

Conflict between mutant viruses is driven by cheater-of-cheater dynamics, leading to the fall of one cheater and rise of another.
Abstract
Cheater viruses, also known as defective interfering viruses, cannot replicate on their own yet replicate faster than the wild type upon coinfection. While there is growing interest in using cheaters as antiviral therapeutics, the mechanisms underlying cheating have been rarely explored. During experimental evolution of MS2 phage, we observed the parallel emergence of two independent cheater mutants. The first, a point deletion mutant, lacked polymerase activity but was advantageous in viral packaging. The second synonymous mutant cheater displayed a completely different cheating mechanism, involving an altered RNA structure. Continued evolution revealed the demise of the deletion cheater and rise of the synonymous cheater. A mathematical model inferred that while a single cheater is expected to reach an equilibrium with the wild type, cheater demise arises from antagonistic interactions between coinfecting cheaters. These findings highlight layers of parasitism: viruses parasitizing cells, cheaters parasitizing intact viruses, and cheaters may parasitize other cheaters.
INTRODUCTION
RNA viruses are notorious for their rapid evolution, which is driven by high mutation rates and short generation times. This rapid evolution is often cited as one of the reasons why viruses adapt so quickly to novel environments, including the ability to jump between host species and create devastating epidemics (1). However, the rapid evolution of RNA viruses comes at a cost to the virus population: Many virus particles that are generated during infection may be defective, i.e., unable to fully complete a cycle of infection (2). One category of defective viruses is particularly intriguing, namely, defective interfering particles (DIPs), which, as their name implies, bear a defect in their ability to replicate yet are able to interfere with the replication of wild-type (WT) viruses. Interference occurs when a DIP coinfects with a WT virus and subverts the WT resources for its own replication, leading to more DIP than WT progeny.
Much interest has recently arisen in DIPs since they may be considered as therapeutic agents: By competing over resources with the pathogenic WT viruses, they markedly reduce the infectious load while stimulating both adaptive and innate immunity (3–7). DIPs also represent a form of an ecological interaction between two viruses. Diverse forms of interactions between microbes exist (8), from communication (9–11) to cooperation (12–14), and even types of “warfare” (15). Recent years have also seen the nascent field of “sociovirology” emerge (16), a field that describes virus-virus interactions and their impact on virus evolution (17–21). DIPs represent an interaction that can be defined as “social cheating.” Cheaters have been observed across a wide array of viruses such as influenza A virus, dengue virus, and hepatitis C virus (2). Notably, while theory has made many leaps forward in understanding cheaters, a mechanistic understanding of virus cheating still lags behind. The exceptions are cheater viruses with markedly truncated genomes, often bearing only the 5′ and 3′ ends of the genome that contain polymerase-binding regions and packaging signals. In these cases, the advantage of the cheater in the presence of WT is the more efficient replication of these short genomic fragments (22–25).
To study the emergence of cheater RNA viruses, we focused on the model virus MS2. MS2 is one of the smallest known viruses with a +ssRNA genome of 3569 bases that encodes merely four proteins: a coat/capsid protein (CP), a maturation protein (A) that binds the pilus of the Escherichia coli host, a viral RNA-dependent RNA polymerase (replicase), and a small lysis protein encoded in an overlapping reading frame (26). Notably, these mere four proteins still allow the phage to replicate efficiently and lyse its host in less than an hour, with a burst size of >5000 infectious viruses exiting each cell on average (27). Because of its tiny genome, overlapping reading frames, and a multitude of functionally important RNA structures (28–30), the genome of MS2 is highly constrained. We observed that different single-base mutations in the phage genome can lead to potent cheating in MS2 and were able to characterize the following: (i) the mechanism that leads to a defect, (ii) the mechanistic advantage of cheaters during coinfection, and (iii) the intergenomic interactions between cheaters. Last, we provide a mathematical model that captures the ecological antagonistic interaction between two virus cheaters during coinfection. The rapid emergence of consecutive cheaters during a short time that is comparable to the duration of an infection of a human host by an RNA virus demonstrates the high unpredictability of RNA virus evolution and reveals new hurdles in the path to using cheater/DIPs as antiviral therapeutics.
RESULTS
Rapid emergence of cheaters during experimental evolution
We used experimental evolution to characterize MS2 evolution under the optimal growth temperature of its E. coli host (37°C). A clonal MS2 stock was propagated from a single plaque, and 15 serial passages were performed at two biological replicates denoted as lines A and B (Methods and fig. S1A). The serial passage protocol used was designed according to the following considerations: (i) We ensured a large population size to avoid, as much as possible, the effects of genetic drift (31, 32). In each passage, we began with an input of 1010 plaque-forming units (PFUs) and 1010 bacterial cells, yielding a multiplicity of infection (MOI) of 1; (ii) we limited host-virus coevolution since the same naїve hosts were provided for each passage, and (iii) we started the serial passaging with a single genotype (a single plaque) to ensure that all ensuing diversity was created de novo. We then determined the frequency of each mutation in the population through repeated deep sequencing (Methods).
We began by focusing on presumably adaptive mutations, inferred as such by the fact that they rapidly increased in frequency across time (Fig. 1, A and B) (33). When we determined the identity of these so-called adaptive mutations, we were surprised to observe a single nucleotide deletion at position 1764 (Δ1764) that reached a frequency of ~50% in both lines. This mutation affects two overlapping reading frames of the MS2 genome (Fig. 1B), and no additional indels were observed upstream or downstream the deletion that could correct for the aberrant reading frame. Δ1764 is expected to have three independent effects (Fig. 1, C and D). First, it will completely abolish the expression of a functional replicase protein since the deletion occurs at the second codon of this gene. Notably, the replicase protein is essential for the MS2 replication cycle. Second, since Δ1764 also affects the overlapping reading frame of the lysis protein, it is expected to yield a truncated version of this protein that includes only the N-terminal end with several additional amino acids. The altered frame of the replicase gene is expected to recreate the C-terminal part of the lysis protein (Fig. 1D). Thus, two “halves” of the lysis protein are generated, rendering it unclear whether these two halves are functional [(34), but see (35)]. Last, the deletion directly affects a critical RNA secondary structure known as the TR loop, which interacts with the coat protein at the final stages of the virus’s replication cycle and initiates the virion self-assembly (Fig. 1C) (26, 36–39).
Fig. 1. High-frequency mutations detected by experimental evolution of MS2 and their genomic effects.
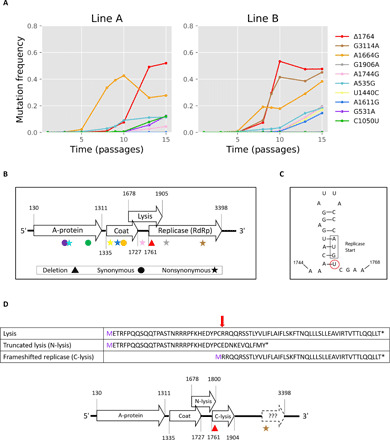
(A) Trajectories of mutations that increased in frequency and attained a frequency of at least 10% in one of the replicate lines A or B. (B) An illustration of the MS2 genome with the genomic location and effect of the mutations in (A). Shapes represent mutation type, and colors represent specific mutations, as in (A). RdRP, RNA-dependent RNA polymerase. (C) The location of Δ1764 (encircled) with respect to the RNA secondary structure of the TR loop. (D) The effect of Δ1764 on the lysis protein results in two peptides of the protein created by the truncated lysis product and by the frameshifted replicase reading frame. The amino acid affected by Δ1764 is marked with a red arrow.
The inferred absence of a functional replicase protein in Δ1764 led us to suspect that this is a “cheater” virus, here defined as having the following characteristics: (i) Cheaters are defective, i.e., cannot replicate in a cell unless a WT virus genome is also present, and (ii) cheaters have an advantage over WT viruses during coinfection, manifested as a higher proportion of cheater progeny compared to WT exiting a coinfected cell (40–42). We set out to test and obtain a mechanistic understanding of these two features.
The mechanism of defection of the deletion mutant
We set out to further validate that Δ1764 is indeed defective and cannot replicate on its own. Notably, no genetic system is available for the particular MS2 strain that we work with, and our attempts at creating a genetic system consistently resulted in death of the bacterial cells bearing a DNA plasmid encoding the full MS2 genome, presumably due to leaky expression of the lysis protein under even the most stringent promoters (43). Thus, to test whether Δ1764 cannot replicate on its own, we seeded virus colonies initiated by a single particle using a standard plaque assay, with viruses derived from passage 15A (p15A) and p15B populations. Our assumption was that a defective particle that does not have all of the requirements for a successful life cycle will not create a plaque in a standard plaque assay. We deep-sequenced a total of 20 plaques (10 from each line), and we did not detect Δ1764 virus in any of these plaques (Fig. 2A). On the basis of a binomial distribution and an average frequency of around 50%, the probability of not observing Δ1764 across 20 plaques if it were not defective is extremely low (P < 10−6). The only mutation that we did observe in the plaques was A535G. This provided statistical support (P < 0.05) that, aside from A535G, every one of the mutations exceeding a mean frequency of 15% across both replicas was either independently defective or resides on the same genome (haplotype) together with another defective mutation.
