://www.cbioportal.org/index.do?cancer_study_list=&cancer_study_id=all&data_priority=0&case_ids=&gene_set_choice=user-defined-list&gene_list=&clinical_param_selection=null&tab_index=tab_visualize)18,19 suggested that the prognosis of GBM patients with elevated DHODH mRNA levels (red line, Z-score >2) was worse than the other (blue line); median survival and median disease-free survival of the patients with elevated DHODH (red line) was 13.11 and 4.01 months, whereas those of the other (blue line) was 14.19 and 7.59 months, respectively (Supplementary Figure 3C). In addition, excess uridine addition (500 µM) was needed to rescue 10580-induced cytotoxicity in GICs (Supplementary Figure 3D). Taken together, these data suggested that pyrimidine synthesis in GICs/GICRs largely depends on the de novo pathway rather than the salvage pathway, despite the expression of UCKs in the cells.
10580. Inhibits GIC Proliferation, Survival, and Stemness
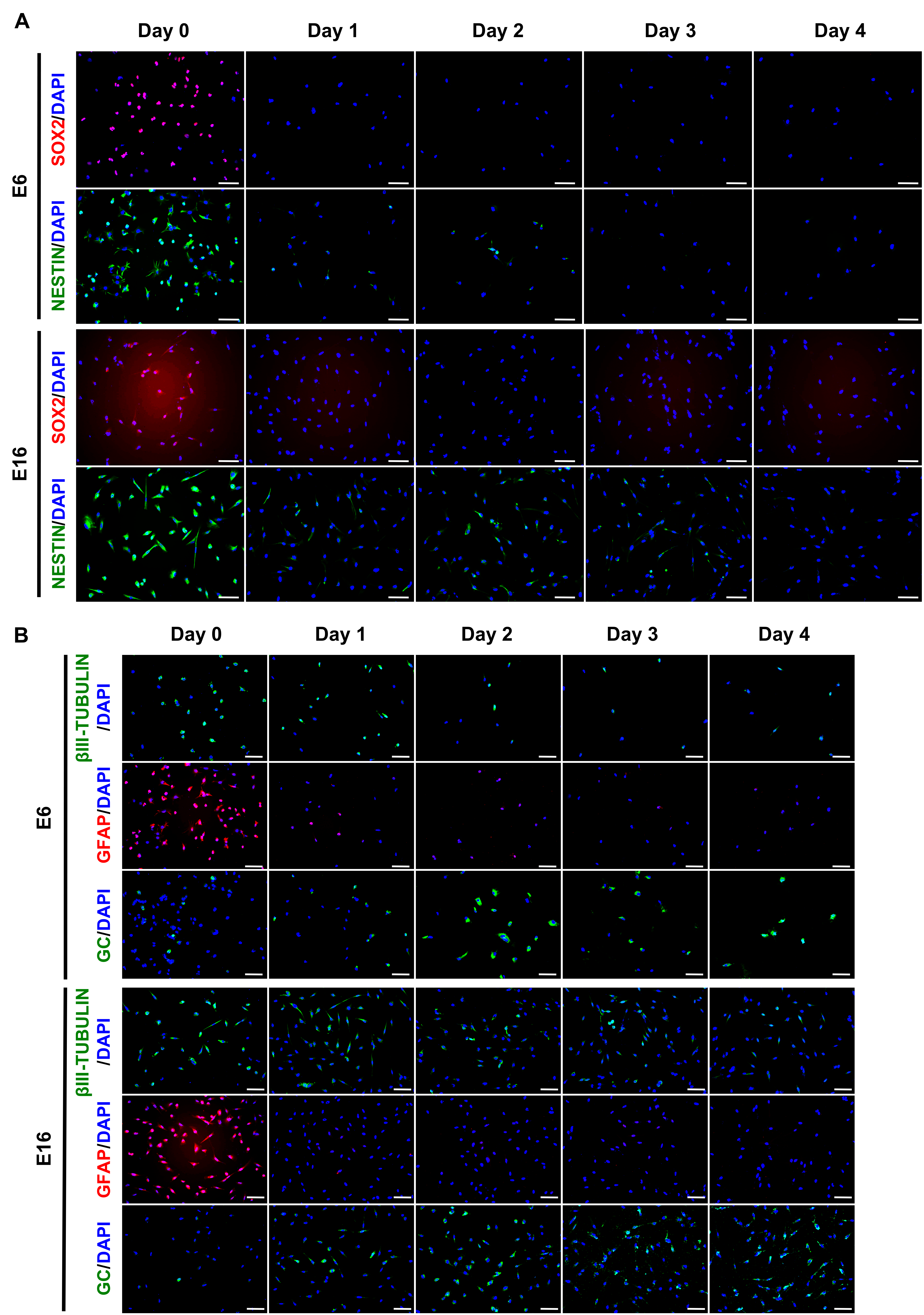
We investigated the proliferation, death, and differentiation of 10580-treated GICs. A BrdU incorporation assay confirmed that 10580 strongly blocked cell proliferation (Fig. 3A). Conversely, the number of active caspase-3 (CASP3)–positive cells increased over time in the presence of 10580 (Fig. 3B, Supplementary Figure 4). To examine cell differentiation, we immunolabeled 10580-treated cells for NSC markers SOX2 and Nestin and for differentiation markers, the neuronal marker βIII tubulin, the astrocyte marker glial fibrillary acidic protein (GFAP), and the oligodendrocyte marker galactocerebroside (GC). SOX2 protein was rapidly undetectable in 10580-treated GICs within 1 day, before the visible changes in cell death and differentiation (Fig. 3C, Supplementary Figure 5A). In parallel, the NESTIN and GFAP signals in the surviving 10580-treated GICs disappeared over time. The number of βIII tubulin–positive cells gradually decreased with time, whereas the population of GC-positive cells transiently increased and then decreased at 4 days after 10580 treatment (Fig. 3C, Supplementary Figure 5).
