

Abstract
Nicotinamide adenine dinucleotide (NAD+) is an essential metabolite that is reported to decline in concentration in tissues of aged animals. Strategies to increase NAD+ availability have shown promise in treating many conditions in rodents, including age-related degeneration, which has in turn driven intense interest in the effects of supplements on human health. However, many aspects of NAD+ metabolism remain poorly understood, and human data are limited. Here, we discuss the state of the evidence for an age-related decline in NAD+, along with potential mechanistic explanations, including increased consumption or decreased synthesis of NAD+ and changes in the composition of cells or tissues with age. Key challenges for the field involve the development of better tools to resolve information on the NAD+ content of specific cells and subcellular compartments as well as determining the threshold levels at which NAD+ depletion triggers physiological consequences in different tissues. Understanding how NAD+ metabolism changes with age in humans may ultimately allow the design of more targeted strategies to maintain its availability, such as inhibition of key consumers in specific tissues or direct delivery of precursors to sites of deficiency. In the meantime, human clinical trials with oral supplements are poised to provide some of the first direct evidence as to whether increasing NAD+ availability can impact human physiology. Thus, it is an exciting time for NAD+ research, with much remaining to be learned in terms of both basic biology and potential therapeutic applications.
NAD+ is critical to life:
Nicotinamide adenine dinucleotide (NAD+) is an electron (hydride) acceptor that is fundamental to life. It is the product of vitamin B3 metabolism and severe deficiency leads to a condition termed pellagra that is characterized by the 4 Ds: dermatitis, dementia, diarrhea, and death. Accepting and donating electrons interconverts NAD+ with its reduced form, NADH, in a process that is essential for central carbon metabolism, including glycolysis, the Krebs cycle, and oxidative phosphorylation, as well as hundreds of other metabolic reactions within the cell. In addition, NAD+ serves as a co-substrate for several classes of enzymes, including the sirtuins, poly ADP-ribose polymerases (PARPs), cyclic ADP-ribose synthases such as CD38 and CD157, mono ADP-ribosyltransferases, and the “executioner” enzyme Sterile alpha and Toll/interleukin-1 receptor motif containing 1 (SARM1). All of these enzymes consume NAD+ and release the nicotinamide (NAM) moiety, necessitating a means to constantly replenish cellular NAD+ pools. In mammals, NAD+ is primarily synthesized via the salvage pathway from nicotinamide (NAM), de novo from tryptophan, or through the Priess-Handler pathway from nicotinic acid (NA). An alternate pathway for NAD synthesis that was more recently discovered is phosphorylation of nicotinamide (or nicotinic acid) riboside by nicotinamide riboside kinases1. Under conditions of stress or disease, these mechanisms may not be sufficient to fully maintain the NAD+ pool, leading to a decline in its concentration. Given its role at the nexus of many biochemical pathways related to redox balance, energy production, and intracellular signaling, the loss of NAD+ homeostasis may have dire consequences. Consistently, supplementation with NAD precursors yields therapeutic benefits in many rodent models of acute or chronic stress, metabolic and age-related diseases2.
Evidence that NAD+ declines with age:
There is substantial evidence that the concentration of NAD+ in rodent tissues decreases with age3–13 (reviewed in Yoshino et al.2). Reductions in NAD+ have been observed during aging in worms7, rodent tissues and human samples. However, the degree of decline and which tissues are affected vary considerably across studies. For instance, the NAD+ decline in skeletal muscle for aged rodents has been reported to be anywhere from ~15-65%. In aged liver, most reports suggest from ~10-50% decline, with the notable exception of one study in which NAD+ concentration increased with age in the livers of female C57BL/6JBomTac mice6. The same authors previously reported that this strain maintains hepatic NAD+ concentration when challenged with high fat diet14, and speculate that it might be particularly resistant to NAD+ decline. The increase in NAD+ with age likely reflects de novo synthesis, since it still occurs when Nampt is deleted in hepatocytes. For other tissues, there are fewer studies available, but some degree of decline is generally reported. Intriguingly, the NAD+ concentration in the brains of mice was found to decrease between weaning and young adulthood, and then to decrease further by middle age (12 months old), effects that precede most of the observations in other tissues13.
In humans, NAD+ concentration has been reported to decrease with age in skin samples. Although the precise degree of decline was not calculated, average concentration appeared to decrease at least 50% over the course of adult aging, and to be several fold-lower in adults as compared to newborns15. NAD(H) was also reported to decline by approximately 14% in the cerebrospinal fluid of subjects > 45 years of age as compared to those ≤ 45 years5. Two MRI-based studies have also provided evidence for NAD+ decline in human brains with age, ranging from ~10% to ~25% between young adulthood and old age10,16. In contrast, a third study that has been made available prior to peer review suggests that there is not a consistent difference in NAD+ content between the brains of young and old humans17. Therefore larger studies may still be required to provide a final definitive answer as to how the NAD+ content of human brain changes with age. Human liver samples from patients > 60 years of age were found to exhibit an approximately 30% decline in NAD+ concentration compared to samples from patients < 45 years of age11. In a small cohort of older individuals NAD+ content was also shown to decrease in monocyte-derived macrophages18. Finally, plasma NAD+ levels (which are at nanomolar concentrations, 3-4 orders of magnitude below tissue concentrations, and are of uncertain significance) were recently shown to decline sharply with age in humans19 in a study that used improved methodology as compared to a prior report that showed only a modest trend5. Therefore, a decline in NAD+ concentration in at least some tissues appears to be a conserved feature of aging across species, including humans.
Why does NAD+ fall?
Many different mechanisms have the potential to contribute to an age-related decline in NAD+. The most straightforward is increased activity of NAD+ consuming enzymes, which may be related to inflammation (CD38) or DNA damage (PARPs). However, it remains unclear whether NAD+ synthesis is fully maintained with age, and more nuanced changes, such as alterations in the cellular composition or redox state of aging tissues may also play a role. Finally, it should not be overlooked that organelles such as mitochondria concentrate NAD+, and a change in the mitochondrial content of a cell could therefore result in a net NAD+ change without necessarily affecting the concentration in any specific compartment. Here, we consider the available evidence for each of these potential contributors.
CD38/CD157
CD38 and CD157 are members of a family of bi-functional enzymes that use NAD+ to generate ADP ribose and cyclic ADP-ribose, which serves as an intracellular second messenger for calcium signaling. CD38 can also generate nicotinic acid adenine dinucleotide phosphate (NAADP), another calcium mobilizing metabolite, in vitro but it remains unclear whether this activity is relevant in vivo20,21. CD38 is the only NAD+ consumer that has been documented to increase in mRNA expression across multiple tissues of aging mice, and is thus a strong candidate to explain an age-related increase in consumption of NAD+ 4. CD38 null mice have elevated NAD+ concentrations in multiple tissues, and exhibit a blunted or absent decline with age. Intriguingly, a CD38 inhibitor given late in life also elevates NAD+ levels in multiple tissues and ameliorates age-related phenotypes in mice22. Despite these straightforward conclusions, there are a number of puzzling aspects to the CD38 story. First, the enzyme is restricted largely to immune cells23–26, with very low expression in hepatocytes27 or myotubes28, making it hard to understand why total NAD+ concentrations in tissues such as liver or muscle are so dramatically affected. On this point, it is worth noting that immune cells have been documented to infiltrate aging tissues, which may account for the observed increases in CD38 mRNA and enzymatic activity. In addition, it has recently been discovered that factors secreted from senescent cells can induce CD38 expression in macrophages and endothelial cells, amplifying the increase in tissue CD3829. A second mystery centers around the configuration of the enzyme. While there is a small amount of intracellular CD38 on vesicles, more than 90% of CD38 functions as an ecto-enzyme, facing the extracellular space and lacking access to intracellular NAD+ stores30. Interestingly, CD38 also has a second substrate; in addition to cleaving NAD+, it can cleave the precursor nicotinamide mononucleotide (NMN)4. NMN is present in the plasma, although the apparent concentration can vary by two orders of magnitude depending on the technique used to detect it31–34. Thus, CD38 could potentially decrease NAD+ by direct consumption and by destruction of extracellular NMN that would otherwise serve as a precursor. Given that the loss of CD38 activity causes a large increase in the NAD+ content of aged tissues, it is also worth considering whether the more subtle decline in NAD+ observed with natural aging is truly prevented, or merely “swamped” by the larger effect in CD38 null animals. Clearly, this is an intriguingly story with details yet to be elucidated.