Fig. 2. Two major lineages of defective mutants.
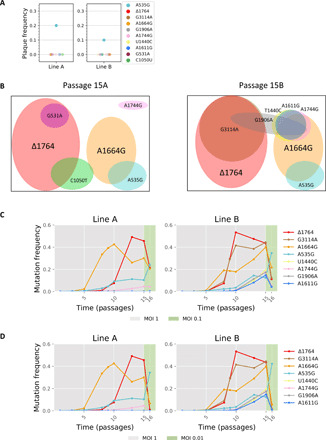
(A) Frequency of mutants in 20 plaques seeded from p15. Of the mutations that arose in the original experiment, the only one observed in the plaques was A535G. (B) Results of haplotype inference based on MinION sequencing for virus populations from p15 line A (left) and line B (right). Each mutation is represented by a circle (color-coded as in Fig. 1A) whose size is roughly proportional to its frequency at p15. Our results suggest that two major cheater lineages are present in both populations with additional different hitchhiking mutations in each line. The full list of inferred haplotypes is given in table S1. (C and D) Details as in Fig. 1A and p1 to p14 are the same as in Fig. 1A. Passaging was reinitiated from a frozen stock of p14. p15 was created at an MOI of 1, yet p16 was created at lower MOIs: MOI of 0.1 (C) and MOI of 0.01 (D), marked in green. All panels show a strong decrease in defective frequencies at lower MOIs.
To test whether the various presumably defective mutants are independent or not, we performed direct long-read RNA sequencing using MinION by Oxford Nanopore. We sequenced more than 15,000 full genomes from p15 of lines A and B and used an approach that we have recently developed to overcome the high error rate of MinION (Methods) (44). Our results revealed that Δ1764 and the second most prominent mutation in the experiment, A1664G, are independent and do not reside on the same genomes. On the other hand, most other mutations were found to be linked to Δ1764 and A1664G. Specifically, G3114A, which rises to high frequencies together with Δ1764, was shown to be linked to Δ1764. We note that it is possible that another reading frame is created in the middle of the original replicase open reading frame (Fig. 1D), and we cannot rule out that G3114A affects this reading frame somehow. However, the lack of plaques bearing G3114A allows us to infer that this mutation does not rescue the Δ1764 phenotype of a nonactive replicase protein (Fig. 2A).
We conclude that there are two major cheater lineages in our populations (Fig. 2B and table S1). Last, our results showed that by p15, there are very few of the original WT genomes left (table S1). The exception is A535G, as suggested by its presence in plaques, since it was inferred to be on a haplotype of its own (table S1) and since this mutation has been previously observed in low-MOI experiments (table S2, top). We conclude that it is the only bona fide adaptive mutation in our experiment and likely acts as “WT like” with respect to the cheaters herein.
One of the characteristics of cheating is its dependence on coinfection (41, 45–47). The lower the MOI, the lower the coinfection rate is, and hence, there is lower probability of coinfection with WT. To this end, we recreated p16 at low MOI values while still maintaining sufficiently large population sizes (Fig. 2, C and D, and Methods). In line with our expectations, at both an MOI of 0.1 and of 0.01, we observed a sharp decrease in all mutation frequencies except for A535G. This effect was more substantial at an MOI of 0.01, as compared to an MOI of 0.1, and was maintained in additional low-MOI passages, ruling out that the drop in mutation frequencies was merely due to a one-time stochastic bottleneck effect (fig. S2). The dependency of the alleged cheaters on the MOI provides further support that they are indeed defective and depend on WT for replication.
We next set out to test which of the three defects that are potentially caused by the point deletion are responsible for its lack of ability to create an infection on its own. We began by expressing a helper plasmid in E. coli that expresses the replicase gene (fig. S3). We then seeded virus colonies initiated by a single particle from the p15B population (the population with the highest relative abundance of both Δ1764 and A1664G) using a standard plaque assay, as described above. We collected viruses from these helper-host strain plaques, selected for viruses that could not create plaques on the WT host strain, and then sequenced the viruses in these plaques. Of five plaques that we sequenced using Sanger sequencing, three contained Δ1764 and two contained A1664G. One of the A1664G plaques also bore a cluster of additional mutations (U1440C, A1611G, A1744G, and G1906A), all of which were completely absent in the 20 plaques isolated from WT E. coli and were indeed identified on a single haplotype (Fig. 2B and table S1). The fact that we succeeded in isolating Δ1764 mutants when providing RNA replicase activity in trans strongly suggests that the two truncated lysis polypeptides are sufficient for lysing the host cells.
We were initially interested in performing experiments with the isolated deletion viruses. When setting out to create a stock of the isolated Δ1764 plaques, we observed that these strains were genetically unstable. Populations generated from the Δ1764 plaque contained many additional proximal mutations (data now shown). We concluded that the expression of the helper replicase is different from the expression of replicase during infection with WT phage and completely changes the selective pressures acting on the phage population (fig. S4). To summarize, we have shown that the lack of a replicase is the major defect of Δ1764, but we could still not isolate pure Δ1764 genotypes that did not contain many additional mutations elsewhere in its genome.
The mechanistic advantage of the deletion mutant
The fact that Δ1764 displayed a marked increase in frequency in both lines is a strong indication that it bears a replicative fitness advantage over WT during coinfection. We thus sought to identify the mechanism that confers a benefit to Δ1764. We set out to test whether the Δ1764 genome is preferentially replicated, possibly through an alteration of RNA secondary structure affecting replication.
We first verified using quantitative polymerase chain reaction (qPCR) that genome replication occurred already after 15 and 30 min and that these time points precede cell lysis (figs. S4 and S5). We infected cells with populations from various passages (5, 8, 10, 13, and 15) from line B and sequenced the intracellular genomes at time points 0, 15, 30, and overnight (Methods). Our results not only did not show a replication advantage for Δ1764 but rather pointed at a strong replicative disadvantage. This was manifested as a strong drop in the frequency of Δ1764 between time points 15 and 30, which we did not observe for either WT/A535G or A1664G (Fig. 3A). When testing the frequency of Δ1764 after overnight growth, we saw that it bounded back, strongly suggesting that the advantage of the deletion mutant is after replication. This led us to the hypothesis that the most likely advantage of Δ1764 is at the stage of packaging.
Fig. 3. The mechanism endowing an advantage to Δ1764.
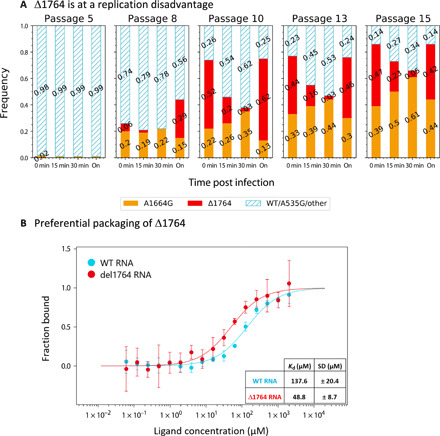
(A) Populations from p5, p8, p10, p13, and p15 of line B were used to infect cells to test the intracellular replication of the different mutants. We tracked the intracellular frequency of the two major mutations found in the experiment versus WT or WT-like variants. Results show similar dynamics for WT/A535G and A1664G, as opposed to a marked decline in frequency of Δ1764 that is compensated after overnight growth (labeled as “on”). (B) In vitro binding assay between the MS2 coat protein and the RNA of the TR loop (fig. S6B) reveal higher affinity of Δ1764 RNA for the coat protein. The initial fluorescence data from the Monolith NT.115 pico instrument (NanoTemper Technologies) was used to calculate a Kd (dissociation constant) of 137.6 ± 20.4 μM for the WT RNA and a Kd of 48.8 ± 8.7 μM for the Δ1764 RNA [n = 4 independent measurements (fig. S6, C and D); error bars represent the SD; y axis is normalized to fraction bound].
To test whether Δ1764 is packaged better, we performed an in vitro binding assay between the MS2 coat protein and the RNA of the TR loop using two synthetic RNA constructs: the WT and the Δ1764 sequences (Methods). WT MS2 coat protein was purified (fig. S6A) and tested for its ability to bind these synthetic RNA constructs using microscale thermophoresis. In line with our hypothesis, the coat protein had a markedly higher affinity for the Δ1764 RNA than for the WT RNA [dissociation constant (KD) = 48 and 137 nM, respectively] (Fig. 3B). We conclude that the difference in binding affinities likely allows for preferential packaging of Δ1764 genomes during coinfection with the WT. Although this has been speculated before as a possible advantage for cheater/DIPs (41), to our knowledge, this is the first time that such preferential packaging as a cheating mechanism has been identified and directly verified.