Fig. 3.

10580 induces cell cycle arrest, cell death, and differentiation in GICs.
(A) Proportion of BrdU+ cells among E6 (black column) and E16 GICs (white column) cultured in the presence of 10580. (B) Proportion of CASP3+ cells among E6 (black column) and E16 GICs (white column) cultured in the presence of 10580. (C) Proportion of cells stained for the NSC markers SOX2 and NESTIN and the differentiation markers βIII tubulin, GFAP, and GC, in E6 (black column) and E16 GICs (white column) cultured in the presence of 10580. (D) Representative photographs of either control (cont)-sh- or DHODH-sh-expressing E6 cells that were immunostained for GFP (green), SOX2 (purple or white), and CASP3 (red). In cont-sh cells (left panels), arrows indicate GFP+/SOX2+/Casp3− cells. In DHODH-sh cells (right panels), arrows indicate GFP+/Sox2+/−/Casp3+ cells, arrowheads indicate GFP+/SOX2−/CASP3− cells. Nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (blue). Scale bar: 30 µm. (E) Proportion of SOX2+ cells among either cont-sh- or DHODH-sh-expressing GICs (GFP+). (F) Proportion of CASP3+ cells among either cont-sh- or DHODH-sh-expressing GICs (GFP+). All experiments were repeated at least 3 times with similar results. Error bar: ±SD. Statistical significance was determined by the t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
Using a DHODH-specific short hairpin RNA, we confirmed that DHODH knockdown decreased SOX2 levels (58% and 55% in E6 and E16, respectively) and induced CASP3 activation in GICs (44% and 47% in E6 and E16, respectively) (Fig. 3D–F), similar to what was observed in 10580-treated GICs. Taken together, these data revealed that 10580 inhibits cell proliferation, survival, and stemness by impeding DHODH activity.
