

Abstract
Purpose:
Low dose computed tomography (CT) screening can reduce lung cancer mortality. However, CT screening has a false discovery rate of nearly 96%. We sought to assess if urine samples can be a source for DNA methylation-based detection of non-small cell lung cancer.
Experimental Design:
This nested case-control study of subjects with suspicious nodules on CT imaging obtained plasma and urine samples pre-operatively. Cases (n=74) had pathological confirmation of non-small cell lung cancer. Controls (n=27) had a non-cancer diagnosis. We detected promoter methylation in plasma and urine samples using Methylation on Beads and quantitative methylation-specific real-time PCR for cancer-specific genes (CDO1, TAC1, HOXA7, HOXA9, SOX17, and ZFP42).
Results:
DNA methylation at cancer specific loci was detected in both plasma and urine, and was more frequent in cancer patients compared to controls for all 6 genes in plasma and in CDO1, TAC1, HOXA9, and SOX17 in urine. Univariate and multivariate logistic regression analysis showed that methylation detection in each one of six genes in plasma and CDO1, TAC1, HOXA9, and SOX17 in urine was significantly associated with the diagnosis of NSCLC, independent of age, race and smoking pack-years. When methylation was detected for 3 or more genes in both plasma and urine, the sensitivity and specificity for lung cancer diagnosis was 73% and 92% respectively.
Conclusion
DNA methylation-based biomarkers in plasma and urine could be useful as an adjunct to CT screening to guide decision-making regarding further invasive procedures in patients with pulmonary nodules.
Keywords: lung cancer screening, epigenetic, gene promoter hypermethylation, molecular biomarkers, liquid biopsy
INTRODUCTION:
Lung cancer is the leading cause of cancer-related mortality among men and women in the United States (1). It comprises one quarter of all cancer related deaths in the United States (1). Non-small cell lung cancer (NSCLC), accounts for 87% of lung cancer cases and has a 18% overall 5-year survival rate (2). This low survival rate is most likely due to the fact that over 40% of NSCLC cases are diagnosed at stage IV, which has a 5-year survival rate of 2–13% (3,4).
The National Lung Screening Trial (NLST) showed that lung cancer screening can reduce lung cancer mortality 20% by using low dose computed tomography (LDCT) (5). However, in that study, baseline LDCT scans were positive in 27.3% of subjects with a false positive rate of 26.3% and a false discovery rate of 96% (5). Altering criteria for designating a scan as positive and the implementation of the Lung-RADS algorithm can reduce this rate (6,7). For example, by raising the criteria for a positive nodule from ≥ 4mm as reported in NLST (5) to ≥ 6 mm, the false positive rate is decreased to 17% (8). However, 7 patients would have had delayed diagnosis (2.7% of detected cancer patients with delay), and the majority of “positive” LDCT findings (4470/4726) continue to be from subjects without cancer (FDR of 94.5%). Lung-RADS criteria further reduces the false positive rate of baseline screening to 12.8% (6,7), but also reduces sensitivity to 84.9% with 25 cancers not detected on the baseline scan (248 vs 273 for NLST cutoff), and 3095/3334 positive scans are from patients without cancer, for a FDR of 93.6%. This demonstrates the need for biomarker approaches for the management of screen detected pulmonary nodules.
Using lung cancer tissue samples from The Cancer Genome Atlas (TCGA), we identified very highly sensitive and specific epigenetic changes for lung cancer (all stages) based on promoter gene methylation being able to discriminate lung cancer tissue from normal lung samples (9). Later we validated the use of these epigenetic markers with the detection of the promoter gene methylation in liquid biopsies using plasma and sputum for NSCLC early stages (10). Urine can be a source for detection of specific somatic mutations in tumor DNA from cancers including urothelial, colon and lung cancer (11–13). In addition, epigenetic changes in tumor DNA from urine sediment has been reported for urothelial (renal) cancer (14). Our aim was to determine if urine samples can be used as a source for DNA methylation detection in NSCLC.
MATERIALS & METHODS
Study Population
The Study population consists of a prospective, observational nested case-control from two institutions: The Johns Hopkins Hospital (within the Johns Hopkins Lung Cancer Specialized Program of Research Excellence (SPORE) and the University of Illinois at Chicago Hospital Health Science System (UIHHSS). This study was conducted in accordance with the Declaration of Helsinki. Institutional Review Board approval was obtained prior to study initiation (NA_00005998 for Johns Hopkins and IRB #2017–1286 and #2018–0755 for UIC). All participants from both institutions signed informed consent. The reporting of this study conforms to the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement (15). Patients selected for inclusion in this study were 50 years and older and had a CT scan for suspicion of lung cancer and referred to surgery for resection. Exclusion criteria comprised having small cell lung cancer pathology or presence of other malignancy or any history of cancer within the past 5 years or an adult lacking the capacity to consent. Surgical resection and pathological records were obtained from lung cancer lesions in patients who met the TNM guidelines classification criteria (3,16). Cases had pathologically confirmed Non-small cell lung cancer. Controls were defined as patients histologically confirmed not to have cancer. Pack-years of cigarette smoking were defined as the average number of packs smoked per day times the number of years smoked. Nodule size and volume were obtained from the pathology report, and nodule volume was calculated using the ellipsoid volume formula (Volume = 4/3 × π × radius A × radius B × radius C). Urine and plasma samples were obtained from all participants. The population composition was comprised of a predominantly Caucasian population from the Johns Hopkins Hospital (n=52) and African Americans from the University of Illinois at Chicago (n=49 for a total of 101 patients examined in this study.
Plasma and Urine Sample Collection
Patients enrolled in the study provided urine and matching blood samples. Blood samples were collected EDTA in tubes (Becton Dickinson, Franklin Lakes, NJ) and processed within 2 hour after sample collection. Plasma was collected and stored in at −80 °C freezer until use for up to 6 months. Longer storage of specimens has not previously affected the ability to detect DNA methylation in plasma, and more critical for DNA degradation is the time from collection to processing and freezing. Urine samples were collected in a 50mL urine collection container (Fisher NC9512383) For processing, 10 mL of urine was transferred into 15 mL conical tubes. To prevent DNA degradation, 200 μL of 0.5 M EDTA (pH 8.0) (Thomas Scientific, Inc.: 351027721) was added and mixed into each tube and spun at 3200 rpm for 15 minutes. Supernatant was collected and stored in at −80° C until use.
DNA Isolation and Bisulfate Conversion
DNA was extracted from urine and plasma samples using an optimized Methylation On Beads (MOB) protocol. MOB is a process that allows DNA extraction and bisulfite conversion in a single tube via the use of silica super magnetic beads (10,17). This approach yields a 1.5- to 5-fold improvement in extraction efficiency compared with traditional techniques (10,18). We optimized the MOB protocol which was previously a 24-hour protocol to a 6-hour protocol. We are newly describing MOB protocol for the isolation of DNA from the urine.
In the improved protocol, plasma samples were incubated with Proteinase K (10 mg/mL) (New England Biolabs Co.: P8107s) and Buffer AL (Qiagen, Co.: 19075) at 55°C for 1 hour. During the DNA bisulfite treatment procedure, CT lightning conversion reagent was added and incubated at 98°C for 10 mins and then at 70°C for 1 hour.
For DNA extraction from urine 150 μL of Proteinase K (10 mg/mL) (New England Biolabs Co.: P8107s) was added to 3mL of urine followed by 3mL of Buffer AL (Qiagen, Co.: 19075) and incubated in a water bath at 55 °C for 1 hour. After digestion 3ml of 100% of isopropanol and 150ul of Magnetic Beads (Promega, Co: magnesi KF-MD1471) were added to the sample to bind the DNA. Plasma and urine samples were prepared with parallel digestion workflows running concurrently (10).