Mathematical modeling predicts an equilibrium
Game theory has been instrumental in studying the interactions between cheaters (termed defectors) and WT (termed cooperators). To gain further insights into the nature of virus cheating in our system, we developed a mathematical model based on game theory that incorporates the process of viral coinfection (Methods). The model assumes Poisson probabilities for coinfection by WT and cheaters, and a fitness matrix that represents whether more or less WT and/or various cheaters are released from a cell during coinfection (Fig. 4A). To infer the parameters of the matrix, we used a rejection approximate Bayesian computation with sequential Monte Carlo (ABC-SMC) framework (Methods). Across all models explored below, we made the following assumptions that were derived from our experimental results: (i) fitness of WT-only infections (WWT∣WT) was defined as one, (ii) fitness of Δ1764 only (WΔ1764∣Δ1764) was set to zero (no replicase), and (iii) the prior distribution for fitness of WT during infection with Δ1764 (WWT∣Δ1764) was assumed to be uniform over [0,1). The latter was inferred on the basis of our measurements of bacterial cell density, which showed that the presence of Δ1764, but not A1664G, negatively affected the number of bacterial cells lysed (Fig. 4D).
Fig. 4. Game theory–based modeling of a single cheater and WT dynamics.
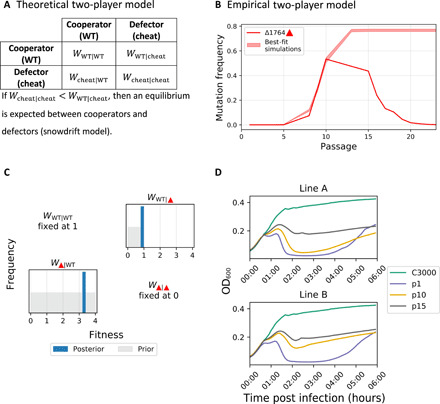
(A) Theoretical fitness matrix of a cooperator-defector system, with notations, as described previously (47). (B) WT and single Δ1764 cheater model, empirical frequency measurements of line B (solid line) with an interval (band) derived from the posterior distribution. (C) Posterior distributions for inferred parameters (dark blue) versus prior distributions (gray). (D) Host cell lysis c3000 bacteria post infection with MS2 populations from p1, p10, and p15 from lines A and B. Notably, passaged populations with high proportions of Δ1764 (p15A, p10B, and p15B) display reduced cell lysis. On the other hand, p10A, which has a high proportion of A1664G, does not display reduced cell lysis.
We began with a simple WT-cheater model, mimicking WT-Δ1764 only. This relatively simple model provided a reasonable fit to the experimental results until p10 in line B and p13 in line A with regard to Δ1764 (Fig. 4, B and C, and fig. S7, A and B). Our results suggested that Δ1764 evolves under the so-called snowdrift model, where both WT and cheater reach a stable equilibrium. This steady state is intuitive: Cheaters increase in frequency because of their advantage during coinfection but cannot take over the population since they are completely dependent on WT. This is in line with previous predictions (12, 47–51).
Consequently, we were interested in examining the stability of Δ1764. To this end, we continued serial passaging while maintaining an MOI of 1 (Fig. 5A). Our results show that A1664G rose steadily in frequency, while Δ1764 decreased in both the A and B lines until it eventually almost disappeared from both populations. This result was completely at odds with our predictions that (i) the two lineages of cheaters Δ1764 and A1664G are independent, and (ii) theoretically, both should reach a steady state, as described above. We hence concluded that there is some form of an antagonistic competition between the two cheaters and set out to examine A1664G more in depth.
Fig. 5. A second partial cheater lineage takes over the viral population.
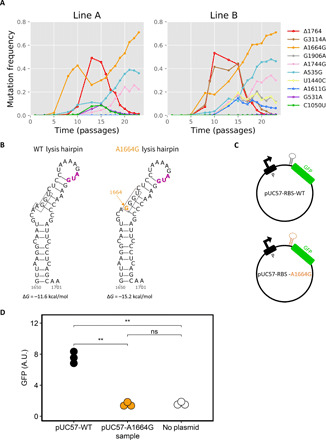
(A) Additional passages initiated from a p14 frozen stock reveal a decline in Δ1764 and increase in A1664G frequencies. Only mutations displayed in Fig. 1 are shown; fig. S1C displays a few additional minor mutations that arose. (B) The position of the A1664G synonymous mutation on the secondary RNA structure lysis hairpin, constructed by Mfold (70). The inferred RBS is boxed, and the start codon is marked in purple. The A1664G mutation creates a stronger stem-loop structure, as evident by an inferred ΔG of −11.6 kcal/mol for the WT RNA and −15.2 kcal/mol for A1664G RNA. (C) Synthetic constructs bearing GFP were generated, one with the RBS of WT virus and one with the A1664G mutant. (D) Fluorescence level comparison of the WT- and A1664G-bearing constructs suggest reduced protein levels for the A1664G RBS. The plasmid bearing the A1664G RBS construct had a GFP expression level much lower than the WT construct and equal to that of the bacteria bearing no plasmid. Fluorescence levels are calculated as the median GFP level of the population. Each construct was tested in three biological replicates (fig. S8), and the group means were compared using t tests with false discovery rate correction for multiple testing (P values of <0.01 are marked with **). ns, not significant; A.U., arbitrary units.
An additional partial cheater rapidly emerges that outcompetes the first cheater
Our results pinpoint A1664G as an additional possible cheating-enabling mutation, which is intriguing given that this is a synonymous mutation. We noted that the A1664G mutants that we isolated from the helper plasmid assay were actually able to repeatedly create tiny plaques that were almost nonvisible when grown on our WT c3000 bacteria, which do not contain the helper plasmid. This, together with the dependency on the WT (Fig. 2, C and D), suggested that A1664G is a partial cheater and not a full cheater like the deletion mutant (52), i.e., its fitness is not zero on its own. We further deduce that the advantage of the A1664G mutant occurs after replication, as its intracellular replication rate is similar to that of the WT (Fig. 3A).
The A1664G synonymous substitution is located in the coat protein, but this position is also the first position in the ribosome-binding site (RBS) of the lysis protein and part of an RNA stem-loop (Fig. 5B). The mutation at the RBS is predicted to decrease ribosome binding (27, 53) and alter the RNA structure (Fig. 5B), leading to lower RBS accessibility (28, 54). To test this, we generated synthetic plasmid constructs bearing the WT or A1664G RBS and surrounding RNA structure, fused to green fluorescent protein (GFP) (Fig. 5C). Our results showed that, indeed, the A1664G RBS led to markedly lower expression of the GFP product (Fig. 5D). Moreover, the A1664G RBS showed an equal expression to that of the bacteria bearing no plasmid, pointing to extremely low RBS accessibility.
Mathematical modeling of intervirus conflict dynamics
We turned back to the mathematical model to understand the interaction between the two cheaters and now focused on a 3 × 3 fitness matrix with parameters for all pairwise interactions and an additional three parameters for triple interactions (Methods). The model incorporated all the assumptions derived from the experiments, as described above, and also assumed that the prior distribution for fitness of A1664G only (WA1664G∣A1664G) was assumed to be uniform over (0,1) (small plaques, damaged RBS). Our results showed that the three-way model with two cheaters provides a better fit than the simple WT-cheater model and successfully recapitulates the rise/demise pattern we see for the two cheaters (Fig. 6A). Next, we focused on the posterior distributions and were able to infer several conclusions. First, both cheaters were correctly inferred as bearing an advantage (w > 1) given the coinfection with WT. The fitness of the deletion mutant was consistently higher than that of A1664G (Fig. 6, B and C). Next, we inferred that Δ1764 is barely able to replicate (or even not at all) in the presence of A1664G, with fitness lower than 0.5 in 92% of its posterior distribution (Fig. 6B). The inference was agnostic regarding how well A1664G replicates in the presence of Δ1764, reflected by a wide posterior distribution, spanning a wide range of values. However, the joint posterior distributions reveal that Δ1764 replicates less well than A1664G in 90% of the joint posterior distribution. This imbalance between the two cheaters was similarly strongly supported by the inference of the model during triple infection (WT, Δ1764, and A1664G), a scenario which is likely prevalent during passages ~13-15 (Fig. 6, B and C).
Fig. 6. Modeling the dynamics of cheater viruses.