PARPs
Poly ADP-ribose polymerase activity is a major driver of NAD+ catabolism and can be further stimulated in response to DNA damage and genotoxic stress. PARPs have been proposed to mediate the age-related decline in NAD+ primarily based on increases in PARP1 activity and PARylation of proteins in aged tissues, which is suggestive of increased NAD+ consumption15,22,35. This model is attractive because DNA damage is a well-established stimulus for PARPs and is known to accumulate with age36. Moreover, animal models with increased DNA damage exhibit NAD+ depletion that can be restored by PARP inhibition37,38, and mutant mice lacking PARP1 have been reported to have increased tissue NAD+ levels39. However, several key points of this model have not yet been tested. First, the activity of PARG, the enzyme that removes PARylation40, is not well-documented in aged tissues. Thus, an alternative explanation for increased PARylation with age is that the rate of removal decreases, rather than increase in synthesis. Second, it has not been established how much PARylation actually contributes to total NAD+ flux in vivo. The contribution of PARPs to NAD+ flux is substantial (~1/3 of total flux) in cultured T47D breast cancer cells31, but whether this holds up across tissue in animals remains to be seen. Third, it remains possible that the PARylation observed in tissue homogenates represents only a small minority of cells that are stressed or apoptotic, in which case it would not explain the loss of a large fraction of total tissue NAD+. In an alternative to the view that DNA damage precedes NAD+ depletion, it was recently shown that low NAD+ levels initiate an interaction between DBC1 and PARP1, leading to decreased PARP activity and subsequent accumulation of DNA damage41. In support of this model, NAD+ precursor supplementation inhibits the interaction between PARP1 and DBC1, restoring PARP1 activity and reducing DNA damage in the livers of aged mice. Thus, further studies are required to fully determine the tissue-specific cause and effect relationships between NAD+ concentration, DNA damage, and PARP activity during aging.
SARM1
SARM1 is protein involved in axonal (Wallerian) degeneration that was recently shown to possess an intrinsic NAD+ cleavage activity, generating NAM, ADP ribose and cyclic ADP ribose42,43. Activation of SARM1 rapidly depletes the NAD+ pool43,44, whereas inhibition of SARM1’s cleavage activity or Sarm1 deletion protects against axonal degeneration, improves survival in mouse model of axonopathy, and attenuates diabetic peripheral neuropathy42–47. Accordingly, SARM1 is now being investigated as a potential therapeutic target in neurodegenerative diseases, although early results from mouse models of Amyotrophic lateral sclerosis have been mixed, with no clear improvement in behavioral deficits48,49. SARM1 is also expressed in peripheral tissues including the liver and kidneys, where its role has not been characterized in detail50. Almost no data are currently available on the expression or activity of SARM1 in neurons or peripheral tissues with age. Thus, its role in age-related NAD+ decline, if any, remains to be determined.
NAD+ Synthesis
Whether NAD+ synthesis changes with age in mammalian tissues is largely unknown. Most tissues synthesize NAD+ primarily from NAM via the salvage pathway, whereas the liver also synthesizes a substantial portion of NAD+ via de novo synthesis from tryptophan, and limited synthesis from tryptophan and nicotinic acid occurs in other tissues, including kidney and macrophages18,31. NAD+ can also be synthesized via nicotinamide riboside kinase (NRK) enzymes51, but the significance of this pathway to endogenous NAD+ turnover is not yet clear, since NR concentrations are low in the absence of supplementation, and tissue NAD+ concentrations are maintained in young mice lacking NRKs52,53.
Decreased expression of the key salvage pathway enzyme nicotinamide phosphoribosyltransferase (Nampt) in with age has been reported in some tissues, including adipose tissues, skeletal muscle, retinal pigment epithelial cells, and certain brain regions4,12,13,54–58. Moreover, circulating levels of extracellular NAMPT packaged in extracellular vesicles decline with age both in rodents and humans58. Adipose-tissue specific overexpression of Nampt and treatment of aged mice with extracellular vesicles from young mice led to improvements in physical activity, sleep quality, glucose homeostasis, and lifespan. The expression of the de novo pathway enzyme quinolate phosphoribosyltransferase (QPRT) is decreased in macrophages isolated from aged humans and mice18, suggesting a potential decrease in NAD+ synthesis from tryptophan. NRK1 and NRK2 protein levels are unchanged in the adipose tissue and skeletal muscle, respectively of aged individuals55. All pathways to NAD+ synthesis required the activity of nicotinamide mononucleotide adenylyltransferases (NMNATs). A significant decline in mRNA for Nmnat1 and trends for Nmnat2 and Nmnat3 were reported in the livers of aged mice4, and expression levels of Nmnat isoforms are also reduced in the kidneys, oocytes and colons59–61. Collectively, these studies support a potential decrease in NAD+ synthetic capacity with age. Ultimately, flux studies with NAD+ precursors will be required to determine whether net synthesis of NAD+ decreases with age, or if tissue concentration is reduced primarily due to increased consumption.
Tissue composition
Changes in the cellular composition of some tissues occur with age. One place where changes in cell type are particularly obvious is in adipose, in which there is a switch from brown/beige to white adipocytes over time62. Thus, care must be taken to separate effects of cell composition from effects of aging per se when studying NAD+ concentration and turnover in aged adipose tissues. Another example of cell-type specific changes that occur with age is fiber type switching in skeletal muscle63. Aging is also characterized by persistent chronic immune cell infiltration that contributes to declines in physiological function at the tissue and organismal level64. In addition, tissues can accumulate fluid, lipid or glycogen that can affect the total weight in a way that does not reflect cell content. For this reason, it may be preferable to normalize to protein or another internal standard, rather than to the wet or dry weight of tissue in many cases. This should be especially considered for animals with obvious differences in adiposity, such as comparisons between lean and western diet-fed mice Advanced mass spectrometry-based technologies such as single cell metabolomics and imaging mass spectrometry will allow a better understanding of the contribution of specific cell types to changes in tissue NAD+ concentration.
Redox status
The majority of studies to date have focused on NAD+, rather than NADH measurements, largely because NADH requires more difficult extraction conditions, is less stable, and is generally present at lower concentrations, decreasing confidence in the measurements. Nevertheless, NADH concentration is critical to the interpretation of lower NAD+ with age, since a redox shift to a more reduced state could lower NAD+ concentration without changing the (NAD+ + NADH) pool. Indeed, an MRI-based study on NAD+ content in human brains determined that NAD+ decreases with age, but NADH increases such that the decline in the total pool size is slower than it would appear from NAD+ alone10. A shift toward a more reduced NAD+/NADH ratio has also been reported in the plasma of aged individuals19. In addition, a study in rats that included both NAD+ and NADH across multiple tissues with age similarly concluded that there is a redox shift in favor of NADH that exaggerates the NAD+ decline3. Improving the quality of NADH measurements and factoring redox changes into tissue NAD+ measurements are important goals for the field moving forward.
