

Abstract
High salt intake (HS) is associated with obesity and insulin resistance. ET-1, a peptide released in response to HS, inhibits the actions of insulin on cultured adipocytes through ET-1 type B (ETB) receptors; however, the in vivo implications of ETB receptor activation on lipid metabolism and insulin resistance is unknown. We hypothesized that activation of ETB receptors in response to HS intake promotes dyslipidemia and insulin resistance. In normal salt (NS) fed rats, no significant difference in body weight or epidydimal fat mass was observed between control and ETB deficient rats. After 2 weeks of HS, ETB def rats had significantly lower body weight and epidydimal fat mass compared to controls. Non-fasting plasma glucose was not different between genotypes, however plasma insulin concentration was significantly lower in ETB deficient rats compared to controls suggesting improved insulin sensitivity. In addition, ETB deficient rats had significantly higher circulating free fatty acids in both NS and HS groups, with no difference in plasma triglycerides between genotypes. In a separate experiment, ETB deficient rats had significantly lower fasting blood glucose and improved glucose and insulin tolerance compared to controls. These data suggest that ET-1 promotes adipose deposition and insulin resistance via the ETB receptor.
Introduction
Global dietary sodium intake as of 2010 has increased to ~3.95 g/day, nearly twice the recommended limit of 2 g/day established by the World Health Organization (Powles et al. 2013). Excess sodium intake raises blood pressure (BP) (Sacks et al. 2001), a major risk factor for cardiovascular disease (Aburto et al. 2013; Lim et al. 2012), stomach cancer (Ferlay et al. 2010), and renal disease. More recently, high dietary sodium has been associated with obesity and insulin resistance, a precursor for type 2 diabetes mellitus, independent of caloric intake (Grimes et al. 2016; Kang et al. 2016). Notably, mechanisms by which high salt (HS) intake may promote obesity and insulin resistance are severely under-studied.
Factors associated with sodium homeostasis contribute to lipid metabolism and overall metabolic balance. Endothelin-1 (ET-1), a 21-amino acid peptide, is upregulated during chronic HS intake (Speed et al. 2015; Tsai et al. 2006). In response to chronic HS intake, ET-1 is thought to maintain vascular tone through vasoconstrictive and vasodilatory actions. This is mediated through endothelin type A and type B receptors (ETA/ETB) on vascular smooth muscle cells and endothelial cells respectively (Ogawa et al. 1991; Rubanyi and Polokoff 1994). In addition, renal ET-1 promotes natriuresis by inhibiting Na+ transport via the ETB receptor. Although ET-1 is secreted mainly by endothelial cells (Kisanuki et al. 2010), it is also produced in a variety of cells, including vascular smooth muscle cells, cardiomyocytes, macrophages, leukocytes, fibroblasts, and adipocytes (Juan et al. 2007; Rubanyi and Polokoff 1994). ET-1 acts through two receptors, ETA and ETB , which are expressed throughout most organs and tissues within the body, including thoses associated with lipid metabolism and inuslin signling, therefore the potential that ET-1 contributes to metabolic function are high.
The endothelin system has recently been implicated in lipid metabolism. High levels of circulating ET-1 have been found in both obese and diabetic individuals (Ferri et al. 1997; Schneider et al. 2002). In vitro studies determined that ET-1 acting on the ETA receptor causes lipolysis in differentiated adipocyte-like 3T3-L1 cells (Eriksson et al. 2009). In addition, ET-1 has been shown to inhibit insulin-stimulated glucose uptake in cultured adipocytes, mediated via the ETB receptor (Chou et al. 1994; Juan et al. 2007; Ottosson-Seeberger et al. 1997). Therefore, we hypothesized that activation of ETB receptors in response to HS intake promotes dyslipidemia and insulin resistance, and the goal of the current study was to determine if loss of ETB receptor function alters metabolic parameters, including body weight (BW), adiposity, plasma insulin, triglycerides, and glucose.
Materials and Methods
The Institutional Animal Care and Use Committee at the University of Alabama at Birmingham approved all protocols in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Male endothelin B deficient (ETB def) or transgenic control (control) littermates (11-12 weeks of age) were obtained from our in-house colony. This colony was produced from the spotting lethal rat, which has a naturally occurring mutation in the ETB receptor gene rendering a non-functional protein. These animals did not survive long after birth due to abnormal formation of the enteric nervous system that lead to megacolon; however, Gariepy et al were able to “rescue” the model by inserting a human ETB receptor transgene under the control of the dopamine β hydroxylase promoter. Therefore, expression of functional ETB receptors is only present in sympathetic tissue (Gariepy et al. 2000). ETB def rats have a 10-fold elevation in plasma ET-1 and a reduced ETA receptor binding, at least in the kidney (Taylor et al. 2003). Animals were housed in temperature and humidity controlled rooms with 12:12 hour light:dark cycles and were allowed food and water ad libitum.
