

Synthetic lethal combination induces ferroptosis.
Abstract
Identification of targeted therapies for TNBC is an urgent medical need. Using a drug combination screen reliant on synthetic lethal interactions, we identified clinically relevant combination therapies for different TNBC subtypes. Two drug combinations targeting the BET family were further explored. The first, targeting BET and CXCR2, is specific for mesenchymal TNBC and induces apoptosis, whereas the second, targeting BET and the proteasome, is effective for major TNBC subtypes and triggers ferroptosis. Ferroptosis was induced at low drug doses and was associated with increased cellular iron and decreased glutathione levels, concomitant with reduced levels of GPX4 and key glutathione biosynthesis genes. Further functional studies, analysis of clinical datasets and breast cancer specimens revealed a unique vulnerability of TNBC to ferroptosis inducers, enrichment of ferroptosis gene signature, and differential expression of key proteins that increase labile iron and decrease glutathione levels. This study identified potent combination therapies for TNBC and unveiled ferroptosis as a promising therapeutic strategy.
INTRODUCTION
Targeted therapy represents a powerful strategy to treat hormone receptors (estrogen and progesterone) or human epidermal growth factor receptor 2 (HER2)–positive breast cancers. However, for triple-negative breast cancer (TNBC), a particularly aggressive and heterogeneous disease, there are no effective targeted therapies and no precision therapeutic options (1, 2). Numerous studies suggest that targeting of specific signaling pathway or epigenetic mechanisms using monotherapeutic agents is not effective, whereas combination therapies of two or more molecular targets substantially improve clinical outcome. Although potent combination therapies for TNBC have been identified in many preclinical settings, limited efficacy in patients has been associated with drug toxicity, drug resistance, and intra- and intertumoral heterogeneity (3). Recent studies suggest that cancer therapies exploiting the concept of synthetic lethality (SL) can overcome these constraints by selectively targeting vulnerable cancer cells, thereby reducing drug toxicity and side effects (4).
Possibly the best example of cancer therapy exploiting SL interaction is related to poly(adenosine diphosphate–ribose) polymerase (PARP)–Breast Cancer 1/2 (BRCA1/2) interaction. Patients harboring BRCA1/2 mutations are deficient in homologous recombination and lack high-fidelity repair mechanisms for DNA double-strand breaks (5). Therefore, they are highly susceptible to DNA damage induced by PARP inhibitors. PARP inhibitors have been approved as a monotherapy for patients with BRCA-mutated ovarian and breast cancer, including TNBC (6). However, only 10 to 15% of patients with TNBC harbor BRCA1/2 mutations, while the others have different mutations or copy number alternations in different oncogenes or tumor suppressor genes (7, 8) and thus need alternative therapeutic options.
Expression profiling analysis of large cohorts of patients with TNBC identified different molecular subtypes with distinct pathophysiological features and responses to treatments, including basal-like (BL), immune-inflamed, mesenchymal, mesenchymal stem–like (MSL), and luminal androgen receptor positive (9, 10). A recent clinical trial showed that TNBC patients with Programmed death ligand 1 (PD-L1)–positive tumors could benefit from combination of immunotherapy targeting PD-L1 with atezolizumab together with paclitaxel chemotherapy (11). PD-L1 blockade in ovarian tumors was recently found to induce lipid reactive oxygen species (ROS) in cancer cells and facilitate ferroptosis cell death (12).
Ferroptosis is a unique cell death pathway driven by iron-dependent lipid peroxidation. The pathway can be triggered by multiple mechanisms that modulate intracellular glutathione (GSH) pool, labile iron levels, polyunsaturated fatty acids (PUFAs), and the activity of GSH peroxidase 4 (GPX4), a selenoprotein that reduces phospholipid hydroperoxides using GSH and prevents ferroptotic cell death (13–15). Many cancer cells use different mechanisms to tolerate iron, certain amino acids, and/or lipid metabolism and, consequently, to prevent ferroptosis. The intrinsic capacity of cancer cells to escape ferroptosis varies between cancer types and confers distinct susceptibility to ferroptosis inducers. Generally, cancers of nonepithelial origin are more sensitive to ferroptosis compared to epithelial carcinomas including breast cancer (16).
In this study, we report that, among the different breast cancer subtypes, TNBC is particularly susceptible to ferroptosis. We show that TNBCs are enriched in ferroptosis gene signature, and we identified a synthetic lethal drug combination that induces ferroptosis in TNBC of both BL and mesenchymal subtypes. The potency of this combination, targeting the bromodomain and extraterminal (BET) proteins and the proteasome, was demonstrated in vitro and in animal models, and its clinical relevance was confirmed by computation analysis. Collectively, our findings demonstrate the power of SL screens to identify effective combination therapies, offer new therapeutic options for patients with TNBC, and highlight ferroptosis as a promising avenue for TNBC therapy.
RESULTS
A drug combination screen identified potent combination therapies for TNBC subtypes
To identify potent drug combinations for different molecular subtypes of TNBC (9, 10), we designed a unique high-throughput screen (HTS) reliant on the concept of SL. The HTS was carried out using 12 different human TNBC cell lines of different molecular subtypes, including BL (BL1 and BL2) and mesenchymal, mesenchymal stem–like (M/MSL), a human normal-like mammary cell line (MCF10A), and 17 selected pharmacological inhibitors (fig. S1A). The 17 inhibitors target predominant cancer-related pathways, DNA damage checkpoints, proteasome, or heat shock proteins (fig. S1A) and were proposed to inhibit TNBC growth in different preclinical settings (1, 17).
In brief, pairwise drug combinations were applied at very low doses to identify highly potent synergistic combinations with minimal toxicity. The half maximal inhibitory concentration (IC50) values of each individual drug in the 13 human cell lines were calculated from cell viability curves using serial dilutions and cell viability assay (CellTiter-Glo viability assay) (table S1). We then applied low doses (~5- to 10-fold lower concentration than the apparent IC50 values) of each drug and confirmed minimal (≤25%) effects on cell viability (fig. S1, B and C, and table S2). Subsequently, 106 drug pairs were applied at the examined low doses (table S2), and their effects on cell viability were assessed (CellTiter-Glo). Drug combinations were tested in triplicates, and potent combinations were defined as those inducing at least 75% death in TNBC lines, with minimal (≤20%) or no effect on MCF10A cell viability. Out of the 106 different combination pairs that were tested, 27 preferentially affected TNBC (Fig. 1A). Among them, 13 were effective in all the TNBC cell lines, whereas 9 were preferentially potent for the mesenchymal/mesenchymal stem-like (M/MSL) subtype and 5 were potent for BL TNBC cell lines (Fig. 1B). Potent combinations were further tested by different cell viability assays (fig. S1D).
Fig. 1. Synthetic lethal combination therapies for TNBC subtypes.
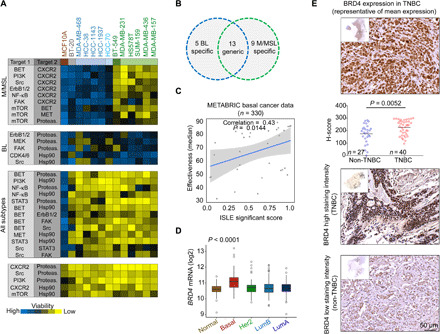
(A) Heatmap representation of drug screen results. Inhibitors of the indicated target pairs (fig. S1A) were applied at very low doses (~2 to 20% of IC50 as a single agent, for most cases), and viability of the indicated cell lines (12 TNBC + MCF10A) was measured 72 hours later. Effects on cell viability were calculated as percentage of control untreated cells and marked by color codes; blue, no/low cell death; yellow, high cell death. Drug pairs that reduced cell viability (≥75%) across several cell lines were clustered together according to cell line subtypes. We defined pairs that were potent for mesenchymal cell lines (M/MSL, green), basal-like (BL1/2, blue), and those that were potent in all subtypes but had low effect on MCF10A (≤20%) viability. Toxic combinations affect all cell lines (bottom). Results are mean values of three experiments. Actual screen data are included in table S3. (B) Venn diagram of hits obtained in the screen; mesenchymal (M/MSL)–specific, BL-specific (BL), and generic, which were effective in all subtypes. (C) Scatterplot between ISLE significance scores and drug combination effectiveness. Spearman correlation and P value are shown. Basal breast cancer data (330 patients; METABRIC) were used in the ISLE pipeline. (D) Box plots of BRD4 expression in patients with breast cancer grouped by PAM50. The difference between the basal patients and any other PAM50 group is significant (t test, P value of <0.0001). (E) IHC analysis of BRD4 expression in TNBC and non-TNBC tumors. Staining intensity of BRD4 was scored as described in Materials and Methods. Representative images of BRD4 staining in TNBC (n = 40) and non-TNBC (n = 27) tissues along with a column scatterplot of the H-score distribution among the specimens are shown. The difference between the groups is significant (t test).
To prioritize clinically promising synergistic combinations, we applied an unbiased computational analysis to identify clinically relevant synthetic lethal pairs as previously described (18). In brief, we mined genomic and transcriptomic data from 330 basal breast cancer patient tumors from the METABRIC dataset (19) and large-scale perturbation studies [short hairpin RNA (shRNA)/CRISPR] in cell lines to predict clinically relevant SL interactions between all possible target gene pairs of drugs used in our screen (see Materials and Methods and fig. S1A). We then estimated the strength of each SL interaction by computing their individual ISLE (Identification of clinically relevant Synthetic Lethality) significance score (fig. S1E) (see Materials and Methods), where high scores represent stronger SL interaction (18). We hypothesize that effective or synergistic drug combinations exhibit higher strength of SL interactions. The ISLE significance scores method highly scored our experimental potent combinations (Fig. 1C and fig. S1E) and revealed a significant correlation between the ISLE significance scores and the results of our drug combination screen (Spearman’s correlation = 0.43, P = 0.0144; see Materials and Methods and Fig. 1C). We could predict highly effective combinations using ISLE significance scores with an accuracy [area under the receiver operating characteristic curve (ROC-AUC)] of 87.76%.
In light of the ISLE scoring (fig. S1E), the relative toxicity of the different combinations (Fig. 1A and fig. S1D), and the high sensitivity of TNBC to BET inhibitor (20), we selected two potent drug combinations targeting the epigenetic readers of the BET family. The first combination was generic for all the tested TNBC subtypes (BL1/2 and M/MSL), using the BET inhibitor JQ1 and the proteasome inhibitor bortezomib (BTZ), while the second was an M/MSL-specific combination of JQ1 and the CXCR2 receptor antagonist SB225022 (21).
JQ1 is a thienodiazepine-based small-molecule inhibitor, which competitively inhibits the binding of bromodomains to acetylated histones and thus the activity of BRD4 (22). BRD4 transcripts are significantly enriched in patients with BL breast cancer (Fig. 1D) as well as in TNBC cell lines relative to other breast cancer lines (fig. S2A), as indicated by dataset analysis of The Cancer Genome Atlas (TCGA) breast cancer tumors [RNA sequencing (RNA-seq) and PAM50] and of the Cancer Cell Line Encyclopedia (CCLE). Furthermore, immunohistochemical (IHC) analysis of 67 breast cancer sections showed that the BRD4 protein is significantly enriched in TNBC compared to non-TNBC tumors (Fig. 1E), thus highlighting BRD4 as a promising therapeutic target for TNBC.
Synthetic lethal combinations targeting BET in TNBC subtypes
Among the generic potent combinations identified in our screen, dual inhibition of BET and the proteasome appeared to be a promising choice. A previous small interfering RNA screen suggested that proteasomal targeting could be beneficial for patients with BL TNBC (23), and our ISLE scoring (fig. S1E) predicted high clinical relevance of BET and proteasome cotargeting. Likewise, the chemokines interleukin-8 (IL8), IL6, and CXCL1, which bind the CXCR1/2 receptors and are implicated in TNBC growth and metastasis (24, 25), are enriched in TNBC compared to other breast cancer subtypes (fig. S2B), implying that inhibition of CXCR2 could be beneficial for patients of the M/MSL subtype.
