

Abstract
Purpose:
Gliomas are brain tumors with dismal prognoses. The standard-of-care treatments for gliomas include surgical resection, radiation, and temozolomide administration; however, they have been ineffective in providing significant increases in median survival. Antigen-specific cancer vaccines and immune checkpoint blockade may provide promising immunotherapeutic therapeutic approaches for gliomas.
Experimental Design:
Here, we have developed immunotherapy delivery vehicles based on synthetic high-density lipoprotein (sHDL) loaded with CpG, a Toll-like receptor 9 agonist, and tumor-specific neoantigens (NeoAgs) to target gliomas and elicit immune-mediated tumor regression.
Results:
We demonstrate that vaccination with NeoAg peptide-sHDL/CpG cocktail in combination with anti-PDL1 immune checkpoint blocker elicits robust NeoAg-specific T cell responses against GL261 cells and eliminated established orthotopic GL261 glioma in 33% of mice. Mice remained tumor free upon tumor cell re-challenge in the contralateral hemisphere, indicating the development of immunological memory. Moreover, in a genetically engineered murine model of orthotopic mutant IDH1 (mIDH1) glioma, sHDL vaccination with mutant IDH1 NeoAg eliminated glioma in 30% of animals and significantly extended the animal survival, demonstrating the versatility of our approach in multiple glioma models.
Conclusions:
Overall, our strategy provides a general roadmap for combination immunotherapy against gliomas and other cancer types.
Keywords: Glioma, HDL Nanodiscs, Nanoparticles, Neoantigens, Vaccination
Introduction
Gliomas are devastating brain cancers with a median survival rate of ~15 months (1). Currently, the standard-of-care for patients diagnosed with glioma includes surgery, radiation, and chemotherapy, but they remain ineffective at significantly increasing medial survival. Treatment effectiveness for glioma has been limited due to tumor heterogeneity, an immunosuppressive tumor microenvironment (TME), and the presence of the blood brain barrier, which hampers the transport of therapeutics to the central nervous system (CNS) (2,3). Despite surgical resection, patients invariably develop disease progression and tumor recurrence due to residual tumor cells (4,5).
Immunotherapy has recently emerged as a novel and attractive therapeutic platform for glioma (6–8). Immune checkpoint blockade designed to reinvigorate immune responses against tumor has shown promising results across multiple types of solid cancer (9,10). There are various ongoing clinical trials assessing therapeutic benefits of anti-PD-1 and anti-CTLA4 therapies in patients with primary and recurring gliomas (11); however, clinical trial results have been unimpressive thus far. Phase III trial NCT02667587 reported in 2019 that nivolumab (anti-PD-1) combined with radiation therapy did not prolong overall survival of glioma patients, compared with temozolomide and radiation (12). Initial results from a phase III trial (NCT02017717) examining the efficacy of nivolumab with or without ipilimumab (anti-CTLA-4) in patients with recurrent glioma have shown that nivolumab alone did not prolong overall survival, but the combination of nivolumab and ipilimumab is still under investigation (13). Hence, there is an urgent need to for developing novel immune-mediated combination approaches for treating glioma.
A complementary approach to immune checkpoint blockade is to vaccinate patients against their own tumor cells using endogenous tumor-specific antigens or neoantigens (14). Initial clinical trials examining neoantigen-based vaccines against advanced melanoma and glioma have shown promising results (15–17). However, current neoantigen delivery methods, such as direct injection of neoantigen peptides admixed with adjuvants such as oil emulsions, often result in precipitation, accumulation, and sustained inflammation at the injection site with minimal lymphatic drainage, which can lead to immune tolerance and deletion of antigen-specific T cells at the injection site (18–20). Thus, new strategies are needed to improve the delivery of neoantigens and adjuvant molecules to antigen-presenting cells (APCs) in lymphoid tissues in order to achieve potent anti-tumor immunity (21). An ideal neoantigen vaccine system should promote stable and efficient transport of neoantigen peptides to APCs in lymph nodes (LNs) while allowing for co-localized delivery of both antigens and adjuvant molecules to the same APCs without causing an unwanted inflammatory response (21,22).
To address these challenges, various “nano-vaccines” are under development for lymphatic trafficking and targeted delivery to APCs (22–25). In particular, we have previously demonstrated that synthetic high-density lipoprotein (sHDL) nanodiscs can effectively deliver neoantigens and a Toll-like receptor-9 (TLR9) agonist CpG to APCs in draining LNs and generate potent cytotoxic CD8α+ T cell lymphocyte (CTL) responses with promising anti-tumor efficacy (26). Here, we have employed the sHDL vaccine platform to generate neoantigen-specific CTLs against glioma and examined their anti-tumor efficacy in syngeneic models of GL261, which is a well-established murine model of glioma (27). We also examined whether neoantigen-based nanodisc vaccination can synergize with anti-PD-L1 immunotherapy. This is motivated by prior reports showing that some gliomas overexpress PD-L1 (28,29) as well as recent encouraging results from an ongoing phase II clinical trial (NCT02336165) studying the combination of durvalumab and radiotherapy in newly diagnosed glioma patients (30). Here, we report that the nanodisc platform in combination with anti-PD-L1 blockade induced robust infiltration of neoantigen-specific CD8α+ T cells into the GL261 tumor microenvironment and achieved potent anti-tumor efficacy with long-term anti-tumor immunity in syngeneic mouse GL261 glioma model. Furthermore, we have also shown the therapeutic efficacy of nanodisc vaccination in a genetically engineered orthotopic model of mutant IDH1 (mIDH1) glioma expressing IDH1-R132H, shATRX, and shTP53 (31).
Materials and Methods
Materials
Neoantigen peptides were synthesized by RS Synthesis (Louisville, KY). Female C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME). Antibodies against mouse CD40 and PD-L1 were purchased from BioXCell (West Lebanon, NH). Polyinosine-polycytidylic acid (polyIC) was purchased from Invivogen (San Diego, CA). 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) was purchased from NOF America (White Plains, NY). 22A Apolipoprotein-A1 mimetic peptide was synthesized by GenScript (Piscataway, NJ). 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[3-(2-pyridyldithio)propionate] (DOPE-PDP) was purchased from Avanti Polar Lipids (Alabaster, AL). Both cholesterol-modified CpG1826 and unmodified CpG1826 were synthesized by Integrated DNA Technologies (Coralville, IA). Interferon-γ (IFN-γ) ELISPOT kits were purchased from Fisher Scientific (Hampton, NH). Cell media was purchased from Invitrogen (Carlsbad, CA). The following antibodies for flow cytometry were purchased from BD Biosciences: rat Anti-Mouse CD107a-APC clone 1D4B; hamster Anti-Mouse CD69-PE clone H1.2F3; rat anti-Mouse CD8α-Brilliant Violet 605, clone 53–6.7; rat anti Mouse Foxp3-PE, clone MF23; and rat Anti-Mouse CD25-PE-Cy7, clone PC61. The following antibodies for flow cytometry were purchased from BioLegend: anti-mouse CD8α- APC, clone 53–6.7; anti-mouse CD279 (PD-1)-PE/Cy7; rat anti-mouse CD4-Brilliant Violet 605, clone: GK1.5; anti-mouse CD3-FITC, clone 17A2; rat anti-mouse CD86-PE/Cy7; anti-mouse CD11c-FITC, clone N418; anti-mouse CD103-APC, clone 2E7; or eBioscience: anti-mouse MHC Class II (I-A/I-E)-PE, clone M5/114.15.2.
Screening of GL261 neoantigen peptides.
Neoantigen peptide sequences chosen from an immunogenomics study on murine glioma models published in 2016 (32) were computationally screened for predicted MHC reactivity using the artificial neural network (ANN) method tool. Six out of ten neoantigen peptides identified in the study were synthesized and screened for in vivo immunogenicity according to the results from predicted MHC binding affinities produced by the Immune Epitope Database Analysis Resource (Supplementary Table 1). Female C57BL/6 mice aged 6–7 weeks were shaved and inoculated with 1 × 106 GL261 cells subcutaneously (s.c.) in the flank. On days 4 and 11 after tumor inoculation, 50 μg of each neoantigen peptide was co-administered intraperitoneally (i.p.) with anti-CD40 (50 μg) and Toll-like receptor-3 (TLR3) agonist polyIC (100 μg). On day 26 after inoculation, all mice were euthanized for spleen extraction and interferon-γ (IFN-γ) ELISPOT analysis to evaluate and compare the immunogenicity of the neoantigen peptide candidates.
Screening of mIDH1 neoantigen peptides.
Peptide epitopes encompassing the mutant IDH1 region were previously reported (33), and from these, we chose two neoantigen epitopes (mIDH1123–132, and mIDH1126–141) according to the results from predicted MHC binding affinities produced by the Immune Epitope Database Analysis Resource.