Protocol 1.
11-12 week old animals were placed on sodium adjusted Envigo diet containing either normal salt (NS; 0.4% NaCl) or high salt intake (HS; 4% NaCl) for 14 days. On the final day, animals were euthanized in 4-hour increments beginning at the time in which the lights turned on (ZT0). Plasma was collected in heparinized tubes and samples were collected and flash frozen in liquid nitrogen. In a subset of animals epidydimal fat pads were excised and weighed to get an estimate of adiposity as a percentage of body weight. Plasma free fatty acids, glucose, and triglycerides were measured by the Metabolic Core at UAB.
Protocol 2.
12-week old ETB def and control littermates that were maintained on normal chow from the animal facilities at UAB were used. Fasted animals were either subjected to an intraperitoneal insulin tolerance test (IPTT) in which 0.5 IU/kg insulin was injected IP at time 0 or an IP glucose tolerance test in which 2 mg/kg dextrose (50% solution) was injected IP at time 0. Blood glucose was measured by a glucometer (Abbott Laboratories) through a drop of blood from a tail prick.
Statistics.
For comparisons of two groups (i.e. fasting blood glucose), data were analyzed by Students t-test. For two variable data (i.e. genotype and diet), data were analyzed by two-way analysis of variance followed by Tukey’s post hoc test. All data were analyzed using GraphPad Prism version 8. Statistical significance was set at α=0.05.
Results.
14-week old ETB def rats maintained on NS had similar body weight compared to littermate controls (Figure 1A). When placed on HS diet for 2 weeks, ETB def rats had significantly lower body weight than littermate controls (Figure 1A, 298 ± 6 vs. 253 ± 4 grams, control vs. ETB def; p<0.0001 by post hoc analysis). In control animals maintained on NS, there was no detectable difference in epidydimal fat mass relative to body weight (Figure 1B, 1.4 ± 0.06 vs. 1.5 ± 0.06 % of body weight, control vs. ETB def, p=not significant). However, ETB def rats fed HS for 2 weeks had significantly lower epidydimal fat mass compared to ETB def rats maintained on NS (Figure 1B, 1.5 ± 0.05 vs. 1.2± 0.06 % of body weight, control vs. ETB def; p<0.05). These data suggest that high salt intake reduces lipid storage or increases lipid utilization in rats lacking ETB receptors.
Figure 1:
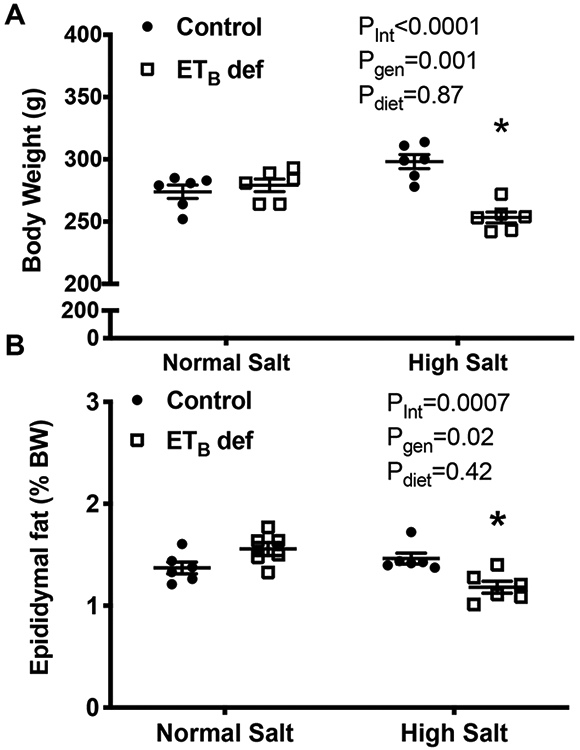
Chronic high salt (HS) feeding reduces adiposity in endothelin B deficient (ETB def) rats. A) Body weight and B) epidydimal fat weight to body weight ratio in transgenic control and ETB def rats fed either a normal salt (NS) or high salt (HS) diet for two weeks. N=6/group; *p<0.05 vs. High Salt ETB def. ANOVA table Key: interaction (int), genotype (gen)
To determine the effects of ETB receptor deficiency on glucose metabolism, plasma glucose and insulin were measured in 4-hour intervals (Figure 2). There were no detectable differences in plasma glucose between control and ETB def rats whether maintained on NS or HS. As expected, NS fed animals had a circadian rhythm in plasma glucose with higher concentration occurring during the active phase at ZT20 (Figure 2A). Interestingly, there was no effect of time on plasma glucose in HS fed animals (Figure 2D). There was no significant effect of time on plasma insulin in NS fed control or ETB def rats (Figure 2B; p=0.08). In contrast there was a significant effect of time on plasma insulin concentration in HS fed animals. In addition, HS fed ETB rats had significantly lower plasma insulin concentration compared to control (Figure 2E). When data was averaged from all time points, no significant difference was found in plasma glucose (Figure 2C); however, plasma insulin was significantly lower in ETB def rats compared to control whether on NS or HS diet (Figure 2F). Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) was significantly lower in ETB def rats and HS fed animals (Figure 2G). These data suggest that loss of ETB function improves control of glucose.