The SL of BET and CXCR2 inhibition (JQ1 + CXCR2in/SB225022) in M/MSL TNBC and of BET and the proteasome (JQ1 + BTZ) in all TNBC subtypes (Fig. 1A) led us to further assess their potency, specificity, and toxicity. Cell viability assays across multiple breast cancer cell lines using increasing drug doses showed a dose-dependent high susceptibility of human M/MSL to BET and CXCR2 inhibition (Fig. 2A), consistent with our screen results. BL TNBC, the nontumorigenic MCF10A, and the luminal MCF7 breast cancer cell lines were not affected by this drug combination at the applied doses, demonstrating its low toxicity and high selectivity to M/MSL TNBC. Furthermore, knockdown of BRD4 expression by shRNA sensitized M/MSL TNBC cell lines to CXCR2 inhibition (fig. S2C). The JQ1 + SB225022 combination markedly reduced the IC50 values of individual drugs in M/MSL cell lines (fig. S3, D and E), and dose-response curves for JQ1 and/or SB225022 revealed high potency of the drug combination (Fig. 2B and fig. S3) with strong synergy, as determined by calculating the combination index (CI) using the Chou-Talalay method (26) (CI < 0.5) (Fig. 2C).
Fig. 2. In vitro validation of drug combinations targeting BET.
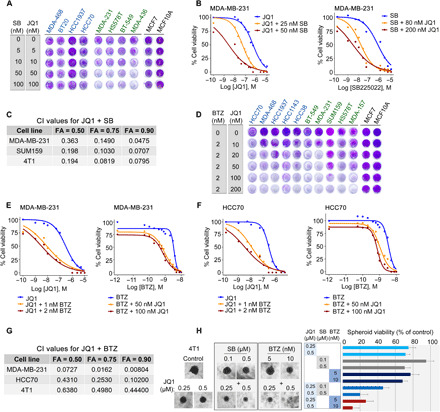
(A) Effects of BET and CXCR2 antagonists on cell viability. The indicated BL1/2 (blue) and M/MSL (green) TNBC cell lines and MCF7 (luminal) and MCF10A (normal-like) lines were treated with the indicated doses of the drugs for 72 hours and stained with crystal violet. Representative pictures of reproducible effects from two to three independent experiments are shown. (B, E, and F) Dose-response curves of single agents and drug combinations in the indicated cell lines treated with varying concentrations of JQ1 and SB225022 (B) or JQ1 and BTZ (E and F) for 72 hours. Dose-response curves are presented as means of four (B) or three (E and F) repeats. (C and G) CI was calculated by the CompuSyn software with the Chou-Talalay equation using multiple doses and response points. CI values for three different indicated FA are shown. (D) Effects of BET and proteasome inhibitors on cell viability were assessed by crystal violet staining as described in (A). (H) Effects of drug combinations on spheroid growth. Representative images and viability assay (CellTiter-Blue, bar graph) of day 15 4T1 spheroids (n = 6 spheroids). Control untreated and treated spheroids with single agents at the indicated doses and with the drug combinations are shown. Scale bar, 100 μm.
In contrast to BET and CXCR2 targeting, the BET and proteasome (JQ1 + BTZ) combination was effective across TNBC cell lines (Fig. 2D). The potency and synergy of the JQ1 + BTZ combination is reflected by the remarkable reduced IC50 values for each individual drug in the presence of the second (Fig. 2, E and F, and fig. S4, A to F) and by the calculated CI values shown for representative cell lines (Fig. 2G). Collectively, these results show high potency and strong synergy of BET and CXCR2 inhibition in M/MSL TNBC and of BET and the proteasome in all TNBC cell lines irrespective of their molecular subtype. The murine breast cancer cell line 4T1, which is considered as a TNBC line (27), was sensitive to the two BET combinations (JQ1 + BTZ and JQ1 + SB225022) in both two-dimensional (2D) (Fig. 2C and figs. S3, C and F, and S4, C and F) and 3D spheroid cultures (Fig. 2H).
Next, we assessed the effects of BET + CXCR2 and BET + proteasome inhibition in vivo using orthotopic tumor models. Because of the short half-life of JQ1 in vivo (28), we used the bromodomain inhibitor OTX015 (MK-8628). OTX015 was previously used in preclinical and clinical trials (29) and behaved similarly to JQ1 in vitro, either as a single agent or in combination with SB225022 or BTZ (fig. S5, A and B). Preliminary pilot experiments using syngeneic orthotopic 4T1 tumor models (fig. S5, C to F) and MDA-MB-231 xenografts in nude mice (fig. S5G) were used to calibrate dose responses. Accordingly, OTX015 was orally administered once a day at 25 mg/kg, whereas SB225022 was intraperitoneally (IP) administered at 5 mg/kg (for both, ~2-fold lower concentrations than previously used) (21, 30). The drug administration scheme is described in fig. S5H. As shown, OTX015 or SB225022 applied as a single agent had modest effects on tumor growth with only ~15% tumor growth inhibition (TGI) (Fig. 3, A to C). In contrast, the OTX015 + SB225022 combination was very effective, drastically reducing tumor size (Fig. 3, A to C), with a TGI close to 100%. This drug combination had minimal toxicity at the applied doses, as indicated by body weight (Fig. 3D). Collectively, these data show the high efficacy of BET and CXCR2 co-inhibition compared to single-agent treatments.
Fig. 3. In vivo validation of drug combinations targeting BET.
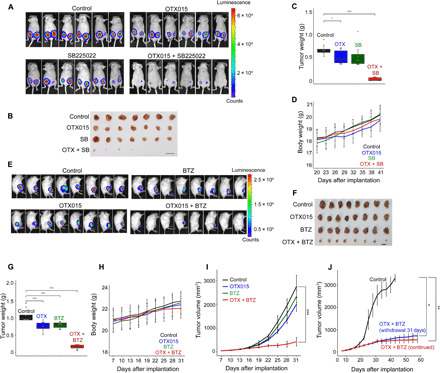
(A to D) Effects of BET and CXCR2 inhibition on tumor growth. Representative bioluminescence imaging of xenograft tumors at day 41 after bilateral implantation of MDA-MB-231 into the mammary fat pad of nude mice is shown in (A). Mice were randomized into four groups (n = 9 per group) at day 23 and then treated with either vehicle, OTX015 (25 mg/kg, oral), SB225022 (5 mg/kg, IP, 5 days/week), or drug combination. The effects of drugs on tumor size at day 41 are shown by the excised tumors (B; scale bar, 1 cm), and the effects on tumor weight are shown in (C). ***P < 0.001 and *P < 0.05 (t test). The different drug treatments had no significant effects on body weight (D). Mean values ± SD of mice body weight at the indicated time points are shown. (E to G) Effects of BET and proteasome inhibition on tumor growth. Representative bioluminescence imaging of 4T1 allograft mice (E) at day 31 after implantation of 4T1 cells into the mammary fat pad of BALB/c mice is shown. Mice were randomized into four groups (n = 10 to 12 per group) at day 13 and then treated with either vehicle, OTX015 (25 mg/kg, oral), BTZ (0.25 mg/kg, IP, every fourth day), or drug combination. The effects of drugs on tumor size at day 31 are shown by the excised tumors (F; scale bar, 1 cm), and the effects on tumor weight are shown in (G), ***P < 0.001 (t test). (H) The different drugs had no significant effects on body weight. Means ± SD from (n = 10 to 12 per group) mice are shown. (I) Tumor volume curves of 4T1 allograft mice treated with vehicle (control), single drugs, or drug combination, ***P < 0.001 (Wilcoxon test). (J) Effects of drug withdrawal at day 31 on tumor growth were measured for 55 days. Data shown are the means ± SD from five to six tumors at each time point, **P < 0.01; *P < 0.05 (Wilcoxon test).
For the generic combination of OTX015 + BTZ, we established orthotopic syngeneic mice models using the 4T1 cells. We applied low doses of both drugs (OTX015: 25 mg/kg per day, orally; BTZ: 0.25 mg/kg, IP; fig. S5I) and examined the effects of single and combined treatments on tumor growth over time. Results clearly showed the profound and significant effects of the combination on TGI compared with monotherapy (Fig. 3, E to G and I). The drug combination had no significant toxicity at the applied doses, as indicated by body weight (Fig. 3H) and by the levels of serological markers for organ damage (table S5). The strong inhibitory effects of the OTX015 + BTZ combination were sustained for at least 55 days and persisted for at least 21 days after cessation of therapy (Fig. 3J). Kaplan-Meier survival curves indicate that the OTX015 + BTZ combination significantly prolonged mice survival for approximately 80 days (fig. S5J). Survival was prolonged even after therapy was discontinued on day 31. Together, these results suggest that cotargeting of BET and the proteasome applied at low doses could be a promising therapeutic approach for TNBC.
BET and proteasome cotargeting robustly triggers ferroptotic cell death
We next examined possible mechanisms underlying the potency of the two drug combinations. We found that co-inhibition of BET and CXCR2 induced apoptotic cell death, as demonstrated by annexin V staining (fig. S6A) and by PARP and caspase 3 cleavage (Fig. 4A). In contrast, co-inhibition of BET and the proteasome, applying low drug doses, induced neither PARP cleavage nor annexin V staining (Fig. 4B and fig. S6B), implying that a non-apoptotic pathway is involved. We therefore systematically examined the involvement of four major cell death pathways—apoptosis, necrosis, autophagy, and ferroptosis—using specific inhibitors and characteristic assays. As seen in Fig. 4C, the pan-caspase inhibitor zVAD-FMK, as well as Necrostatin-1 and 3-methyladenine (3MA), which inhibit apoptosis, necrosis, and autophagic cell death (14), respectively, only marginally rescued cell death in response to the JQ1 + BTZ treatment but effectively rescued cell death in response to their cognate pathway-specific inducers (fig. S6C). In contrast, GSH, the iron chelator 2,2-bipyridyl (2,2-BP), and the antioxidant ferrostatin-1, which are known to inhibit ferroptosis (31), could effectively rescue cell death induced by JQ1 + BTZ, as assessed by different cell viability assays in multiple TNBC cell lines (Fig. 4, C and D, and fig. S6, D and E). Ferroptosis was obtained at a specific range of low doses of JQ1 + BTZ based on liproxstatin-1 rescue experiment (fig. S6F), consistent with their SL interactions. These results strongly suggest that the JQ1 + BTZ combination applied at low doses induces ferroptosis cell death.
Fig. 4. Inhibition of BET and the proteasome triggers ferroptosis cell death.