Formulation and characterization of sHDL nanodiscs carrying neoantigen peptides.
sHDL nanodiscs were prepared by dissolving DMPC and 22A in acetic acid, lyophilizing the mixture, and rehydrating the mixture in 10 mM sodium phosphate buffer, followed by thermocycling (34). Size of nanodisc was measured by dynamic light scattering (DLS). For incorporating neoantigen peptides into nanodiscs, neoantigen peptides were modified with a cysteine-serine-serine sequence at the N-terminus for conjugation to thiol-modified lipids. Modified neoantigen peptides were mixed with pyridyl disulfide-modified phospholipid (DOPE-PDP) for 2–3 hours on an orbital shaker to form a lipid-peptide conjugate. To incorporate lipid-peptide conjugates into sHDL, their mixture was incubated on an orbital shaker at 200 rpm for 1 hour. Cholesterol-modified CpG1826 (cho-CpG) was added by simple mixing of antigen-loaded nanodiscs and cho-CpG at a DMPC:cho-CpG weight ratio of 50:1. All nanodisc formulations were analyzed by Zetasizer to measure hydrodynamic size and zeta potential; by reverse-phase UPLC/MS and HPLC to measure the extent of lipid-peptide conjugation and incorporation; and by GPC to assess incorporation of cho-CpG.
Combination immunotherapy in a subcutaneous model of GL261 tumor.
All animal experiments were in accordance with the approved by the Institutional Animal Care & Use Committee (IACUC) at the University of Michigan, Ann Arbor. Immunocompetent female C57BL/6 mice (6–8 weeks old, Jackson Laboratories) were inoculated s.c. with 1.2 × 106 GL261 cells in the flank. When tumors were palpable, mice were administered s.c. at the tail base with neoantigen peptide-sHDL/CpG cocktail, soluble neoantigen peptide cocktail + CpG, or PBS. Vaccines were given with a 7-day interval. A subset of animals received anti-PD-L1 i.p. on days 1 and 4 after each vaccination. Injection dose was 15 μg for each peptide, 15 μg for CpG, and 100 μg for anti-PD-L1 IgG. Mice were euthanized when tumors reached 1.5 cm in diameter. Long-term survivors that exhibited complete tumor regression were re-challenged 72 days after the boost vaccination with 1.2 × 106 GL261 cells at the contralateral flank. For analysis of the tumor microenvironment, mice were treated as indicated and on day 8 after vaccination, tumors were harvested for flow cytometric analysis on a BioRad Zeti flow cytometer. Tumors were digested into single cell suspensions using a cocktail of DNase I and collagenase, followed by antibody staining and flow cytometry analyses.
IFN-γ ELISPOT analysis.
Six days after either prime or boost vaccination, blood samples were taken from the submandibular vein, or spleens were excised. Red blood cells were lysed and removed from the samples. For analysis of peripheral blood mononuclear cells(35), 0.1 × 106 PBMCs were plated in each well of an anti-IFN-γ-coated 96-well Immunospot plate in RPMI media + 10% FBS + 1% penicillin/streptomycin. For analysis of splenocytes, 0.5 × 106 splenocytes were plated in each well of 96-well Immunospot plate coated with anti-IFN-γ IgG in RPMI media + 10% FBS + 1% Penicillin/streptomycin. Neoantigen peptides were dissolved in water, diluted in RPMI media, and incubated with PBMCs or splenocytes for 18 hours at 37C. Plates were then processed according to the manufacturer’s instructions and later read at Cancer Center Immunology Core at the University of Michigan. The maximum detectable signal was 3000 spots in each well.
Combination immunotherapy in an orthotopic model of GL261 tumor
Immunocompetent female C57BL/6 or immunocompromised CD4−/− and CD8−/− knockout (KO) mice (Jackson Laboratory) were stereotactically injected with 20,000 GL261 cells into the right striatum using a 22-gauge Hamilton syringe (1 μL over 1 minute) with the following coordinates: +1.00 mm anterior, 2.5 mm lateral, and 3.00 mm deep to establish brain tumors (36–39). Mice were vaccinated s.c. at the tail base with the nanodisc vaccine or free neoantigen peptides and administered with anti-PD-L1 IgG i.p. at indicated time points. Long-term survivors in the nanodisc treatment group were re-challenged by inoculating mice with GL261 cells in the contralateral (left) hemisphere. To assess the immune cell population within the GL261 tumor microenvironment in the brain, mice were euthanized two days after the third vaccination, and brains were extracted. Tumor mass was dissected and homogenized using Tenbroeck (Corning) homogenizer in DMEM media containing 10% FBS. Immune cell populations in the tumor microenvironment were enriched with 30%−70% Percoll (GE Lifesciences) density gradient. Live/dead staining was carried out using fixable viability dye (eBioscience). Non-specific antibody binding was blocked with CD16/CD32, followed by staining with the following antibodies. Macrophages were labeled with CD45, F4/80, and CD206 antibodies. T cells were labeled with CD45, CD3, CD8α and CD4 antibodies. All antibodies were purchased from BioLegend. M1 macrophages were identified as CD45+/F4/80+/CD206low, and M2 macrophages were identified as CD45+/F4/80+/CD206high. Effector CD8α+ T cells were identified as CD45+/CD3+/CD8α+ and helper CD4+ T cells were identified as CD45+/CD3+/CD4+. T cell exhaustion was assessed by staining for PD-1. Regulatory T cells (Tregs) were identified as CD45+/CD3+/CD4+/CD25+/Foxp3. Antibody staining was carried out for 30 min at 4°C. Flow cytometry was performed using FACSAria flow cytometer (BD Biosciences) and analyzed using Flow Jo version 10 (Treestar) (40).
Nanodisc vaccination in an orthotopic model of mIDH1 tumor.
Immunocompetent female C57BL/6 mice were stereotactically injected with 25,000 mIDH1 neurospheres into the right striatum using a 22-gauge Hamilton syringe (1 μL over 1 minute) with the following coordinates: +1.00 mm anterior, 2.5 mm lateral, and 3.00 mm deep to establish brain tumors (36–39). Mice were vaccinated s.c. at the tail base with the nanodisc vaccine or free neoantigen peptides at indicated time points. Long-term survivors in the nanodisc treatment group were re-challenged by inoculating mice with mIDH1 cells in the contralateral (left) hemisphere.
Statistical analysis.
Sample sizes were chosen based on preliminary data from pilot experiments. For animal studies, the mice were randomized to match similar primary tumor volume, and all procedures were repeated at least twice in a non-blinded fashion. The results are expressed as mean ± S.E.M. Statistical analysis was performed with two-tailed t-tests for individual group comparisons or one-way ANOVA, followed by Tukey’s post-hoc analyses for multiple comparison tests with Prism 8.0 software (GraphPad Software). Analyses of survival differences were performed using Kaplan-Meier survival analyses with Log-rank Mantel-Cox. Statistical significance is indicated as *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
RESULTS
Selection and validation of GL261 neoantigens
Employing a recently published twelve neoantigen sequences from GL261 murine tumors by Johanns et al. (32), we first subjected these neoantigen peptide sequences for predicted binding to major histocompatibility complex-I (MHC-I) using a MHC-binding prediction tool (IEBD) and selected six neoantigens with low predicted IC50 values and mutated residues residing between the third and seventh peptides in the epitope sequence (41,42) (Supplementary Table 1). To further narrow down neoantigen candidates, we examined immunogenicity of the top six neoantigens in GL261-tumor bearing mice. C57BL/6 mice were inoculated subcutaneously at flank with 106 GL261 cells. On days 4 and 11 after tumor inoculation, 50 μg of each neoantigen peptide was administered intraperitoneally (i.p.) with 50 μg anti-CD40 IgG and 100 μg polyinosine-polycytidylic acid (polyIC). Anti-CD40 IgG and polyIC are a potent adjuvant combination known to amplify antigen-specific T cell responses (43,44). Anti-CD40 IgG enhances DC survival, cytokine release, and upregulation of costimulatory receptors (45), while a Toll-like receptor-3 (TLR3) agonist polyIC promotes cytokine release from DCs. Three out of six peptides co-administered with anti-CD40 IgG and polyIC elicited strong T cell responses, as evidenced by high IFN-γ ELISPOT counts, comparable to the positive control (Supplementary Table 1). In particular, we have identified 3 neoantigen epitopes with robust in vivo immunogenicity, namely AALLNKYLA (NeoAg1, H2-Db-restricted), MSLQFMTL (NeoAg2, H2-Kb-restricted), and GAIFNGFTL (NeoAg3, H2-Db-restricted).