Figure 2:
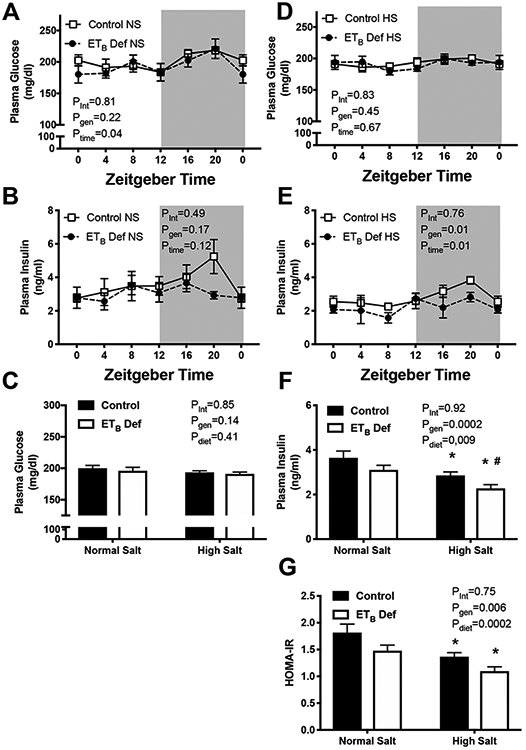
Chronic high salt (HS) intake reduces circulating insulin in endothelin B deficient (ETB def) rats. Transgenic control and ETB def rats were fed either normal salt (NS) or HS diet for 2 weeks, and tissues were collected in non-fasted animals under anesthesia in 4 hour intervals. A & D) plasma glucose (mg/dL; n=5-7/group), B & E) circulating insulin (ng/ml; n=5-7/group), C) average plasma glucose of all time points (n=36-40/group), and F) average plasma insulin concentration of all time points (n=36-40/group) and and G) HOMA-IR caculated from non-fasting insulin and glucose. *p<0.05 vs. NS control; # p<0.05 vs NS ETB def. ANOVA table Key: interaction (int), genotype (gen), time of day (time)
We next sought to determine if ETB def rats had alterations in plasma lipids. There were no detectable differences in plasma triglycerides between control or ETB def rats at any time point of NS fed animals (Figure 3A). There was a significant reduction in triglycerides at ZT12 (beginning of active phase) compared to other time points (Figure 3A). Interestingly, the only significant difference between genotypes in plasma triglyceride concentration of HS fed animals was at ZT12, where the dip in triglyceride concentration was attenuated in control rats but not ETB def rats (Figure 3B; 93.8 ± 10.4 vs. 64.4± 6.2 mg/dl respectively). Average of all time points indicated that HS feeding significantly reduced plasma triglyceride concentration in both genotypes compared to NS fed animals (Figure 3C). Next, we determined the effect of ETB deficiency on circulating free fatty acids. There were no detectable contribution of time on plasma free fatty acids (Figures 3D and 3E); however ETB def rats, whether fed NS or HS, had a small increase in free fatty acid concentration, although not significant (Figure 3F; p=0.08).