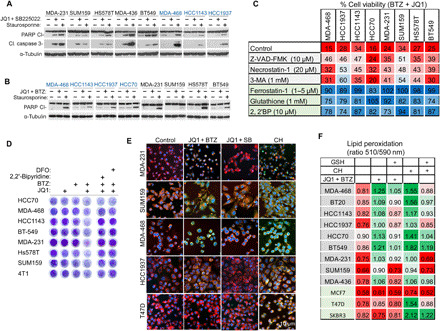
(A and B) Effects of BET and CXCR2 inhibition (A) or BET and proteasome inhibition (B) on PARP (A and B) and caspase-3 cleavage (A) in the indicated cell lines (blue, BL1; black, M/MSL). Staurosporine (150 nM, 16 hours) was used as a positive control. The cells were treated with low doses of JQ1 and either SB225022 (A) or BTZ (B) for 24 hours as described in Materials and Methods, lysed, and assessed by Western blot (WB) for the indicated proteins. (C) Ferroptosis inhibitors rescue cell death induced by JQ1 and BTZ combination. The indicated cell lines were pretreated with the indicated cell death inhibitors for 1 hour and then for an additional 72 hours in the absence or presence of JQ1 and BTZ (see Materials and Methods). Cell viability (CellTiter-Blue) is presented as percentage of untreated cells. The effects of the inhibitors on their cognate death pathways are shown in fig. S6C. Mean values of three experiments are shown (means ± SD; table S6). (D) Iron chelators rescued cell death induced by JQ1 and BTZ treatment. The indicated TNBC cell lines were treated with JQ1 and BTZ (see Materials and Methods) for 72 hours in the absence or presence of deferoxamine (DFO) (100 μM) or 2,2′-dipyridyl (10 μM), and cell viability was assessed by crystal violet staining. (E) Representative confocal images of the indicated TNBC cell lines and T47D cells stained with C11-BODIPY (10 μM) as described in Materials and Methods. The cells were treated with cumene hydroperoxide (CH) (100 μM, 3 hours) as a positive control, and either with JQ1 + SB225022 or with JQ1 and BTZ (see Materials and Methods) for 16 hours. Scale bar, 10 μm. (F) Assessment of lipid peroxidation in breast cancer cells in response to BET and proteasome inhibition. TNBC (gray) and non-TNBC (green) cell lines were incubated with JQ1 and BTZ (see Materials and Methods) for 16 hours or with CH (100 μM, 3 hours). Where indicated, glutathione (1 mM; see Materials and Methods) was applied. Lipid peroxidation is shown as the ratio between fluorescence emission at 510 nm (green) and 590 nm (red) (see Materials and Methods). Mean values of three experiments are shown (mean values ± SD; table S7).
To further corroborate these results, we examined the effects of the JQ1 + BTZ combination on necrotic and ferroptotic specific markers. Necrosis was induced by a combination of tumor necrosis factor–α, Smac mimetic, and zVAD-FMK and monitored by MLKL (mixed lineage kinase domain-like protein) phosphorylation at Ser358 as previously described (32). As shown in fig. S6G, the JQ1 + BTZ combination had a slight or no effect on MLKL phosphorylation, suggesting that necrosis is not the death-induced pathway. Ferroptosis, the iron-dependent program cell death driven by lipid peroxidation, was monitored by cellular lipid peroxidation levels applying the BODIPY 581/591 C11 dye. The emission of the BODIPY dye shifts from 590 nm (red) to 510 nm (green) upon oxidation (33). As shown in Fig. 4E, in contrast to JQ1 + SB225022, the JQ1 + BTZ combination markedly increased C11-BODIPY oxidation in TNBC cell lines, as demonstrated by the representative fluorescent images and by the quantitative analysis of the 510/590-nm (green/red) fluorescence ratio (Fig. 4F). Induction of lipid peroxidation by the JQ1 + BTZ combination was observed only in TNBC and not in the luminal breast cancer cell lines, was rescued by GSH pretreatment in most of the examined TNBC lines (Fig. 4F), and was further validated by the TBARS (thiobarbituric acid reactive substances) assay (fig. S6H). These results strongly indicate that TNBC cells are selectively more susceptible to ferroptosis induced by the JQ1 + BTZ combination.
As iron is required for lipid peroxide accumulation and ferroptosis execution (13), we examined the effect of JQ1 + BTZ cotreatment on total cellular iron levels using a colorimetric assay (QuantiChrom Iron Assay Kit, BioAssay Systems). As shown, the JQ1 + BTZ combination increased the levels of cellular iron selectively in TNBC cell lines and not in the examined luminal cell lines (Fig. 5A). Concomitantly, we observed a significant decrease in cellular reduced GSH levels (Fig. 5B) and an increase in reactive oxidative species (ROS) as demonstrated by CM-H2DCFDA fluorescent images (fig. S6I) and quantified by fluorescence measurements (488 nm) (Fig. 5C). Pretreatment with GSH reduced the effect of JQ1 + BTZ on both H2DCFDA fluorescence (Fig. 5C and fig. S6I) and cell death (Fig. 4C and fig. S6E). Collectively, these results indicate that the JQ1 + BTZ combination has significant effects on key ferroptosis regulators in TNBC, including cellular iron and antioxidant GSH levels.
Fig. 5. Effects of BET and proteasome co-inhibition on cellular iron, GSH, and ROS levels.
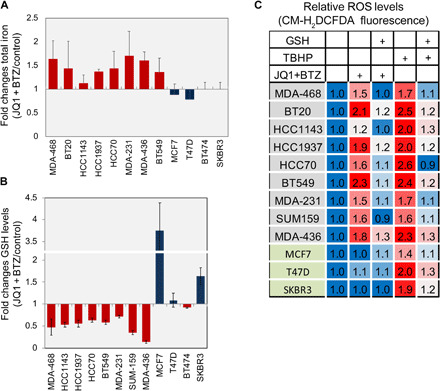
(A and B) Inhibition of BET and the proteasome increased iron levels (A) and decreased GSH levels (B) in TNBC cell lines. TNBC and non-TNBC (MCF7, SKBR3, T47D, and BT474) cells were incubated with JQ1 and BTZ (see Materials and Methods) for 16 hours, and total iron (A) and reduced GSH (B) levels were measured as described in Materials and Methods. Mean values of three experiments are shown (mean values ± SD; table S8). (C) Inhibition of BET and the proteasome increased ROS in TNBC cells. The indicated breast cancer cells were treated with JQ1 and BTZ (see Materials and Methods) for 12 hours or with tert-butyl hydrogen peroxide (TBHP; 100 μM) for 2 hours. Where indicated, GSH (1 mM) was applied 1 hour before drug treatment. Cells were then incubated with CM-H2DCFDA to measure ROS as described in Materials and Methods. Results are expressed as folds of control in at least three experiments (mean values ± SD; table S9).
BET and proteasome cotargeting modulates GPX4 and ferroptosis-associated genes in TNBC
Previous studies suggested that GSH levels are reduced in TNBC (34). As GSH is a necessary cofactor of GPX4, a major regulator of ferroptosis (33, 35), we examined its mRNA expression levels across 956 breast cancer and normal-like samples from TCGA dataset. As shown, the GPX4 level was significantly lower in basal breast cancer compared to normal or other breast cancer subtypes (Fig. 6A).
Fig. 6. BET and proteasome inhibition strongly affects GPX4 level and transcription of key ferroptotic genes.
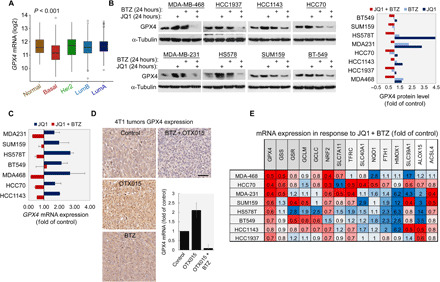
(A) Box plot showing the expression of GPX4 in patients with breast cancer grouped by PAM50. The differences between the BL patients and any other PAM50 groups are significant (t test, P < 0.001). (B and C) Effects of BET and proteasome inhibition on the levels of GPX4 protein (B) and transcript (C). The indicated TNBC cell lines were treated with JQ1, BTZ, or both (see Materials and Methods) for 24 hours. Levels of GPX4 protein were assessed by WB. Intensities of GPX4 bands were quantified, normalized, and presented as fold of control in the bar graph (B). GPX4 mRNA levels were assessed by qPCR. Mean values ± SD of at least two repeats are shown (C). (D) Effects of BET and proteasome inhibition on level of GPX4 protein and transcript in 4T1 tumors. Representative IHC staining of 4T1 tumors treated with OTX015 (25 mg/kg per day, orally), BTZ (0.25 mg/kg, IP, every fourth day), or both to detect protein expression is shown. GPX4 mRNA levels were evaluated by qPCR. Results are mean values ± SD of at least six mice per group. (E) Effects of BET and proteasome inhibition on level of GPX4 transcript and additional key ferroptosis genes. The indicated TNBC cell lines were treated with JQ1 and BTZ (see Materials and Methods) for 24 hours, and mRNA levels of the indicated genes were assessed by qPCR. The results are reported as fold of control. Mean values of at least three independent experiments are shown (mean values ± SD; table S11).
We then examined the influence of the JQ1 + BTZ combination on the protein and mRNA levels of GPX4 in multiple TNBC cell lines using Western blotting (WB) (Fig. 6B) and quantitative polymerase chain reaction (qPCR) analysis (Fig. 6C). As shown, JQ1, applied at a low concentration, increased GPX4 protein (up to ~4.5-fold; Fig. 6B and fig. S7A) and transcript (Fig. 6C and fig. S7B) levels in all the examined TNBC cell lines as well as OTX015 in 4T1 tumors (Fig. 6D), whereas the JQ1 + BTZ combination markedly reduced GPX4 protein levels (Fig. 6B). The JQ1 + BTZ combination also suppressed the transcription of GPX4 in multiple TNBC cell lines in vitro (Fig. 6C), while the OTX015 + BTZ combination reduced both the protein and mRNA levels of GPX4 in 4T1 tumors, as shown by IHC staining of tumor sections and by qPCR (Fig. 6D).
The profound effects of the JQ1 + BTZ combination on GPX4 expression led us to examine its influence on other ferroptosis-associated genes. We selected 15 key ferroptosis-related genes (Fig. 6E and fig. S7C), of which 12 belong to the ferroptosis gene signature (www.kegg.jp/kegg-bin/show_pathway?hsa04216), and assessed their transcript levels in response to the JQ1 + BTZ combination in multiple TNBC cell lines. As shown in Fig. 6E, we observed a down-regulation of NRF2 (Nuclear factor erythroid 2–related factor 2) in the majority of TNBC cell lines. GSS (GSH synthetase), GCLM [the modifier subunit of γ-glutamylcysteine ligase (γGCL)], GCLC (the catalytic subunit of γGCL), and/or GSR (GSH disulfide reductase) were also reduced in many TNBC cell lines upon JQ1 + BTZ treatment.
γGCL and GSS are two critical GSH biosynthesis enzymes in mammalian cells (36), whereas GSR catalyzes the reduction of GSH disulfide (GSSG) to GSH. The reduced expression of these enzymes in response to the JQ1 + BTZ combination is consistent with the decreased levels of cellular GSH (Fig. 5B) and ferroptosis cell death. Likewise, low levels of NRF2, the master regulator of the cellular antioxidant response, are consistent with reduced levels of GSH and increased levels of cellular ROS (Fig. 5C and fig. S6I) and, possibly, the reduced levels of GPX4, a putative NRF2 target gene (fig. S7C) (37). Elevated ROS levels may induce up-regulation of HMOX1 (heme oxygenase-1) and NQO1 (reduced form of nicotinamide adenine dinucleotide phosphate quinone dehydrogenase 1) antioxidant protective enzymes (38). ALOX15, which was strongly up-regulated in M/MSL cell lines, is involved in peroxidation of PUFAs and thus accelerates ferroptosis (39). Other proteins/enzymes were distinctly modified among the different cell lines. Nevertheless, it appears that GPX4 and the key enzymes/proteins regulating GSH levels are markedly affected by the JQ1 + BTZ combination.