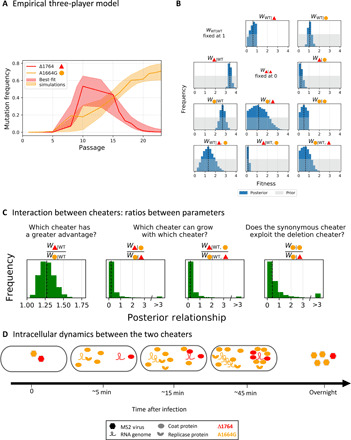
(A) Empirical frequency measurements of line B (solid line/s) with an interval (band) derived from simulations based on the posterior distribution. (B) Posterior distributions for inferred parameters (dark blue) versus prior distributions (gray). The dashed lines represent the median of the posterior distribution. (C) Relationships between pairs of parameters across joint posterior distributions. We conclude that the advantage of Δ1764 is greater than A1664G in the presence of the WT (left) yet that A1664G is able to replicate better in the presence of Δ1764 than vice versa (middle and right). Results are shown for line B; results for line A lead to similar conclusions (fig. S7, C to E). (D) Proposed mechanistic model for the antagonism between both cheaters. During coinfection of a cell with both cheaters, A1664G produces abundant coat products. Because of its efficient binding of coats, Δ1764 is packaged before its genome is replicated. Accordingly, more A1664G genomes will be released from the cell than Δ1764 genomes.
To summarize, our results suggest that the interaction between the two cheaters is the factor responsible for the unexpected demise-and-rise pattern that we see in both replicas. Our inference strongly suggests that A1664G is much more successful in replication than Δ1764 when the two coinfect a cell. In other words, A1664G successfully cheats Δ1764, whereas vice versa is not true.
DISCUSSION
In this work, we have found the incredibly rapid emergence of two potent point-mutant cheaters, which are both defective and, at the same time, bear a selective advantage over WT during coinfection. The mechanisms underlying the defects and the advantages of both cheaters are completely different. Notably, various mechanisms of cheating have been suggested in theory but have not yet been observed and proven conclusively (55). Intriguingly, it only takes a point mutation to create a marked double change in function (a defect and an advantage). We further infer that there is an antagonistic interaction between the two cheaters, supported by the dynamics of the virus at later passages (Fig. 5A).
What could explain this antagonistic interaction and, in particular, the predictions of our model that the deletion cheater cannot replicate in the presence of the synonymous cheater? To explain this, we need to first hypothesize that the advantage of the synonymous cheater is during coinfection with the WT. We suggest that this advantage occurs from an increase in coat protein production, as ribosomes are more available since they are not actively translating the lysis protein in the overlapping reading frame. This is further supported by the fact that (i) A1664G mutant creates a preferable codon at this position (27, 53) and (ii) by the results of our replication rate assay (Fig. 3A), which show that the A1664G advantage occurs after replication. We further suggest that during coinfection with WT, the A1664G is complemented by the lysis protein generated by WT. Thus, in the presence of the “helper” replicase protein, this defect is overcome because of production of more virus genomes and hence more lysis proteins.
We next focus on the interaction between the two cheaters during coinfection. We suggest a spatial model whereby replicase protein products tend to remain near the RNA genomes that created them. Accordingly, during coinfection of the deletion mutant with any genome that encodes for a replicase (the WT or the synonymous cheater), only a small proportion of replicase products diffuse toward it. This is strongly supported by our measurements of rate of replication (Fig. 3A). On the other hand, the deletion cheater is packaged extremely efficiently, giving it an advantage during coinfection exclusively with the WT. However, we suggest that during coinfection with the synonymous cheater, the latter creates many more coat protein products (as compared to WT), and thus, the deletion mutant is packaged early, before it succeeds in replicating its genome to many copies. This puts the deletion cheater at a numerical disadvantage versus the synonymous cheater that undergoes exponential genome replication (Fig. 6D).
We did not find evidence for the presence of our cheaters in previous reports of naturally isolated Leviviridae strains or in laboratory-derived populations (table S2, top) with the exception of an additional high-MOI experiment that we have performed (table S2, bottom). This absence is most likely since all the previous experiments that led to the deposition of MS2 sequences into databases were based on plaque sequencing or low-MOI passaging. The lack of cheaters in naturally isolated strains suggests that these viruses are not detected when the consensus sequence is reported for viruses. In other words, it is possible that for many RNA viruses, cheaters can be present as a minority that is not sequenced and/or is not reported. In line with this, when performing deep sequencing of the American Type Culture Collection (ATCC) stock of MS2 viruses that we purchased, we found A1664G at a frequency of 7%, suggesting that the stock was created at high MOI. This suggests that cheaters may be present in any population that has undergone high-MOI replication.
Molecular parasites and their effects on evolution
The cheaters that we detected herein are an example of a more general interaction found in the natural world, that of molecular parasites, which are particularly prevalent in microbes, in the form of selfish mobile genetic elements (MGEs). Similar to what has been observed herein, MGEs can have their own parasites, resulting in complex ecological and evolutionary interactions, as has been observed for various MGEs, including satellite plasmids and satellite viruses (56–58). One particularly interesting effect of molecular parasites on evolution is that they may allow the crossing of so-called fitness valleys in the context of fitness landscapes: Parasites may bear genes with mutations that are deleterious to their hosts but that are advantageous to the parasite. This is also the case herein for Δ1764 for example. However, one may envisage an additional compensatory mutation that occurs and rescues Δ1764, creating a fully viable phage, and this constitutes the crossing of a fitness valley.
Virus cheaters may have several short- and long-term effects on viral replication in natural settings. First, cheaters reduce the mean fitness of the virus population, a fact that should be carefully considered when considering phage therapy (59). Our populations, for example, bore either one or two cheaters at very high frequencies throughout the entire experiment. Second, cheaters may promote the emergence of a new WT strain that is resistant to cheaters. This would occur in our settings, for example, if the WT virus would evolve to avoid having its proteins hijacked by cheaters. It seems that this naturally occurs on the basis of our proposed model whereby a viral polymerase is spatially associated with the genome that produced it; it is tempting to speculate that natural selection may even further promote such a trait. Third, we observed that while the deletion cheater theoretically represents a more potent antiviral agent, it did not survive and was replaced by the synonymous cheater that is potentially more virulent.
In general, what is the likelihood that virus cheaters are present in natural settings? This depends on the MOI and the probability of coinfection and as such will vary by the characteristics of viral infection. For viruses that infect dense host cell populations (e.g., a human organ or a bacterial biofilm), high MOI or fluctuating MOI is expected, promoting the perseverance or cyclic emergence and disappearance of cheaters. In typical acute infections of human RNA viruses, high MOI is likely to be achieved very rapidly, as in acute infection of HIV where 109 cells are infected every day (60). Given this potential for high MOI, how quickly do cheaters emerge? Here, we have shown it takes one single point mutation, which generates both a defect and an advantage. How likely will this occur in other viruses? We propose that regions in the genomes with overlapping functions, which are often considered to be under strong purifying selection, are actually regions with a potential for mutations to create these double effects. Notably, most RNA viruses have short dense genomes with multiple overlapping information, including overlapping reading frames, and/or important RNA structures that overlap with reading frames as well. We thus suggest that point-mutant cheaters may exist during natural infections of all viruses yet may often be overlooked or erroneously classified as adaptive mutations. Our results suggest that an in-depth understanding of the mechanisms that allow the formation and disappearance of virus cheaters is necessary to further our understanding of their impact on evolution, on infection outcome, and on how they may be manipulated to combat disease.
METHODS
MS2 phage stock preparation
MS2 phage (from ATCC 15597-B1) and log-phase donor E. coli c-3000 were cultured in LB medium at 37°C with shaking overnight. Cells were removed by centrifugation at 4000 rpm for 20 min at 25°C. The supernatant was filtered with a 0.22-μm filter (Stericup Filter, EMD Millipore) to remove any remaining residues. The phage was then stored at 4°C. Typically, a stock concentration of 1011 to 1012 PFU/ml was obtained. A single plaque of MS2 (grown on the host E. coli c-3000) was picked into an Eppendorf tube containing 1 ml of NaCl (0.85%) using a sterile Pasteur pipette. The tube was stored overnight at 4°C to allow the release of the viral particles from the agarose. Ten microliters of chloroform were then added to the sample; the tube was vortexed and centrifuged for 3 min at 13,000 rpm at room temperature. The supernatant was filtered with a 0.22-μm Minisart syringe filter (Sartorius) to remove any residues.