Assessment of cell free DNA tumor fraction
DNA isolated from plasma and urine from patients with mutations in EGFR and KRAS detected in tumor DNA was sent to our Genome Research Core facility for digital droplet quantitative PCR (ddPCR) for these loci. ddPCR™ Supermix was used for probes (1st line - ddPCR SMX PRBS nodUTP 500RXN) from BioRad with the restriction enzyme from LifeTechnologies to set up PCR before generating droplets. The assays used were: 1. ddPCR™ Mutation Assay: KRAS p.G12Vc.35G>T, Human and 2. ddPCR™ Mutation Assay: EGFR p.L858Rc.2573T>G, Human. Tumor DNA fraction in cfDNA was defined as the number of mutated DNA copies detected by ddPCR divided by the total number of DNA copies (mutant + wildtype).
DNA Methylation Analysis
Primers and hybridization probes for methylation analysis were designed using Primer3 (v.0.4.0) (19,20). For this study, primers generating shorter amplicons were designed within the same genomic regions of the primers we previously used for plasma methylation detection (10). All primer and probe sequences are listed in supplementary table S1. The analysis was performed using quantitative real-time Methylation specific PCR, and β-Actin was used as reference gene for normalization of methylation levels (21). The PCR reaction mix and 2−ΔCT for each methylation detection replicate was performed as previously described (10). Positive promoter gene methylation was defined as 2−ΔCT values ≧ 10−10 and negative promoter gene methylation as 2−ΔCT values < 10−10. This bimodal distribution of 2−ΔCT values, as shown previously in sputum and plasma detection (10), is in essence detectable vs. undetectable, and all lower quantities (< 10−10) are actually zero, with real-time cycle thresholds of infinity designated as CT of 100 to allow calculation of 2−ΔCT. The variation in 2−ΔCT for undetectable methylation is the result of different control (β-actin) cycle thresholds. In this study, the lowest positive methylation 2−ΔCT value was 1.66 × 10−5 and the highest negative methylation 2−ΔCT value was 1.85 × 10−19.
Statistical analysis
Quantitative data are expressed as median (interquartile range) for continuous variables and frequency (percentage) for categorical variables. Baseline demographic characteristics of the cases and controls were compared with the Wilcoxon rank sum test for continuous data and the Fisheŕs exact test for categorical data. Two-sided statistical tests were used. Pearson correlation analysis was obtained for the ΔCT values from DNA methylation from plasma and urine.
We determined the sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV) and false positive rate (FPR) in this cohort using the presence or absence of detectable methylation, using the methylation ΔCT values. We obtained receiver operator characteristic (ROC) analysis using the 2−ΔCT values for individual genes to determine the performance of each individual gene. The area under the curve (AUC) was reported with 95% confidence interval (CI). Additionally, we tested whether having at least 1 positive methylated gene vs 2 vs 3 vs 4 vs 5 vs all simultaneously positively methylated has a better area under the curve. We found that looking into plasma, urine and both the best performing option was the one with having at least 3 genes with positive methylation. Univariate and multivariate logistic regression models adjusted by age, race and pack-years were used to assess the association of methylated genes with lung cancer diagnosis by measuring the odds ratio (ORs) with 95 % CI. P values < 0.05 were considered statistically significant. Bonferroni correction was used on all performed analysis to adjust p values for multiple tests. Supplementary table S2 provides the Univariate logistic regression analysis for risk of having NSCLC for each of the covariates. Only age was significantly associated with NSCLC risk (p=0.03) among baseline characteristics (uncorrected analysis), however after Bonferroni correction for multiple comparisons there were no significant associated variables with NSCLC risk (Supplemental table S2). But given the importance of age, race and pack-years, these were included as covariates in the multivariate analysis. All statistical analyses were performed using R statistic software, version 3.4.0, Vienna, Austria (22).
RESULTS
Characteristics of the Patients
One hundred and one patients fulfilled inclusion criteria, with 74 NSCLC subjects and 27 controls with non-cancerous lung lesions (Table 1). Clinical and demographic variables were balanced in cases and controls. Overall the 101 subjects median age was 64 years old; with 51% males and 49% females; 51% Caucasian, 38% African Americans, and 11% others; median BMI of 27; 19% current smokers, 66% former smokers, 15% never smoker; with a median 30 pack-year history.
Table 1.
Baseline Characteristics of the 101 Subjects.
| Patient Characteristics | Cancer (N=74) | Control (N=27) | p Value |
|---|---|---|---|
| Age at diagnosis (years) (IQR) | 64 (59–70) | 62 (50–67) | 0.10 |
| Gender | |||
| Male (%) | 34 (46%) | 18 (67%) | 0.08 |
| Female (%) | 40 (54%) | 9 (33%) | |
| Race | |||
| White (%) | 41 (55%) | 11 (41%) | 0.27 |
| African American (%) | 24 (32%) | 14 (52%) | |
| Hispanic (%) | 1 (1%) | 1 (4%) | |
| Asian (%) | 4 (5%) | 0 (0%) | |
| Other (%) | 4 (5%) | 1 (4%) | |
| BMI (IQR) | 26 (22–30) | 27 (24–32) | 0.26 |
| Smoking status | |||
| Current (%) | 13 (18%) | 6 (22%) | 0.75 |
| Former (%) | 49 (66%) | 18 (67%) | |
| Never (%) | 12 (16%) | 3 (11%) | |
| Pack-year (IQR) | 30 (15–46) | 30 (10–46) | 0.93 |
| COPD (%) | 23 (31%) | 5 (19%) | 0.31 |
| FEV1 % Predicted (IQR) | 82 (72–95) | 69 (60–81) | 0.07 |
| FVC % Predicted (IQR) | 87 (77–103) | 80 (57–103) | 0.29 |
| FEV1/FVC % Ratio (IQR) | 78(77–80) | 79 (77–82) | 0.66 |
| Histology | |||
| Adenocarcinoma (%) | 65 (88%) | NA | NA |
| Squamous-cell (%) | 9 (12%) | NA | |
| Stage | |||
| I (%) | 34 (46%) | NA | NA |
| II (%) | 14 (19%) | NA | |
| III (%) | 12 (16%) | NA | |
| IV (%) | 14 (19%) | NA | |
| Nodule size (cm) | 2.1 (1.6–3.7) | 3 (2.3–4) | 0.70 |
| < 1cm | 4 (6%) | 1 (11%) | 0.28 |
| 1–2 cm | 22 (35%) | 1 (11%) | |
| > 2 cm | 37 (59%) | 7 (78%) |
Abbreviations: Chronic obstructive pulmonary disease: COPD, Forced Expiratory Volume in one second: FEV1, Forced vital capacity: FVC, Interquartile range: IQR.
Detection of DNA Methylation
To detect DNA methylation in free DNA present in the urine, we modified our previous approach to detect shorter fragments of DNA, as described in the methods section. We first measured methylation using ΔCT values. Methylation was detected more frequently in all 6 genes in cancer patients compared to controls in plasma (Figure 1A & 2A). In urine, CDO1, TAC1, HOXA9, and SOX17 showed significantly more cancer patients having positive methylation compared to controls (Figure 1B & 2B), and the quantitation of methylation was similar in urine and plasma when detectable. A summary of the detection of methylation (Table 2), demonstrates the sensitivity and specificity for lung cancer diagnosis using individual genes from plasma ranged from 58–93% and 28–84% respectively. The frequency of methylation detection in urine vs. plasma for cancer patients was similar for CDO1, SOX17, and ZFP42, but in urine was slightly lower for TAC1, HOXA7, and HOXA9. FPR in plasma ranged from 16–72%. When at least 3 genes had positive methylation in plasma the sensitivity and specificity for lung cancer diagnosis was 88% and 60% respectively with FPR 40%. Sensitivity and specificity for lung cancer diagnosis using individual genes from urine ranged from 48–92% and 22–81% respectively with an FPR 19–78%. When at least 3 genes had positive methylation in urine, the sensitivity and specificity for lung cancer diagnosis was 93% and 30% respectively with FPR 70%. When both urine and plasma results were combined, the sensitivity and specificity for lung cancer diagnosis for simultaneously methylated genes in plasma and urine ranged from 27–85% and 32–96% respectively for individual genes, with a FPR 4–68% (4–15% when not considering ZFP42). When at least 3 genes had simultaneous positive methylation both in plasma and urine the sensitivity and specificity for lung cancer diagnosis was 73% and 92% respectively with an FPR 8%.