Synthesis of nanodiscs carrying GL261 neoantigens
Having identified 3 top candidate neoantigens, we next synthesized sHDL nanodiscs incorporated with each neoantigen. Figure 1A shows the overall schematic for the synthesis of sHDL nanodiscs co-loaded with neoantigens and CpG. Blank sHDL nanodiscs were first prepared using 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 22A apolipoprotein-A1 mimetic peptide. Next, neoantigen peptides pre-modified with a cysteine-serine-serine (CSS) linker were conjugated with 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[3-(2-pyridyldithio)propionate] (DOPE-PDP), and the peptide-lipid conjugate was added to blank nanodiscs (Fig. 1A). Each nanodisc formulation was analyzed with dynamic light scattering (DLS) to assess the particle size. Blank nanodiscs as well as nanodiscs carrying each neoantigen peptide all had similar particle sizes ranging 9–13 nm, indicating that the addition of neoantigens did not significantly change the size of sHDL. NeoAg1-Nanodisc had a positive charge of 3.1 ± 2.3 mV, while NeoAg2-Nanodisc and NeoAg3-Nanodisc had negative charges of −1.8 ± 3.1 mV and −3.4 ± 4.3 mV, respectively (Fig. 1C). We also examined whether nanodiscs carrying each neoantigen were compatible when mixed all together. Three nanodisc formulations combined into one cocktail yielded a stable mixture of nanodiscs with an average diameter of 12.2 ± 2.7 nm and a negative charge of −2.3 ± 3.9 mV (Fig. 1C).
Figure 1. Development and characterization of neoantigen-loaded sHDL nanodiscs.
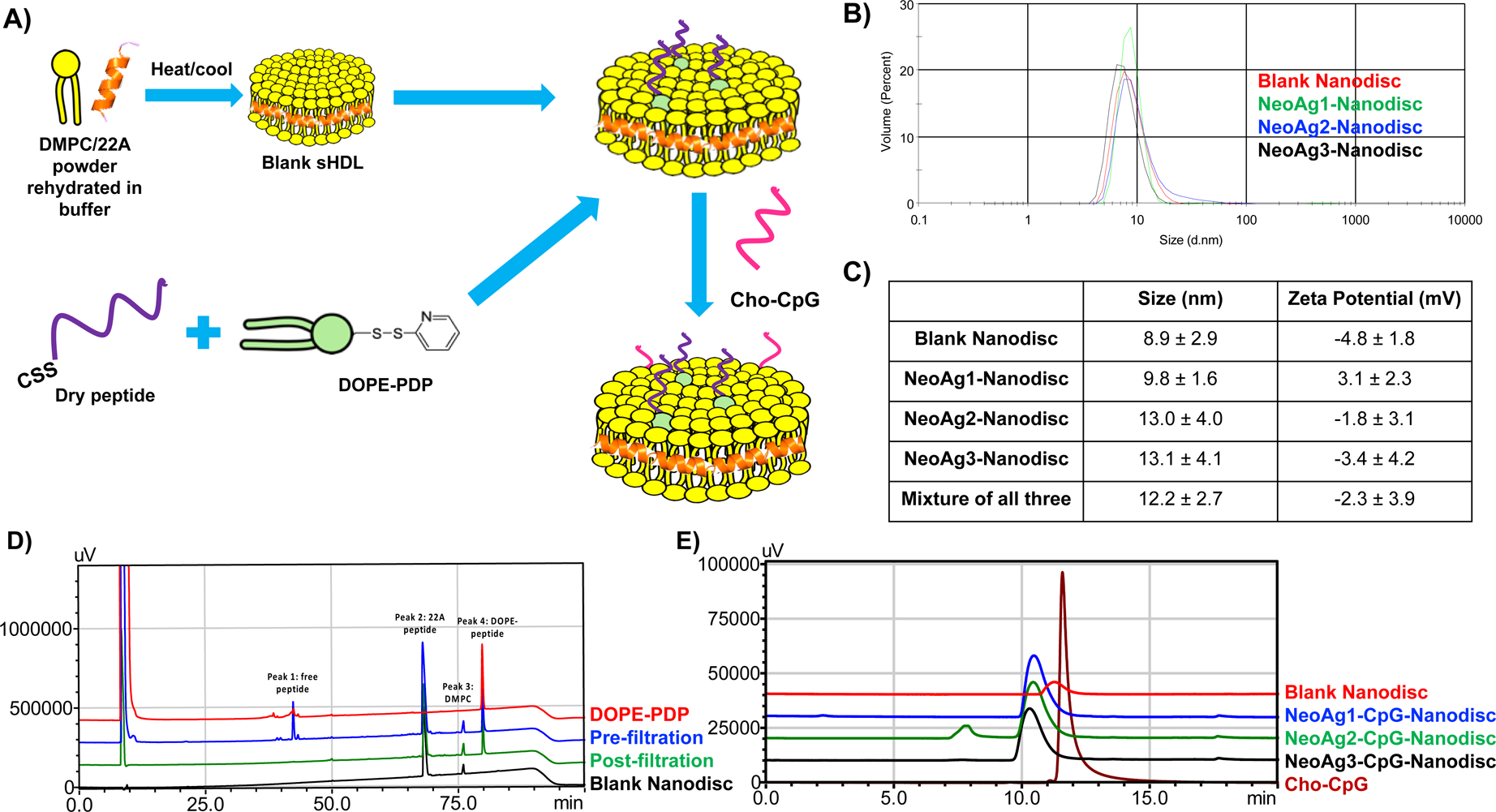
A) Schematic of the formulation process for NeoAg-loaded sHDL nanodiscs. B) Representative DLS profiles for each glioma NeoAg-sHDL formulation. C) The hydrodynamic size and zeta potential of each glioma NeoAg-sHDL formulation. D) Representative HPLC chromatograms for one glioma NeoAg-sHDL formulation. E) GPC chromatogram set for all glioma NeoAg-sHDL formulations after loading cholesterol-modified CpG.
We quantified the amount of neoantigen peptides loaded into nanodiscs using UPLC/MS and HPLC. We observed successful conjugation of all three neoantigen peptides to DOPE lipid with > 99% efficiency and > 90% incorporation efficiency of neoantigen-lipid conjugates into nanodiscs (Fig. 1D), as quantified by the amount of neoantigen-lipid conjugates remaining before and after filtration of nanodiscs. HPLC chromatograms also showed disappearance of the free peptide peaks after filtration, which indicated efficient removal of free peptide from neoantigen-loaded nanodiscs. Nanodiscs were subsequently incubated with cholesterol-modified CpG1826 (cho-CpG), a TLR9 agonist, by simple mixing at a DMPC:cho-CpG weight ratio of 50:1. GPC analysis confirmed > 99% incorporation of cho-CpG, resulting in nanodiscs co-loaded with neoantigens and CpG (NeoAg-CpG-Nanodisc) (Fig. 1E).
Therapeutic efficacy of nanodisc vaccination combined with immune checkpoint therapy
To identify the optimum neoantigen dose and combination, we next evaluated the immunogenicity and anti-tumor effects of sHDL nanodisc vaccination combined with anti-PD-L1 IgG immune checkpoint therapy. C57BL/6 mice were inoculated with 1.2 × 106 GL261 tumor cells at s.c. flank and vaccinated on days 8 and 15 with NeoAg-1, NeoAg-2, and NeoAg-3 in either soluble or nanodisc forms. A subset of animals also received i.p. administration of 100 μg anti-PD-L1 IgG on days 1 and 4 after each vaccination (Fig. 2A). Soluble peptide vaccination with NeoAgs + CpG induced detectable levels of T-cell responses after the boost immunization as shown by IFN-γ+ ELISPOT assay performed with PBMCs (Fig. 2B). Notably, compared with soluble vaccination with NeoAgs + CpG, NeoAgs-CpG-Nanodisc vaccination significantly improved T-cell responses against all 3 NeoAgs, generating approximately 3-fold (P < 0.01), 6-fold (P < 0.0001), and 4-fold (P < 0.01) higher IFN-γ+ T-cell responses to NeoAg-1, NeoAg-2, and NeoAg-3, respectively (Fig. 2B). ELISPOT assay performed with splenocytes also indicated potent IFN-γ+ T-cell responses against all 3 NeoAgs (Fig. 2C). Addition of anti-PD-L1 IgG therapy to nanodisc vaccination further augmented NeoAg-specific T-cell responses, as shown by the splenocyte ELISPOT assay (Fig. 2C).
Figure 2. Neoantigen-specific T cell responses and anti-tumor efficacy exerted by NeoAgs-CpG-Nanodiscs and anti-PD-L1 therapy in s.c. flank GL261 tumor model.