Serial passaging
Clonal MS2 stock was propagated from a single plaque to ensure that the experiment began with a phage population as homogeneous as possible. This clone was the precursor of all the evolutionary lines established in this work. We performed 15 serial passages at the optimal temperature of 37°C with two biological replicates (A and B). The serial passages were performed as follows: Cultures of naїve E. coli c-3000 were grown at 37°C to an OD600 (optical density at 600 nm) of 0.4. Each passage was infected with phages from the previous passage, keeping the MOI around 1 PFU per cell. The cultures were grown for 16 hours with shaking, and the E. coli cells were then removed by centrifugation. The supernatant was subjected to filtration with a 0.22-μm filter (Stericup Filter, EMD Millipore) to remove any remaining residues. Naїve hosts were provided for each passage. Each passage consists of between one and two replication cycles (fig. S1A). The new phage stock was then stored at 4°C. Aliquots of these phage stocks were used for measuring the concentration of phages by plaque assays, infecting the next serial passage, isolating RNA for whole-genome deep sequencing (as described below), and maintaining a frozen stock of the evolving lines in 15% glycerol at −80°C.
It is important to note that the MOI does not directly reflect the number of viruses in the culture but rather the number of PFUs capable of a successful replication cycle on their own (61). This means that in our experiment, because of the presence of the cheaters, the actual number of viruses increases over time, while the number of PFUs is constant across passages.
We performed additional passaging using the same protocol, using a frozen stock of p14 as a starting point. Sequencing of the new p15 showed practically the same results as the old p15, reassuring us of the reproducibility of our results. The effect of lower MOIs was tested on this new p15 (Fig. 2, C and D).
Plaque assay
The plaque assay procedure was used to determine the concentration of the MS2 phage (to measure the virus titer). The plaque assay was conducted according to the method described in (62) with some modifications. E. coli was grown to mid-logarithmic phase (OD600 of 0.3 to 0.4) in LB at 37°C with shaking. Serial dilutions of the MS2 samples were prepared in 0.85% NaCl to reduce the MS2 phage concentration to below 200 PFU/ml. This MS2 phage concentration could easily be counted on each plate with the naked eye. Approximately 5 ml of soft agar (70%) was dispensed into each test tube with 1 ml of E. coli. Then, 0.1 ml of each MS2 phage sample was added to each tube. The contents of the tubes were then emptied onto petri dishes containing solid base agar and allowed to harden. The plates were incubated overnight at 37°C. Next, the plaques were enumerated, and the concentration of MS2 phage in PFU/ml was established.
MS2 RNA isolation
RNA was isolated by three methods. The first was used in serial passaging experiments and the second two methods were used for replication assays:
1) RNA from all serial passage stocks was isolated using the Bio Tri RNA. MS2-concentrated stock in phosphate-buffered saline (500 μl) was mixed with 500 μl of Bio Tri RNA (Bio-Lab) and 100 μl of chloroform. The sample was vortexed for 5 min and then centrifuged for 5 min at 13,000 rpm at room temperature. The upper layer (700 μl) was taken for ethanol precipitation. Then, 700 μl of isopropanol was added to the sample. The sample was stored overnight at −20°C. The sample was centrifuged for 20 min at 13,000 rpm at 4°C. One thousand microliters of 70% cold ethanol was added to the pellet. The sample was centrifuged again for 20 min at 13,000 rpm at 4°C. The pellet was dried under vacuum for 10 min and dissolved in 30 μl of ribonuclease-free water.
2) RNA from time points 0 and overnight (On) was isolated using the ZR viral RNA kit (Zymo Research) according to the manufacturer’s instructions.
3) Total RNA from E. coli cells from time points 15 and 30 was isolated using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions.
Illumina MiSeq library preparation
Illumina MiSeq library preparation and read alignment were performed as described in (44). Briefly, the MS2 RNA was reverse-transcribed using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific), using 3R primer (TGGGTGGTAACTAGCCAAGCAG). complementary DNA (cDNA) from the reverse transcription reaction was directly used as a template for the PCR amplification of the full MS2 genome in two or three overlapping fragments, and PCR reactions were performed using Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific) according to the manufacturer’s instructions. When amplifying in three amplicons, the primers used were as follows: 1F (GGGTGGGACCCCTTTCGG), 1R (TTTTTCTAGAGAGCCGTTGCCT), 2F (GGCCCAAATCTCAGCCATGC), 2R (CGTGTCTGATCCACGGC), 3F (GGCACAAGTTGCAGGATGCA), and 3R (TGGGTGGTAACTAGCCAAGCAG). When amplifying in two amplicons, primers used were 1F, 4R (AAGCAATTGCTGTAAAGTCGTCAC), 4F (CCCAAGCAACTTCGCTAACG), and 3R. As quality control, we sequenced samples both in two and three amplicons, and sequencing results were reproducible.
Purified amplicons were diluted and pooled in equimolar concentrations. The Illumina Nextera XT library preparation protocol and kit were used to produce DNA libraries, according to the manufacturer’s instructions. Libraries were sequenced on Illumina MiSeq using various MiSeq reagent kits for paired-end reads.
Illumina MiSeq deep-sequencing read alignment and analysis
Analysis of the data was performed using the AccuNGS pipeline (63) with the parameters minimal percent identity of 85, E-value threshold of 1 × 10−7, and q score cutoff of 30. The pipeline uses the basic local alignment search tool (BLAST) to map the reads to the reference genome, searches for variants that appears on both overlapping reads, and calls variants with a given q score threshold. Last, it infers the frequency of each variant in the population. The reference genome was determined by comparing the consensus of p1 to GenBank ID V00642.1 (differences noted in fig. S1B). Libraries attained an average coverage of ~10,000 reads per base. Positions at the two ends of the genome (at the primer positions and five adjacent positions) were removed from analysis because they had low coverage and higher variability. This was also verified by a control plasmid containing the MS2 genome bearing eight single-nucleotide polymorphisms that differentiate it from the p1 consensus [provided by D. Peabody (64)]. Three mutations that were present at ~10% in the population from p1 and whose frequency did not change (C3299T and C224T) or decreased (G1554A) were removed from the analysis.
Linkage among mutations using long-read Oxford Nanopore MinION sequencing
The Oxford Nanopore MinION was used to directly sequence the MS2 RNA genome. We sequenced p15A and p15B in separate runs, as described in detail in (44). Alignment was performed using BLAST with parameters of minimal percent identity of 60 and E-value threshold of 1 × 10−7 to allow the highly variable MinION reads to map. We inferred strains and divided the population accordingly, as described in (44), by haplotype/strain identification analysis using the mutations appearing in Fig. 1A.
Inflection point calculation
We performed an experiment to study the relative amounts of progeny phage released, as described in (65) with some modifications. E. coli c-3000 was grown to mid-logarithmic phase (OD600 of 0.3 to 0.4) in LB at 37°C with shaking. E. coli cultures (1.8 ml) were infected with 200 μl of 107 PFU/ml WT viruses for 4 hours with shaking. The infection was stopped by centrifuging the sample at different time points, and the supernatant was subsequently filtered with a 0.22-μm filter to remove any residues. The plaque assay procedure was performed to determine the quantity of new phages. The inflection point was estimated by fitting a four-parameter logistic growth model using JMP (JMP, version 12; SAS Institute Inc., Cary, NC, 1989 to 2007). The inflection point was around 67 min.
The creation of helper E. coli strain
Plasmid pBAD-rep was constructed by cloning a 1638–base pair (bp) fragment of the complete replicase gene into pBAD24 plasmid. The replicase gene was amplified from the cDNA template of the MS2 genome from p1 using PCR with Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific) according to the manufacturer’s instructions with primers F5 (GCTAGCAGGAGGAATTCCATATGTCGAAGACAACAAAGAAGTTC) and R5 (ATCCGCCAAAACAGCCAAGCTTACCGAGGAGAGCTCGCTG). The purified vector and insert were mixed with 10 μl of Gibson Assembly Master Mix (New England Biolabs) and incubated at 37°C for 15 min according to the manufacturer’s instructions. The pBAD-rep plasmid was transformed into XL1 E. coli electrocompetent cells, and the resulting transformants were verified by PCR. To induce replicase expression, 0.2% arabinose was added to the culture (fig. S3).
Bacteria growth assays
Bacteria were grown in microplates at 37°C in LB with medium shaking. Absorbance of the culture was measured using a BioTek plate reader at a wavelength of 600 nm (OD600) every 5 min over 4 hours. Virus infections were performed at an MOI of 1 unless otherwise noted. Experiments were performed in triplicate.