Figure 1. Quantitative Methylation Detection in Plasma and Urine.

Scatter plots with boxplots showing the converted ΔCT methylation values in a logarithmic scale for the studied genes from plasma (A) and urine (B). These values show a bimodal distribution with the lower group the values corresponding to those samples with no detectable amplification. Most plasma and urine samples from cancer patients have detectable methylation, while a few patients are undetectable. The majority of controls have undetectable methylation at these loci, although some patients do have detectable methylation that is quantitatively similar to cancer patients. Significance values: ‘****’ p < 0.0001; ‘***’ p <0.001: ‘**’p < 0.01: ‘*’p <0.05: ‘.’p <0.1; ‘NS’ non-significant (p>0.1)
Figure 2. Methylation Detection Frequency in plasma and urine, comparing patients with NSCLC vs controls and early stages (I-II) vs late stages (III-IV).
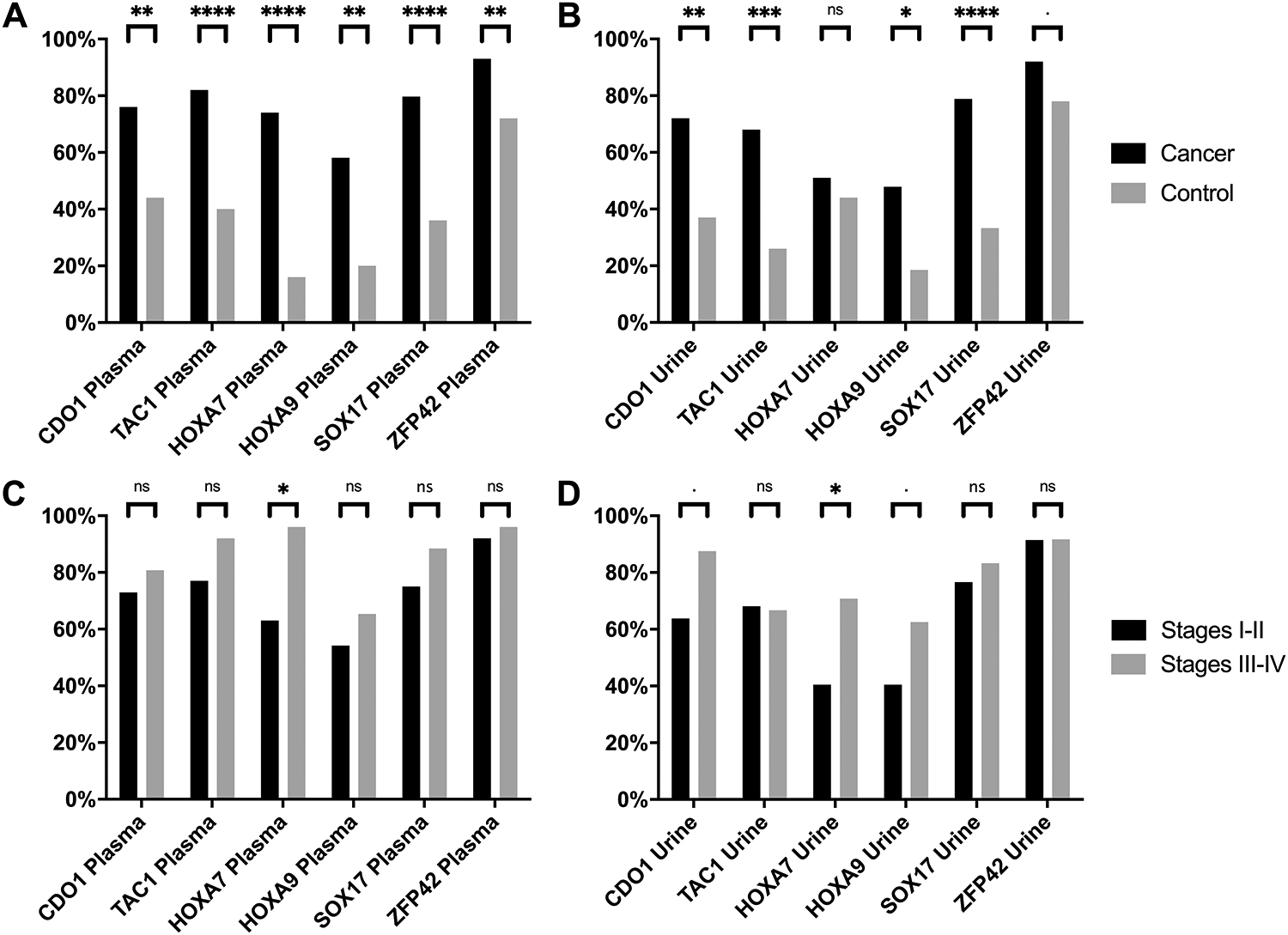
This bar plot shows the percentage of positive methylation detected in plasma (A& C) and urine (B & D) comparing NSCLC patients (black) vs controls (grey) in A and B and comparing NSCLC early stage NSCLC patients (black) vs late stage NSCLC patients (grey) in C and D. Significance values: ‘****’ p < 0.0001; ‘***’ p <0.001: ‘**’p < 0.01: ‘*’p <0.05: ‘.’p <0.1; ‘NS’ non-significant (p>0.1). Source data can be found in Table 2 and supplementary tables S4 and S5 respectively, where “n” is the number of samples positive for methylation detection in cancer and controls in plasma and urine.
Table 2.