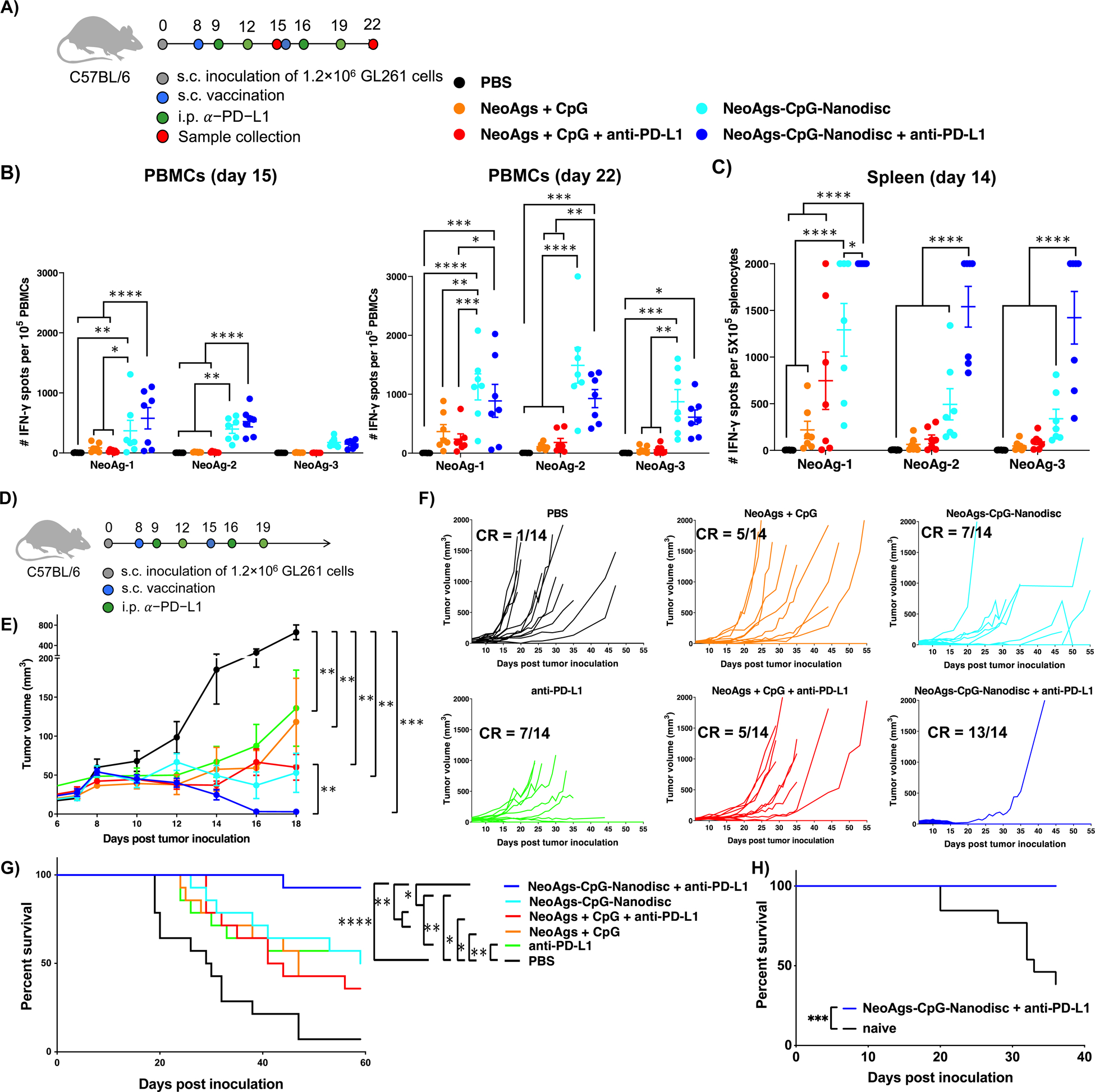
A) Treatment regimen and study timeline (n = 7 per group). B) IFN-γ ELISPOT assays were performed using PBMCs on 7 days after prime (left) and boost (right) vaccinations. C) IFN-γ ELISPOT assays were performed using splenocytes on 6 days after prime vaccination. D) Treatment regimen and study timeline for combination therapy. E) Tumor growth summary for all treatment groups (n = 14 per group). F) Individual tumor growth curves for all animals in the study. G) Kaplan-Meier overall survival curves. H) On day 90, surviving mice from the NeoAgs-CpG-Nanodisc + anti-PD-L1 group were re-challenged with GL261 cells in the contralateral flank.
We next examined the therapeutic efficacy of nanodisc vaccination combined with anti-PD-L1 IgG therapy. C57BL/6 mice bearing GL261 at s.c. flank were treated as above (Fig. 2D). Nanodisc vaccination alone efficiently slowed tumor growth (Fig. 2E) and eliminated tumors in 7 out of 14 animals (Fig. 2F). As T cell exhaustion is widely reported in GL261 tumors (46), we combined nanodisc vaccination with anti-PD-L1 IgG therapy, which led to stronger anti-tumor efficacy (P < 0.01, Fig. 2E) and elimination of established tumors in 13 out of 14 animals (Fig. 2F). On the other hand, soluble neoantigen vaccination with or without anti-PD-L1 IgG therapy had only 5 out of 14 mice with complete response (Fig. 2F). Overall, nanodisc vaccination plus anti-PD-L1 IgG therapy resulted in ~90% animal survival rate (Fig. 2G), representing a significant improvement over all other treatment conditions. When re-challenged with GL261 tumor cells on day 72 after the boost vaccination, all surviving animals from the nanodisc plus anti-PD-L1 IgG group were protected against tumor growth (Fig. 2H), indicating long-term anti-tumor memory response. Taken together, these results demonstrated that nanodisc vaccination combined with ICB therapy exerted potent and durable T-cell responses with robust anti-tumor efficacy against s.c. GL261 tumors.
Immune activation within the tumor microenvironment.
We next performed flow cytometry analyses on s.c. GL261 tumor-bearing mice and examined the impact of combination immunotherapy on the tumor microenvironment. Nanodisc vaccination combined with anti-PD-L1 IgG therapy promoted robust intratumoral infiltration of CD8α+ T cells into s.c. GL261 tumors (Fig. 3A). Intratumoral CD8α+ T cells in the nanodisc + anti-PD-L1 IgG group exhibited 1.5-fold decrease in PD-1 expression (P < 0.01, compared with PBS, Fig. 3B) and 2.5-fold increase expression of degranulation marker CD107α, compared with soluble neoantigen vaccination (P < 0.0001) or PBS control (P < 0.001) (Fig. 3C). Nanodisc + anti-PD-L1 IgG also increased the absolute number of CD3+CD8α+ T cells, PD-1+CD3+CD8α+ T cells, and CD107α+CD3+CD8α+ T cells within the tumor tissues, compared with the PBS control (P < 0.05, Supplementary Fig. 1). We also observed a 6-fold increase in the ratio of CD8α+ T cells to CD4+Foxp3+ regulatory T cells (Tregs) in animals treated with nanodisc + anti-PD-L1 IgG, compared with soluble vaccine + anti-PD-L1 IgG (P < 0.05) or PBS control (P < 0.01) (Fig. 3D). Moreover, intratumoral DCs in animals treated with nanodisc + anti-PD-L1 IgG treatment exhibited an activated phenotype with increased expression of CD86 and CD103 (Fig. 3E–F).
Figure 3. Immune activation within the tumor microenvironment after NeoAgs-CpG-Nanodisc plus anti-PD-L1 therapy.