TNBCs are enriched in a ferroptosis gene signature and vulnerable to ferroptosis inducers
Thus far, our findings indicate that TNBCs, compared to other breast cancers, are more susceptible to ferroptotic cell death induced by the JQ1 + BTZ combination (Fig. 4) and that low doses of the JQ1 + BTZ treatment affects the expression level of GPX4 and several additional ferroptosis-associated genes (Fig. 6, B to E). We also showed that GPX4 levels are significantly lower in basal patients (Fig. 6A), and a previous report suggested that GSH levels are particularly low in basal breast cancers (34). These observations suggest that TNBCs could be more susceptible to ferroptosis and thus more vulnerable to ferroptosis-inducing agents. To explore this possibility, we examined the susceptibility of 10 TNBC and 4 to 7 non-TNBC cell lines to the ferroptosis inducers Fin56 and erastin (Fig. 7A and fig. S8A). As shown, Fin56 and erastin substantially reduced viability of all TNBC cell lines tested but weakly affected non-TNBC cell lines at the indicated doses (Fig. 7A). Area under the viability curves (AUCs) of multiple repeats using 16 different breast cancer cell lines indicated that TNBCs are significantly more susceptible to ferroptosis compared to non-TNBC cell lines (fig. S8A).
Fig. 7. TNBCs are vulnerable to ferroptosis and enriched in ferroptosis signature.
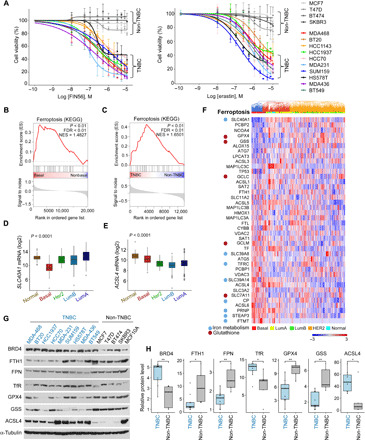
(A) Cell viability of the indicated TNBC and non-TNBC cell lines in response to increasing concentrations of Fin56 or erastin. The indicated cells were treated with FIN56 or erastin for 72 hours, and cell viability was measured by MTT assays. Mean values ± SD of three independent experiments are shown. The differences in the area under the curve between the TNBC and non-TNBC are significant (fig. S8A). (B and C) GSEA plot of normalized enrichment score of ferroptosis pathway for either basal versus non-basal patients in the TCGA dataset (B) or TNBC versus non-TNBC cell lines in the CCLE dataset (C). Enrichment is significant, P value/FDR < 0.01. (D and E) Box plots showing the expression of SLC40A1 (D) and ACSL4 (E) in patients with breast cancer grouped by PAM50. The differences between the basal patients and any other PAM50 group are significant (t test, P < 0.001). (F) Heatmap of normalized expression of the ferroptosis gene signature (KEGG, 40 genes) in patients with breast cancer from the TCGA dataset (n = 956). Patients (in columns) are arranged by unsupervised clustering of gene expression. Bar at the top of the heatmap indicates the subtype of each patient (PAM50). Ferroptotic genes that also belong to the iron metabolism or GSH signature are marked. (G and H) Levels of ferroptosis proteins and BRD4 in TNBC and non-TNBC cell lines. The expression levels of the indicated proteins were assessed by WB, and band intensities were quantified by ImageJ software. The relative expression in TNBC compared to non-TNBC is shown in the box plots (H) *P < 0.05, **P < 0.01.
Although these inhibitory effects could be attributed to the intrinsic low levels of GPX4 in TNBC (Fig. 6A), ferroptosis is regulated by additional proteins involved in intracellular GSH homeostasis as well as in iron and lipid metabolism (40) and is characterized by a specific 40-gene signature (www.kegg.jp/kegg-bin/show_pathway?hsa04216). We used the ferroptosis gene signature to examine the expression profile of ferroptotic genes in TNBC versus non-TNBC cell lines and in clinical samples of different breast cancer subtypes (PAM50 classification). We first performed Gene Set Enrichment Analysis (GSEA) of differentially regulated genes in basal versus non-basal breast cancer samples using TCGA datasets (n = 956). We found a significant enrichment of the ferroptosis signature in the basal relative to other breast cancers subtypes [false discovery rate (FDR) q value < 0.01] (Fig. 7B). Significant enrichment of ferroptosis was also obtained in the TNBC versus non-TNBC cell lines from the CCLE (FDR q value < 0.01; Fig. 7C).
Of the 40 ferroptotic genes, 31 were significantly differentially expressed (FDR < 0.05) in basal versus non-basal tumors, of which 12 were down-regulated and 19 were up-regulated. Among the different ferroptotic genes, ferroportin (SLC40A1) and ACSL4 (acyl–coenzyme A synthetase long chain family member 4) displayed the most prominent differences between TNBC and non-TNBC cell lines. Ferroportin is the major cellular iron exporter, while ACSL4 plays a key role in arachidonic acid metabolism and is essential for ferroptosis execution (41). The relative expression of these two genes in each of the 55 CCLE breast cancer cell lines is shown in fig. S8 (B and C). Significant low levels of Ferroportin and relatively high levels of ACSL4 transcripts were also obtained in basal breast cancer compared to other breast cancer subtypes (PAM50) (Fig. 7, D and E). Unsupervised hierarchical clustering analysis of 956 breast cancer tissues (142 basal, 434 LumA, 194 LumB, 67 HER2, and 119 normal-like) showed that basal tumors form a unique cluster separated from the non-basal tumors (Fig. 7F), further strengthening the association of ferroptosis with BL tumors relative to other breast cancer subtypes and highlight the clinical relevance of this pathway for TNBC.
As a complementary approach to the transcriptomic analysis described above, we examined whether ferroptotic proteins are also differentially expressed in TNBC. We first assessed the levels of six selected proteins in multiple breast cancer cell lines by WB. As shown in Fig. 7 (G and H), the levels of ferroportin, GPX4, GSS, and ferritin heavy chain 1 (FTH1) were significantly lower in TNBC relative to non-TNBC cell lines, whereas the levels of the iron importer transferrin receptor (TfR) were higher in TNBC cell lines. ACSL4 was also significantly higher in TNBC cell lines, consistent with its gene expression pattern (Fig. 7G and fig. S8B).
To further corroborate the link between ferroptosis and TNBC and to highlight its clinical relevance, we selected four key ferroptotic proteins (GPX4, GSS, ferroportin, and TfR) and evaluated their expression level by immunohistochemistry in clinical samples of TNBC and non-TNBC patients. Approximately 70 (66 to 74) human breast tumor sections were immunostained for GPX4, GSS, ferroportin, and TfR. The samples were evaluated semiquantitatively, and their calculated H-scores were plotted in column scatter graphs. We also calculated how many tumors with high and low staining intensity belong to TNBC relative to non-TNBC. As shown in Fig. 8, while TfR staining was significantly higher in TNBC tumors, immunostaining for GPX4, GSS, and ferroportin showed significantly lower expression levels in TNBC sections.
Fig. 8. IHC analysis of ferroptotic proteins in breast cancer samples.
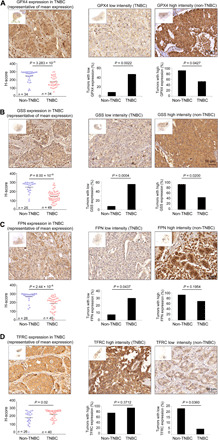
(A to D) Representative images of IHC analysis of breast cancer specimens from patients with TNBC and non-TNBC patients immunostained with antibodies against GPX4 (A), GSS (B), Ferroportin (FPN) (C), and TfR (TFRC) (D). Approximately 65 breast cancer tissues were immunostained for each protein, and staining intensity was scored as described in Materials and Methods. The H-score of patients with TNBC relative to non-TNBC patients is shown in the scatter graphs. Percentages of tumors with low- or high-intensity scores are shown in the middle or right graphs along with representative images. Overall view of the entire section is shown as an insert for each image. Scale bars, 50 μm. Unpaired two-tailed t test was used to compare differences between H-scores of patients with TNBC and non-TNBC patients. Fisher’s exact test was applied to compare percentage of tumor sections with high or low protein expression between TNBC and non-TNBC groups.
These results are consistent with the mRNA expression profiles from TCGA dataset and strongly suggest that TNBCs distinctly express key proteins that regulate ferroptosis. The high TfR and low ferroportin, which mediate iron import and export, respectively, may contribute to an increase in cellular labile iron (42), while low GPX4 and GSS may reduce antioxidation defense capacity and increase the probability of lipid peroxidation and consequently the susceptibility of patients with TNBC to ferroptosis inducers.
In light of the analysis described above, we incorporated the differential expression data of ferroptotic genes in BL breast cancer into the KEGG ferroptosis pathway. We included the transsulfuration pathway that mediates the transfer of sulfur atom of methionine into the sulfur atom of cysteine (43) and the pentose phosphate pathway (fig. S8D), which both influence ferroptosis (13). This illustration highlights the interplay between metabolic pathways that regulate ferroptosis and the differential expression of key regulators in BL tumors. The most prominent is the substantial low level of ferroportin concomitant with the reduced levels of GSH biosynthesis enzymes and of GPX4, which collectively suggests that TNBCs are intrinsically prone to have high labile iron, low GSH antioxidant defense capacity, and high PUFAs and thus are particularly vulnerable to ferroptosis execution.
DISCUSSION
In this study, we applied an SL screen and identified clinically relevant combination therapies for TNBC subtypes (Fig. 1, A to C, and fig. S1E). As targeting of epigenetic alterations provides a new opportunity for therapeutic intervention (44) and BRD4 has emerged as a promising anticancer therapeutic target for TNBC (20, 45), we further validated combination therapies targeting BET. First, we showed that BRD4 transcript and protein levels are highly enriched in TNBC tumors and cell lines (Fig. 1, D and E, and fig. S2A). Subsequently, we validated the high potency and low toxicity of the two combinations targeting BET—BET and CXCR2 inhibitor and BET and the proteasome inhibitor—using multiple cell-based assays, applying 2D cell culture and 3D spheroids (Fig. 2). In vivo validation was carried out using orthotopic tumor models, applying immunocompetent and immunodeficient mice (Fig. 3) and OTX015 as a BET inhibitor.
By systematic analysis of the mechanisms underlying the high potency of BET + CXCR2 inhibition in mesenchymal TNBC and of BET + proteasome in all major TNBC subtypes (BL1/2 and M/MSL), we found that, in contrast to apoptotic cell death induced by low doses of BET + CXCR2 inhibition, BET + proteasome inhibition induced ferroptotic cell death (Fig. 4 and fig. S6). It was previously shown that inhibition of BET and B cell lymphoma extra large B cell lymphoma-extra large (BCL-xL) in TNBC also induced apoptosis (46), while targeting of BET alone was insufficient and resulted in up-regulation of B cell lymphoma-extra large (BCL-xL) and drug resistance (20). Likewise, we found an up-regulation of GPX4 in response to BET inhibition alone at the applied doses but a substantial down-regulation of GPX4 in response to the BET + proteasome inhibition (Fig. 6, B to D), possibly due to the concurrent down-regulation of NRF2 (Fig. 6E). This possibility, however, needs further investigation. Down-regulation of GPX4 was accompanied by an increase in intracellular ROS (Fig. 5C and fig. S6I) and a decrease in the expression of several GSH biosynthesis genes (Fig. 6E), concomitant with reduced intracellular GSH levels (Fig. 5B). These effects of BET inhibition alone or together with either BTZ or CXCR2 were obtained at low drug concentrations consistent with the SL concept and dose-dependent transcriptional reprogramming. It was previously shown that high versus low concentrations of JQ1 induced distinct gene expression profiles (47) and that high concentrations of JQ1 (10 μM) can induce ferroptosis in a few mesenchymal TNBC cell lines (48), whereas high doses of BTZ (50 to 500 nM) induced apoptosis in several TNBC cell lines (49). Together, these studies suggest that a cocktail of anticancer drugs can induce a specific cell death pathway through unique transcriptional and signaling reprogramming, which is largely dependent on the applied doses and the intrinsic properties of the cancer type.