Gene Methylation Detection, Sensitivity, Specificity Using Detectable vs. Non-detectable cutoff
| Cancer (n=74) | Control (n=25) | |||||||
|---|---|---|---|---|---|---|---|---|
| Plasma | n | Sensitivity | n | Specificity | PPV | NPV | AUC | AUC 95% CI |
| CDO1 | 56 | 76% | 11 | 56% | 84% | 44% | 0.68 | 0.55–0.80 |
| TAC1 | 61 | 82% | 10 | 60% | 86% | 54% | 0.73 | 0.61–0.86 |
| HOXA7 | 55 | 74% | 4 | 84% | 93% | 53% | 0.79 | 0.69–0.90 |
| HOXA9 | 43 | 58% | 5 | 80% | 90% | 39% | 0.66 | 0.54–0.77 |
| SOX17 | 59 | 80% | 9 | 64% | 87% | 52% | 0.75 | 0.63–0.86 |
| ZFP42 | 69 | 93% | 18 | 28% | 79% | 58% | 0.70 | 0.58–0.82 |
| All (at least 3 positive) | 65 | 88% | 10 | 60% | 87% | 63% | 0.68 | 0.56–0.80 |
| Cancer (n=71) | Control (n=27) | |||||||
| Urine | n | Sensitivity | n | Specificity | PPV | NPV | AUC | AUC 95% CI |
| CDO1 | 51 | 72% | 10 | 63% | 84% | 46% | 0.70 | 0.58–0.82 |
| TAC1 | 48 | 68% | 7 | 74% | 87% | 47% | 0.70 | 0.58–0.83 |
| HOXA7 | 36 | 51% | 12 | 56% | 75% | 30% | 0.54 | 0.41–0.67 |
| HOXA9 | 34 | 48% | 5 | 81% | 87% | 37% | 0.66 | 0.54–0.77 |
| SOX17 | 56 | 79% | 9 | 67% | 86% | 55% | 0.76 | 0.65–0.88 |
| ZFP42 | 65 | 92% | 21 | 22% | 76% | 50% | 0.65 | 0.52–0.77 |
| All (at least 3 positive) | 66 | 93% | 19 | 30% | 78% | 62% | 0.70 | 0.58–0.81 |
| Cancer (n=71) | Control (n=27) | |||||||
| Plasma and Urine | n | Sensitivity | n | Specificity | PPV | NPV | AUC | AUC 95% CI |
| CDO1 | 42 | 58% | 4 | 85% | 91% | 42% | 0.69 | 0.5–0.82 |
| TAC1 | 39 | 53% | 2 | 92% | 95% | 41% | 0.72 | 0.59–0.85 |
| HOXA7 | 32 | 45% | 4 | 85% | 89% | 37% | 0.70 | 0.58–0.82 |
| HOXA9 | 20 | 27% | 1 | 96% | 95% | 33% | 0.77 | 0.66–0.87 |
| SOX17 | 47 | 65% | 3 | 88% | 94% | 48% | 0.78 | 0.67–0.89 |
| ZFP42 | 60 | 85% | 17 | 32% | 78% | 42% | 0.72 | 0.60–0.84 |
| All (at least 3 positive) | 52 | 73% | 2 | 92% | 96% | 55% | 0.72 | 0.61–0.84 |
Abbreviations: Area under the curve: AUC; positive predictive value: PPV; negative predictive value: NPV.
Circulating cell free DNA tumor fraction
To compare methylation detection in plasma and urine to the tumor fraction in these samples, we identified 16 patients with known driver mutations detected in their lung cancer tissue specimens, (11 patients with KRAS mutations and 5 with EGFR mutations). We used a standardized digital droplet quantitative PCR (ddPCR) on DNA isolated from plasma and urine to assess the number of copies (and mutant fraction) of circulating DNA containing the known mutation. The mutant fraction for each patient in plasma (Figure 3A) and urine (Figure 3B) is compared to the methylation quantitation. For patients with detectable DNA mutations, methylation levels are similar or higher than mutational quantities in plasma, and similar in urine. The detection frequency for mutations, which in plasma was 9/16 (56%) and in urine was 3/16 (19%), is compared to the methylation detection frequency for these same 16 patients (Figure 3C). This shows similar frequency of detection in plasma, but a greater frequency of methylation detection in urine.
Figure 3. Circulating DNA mutation fraction and circulating DNA methylation detection values in plasma and urine and the Frequency of circulating DNA mutation detection and circulating DNA methylation detection in plasma and urine.
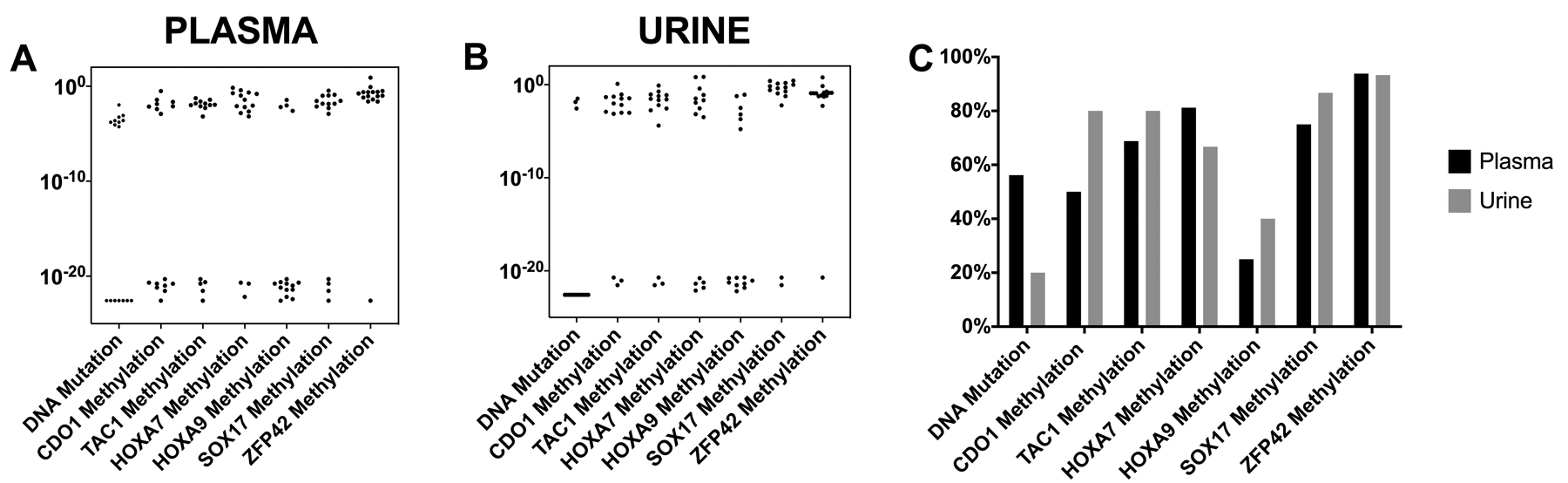
A & B. Scatter plots show the circulating DNA mutation fraction plotted on logarithmic scale for 16 patients with known driver mutations (KRAS and EGFR) from plasma (A) and urine (B) compared to quantitative ΔCt methylation values for each gene on the same 16 patients. C. Bar plot showing the frequency of circulating DNA mutation detection in plasma and urine compared to the percentage of positive methylation detected in plasma and urine for the 16 samples with known driver mutations.
Plasma and urine methylation correlation
Genes were simultaneously methylated in both plasma and urine (positive concordance) in a minimum of 22% of patients (e.g. SOX17) to a max 80% of patients (e.g. ZFP42) when all subjects were included (Supplemental Table S3), and non-methylated status was matched in plasma and urine (negative concordance) ranging from 5% (e.g. ZFP42) to 31% (e.g. HOX9) for all subjects. Total concordance of simultaneously methylated and simultaneously non-methylated in both plasma and urine ranged from 53% (e.g. HOX9) to 85% (e.g. ZFP42). The level of methylation was also concordant, and the ΔCT values for TAC1, HOXA7 and SOX17 showed significant correlation between the methylation detection in plasma and urine when looking all patients (Supplemental Table S3). However, there was a difference in relative importance of positive and negative concordance between cancer patients and controls. For NSCLC, the positive methylation concordance increased to a range 28–85% and the negative methylation concordance decreased to a range 0 to 21% with a total concordance of 48 to 85% among NSCLC patients. Among cancer-free controls, the positive methylation concordance decreased to a range 4 to 68% and the negative methylation concordance increased to a range 20 to 64% with a total concordance of 52 to 88% among cancer-free controls. For discordant samples, there was a similar frequency of positive plasma and negative urine compared to positive urine and negative plasma.