Figure 3:
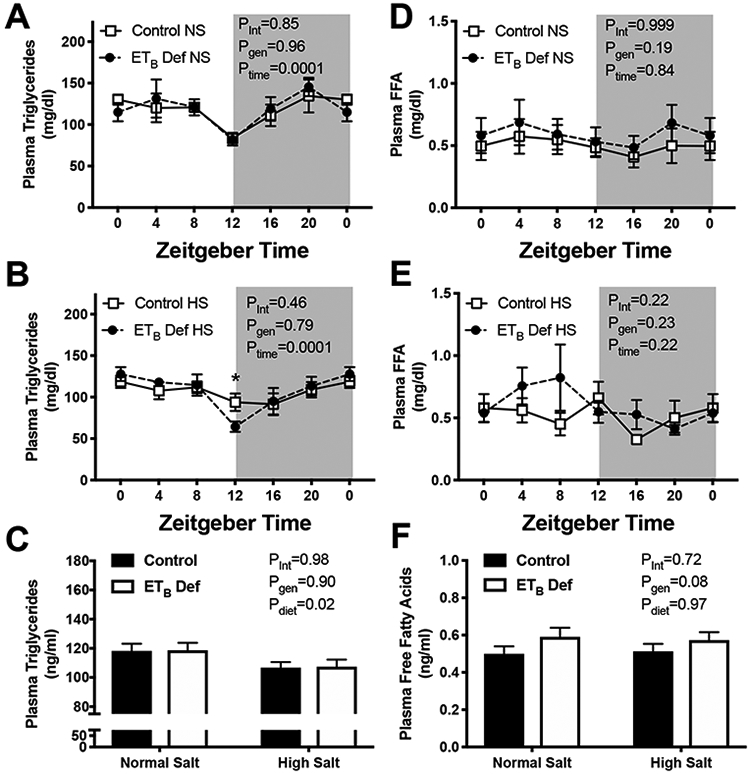
Endothelin B deficient (ETB def) rats have increased circulating free fatty acids. Transgenic control and ETB def rats were fed either normal salt (NS) or high salt (HS) diet for 2 weeks, and tissues were collected in non-fasted animals under anesthesia in 4 hour intervals. A & B) plasma triglycerides (mg/dL; n=5-7/group), D & E) plasma free fatty acids (mg/dl; n=5-7/group), C) average plasma triglycerides of all time points (n=36-40/group), F) average plasma free fatty acid concentration of all time points (n=36-40/group). ANOVA table Key: interaction (int), genotype (gen), time of day (time)
The previous experiment suggests that ETB deficient rats have an improvement in insulin sensitivity and glucose control; however, samples were collected under non fasting conditions and under anesthesia, thereby artificially inflating plasma glucose concentrations. Therefore, we next wanted to determine if ETB deficiency improves glucose control using more direct measures of insulin sensitivity including glucose and insulin tolerance tests. Our results indicate that ETB def rats have significantly lower fasting blood glucose (89.3±2.5 vs. 105.9±3.7 mg/dL respectively). In addition, ETB def rats had improved glucose tolerance (33418±1891 vs 24715±3161 AUC; control vs. ETB def rats) and insulin tolerance (3849±769 vs. 4669±976 AUC; control vs. ETB def rats). These data indicate that even under normal salt feeding, ETB deficiency improves insulin sensitivity.
Discussion
HS stimulation of ET-1 plays a major role in maintaining blood pressure by promoting the excretion of Na+ by the kidney and extracellular Na+; however, less is known about the contribution of ET-1/ETB signaling to lipid metabolism and insulin signaling . The current study indicates that loss of ETB receptor function improves insulin signaling and in HS fed animals, reduces adiposity. In rats lacking functional ETB receptors, two weeks of HS intake significantly reduced body weight and epidydimal fat mass compared to control rats. In addition, ETB def rats have lower circulating insulin with no difference in plasma glucose (non-fasting) resulting in a lower HOMA-IR, suggesting that improved insulin sensitivity in ETB def rats. Interestingly, ETB def rats maintained on NS diet have lower fasting blood glucose and improved glucose tolerance independent of body weight differences. These data suggest that ETB receptor activation may play a role in pathophysiology associated with obesity such as adiposity and insulin resistance.
HS intake is associated with dyslipidemia and insulin resistance in human populations independent of caloric intake;(Kang et al. 2016) however, this is not the case in rodent models, including the current study in which HS fed animals had lower circulating triglycerides and improved HOMA-IR compared to NS fed rats. Even more puzzling is that HS feeding had no effect on circulating free fatty acids, yet lowered circulating triglycerides. The reduction in triglycerides may be explained by a reduction in absorption of fats from the gut. It has been shown that HS feeding reduces the uptake of fatty acids by the intestine (Weidemann et al. 2015). One would expect compensation from the adipose tissue and liver to maintain free fatty acids for energy, possibly at the expense of lower triglycerides.
Several lines of evidence suggest that ETB receptor activation may promote insulin resistance and adiposity. A number of groups have shown a positive correlation between plasma ET-1 levels, obesity and insulin resistance (Cardillo et al. 2004; Ferri et al. 1995; Weil et al. 2011). In addition, visceral adipose from obese individuals produced 2.5 fold more ET-1 than adipose from lean counterparts. The same study showed that ET-1, via the ETB receptor, blocks insulin mediated anti-lipolysis. This appears to be related to activation of phosphokinase C (PKC) activity (van Harmelen et al. 2008). Finally, a single nucleotide polymorphism in the Ednrb gene is associated with reduced risk of developing obesity in 2 separate populations in Spain (Martinez-Barquero et al. 2015). It is unclear if this SNP is associated with a gain or loss in function, but provides evidence that the ETB receptor may be associated with obesity in humans. Our data indicates that loss of ETB receptor function can reduce adiposity when endogenous production of ET-1 is stimulated.