10580 Prevents GICR Tumorigenesis In Vivo
We examined the anti-tumor activity of 10580 in vivo. Since 10580 did not enter the brain parenchyma through the blood–brain barrier (BBB), we transplanted GICRs, E6R and E16R cells, subcutaneously into the hip, instead of the brain, of NOD/SCID mice and allowed the tumors to reach a size greater than 100 mm3 before initiating the daily oral administration of 10580 for 11 days. As shown in Fig. 4A, 10580 administration markedly blocked tumorigenesis of GICRs in vivo compared with control, without any visible toxicity to the mice. We removed tumors from the mice at day 12 after drug treatment and sectioned and analyzed the tumor tissues. The histopathological examination of similar-sized (E6R) and representative (E16R) tumors from control and 10580-treated mice showed that 10580 treatment significantly decreased hypercellularity and the tumor mass (Supplementary Fig. 6A). Immunolabeling confirmed the decreased number of SOX2-positive cells and the increased number of active CASP3-positive cells in 10580-treated tumors compared with control tumors (Fig. 4B, C; Supplementary Figure 6B, C). We also examined the anti-GICR activity of BRQ, which was recently demonstrated as an antitumor drug,20–22 and found that BRQ also prevented tumorigenesis of GICRs similarly to 10580 (Fig. 4A). These data indicated that DHODH inhibitors are effective against GICR tumorigenesis and suggested 10580 as a potential new drug for GBM therapy.
Fig. 4.

Antitumorigenic effect of 10580 against GICRs. (A) E6R and E16R tumor volumes in mice treated daily with saline (open circles), 10580 (closed circles), or BRQ (closed triangles) (n = 6). Statistical significance was determined by the t-test. Error bar: ±SEM. (B) Proportion of Sox2+ cells among human mitochondria+ cells in the tumors. Error bar: ±SD. (C) Proportion of CASP3+ cells in the tumors. Error bar: ±SD. *P < 0.05, **P < 0.01, ***P < 0.001.
UDP-GlcNAc Rescues SOX2 Reduction and Cell Death in 10580-Treated GICs
We addressed how 10580 inhibits the stemness of GICs by blocking the de novo pyrimidine synthesis pathway. Since SOX2, an essential stem cell factor, quickly disappeared in 10580-treated GICs before they died, we investigated the mechanism by which DHODH inhibition decreases SOX2 levels. Histopathological analysis showed SOX2 immunoreactivity in the cytoplasm rather than in the nucleus in active CASP3-positive GICs (Supplementary Figure 6B, C), suggesting that 10580 induced the nuclear export of SOX2. Acetylation of SOX2 at lysine 75 (K75) in the sex determining region Y–related high mobility group domain has been shown to be essential for its nuclear export through interaction with chromosomal maintenance 1 (CRM1, also known as exportin 1).23,24 Indeed, we observed SOX2 accumulation in the nucleus of 10580-treated GICs in the presence of the CRM1 inhibitor leptomycin B (LMB) (Supplementary Figure 7A), indicating that 10580 induced SOX2 nuclear export in a CRM1-dependent manner.
We focused on O-GlcNAcylation, an O-linked N-acetylglucosamine (O-GlcNAc) modification, for the following reasons. O-GlcNAcylation was shown to regulate the nuclear localization of transcription factors, such as β-catenin,25,26 and to play essential roles in cancer and stem cells.27–30 Our metabolite analysis showed a significant reduction in uridine diphosphate N-acetylglucosamine (UDP-GlcNAc), the substrate of O-GlcNAcylation, in 10580-treated GICs (Fig. 5A, Supplementary Figure 2D). Furthermore, O-GlcNAcylation at serine 248 (S248) of SOX2 markedly decreased the differentiation of embryonic stem (ES) cells, while substitution of serine for alanine at residue 248 (Sox2 S248A) changed its interactors and modulated gene expression in ES cells: Wild-type SOX2 associated with histone-deacetylase 2 and DNA methyltransferase 1, whereas SOX2 S248A interacted with base excision and repair factors.31 Taken together, these findings suggest that O-GlcNAcylation regulates SOX2 localization and function in GICs.
Fig. 5.