Mitochondrial/organellar NAD
NAD+ is highly compartmentalized within the cell. Total and free NAD+ concentrations are higher in the mitochondria than in the cytosol65,66 and mitochondrial NAD can be preserved under conditions that deplete total tissue concentrations67,68. The same may be true for multiple other organelles such as peroxisomes, which also contain NAD+ and possess a transporter69, but these concentrations have not been measured to date. The mitochondrial NAD+ redox state is also more reduced than that of cytosolic NAD+ 70. Thus, changes in the number of mitochondria could lead to changes in total tissue NAD+ and apparent redox state (based on total extractable NAD+ and NADH) without actually changing NAD+ concentration or redox state in any subcellular compartment, i.e., only changing the fraction of the tissue that is composed of mitochondria. A crude assessment of the mitochondrial NAD+ pool can be made by extracting the organelles, but this technique cannot provide reliable information on redox state, and it remains unknown whether NAD+ in the mitochondrial matrix is recovered quantitatively. Fluorescent NAD+ sensors can provide a more reliable indication of mitochondrial free NAD+ content and redox state in live cells66,70, but have not yet been applied to animal models.
Whether nuclear NAD+ content varies from that in the cytosol is less clear. The nuclear pore is large enough to allow diffusion of NAD+, cells can tolerate loss of the nuclear or cytosolic NMNAT isoforms, and genetically encoded NAD+ biosensors indicate similar free NAD+ concentrations in the nucleus and cytosol of 293T cells at rest and throughout the time course of NAD+ depletion after NAMPT inhibition66,71. However, recent observations suggest that nuclear and cytoplasmic NMNATs can compete for NMN to drive local NAD+ synthesis65, and the recruitment of NMNAT1 to specific promoter regions to support gene transcription72 highlights the potential importance of microdomains within organelles. Biosensor studies have also suggested the possibility of slight differences in steady state concentration between nuclear and cytoplasmic NAD+ pools in U2OS cells70. Understanding organellar NAD+ pools in vivo, including the use of organelle-specific NAD+ biosensors, will be a high priority in future studies on the roles of NAD+ in health and aging.
How much NAD+ is enough?
Knowing that measured NAD+ concentrations in tissues fall with age or disease, it becomes important to understand how much NAD+ is actually required for normal tissue function. Given the essential nature of NAD+ in hundreds of biochemical reactions, including glycolytic and mitochondrial energy production, it seems intuitive that any change in concentration could have major consequences. However, mice with a drastic (~85%) reduction in skeletal muscle NAD+ content have surprisingly mild phenotypes into early adulthood, and increasing skeletal muscle NAD+ content is almost without effect in young adult animals, requiring a prolonged exercise training regimen to tease out differences in performance8,73,74. Similarly, the respiratory capacity of mitochondria isolated from cultured myotubes is maintained until NAD+ is depleted by ~80% or more. These observations raise a number of questions: Is NAD+ concentration in excess under normal circumstances? Is it more important or limiting in some tissues than in others? If large changes in NAD+ concentration can be tolerated, then do the milder changes in aging and disease states actually matter? We will consider each of these questions in turn.
Is “normal” NAD+ in excess of actual need?
This question has proven surprisingly difficult to answer, in part due to the compartmentalization and protein binding properties of NAD+. Because the concentration of the free nucleotide varies throughout the cell, and the activities of many NAD+ consuming enzymes may be modulated by other factors including local NAD+/NADH redox state, feedback inhibition from nicotinamide, binding partners, or post-translational modifications, simply measuring the in vitro Km of specific consumers for NAD+ and comparing to the concentration measured in tissue homogenates is not sufficient to understand whether their activities will be affected by small concentration changes in intact cells or tissues. Recent advances using targeted fluorescent biosensors have allowed approximate concentrations to be assigned for free NAD+ in the nucleus, cytosol, and mitochondria (e.g., 109, 106, and 230 μM, respectively in HeLa cells)66. These values suggest that enzymes such as GAPDH (Km for NAD: 11 μM)75 and dehydrogenases of the Krebs cycle (Km for NAD dependent reactions catalyzed by isocitrate dehydrogenase, malate dehydrogenase and alpha-ketoglutarate dehydrogenase are 40, 114 and 25 μM, respectively)76–78 should be able to function unless a substantial degree of depletion occurs, whereas some NAD+ consumers, such as sirtuins (Km for NAD+ ranges from 26 μM for Sirt6, to 880 μM for Sirt3 and 980 μM for Sirt5, reviewed in79) and PARP1 (Km ~100 μM)80, may be more responsive to small changes in NAD+. Accordingly, a study in T47D breast cancer cells showed that NAD+ depletion occurs with approximately first-order kinetics, indicating that the major consumers are directly responsive to NAD+ concentration31. In the same cells, inhibition of either SIRT1/2 or PARPs was able to reduce NAD+ turnover by about one third. Although it has been suggested that the age-dependent decline in NAD+ limits Sirt3 and PARP1 activity in tissues4,22, analogous experiments looking at NAD+ turnover rates during aging have not yet been performed in vivo. In this regard, it is worth noting that the half turnover time of NAD+ in some tissues is as little as 20 minutes, as compared to 6-8 hours in cells (and other tissues), suggesting that there are important NAD+-intensive processes that are not replicated in cultured cells31.
Are specific tissues more sensitive to NAD+ levels?
Addressing the effects of incremental NAD+ depletion in specific tissues is challenging. Eliminating NAD+ salvage by deleting Nampt throughout the body causes embryonic lethality, and heterozygous mice have normal hepatic NAD+ levels and a mild phenotype; glucose-stimulated insulin secretion is decreased only in females34. Inducible knockout of Nampt in the whole body of adult mice results in reduced NAD+ levels in the liver and intestine, atrophy of intestinal villi, depletion of visceral adipose depots, a decrease in nutrient absorption, weight loss and death within 7-10 days after induction81. Pellagra, the disease triggered by nutritional NAD+ deficiency, prominently affects skin, the gastrointestinal tract, and neurologic function, suggesting that these tissues may be prone to NAD+ loss. In fact, NAD+ levels are reported to fall with age in human skin15. However, direct measurements of NAD+ in humans that have or are at risk for pellagra have generally been made only on blood samples, making it difficult to judge the threshold concentration required to prevent dysfunction. As noted above, deletion of Nampt in skeletal muscle leads to severe depletion of NAD+ that is surprisingly well tolerated8. Below, we consider evidence from some additional tissues.
Nampt deletion in hepatocytes causes a milder NAD+ depletion than is seen in skeletal muscle (0-50% loss), probably because the liver is able to generate NAD+ through the Preiss-Handler pathway and de novo synthesis from tryptophan6,82. Nampt deletion in hepatocytes is sufficient to cause a defect in liver regeneration82, which requires substantial NAD+ biosynthesis, but has little effect on basal mitochondrial function or hepatic metabolism in chow or high fat diet fed mice6,82. At the same time, the harmful effects of miR-34a in high fat diet fed mice have been attributed to suppression of Nampt-dependent NAD+ biosynthesis, and enhancing hepatic Nampt expression has been shown to protect against acetaminophen toxicity and alcoholic steatosis, and to promote liver regeneration11,82–84. Finally, the oncogenic effect of unconventional prefoldin RPB5 interactor in hepatocytes has been attributed to suppression of de novo NAD+ biosynthesis, which results in a > 50% loss of NAD+ and increased DNA damage85. Thus, the available evidence suggests that the liver can maintain basal function with up to ~50% NAD+ depletion, but is more sensitive to NAD+ loss under stress.