Gene Methylation and Lung Cancer Diagnostic Accuracy
ROC curves for lung cancer detection were obtained for each single gene; using the normalized methylation ΔCT values calculated as described in methods. The AUC values were 0.66–0.79 in plasma samples and 0.54–0.76 in urine samples. The genes with the largest AUC values in plasma were TAC1 AUC: 0.73 95% CI (0.61–0.86); HOXA7 AUC: 0.79 95% CI (0.69–0.90); and SOX17 AUC: 0.75 95% CI (0.63–0.86) (Table 2). When examining multigene methylation, having at least 3 positive methylated genes in plasma has an AUC of 0.68 95% CI (0.56–0.80). The genes with the largest AUC in urine were: CDO1 AUC: 0.70 95% CI (0.58–0.82); TAC1 AUC: 0.70 95% CI (0.58–0.83); and SOX17 AUC: 0.76 95% CI (0.65–0.88) (Table 2). When 3 positive methylated genes in urine is used, the AUC was 0.70 95% CI (0.58–0.81). The genes with the largest AUC when simultaneously methylated both in plasma and urine were: TAC1 AUC: 0.72 95% CI (0.59–0.85); HOXA9 AUC: 0.77 95% CI (0.66–0.87); SOX17 AUC: 0.78 95% CI (0.67–0.89); and ZFP42 AUC: 0.72 95% CI (0.60–0.84).
Gene methylation and lung cancer risk association
Univariate logistic regression for risk of having NSCLC was obtained for each one of the baseline characteristics to assess for confounders (Supplemental table S2). In order to seek for other confounders for the multivariate analysis we looked at the differences in methylation between Caucasians and African Americans given that in our study we had 51% Caucasians and 38% African Americans and that African American have been underrepresented in the scientific literature and carry a disproportionate frequency of the lung cancer burden, with 11% higher incidence rate compared with their Caucasian counterparts, later stage diagnosis and poorer 5-year overall survival rate (16% in AA Vs 19% in Caucasian) (5,23–27). We did not find any difference in the percentage of positive methylation in any of the genes when comparing Caucasians with African Americans (more details about the sensitivity, specificity, PPV, NPV, FPR and AUC for each gene in plasma, urine and both for Caucasian patients and African Americans in supplemental materials). Also, in our study, only age was significantly associated with NSCLC risk (p=0.03) among all baseline characteristics however after Bonferroni correction for multiple comparisons there were no significant associated variables with NSCLC risk (Supplemental table S2).
Univariate logistic regression analysis showed that the ΔCt methylation value of each one of the genes in plasma was significantly associated with NSCLC risk (Table 3). This association remained statistically significant for CDO1, TAC1, HOXA7, HOXA9, and SOX17 after adjusting by age, race and smoking pack-years (Bonferroni corrected for multiple comparisons). The results shown by logistic regression analysis confirm those found by the direct comparison on the differences on percentage of methylation of cancer vs controls. Additionally, having at least 3 genes with positive methylation in plasma was significantly associated with NSCLC risk both in univariate and multivariate analysis.
Table 3.
Univariate and multivariate logistic regression analysis for risk of having NSCLC.
| Univariate analysis | Multivariate analysis | ||||||
|---|---|---|---|---|---|---|---|
| OR | OR 95% CI | p Value | OR | OR 95% CI | p Value Non corrected | p Value Bonferroni Correction | |
| CDO1 methylated in plasma | 3.96 | 1.54–10.49 | 0.005 | 4.90 | 1.73–4.77 | 0.003 | 0.023 |
| TAC1 methylated in plasma | 7.04 | 2.65–19.79 | <0.001 | 9.22 | 3.11–230.63 | <0.001 | <0.001 |
| HOXA7 methylated in plasma | 15.20 | 5.05–57.34 | <0.001 | 17.02 | 5.16–71.84 | <0.001 | <0.001 |
| HOXA9 methylated in plasma | 5.55 | 2.00–18.14 | 0.002 | 6.05 | 2.05–21.22 | 0.002 | 0.015 |
| SOX17 methylated in plasma | 6.99 | 2.65–19.65 | <0.001 | 7.03 | 2.53–21.19 | <0.001 | 0.002 |
| ZFP42 methylated in plasma | 5.37 | 1.54–20.09 | 0.009 | 5.13 | 1.32–021.49 | 0.019 | 0.135 |
| At least 3 genes methylated in plasma | 10.83 | 3.87–32.80 | <0.001 | 13.56 | 4.33–549.21 | <0.001 | <0.001 |
| CDO1 methylated in urine | 4.34 | 1.73–11.41 | 0.002 | 6.69 | 2.31–22.02 | <0.001 | 0.006 |
| TAC1 methylated in urine | 5.96 | 2.29–17.11 | <0.001 | 5.30 | 1.87–16.84 | 0.003 | 0.018 |
| HOXA7 methylated in urine | 1.29 | 0.59–3.18 | 0.580 | 1.40 | 0.52–3.88 | 0.503 | 1.000 |
| HOXA9 methylated in urine | 4.04 | 1.47–13.15 | 0.011 | 4.33 | 1.46–15.17 | 0.013 | 0.088 |
| SOX17 methylated in urine | 7.47 | 2.87–20.78 | <0.001 | 8.69 | 3.05–27.81 | <0.001 | <0.001 |
| ZFP42 methylated in urine | 3.10 | 0.88–10.92 | 0.073 | 3.82 | 0.98–15.57 | 0.053 | 0.374 |
| At least 3 genes methylated in urine | 5.56 | 1.66–20.33 | 0.006 | 5.76 | 1.60–22.73 | 0.008 | 0.059 |
| CDO1 Plasma and Urine | 7.45 | 2.55–27.45 | <0.001 | 12.68 | 3.55–61.04 | <0.001 | 0.003 |
| TAC1 Plasma and Urine | 13.76 | 3.71–89.64 | <0.001 | 13.81 | 3.38–99.40 | <0.001 | 0.010 |
| HOXA7 Plasma and Urine | 4.72 | 1.61–17.34 | 0.009 | 4.90 | 1.55–19.24 | 0.012 | 0.081 |
| HOXA9 Plasma and Urine | 9.63 | 1.85–177.47 | 0.031 | 10.19 | 1.83–192.57 | 0.031 | 0.217 |
| SOX17 Plasma and Urine | 14.41 | 4.47–65.06 | <0.001 | 18.17 | 5.12–91.16 | <0.001 | <0.001 |
| ZFP42 Plasma and Urine | 2.57 | 0.87–7.41 | 0.081 | 2.76 | 0.86–8.82 | 0.083 | 0.581 |
| At least 3 genes in Plasma and Urine | 31.47 | 8.27–208.29 | <0.001 | 69.34 | 13.21–721.89 | <0.001 | <0.001 |
Abbreviations: CI confidence Interval; OR Odds Ratio; NA non-applicable
Multivariate analysis adjusted by age, race and pack years
In urine, univariate logistic regression analysis showed methylation of CDO1, TAC1, HOXA9, and SOX17 was significantly associated with lung cancer risk. ZFP42 had a trend towards significance (p=0.07), but the low specificity of this gene reduced the significant difference. These findings remained statistically significant for CDO1, TAC1, and SOX17 after adjusting for age, race and pack-years (Bonferroni corrected for multiple comparisons). Having at least 3 genes with positive methylation in urine was also significantly associated with NSCLC risk in univariate analysis.
Univariate logistic regression analysis showed that having the gene simultaneously methylated in both plasma and urine in CDO1, TAC1, HOXA7, HOXA9, and SOX17 was significantly associated with lung cancer risk. These findings remained statistically significant for CDO1, TAC1, and SOX17 after adjusting by age, race and pack-years (Bonferroni corrected for multiple comparisons). Finally, having at least 3 genes with simultaneous positive methylation in both plasma and urine was significantly associated with NSCLC risk both in univariate and multivariate analysis.
Subset analysis by stage
When comparing the frequency of detection of early (Stage I & II) vs late stages (III & IV) in plasma and urine, for many loci there was a higher percentage of positive methylation in late stages (III and IV) compared to early stages (I and II) (Figure 2C and 2D and Supplemental Figure S1 showing quantitative methylation). In early stage patients vs controls, methylation was detected more frequently in all genes in cancer patients compared to controls in plasma (Supplemental Figure S2A). In urine, CDO1, TAC1, and SOX17 showed significantly more people having positive methylation among those with cancer compared to controls (Supplemental Figure S2B). When comparing late stage patients vs controls, methylation was detected more frequently in all genes in cancer patients compared to controls in plasma (Supplemental Figure S3A). In urine, 5 genes showed significantly more people having positive methylation among those with cancer compared to controls (Supplemental Figure S3B).