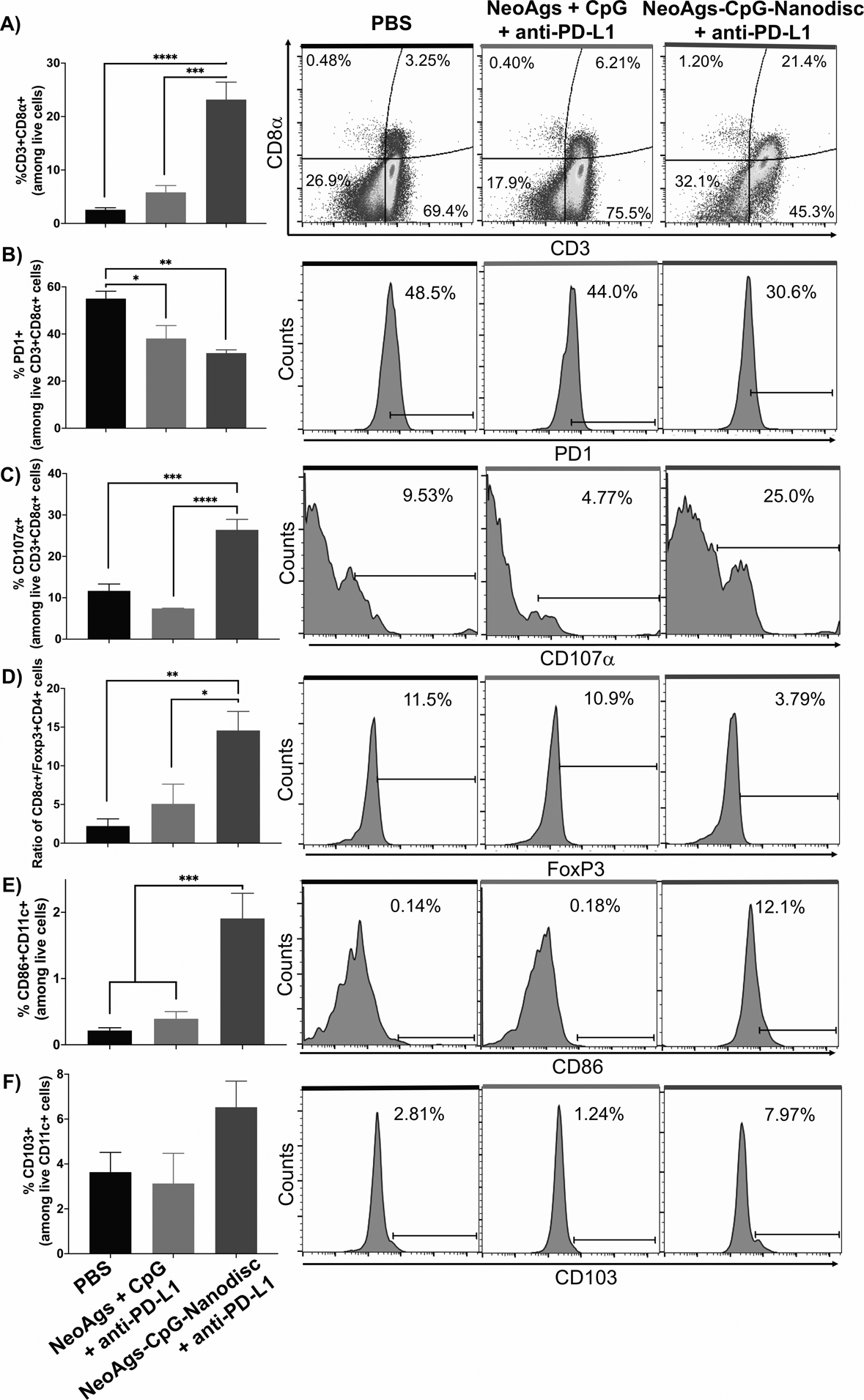
A-E) C57BL/6 mice inoculated s.c. with 1.2 × 106 GL261 tumor cells were vaccinated on day 20 and administered with anti-PD-L1 on days 21 and 24. On day 26, tumor tissues were stained with antibodies and analyzed by flow cytometry for CD8a+ T-cells, Tregs, and DCs (n = 4 per group).
Therapeutic efficacy of nanodisc vaccination in an orthotopic GL261 glioma model
Having shown immunogenicity and potency of nanodisc vaccination in the s.c. flank model, we proceeded to assess the therapeutic efficacy of nanodisc vaccination in an orthotopic glioma model. C57BL/6 mice were inoculated with 20,000 GL261 cells via stereotactic injection into the right striatum on day 0. Animals received 4 weekly immunizations of soluble or nanodisc vaccines at s.c. tail base, starting day 7 post tumor implantation. Animals also received i.p. administration of 100 μg anti-PD-L1 on days 0, 1, and 4 after each vaccination (Fig. 4A).
Figure 4. Anti-tumor efficacy exerted by NeoAgs-CpG-Nanodiscs and anti-PD-L1 therapy in an orthotopic GL261 tumor model.
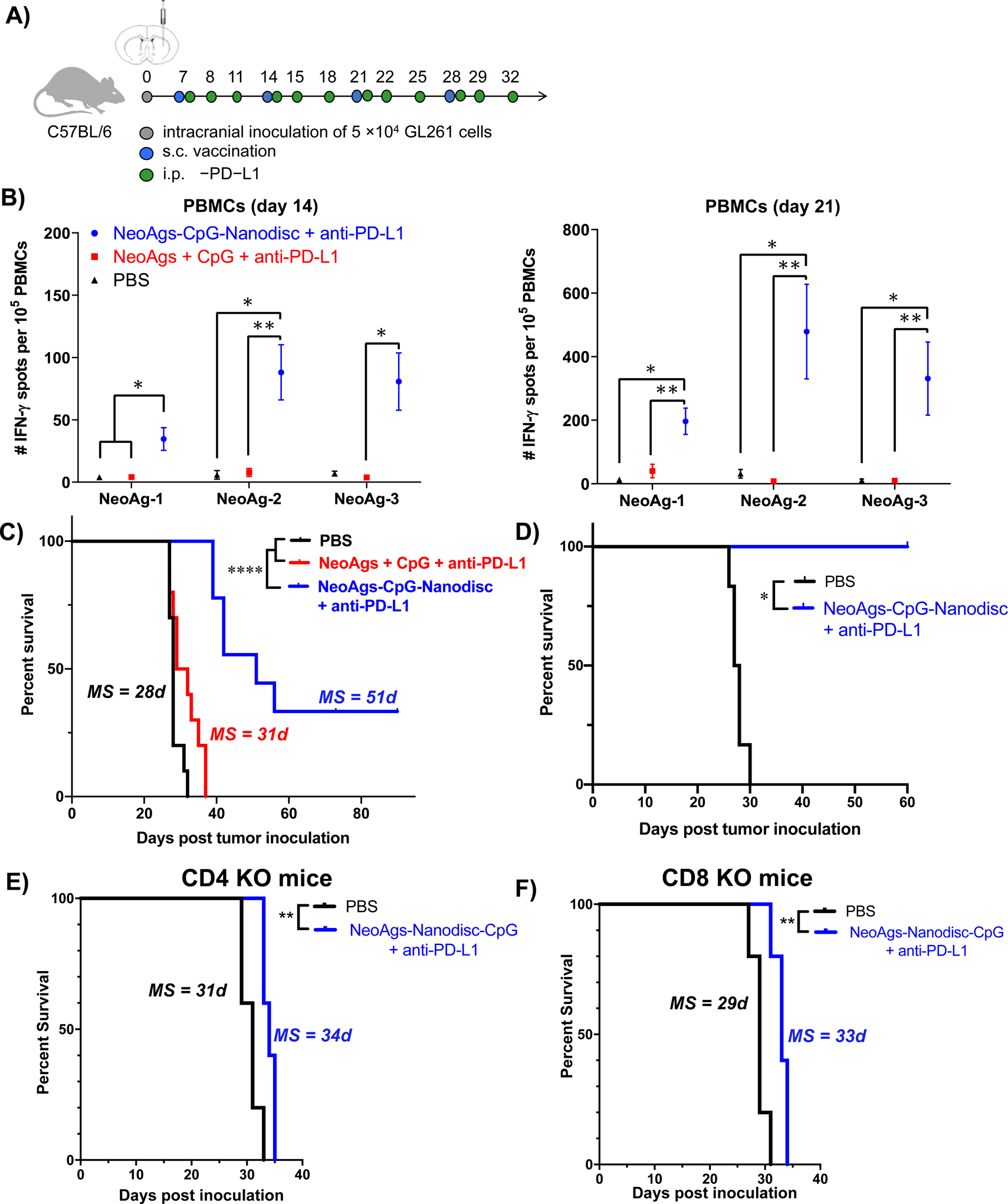
A) Treatment regimen and study timeline. B) IFN-γ ELISPOT assays were performed using PBMCs on day 7 after prime (left) and boost (right) vaccinations (n = 3 per group). C) Kaplan-Meier overall survival curves for all treatment groups (n = 10 per group). D) Kaplan-Meier survival analysis of long-term survivors from (C) re-challenged with GL261 cells in the contralateral hemisphere. E) CD4−/− KO mice or F) CD8−/− KO mice carrying orthotopic GL261 tumors were treated with nanodisc vaccine + anti-PD-L1 therapy as in A) and monitored for survival (n = 5 per group).
ELISPOT assay performed on PBMCs indicated that nanodisc vaccine + anti-PD-L1 therapy elicited potent IFN-γ+ T-cell responses against all 3 NeoAgs (Fig. 4B). A single cycle of nanodisc vaccination and anti-PD-L1 therapy improved IFN-γ+ T-cell responses against NeoAg-1, NeoAg-2, and NeoAg-3 by 7-fold (P < 0.05), 8-fold (P < 0.01), and 15-fold (P < 0.05), compared with soluble vaccine + anti-PD-L1 (Fig. 4B). NeoAg-specific T-cell responses were further augmented after the 2nd cycle of nanodisc vaccine + anti-PD-L1 therapy, as shown by 5-fold, 100-fold, and 30-fold higher IFN-γ+ T-cell responses (NeoAg-1, NeoAg-2, and NeoAg-3, respectively, P < 0.01, Fig. 4B), compared with the soluble vaccine + anti-PD-L1 group.