The role of Nampt in adipose tissue is more complex because it not only plays a direct role in NAD+ biosynthesis within adipocytes, but is also secreted and reported to influence NAD+ concentration in certain tissues including the hypothalamus58,86. After deletion of Nampt in adipocytes, 72-93% loss of NAD+ was reported across adipose tissue depots, whereas in the hypothalamus, but not the hippocampus, NAD+ was decreased by ~20%. These mice exhibited decreased physical activity that was attributed to the change in hypothalamic NAD+, and overexpression of NAMPT caused reciprocal phenotypes, increasing hypothalamic NAD+ (~10%) and physical activity. In addition, these mice exhibited a decline in whole-body insulin sensitivity that was associated with altered phosphorylation of CDK5 and PPARγ in adipose tissue, and was improved by rosiglitazone (a PPARγ agonist)87. Similarly generated mice were also found to have impaired mitochondrial respiration in brown fat and to be unable to expand adipose mass in response to a high fat diet88. Instead, adipose depots became fibrotic and mice maintained a lower body weight with improved glucose tolerance. In a separate study, intracerebroventricular injection of intact NAD+ was shown to increase NAD+ by 30-50% in the mediobasal hypothalamus and to suppress fasting-induced hyperphagia89. This NAD+-mediated anorexic effect was dependent on astrocyte expression of connexin 43, which can transport NAD+ across the plasma membrane90. Contrarily, intracerebroventricular injection of FK866 to deplete the NAD+ pool was shown to suppress fasting and ghrelin induced food intake91. The ability of both increased and decreased NAD+ to suppress food intake could be due to differences in timing or competing effects of different cell types and/ or hypothalamic regions. Thus, severe depletion of NAD+ in adipose tissue has mainly negative, but also some positive effects, whereas more subtle changes in hypothalamic NAD+ appear to have functional consequences. Interestingly, transfer of NAMPT-containing vesicles was reported to be sufficient to improve physical function and longevity in aged mice58.
In a number of other cell types, including projection neurons, adult neural stem cells, Schwann cells, immune cells, and retinal pigmented epithelial cells, Nampt deletion leads to overt dysfunction or death13,54,92,93. Inducible deletion of Nampt in projection neurons lowers cortical NAD+ by ~70% at 21 days post deletion (median survival 22 days). Other cell populations are either absent or too small or scattered to allow accurate NAD+ quantitation after Nampt deletion, but the loss of these cells presumably indicates that NAD+ depletion is more severe than in normal aging, and highlights the need for better fine-tuning of NAD+ availability to elucidate the relevance of modest declines. Further studies are required to establish the thresholds of NAD+ depletion that have functional consequences in each tissue and cell type and to determine whether these thresholds change over the lifespan.
How can small decreases in NAD+ with age matter if large changes are tolerated?
Another important question to consider is whether the small decreases in NAD+ reported to occur during aging (generally < 30%) are likely to matter, given that much larger changes can have subtle consequences. This is particularly puzzling in the case of skeletal muscle, where a 20-65% decline in NAD+ content has been reported with age and functional benefits have been reported from supplementation of the NAD+ pool, yet genetically altered mice initially tolerate a greater degree of NAD+ loss2,8,94. Skeletal muscle specific Nampt KO mice have an approximately 85% loss of NAD+ and have almost no observable phenotypes as young adults8. Although they develop myopathy by 5-7 months of age, this can be reversed by a nicotinamide riboside supplementation regimen that only modestly increases skeletal muscle NAD+ content, leaving the rescued animals with much less NAD+ than is present in aged wild type mice. This might suggest that the “rescue” comes from effects of NR in another tissue, however benefits are observed in ex vivo contraction assays and even in mitochondria isolated from the muscles. Moreover, preventing the age-related decline in skeletal muscle NAD+ content by overexpression of Nampt was sufficient to attenuate the age-related decline in endurance, supporting the hypothesis that NAD+ decline is functionally relevant8. One possibility is that something else changes in aged muscle that renders it more reliant on NAD+-dependent processes. Interestingly, genetic loss of ~95% of mitochondrial respiratory capacity in skeletal was reported to have almost no consequences until about 4 months of age, a time course that is reminiscent of the effect of severe NAD+ depletion and suggests a fundamental metabolic switch95. Moreover, aging skeletal muscle was reported to exhibit an NAD+-dependent “pseudohypoxic state” that was not observed in young muscles depleted of NAD+, again suggesting age- or time-dependent differences in the consequences of NAD+ deficiency9. A second possible way to reconcile consequences of age-related NAD+ decline with the lack of consequences of more severe depletion in young animals is to hypothesize that the NAD+ in aged tissues is not distributed evenly; NAD+ may be heterogeneous such that some cells have normal NAD+ content while others have severe deficiencies. To date, it has been very difficult to measure NAD+ in tissues with cellular resolution, but new technologies such as imaging mass spectrometry may soon change this. In addition, NADH autofluorescence can be measured at high resolution (e.g.,96,97), and the generation of mice using recently developed florescent sensors66 may even allow subcellular compartment-specific information on NAD+ content to be obtained. These types of experiments represent and important frontier in NAD+ research.
Will NAD+ supplementation delay aging?
There is now an extensive body of literature supporting health benefits of NAD+ supplementation in rodents, although the number of studies examining aging per se is much more limited and human data are generally absent or negative for most indications. Nicotinamide riboside initiated at 2 years of age improved stem cell function in C57BL/6 mice and modestly extended their remaining lifespan94. Nicotinamide mononucleotide administered between the ages of 5 and 17 months attenuated multiple age-related phenotypes in the same strain; the treated mice exhibited improvements in body composition, insulin sensitivity, mitochondrial respiration, eye function, and bone mineral density98, although the effect of NMN on lifespan has not been reported to date. NAM supplementation beginning at one year of age reduced hepatic steatosis and improved glucose homeostasis in high fat diet fed mice without extending lifespan99. To-date 46 clinical trials are registered for NR, and five for NMN (ClinicalTrials.gov, accessed December 2019) to evaluate the safety and potential benefits of NAD+ supplementation in metabolic, cardiovascular, and neurological diseases. Most have yet to report results. However, a study of 12 weeks of NR supplementation did not detect any improvement in insulin sensitivity or skeletal muscle mitochondrial function in obese, insulin resistant men100–102. Another trial with NR also failed to find evidence of improvements in glucose homeostasis or exercise performance, but provided preliminary data that are suggestive of modest benefits on blood pressure and arterial stiffness103. Three weeks of NR treatment boosted NAD+ level in the blood but not skeletal muscle of aged individuals104. However, the levels of NAM catabolites, N-methyl nicotinamide, N1-methyl-2-pyridone-5-carboxamide, and N1-methyl-2-pyridone-5-carboxamide were increased in skeletal muscle, blood and urine. Unexpectedly, NR supplementation decreased the expression of genes involved in energy metabolism in skeletal muscle, although mitochondrial bioenergetics remained unchanged. NR also appears to decrease circulating levels of several pro-inflammatory cytokines in humans, which could have long-term benefits104. Confirmation of these effects in longer studies with greater statistical power would indicate mechanisms by which NR could potentially have a positive impact on human lifespan. In addition, a preliminary study on ALS, a progressive age-related disease, indicated improvement in patients treated with a proprietary combination of NR and pterostilbene105. Larger studies will be required to confirm these effects and elucidate whether NR per se is playing an important role. Nicotinamide mononucleotide has lagged behind NR in terms of human trials, but at least one study has been completed (NCT03151239) and is expected to report results in the near future. On the whole, it is simply too early to state with confidence whether NAD+ supplementation will have a beneficial effect on human lifespan, or which of the findings from rodent models will translate into clinically relevant changes in human patients. In addition, it is important to keep in mind that while the available studies with NR uniformly support its safety, longer-term studies with large populations have not been performed.