Nuclear export of SOX2 is a part of 10580-dependent GIC death. (A) O-GlcNAcylation of SOX2 through the OGT/UDP-GlcNAc reaction. (B) Proportion of SOX2+ cells among E6 and E16 GICs cultured in the presence of 10580 alone or in combination with UDP-GlcNAc. (C) Dose-dependent effects of UDP (open circle) and UDP-GlcNAc (closed circle) in 10580 (10 µM)-treated E6 and E16 GICs. (D) O-GlcNAcylation analysis of SOX2 in E6 GIC that were cultured in DMSO alone (N), 10580 alone (–), 10580+UDP-GlcNAc (GlcNAc), or 10580+UDP. Immunoprecipitated cell lysates using anti-FLAG M2 antibody were analyzed by western blotting (upper panel). O-GlcNAcylated SOX2 level shown in upper panels was presented as fold change against FLAG (lower panel). Protein quantification was performed using ImageJ software. (E) Proportion of nuclear SOX2-retaining GICs expressing control, SOX2 wt, SOX2 K75A, or SOX2 S248A. (F) Proportion of survival/proliferating 10580-treated GICs in the presence (black column) or absence (white column) of the pan-caspase inhibitor Z-VAD-FMK. (G) Proportion of nuclear SOX2+ GICs that were cultured in DMSO, 10580, or 10580+Z-VAD for 2 days. ns: not significant. All experiments were repeated at least 3 times with similar results. Error bar: ±SD. Statistical significance was determined by t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
To examine this possibility, we cultured GICs with 10580 alone or 10580 plus UDP-GlcNAc for 2 days, and immunolabeled the cells for SOX2 and NESTIN. The addition of UDP-GlcNAc to 10580-treated GICs rescued not only the reduction in SOX2 (E6 cells: 1.5% for 10580 vs 49.5% for 10580+UDP-GlcNAc; E16 cells: 4.6% for 10580 vs 45.4% for 10580+UDP-GlcNAc) but also the cell death (Fig. 5B, C; Supplementary Figure 7B). Because UDP is released from UDP-GlcNAc in the process of O-GlcNAcylation, we examined whether UDP neutralizes 10580 cytotoxicity in GICs. As shown in Figure 5C, UDP addition also prevented 10580 cytotoxicity in a dose-dependent manner, although the rescue ability of UDP-GlcNAc was greater. UDP-GlcNAc releases UDP in the process of O-GlcNAcylation, while UDP is a component of UDP-GlcNAc, indicating that it is currently impossible to pinpoint an O-GlcNAc–specific function during 10580 treatment. Nonetheless, the difference between the addition of UDP and UDP-GlcNAc is likely a direct function of O-GlcNAcylation.
We next examined O-GlcNAcylation of SOX2 in 10580-, 10580+UDP-, and 10580+UDP-GlcNAc–treated cells. We cultured FLAG-tagged SOX2-expressing GICs in the presence of DMSO, 10580, 10580+UDP, or 10580+UDP-GlcNAc and then analyzed O-GlcNAcylation of SOX2 by the immunoprecipitation–western blotting method. We found that 10580 alone decreased O-GlcNAcylation of SOX2 in GICs, whereas addition of either UDP or UDP-GlcNAc recovered O-GlcNAcylation of SOX2 in 10580-treated GICs (Fig. 5D).
To further examine the relationship between O-GlcNAcylation and SOX2, we cultured GICs in the presence of OSMI-1, an inhibitor of O-GlcNAc transferase (OGT), and examined cell proliferation and SOX2 immunoreactivity. OSMI-1 inhibited GIC proliferation in a dose-dependent manner and decreased the percentage of SOX2-positive cells (E6 cells: 71% for control vs 32% for OSMI-1; E16 cells: 65% for control vs 30% for OSMI-1) (Supplementary Figure 8). Collectively, these data suggested that O-GlcNAcylation is essential for SOX2 nuclear accumulation and GIC proliferation.