Overall sensitivity and AUC were slightly higher for late stages (Supplemental table S5) compared with early stages (Supplemental table S4). Sensitivity for early stages NSCLC diagnosis using individual genes from plasma ranged from 54–92% with AUC ranging from 64–73%. Sensitivity and specificity for late stages NSCLC using individual genes from plasma ranged from 65–100% with AUC ranging from 69–91%. Sensitivity and specificity for early stages NSCLC diagnosis using individual genes from urine ranged from 40–91% with AUC ranging from 49–74%. Sensitivity and specificity for late stages NSCLC using individual genes from urine ranged from 63–96% with AUC ranging from 62–86%.
DISCUSSION
The results from this study show that methylation can be detected more frequently in cancer patients compared to controls in all 6 genes in plasma and in CDO1, TAC1, HOXA9, and SOX17 in urine. When at least 3 genes had simultaneous positive methylation both in plasma and urine the sensitivity and specificity for lung cancer diagnosis was 73% and 92% respectively with FPR 8%. These data suggest that epigenetic biomarkers from liquid biopsies based on methylation detection from plasma and urine may compliment LDCT screening to help in the decision process to proceed with further invasive diagnostic/treatment procedures. The FPR in the current study are significantly lower than reported for LDCT screening from the NLST trial (NLST FPR 24%) (5) and in Lung Cancer Screening in the Veterans Health Administration with FDR 98% (28).
The use of non-invasive liquid biopsies for cancer monitoring and most recently for early cancer diagnosis is becoming increasingly accepted in oncology given the recent developments overcoming the detection challenges for minute quantities of DNA from low quantity DNA yielding body fluids (29,30). Previous studies sought to improve lung cancer detection accuracy by the use of molecular biomarkers obtained from non-invasive liquid biopsies (31–37). Additionally, several studies found that DNA methylation could be associated with lung cancer independently of pack years. (38–41). However, none of these tests achieved adequate sensitivity and specificity (31–37,42–44) to adopt for lung cancer screening. With improvements in DNA extraction methods and processing for methylation detection, along with the use of highly prevalent cancer specific methylation targets, we believe we overcame these limitations and optimized it for methylation detection in liquid biopsies (10,17). In previous studies, we showed that MOB can reduce sample loss thereby increasing DNA methylation detection sensitivity. In the present study we further optimized an ultrasensitive detection strategy based on MOB and real time quantitative methylation specific PCR (qMSP) for DNA methylation detection from plasma and urine. The real-time qMSP assay used in this study can detect single molecules containing dense hypermethylation with the pattern for which the assay has been designed (fully methylated). It is less efficient for incomplete methylation in this region but may also detect partial methylation. Other methods able to distinguish partial methylation (including bisulfite sequencing) lack sensitivity for the rare molecules present in plasma or urine and without extremely deep reads (100,000x coverage) would not approach the necessary level of sensitivity. We have developed approaches that can detect partial methylation, and detect epigenetic heterogeneity in difficult samples such as liquid biopsies that contain low fractional concentrations of circulating tumor DNA (ctDNA) and rare epigenetic sub-clonal populations (45,46), but are not yet capable of analyzing a large number of samples and would not be suitable for the very short DNA fragments found in urine. Our previous publications provided additional data to address this question. (10) However, it is seen that there are varying levels of methylation in the tumor and that most plasma levels are lower than the tissue, expected since the fraction of cfDNA in plasma from the tumor is much lower than the tumor cellularity of the tumor itself. We have also recently published (47) quantitative methylation of tumor vs benign nodules for many of these genes, which indeed show some differences in methylation between cancer and normal, and quantitively between different tumors (47). The majority of tumors have high levels of methylation, while the majority of benign lesions have no detectable methylation. For benign nodules, the quantity of methylation is similar, but in some cases, lower than tumors. We can confirm the effects dilution of tumor DNA in the plasma has on the quantity of methylation detected in plasma. As previously published, the quantity of methylation, when detected in the plasma, is lower than that observed in tumor tissue (10).
Urine has been shown to be a source for detection of specific tumor genetic mutations in circulating tumor DNA from different types of cancer including urothelial, colon and lung cancer (11–13). Because of this, we further optimized our previously published approach by using shorter amplicons in order to be able to detect methylation changes from the smaller DNA fragments present in urine. Urine samples have advantages compared to other sources of body fluids. They are non-invasive and easy to obtain, larger volumes can be easily collected, they have less processing limitations compared to other samples, can be stored at room temperature, and the DNA content is stable for longer periods of time than other body fluids. Because of that, urine samples have the potential to be easily implemented in primary care practice.
We observed some differences in sensitivity for detection according to stage, which for most genes was greater for late stage (III-IV) than early stage (I-II). This is not surprising given other studies showing greater sensitivities for detecting late stage cancers compared to early, primarily related to increased levels of ctDNA with larger tumor burden (29). While this is one explanation, an additional factor could be increased tumor DNA methylation associated with lung cancer progression. However, for these loci, there is no evidence for a stage increase in the presence of DNA methylation at these loci, and indeed, these were chosen since they are frequently methylated in early stage lung cancer (9,10), making them optimal as early detection biomarkers. This suggests that tumor burden, and increased levels of tumor derived cell free DNA, is the likely reason for differences in detection of DNA methylation.
Additionally, in this study, we note some differences in methylation detection between plasma and urine, with a trend towards better methylation detection in plasma compared to urine. This was also reflected on the circulating free DNA mutation tumor fraction which was detected in 56% of plasma samples and 19% of urine samples. The differences in methylation detection between plasma and urine may be multifactorial. First, urine is a plasma ultrafiltrate, and thus only a portion of plasma DNA may be filtered into the urine. In addition, only small DNA molecules make it into the urine stream. In order to detect DNA methylation in urine, we redesigned this assay to detect even shorter amplicons, but even with this approach some methylated cfDNA molecules may be too small to be detected. Finally, there could be stability issues from exposure of cfDNA to the kidney or urothelial tract environments that could result in loss of tumor DNA in urine compared to plasma. However, despite these differences, urine should be further explored for its utility for cancer detection as discussed in the introduction.
For our approach, we report a relatively quantitative (normalized to input amplifiable DNA) measure of the abnormal methylation among cfDNA, and this regional DNA methylation was previously shown to be a cancer specific change (9,10). In lung cancer, detection of EGFR mutations in urine has been shown to be comparable to blood-based detection (13,48–50). This also appears to be from cfDNA, and sensitivities for detecting EGFR mutations in urine are similar to what we observe for metastatic lung cancer patients where urine mutation detection has been reported. In the majority of patients, there was concordance for methylation detection between plasma and urine. Discordance in cancer patients was split between positive methylation detection in urine and plasma, and most likely reflects stochastic detection from sampling very rare tumor cfDNA fragments. If true, additional sampling time points or greater volume of plasma or urine may increase sensitivity.
This study strongly suggests that methylation of specific genes hypermethylated in NSCLC can be detected in urine by using assay modifications for the more fragmented cfDNA found in urine. While this study did not fully address the clinical utility of this approach, as discussed in the methods section, patients selected for inclusion were 50 years and older and had a CT scan for suspicion of lung cancer referred to surgery for resection. The utility of methylation detection would likely not be for the smallest nodules (which are lowest risk and can be followed) or large lesions (where risk is high, and evaluation needed). Rather, the indeterminate nodule (8 to 20–30 mm) has a cancer risk where further discrimination is most needed where potentially this epigenetic liquid biopsy test could help.