Conclusion
NAD+ plays fundamental roles in the metabolic reactions that underlie all life. The observations that: 1) NAD+ concentration declines with age in multiple tissues, and 2) NAD+ precursor supplements are beneficial in rodents, create an attractive narrative that restoring NAD+ concentration to youthful levels might stave off some of the damaging effects of age. Thus, NAD+ biology is of interest for identifying therapeutics and/or nutraceuticals that promote healthier aging. However, much remains to be understood about the causes and consequences of age-related NAD+ decline. Human clinical data on NAD+ restoration remain sparse, but have provided weak evidence for beneficial effects on vascular health and inflammation while providing relatively robust evidence against a major benefit for insulin sensitivity or muscle performance. Further studies are clearly needed and improving our understanding of NAD+ metabolism in specific tissues and subcellular compartments, as well as the potential for cross talk with other pathways may offer new opportunities for more targeted therapeutic interventions.
Figure 1: NAD+ as a redox cofactor and cosubstrate for signaling enzymes.
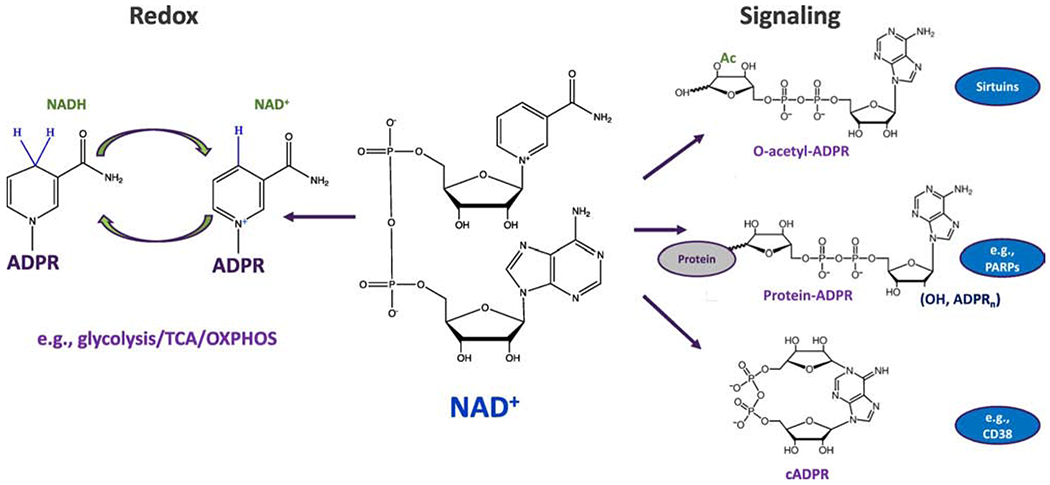
NAD+ (center) can accept electrons (in the form of a hydride anion, H−), converting the cofactor to its reduced form, NADH, and facilitating the oxidation of substrates. Subsequently, the electrons can be donated to facilitate reduction reactions with concomitant oxidation of NADH back to NAD+. This process is critical to hundreds of reactions, including those of central carbon metabolism, driving energy production (i.e., glycolysis, the TCA cycle and oxidative phosphorylation). NAD+ also serves as a co-substrate for several families of enzymes that regulate key biological processes via changes in protein modification or the generation of signaling molecules (e.g., Sirtuins, PARPs and CD38/CD157).
Figure 2: Potential mechanisms resulting in lower tissue NAD+ concentrations with age.
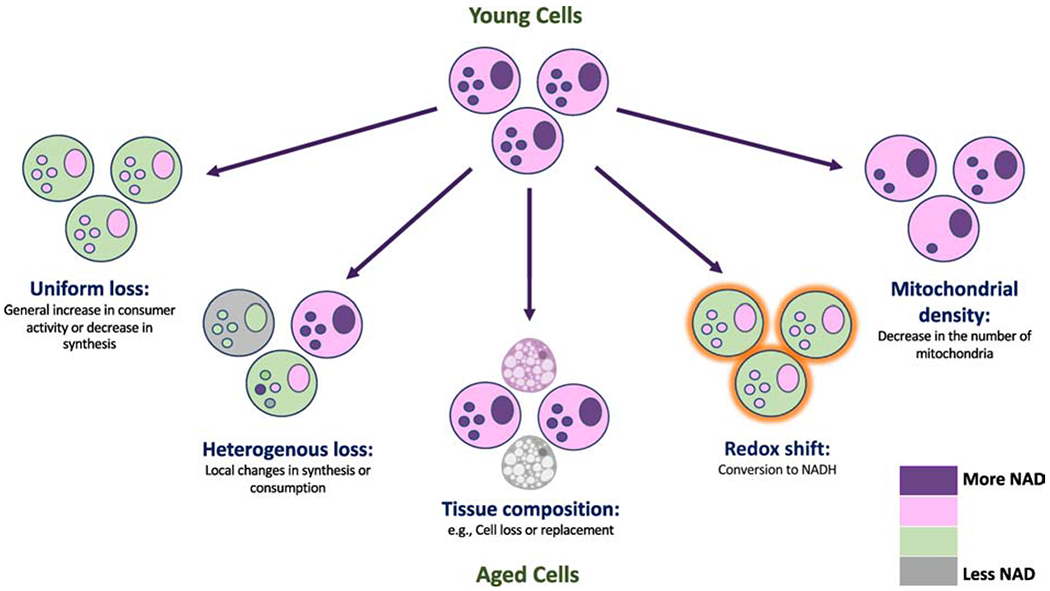
Schematic illustrating distinct scenarios that could result in lower measured NAD+ in extracts of tissues from aged animals. Uniform loss involves all cells experiencing a similar NAD+ deficit. Heterogenous loss suggests local defects resulting in impaired synthesis or excess consumption that could affect a subset of cells disproportionately. Tissue composition may also change with age, resulting in decreased cellularity or the appearance of cells with less NAD+ (e.g., adipocytes). A shift in the redox balance could lower NAD+ without any change in the total (NAD+ + NADH) pool. A decrease in the number of mitochondria (or other NAD+-rich organelles) could decrease the whole-tissue NAD+ concentration and apparent redox state (i.e., whole tissue NAD+:NADH ratio) without actually changing NAD+ concentration or redox state in any given compartment.