Replication rate test
As in the serial passaging experiment, naїve E. coli c-3000 were grown at 37°C to an OD600 of 0.3 to 0.4. Fifteen milliliters of bacteria were infected with 3 ml of different viral populations: p5B, p8B, p10B, p13B, and p15B, each one diluted to produce an MOI of 1. The cultures were grown at 37°C with shaking. The cultures (1.5 ml) were removed at 15 and 30 min post infection and after growth overnight. Infection was stopped, and the bacteria were separated from the supernatant by centrifugation at 13,000 rpm for 1 min at 4°C. The cells were lysed by resuspension in 0.2-mg lysozyme and incubated at room temperature for 10 min. Viral RNA from these three samples and from the original viral populations was extracted, as described above (MS2 RNA isolation). Illumina MiSeq library was prepared and analyzed, as described above. In Fig. 3A, the frequency of group WT/A535G/other was calculated as 1 − (f(A1664G) + f(Δ1764 )), as A1664G and Δ1764 are mutually exclusive.
Expression and purification of coat protein
Plasmid pET28-coat was constructed by cloning a 392-bp fragment of the complete coat gene into pET28a plasmid. The coat gene was amplified from the cDNA template of the MS2 genome from p1 using PCR with Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific) according to the manufacturer’s instructions with primers F6 (TAAGAAGGAGATATACCATGGCTTCTAACTTTACTCAGTTCG) and R6 (TCAGTGGTGGTGGTGGTGGTGGTAGATGCCGGAGTTTGCT). The purified vector and insert were mixed with 10-μl Gibson Assembly Master Mix (New England Biolabs) and incubated at 37°C for 15 min according to the manufacturer’s instructions. The pET28-coat plasmid transformed into BL21 E. coli electrocompetent cells, and the resulting transformants were verified by PCR and Sanger sequencing.
The MS2 His-tagged coat protein was expressed from the pET28-coat expression plasmid in E. coli BL21. Overnight bacterial culture was diluted in 1 liter of LB medium, grown until OD600 of 0.3, and then induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 4 hours. Then, the bacteria were harvested by centrifugation at 4000 rpm for 10 min at 4°C. The cells were resuspended in 20 ml of binding buffer [1 M NaCl and 20 mM tris-HCl (pH8)] with 0.1% Triton X-100. Bacteria were lysed by three pulses of short 30-s sonication. Cell debris was removed by centrifugation at 11,000 rpm for 30 min. The HiTrap column (GE Healthcare Life Sciences) was washed with six column volumes of double-distilled water (DDW) followed by six column volumes of binding buffer with 5 mM imidazole. The column was loaded with 20 ml of the sample followed by one column volume of wash buffer [1 M NaCl, 20 mM tris-HCl, and 5 mM imidazole (pH8)]. Last, His-tagged coat protein was eluted with 20 ml of elution buffer [1 M NaCl, 20 mM tris-HCl, and 100 mM imidazole (pH8)]; and four fractions of 5 ml were collected. A small sample from each fraction was separated on 4 to 20% SDS–polyacrylamide gel electrophoresis followed by Coomassie staining to test protein purity (fig. S6A, left). Buffer exchange to Hepes buffer (50 mM Hepes, 100 mM NaCl, and 0.5 mM EDTA) was performed using Amicon Ultracel 3K according to the manufacturer’s instructions (fig. S6A, right).
Coat RNA binding assay
The purified coat protein was labeled with a reactive dye using a protein labeling kit, RED-NHS (Amine Reactive) (NanoTemper Technologies, Munich, Germany), which was used according to the manufacturer’s instructions. Unreacted dye was removed with the supplied dye removal column equilibriated with MST (microscale thermophoresis) buffer [50 mM tris-HCl (pH 7.8), 150 mM NaCl, 10 mM MgCl2]. The degree of labeling was determined using utraviolet/visible (UV/VIS) spectrophotometry at 650 and 280 nm.
Synthetic 26-bp RNA fragments containing positions 1745 to 1771 from the MS2 genome, one identical to the WT sequence and one containing the deletion at Δ1764, were purchased from Hylabs. In this MST experiment, the concentration of the NHS-labeled coat protein was kept constant (1 nM), while the concentration of the binding RNA (TR loop) was varied between 0.061 and 2000 μM. The WT and Δ1764 RNA at various concentrations were incubated for 5 min with labeled coat (1 nM) in a binding buffer (50 mM Hepes, 100 mM NaCl, and 0.5 mM EDTA). The samples were loaded into Monolith NT.115 capillaries (NanoTemper Technologies), and the binding was measured by the microscale thermophoresis technology in a Monolith NT.115 pico instrument (NanoTemper Techhnologies) according to the manufacturer’s instructions. Instrument parameters were adjusted to 60% light-emitting diode power and medium MST power. Data of four independently pipetted measurements were analyzed (MO.Affinity Analysis software version 2.3, NanoTemper Technologies), and the Kd (dissociation constant) was calculated using the initial fluorescence data from the MST. Data for the four repeats are shown in fig. S6 (C and D). We used the denatured coat protein as a negative control, where no binding to the RNA was observed.
Real-time PCR
Bacterial cultures (1.5 ml) were grown until OD600 of 0.3 and infected by WT MS2 phages at an MOI of 1, and after 0, 7, 15, 30, and 45 min, the cells were harvested by centrifugation at 13,000 rpm for 1 min at room temperature. The cells were resuspended in 0.2 mg of lysozyme and incubated at room temperature for 10 min. Total RNA from the E. coli cells was isolated using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. The RNA samples were quantified with Qubit (Q33216, Life Technologies). The reverse transcription reaction was performed on 5 μl of RNA phage (500 ng) with random hexamers (0.5 μg/μl). The mixture was incubated for 5 min at 70°C followed by incubation on ice. After brief centrifugation, 4 μl of running buffer were added along with 1 μl of deoxynucleotide triphosphates (dNTPs) (10 nM), 2 μl of Mg2+, 3 μl of DDW, and 1 μl of reverse transcriptase (200 U/μl; Promega). The mixture was incubated for 5 min at 25°C and then 1 hour at 42°C followed by inactivation for 15 min at 70°C. qPCR was performed using an SYBR green PCR master mix (Tamar) in a total volume of 20 μl with the primers F3 (GGCACAAGTTGCAGGATGCA) and R2 (CGTGTCTGATCCACGGC). Reactions were carried out on a Bio-Rad using the standard cycling parameters. Every sample was examined in triplicate, and the results were normalized to time point 7 (fig. S5B).
GFP gene expression analysis
Constitutive pUC57-GFP reporter plasmids were constructed under the control of the PA1/04/03 promoter (66). The plasmids were synthetically designed and obtained from GENEWIZ (Plainfield, USA). They were then transformed into DH12S E. coli electrocompetent cells, and the resulting transformants were verified by PCR and Sanger sequencing. A single colony of each relevant strain was picked and inoculated into 2 ml of LB medium supplemented with ampicillin, and the colonies were cultured overnight at 37°C with agitation. The following morning, cultures were diluted to LB with 1 mM IPTG and then grown to an OD600 of 0.1, when they were taken to the flow cytometer. Flow cytometry (Gallios flow cytometer; Beckman Coulter) was used to identify and quantify the number of GFP-expressing cells and the expression level. The emission filters used were GFP 525/40. All samples were measured at the “low” acquisition rate to reduce intrasample variability. Events were discriminated upon the forward-scatter parameter, and for each run, 1,500 to 47,000 events were analyzed.
Mathematical model
Let us denote N as the number of E. coli host cells and the set of genotypes that can infect these cells as G = (g1, …, gm).We denote ni and fi as the number and frequency of a given genotype,i ∈ (1…m), respectively. Here, we explore m = 2,3, and below, exemplify m = 3. We define Pe as the probability of an infection event e, which defines which of the genotypes infected a cell. There are possible events, which includes a null event where no infection occurred. We assume that a Poisson process describes the number of genomes that will infect a cell. Accordingly, cells and virions are well mixed (which is guaranteed by our experimental procedure), and the relative MOI of a genotype at time t determines the expected number of cells infected by genome gi (akin to the λ parameter of a Poisson process). We further note that our experimental setup guaranteed that since we ensured an MOI of 1 for nondefective viruses. Without loss of generality, we define the probability of an event , defined as infection by one or more genomes of type gi and zero genome of types gj and gk,where i, j, k ∈ (1…3), as follows
where c is chosen so that we traverse all possible infection states (in practice, c = 1000 was sufficient). It follows that the probability of infection by more than one genome for two different genotypes and none of the third is
Last, the probability of a triple infection and the probability of a null infection are defined as
We next define a fitness matrix W that determines the relative contribution of each infecting genotype to progeny of the same genotype in the next generation, depending on whether coinfection occurs. Thus, entry wij = wi∣j reflects the fitness of genotype gi during coinfection with genotype gj. To model triple infections, i.e., infections with three different genotypes, we further define three additional parameters in the matrix W: witriple = wi∣j, k, reflecting the fitness of genotype gi during coinfection with genotypes gj and gk (see Fig. 6C). This allows us to then calculate the relative abundance of genotype gi over time as follows
Inferring posterior distributions for W using ABC-SMC
Simulations under the model were run with N, nWT = 1010 held fixed, as in the experimental protocol. Cheater frequencies were initiated both at 105. We further allowed one to two replication cycles per passage. We noted that changing the initial cheater frequencies or the number of cycles per night did not change the dynamics that we obtained but only the particular timing where a mutant increased or decreased. We formally estimated the posterior distributions of all parameters in W using an ABC-SMC (67) [as implemented in astroABC (68)]. As a summary statistic, we used the mean ℓ1distance between the simulated trajectories of cheater frequencies and the empirical trajectories of the cheater frequencies of either line A or line B , similar to (69), where T is the number of passages that were sequenced. Prior distributions for all parameters were assumed to be uniform over [0,4) unless mentioned otherwise. ABC-SMC was run first for line B with a threshold ε that decreased exponentially from 0.3 to 0.07 over 15 iterations and with 10,000 particles (values of W) per iteration. When inference was run for line A, the final ε obtained was 0.11; for line B, the final ε obtained was 0.07. We used the 10,000 particles from the final iteration to generate posterior distributions. As the single-cheater model was not able to capture the rise and demise of Δ1764, was calculated only until p15 in the single-cheater model.