This study as a first proof of principle has limitations. These include a relatively small sample size, with its associated limited power to accurately assess the sensitivity and specificity of methylation detection in urine. Additionally, since the current study included NSCLC patients of all stages, with only 65% of patients diagnosed at early stages and a relatively small number of controls, future prospective studies with larger sample sizes and limited to early cancer stage are needed to fully explore and validate this approach for early detection use.
CONCLUSION
This study suggests that epigenetic biomarkers from liquid biopsies based on methylation detection from plasma and urine could be used as an adjunct to CT screening to guide the decision-making regarding further invasive procedures in patients with pulmonary nodules. Plasma and urine methylation detection yield low false positive rates and the methylation of these genes is associated with a high NSCLC risk independent of age, race and smoking pack-year.
Supplementary Material
Translational relevance.
The National Lung Screening Trial showed that lung cancer screening can reduce lung cancer mortality 20% using low dose computed tomography. However, CT screening has a false discovery rate of nearly 96%. Biomarkers from liquid biopsy assays hold promise for enhancing the diagnostic accuracy of early stage lung cancer screening in conjunction with CT imaging. Urine samples have the potential to be easily implemented in a primary care practice. This study suggests that liquid biopsy biomarkers based on methylation detection from plasma and urine could be used as an adjunct to CT screening to help guide the decision to proceed with further invasive procedures, since plasma and urine yield low false positive rates and the methylation of these genes is associated with a high lung cancer risk independent of age, race and pack-year.
ACKNOWLEDGEMENTS
The University of Illinois at Chicago Cancer Center, EDRN U01CA214165-03, DOD W81XWH-12-1-0323, and in part, under a Grant with the Pennsylvania Department of Health. The Department specifically disclaims responsibility for any analyses, interpretations or conclusions.
Footnotes
Conflict of Interest Statement:
The authors of this manuscript report no relationship to disclose.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69(1):7–34 doi 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Noone AM, Cronin KA, Altekruse SF, Howlader N, Lewis DR, Petkov VI, et al. Cancer Incidence and Survival Trends by Subtype Using Data from the Surveillance Epidemiology and End Results Program, 1992–2013. Cancer Epidemiol Biomarkers Prev 2017;26(4):632–41 doi 10.1158/1055-9965.EPI-16-0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Detterbeck FC, Boffa DJ, Kim AW, Tanoue LT. The Eighth Edition Lung Cancer Stage Classification. Chest 2017;151(1):193–203 doi 10.1016/j.chest.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 4.Cronin KA, Lake AJ, Scott S, Sherman RL, Noone AM, Howlader N, et al. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 2018;124(13):2785–800 doi 10.1002/cncr.31551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.National Lung Screening Trial Research Team, Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011;365(5):395–409 doi 10.1056/NEJMoa1102873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinsky PF, Gierada DS, Black W, Munden R, Nath H, Aberle D, et al. Performance of Lung-RADS in the National Lung Screening Trial: a retrospective assessment. Ann Intern Med 2015;162(7):485–91 doi 10.7326/M14-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White CS, Dharaiya E, Dalal S, Chen R, Haramati LB. Vancouver Risk Calculator Compared with ACR Lung-RADS in Predicting Malignancy: Analysis of the National Lung Screening Trial. Radiology 2019;291(1):205–11 doi 10.1148/radiol.2018181050. [DOI] [PubMed] [Google Scholar]
- 8.Gierada DS, Pinsky P, Nath H, Chiles C, Duan F, Aberle DR. Projected outcomes using different nodule sizes to define a positive CT lung cancer screening examination. Journal of the National Cancer Institute 2014;106(11) doi 10.1093/jnci/dju284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wrangle J, Machida EO, Danilova L, Hulbert A, Franco N, Zhang W, et al. Functional identification of cancer-specific methylation of CDO1, HOXA9, and TAC1 for the diagnosis of lung cancer. Clin Cancer Res 2014;20(7):1856–64 doi 10.1158/1078-0432.CCR-13-2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hulbert A, Jusue-Torres I, Stark A, Chen C, Rodgers K, Lee B, et al. Early Detection of Lung Cancer Using DNA Promoter Hypermethylation in Plasma and Sputum. Clin Cancer Res 2017;23(8):1998–2005 doi 10.1158/1078-0432.CCR-16-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Su YH, Wang M, Brenner DE, Ng A, Melkonyan H, Umansky S, et al. Human urine contains small, 150 to 250 nucleotide-sized, soluble DNA derived from the circulation and may be useful in the detection of colorectal cancer. The Journal of molecular diagnostics : JMD 2004;6(2):101–7 doi 10.1016/S1525-1578(10)60497-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Millholland JM, Li S, Fernandez CA, Shuber AP. Detection of low frequency FGFR3 mutations in the urine of bladder cancer patients using next-generation deep sequencing. Res Rep Urol 2012;4:33–40 doi 10.2147/RRU.S32736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reckamp KL, Melnikova VO, Karlovich C, Sequist LV, Camidge DR, Wakelee H, et al. A Highly Sensitive and Quantitative Test Platform for Detection of NSCLC EGFR Mutations in Urine and Plasma. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2016;11(10):1690–700 doi 10.1016/j.jtho.2016.05.035. [DOI] [PubMed] [Google Scholar]
- 14.Hoque MO, Begum S, Topaloglu O, Jeronimo C, Mambo E, Westra WH, et al. Quantitative detection of promoter hypermethylation of multiple genes in the tumor, urine, and serum DNA of patients with renal cancer. Cancer research 2004;64(15):5511–7 doi 10.1158/0008-5472.CAN-04-0799. [DOI] [PubMed] [Google Scholar]
- 15.von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP, et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Ann Intern Med 2007;147(8):573–7 doi 10.7326/0003-4819-147-8-200710160-00010. [DOI] [PubMed] [Google Scholar]
- 16.Ettinger DS, Aisner DL, Wood DE, Akerley W, Bauman J, Chang JY, et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 5.2018. J Natl Compr Canc Netw 2018;16(7):807–21 doi 10.6004/jnccn.2018.0062. [DOI] [PubMed] [Google Scholar]
- 17.Bailey VJ, Zhang Y, Keeley BP, Yin C, Pelosky KL, Brock M, et al. Single-tube analysis of DNA methylation with silica superparamagnetic beads. Clinical chemistry 2010;56(6):1022–5 doi 10.1373/clinchem.2009.140244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keeley B, Stark A, Pisanic TR 2nd, Kwak R, Zhang Y, Wrangle J, et al. Extraction and processing of circulating DNA from large sample volumes using methylation on beads for the detection of rare epigenetic events. Clinica chimica acta; international journal of clinical chemistry 2013;425(C):169–75 doi 10.1016/j.cca.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brandes JC, Carraway H, Herman JG. Optimal primer design using the novel primer design program: MSPprimer provides accurate methylation analysis of the ATM promoter. Oncogene 2007;26(42):6229–37 doi 10.1038/sj.onc.1210433. [DOI] [PubMed] [Google Scholar]
- 20.Untergrasser ACI, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG 2012 Primer3web. <http://primer3.ut.ee/>. [DOI] [PMC free article] [PubMed]