All animals treated with soluble vaccine + anti-PD-L1 succumbed to tumor growth within 40 days without any statistical difference from the PBS control group (Fig. 4C). In stark contrast, nanodisc vaccine + anti-PD-L1 therapy exerted significantly enhanced anti-tumor efficacy, resulting in complete response in 3 out of 9 mice (33% CR) without any signs of recurrence until day 90 (P < 0.0001, Fig. 4C). To assess for long-term immunity, survivors in the nanodisc vaccine + anti-PD-L1 group were re-challenged on day 90 by stereotactic injection of GL261 cells into the contralateral hemisphere; the animals did not show any signs of neurological deficits during 60 days of observation (Fig. 4D). Moreover, we also tested an abbreviated treatment regimen (3 cycles of vaccination plus 3 administration of anti-PD-L1 therapy). Nanodisc vaccine + anti-PD-L1 group had a slightly reduced complete response rate of ~15%, which still represented a significant improvement over the soluble vaccine + anti-PD-L1 therapy group (P < 0.001, Supplementary Fig. 2). Overall, nanodisc vaccine combined with anti-PD-L1 therapy exerted strong anti-tumor efficacy in a murine model of orthotopic glioma. To evaluate whether the efficacy of this treatment was dependent on the host’s T cells, we inoculated GL261 cells in the brains of CD4−/− or CD8−/− KO mice and then treated the animals with nanodisc vaccine + anti-PD-L1 as above. Nanodisc vaccine + anti-PD-L1 therapy had minimal impact on animal survival (Fig. 4E–F). These results indicate the critical role played by the CD4+ and CD8α+ T cells in mediating a therapeutic response for NeoAgs-CpG-Nanodiscs + anti-PD-L1 therapy.
Intratumoral infiltration of CD8α+ T cells within CNS.
We performed flow cytometric analyses on GL261 glioma tumors isolated from CNS on day 23 from the above experiment. Nanodisc vaccine plus anti-PD-L1 therapy promoted a significant (~3.4 fold) increase in the frequency of intratumoral CD8α+ T cells (P < 0.0001) with ~2-fold lower expression level of PD-1 (P < 0.0001), compared with the PBS control group (Fig. 5A, B, Supplementary Fig. 3A). Nanodisc + anti-PD-L1 therapy also significantly decreased the frequency of CD25+Foxp3+ Tregs (P < 0.001, Fig. 5C, Supplementary Fig. 3B), resulting in 6.7-fold increase in the ratio of CD8α+ T cells to Tregs (P < 0.05, Fig. 5D). We also observed 4-fold higher ratio of M1-like macrophages (CD45+F4/80+CD206-) to M2-like macrophages (CD45+F4/80+CD206+) in the TME of GL261 tumor bearing mice treated with nanodisc + anti-PD-L1 combination immunotherapy (P < 0.001, Fig. 5E). On the other hand, we did not observe difference in the levels of activation markers (CD80, CD86, and MHC-II) on intratumoral DCs in nanodisc vaccine plus anti-PD-L1 versus PBS treatment groups. (Supplementary Fig. 3C).
Figure 5. Tumor microenvironment analysis of GL261 tumors in CNS.

A-I) C57BL/6 mice were inoculated with GL261 cells and treated as in Figure 5, and tumors were isolated on day 23 and analyzed by flow cytometry (n = 5 per group). Shown are A) CD8α+ T cells among all T cells, B) PD-1 receptor expression on CD8α+ T cells, C) CD25+Foxp3+ regulatory T cells among CD4+ T cells, D) ratio of CD8α+ T cells to regulatory T cells, and E) the ratio of M1 (CD206- F4/80+) to M2 (CD206+ F4/80+) macrophages.
Therapeutic efficacy of nanodisc vaccination in an orthotopic mIDH1 glioma model
Having shown immunogenicity and potency of nanodisc vaccination in an orthotopic GL261 glioma model, we proceeded to assess the therapeutic efficacy of nanodisc vaccination in a genetically engineered murine model of mIDH1 glioma (31). C57BL/6 mice were inoculated with 25,000 mIDH1 neurospheres via stereotactic injection into the right striatum on day 0. Animals received 3 weekly immunizations of soluble or nanodisc vaccines at s.c. tail base, starting day 7 post tumor implantation (Fig. 6A). We first tested nanodiscs carrying CpG with either mIDH1123–132 or mIDH1126–141 neoantigens, which are predicted to be good binders for MHC-I in C57Bl/6. Nanodisc vaccination with either mIDH1123–132 or mIDH1126–141 significantly extended the survival with the media survival (MS) of 65 days and 59 days (P < 0.01, Fig. 6B), compared with the MS of 35 days for PBS and 45 days for Nanodisc-CpG. Moreover, when the survivors from both nanodisc groups were re-challenged with mIDH1 neurospheres in the contralateral hemisphere, 100% of the animals resisted tumor recurrence (P < 0.05, Fig. 6C), suggesting nanodisc-mediated immune memory against mIDH1 glioma. However, the combination of nanodiscs delivering both mIDH1123–132 and mIDH1126–141 did not further extend the animal survival (MS = 63 days, Supplementary Fig. 4), potentially due to the overlapping T cell responses against mIDH1123–132 and mIDH1126–141 epitopes.
Figure 6. Survival analysis of mIDH1tumor-bearing mice vaccinated with mIDH1 neoantigens.
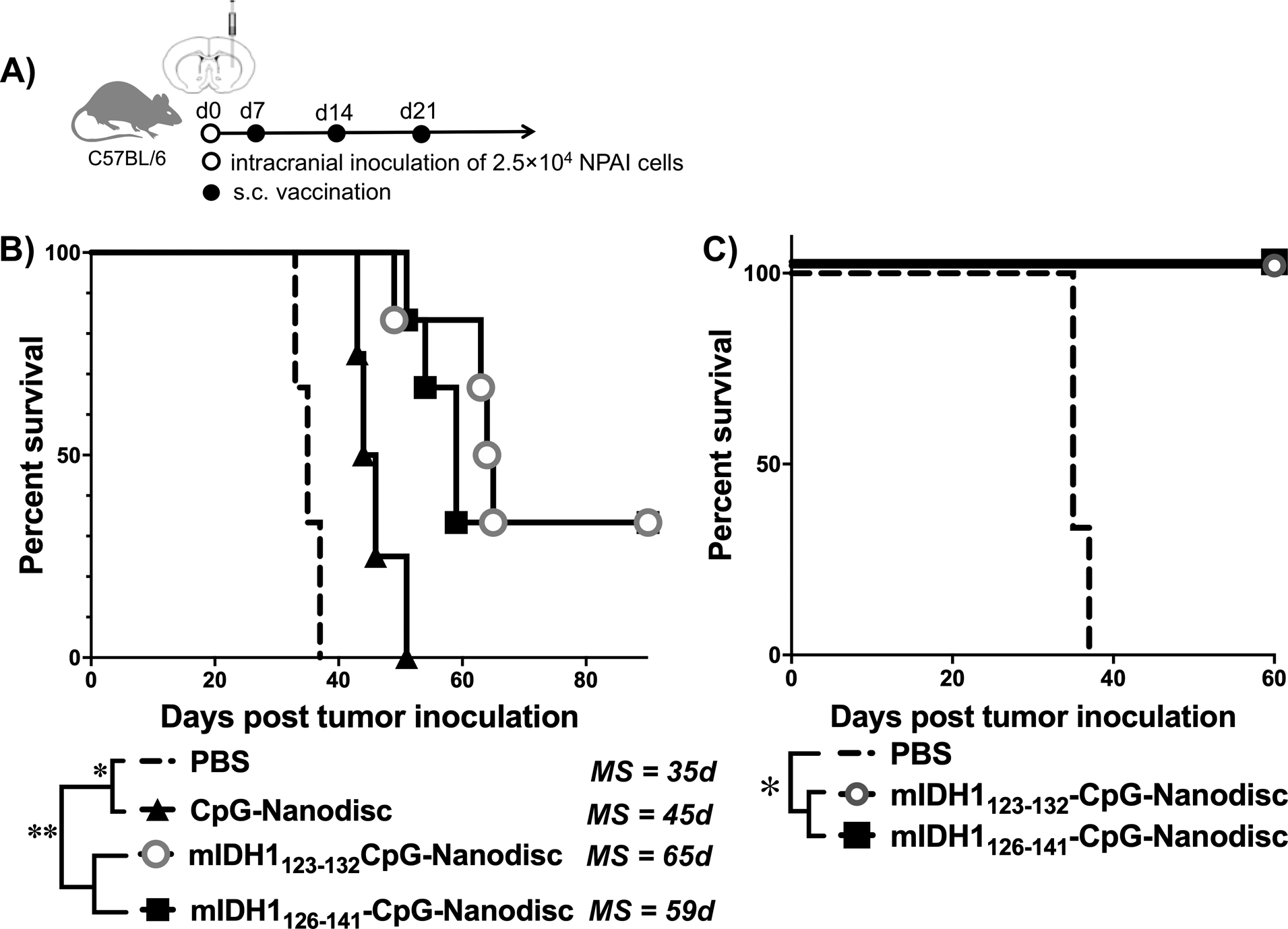
A) Treatment regimen and study timeline. B) Kaplan-Meier overall survival curves for mice treated with the indicated vaccine formulations. C) On day 90, surviving mice were re-challenged with mIDH1 neurospheres in the contralateral hemisphere and monitored for survival. (n = 3 for PBS; n = 4 for CpG-Nanodisc; n = 6 for mIDH1-CpG-Nanodiscs).