It was previously shown that TNBC cells are particularly sensitive to GSH depletion (34) and that BL breast cancer cells, as opposed to luminal cells, display cystine addiction and rapidly die in response to cystine deprivation (50). The xCT cystine/glutamate antiporter mediates uptake of cystine, which is rapidly reduced to cysteine, the rate-limiting amino acid for GSH biosynthesis. Inhibition of xCT system as well as glutamine restriction attenuated the growth of a subset of BL TNBC. Therefore, the xCT antiporter (51) and the ASCT2 (Alanine Serine Cysteine Transporter 2) glutamine transport (52) were proposed as therapeutic targets for TNBC. However, xCT inhibitors, such as sulfasalazine (SASP), are not effective under physiological conditions. SASP is labile and insoluble and induces multiple side effects (anti-inflammatory), whereas clinically approved ASCT2 inhibitors are currently not available. Other ferroptosis inducers including erastin, sorafenib, and glutamate are not potent, not selective, and/or metabolically unstable in animal models (53). Similarly, GSH biosynthesis inhibitors are highly toxic and failed in clinical trials (54). Our combination, on the other hand, of BET and proteasome inhibition is generic (Fig. 1, A and B) and very potent in animal models (Fig. 3), thus highlighting its clinical potential and possible unique advantage over other approaches that induce ferroptosis.
The effects of BET and proteasome inhibition on ferroptosis were preferentially obtained in TNBC compared to other breast cancer cell lines (Fig. 4), similar to the effects of erastin and Fin56 (Fig. 7A). This high vulnerability of TNBC to ferroptosis is associated with the distinct expression pattern of ferroptosis gene signature (Fig. 7, B to F, and fig. S8D), particularly of key regulatory genes. In addition to the low levels of GPX4 in TNBC versus non-TNBC tumors (Figs. 6A, 7F, and 8A) or cell lines (Fig. 7, G and H), significant high expressions of ACSL4 in TNBC tumors and cell lines were observed (Fig. 7, E and G, and fig. S8C). ACSL4 is essential for ferroptosis execution and is considered to be a ferroptosis biomarker (55), which dictates ferroptosis sensitivity (41), consistent with our results.
Genes and proteins that regulate intracellular iron levels are also distinctly expressed in TNBC versus non-TNBC tumors and cell lines (Fig. 7G and fig. S8D). In particular, a substantial low level of the iron exporter ferroportin (Figs. 7, D, G, and F, and 8C and fig. S8B), concomitant with the high expression level of the iron importer TfR (Figs. 7, G and H, and 8D), may increase intracellular labile iron pool to facilitate iron-dependent lipid peroxidation. An increase in intracellular iron levels was obtained in TNBC cell lines in response to BET + proteasome inhibition (Fig. 5A), implying that this drug combination, applied at low doses, not only is effective as a therapeutic strategy but also can be used as an experimental tool to investigate molecular mechanisms that regulate ferroptosis, at least in TNBC. Collectively, these analyses suggest that TNBCs have a unique metabolic state of iron and GSH homeostasis that enhance their susceptibility to ferroptosis.
In summary, our study shows that an SL screen is a powerful approach to identify clinically relevant potent combinations for cancer therapy. We validated two promising therapeutic strategies mediated by epigenetic targeting toward precision medicine in TNBC. Our findings uncovered an intrinsic susceptibility of TNBC to ferroptosis and highlight this death pathway as a TNBC druggable target.
MATERIALS AND METHODS
Chemicals and drugs
The small-molecule inhibitors used in the screen were purchased from different sources. Mammalian target of rapamycin (mTOR) inhibitor (Rad00, S1120), SRC inhibitor (dasatinib, S1021), mitogen-activated protein kinase kinase (MEK) inhibitor (GSK1120212B, S2673), cyclin-dependent kinase 4/6 (CDK4/6) inhibitor (PD0332991, S1116), BCL2 inhibitor (ABT737, S1002), epidermal growth factor receptor (EGFR) inhibitor (neratinib, S2150), phosphatidylinositol 3-kinase (PI3K) inhibitor (GDC-0941, S1065), and PARP inhibitor (BMN673, S7048) were obtained from Selleck Chemicals. STAT3 inhibitor (Stattic, 573099) was purchased from Calbiochem. Proteasome inhibitor (BTZ) and BET inhibitor (JQ1) were purchased from AdooQ BioScience (CA, USA). Focal adhesion kinase (FAK) inhibitor (PF-573228), CXCR2 inhibitor (SB225022), heat shock protein 90 (HSP90) inhibitor (17-DMAG), and nuclear factor κB (NF-κB) inhibitor [pyrrolidine dithiocarbamate (PDTC)] were from Sigma-Aldrich. Janus kinase (JAK) inhibitor (ruxolitinib) was from Santa Cruz Biotechnology, and cMet inhibitor (EMD 1214063) was from MedChemExpress. OTX015 was purchased from Changzhou Medi-tech Bioscientific Co. (China). The apoptotic inhibitor zVAD-FMK (sc-3067) and the autophagy inhibitor 3-MA (sc-205596) were purchased from Santa Cruz Biotechnology. Liproxstatin-1 (17730) was from Cayman Chemical. Necrostatin-1 (N9037), erastin (E7781), staurosporine (S5921), rapamycin (553210), Ferrostatin-1 (SML0583), reduced l-GSH (G6013), 2,2′-bipyridyl (D216305), thiazolyl blue tetrazolium bromide (MTT) (M5655), and deferoxamine mesylate (DFO) (D9533) were from Sigma-Aldrich.
Antibodies
The following antibodies were purchased from Cell Signaling Technology (USA): rabbit anti-MLKL (14993), rabbit anti-phospho Ser358 MLKL (74921), rabbit anti–cleaved caspase-3 (9664), and rabbit anti-PARP antibody (9542). Mouse anti–α-tubulin (T6074) and rabbit anti–ferritin heavy chain (F5012) were supplied by Sigma-Aldrich. Rabbit anti-GPX4 (ab125066, clone EPNCIR 144) was purchased from Abcam Inc. (USA), and rabbit anti-BRD4 (A700-004, clone BL-149-2H5) was from Bethyl Laboratories (USA). Rabbit anti-Ferroportin (NBP1-21502) was from Novus Biologicals. Mouse anti-ACSL4 (sc-365230, clone F4) and GSS (sc-166882) were obtained from Santa Cruz Biotechnology. Mouse anti-TfR (13-6800, clone H68.4) was from Invitrogen.
Cell culture
All the breast cancer cell lines, immortalized normal breast epithelial cell line MCF10A, and human embryonic kidney (HEK) 293T cells were originally obtained from the American Type Culture Collection (USA). HCC38 was obtained from M. Virginie (Institute Curie, Research Centre, Paris, France; 2014). MDA-MB-468, HCC1143, HCC1937, HCC70, HCC38, MDA-MB-231, BT549, SUM159, and Hs578T cells were grown in RPMI, while HEK293T cells were grown in Dulbecco’s modified Eagle medium (DMEM) (Gibco BRL, USA). Unless otherwise indicated, all cell lines were cultured in medium containing 10% fetal bovine serum (Gibco BRL, USA) and penicillin/streptomycin. l-Glutamine (2 mM) was added to Hs578T and HCC38 medium, and sodium bicarbonate (1.5 g/liter), 10 mM Hepes (pH 7.4), and 1.0 mM sodium pyruvate were added to HCC38 medium. BT20 cells were grown in Eagle’s minimum essential medium plus 1 mM sodium pyruvate and 2 mM l-glutamine. MCF10A cells were grown in DMEM∶F12 (1∶1) medium supplemented with EGF (20 ng/ml; PeproTech), insulin (10 μg/ml; Sigma-Aldrich), cholera toxin (100 ng/ml; Sigma-Aldrich), hydrocortisone (1 μg/ml; Sigma-Aldrich), and 5% heat-inactivated horse serum. Cells were cultured at 37°C in a humidified incubator of 5% CO2. Cell lines were routinely (once a month) checked for mycoplasma using a commercially available kit (Biological Industries, Israel).
shRNA lentivirus infection
Lentiviral constructs of human BRD4 shRNAs were obtained from M. A. Esteban (Guangzhou Institutes of Biomedicine and Health, Guangzhou, China). Lentivirus production and infection were conducted essentially as previously described (56). Infected cells (control and BRD4 shRNA) were grown in selection medium containing puromycin (1 μg/ml) for 72 hours. Knockdown of BRD4 was assessed by WB.
Drug screen, cell viability assays, and drug synergy
To evaluate the effects of the 17 selected small-molecule inhibitors used in the screen (fig. S1A) on the viability of the 13 human cell lines, dose-response experiments were carried out using 7 to 10 drug concentrations applied at threefold serial dilution within a range of 13 nM to 10 μM (17-DMAG and BTZ were used at a range of 0.5 to 100 nM). Cells in 96-well plates were treated with the described drug doses or with dimethyl sulfoxide (DMSO) as control for 72 hours, and cell viability was measured by MTT assays as previously described (57). Cell viability was presented as percentage of control, and dose-response curves were generated using the drc package in R. IC50 values were calculated from the curves for each drug in each cell line. The drug screen was performed in 96-well plates, and cell viability was determined by CellTiter-Glo (Promega) 72 hours after adding the drugs according to the manufacturer’s instructions. Luminescence was measured by a PHERAStar Plus plate reader (BMG Labtech, Germany). Where indicated, CellTiter-Blue (Promega) instead of MTT assay was used. Changes in cell viability are presented as the ratio of viable cells between drug-treated and the respective mock-treated control cells. Where indicated, the fraction of cells that were inhibited by the drugs was described as fraction affected (FA). Synergy of drug pairs was calculated by the CompuSyn software using the Chou-Talalay equation. CI < 0.5 indicates synergism. For crystal violet staining, the cells were washed with phosphate-buffered saline (PBS) and then fixed and stained with 0.2% crystal violet solution in 4% formalin for 10 min. The cells were washed with distilled water, dried, and scanned using an HP Scanjet G4010.
Cell treatments with drug combinations, ferroptosis inducers, and cell death rescue experiments
Drug combinations were routinely applied at a low cell density of ~40 to 50% confluency approximately 16 to 20 hours following seeding. Cell numbers were calibrated for each cell line (TNBC and non-TNBC, ~16 cell lines) considering cell size, growth rate, and general morphology (cell-cell contact), and similar conditions were used for most functional assays. Drug combinations were freshly prepared at 10× concentration in growth medium before use. All the drugs used in the study were aliquoted in small amounts to avoid repeated freeze-thaw cycles and were protected from light. For most assays, including cell viability assay, colony formation assays, mRNA and protein expression assays, and ferroptosis assays, the human TNBC cells (up to 12 cell lines) were treated with BTZ at 2 to 3 nM (2 nM for most cell lines) and/or JQ1 at 50 to 100 nM (50 nM for most cell lines). For the 4T1 murine cell line, JQ1 was used at 250 nM and BTZ was used at 8 nM. JQ1 (20 mM stock in DMSO) and BTZ (50 μM stock in DMSO) were used to prepare a serial dilution in growth medium by extensive vortexing. Similarly, for the M/MSL TNBC cell lines, we used JQ1 and SB225022 (15 mM stock in DMSO diluted to 1 mM in growth medium) at 50 to 100 nM (50 nM for most cell lines). For 4T1 cells, JQ1 and SB225022 were used at 250 nM concentrations. Drug combinations were applied to maintain low DMSO concentration (below 0.1%) in all assays. Batch effects were obtained during the time course of this study, particularly for JQ1 and BTZ. Therefore, the applied effective doses maintained the range described in our screen, based on individual drug (JQ1, BTZ, and SB225022) response of less than 25% cell death (>75% cell viability) and at least 65 to 85% cell death in response to drug combination. The effects of various cell death pathway inhibitors were calibrated for each cell line for doses and effects. Ferroptosis inhibitor Ferrostatin-1 was calibrated and used in a concentration range of 1 to 5 μM for pretreatment in different cell lines. Cell death inhibitors were used to inhibit apoptosis (zVAD-FMK), necrosis (Necrostatin-1), autophagy (3-MA), and ferroptosis (Ferrostatin-1, GSH, and 2,2-bipyridyl, and DFO) at indicated concentrations 1 hour before the treatment with combination therapy.