10580 Separately Induces Cytotoxicity and SOX2 Nuclear Export in GICs
In order to address the relationship between 10580-dependent nuclear export of SOX2 and the cytotoxicity, we established 3 GIC lines expressing SOX2 wild-type (wt) or 2 mutant forms of SOX2, SOX2 K75A and SOX2 S248A, which lack CRM1 binding and O-GlcNAcylation, respectively. As expected, SOX2 K75A remained in the nucleus, even in the presence of 10580 (63% and 58% in E6 and E16 cells, respectively), whereas SOX2 wt (36% and 22% in E6 and E16 cells, respectively) and SOX2 S248A (18% and 15% in E6 and E16 cells, respectively) were exported (Fig. 5E, Supplementary Figure 9A). SOX2 K75A–expressing GICs were partially resistant to 10580-induced cytotoxicity compared with control GICs and those expressing SOX2 wt or SOX2 S248A, although overexpression of SOX2 K75A did not completely rescue cell death (Supplementary Figure 9B). These results indicated that the de novo pyrimidine synthesis blockage, but not SOX2 nuclear export, is the main cause of the 10580-dependent cell death. Moreover, addition of the pan-caspase inhibitor Z-VAD-FMK transiently prevented 10580-dependent cytotoxicity against GICs but did not block SOX2 nuclear export (Fig. 5F, G). These data indicated that 10580-induced SOX2 nuclear export and cytotoxicity are independent phenomena and SOX2 nuclear export is unlikely a consequence of cell death.
Discussion
We have identified a potential candidate drug, 10580, that inhibits GIC tumorigenesis by directly killing the cells but does not show any toxicity to mice when orally administered for 11 days. We demonstrated that 10580 eradicated GICs/GICRs by inhibiting the activity of DHODH, which has been shown to be a potential therapeutic target for malignant tumors, including acute myeloid leukemia, pancreatic cancer, and melanoma.20–22 To the best of our knowledge, 10580 is the most potent DHODH inhibitor, with an IC50 value of 9 nM, below those of other well-known DHODH inhibitors, including BRQ (12 nM), teriflunomide (262 nM), and vidofludimus (141 nM). Thus, these data suggest that 10580 is a new promising anticancer drug for many types of malignant tumors.
Several DHODH inhibitors are used as antirheumatic drugs in the clinical setting by suppressing the immune system, indicating that immunosuppression may be a side effect of these drugs. However, hematopoietic stem cells from mice treated with BRQ were functionally equivalent to those of untreated mice.32 Additionally, since 10580 cytotoxicity against GICs/GICRs was very strong, it may not require long-term application. These results suggest that the side effects of 10580 may be considerably minimal.
The finding that 10580 specifically killed GICs/GICRs but relatively spared normal cells, including NSCs, unveiled a novel mechanism in which GICs/GICRs largely depend on the de novo pyrimidine synthesis pathway for survival and maintenance: DHODH is more highly expressed in GICs/GICRs than in normal cells, including NSCs. DHODH knockdown killed GICs. Whereas small amounts of UDP rescued 10580-induced cytotoxicity in GICs/GICRs, large amounts of uridine were required to achieve the same results. In addition, pyrimidine is synthesized mainly through the salvage pathway in many types of normal cells and tissues, including those in the brain in particular.33 Collectively, these data also suggest that DHODH inhibitors exert strong antitumor function with fewer side effects.
We unexpectedly found that 10580 dramatically decreased Sox2 levels in GICs in vitro and in vivo. This decrease was caused by the acceleration of CRM1-dependent Sox2 nuclear export: Addition of the CRM1-specific inhibitor LMB completely blocked 10580-dependent Sox2 reduction in GICs. Sox2 K75A, which lacks CRM1 binding, was retained in the nucleus of 10580-treated cells. Furthermore, we found that O-GlcNAcylation was involved in Sox2 nuclear retention: OSMI-1, an OGT inhibitor, decreased Sox2 levels in GICs, while Sox2 S248A, an O-GlcNAc–deficient form of Sox2, was less in the nucleus of 10580-treated GICs. O-GlcNAcylation of Sox2 was decreased by 10580, whereas UDP-GlcNAc addition maintained O-GlcNAcylation of Sox2 and its nuclear localization in 10580-treated GICs. Collectively, these findings suggest that one of the essential O-GlcNAc functions is to retain Sox2 in the nucleus by blocking either K75 acetylation, which is essential for CRM1 binding, or the association between Sox2 and CRM1. It is currently impossible to investigate this hypothesis, as there is no amino acid that can mimic O-GlcNAcylated serine.