Supplementary Material
Acknowledgments
We thank A. Herskovits, O. Kobiler, R. Sorek, T. Hagai, and Y. Friedman for critical reading of the manuscript. We also thank J. Hirsch, R. Twarock, E. Dykeman, and P. Stockley for productive advice and R. Oshri, T. Bareia, and Y. Diesendruck for technical help. We thank the Technion Center for Structural Biology and the Protein Analysis Unit from the Weizmann Institute for providing MST and pico-MST facility and services. Funding: A.S. was supported by the Israel Science Foundation (grant number 1333/16). U.G. was supported by H2020 European Research Council Advanced Grant no. 787514 CRISPR-Evol and the Volkswagen Foundation “Experiment!” grant. D.M. and M.G. were supported by a Safra fellowship from the Edmond J. Safra Center for Bioinformatics at Tel Aviv University. Author contributions: Conceptualization: M.M., U.G., and A.S. Experiments: M.M. and N.H. Data analysis: N.H., D.M., M.M., and M.G. Mathematical modeling: N.H., D.M., A.E., and A.S. Writing, reviewing, and editing: M.M., N.H., D.M., M.G., A.E., U.G., and A.S. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The sequencing data created and used in this study are available in the Sequencing Read Archive (www.ncbi.nlm.nih.gov/sra), under BioProject PRJNA575138, except for sequencing data for p1A, p15A, and p15B, which are available under BioProject PRJNA547685. Further analysis frequency files are available in Zenodo: https://doi.org/10.5281/zenodo.3466682. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/34/eabb7990/DC1
REFERENCES AND NOTES
- 1.Duffy S., Shackelton L. A., Holmes E. C., Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 9, 267–276 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Rezelj V. V., Levi L. I., Vignuzzi M., The defective component of viral populations. Curr. Opin. Virol. 33, 74–80 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Bdeir N., Arora P., Gärtner S., Hoffmann M., Reichl U., Pöhlmann S., Winkler M., A system for production of defective interfering particles in the absence of infectious influenza A virus. PLOS ONE 14, e0212757 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rast L. I., Rouzine I. M., Rozhnova G., Bishop L., Weinberger A. D., Weinberger L. S., Conflicting selection pressures will constrain viral escape from interfering particles: Principles for designing resistance-proof antivirals. PLOS Comput. Biol. 12, e1004799 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dimmock N. J., Easton A. J., Defective interfering influenza virus RNAs: Time to reevaluate their clinical potential as broad-spectrum antivirals? J. Virol. 88, 5217–5227 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanner E. J., Jung S.-Y., Glazier J., Thompson C., Zhou Y., Martin B., Son H.-I., Riley J. L., Weinberger L. S., Discovery and engineering of a therapeutic interfering particle (TIP): A combination self-renewing antiviral. bioRxiv , 820456 (2019). [Google Scholar]
- 7.Sun Y., López C. B., The innate immune response to RSV: Advances in our understanding of critical viral and host factors. Vaccine 35, 481–488 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.West S. A., Diggle S. P., Buckling A., Gardner A., Griffin A. S., The social lives of microbes. Annu. Rev. Ecol. Evol. Syst. 38, 53–77 (2007). [Google Scholar]
- 9.Erez Z., Steinberger-Levy I., Shamir M., Doron S., Stokar-Avihail A., Peleg Y., Melamed S., Leavitt A., Savidor A., Albeck S., Amitai G., Sorek R., Communication between viruses guides lysis–lysogeny decisions. Nature 541, 488–493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pollak S., Omer-Bendori S., Even-Tov E., Lipsman V., Bareia T., Ben-Zion I., Eldar A., Facultative cheating supports the coexistence of diverse quorum-sensing alleles. Proc. Natl. Acad. Sci. U.S.A. 113, 2152–2157 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doğaner B. A., Yan L. K. Q., Youk H., Autocrine signaling and quorum sensing: Extreme ends of a common spectrum. Trends Cell Biol. 26, 262–271 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Chao L., Elena S. F., Nonlinear trade-offs allow the cooperation game to evolve from Prisoner's Dilemma to Snowdrift. Proc. Biol. Sci. 284, 20170228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xue K. S., Hooper K. A., Ollodart A. R., Dingens A. S., Bloom J. D., Cooperation between distinct viral variants promotes growth of H3N2 influenza in cell culture. eLife 5, e13974 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ciota A. T., Ehrbar D. J., Van Slyke G. A., Willsey G. G., Kramer L. D., Cooperative interactions in the West Nile virus mutant swarm. BMC Evol. Biol. 12, 58 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Granato E. T., Meiller-Legrand T. A., Foster K. R., The evolution and ecology of bacterial warfare. Curr. Biol. 29, R521–R537 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Díaz-Muñoz S. L., Sanjuán R., West S., Sociovirology: Conflict, cooperation, and communication among viruses. Cell Host Microbe 22, 437–441 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DaPalma T., Doonan B. P., Trager N. M., Kasman L. M., A systematic approach to virus–virus interactions. Virus Res. 149, 1–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Díaz-Muñoz S. L., Viral coinfection is shaped by host ecology and virus–virus interactions across diverse microbial taxa and environments. Virus Evol. 3, vex011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanjuán R., Thoulouze M.-I., Why viruses sometimes disperse in groups. Virus Evol. 5, vez014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altan-Bonnet N., Chen Y.-H., Intercellular transmission of viral populations with vesicles. J. Virol. 89, 12242–12244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santiana M., Ghosh S., Ho B. A., Rajasekaran V., Du W.-L., Mutsafi Y., De Jésus-Diaz D. A., Sosnovtsev S. V., Levenson E. A., Parra G. I., Takvorian P. M., Cali A., Bleck C., Vlasova A. N., Saif L. J., Patton J. T., Lopalco P., Corcelli A., Green K. Y., Altan-Bonnet N., Vesicle-cloaked virus clusters are optimal units for inter-organismal viral transmission. Cell Host Microbe 24, 208–220.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nayak D. P., Defective interfering influenza viruses. Annu. Rev. Microbiol. 34, 619–644 (1980). [DOI] [PubMed] [Google Scholar]
- 23.Marriott A. C., Dimmock N. J., Defective interfering viruses and their potential as antiviral agents. Rev. Med. Virol. 20, 51–62 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Tapia K., Kim W.-K., Sun Y., Mercado-López X., Dunay E., Wise M., Adu M., López C. B., Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLOS Pathog. 9, e1003703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vignuzzi M., López C. B., Defective viral genomes are key drivers of the virus–host interaction. Nat. Microbiol. 4, 1075–1087 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rolfsson Ó., Middleton S., Manfield I. W., White S. J., Fan B., Vaughan R., Ranson N. A., Dykeman E., Twarock R., Ford J., Kao C. C., Stockley P. G., Direct evidence for packaging signal-mediated assembly of bacteriophage MS2. J. Mol. Biol. 428, 431–448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grosjean H., Fiers W., Preferential codon usage in prokaryotic genes: The optimal codon-anticodon interaction energy and the selective codon usage in efficiently expressed genes. Gene 18, 199–209 (1982). [DOI] [PubMed] [Google Scholar]
- 28.Klovins J., Tsareva N. A., De Smit M. H., Berzins V., Van Duin J., Rapid evolution of translational control mechanisms in RNA genomes. J. Mol. Biol. 265, 372–384 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Olsthoorn R. C., Licis N., Van Duin J., Leeway and constraints in the forced evolution of a regulatory RNA helix. EMBO J. 13, 2660–2668 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Himbergen J., van Geffen B., van Duin J., Translational control by a long range RNA–RNA interaction; a basepair substitution analysis. Nucleic Acids Res. 21, 1713–1717 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otto S. P., Whitlock M. C., The probability of fixation in populations of changing size. Genetics 146, 723–733 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lande R., Natural selection and random genetic drift in phenotypic evolution. Evolution 30, 314–334 (1976). [DOI] [PubMed] [Google Scholar]