DISCUSSION
In this work, we have successfully developed a nanodisc platform for neoantigen-based personalized vaccination against gliomas. We have demonstrated that (1) neoantigens with different physicochemical properties can be loaded onto sHDL nanodiscs with CpG (26); (2) neoantigen-loaded sHDL nanodiscs in combination with anti-PD-L1 immune checkpoint blockade can elicit significantly greater systemic neoantigen-specific CD8α+ T cell expansion when compared with soluble neoantigen peptides in both flank and orthotopic GL261 tumor models; and (3) neoantigen-loaded sHDL nanodiscs when used in combination with anti-PD-L1 immune checkpoint blockade drive intratumoral infiltration of neoantigen-specific CD8α+ T cells and exert potent anti-tumor effects in an orthotopic model of glioma.
In particular, our initial screening of C57BL/6-derived GL261 neoantigen candidates narrowed our choices of neoantigens from the previously identified pool (32) to three unique peptides based on their significant immunogenicity in GL261 tumor-bearing mice. During optimization of the vaccine formulations, we found that all three neoantigen-loaded nanodisc formulations could be combined into one cocktail without precipitation (Fig. 1), which allowed us to test the anti-tumor potential of these neoantigens using a single formulation. In both flank and orthotopic glioma tumor models, we found that administration of a prime-boost NeoAgs-CpG-Nanodisc vaccination induced robust systemic expansion of IFN-γ producing, neoantigen-specific CD8α+ T cells (Fig. 2B, 4B). We have previously shown that the nanodisc platform allowed for efficient co-delivery of neoantigens and CpG to DCs in draining LNs in various models of s.c. flank tumors (26,47,48); thus, based on these, we hypothesized that this general phenomenon would also induce CTL responses in orthotopic tumors located in the brain. Herein, we report that sHDL nanodisc platform provides a powerful and convenient strategy to generate neoantigen-specific T-cell responses against glioma. While nanodisc vaccine alone was not effective at extending the survival of GL261 tumor-bearing mice, potentially due to T cell exhaustion (46), nanodisc vaccination in combination with anti-PD-L1 immune checkpoint blockade produced a complete response rate of 93% in mice bearing s.c. flank GL261 glioma tumor (Fig. 2D). Nanodisc combined with anti-PD-L1 induced the maturation of intratumorally DCs, followed by intratumoral infiltration of CD8α+ T cells with CD107α effector phenotype into the TME (Fig. 4). This led to tumor regression by promoting neoantigen presentation by DCs to CD8α+ T cells and subsequent CD8α+ T cell-dependent tumor cell killing. NeoAgs-CpG-Nanodiscs also promoted a significant increase in the ratio of cytotoxic (CD8α+) T cells to Tregs (Foxp3+CD4+) (Fig. 4). These results indicated vaccine-induced shifts in the balance of effector T cells in the TME, leading to improved survival outcomes and protective immunity against tumor relapse.
Extending our findings from the s.c. flank model, we have also demonstrated the therapeutic efficacy of NeoAgs-CpG-Nanodisc + anti-PD-L1 treatment in a murine model of GL261 orthotopic glioma. IFN-γ ELISPOT analysis of PBMCs revealed significantly higher frequencies of neoantigen-specific CD8α+ T cells across all three neoantigens in mice treated with nanodisc vaccination, compared with soluble neoantigen peptides (Fig. 4B). This increased frequency of circulating neoantigen-specific CD8α+ T cells in turn led to their robust infiltration into CNS with glioma tumors (Fig. 5A). Nanodisc vaccine combined with anti-PD-L1 therapy also significantly decreased the frequencies of immunosuppressive Tregs, TAMs, and PD1+ exhausted T cells in the TME (Fig. 5). Additionally, when the long-term survivors from the Nanodisc + anti-PD-L1 group were re-challenged with GL261 tumors in the contralateral hemisphere, they remained tumor-free without further treatment (Fig. 4D), suggesting establishment of immunological memory. Moreover, we have also demonstrated the therapeutic efficacy of nanodisc vaccination in a second genetically engineered mouse glioma model. Using a genetically engineered murine mIDH1 glioma model (31), we have shown that nanodisc vaccination against mIDH1123–132 or mIDH1126–141 significantly extended animal survival and established long-term immunity against mIDH1 tumors (Fig. 6). To the best of our knowledge, our work is the first to show a personalized neoantigen vaccine platform that can elicit effective anti-tumor immunity and promote lasting immunological memory to prevent tumor recurrence in murine orthotopic models of glioma.
Despite these exciting results, there are still challenges to overcome before clinical translation of personalized nanodisc vaccination against gliomas. First, more in-depth cytokine and chemokine analyses should be conducted to elucidate the mechanisms by which immunosuppression was reversed and immune memory was generated to improve the overall survival after treatment with NeoAgs-CpG-Nanodisc + anti-PD-L1. Second, we have only tested only one immune checkpoint blockade antibody in our study. Combinations of different or multiple immune checkpoint blockade antibodies with the nano-vaccine should be tested to determine how multi-pronged approach could be optimized to treat tumors as aggressive as glioma. Clinical trials studying anti-PD-1 and anti-CTLA4 therapies in patients with both primary and recurring glioma are ongoing but have no conclusive results yet. Due to the possibility that certain tumors might be unresponsive to immune checkpoint blockade, future work on our combinatorial approach to immunotherapy should include investigation of the mechanisms of resistance to immune checkpoint blockade and elucidation of immune cell activation, which will facilitate identification of ideal therapeutics and their treatment regimens. Third, as genomic profiling of primary and recurrent gliomas has shown that recurrent tumors possess significantly more mutations (49), it would be interesting to evaluate our neoantigen-based approach for combination immunotherapy in the setting of recurrent gliomas. Lastly, the current rate-limiting step of the nano-vaccine formulation is neoantigen peptide identification and synthesis. It is estimated that 6–8 weeks would be required for neoantigen identification and production of GMP-grade peptides. We are currently streamlining the sHDL formulation process so that antigen loading and characterization can be performed in one week after completion of peptide synthesis. Overall, the sHDL nanodisc provides a promising and versatile platform for minimally invasive (s.c.) delivery of neoantigens and adjuvant molecules. As the sHDL formulation process has been proven scalable and safe in prior phase I trials (34), we anticipate that our strategy outlined here will provide a general roadmap for personalized vaccination for immunotherapy against glioma and other cancer types.
Supplementary Material
Translational Relevance:
Only a subset of patients currently benefits from immune checkpoint inhibitors, thus highlighting an urgent need to improve cancer immunotherapy. Combination immunotherapies, including cancer vaccines, could boost T-cell immunity, but the efficacy of cancer vaccines has been limited, especially for gliomas. Here, we present a new strategy for personalized cancer vaccination against gliomas. Briefly, we have developed synthetic high-density lipoprotein (sHDL) loaded with CpG, a Toll-like receptor 9 agonist, and tumor-specific neoantigens to target gliomas and elicit immune-mediated tumor regression. We report that sHDL vaccination in combination with anti-PD-L1 immunotherapy elicits potent neoantigen-specific T-cell responses and leads to tumor regression, long term survival, and immunological memory. Our strategy provides a general roadmap for personalized vaccination for immunotherapy against gliomas and other cancer types.
Synopsis:
We have developed immunotherapy delivery vehicles based on synthetic high-density lipoprotein loaded with CpG, a Toll-like receptor 9 agonist, and tumor-specific neoantigens to target gliomas and elicit immune-mediated tumor regression.
Acknowledgements
This work was supported in part by NIH (R01EB022563, R01CA210273, R01CA223804, R01AI127070, R21NS091555, R01HL134569, U01CA210152, R37-NS094804, R01-NS105556, R01-NS076991 and 1R21NS107894). J.J.M. is supported by DoD/CDMRP Peer Reviewed Cancer Research Program (W81XWH-16-1-0369) and NSF CAREER Award (1553831). L.S. acknowledges financial support from the UM Pharmacological Sciences Training Program (PSTP) (GM007767 from NIGMS). MGC is supported by the Rogel Cancer Centre Research Scholar Award. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
Footnotes
Conflict of Interest:
A patent application for the nanodisc vaccines has been filed, with J.J.M. and A.S. as inventors, and J.J.M. and A.S. as co-founders of EVOQ Therapeutics, LLC., which develops the technology.