O-GlcNAcylation has been shown to regulate many biological functions by modifying key molecules involved in signaling, transcription, epigenetics, and other processes. It is well known that cancer cells have increased uptake of glucose, amino acids, fatty acids, and nucleotides, resulting in the increased levels of UDP-GlcNAc and accelerated O-GlcNAcylation of key factors, such as p53, nuclear factor-kappaB, Myc, hypoxia-inducible factor 1, SOX2, histone deacetylase 1, sirtuin 1, and phosphofructokinase 1, which play important roles in stemness and tumorigenesis.23–28,34 Notably, the O-GlcNAcylation of phosphofructokinase 1 increases UDP-GlcNAc by escalating hexosamine biosynthesis, creating a positive feedback loop for UDP-GlcNAc production. Thus, it is essential that future research elucidate the detailed mechanism by which O-GlcNAcylation regulates the functions of these key factors.
Here, we highlight the DHODH inhibitor 10580 as a new potential compound that specifically killed GICs/GICRs without any obvious side effects. Since 10580 did not pass through the BBB, the next challenge is to develop a 10580 delivery system or to find novel DHODH inhibitors that can cross the BBB for GBM therapy.
Funding
This work was partly supported by collaborative research grants from Fujifilm Corporation (to T.K. and T.N.) and was carried out under the Joint Research Program of the Institute for Genetic Medicine, Hokkaido University (to T.K.).
Conflict of interest statement. No potential conflicts of interest were disclosed.
Authorship statement. Y.I., S.W., Y.M., and T.K. designed the study. S.E., Y.I., and T.K. performed experimental work. S.E., Y.I., S.K., T.T., S.M., E.W., M.U., T.H., Y.M., S.W., and T.K. performed data analysis. Y.I. and T.K. produced the text and the figures. Y.M., S.W., S.T., K.H., T.N., and S.H. provided patient materials and data. T.K. provided leadership for the project.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Daisuke Yamashita and Takanori Ohnishi for providing hGICs, and Yoshihiro Tsukamoto, Yumiko Shinohe, and Kanako Ohyama for technical assistance.
References
- 1. Stupp R, Mason WP, van den Bent MJ, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 2. Stupp R, Hegi ME, Mason WP, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. [DOI] [PubMed] [Google Scholar]
- 3. Singh SK, Clarke ID, Hide T, Dirks PB. Cancer stem cells in nervous system tumors. Oncogene. 2004;23(43):7267–7273. [DOI] [PubMed] [Google Scholar]
- 4. Kondo T. Brain cancer stem-like cells. Eur J Cancer. 2006;42(9):1237–1242. [DOI] [PubMed] [Google Scholar]
- 5. Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6(6):425–436. [DOI] [PubMed] [Google Scholar]
- 6. Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29(12):1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blough MD, Westgate MR, Beauchamp D, et al. Sensitivity to temozolomide in brain tumor initiating cells. Neuro Oncol. 2010;12(7):756–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mazzio E, Badisa R, Mack N, Deiab S, Soliman KF. High throughput screening of natural products for anti-mitotic effects in MDA-MB-231 human breast carcinoma cells. Phytother Res. 2014;28(6):856–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gupta PB, Onder TT, Jiang G, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138(4):645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsukamoto Y, Ohtsu N, Echizenya S, et al. Chemical screening identifies EUrd as a novel inhibitor against temozolomide-resistant glioblastoma-initiating cells. Stem Cells. 2016;34(8):2016–2025. [DOI] [PubMed] [Google Scholar]
- 11. Hide T, Takezaki T, Nakatani Y, Nakamura H, Kuratsu J, Kondo T. Sox11 prevents tumorigenesis of glioma-initiating cells by inducing neuronal differentiation. Cancer Res. 2009;69(20):7953–7959. [DOI] [PubMed] [Google Scholar]