RNA extraction and real-time PCR
Total RNA from cell lines and mouse tumor tissues was extracted and purified using TRI Reagent (Sigma-Aldrich). RNA was reverse-transcribed into complementary DNA (cDNA) using the Applied Biosystems High-Capacity cDNA Reverse Transcription Kit with random primers according to the manufacturer’s instructions.
Real-time PCR analysis was performed on an ABI StepOnePlus 7500 Real-Time PCR system (Applied Biosystems, Invitrogen) using SYBR Green Master Mix reagents (Roche) according to the manufacturer’s guidelines. Real-time PCR primers were designed using the Primer Express software of Applied Biosystems (Invitrogen) and were calibrated before use. Expression levels of actin were used for normalization. The relative levels of mRNA were calculated using the ΔΔCT method. The primer sequences used in this study are listed in table S12.
Annexin V staining
Cells in 24-well plates were treated with drugs as indicated. Before staining, cells were washed twice with Cell Staining Buffer (catalog no. 420201, BioLegend, CA) and then incubated in Annexin V Binding Buffer (catalog no. 422201, BioLegend, CA) for 5 min at room temperature (RT). Phycoerythrin annexin V (catalog no. 640908, BioLegend, CA) was diluted 1:20 in Annexin V Binding Buffer and incubated with the cells for 15 min at RT in the dark. Cells were then washed twice with Annexin V Binding Buffer, stained with 1 mM Hoechst 33342 (Sigma-Aldrich) in the same buffer, washed, and analyzed by an Axio Imager 2 microscope (Zeiss) using appropriate filters.
Immunoblotting
Total protein from cell lines was extracted in lysis buffer containing 0.2% Triton X-100, 50 mM Hepes (pH 7.5), 100 mM NaCl, 1 mM MgCl2, 50 mM NaF, 0.5 mM NaVO3, 20 mM β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride, leupeptin (10 μg/ml), and aprotinin (10 μg/ml). For lysis of tumor tissues, 1% Triton X-100 was used in the same buffer. Cell lysates were centrifuged at 14,000 rpm for 15 min at 4°C, protein concentration of the supernatants was determined by Bradford assay (Bio-Rad, Hercules, CA), and the samples were normalized accordingly. Equal amounts of protein (60 to 80 μg) were analyzed by SDS–polyacrylamide gel electrophoresis and WB as previously described (58). Primary antibodies for cleaved PARP and caspase-3, pMLKL, MLKL, and TfR (Transferrin Receptor) were diluted 1:00; anti-BRD4, Ferroportin, GPX4, and ferritin antibodies were diluted 1:2000; and anti-GSS and ACSL4 were diluted 1:500, whereas α-tubulin antibody was diluted 1:10,000. Densitometric analysis was performed using the ImageJ software (National Institutes of Health, USA), and the intensity of each band was normalized to its respective α-tubulin loading.
Immunohistochemistry, image analysis, and scoring
Formalin-fixed tumor sections derived from mouse allografts as well as from patients with breast cancer were analyzed by immunohistochemistry. In brief, anonymized breast cancer tissue samples were collected from patients undergoing surgical resection at the Department of Gynecology and Obstetrics at the Goethe University in Frankfurt, Germany. The tissue samples were obtained with the Institutional Review Board approval (Ethik-Kommission Fachbereich Medizin der Goethe-Universität Frankfurt) and patient’s written informed consent. The samples were characterized according to standard pathology, including estrogen receptor, progesterone receptor, and HER2 status. Immunohistochemistry was performed on 3-μm serial sections from formalin-fixed, paraffin-embedded tumor sections essentially as described previously (57). In brief, the sections were deparaffinized (10 min at 60°C and then 15 min of immersion in xylene), rehydrated (incubation in 100, 96, and 70% ethanol, 10 min each), washed (PBS for 3 min and then distilled water, 2 × 3 min), and then incubated for 30 min in methanol/H2O2 (0.9%) solution to quench endogenous peroxidases. Antigen retrieval was performed by microwave irradiation for 10 min in citric acid (pH 6.1 for anti-BRD4 and anti-GSS antibodies) or in tris-EDTA buffer (10 mM tris-HCl and 1 mM EDTA; pH 9.0 for anti-GPX4, anti-ferroportin, anti-TfR antibodies). Following 1.5 hours of blocking [20% normal horse serum and 0.2% Triton X-100 (0.5% for anti-BRD4) in PBS], tissue sections were immunostained with antibodies to BRD4 (1:400), GPX4 (1:400), GSS (1:150), FPN (1:150), and TFRC (1:300). Negative controls were treated similarly without a primary antibody. Immunostained slides were counterstained with hematoxylin (Sigma-Aldrich).
Immunohistochemically stained sections were digitally scanned at ×200 magnification using a Pannoramic 250 Flash III scanner (3DHISTECH Ltd.). The images were visualized and analyzed using CaseViewer software (3DHISTECH Ltd.). The staining intensity of each section was scored as 0 (no staining), 1+ (weak staining), 2+ (moderate staining), or 3+ (strong staining), and the percentage of positive cells was determined from at least five different random regions of tumor sections to define reactivity extension values. The H-scores for tumor tissues were calculated by multiplying the staining intensity and reactivity extension values (range, 0 to 300). Tumor grades and subtypes were evaluated according to clinical and pathological data. Statistics were performed using unpaired two-tailed t test to compare differences between H-scores of patients with TNBC and non-TNBC patients. Fisher’s exact test was applied to compare percentage of tumor in high or low scores, where an H-score from 0 to 150 was considered as low and an H-score from 150 to 300 was assigned as high.
In vivo studies
All animal studies were performed according to protocols approved by the Weizmann Institutional Animal Care and Use Committee. For mouse xenograft models, MDA-MB-231 cells (2 × 106 per mouse) expressing green fluorescent protein (GFP)–luciferase were implanted bilaterally into the fat pads of the fourth inguinal mammary gland of 6-week-old female athymic nude-Foxn1nu mice. Twenty-three days later, mice were randomized into four groups (n = 10 mice per group) and treated with the following: (i) vehicle; (ii) OTX015, daily through oral gavage (25 mg/kg); OTX015 (100 mg/ml stock in DMSO) was diluted in vehicle solution containing 2% DMSO, 30% polyethylene glycol (PEG)–300, and 5% Tween 80; (iii) SB225022, which was intraperitoneally administered 5 days a week, at 5 mg/kg prepared in PBS; or (iv) OTX015 + SB225022 combination. Mice were treated for 18 days, and mouse body weights were measured every 3 days. Tumor volumes (in cubic millimeter) were measured every 2/3 days by a digital Vernier caliper and calculated according to the width2 × length/2 formula. Tumor progression was monitored using Xenogen IVIS Spectrum in vivo bioluminescence. Mice were anesthetized by isoflurane inhalation, and then d-luciferin (100 μl; 7.5 mg/ml; Regis Technologies) was intraperitoneally injected. Bioluminescence images were acquired within 10 min after injection by the charge-coupled device camera of the IVIS instrument with the Living Image 3.0 software (Xenogen Caliper Life Sciences). At the end of the study, mice were sacrificed, tumors were excised, and tumor weights were measured. Tumor samples were preserved in either liquid nitrogen or 10% buffered formalin for further analysis. Similar to the xenograft models, syngeneic tumor models were established by bilateral implantation of 4T1 murine cells (GFP-luciferase expressed, 1.5 × 105 per mouse) into the fourth inguinal mammary gland of BALB/c mice. Two weeks later, mice were randomized into four groups and treated with vehicle, OTX015 (25 mg/kg per day, orally), BTZ (0.25 mg/kg, IP, prepared in PBS, every fourth day), or the combination of OTX015 + BTZ. Animal body weight was measured every 2 days, and tumor progression was monitored as described above. Toxicity of the OTX015 + BTZ combination was further assessed by measuring the levels of common biomarkers for organ damage, including alkaline phosphatase (ALP), alkaline aminotransferase (ALT), blood urea nitrogen (BUN), and creatinine. In brief, mice were anesthetized, and blood was extracted by cardiac puncture. Blood was coagulated by incubation at RT for 20 min and then centrifuged at 1000g for 10 min to separate the serum. The levels of ALP, ALT, BUN, and creatinine in the serum were measured by the Vetscan VS2 Chemistry Analyzer (Abaxis, CA) following the manufacturer’s instructions.
ROS measurements
Levels of intracellular ROS were measured by the cell-permeable dye CM-H2DCFDA [5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester] (Invitrogen, LSC6827). Cells were cultured in 96-well plates for 16 hours and treated with drugs as indicated. To assess the ROS level, cells were loaded with 10 μM CM-H2DCFDA for 30 min at 37°C in the dark. Fluorescence of CM-H2DCFDA was measured by a fluorescence microplate reader (Molecular Devices, Sunnyvale, CA, USA) using 495 nm for excitation and 527 nm for emission. Data are presented as percentage of ROS relative to untreated controls. The values were normalized to the number of living cells in each well. Cells treated with 50 μM tetrabutyl hydrogen peroxide (TBHP) for 30 min were used as positive control. To detect intracellular ROS by live cell imaging, cells were grown in 24-well plates and treated as described above. Fluorescence images were taken from three different areas of the 24-well plate using appropriate filters and an LSM800 confocal microscope (Carl Zeiss GmbH).
GSH measurements
The level of GSH was measured using a Glutathione Colorimetric Assay kit (catalog no. K261, BioVision, CA, USA). Cells (0.75 × 106 to 1 × 106) were treated with drugs as indicated and lysed in GSH assay buffer, and GSH levels were measured according to the manufacturer’s instructions. Colorimetric signals were measured by absorbance at 405 nm using a microplate reader (Molecular Devices, Sunnyvale, CA, USA). The GSH concentration was calculated using a standard curve. The values were then normalized to total protein quantity per lysate samples. Results are expressed as means ± SD of fold changes relative to untreated controls.
Iron measurements
Total cellular iron was measured using the colorimetric QuantiChrom Iron Assay Kit (DIFE-250, BioAssay Systems, CA). In brief, cells cultured in six-well plates were washed with PBS and lysed in 50 μl of lysis buffer containing 1% Triton X-100. Cell lysates were centrifuged at 15,000 rpm for 10 min, and supernatants were used to measure total iron labile (Fe2+ and Fe3+) according to the manufacturer’s instructions. The method uses a chromogen that forms a blue-colored complex specifically with Fe2+ and thus can also be used to measure the level of reduced Fe3+ to Fe2+ to monitor total iron. Colorimetric signals were measured by absorbance at 590 nm using a microplate reader (Infinite 200 PRO, Tecan Trading AG, Switzerland). Results were normalized to protein content in cell lysates and expressed as means ± SD of fold changes relative to untreated controls.