One of the major findings of this study is that loss of ETB receptor function lowers circulating insulin and improves insulin sensitivity as measured by fasting blood glucose and glucose and insulin tolerance. One potential mechanism of reduced insulin is loss of ETB mediated insulin release from pancreatic beta cells. It is well documented that ET-1 promotes insulin release via the ETB receptor;(Brock et al. 1999; De Carlo et al. 2000) however, without improvement in insulin signaling in peripheral tissues, this would be expected to promote hyperglycemia. Therefore, whole body loss of ETB receptor function appears to reduce insulin secretion, while also improving insulin signaling in insulin sensitive tissues. Polak et. al. recently showed that blocking ETB receptors improves insulin sensitivity in a mouse model of sleep apnea (Polak et al. 2018). Several mechanisms by which activation of ETB could promote insulin resistance exists. First, PKC is a known second messenger through which ETB receptors signal (Takuwa et al. 1990). PKC inhibits insulin receptor substrate-1 (IRS1) and is thought to be a major factor in the development of insulin resistance in muscle and liver of obese individuals (Li et al. 2015). Another potential mechanism by which ETB receptor activation could cause insulin resistance is through activation of c-Jun N-terminal kinase, which leads to activation of MAPK (Aquilla et al. 1996). MAPK can also inhibits IRS-1 (Fujishiro et al. 2003). Another potential mechanism by which ETB activation may promote insulin resistance occurs at the level of the adipocyte. ETB receptor activation has been shown to inhibit peroxisome proliferator-activated receptor gamma (PPAR-λ) (Wolf et al. 2014). PPAR-λ is a transcription factor that promotes production of adipokines such as adiponectin by adipose. Adiponectin has profound insulin sensitizing effects on both liver and muscle, and adiponectin null mice have severe insulin resistance (Nawrocki et al. 2006). More work is needed to uncover complete mechanisms by which loss of ETB receptor function improves glucose control.
One potential confounder with the ETB def rat is that loss of ETB receptor function leaves the ETA receptor unopposed, therefore, it is possible that loss of adiposity is due to activation of the ETA receptor. In vitro activation of the ETA receptor causes increased lipolysis in differentiated 3T3-L1 adipocytes suggesting this as a potential mechanism for reduced adiposity (Eriksson et al. 2009). In addition, increased lipolysis via ETA activation may also be the cause of elevated free fatty acids in ETB def rats, although they were not elevated to a pathophysiological level. This would agree with a recent study published by Farrah et. al. indicating that blockade of ETA receptors improves plasma lipids in patients with chronic kidney disease. Surprisingly, the increase in free fatty acids was in the absence of changes in circulating triglycerides in ETB def animals. This may occur through increased lipoprotein lipase or hepatic lipase activity. More work, potentially with tissue specific knockout animals, in this area is needed to determine the role of and mechanisms by which ETA receptors may promote dyslipidemia in obesity.
Several limitations of the current study exist. First, plasma for protocol 1 was collected while animals were under anesthesia and the animals were not fasted. This artificially inflates plasma glucose concentrations. Insulin resistance was only estimated in HS fed animals under these circumstances. In addition, HS diet was only give for two weeks, and the difference in epidydimal tissue weight was only about 15 percent. Longer duration of HS feeding may have created a larger effect size. This is somewhat supported by a study in which increased body weight was attenuated by HS in mice chronically fed high fat diet (Weidemann et al. 2015), although this was attributed to reduced digestive efficiency.
Figure 4:
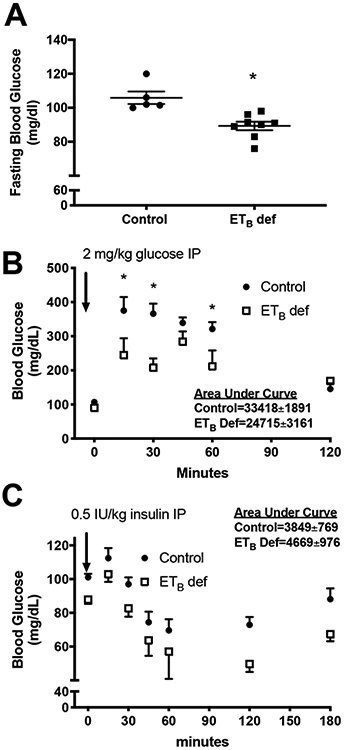
Endothelin B deficient (ETB def) rats have improved insulin sensitivity. A) Fasting plasma glucose (mg/dl; *p<0.05 vs. control), B) glucose tolerance test, and C) IP insulin tolerance test in transgenic control or ETB def rats maintained on normal salt (NS) diet. *p<0.05 by post hoc analysis.
Acknowledgements
This work was supported by National Heart, Lung, and Blood Institute grant (R00 HL127178 to JSS and National Institute of General Medicine Sciences grant P20 GM104357 to JSS.
Footnotes
Disclosures
The authors have no disclosures.