- 12. Kondo T, Raff M. The Id4 HLH protein and the timing of oligodendrocyte differentiation. EMBO J. 2000;19(9):1998–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hayakawa N, Noguchi M, Takeshita S, et al. Structure-activity relationship study, target identification, and pharmacological characterization of a small molecular IL-12/23 inhibitor, APY0201. Bioorg Med Chem. 2014;22(11):3021–3029. [DOI] [PubMed] [Google Scholar]
- 14. Araki K, Kusano H, Sasaki N, et al. Redox sensitivities of global cellular cysteine residues under reductive and oxidative stress. J Proteome Res. 2016;15(8):2548–2559. [DOI] [PubMed] [Google Scholar]
- 15. Nishide K, Nakatani Y, Kiyonari H, Kondo T. Glioblastoma formation from cell population depleted of prominin1-expressing cells. PLoS One. 2009;4(8):e6869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takanaga H, Tsuchida-Straeten N, Nishide K, Watanabe A, Aburatani H, Kondo T. Gli2 is a novel regulator of sox2 expression in telencephalic neuroepithelial cells. Stem Cells. 2009;27(1):165–174. [DOI] [PubMed] [Google Scholar]
- 17. Rasband WS. ImageJ. Bethesda, Maryland: National Institutes of Health; 1997–2012. [Google Scholar]
- 18. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. White RM, Cech J, Ratanasirintrawoot S, et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471(7339):518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sykes DB, Kfoury YS, Mercier FE, et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. 2016;167(1):171–186.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koundinya M, Sudhalter J, Courjaud A, et al. Dependence on the pyrimidine biosynthetic enzyme DHODH is a synthetic lethal vulnerability in mutant KRAS-driven cancers. Cell Chem Biol. 2018;25(6):705–717.e11. [DOI] [PubMed] [Google Scholar]
- 23. Baltus GA, Kowalski MP, Zhai H, et al. Acetylation of sox2 induces its nuclear export in embryonic stem cells. Stem Cells. 2009;27(9):2175–2184. [DOI] [PubMed] [Google Scholar]
- 24. Kamachi Y, Kondoh H. Sox proteins: regulators of cell fate specification and differentiation. Development. 2013;140(20):4129–4144. [DOI] [PubMed] [Google Scholar]
- 25. Sayat R, Leber B, Grubac V, Wiltshire L, Persad S. O-GlcNAc-glycosylation of beta-catenin regulates its nuclear localization and transcriptional activity. Exp Cell Res. 2008;314(15):2774–2787. [DOI] [PubMed] [Google Scholar]
- 26. Yang X, Qian K. Protein O-GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol. 2017;18(7):452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jang H, Kim TW, Yoon S, et al. O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network. Cell Stem Cell. 2012;11(1):62–74. [DOI] [PubMed] [Google Scholar]
- 28. Fardini Y, Dehennaut V, Lefebvre T, Issad T. O-GlcNAcylation: a new cancer hallmark? Front Endocrinol (Lausanne). 2013;4:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ferrer CM, Lynch TP, Sodi VL, et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell. 2014;54(5):820–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bond MR, Hanover JA. A little sugar goes a long way: the cell biology of O-GlcNAc. J Cell Biol. 2015;208(7):869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Myers SA, Peddada S, Chatterjee N, et al. SOX2 O-GlcNAcylation alters its protein-protein interactions and genomic occupancy to modulate gene expression in pluripotent cells. Elife. 2016;5:e10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sykes DB, Kfoury YS, Mercier F, et al. Inhibition of the enzyme dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Blood. 2016;128(22):1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Balestri F, Barsotti C, Lutzemberger L, Camici M, Ipata PL. Key role of uridine kinase and uridine phosphorylase in the homeostatic regulation of purine and pyrimidine salvage in brain. Neurochem Int. 2007;51(8):517–523. [DOI] [PubMed] [Google Scholar]
- 34. Hanover JA, Chen W, Bond MR. O-GlcNAc in cancer: an oncometabolism-fueled vicious cycle. J Bioenerg Biomembr. 2018;50(3):155–173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.