Lipid peroxidation
The Image-iT Lipid Peroxidation Kit (catalog no. C10445, Thermo Fisher Scientific) was used to detect lipid peroxidation in live cells using the BODIPY 581/591 C11 fluorescent reporter. In brief, cells were plated in High-Content Imaging Glass Bottom 96-well Microplates (Corning) for 16 hours and then treated with drugs as indicated. The lipid peroxidation sensor C11-BODIPY (581/591) (10 μM) was added for 30 min and then 1 mM Hoechst 33342 (Sigma-Aldrich) in PBS. Cells were washed with PBS, and fluorescence was measured at two wavelengths, one at an excitation/emission of 581/590 nm (Texas Red filter set) for the reduced dye, and the other at an excitation/emission of 488/510 nm (traditional fluorescein isothiocyanate filter set) for the oxidized dye. Hoechst was measured at an excitation/emission of 350/461 nm (4′,6-diamidino-2-phenylindole filter set) using the Infinite 200 PRO Tecan microplate reader (Tecan Inc., Switzerland). The green-to-red fluorescence intensity ratio was used to measure lipid peroxidation. The values were normalized to the number of living cells in each well using Hoechst staining values. For live cell imaging, the cells were treated as described above, and fluorescence images were acquired using a laser confocal microscope (LSM800, Zeiss) and the ZEN Imaging Software with the abovementioned excitation and emission wavelengths.
TBARS assay for lipid peroxidation was performed to quantitate cellular lipid peroxidation as described earlier with minor modifications (59). In brief, cells were treated with drugs for 16 hours and then washed with ice-cold PBS, lysed in PBS containing 0.5% Triton X-100 (200 μl volume), vortexed, and incubated on ice for 15 min. Subsequently, 1.5 μl of butylated hydroxytoluene (100 mM) was added to 150 μl of cell lysate, followed by 75 μl of 15% trichloroacetic acid to precipitate proteins. Then, 75 μl of HCl (0.25 M), 75 μl of 2-thiobarbituric acid (0.375%), and 30 μl of 8.5% SDS were added. The samples were vortexed and incubated at 95°C for 60 min for color development. Reactions were stopped by a 5-min incubation on ice and centrifuged for 15 min at 1500g, and then 200 μl of the supernatant was transferred to a 96-well plate. Absorbance at 532 nm was measured using a microplate reader (Infinite 200 PRO, Tecan Trading AG, Switzerland). Blank reaction of lysis buffer was used as reference, and the relative TBARS concentration was determined. The final TBARS values were normalized according to the protein concentration of the samples (measured by Bradford protein assay reagent). Lipid peroxidation was calculated as folds of untreated control and plotted as means ± SD values.
Generation of 3D spheroids
Spheroids were generated using a liquid overlay cultivation technique, using nonadherent, U-shaped 96-well plates precoated with 1% (w/v) agar in PBS (60 μl per well). Single-cell suspension of 4T1 cells (7 × 103 cells in 100 μl of medium) was loaded into each well. Optimal 3D structures were enhanced by microplate centrifugation at 1000g for 5 min. Plates were then incubated for 3 days at 37°C, 5% CO2, and 90% humidity to form a single spheroid per well. Three-day-old spheroids were treated with the indicated drugs for 15 days (replaced every 3 days). Bright-field pictures of spheroids were acquired by an inverted microscope at ×5 magnification. At the end of the experiment, spheroid viability was measured by the CellTiter-Blue viability assay (Promega) according to the manufacturer’s instructions. Results were expressed as percentage of cell viability compared to untreated controls.
SL analysis
We used the pipeline ISLE (18), a data-driven approach to identify the clinically relevant SL gene pairs by mining 330 basal-subtype breast cancer patient tumor data from the METABRIC dataset (19). In brief, ISLE was proceeded with the following four steps: (i) It first identified potential candidate SL pairs by mining large-scale in vitro shRNA/CRISPR knockout datasets. (ii) ISLE then uses the patients’ gene expression (RNA-seq) and somatic copy number alterations data from the METABRIC patient dataset (19) and integrates clinical data. Among the candidate pairs in step (i), it identifies candidate SL pairs whose co-inactivation (based on gene expression and copy number) is underrepresented among the tumors due to negative selection. (iii) ISLE then further filters the candidate SLs by selecting those whose co-inactivation is associated with better prognosis in patients with basal-subtype breast cancer. (iv) Last, ISLE uses evolutionary evidence to predict candidate SLs, as SL pairs are likely to have a similar evolutionary history. More details of the ISLE pipeline can be found in Lee et al. (18).
To estimate the strength of candidate SL interactions, the ISLE significance scores were calculated (18). For every drug combination, we computed the ISLE significance score between the drug target gene pairs of the corresponding drugs. For drugs that target multiple targets, we have multiple SL pairs between two drugs, and we choose the sum value as the ISLE significance score. Once these significance scores are computed for all drug combinations, we take its rank and normalize it by the number of total combinations to compute the ISLE significance score.
The targets of the drugs are provided in fig. S1A. We also manually searched each of the drugs and their associated drug targets using DrugBank and other important websites/literature. Drugs that target a gene family or pathway may have many genes associated with it. For each drug combination, we compute the effectiveness in each cell line (effectiveness = % cell death; 100% − % viability). We then take the median value of effectiveness across all 13 cell lines to compute the overall effectiveness for each drug combination. This was used in Fig. 1C. We calculated the Spearman correlation between ISLE significance scores and measured the effectiveness across all combinations. The accuracy of predicting effectiveness by the ISLE significance scores was measured by the ROC-AUC, generated by comparing the ISLE scores of the 21% most effective combinations versus the 21% least effective combinations.
Bioinformatic analysis
Datasets
Gene expression analysis of patients with breast cancer was carried out on the basis of TCGA breast cancer dataset (version 2017-10-13), which includes 142 basal, 434 LumA, 194 LumB, 67 HER2, and 119 normal-like patients (determined by PAM50 based on the RNA-seq). Results were verified in the METABRIC dataset, which includes 199 basal, 218 HER2, 675 LumA, 460 LumB, and 140 normal-like patients (table S13). Gene expression data of breast cancer cell lines were taken from the 8-11-2017 version of the CCLE (Broad Institute), which includes 23 TNBC lines and 33 non-TNBC lines plus 1 normal-like. All data are log2-transformed before analysis.
Gene set enrichment analysis
GSEA was performed using GSEA software (Broad Institute). Genes were ranked according to the signal-to-noise ratio of each gene between basal and non-basal patients (TCGA dataset) or between TNBC versus non-TNBC cell lines (CCLE dataset). The overrepresentation of certain gene signatures in these ranked gene lists is given by the normalized enrichment score (NES).
Dataset analysis
For comparisons of gene expression in the CCLE dataset, gene expression values (log2) were normalized across all breast cancer lines (so that the average expression is 0 and the SD is 1). For TCGA dataset, gene expression was not normalized. One-way analysis of variance (ANOVA) between groups of patients or cell lines, based on their PAM50, was performed using Partek Genomic Suite (Partek, MO). A step-up P value (FDR) below 0.05 is considered significant. Heatmap of signature gene expression in patients from the TCGA dataset was performed using the Partek Genomic Suite. Patients were clustered unsupervised, and the partition of the PAM50 phenotype to different clusters was visualized.
Statistical analysis
Data are presented as means ± SD. To compare between experimental groups, we used Student’s t test (two-sided) for most experiments, Wilcoxon test (ranked, two-sided) for tumor volumes in vivo (Fig. 3), and Fisher’s exact test for IHC staining (Fig. 8). For dataset analysis, one-way ANOVA was used to compare the means of different PAM50 groups. t test was used to evaluate the means between basal and each one of the other PAM50 groups. The reported P values associated with dataset analysis (TCGA or CCLE) are P values corrected for false discovery rate, measured by the step-up method. A P value of <0.05 is considered significant.
Supplementary Material
Acknowledgments
S.L. is the incumbent of the Joyce and Ben B. Eisenberg Chair of Molecular Biology and Cancer Research. Funding: This work was supported by the Israel Science Foundation (ISF) grant no. 1530/17 and by the ISF-NSFC joint research program (grant no. 2526/16), by the MDACC-SINF grant, and by a research grant from D. E. Stone. This work was also supported in part by the Intramural Research Program of the NIH, NCI, and used the NIH HPC Biowulf cluster resource. Author contributions: N.V. designed and performed most of the experiments. Y.V. was involved in data analysis, bioinformatics, and statistic tests and reviewed all raw data of the study. A.S. helped with IHC staining. E.R. and N.U.N. performed the computation analysis for SL. T.K. provided the breast cancer sections. G.M. contributed to the discussion and reverse phase protein array analysis (not shown). V.D., L.K., and H.R. were involved in the revision of the manuscript. F.M. was involved in discussion, reviewing, and editing the manuscript. S.L. designed the study, supervised the experimental team, and wrote the manuscript. All authors read and approved the final manuscript. Competing interests: G.M. has the following relationships: Consultant/scientific advisory board companies: AstraZeneca, Chrysalis (travel reimbursement only), GSK, ImmunoMet, Ionis, Lilly USA LLC, PDX Pharma, SignalChem Lifesciences, Symphogen, Tarveda, Turbine, and Zentalis. Stock/options/financial companies: Catena Pharmaceuticals, ImmunoMet, SignalChem Lifesciences, and Tarveda. Licensed technology: HRD assay to Myriad Genetics, DSP to NanoString. Sponsored research: NanoString Center of Excellence, Ionis (provision of tool compounds). E.R. is a nonpaid scientific advisor to Pangea Therapeutics, a cancer SL–based startup company that he had cofounded in Israel and divested from after joining the NIH. Israel patent application pending (Yeda Research and Development Co. Ltd., coinventors: S.L., N.V., A.S., and Y.V.). The authors declare that they have no other competing interests. Data availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/34/eaba8968/DC1
REFERENCES AND NOTES
- 1.Kalimutho M., Parsons K., Mittal D., López J. A., Srihari S., Khanna K. K., Targeted therapies for triple-negative breast cancer: Combating a stubborn disease. Trends Pharmacol. Sci. 36, 822–846 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Perou C. M., Molecular stratification of triple-negative breast cancers. Oncologist 15 ( Suppl 5), 39–48 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Saputra E. C., Huang L., Chen Y., Tucker-Kellogg L., Combination therapy and the evolution of resistance: The theoretical merits of synergism and antagonism in cancer. Cancer Res. 78, 2419–2431 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Brunen D., Bernards R., Drug therapy: Exploiting synthetic lethality to improve cancer therapy. Nat. Rev. Clin. Oncol. 14, 331–332 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Bryant H. E., Schultz N., Thomas H. D., Parker K. M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N. J., Helleday T., Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Faraoni I., Graziani G., Role of BRCA mutations in cancer treatment with poly(ADP-ribose) polymerase (PARP) inhibitors. Cancer 10, 487 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sporikova Z., Koudelakova V., Trojanec R., Hajduch M., Genetic markers in triple-negative breast cancer. Clin. Breast Cancer 18, e841–e850 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Stevens K. N., Vachon C. M., Couch F. J., Genetic susceptibility to triple-negative breast cancer. Cancer Res. 73, 2025–2030 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehmann B. D., Bauer J. A., Chen X., Sanders M. E., Chakravarthy A. B., Shyr Y., Pietenpol J. A., Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 121, 2750–2767 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lehmann B. D., Jovanović B., Chen X., Estrada M. V., Johnson K. N., Shyr Y., Moses H. L., Sanders M. E., Pietenpol J. A., Refinement of triple-negative breast cancer molecular subtypes: Implications for neoadjuvant chemotherapy selection. PLOS ONE 11, e0157368 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmid P., Adams S., Rugo H. S., Schneeweiss A., Barrios C. H., Iwata H., Diéras V., Hegg R., Im S.