References
- 1.Hanif F, Muzaffar K, Perveen K, Malhi SM, Simjee Sh U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac J Cancer Prev 2017;18:3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fan C-H, Chang E-L, Ting C-Y, Lin Y-C, Liao E-C, Huang C-Y, et al. Folate-conjugated gene-carrying microbubbles with focused ultrasound for concurrent blood-brain barrier opening and local gene delivery. Biomaterials 2016;106:46–57 [DOI] [PubMed] [Google Scholar]
- 3.Chen P-Y, Hsieh H-Y, Huang C-Y, Lin C-Y, Wei K-C, Liu H-L. Focused ultrasound-induced blood–brain barrier opening to enhance interleukin-12 delivery for brain tumor immunotherapy: a preclinical feasibility study. Journal of translational medicine 2015;13:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weigel BJ, Rodeberg DA, Krieg AM, Blazar BR. CpG oligodeoxynucleotides potentiate the anti-tumor effects of chemotherapy or tumor resection in an orthotopic murine model of rhabdomyosarcoma. Clinical Cancer Research 2003;9:3105–14 [PubMed] [Google Scholar]
- 5.Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, et al. Phase Ib Trial of Mutant Herpes Simplex Virus G207 Inoculated Pre-and Post-tumor Resection for Recurrent GBM. Mol Ther 2008;17:199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ali S, King GD, Curtin JF, Candolfi M, Xiong W, Liu C, et al. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer Res 2005;65:7194–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curtin JF, Liu N, Candolfi M, Xiong W, Assi H, Yagiz K, et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS medicine 2009;6:e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamran N, Kadiyala P, Saxena M, Candolfi M, Li Y, Moreno-Ayala MA, et al. Immunosuppressive Myeloid Cells’ Blockade in the Glioma Microenvironment Enhances the Efficacy of Immune-Stimulatory Gene Therapy. Mol Ther 2017;25:232–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer cell 2015;27:450–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer 2012;12:252–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang J, Liu F, Liu Z, Tang H, Wu H, Gong Q, et al. Immune Checkpoint in Glioblastoma: Promising and Challenging. Frontiers in pharmacology 2017;8:242- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.BMS. Bristol-Myers Squibb Announces Phase 3 CheckMate −498 Study Did Not Meet Primary Endpoint of Overall Survival with Opdivo (nivolumab) Plus Radiation in Patients with Newly Diagnosed MGMT-Unmethylated Glioblastoma Multiforme. news.bms.com2019. [Google Scholar]
- 13.Filley AC, Henriquez M, Dey M. Recurrent glioma clinical trial, CheckMate-143: the game is not over yet. Oncotarget 2017;8:91779–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348:69–74 [DOI] [PubMed] [Google Scholar]
- 15.Ott PA, Hu ZT, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017;547:217–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019;565:234–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanovic S, Gouttefangeas C, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019;565:240–5 [DOI] [PubMed] [Google Scholar]
- 18.Redmond WL, Sherman LA. Peripheral tolerance of CD8 T lymphocytes. Immunity 2005;22:275–84 [DOI] [PubMed] [Google Scholar]
- 19.Melief CJ, van der Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nature reviews Cancer 2008;8:351–60 [DOI] [PubMed] [Google Scholar]
- 20.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, et al. Persistent antigen at vaccination sites induces tumor-specific CD8(+) T cell sequestration, dysfunction and deletion. Nat Med 2013;19:465–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheetz L, Park KS, Li Q, Lowenstein PR, Castro MG, Schwendeman A, et al. Engineering patient-specific cancer immunotherapies. Nat Biomed Eng 2019;3:768–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Irvine DJ, Hanson MC, Rakhra K, Tokatlian T. Synthetic Nanoparticles for Vaccines and Immunotherapy. Chemical reviews 2015;115:11109–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nature biotechnology 2007;25:1159–64 [DOI] [PubMed] [Google Scholar]
- 24.Zhu G, Mei L, Vishwasrao HD, Jacobson O, Wang Z, Liu Y, et al. Intertwining DNA-RNA nanocapsules loaded with tumor neoantigens as synergistic nanovaccines for cancer immunotherapy. Nature communications 2017;8:1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nam J, Son S, Park KS, Zou W, Shea LD, Moon JJ. Cancer nanomedicine for combination cancer immunotherapy. Nature Reviews Materials 2019;4:398–414 [Google Scholar]
- 26.Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nature materials 2017;16:489–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ueda R, Fujita M, Zhu X, Sasaki K, Kastenhuber ER, Kohanbash G, et al. Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin Cancer Res 2009;15:6551–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xue S, Song G, Yu J. The prognostic significance of PD-L1 expression in patients with glioma: a meta-analysis. Scientific reports 2017;7:4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer immunology research 2016;4:124–35 [DOI] [PubMed] [Google Scholar]
- 30.Reardon DA, Kaley TJ, Dietrich J, Clarke JL, Dunn G, Lim M, et al. Phase II study to evaluate safety and efficacy of MEDI4736 (durvalumab) + radiotherapy in patients with newly diagnosed unmethylated MGMT glioblastoma (new unmeth GBM). Journal of Clinical Oncology 2019;37:2032 [Google Scholar]
- 31.Nunez FJ, Mendez FM, Kadiyala P, Alghamri MS, Savelieff MG, Garcia-Fabiani MB, et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic upregulation of the DNA damage response. Sci Transl Med 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johanns TM, Ward JP, Miller CA, Wilson C, Kobayashi DK, Bender D, et al. Endogenous neoantigen-specific CD8 T cells identified in two glioblastoma models using a cancer immunogenomics approach. Cancer immunology research 2016;4:1007–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pellegatta S, Valletta L, Corbetta C, Patane M, Zucca I, Riccardi Sirtori F, et al. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol Commun 2015;3:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuai R, Li D, Chen YE, Moon JJ, Schwendeman A. High-Density Lipoproteins: Nature’s Multifunctional Nanoparticles. ACS nano 2016;10:3015–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geiger JD, Hutchinson RJ, Hohenkirk LF, McKenna EA, Yanik GA, Levine JE, et al. Vaccination Of Pediatric Solid Tumor Patients with Tumor Lysate-pulsed Dendritic Cells Can Expand Specific T Cells and Mediate Tumor Regression. Cancer Research 2001;61:8513–9 [PubMed] [Google Scholar]
- 36.Candolfi M, Curtin JF, Nichols WS, Muhammad AG, King GD, Pluhar GE, et al. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. Journal of Neuro-Oncology 2007;85:133–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Candolfi M, Xiong W, Yagiz K, Liu C, Muhammad AK, Puntel M, et al. Gene therapy-mediated delivery of targeted cytotoxins for glioma therapeutics. Proc Natl Acad Sci U S A 2010;107:20021–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Candolfi M, Yagiz K, Wibowo M, Ahlzadeh GE, Puntel M, Ghiasi H, et al. Temozolomide does not impair gene therapy-mediated anti-tumor immunity in syngeneic brain tumor models. Clin Cancer Res 2014;20:1555–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kadiyala P, Li D, Nunez FM, Altshuler D, Doherty R, Kuai R, et al. High-Density Lipoprotein-Mimicking Nanodiscs for Chemo-immunotherapy against Glioblastoma Multiforme. ACS nano 2019;13:1365–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang J, Sanderson NS, Wawrowsky K, Puntel M, Castro MG, Lowenstein PR. Kupfer-type immunological synapse characteristics do not predict anti-brain tumor cytolytic T-cell function in vivo. Proc Natl Acad Sci U S A 2010;107:4716–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engelhard VH. Structure of peptides associated with MHC class I molecules. Current opinion in immunology 1994;6:13–23 [DOI] [PubMed] [Google Scholar]
- 42.Rötzschke O, Falk K, Mack J, Lau J, Jung G, Strominger J. Conformational variants of class II MHC/peptide complexes induced by N-and C-terminal extensions of minimal peptide epitopes. Proceedings of the National Academy of Sciences 1999;96:7445–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho HI, Barrios K, Lee YR, Linowski AK, Celis E. BiVax: a peptide/poly-IC subunit vaccine that mimics an acute infection elicits vast and effective anti-tumor CD8 T-cell responses. Cancer Immunol Immunother 2013;62:787–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buhtoiarov IN, Sondel PM, Wigginton JM, Buhtoiarova TN, Yanke EM, Mahvi DA, et al. Anti‐tumour synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN immunotherapy through repolarization of tumour-associated macrophages. Immunology 2011;132:226–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hashem AM, Gravel C, Chen Z, Yi Y, Tocchi M, Jaentschke B, et al. CD40 Ligand Preferentially Modulates Immune Response and Enhances Protection against Influenza Virus. The Journal of Immunology 2014;193:722–34 [DOI] [PubMed] [Google Scholar]
- 46.Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin Cancer Res 2017;23:124–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuai R, Sun X, Yuan W, Xu Y, Schwendeman A, Moon JJ. Subcutaneous Nanodisc Vaccination with Neoantigens for Combination Cancer Immunotherapy. Bioconjug Chem 2018;29:771–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuai R, Sun X, Yuan W, Ochyl LJ, Xu Y, Hassani Najafabadi A, et al. Dual TLR agonist nanodiscs as a strong adjuvant system for vaccines and immunotherapy. J Control Release 2018;282:131–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang L, Liu Z, Li J, Huang T, Wang Y, Chang L, et al. Genomic analysis of primary and recurrent gliomas reveals clinical outcome related molecular features. Scientific reports 2019;9:16058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.