References
- Aburto NJ, Ziolkovska A, Hooper L, Elliott P, Cappuccio FP, and Meerpohl JJ 2013. Effect of lower sodium intake on health: systematic review and meta-analyses. BMJ 346: f1326. doi: 10.1136/bmj.f1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquilla E, Whelchel A, Knot HJ, Nelson M, and Posada J 1996. Activation of multiple mitogen-activated protein kinase signal transduction pathways by the endothelin B receptor requires the cytoplasmic tail. J Biol Chem 271(49): 31572–31579. doi: 10.1074/jbc.271.49.31572. [DOI] [PubMed] [Google Scholar]
- Brock B, Gregersen S, Kristensen K, Thomsen JL, Buschard K, Kofod H, and Hermansen K 1999. The insulinotropic effect of endothelin-1 is mediated by glucagon release from the islet alpha cells. Diabetologia 42(11): 1302–1307. doi: 10.1007/s001250051442. [DOI] [PubMed] [Google Scholar]
- Cardillo C, Campia U, Iantorno M, and Panza JA 2004. Enhanced vascular activity of endogenous endothelin-1 in obese hypertensive patients. Hypertension 43(1): 36–40. doi: 10.1161/01.HYP.0000103868.45064.81. [DOI] [PubMed] [Google Scholar]
- Chou YC, Perng JC, Juan CC, Jang SY, Kwok CF, Chen WL, Fong JC, and Ho LT 1994. Endothelin-1 inhibits insulin-stimulated glucose uptake in isolated rat adipocytes. Biochem Biophys Res Commun 202(2): 688–693. doi: 10.1006/bbrc.1994.1985. [DOI] [PubMed] [Google Scholar]
- De Carlo E, Milanesi A, Martini C, Maffei P, Sicolo N, and Scandellari C 2000. Endothelin-1 and endothelin-3 stimulate insulin release by isolated rat pancreatic islets. J Endocrinol Invest 23(4): 240–245. doi: 10.1007/BF03343715. [DOI] [PubMed] [Google Scholar]
- Eriksson AK, van Harmelen V, Stenson BM, Astrom G, Wahlen K, Laurencikiene J, and Ryden M 2009. Endothelin-1 stimulates human adipocyte lipolysis through the ET A receptor. Int J Obes (Lond) 33(1): 67–74. doi: 10.1038/ijo.2008.212. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, and Parkin DM 2010. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127(12): 2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- Ferri C, Bellini C, Desideri G, Baldoncini R, Properzi G, Santucci A, and De Mattia G 1997. Circulating endothelin-1 levels in obese patients with the metabolic syndrome. Exp Clin Endocrinol Diabetes 105 Suppl 2: 38–40. doi: 10.1055/s-0029-1211794. [DOI] [PubMed] [Google Scholar]
- Ferri C, Bellini C, Desideri G, Di Francesco L, Baldoncini R, Santucci A, and De Mattia G 1995. Plasma endothelin-1 levels in obese hypertensive and normotensive men. Diabetes 44(4): 431–436. doi: 10.2337/diab.44.4.431. [DOI] [PubMed] [Google Scholar]
- Fujishiro M, Gotoh Y, Katagiri H, Sakoda H, Ogihara T, Anai M, Onishi Y, Ono H, Abe M, Shojima N, Fukushima Y, Kikuchi M, Oka Y, and Asano T 2003. Three mitogen-activated protein kinases inhibit insulin signaling by different mechanisms in 3T3-L1 adipocytes. Mol Endocrinol 17(3): 487–497. doi: 10.1210/me.2002-0131. [DOI] [PubMed] [Google Scholar]
- Gariepy CE, Ohuchi T, Williams SC, Richardson JA, and Yanagisawa M 2000. Salt-sensitive hypertension in endothelin-B receptor-deficient rats. J. Clin. Invest. 105(7): 925–933. doi: 10.1172/JCI8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes CA, Riddell LJ, Campbell KJ, He FJ, and Nowson CA 2016. 24-h urinary sodium excretion is associated with obesity in a cross-sectional sample of Australian schoolchildren. Br J Nutr 115(6): 1071–1079. doi: 10.1017/S0007114515005243. [DOI] [PubMed] [Google Scholar]
- Juan CC, Chuang TY, Chang CL, Huang SW, and Ho LT 2007. Endothelin-1 regulates adiponectin gene expression and secretion in 3T3-L1 adipocytes via distinct signaling pathways. Endocrinology 148(4): 1835–1842. doi: 10.1210/en.2006-0654. [DOI] [PubMed] [Google Scholar]