-A., Wright G. S., Henschel V., Molinero L., Chui S. Y., Funke R., Husain A., Winer E. P., Loi S., Emens L. A.; IMpassion Trial Investigators , Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 379, 2108–2121 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Wang W., Green M., Choi J. E., Gijón M., Kennedy P. D., Johnson J. K., Liao P., Lang X., Kryczek I., Sell A., Xia H., Zhou J., Li G., Li J., Li W., Wei S., Vatan L., Zhang H., Szeliga W., Gu W., Liu R., Lawrence T. S., Lamb C., Tanno Y., Cieslik M., Stone E., Georgiou G., Chan T. A., Chinnaiyan A., Zou W., CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stockwell B. R., Angeli J. P. F., Bayir H., Bush A. I., Conrad M., Dixon S. J., Fulda S., Gascón S., Hatzios S. K., Kagan V. E., Noel K., Jiang X., Linkermann A., Murphy M. E., Overholtzer M., Oyagi A., Pagnussat G. C., Park J., Ran Q., Rosenfeld C. S., Salnikow K., Tang D., Torti F. M., Torti S. V., Toyokuni S., Woerpel K. A., Zhang D. D., Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dixon S. J., Lemberg K. M., Lamprecht M. R., Skouta R., Zaitsev E. M., Gleason C. E., Patel D. N., Bauer A. J., Cantley A. M., Yang W. S., Morrison B. III, Stockwell B. R., Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conrad M., Kagan V. E., Bayir H., Pagnussat G. C., Head B., Traber M. G., Stockwell B. R., Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 32, 602–619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu T., Ding W., Ji X., Ao X., Liu Y., Yu W., Wang J., Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell. Mol. Med. 23, 4900–4912 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bayraktar S., Gluck S., Molecularly targeted therapies for metastatic triple-negative breast cancer. Breast Cancer Res. Treat. 138, 21–35 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Lee J. S., Das A., Jerby-Arnon L., Arafeh R., Auslander N., Davidson M., Garry L. M., James D., Amzallag A., Park S. G., Cheng K., Robinson W., Atias D., Stossel C., Buzhor E., Stein G., Waterfall J. J., Meltzer P. S., Golan T., Hannenhalli S., Gottlieb E., Benes C. H., Samuels Y., Shanks E., Ruppin E., Harnessing synthetic lethality to predict the response to cancer treatment. Nat. Commun. 9, 2546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Curtis C., Shah S. P., Chin S.-F., Turashvili G., Rueda O. M., Dunning M. J., Speed D., Lynch A. G., Samarajiwa S., Yuan Y., Gräf S., Ha G., Haffari G., Bashashati A., Russell R., Kinney S. M.; METABRIC Group, Langerød A., Green A., Provenzano E., Wishart G., Pinder S., Watson P., Markowetz F., Murphy L., Ellis I., Purushotham A., Børresen-Dale A.-L., Brenton J. D., Tavaré S., Caldas C., Aparicio S., The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shu S., Lin C. Y., He H. H., Witwicki R. M., Tabassum D. P., Roberts J. M., Janiszewska M., Huh S. J., Liang Y., Ryan J., Doherty E., Mohammed H., Guo H., Stover D. G., Ekram M. B., Peluffo G., Brown J., D’Santos C., Krop I. E., Dillon D., Keown M. M., Ott C., Qi J., Ni M., Rao P. K., Duarte M., Wu S.-Y., Chiang C.-M., Anders L., Young R. A., Winer E. P., Letai A., Barry W. T., Carroll J. S., Long H. W., Brown M., Liu X. S., Meyer C. A., Bradner J. E., Polyak K., Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 529, 413–417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Devapatla B., Sharma A., Woo S., CXCR2 inhibition combined with sorafenib improved antitumor and antiangiogenic response in preclinical models of ovarian cancer. PLOS ONE 10, e0139237 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi J., Bromodomain and extraterminal domain inhibitors (BETi) for cancer therapy: Chemical modulation of chromatin structure. Cold Spring Harb. Perspect. Biol. 6, a018663 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrocca F., Altschuler G., Tan S. M., Mendillo M. L., Yan H., Jerry D. J., Kung A. L., Hide W., Ince T. A., Lieberman J., A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell 24, 182–196 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hartman Z. C., Poage G. M., den Hollander P., Tsimelzon A., Hill J., Panupinthu N., Zhang Y., Mazumdar A., Hilsenbeck S. G., Mills G. B., Brown P. H., Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 73, 3470–3480 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dominguez C., McCampbell K. K., David J. M., Palena C., Neutralization of IL-8 decreases tumor PMN-MDSCs and reduces mesenchymalization of claudin-low triple-negative breast cancer. JCI Insight 2, e94296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chou T.-C., Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Miller F. R., Miller B. E., Heppner G. H., Characterization of metastatic heterogeneity among subpopulations of a single mouse mammary tumor: Heterogeneity in phenotypic stability. Invasion Metastasis 3, 22–31 (1983). [PubMed] [Google Scholar]
- 28.Doroshow D. B., Eder J. P., LoRusso P. M., BET inhibitors: A novel epigenetic approach. Ann. Oncol. 28, 1776–1787 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Alqahtani A., Choucair K., Ashraf M., Hammouda D. M., Alloghbi A., Khan T., Senzer N., Nemunaitis J., Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Future Sci. OA 5, FSO372 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vázquez R., Riveiro M. E., Astorgues-Xerri L., Odore E., Rezai K., Erba E., Panini N., Rinaldi A., Kwee I., Beltrame L., Bekradda M., Cvitkovic E., Bertoni F., Frapolli R., D’Incalci M., The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget 8, 7598–7613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao J. Y., Dixon S. J., Mechanisms of ferroptosis. Cell. Mol. Life Sci. 73, 2195–2209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun L., Wang H., Wang Z., He S., Chen S., Liao D., Wang L., Yan J., Liu W., Lei X., Wang X., Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Ingold I., Berndt C., Schmitt S., Doll S., Poschmann G., Buday K., Roveri A., Peng X., Freitas F. P., Seibt T., Mehr L., Aichler M., Walch A., Lamp D., Jastroch M., Miyamoto S., Wurst W., Ursini F., Arnér E. S. J., Fradejas-Villar N., Schweizer U., Zischka H., Angeli J. P. F., Conrad M., Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422.e21 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Beatty A., Fink L. S., Singh T., Strigun A., Peter E., Ferrer C. M., Nicolas E., Cai K. Q., Moran T. P., Reginato M. J., Rennefahrt U., Peterson J. R., Metabolite profiling reveals the glutathione biosynthetic pathway as a therapeutic target in triple-negative breast cancer. Mol. Cancer Ther. 17, 264–275 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang W. S., Ramaratnam R. S., Welsch M. E., Shimada K., Skouta R., Viswanathan V. S., Cheah J. H., Clemons P. A., Shamji A. F., Clish C. B., Brown L. M., Girotti A. W., Cornish V. W., Schreiber S. L., Stockwell B. R., Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hao S., Liang B., Huang Q., Dong S., Wu Z., He W., Shi M., Metabolic networks in ferroptosis. Oncol. Lett. 15, 5405–5411 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Salazar M., Rojo A. I., Velasco D., de Sagarra R. M., Cuadrado A., Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 281, 14841–14851 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Mou Y., Wang J., Wu J., He D., Zhang C., Duan C., Li B., Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 12, 34 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shintoku R., Takigawa Y., Yamada K., Kubota C., Yoshimoto Y., Takeuchi T., Koshiishi I., Torii S., Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 108, 2187–2194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirschhorn T., Stockwell B. R., The development of the concept of ferroptosis. Free Radic. Biol. Med. 133, 130–143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doll S., Proneth B., Tyurina Y. Y., Panzilius E., Kobayashi S., Ingold I., Irmler M., Beckers J., Aichler M., Walch A., Prokisch H., Trümbach D., Mao G., Qu F., Bayir H., Füllekrug J., Scheel C. H., Wurst W., Schick J. A., Kagan V. E., Angeli J. P. F., Conrad M., ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pinnix Z. K., Miller L. D., Wang W., D’Agostino R. Jr., Kute T., Willingham M. C., Hatcher H., Tesfay L., Sui G., Di X., Torti S. V., Torti F. M., Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2, 43ra56 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayano M., Yang W. S., Corn C. K., Pagano N. C., Stockwell B. R., Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 23, 270–278 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohammad H. P., Barbash O., Creasy C. L., Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 25, 403–418 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Pedregal M., Cristobal I., Lobo F., Izarzugaza Y., Sanchez C. C., Moreno V., Hernández T., Rubio J., Zazo S., Torrejon B., Rojo F., Garcia-Foncillas J., Linking pBRD4 and PP2A inhibition in triple negative breast cancer. J. Clin. Oncol. 36, e13011 (2018). [Google Scholar]
- 46.Bai L., Zhou B., Yang C.-Y., Ji J., Eachern D. M., Przybranowski S., Jiang H., Hu J., Xu F., Zhao Y., Liu L., Fernandez-Salas E., Xu J., Dou Y., Wen B., Sun D., Meagher J., Stuckey J., Hayes D. F., Li S., Ellis M. J., Wang S., Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 77, 2476–2487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khoueiry P., Gahlawat A. W., Petretich M., Michon A. M., Simola D., Lam E., Furlong E. E., Benes V., Dawson M. A., Prinjha R. K., Drewes G., Grandi P., BRD4 bimodal binding at promoters and drug-induced displacement at Pol II pause sites associates with I-BET sensitivity. Epigenetics Chromatin 12, 39 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sui S., Zhang J., Xu S., Wang Q., Wang P., Pang D., Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. 10, 331 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tseng L.-M., Liu C.-Y., Chang K.-C., Chu P.-Y., Shiau C.-W., Chen K.-F., CIP2A is a target of bortezomib in human triple negative breast cancer cells. Breast Cancer Res. 14, R68 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang X., Ding C.-K., Wu J., Sjol J., Wardell S., Spasojevic I., George D., Donnell D. P. M., Hsu D. S., Chang J. T., Chi J.-T., Cystine addiction of triple-negative breastcancer associated with EMT augmented death signaling. Oncogene 36, 4235–4242 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Timmerman L. A., Holton T., Yuneva M., Louie R. J., Padró M., Daemen A., Hu M., Chan D. A., Ethier S. P., van’t Veer L. J., Polyak K., Cormick F. M., Gray J. W., Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 24, 450–465 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Geldermalsen M., Wang Q., Nagarajah R., Marshall A. D., Thoeng A., Gao D., Ritchie W., Feng Y., Bailey C. G., Deng N., Harvey K., Beith J. M., Selinger C. I., O’Toole S. A., Rasko J. E. J., Holst J., ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 35, 3201–3208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y., Tan H., Daniels J. D., Uchida K., O’Connor O. A., Stockwell B. R., Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem. Biol. 26, 623–633.e9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamilton D., Batist G., Glutathione analogues in cancer treatment. Curr. Oncol. Rep. 6, 116–122 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Yuan H., Li X., Zhang X., Kang R., Tang D., Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 478, 1338–1343 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Kim S., Leal S. S., Ben Halevy D., Gomes C. M., Lev S., Structural requirements for VAP-B oligomerization and their implication in amyotrophic lateral sclerosis-associated VAP-B(P56S) neurotoxicity. J. Biol. Chem. 285, 13839–13849 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Verma N., Müller A.-K., Kothari C., Panayotopoulou E., Kedan A., Selitrennik M., Mills G. B., Nguyen L. K., Shin S., Karn T., Holtrich U., Lev S., Targeting of PYK2 synergizes with EGFR antagonists in basal-like TNBC and circumvents HER3-associated resistance via the NEDD4-NDRG1 axis. Cancer Res. 77, 86–99 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Kim S., Kedan A., Marom M., Gavert N., Keinan O., Selitrennik M., Laufman O., Lev S., The phosphatidylinositol-transfer protein Nir2 binds phosphatidic acid and positively regulates phosphoinositide signalling. EMBO Rep. 14, 891–899 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ding W.-Q., Liu B., Vaught J. L., Palmiter R. D., Lind S. E., Clioquinol and docosahexaenoic acid act synergistically to kill tumor cells. Mol. Cancer Ther. 5, 1864–1872 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/34/eaba8968/DC1