- 33.Betancourt A. J., Genomewide patterns of substitution in adaptively evolving populations of the RNA bacteriophage MS2. Genetics 181, 1535–1544 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chamakura K. R., Edwards G. B., Young R., Mutational analysis of the MS2 lysis protein L. Microbiology 163, 961–969 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berkhout B., de Smit M. H., Spanjaard R. A., Blom T., van Duin J., The amino terminal half of the MS2-coded lysis protein is dispensable for function: Implications for our understanding of coding region overlaps. EMBO J. 4, 3315–3320 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Valegård K., Murray J. B., Stockley P. G., Stonehouse N. J., Liljas L., Crystal structure of an RNA bacteriophage coat protein–operator complex. Nature 371, 623–626 (1994). [DOI] [PubMed] [Google Scholar]
- 37.Peabody D. S., Role of the coat protein-RNA interaction in the life cycle of bacteriophage MS2. Mol. Gen. Genet. 254, 358–364 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Stonehouse N. J., Scott D. J., Fonseca S., Murray J., Adams C., Clarke A. R., Valegård K., Golmohammadi R., van den Worm S., Liljas L., Stockley P. G., Molecular interactions in the RNA bacteriophage MS2. Biochem. Soc. Trans. 24, 412S (1996). [DOI] [PubMed] [Google Scholar]
- 39.Koning R., van den Worm S., Plaisier J. R., van Duin J., Abrahams J. P., Koerten H., Visualization by cryo-electron microscopy of genomic RNA that binds to the protein capsid inside bacteriophage MS2. J. Mol. Biol. 332, 415–422 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Montville R., Froissart R., Remold S. K., Tenaillon O., Turner P. E., Evolution of mutational robustness in an RNA virus. PLOS Biol. 3, e381 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dennehy J. J., Turner P. E., Reduced fecundity is the cost of cheating in RNA virus ϕ6. Proc. Biol. Sci. 271, 2275–2282 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gutiérrez S., Yvon M., Thébaud G., Monsion B., Michalakis Y., Blanc S., Dynamics of the multiplicity of cellular infection in a plant virus. PLOS Pathog. 6, e1001113 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill H. R., Stonehouse N. J., Fonseca S. A., Stockley P. G., Analysis of phage MS2 coat protein mutants expressed from a reconstituted phagemid reveals that proline 78 is essential for viral infectivity. J. Mol. Biol. 266, 1–7 (1997). [DOI] [PubMed] [Google Scholar]
- 44.Harel N., Meir M., Gophna U., Stern A., Direct sequencing of RNA with MinION Nanopore: Detecting mutations based on associations. Nucleic Acids Res. 47, e148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turner P. E., Parasitism between co-infecting bacteriophages. Adv. Ecol. Res. 37, 309–332 (2005). [Google Scholar]
- 46.Lenski R. E., Velicer G. J., Games microbes play. Selection 1, 89–96 (2001). [Google Scholar]
- 47.Turner P. E., Chao L., Prisoner's dilemma in an RNA virus. Nature 398, 441–443 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Szathmáry E., Natural selection and dynamical coexistence of defective and complementing virus segments. J. Theor. Biol. 157, 383–406 (1992). [DOI] [PubMed] [Google Scholar]
- 49.Wilke C. O., Reissig D. D., Novella I. S., Replication at periodically changing multiplicity of infection promotes stable coexistence of competing viral populations. Evolution 58, 900–905 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Leeks A., Segredo-Otero E. A., Sanjuán R., West S. A., Beneficial coinfection can promote within-host viral diversity. Virus Evol. 4, vey028 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Turner P. E., Chao L., Escape from prisoner’s dilemma in RNA phage Φ6. Am. Nat. 161, 497–505 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Fonville J. M., Marshall N., Tao H., Steel J., Lowen A. C., Influenza virus reassortment is enhanced by semi-infectious particles but can be suppressed by defective interfering particles. PLOS Pathog. 11, e1005204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lucks J. B., Nelson D. R., Kudla G. R., Plotkin J. B., Genome landscapes and bacteriophage codon usage. PLOS Comput. Biol. 4, e1000001 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt B. F., Berkhout B., Overbeek G. P., van Strien A., van Duin J., Determination of the RNA secondary structure that regulates lysis gene-expression in bacteriophage-MS2. J. Mol. Biol. 195, 505–516 (1987). [DOI] [PubMed] [Google Scholar]
- 55.A. Leeks, M. Ghoul, S. West, Cheating in the Viral World. Preprints 2019060106 (2019).
- 56.Zhang X., Deatherage D. E., Zheng H., Georgoulis S. J., Barrick J. E., Evolution of satellite plasmids can prolong the maintenance of newly acquired accessory genes in bacteria. Nat. Commun. 10, 5809 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.A. San Millan, R. C. Maclean, Fitness costs of plasmids: A limit to plasmid transmission, in Microbial Transmission, F. Baquero, E. Bouza, J. Gutiérrez-Fuentes, T. Coque, Eds. (ASM Press, Washington, DC, 2019), pp. 65–79. [Google Scholar]
- 58.Krupovic M., Kuhn J. H., Fischer M. G., A classification system for virophages and satellite viruses. Arch. Virol. 161, 233–247 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Gurney J., Brown S. P., Kaltz O., Hochberg M. E., Steering phages to combat bacterial pathogens. Trends Microbiol. 28, 85–94 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coffin J. M., HIV population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science 267, 483–489 (1995). [DOI] [PubMed] [Google Scholar]
- 61.Williams E. S. C. P., Morales N. M., Wasik B. R., Brusic V., Whelan S. P. J., Turner P. E., Repeatable population dynamics among vesicular stomatitis virus lineages evolved under high co-infection. Front. Microbiol. 7, 370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pierre G., Causserand C., Roques C., Aimar P., Adsorption of MS2 bacteriophage on ultrafiltration membrane laboratory equipments. Desalination 250, 762–766 (2010). [Google Scholar]
- 63.Gelbart M., Harari S., Ben-Ari Y., Kustin T., Meir M., Miller D., Mor O., Stern A., AccuNGS: Detecting ultra-rare variants in viruses from clinical samples. bioRxiv , 349498 (2018). [Google Scholar]
- 64.Shaklee P. N., Negative-strand RNA replication by Qβ and MS2 positive-strand RNA phage replicases. Virology 178, 340–343 (1990). [DOI] [PubMed] [Google Scholar]
- 65.Woody M. A., Cliver D. O., Effects of temperature and host cell growth phase on replication of F-specific RNA coliphage Q beta. Appl. Environ. Microbiol. 61, 1520–1526 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oshri R. D., Zrihen K. S., Shner I., Bendori S. O., Eldar A., Selection for increased quorum-sensing cooperation in Pseudomonas aeruginosa through the shut-down of a drug resistance pump. ISME J. 12, 2458–2469 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Toni T., Welch D., Strelkowa N., Ipsen A., Stumpf M. P. H., Approximate Bayesian computation scheme for parameter inference and model selection in dynamical systems. J. R. Soc. Interface 6, 187–202 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jennings E., Madigan M., astroABC: An approximate bayesian computation sequential Monte Carlo sampler for cosmological parameter estimation. Astron. Comput. Secur. 19, 16–22 (2017). [Google Scholar]
- 69.Zinger T., Gelbart M., Miller D., Pennings P. S., Stern A., Inferring population genetics parameters of evolving viruses using time-series data. Virus Evol. 5, vez011 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zuker M., Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., Ben-Tal N., ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Friedman S. D., Snellgrove W. C., Genthner F. J., Genomic sequences of two novel Levivirus single-stranded RNA coliphages (family Leviviridae): Evidence for recombination in environmental strains. Viruses 4, 1548–1568 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Friedman S. D., Cooper E. M., Casanova L., Sobsey M. D., Genthner F. J., A reverse transcription-PCR assay to distinguish the four genogroups of male-specific (F+) RNA coliphages. J. Virol. Methods 159, 47–52 (2009). [DOI] [PubMed] [Google Scholar]
- 74.Domingo-Calap P., Cuevas J. M., Sanjuán R., The fitness effects of random mutations in single-stranded DNA and RNA bacteriophages. PLOS Genet. 5, e1000742 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Domingo-Calap P., Sanjuán R., Experimental evolution of RNA versus DNA viruses. Evolution 65, 2987–2994 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/34/eabb7990/DC1