Highlights:
NAD+ is an essential metabolite that declines with age
Changes in metabolism and tissue composition may contribute to NAD+ decline
Lack of cellular/subcellular resolution is a limitation of most studies on NAD+
Supplementation of NAD+ is beneficial in aged rodents
Acknowledgements
This work was supported by a grant from the National Institutes of Health (DK098656 to J.A.B.). K.C. is supported by a fellowship from the Crohn’s and Colitis Foundation. M.R.M. is supported by the Burroughs Wellcome Fund and Howard Hughes Medical Institute via the PDEP and Hanna H. Gray Fellows Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007. doi: 10.1016/j.tibs.2006.11.006 [DOI] [PubMed] [Google Scholar]
- 2.Yoshino J, Baur JA, Imai S ichiro. NAD + Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018. doi: 10.1016/j.cmet.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R. Age related changes in NAD+ metabolism oxidative stress and sirt1 activity in wistar rats. PLoS One. 2011. doi: 10.1371/journal.pone.0019194 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Camacho-Pereira J, Tarragó MG, Chini CCS, et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016. doi: 10.1016/j.cmet.2016.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guest J, Grant R, Mori TA, Croft KD. Changes in oxidative damage, inflammation and [NAD(H)] with age in cerebrospinal fluid. PLoS One. 2014. doi: 10.1371/journal.pone.0085335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dall M, Trammell SAJ, Asping M, et al. Mitochondrial function in liver cells is resistant to perturbations in NAD+ salvage capacity. J Biol Chem. 2019. doi: 10.1074/jbc.RA118.006756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mouchiroud L, Houtkooper RH, Moullan N, et al. XThe NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013. doi: 10.1016/j.cell.2013.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frederick DW, Loro E, Liu L, et al. Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab. 2016. doi: 10.1016/j.cmet.2016.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gomes AP, Price NL, Ling AJY, et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013. doi: 10.1016/j.cell.2013.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A. 2015. doi: 10.1073/pnas.1417921112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou CC, Yang X, Hua X, et al. Hepatic NAD+deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br J Pharmacol. 2016. doi: 10.1111/bph.13513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshino J, Mills KF, Yoon MJ, Imai SI. Nicotinamide mononucleotide, a key NAD + intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011. doi: 10.1016/j.cmet.2011.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stein LR, Imai SI. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 2014. doi: 10.1002/embj.201386917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dall M, Penke M, Sulek K, et al. Hepatic NAD + levels and NAMPT abundance are unaffected during prolonged high-fat diet consumption in C57BL/6JBomTac mice. Mol Cell Endocrinol. 2018. doi: 10.1016/j.mce.2018.01.025 [DOI] [PubMed] [Google Scholar]
- 15.Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One. 2012. doi: 10.1371/journal.pone.0042357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bagga P, Hariharan H, Wilson NE, et al. Single-Voxel 1H MR spectroscopy of cerebral nicotinamide adenine dinucleotide (NAD+) in humans at 7T using a 32-channel volume coil. Magn Reson Med. 2019. doi: 10.1002/mrm.27971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elhassan YS, Kluckova K, Fletcher RS, et al. Nicotinamide riboside augments the human skeletal muscle NAD + metabolome and induces transcriptomic and anti-inflammatory signatures in aged subjects: a placebo-controlled , randomized trial. bioRxiv. 2019. doi: 10.1101/680462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minhas PS, Liu L, Moon PK, et al. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat Immunol. 2019. doi: 10.1038/s41590-018-0255-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clement J, Wong M, Poljak A, Sachdev P, Braidy N. The Plasma NAD + Metabolome Is Dysregulated in “normal” Aging. Rejuvenation Res. 2019. doi: 10.1089/rej.2018.2077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang C, Li T, Li Y, et al. CD38 produces nicotinic acid adenosine dinucleotide phosphate in the lysosome. J Biol Chem. 2018. doi: 10.1074/jbc.RA118.002113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmid F, Bruhn S, Weber K, Mittrücker HW, Guse AH. CD38: A NAADP degrading enzyme. FEBS Lett. 2011. doi: 10.1016/j.febslet.2011.10.017 [DOI] [PubMed] [Google Scholar]
- 22.Tarragó MG, Chini CCS, Kanamori KS, et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD+ Decline. Cell Metab. 2018. doi: 10.1016/j.cmet.2018.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amici SA, Young NA, Narvaez-Miranda J, et al. CD38 is robustly induced in human macrophages and monocytes in inflammatory conditions. Front Immunol. 2018. doi: 10.3389/fimmu.2018.01593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Musso T, Deaglio S, Franco L, et al. CD38 expression and functional activities are up-regulated by IFN-gamma on human monocytes and monocytic cell lines. J Leukoc Biol. 2001. doi: 10.1189/jlb.69.4.605 [DOI] [PubMed] [Google Scholar]
- 25.Kang BN, Tirumurugaan KG, Deshpande DA, et al. Transcriptional regulation of CD38 expression by tumor necrosis factor-α in human airway smooth muscle cells: Role of NF-κB and sensitivity to glucocorticoids. FASEB J. 2006. doi: 10.1096/fj.05-4585fje [DOI] [PubMed] [Google Scholar]
- 26.Matalonga J, Glaria E, Bresque M, et al. The Nuclear Receptor LXR Limits Bacterial Infection of Host Macrophages through a Mechanism that Impacts Cellular NAD Metabolism. Cell Rep. 2017. doi: 10.1016/j.celrep.2017.01.007 [DOI] [PubMed] [Google Scholar]
- 27.Rah SY, Kim UH. CD38-mediated Ca 2+ signaling contributes to glucagon-induced hepatic gluconeogenesis. Sci Rep. 2015. doi: 10.1038/srep10741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park DR, Nam TS, Kim YW, Lee SH, Kim UH. CD38-cADPR-SERCA Signaling Axis Determines Skeletal Muscle Contractile Force in Response to β-Adrenergic Stimulation. Cell Physiol Biochem. 2018. doi: 10.1159/000489441 [DOI] [PubMed] [Google Scholar]
- 29.Chini C, Hogan KA, Warner GM, et al. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD+ decline. Biochem Biophys Res Commun. 2019. doi: 10.1016/j.bbrc.2019.03.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shrimp JH, Hu J, Dong M, et al. Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe. J Am Chem Soc. 2014. doi: 10.1021/ja411046j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L, Su X, Quinn WJ, et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018. doi: 10.1016/j.cmet.2018.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grozio A, Mills KF, Yoshino J, et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat Metab. 2019. doi: 10.1038/s42255-018-0009-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hara N, Yamada K, Shibata T, Osago H, Tsuchiya M. Nicotinamide phosphoribosyltransferase/visfatin does not catalyze nicotinamide mononucleotide formation in blood plasma. PLoS One. 2011. doi: 10.1371/journal.pone.0022781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Revollo JR, Körner A, Mills KF, et al. Nampt/PBEF/Visfatin Regulates Insulin Secretion in β Cells as a Systemic NAD Biosynthetic Enzyme. Cell Metab. 2007. doi: 10.1016/j.cmet.2007.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ubaida-Mohien C, Lyashkov A, Gonzalez-Freire M, et al. Discovery proteomics in aging human skeletal muscle finds change in spliceosome, immunity, proteostasis and mitochondria. Elife. 2019. doi: 10.7554/eLife.49874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang G, Han T, Nijhawan D, et al. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell. 2014. doi: 10.1016/j.cell.2014.07.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fang EF, Scheibye-Knudsen M, Brace LE, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD +/SIRT1 reduction. Cell. 2014. doi: 10.1016/j.cell.2014.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheibye-Knudsen M, Mitchell SJ, Fang EF, et al. A high-fat diet and NAD+ activate sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014. doi: 10.1016/j.cmet.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bai P, Cantó C, Oudart H, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011. doi: 10.1016/j.cmet.2011.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gogola E, Duarte AA, de Ruiter JR, et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell. 2018. doi: 10.1016/j.ccell.2018.05.008 [DOI] [PubMed] [Google Scholar]