- Kang YJ, Wang HW, Cheon SY, Lee HJ, Hwang KM, and Yoon HS 2016. Associations of Obesity and Dyslipidemia with Intake of Sodium, Fat, and Sugar among Koreans: a Qualitative Systematic Review. Clin Nutr Res 5(4): 290–304. doi: 10.7762/cnr.2016.5.4.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki YY, Emoto N, Ohuchi T, Widyantoro B, Yagi K, Nakayama K, Kedzierski RM, Hammer RE, Yanagisawa H, Williams SC, Richardson JA, Suzuki T, and Yanagisawa M 2010. Low blood pressure in endothelial cell-specific endothelin 1 knockout mice. Hypertension 56(1): 121–128. doi: 10.1161/HYPERTENSIONAHA.109.138701. [DOI] [PubMed] [Google Scholar]
- Li M, Vienberg SG, Bezy O, O'Neill BT, and Kahn CR 2015. Role of PKCdelta in Insulin Sensitivity and Skeletal Muscle Metabolism. Diabetes 64(12): 4023–4032. doi: 10.2337/db14-1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, Amann M, Anderson HR, Andrews KG, Aryee M, Atkinson C, Bacchus LJ, Bahalim AN, Balakrishnan K, Balmes J, Barker-Collo S, Baxter A, Bell ML, Blore JD, Blyth F, Bonner C, Borges G, Bourne R, Boussinesq M, Brauer M, Brooks P, Bruce NG, Brunekreef B, Bryan-Hancock C, Bucello C, Buchbinder R, Bull F, Burnett RT, Byers TE, Calabria B, Carapetis J, Carnahan E, Chafe Z, Charlson F, Chen H, Chen JS, Cheng AT, Child JC, Cohen A, Colson KE, Cowie BC, Darby S, Darling S, Davis A, Degenhardt L, Dentener F, Des Jarlais DC, Devries K, Dherani M, Ding EL, Dorsey ER, Driscoll T, Edmond K, Ali SE, Engell RE, Erwin PJ, Fahimi S, Falder G, Farzadfar F, Ferrari A, Finucane MM, Flaxman S, Fowkes FG, Freedman G, Freeman MK, Gakidou E, Ghosh S, Giovannucci E, Gmel G, Graham K, Grainger R, Grant B, Gunnell D, Gutierrez HR, Hall W, Hoek HW, Hogan A, Hosgood HD 3rd, Hoy D, Hu H, Hubbell BJ, Hutchings SJ, Ibeanusi SE, Jacklyn GL, Jasrasaria R, Jonas JB, Kan H, Kanis JA, Kassebaum N, Kawakami N, Khang YH, Khatibzadeh S, Khoo JP, Kok C, Laden F, Lalloo R, Lan Q, Lathlean T, Leasher JL, Leigh J, Li Y, Lin JK, Lipshultz SE, London S, Lozano R, Lu Y, Mak J, Malekzadeh R, Mallinger L, Marcenes W, March L, Marks R, Martin R, McGale P, McGrath J, Mehta S, Mensah GA, Merriman TR, Micha R, Michaud C, Mishra V, Mohd Hanafiah K, Mokdad AA, Morawska L, Mozaffarian D, Murphy T, Naghavi M, Neal B, Nelson PK, Nolla JM, Norman R, Olives C, Omer SB, Orchard J, Osborne R, Ostro B, Page A, Pandey KD, Parry CD, Passmore E, Patra J, Pearce N, Pelizzari PM, Petzold M, Phillips MR, Pope D, Pope CA 3rd, Powles J, Rao M, Razavi H, Rehfuess EA, Rehm JT, Ritz B, Rivara FP, Roberts T, Robinson C, Rodriguez-Portales JA, Romieu I, Room R, Rosenfeld LC, Roy A, Rushton L, Salomon JA, Sampson U, Sanchez-Riera L, Sanman E, Sapkota A, Seedat S, Shi P, Shield K, Shivakoti R, Singh GM, Sleet DA, Smith E, Smith KR, Stapelberg NJ, Steenland K, Stockl H, Stovner LJ, Straif K, Straney L, Thurston GD, Tran JH, Van Dingenen R, van Donkelaar A, Veerman JL, Vijayakumar L, Weintraub R, Weissman MM, White RA, Whiteford H, Wiersma ST, Wilkinson JD, Williams HC, Williams W, Wilson N, Woolf AD, Yip P, Zielinski JM, Lopez AD, Murray CJ, Ezzati M, AlMazroa MA, and Memish ZA 2012. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380(9859): 2224–2260. doi: 10.1016/S0140-6736(12)61766-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Barquero V, de Marco G, Martinez-Hervas S, Rentero P, Galan-Chilet I, Blesa S, Morchon D, Morcillo S, Rojo G, Ascaso JF, Real JT, Martin-Escudero JC, and Chaves FJ 2015. Polymorphisms in endothelin system genes, arsenic levels and obesity risk. PLoS One 10(3): e0118471. doi: 10.1371/journal.pone.0118471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, Rossetti L, and Scherer PE 2006. Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem 281(5): 2654–2660. doi: 10.1074/jbc.M505311200. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Nakao K, Arai H, Nakagawa O, Hosoda K, Suga S, Nakanishi S, and Imura H 1991. Molecular cloning of a non-isopeptide-selective human endothelin receptor. Biochem Biophys Res Commun 178(1): 248–255. doi: 10.1016/0006-291x(91)91806-n. [DOI] [PubMed] [Google Scholar]