- 21.Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic acids research 2000;28(8):E32 doi 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; 2019. [Google Scholar]
- 23.Bach PB, Schrag D, Brawley OW, Galaznik A, Yakren S, Begg CB. Survival of blacks and whites after a cancer diagnosis. Jama 2002;287(16):2106–13 doi 10.1001/jama.287.16.2106. [DOI] [PubMed] [Google Scholar]
- 24.Blackstock AW, Herndon JE 2nd, Paskett ED, Perry MC, Graziano SL, Muscato JJ, et al. Outcomes among African-American/non-African-American patients with advanced non-small-cell lung carcinoma: report from the Cancer and Leukemia Group B. Journal of the National Cancer Institute 2002;94(4):284–90 doi 10.1093/jnci/94.4.284. [DOI] [PubMed] [Google Scholar]
- 25.DeSantis CE, Siegel RL, Sauer AG, Miller KD, Fedewa SA, Alcaraz KI, et al. Cancer statistics for African Americans, 2016: Progress and opportunities in reducing racial disparities. CA Cancer J Clin 2016;66(4):290–308 doi 10.3322/caac.21340. [DOI] [PubMed] [Google Scholar]
- 26.Richards TB, Henley SJ, Puckett MC, Weir HK, Huang B, Tucker TC, et al. Lung cancer survival in the United States by race and stage (2001–2009): Findings from the CONCORD-2 study. Cancer 2017;123 Suppl 24:5079–99 doi 10.1002/cncr.31029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeSantis CE, Miller KD, Goding Sauer A, Jemal A, Siegel RL. Cancer statistics for African Americans, 2019. CA Cancer J Clin 2019;69(3):211–33 doi 10.3322/caac.21555. [DOI] [PubMed] [Google Scholar]
- 28.Kinsinger LS, Anderson C, Kim J, Larson M, Chan SH, King HA, et al. Implementation of Lung Cancer Screening in the Veterans Health Administration. JAMA Intern Med 2017;177(3):399–406 doi 10.1001/jamainternmed.2016.9022. [DOI] [PubMed] [Google Scholar]
- 29.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6(224):224ra24 doi 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phallen J, Sausen M, Adleff V, Leal A, Hruban C, White J, et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med 2017;9(403) doi 10.1126/scitranslmed.aan2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kennedy TC, Proudfoot SP, Franklin WA, Merrick TA, Saccomanno G, Corkill ME, et al. Cytopathological analysis of sputum in patients with airflow obstruction and significant smoking histories. Cancer research 1996;56(20):4673–8 doi papers2://publication/uuid/270AD142-0700-4EB0-A6E1-662170FFC855. [PubMed] [Google Scholar]
- 32.Kennedy TC, Proudfoot SP, Piantadosi S, Wu L, Saccomanno G, Petty TL, et al. Efficacy of two sputum collection techniques in patients with air flow obstruction. Acta Cytol 1999;43(4):630–6 doi 10.1159/000331157. [DOI] [PubMed] [Google Scholar]
- 33.Prindiville SA, Byers T, Hirsch FR, Franklin WA, Miller YE, Vu KO, et al. Sputum cytological atypia as a predictor of incident lung cancer in a cohort of heavy smokers with airflow obstruction. Cancer Epidem Biomar 2003;12(10):987–93 doi papers2://publication/uuid/1DA01259-AC9B-4F24-BF1E-C75E39766623. [PubMed] [Google Scholar]
- 34.Belinsky SA, Klinge DM, Dekker JD, Smith MW, Bocklage TJ, Gilliland FD, et al. Gene promoter methylation in plasma and sputum increases with lung cancer risk. Clin Cancer Res 2005;11(18):6505–11 doi 10.1158/1078-0432.CCR-05-0625. [DOI] [PubMed] [Google Scholar]
- 35.Belinsky SA, Liechty KC, Gentry FD, Wolf HJ, Rogers J, Vu K, et al. Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer research 2006;66(6):3338–44 doi 10.1158/0008-5472.CAN-05-3408. [DOI] [PubMed] [Google Scholar]
- 36.Ostrow KL, Hoque MO, Loyo M, Brait M, Greenberg A, Siegfried JM, et al. Molecular analysis of plasma DNA for the early detection of lung cancer by quantitative methylation-specific PCR. Clin Cancer Res 2010;16(13):3463–72 doi 10.1158/1078-0432.CCR-09-3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leng S, Do K, Yingling CM, Picchi MA, Wolf HJ, Kennedy TC, et al. Defining a gene promoter methylation signature in sputum for lung cancer risk assessment. Clin Cancer Res 2012;18(12):3387–95 doi 10.1158/1078-0432.CCR-11-3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pulling LC, Divine KK, Klinge DM, Gilliland FD, Kang T, Schwartz AG, et al. Promoter hypermethylation of the O6-methylguanine-DNA methyltransferase gene: more common in lung adenocarcinomas from never-smokers than smokers and associated with tumor progression. Cancer research 2003;63(16):4842–8. [PubMed] [Google Scholar]
- 39.Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nature reviews Cancer 2004;4(9):707–17 doi 10.1038/nrc1432. [DOI] [PubMed] [Google Scholar]
- 40.Gao X, Zhang Y, Breitling LP, Brenner H. Tobacco smoking and methylation of genes related to lung cancer development. Oncotarget 2016;7(37):59017–28 doi 10.18632/oncotarget.10007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Elgizouli M, Schottker B, Holleczek B, Nieters A, Brenner H. Smoking-associated DNA methylation markers predict lung cancer incidence. Clin Epigenetics 2016;8(1):127 doi 10.1186/s13148-016-0292-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahrendt SA, Chow JT, Xu LH, Yang SC, Eisenberger CF, Esteller M, et al. Molecular detection of tumor cells in bronchoalveolar lavage fluid from patients with early stage lung cancer. Journal of the National Cancer Institute 1999;91(4):332–9 doi 10.1093/jnci/91.4.332. [DOI] [PubMed] [Google Scholar]
- 43.Sanchez-Cespedes M, Esteller M, Wu L, Nawroz-Danish H, Yoo GH, Koch WM, et al. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer research 2000;60(4):892–5. [PubMed] [Google Scholar]
- 44.Silvestri GA, Vachani A, Whitney D, Elashoff M, Porta Smith K, Ferguson JS, et al. A Bronchial Genomic Classifier for the Diagnostic Evaluation of Lung Cancer. N Engl J Med 2015;373(3):243–51 doi 10.1056/NEJMoa1504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Keefe CM, Pisanic TR 2nd, Zec H, Overman MJ, Herman JG, Wang TH. Facile profiling of molecular heterogeneity by microfluidic digital melt. Sci Adv 2018;4(9):eaat6459 doi 10.1126/sciadv.aat6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pisanic TR 2nd, Athamanolap P, Poh W, Chen C, Hulbert A, Brock MV, et al. DREAMing: a simple and ultrasensitive method for assessing intratumor epigenetic heterogeneity directly from liquid biopsies. Nucleic acids research 2015;43(22):e154 doi 10.1093/nar/gkv795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen C, Huang X, Yin W, Peng M, Wu F, Wu X, et al. Ultrasensitive DNA hypermethylation detection using plasma for early detection of NSCLC: a study in Chinese patients with very small nodules. Clin Epigenetics 2020;12(1):39 doi 10.1186/s13148-020-00828-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tchekmedyian N, Mudad R, Blanco FF, Raymond VM, Garst J, Erlander MG, et al. Longitudinal monitoring of ctDNA EGFR mutation burden from urine correlates with patient response to EGFR TKIs: A case series. Lung cancer 2017;108:22–8 doi 10.1016/j.lungcan.2017.02.010. [DOI] [PubMed] [Google Scholar]
- 49.Husain H, Melnikova VO, Kosco K, Woodward B, More S, Pingle SC, et al. Monitoring Daily Dynamics of Early Tumor Response to Targeted Therapy by Detecting Circulating Tumor DNA in Urine. Clin Cancer Res 2017;23(16):4716–23 doi 10.1158/1078-0432.CCR-17-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berz D, Raymond VM, Garst JH, Erlander MG. Non-invasive urine testing of EGFR activating mutation and T790M resistance mutation in non-small cell lung cancer. Exp Hematol Oncol 2015;5:24 doi 10.1186/s40164-016-0052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.