- 41.Li J, Bonkowski MS, Moniot S, et al. A conserved NAD + binding pocket that regulates protein-protein interactions during aging. Science (80- ). 2017. doi: 10.1126/science.aad8242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Summers DW, Gibson DA, DiAntonio A, Milbrandt J. SARM1-specific motifs in the TIR domain enable NAD+ loss and regulate injury-induced SARM1 activation. Proc Natl Acad Sci USA. 2016. doi: 10.1073/pnas.1601506113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron. 2017. doi: 10.1016/j.neuron.2017.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD+ destruction. Science (80- ). 2015. doi: 10.1126/science.1258366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gilley J, Ribchester RR, Coleman MP. Sarm1 Deletion, but Not WldS, Confers Lifelong Rescue in a Mouse Model of Severe Axonopathy. Cell Rep. 2017. doi: 10.1016/j.celrep.2017.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geisler S, Doan RA, Cheng GC, et al. Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight. 2019. doi: 10.1172/jci.insight.129920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheng Y, Liu J, Luan Y, et al. SARM1 gene deficiency attenuates diabetic peripheral neuropathy in mice. Diabetes. 2019. doi: 10.2337/db18-1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peters OM, Lewis EA, Osterloh JM, et al. Loss of Sarm1 does not suppress motor neuron degeneration in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Hum Mol Genet. 2018. doi: 10.1093/hmg/ddy260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.White MA, Lin Z, Kim E, et al. Sarm1 deletion suppresses TDP-43-linked motor neuron degeneration and cortical spine loss. Acta Neuropathol Commun. 2019. doi: 10.1186/s40478-019-0800-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mink M, Fogelgren B, Olszewski K, Maroy P, Csiszar K. A novel human gene (SARM) at chromosome 17q11 encodes a protein with a SAM motif and structural similarity to Armadillo/β-catenin that is conserved in mouse, Drosophila, and Caenorhabditis elegans. Genomics. 2001. doi: 10.1006/geno.2001.6548 [DOI] [PubMed] [Google Scholar]
- 51.Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a preiss-handler independent route to NAD+ in fungi and humans. Cell. 2004. doi: 10.1016/S0092-8674(04)00416-7 [DOI] [PubMed] [Google Scholar]
- 52.Fletcher RS, Ratajczak J, Doig CL, et al. Nicotinamide riboside kinases display redundancy in mediating nicotinamide mononucleotide and nicotinamide riboside metabolism in skeletal muscle cells. Mol Metab. 2017. doi: 10.1016/j.molmet.2017.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ratajczak J, Joffraud M, Trammell SAJ, et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat Commun. 2016. doi: 10.1038/ncomms13103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jadeja RN, Powell FL, Jones MA, et al. Loss of NAMPT in aging retinal pigment epithelium reduces NAD+ availability and promotes cellular senescence. Aging (Albany NY). 2018. doi: 10.18632/aging.101469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Guia RM, Agerholm M, Nielsen TS, et al. Aerobic and resistance exercise training reverses age-dependent decline in NAD+ salvage capacity in human skeletal muscle. Physiol Rep. 2019. doi: 10.14814/phy2.14139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma C, Pi C, Yang Y, et al. Nampt expression decreases age-related senescence in rat bone marrow mesenchymal stem cells by targeting Sirt1 PLoS One. 2017. doi: 10.1371/journal.pone.0170930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu LY, Wang F, Zhang XY, et al. Nicotinamide Phosphoribosyltransferase May Be Involved in Age-Related Brain Diseases. PLoS One. 2012. doi: 10.1371/journal.pone.0044933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoshida M, Satoh A, Lin JB, et al. Extracellular Vesicle-Contained eNAMPT Delays Aging and Extends Lifespan in Mice. Cell Metab. 2019. doi: 10.1016/j.cmet.2019.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guan Y, Wang SR, Huang XZ, et al. Nicotinamide mononucleotide, an NAD+ precursor, rescues age-associated susceptibility to AKI in a sirtuin 1-dependent manner. J Am Soc Nephrol. 2017. doi: 10.1681/ASN.2016040385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu X, Hu F, Zeng J, et al. NMNAT2-mediated NAD+ generation is essential for quality control of aged oocytes. Aging Cell. 2019. doi: 10.1111/acel.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu X, Shen W, Wang Y, Jaiswal A, Ju Z, Sheng Q. Nicotinamide adenine dinucleotide replenishment rescues colon degeneration in aged mice. Signal Transduct Target Ther. 2017. doi: 10.1038/sigtrans.2017.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schosserer M, Grillari J, Wolfrum C, Scheideler M. Age-Induced Changes in White, Brite, and Brown Adipose Depots: A Mini-Review. Gerontology. 2018. doi: 10.1159/000485183 [DOI] [PubMed] [Google Scholar]
- 63.Murgia M, Toniolo L, Nagaraj N, et al. Single Muscle Fiber Proteomics Reveals Fiber-Type-Specific Features of Human Muscle Aging. Cell Rep. 2017. doi: 10.1016/j.celrep.2017.05.054 [DOI] [PubMed] [Google Scholar]
- 64.Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A. Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nat Rev Endocrinol. 2018. doi: 10.1038/s41574-018-0059-4 [DOI] [PubMed] [Google Scholar]
- 65.Ryu KW, Nandu T, Kim J, Challa S, DeBerardinis RJ, Lee Kraus W. Metabolic regulation of transcription through compartmentalized NAD + biosynthesis. Science (80- ). 2018. doi: 10.1126/science.aan5780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cambronne XA, Stewart ML, Kim D, et al. Biosensor reveals multiple sources for mitochondrial NAD+. Science (80- ). 2016. doi: 10.1126/science.aad5168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pittelli M, Formentini L, Faraco G, et al. Inhibition of nicotinamide phosphoribosyltransferase: Cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J Biol Chem. 2010. doi: 10.1074/jbc.M110.136739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sims CA, Guan Y, Mukherjee S, et al. Nicotinamide mononucleotide preserves mitochondrial function and increases survival in hemorrhagic shock. JCI insight. 2018. doi: 10.1172/jci.insight.120182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Agrimi G, Russo A, Scarcia P, Palmieri F. The human gene SLC25A17 encodes a peroxisomal transporter of coenzyme A, FAD and NAD +. Biochem J. 2012. doi: 10.1042/BJ20111420 [DOI] [PubMed] [Google Scholar]