- Ottosson-Seeberger A, Lundberg JM, Alvestrand A, and Ahlborg G 1997. Exogenous endothelin-1 causes peripheral insulin resistance in healthy humans. Acta Physiol Scand 161(2): 211–220. doi: 10.1046/j.1365-201X.1997.00212.x. [DOI] [PubMed] [Google Scholar]
- Polak J, Punjabi NM, and Shimoda LA 2018. Blockade of Endothelin-1 Receptor Type B Ameliorates Glucose Intolerance and Insulin Resistance in a Mouse Model of Obstructive Sleep Apnea. Front Endocrinol (Lausanne) 9: 280. doi: 10.3389/fendo.2018.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles J, Fahimi S, Micha R, Khatibzadeh S, Shi P, Ezzati M, Engell RE, Lim SS, Danaei G, Mozaffarian D, Global Burden of Diseases, N., and Chronic Diseases Expert, G. 2013. Global, regional and national sodium intakes in 1990 and 2010: a systematic analysis of 24 h urinary sodium excretion and dietary surveys worldwide. BMJ Open 3(12): e003733. doi: 10.1136/bmjopen-2013-003733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubanyi GM, and Polokoff MA 1994. Endothelins: molecular biology, biochemistry, pharmacology, physiology, and pathophysiology. Pharmacol Rev 46(3): 325–415. [PubMed] [Google Scholar]
- Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER 3rd, Simons-Morton DG, Karanja N, Lin PH, and Group DA-SCR 2001. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med 344(1): 3–10. doi: 10.1056/NEJM200101043440101. [DOI] [PubMed] [Google Scholar]
- Schneider JG, Tilly N, Hierl T, Sommer U, Hamann A, Dugi K, Leidig-Bruckner G, and Kasperk C 2002. Elevated plasma endothelin-1 levels in diabetes mellitus. Am J Hypertens 15(11): 967–972. doi: 10.1016/s0895-7061(02)03060-1. [DOI] [PubMed] [Google Scholar]
- Speed JS, Heimlich JB, Hyndman KA, Fox BM, Patel V, Yanagisawa M, Pollock JS, Titze JM, and Pollock DM 2015. Endothelin-1 as a master regulator of whole-body Na+ homeostasis. FASEB J 29(12): 4937–4944. doi: 10.1096/fj.15-276584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuwa Y, Kasuya Y, Takuwa N, Kudo M, Yanagisawa M, Goto K, Masaki T, and Yamashita K 1990. Endothelin receptor is coupled to phospholipase C via a pertussis toxin-insensitive guanine nucleotide-binding regulatory protein in vascular smooth muscle cells. J Clin Invest 85(3): 653–658. doi: 10.1172/JCI114488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TA, Gariepy CE, Pollock DM, and Pollock JS 2003. Unique endothelin receptor binding in kidneys of ETB receptor deficient rats. Am. J. Physiol. Regul. Integr. Comp. Physiol 284(3): R674–681. [DOI] [PubMed] [Google Scholar]
- Tsai YH, Ohkita M, and Gariepy CE 2006. Chronic high-sodium diet increases aortic wall endothelin-1 expression in a blood pressure-independent fashion in rats. Exp Biol Med (Maywood) 231(6): 813–817. [PubMed] [Google Scholar]
- van Harmelen V, Eriksson A, Astrom G, Wahlen K, Naslund E, Karpe F, Frayn K, Olsson T, Andersson J, Ryden M, and Arner P 2008. Vascular peptide endothelin-1 links fat accumulation with alterations of visceral adipocyte lipolysis. Diabetes 57(2): 378–386. doi: 10.2337/db07-0893. [DOI] [PubMed] [Google Scholar]
- Weidemann BJ, Voong S, Morales-Santiago FI, Kahn MZ, Ni J, Littlejohn NK, Claflin KE, Burnett CM, Pearson NA, Lutter ML, and Grobe JL 2015. Dietary Sodium Suppresses Digestive Efficiency via the Renin-Angiotensin System. Sci Rep 5: 11123. doi: 10.1038/srep11123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil BR, Westby CM, Van Guilder GP, Greiner JJ, Stauffer BL, and DeSouza CA 2011. Enhanced endothelin-1 system activity with overweight and obesity. Am J Physiol Heart Circ Physiol 301(3): H689–695. doi: 10.1152/ajpheart.00206.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D, Tseng N, Seedorf G, Roe G, Abman SH, and Gien J 2014. Endothelin-1 decreases endothelial PPARgamma signaling and impairs angiogenesis after chronic intrauterine pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 306(4): L361–371. doi: 10.1152/ajplung.00277.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]