- 70.Sallin O, Reymond L, Gondrand C, Raith F, Koch B, Johnsson K. Semisynthetic biosensors for mapping cellular concentrations of nicotinamide adenine dinucleotides. Elife. 2018. doi: 10.7554/eLife.32638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Davila A, Liu L, Chellappa K, et al. Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. Elife. 2018. doi: 10.7554/eLife.33246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang T, Berrocal JG, Yao J, et al. Regulation of poly(ADP-ribose) polymerase-1-dependent gene expression through promoter-directed recruitment of a nuclear NAD + synthase. J Biol Chem. 2012. doi: 10.1074/jbc.M111.304469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frederick DW, Davis JG, Dávila A, et al. Increasing NAD synthesis in muscle via nicotinamide phosphoribosyltransferase is not sufficient to promote oxidative metabolism. J Biol Chem. 2015. doi: 10.1074/jbc.M114.579565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Costford SR, Brouwers B, Hopf ME, et al. Skeletal muscle overexpression of nicotinamide phosphoribosyl transferase in mice coupled with voluntary exercise augments exercise endurance. Mol Metab. 2018. doi: 10.1016/j.molmet.2017.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Phadke M Disruption of NAD + binding site in glyceraldehyde 3-phosphate dehydrogenase affects its intranuclear interactions . World J Biol Chem. 2015. doi: 10.4331/wjbc.v6.i4.366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hartong DT, Dange M, McGee TL, Berson EL, Dryja TP, Colman RF. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat Genet. 2008. doi: 10.1038/ng.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Crow KE, Braggins TJ, Batt RD, Hardman MJ. Rat liver cytosolic malate dehydrogenase: Purification, kinetic properties, role in control of free cytosolic NADH concentration. Analysis of control of ethanol metabolism using computer simulation. J Biol Chem. 1982. [PubMed] [Google Scholar]
- 78.Strumilo SA, Vinogradov VV., Senkevich SB. Kinetic and regulatory properties of alpha-ketoglutarate dehydrogenase complex from bovine adrenals. Ukr Biokhim Zh. 1980. [PubMed] [Google Scholar]
- 79.Cantó C, Menzies KJ, Auwerx J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015. doi: 10.1016/j.cmet.2015.05.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Langelier MF, Ruhl DD, Planck JL, Kraus WL, Pascal JM. The Zn3 domain of human poly(ADP-ribose) polymerase-1 (PARP-1) functions in both DNA-dependent poly(ADP-ribose) synthesis activity and chromatin compaction. J Biol Chem. 2010. doi: 10.1074/jbc.M110.105668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang LQ, Haandel L Van, Xiong M, et al. Metabolic and molecular insights into an essential role of nicotinamide phosphoribosyltransferase. Cell Death Dis. 2017. doi: 10.1038/cddis.2017.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mukherjee S, Chellappa K, Moffitt A, et al. Nicotinamide adenine dinucleotide biosynthesis promotes liver regeneration. Hepatology. 2017. doi: 10.1002/hep.28912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Choi SE, Fu T, Seok S, et al. Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell. 2013. doi: 10.1111/acel.12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xiong X, Yu J, Fan R, et al. NAMPT overexpression alleviates alcohol-induced hepatic steatosis in mice. PLoS One. 2019. doi: 10.1371/journal.pone.0212523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tummala KS, Gomes AL, Yilmaz M, et al. Inhibition of De Novo NAD+ Synthesis by Oncogenic URI Causes Liver Tumorigenesis through DNA Damage. Cancer Cell. 2014. doi: 10.1016/j.ccell.2014.10.002 [DOI] [PubMed] [Google Scholar]
- 86.Yoon MJ, Yoshida M, Johnson S, et al. SIRT1-Mediated eNAMPT Secretion from Adipose Tissue Regulates Hypothalamic NAD+ and Function in Mice. Cell Metab. 2015. doi: 10.1016/j.cmet.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stromsdorfer KL, Yamaguchi S, Yoon MJ, et al. NAMPT-Mediated NAD+ Biosynthesis in Adipocytes Regulates Adipose Tissue Function and Multi-organ Insulin Sensitivity in Mice. Cell Rep. 2016. doi: 10.1016/j.celrep.2016.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nielsen KN, Peics J, Ma T, et al. NAMPT-mediated NAD + biosynthesis is indispensable for adipose tissue plasticity and development of obesity. Mol Metab. 2018. doi: 10.1016/j.molmet.2018.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Roh E, Park JW, Kang GM, et al. Exogenous nicotinamide adenine dinucleotide regulates energy metabolism via hypothalamic connexin 43. Metabolism. 2018. doi: 10.1016/j.metabol.2018.08.005 [DOI] [PubMed] [Google Scholar]
- 90.Bruzzone S, Guida L, Zocchi E, Franco L, De Flora A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001. doi: 10.1096/fj.00-0566fje [DOI] [PubMed] [Google Scholar]
- 91.De Guia RM, Hassing AS, Skov LJ, et al. Fasting- and ghrelin-induced food intake is regulated by NAMPT in the hypothalamus. Acta Physiol. 2020;Jan 3:e134. doi: 10.1111/apha.13437 [DOI] [PubMed] [Google Scholar]
- 92.Rongvaux A, Galli M, Denanglaire S, et al. Nicotinamide Phosphoribosyl Transferase/Pre-B Cell Colony-Enhancing Factor/Visfatin Is Required for Lymphocyte Development and Cellular Resistance to Genotoxic Stress. J Immunol. 2008. doi: 10.4049/jimmunol.181.7.4685 [DOI] [PubMed] [Google Scholar]
- 93.Sasaki Y, Hackett AR, Kim S, Strickland A, Milbrandt J. Dysregulation of NAD+ metabolism induces a Schwann cell dedifferentiation program. J Neurosci. 2018. doi: 10.1523/JNEUROSCI.3304-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang H, Ryu D, Wu Y, et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science (80- ). 2016. doi: 10.1126/science.aaf2693 [DOI] [PubMed] [Google Scholar]
- 95.Diaz F, Thomas CK, Garcia S, Hernandez D, Moraes CT. Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum Mol Genet. 2005. doi: 10.1093/hmg/ddi307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li LZ, Marissa M, Wang T, et al. Two-Photon Autofluorescence Imaging of Fixed Tissues: Feasibility and Potential Values for Biomedical Applications. Adv Exp Med Biol. 2020;1232:375–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xu HN, Zhao H, Chellappa K, et al. Optical Redox Imaging of Fixed Unstained Muscle Slides Reveals Useful Biological Information. Mol Imaging Biol. 2019. doi: 10.1007/s11307-019-01348-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mills KF, Yoshida S, Stein LR, et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016. doi: 10.1016/j.cmet.2016.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mitchell SJ, Bernier M, Aon MA, et al. Nicotinamide Improves Aspects of Healthspan, but Not Lifespan, in Mice. Cell Metab. 2018. doi: 10.1016/j.cmet.2018.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dollerup OL, Chubanava S, Agerholm M, et al. Nicotinamide riboside does not alter mitochondrial respiration, content or morphology in skeletal muscle from obese and insulin-resistant men. J Physiol. 2019. doi: 10.1113/JP278752 [DOI] [PubMed] [Google Scholar]
- 101.Dollerup OL, Christensen B, Svart M, et al. A randomized placebo-controlled clinical trial of nicotinamide riboside in obese men: Safety, insulin-sensitivity, and lipid-mobilizing effects. Am J Clin Nutr. 2018. doi: 10.1093/ajcn/nqy132 [DOI] [PubMed] [Google Scholar]
- 102.Dollerup OL, Trammell SAJ, Hartmann B, et al. Effects of Nicotinamide Riboside on Endocrine Pancreatic Function and Incretin Hormones in Nondiabetic Men With Obesity. J Clin Endocrinol Metab. 2019. doi: 10.1210/jc.2019-01081 [DOI] [PubMed] [Google Scholar]
- 103.Martens CR, Denman BA, Mazzo MR, et al. Chronic nicotinamide riboside supplementation is well-Tolerated and elevates NAD+ in healthy middle-Aged and older adults. Nat Commun. 2018. doi: 10.1038/s41467-018-03421-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Elhassan YS, Kluckova K, Fletcher RS, et al. Nicotinamide Riboside Augments the Aged Human Skeletal Muscle NAD+ Metabolome and Induces Transcriptomic and Anti-inflammatory Signatures. Cell Rep. 2019. doi: 10.1016/j.celrep.2019.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.de la Rubia JE, Drehmer E, Platero JL, et al. Efficacy and tolerability of EH301 for amyotrophic lateral sclerosis: a randomized, double-blind, placebo-controlled human pilot study. Amyotroph Lateral Scler Front Degener. 2019. doi: 10.1080/21678421.2018.1536152 [DOI] [PubMed] [Google Scholar]