

Abstract
Purpose:
Radiation and cetuximab are therapeutics used in management of head and neck squamous cell carcinoma (HNSCC). Despite clinical success with these modalities, development of both intrinsic and acquired resistance is an emerging problem in the management of this disease. The purpose of this study was to investigate signaling of the receptor tyrosine kinase AXL in resistance to radiation and cetuximab treatment.
Experimental Design:
To study AXL signaling in the context of treatment-resistant HNSCC, we used patient-derived xenografts (PDXs) implanted into mice and evaluated the tumor response to AXL inhibition in combination with cetuximab or radiation treatment. To identify molecular mechanisms of how AXL signaling leads to resistance, three tyrosine residues of AXL (Y779, Y821, Y866) were mutated and examined for their sensitivity to cetuximab and/or radiation. Furthermore, reverse phase protein array (RPPA) was employed to analyze the proteomic architecture of signaling pathways in these genetically altered cell lines.
Results:
Treatment of cetuximab- and radiation-resistant PDXs with AXL inhibitor R428 was sufficient to overcome resistance. RPPA analysis revealed that such resistance emanates from signaling of tyrosine 821 of AXL via the tyrosine kinase c-ABL. In addition, inhibition of c-ABL signaling resensitized cells and tumors to cetuximab or radiation therapy even leading to complete tumor regression without recurrence in head and neck cancer models.
Conclusions:
Collectively, the studies presented herein suggest that tyrosine 821 of AXL mediates resistance to cetuximab by activation of c-ABL kinase in HNSCC and that targeting of both EGFR and c-ABL leads to a robust anti-tumor response.
Keywords: AXL, c-ABL Kinase, Head and Neck Cancer, Cetuximab, Radiation, Resistance
Introduction
Globally, head and neck squamous cell carcinoma (HNSCC) represents about 5% of all cancer deaths with almost 900,000 people being newly diagnosed each year (1). HNSCCs arise from the mucosal lining of the aerodigestive tract and are typically treated with surgery, radiation, chemotherapy, and/or molecular-targeted therapy. Because HNSCC characteristically overexpresses the epidermal growth factor receptor (EGFR), a monoclonal antibody for this receptor called cetuximab is commonly used as a treatment option. The addition of cetuximab to radiotherapy or chemotherapy regimens improved overall survival of HNSCC patients as compared to radiotherapy or chemotherapy alone (2,3).
Although cetuximab remains a viable therapeutic option for HNSCC patients, resistance—both intrinsic and acquired—is a major clinical hurdle. Many mechanisms of resistance to cetuximab have been identified including signaling by the receptor tyrosine kinase AXL (4). AXL is a member of the TAM family of receptors and has been shown to mediate resistance to several treatment modalities including radiation (5–7), chemotherapy (6,8–10), and molecular-targeted therapy (11–23). Because of AXL’s prominence in mediating resistance, strategies to inhibit AXL signaling have developed rapidly in order to resensitize cancers to treatment approaches.
Based on previous studies of AXL, we investigated the role of AXL signaling in cetuximab- and radiation-resistant HNSCC with a focus on understanding the signaling cascades emanating from the C-terminal tail of AXL. Using the small molecule inhibitor R428, AXL blockade resensitized tumors to both cetuximab and radiation treatment. In addition, mapping of C-terminal tyrosines of AXL, indicated that tyrosine 821 (Y821) confers resistance to cetuximab and radiation. Specifically, mutation at Y821, but not Y779 or Y866, reduced c-ABL tyrosine kinase expression. Furthermore, inhibition of c-ABL signaling—both in intrinsic and acquired models of resistance—was able to resensitize cells and tumors to cetuximab and radiation treatment. Collectively, these results indicate that AXL plays a role in cetuximab and radiation resistance via signaling through Y821 to c-ABL kinase. This data provides rationale for clinical evaluation of therapeutics targeting AXL and c-ABL in the context of cetuximab- and/or radiation-resistant disease.
Materials and Methods:
Materials
Cetuximab (IMC-225, Erbitux) was purchased from the University of Wisconsin Pharmacy. R428 (BGB324, Bemcentinib) and imatinib were purchased from Selleck Chemicals (Houston, TX), Medchemexpress LLC (Monmouth Junction, NJ) or Apexbio (Houston, TX). PPY-A was obtained from R&D systems (Minneapolis, MN). DMSO was used as the vehicle control in vitro. In vivo human IgG (MilliporeSigma, St. Louis, MO) was the control for cetuximab, hydroxypropyl methylcellulose (0.5%)/Tween80(0.1%) was the vehicle for R428, and DMSO(2%)/PEG300(30%)/Tween80(5%) was the vehicle for imatinib.
Cell line
HN30 cell line was a gift from Dr. Ravi Salgia and cultured in DMEM with 4.5g/dL glucose, 10%FBS, penicillin (100units/mL), streptomycin (100mg/mL). Cell line identity was confirmed using short tandem repeat (STR) analysis and publicly available databases by TRIP lab at University of Wisconsin. Mycoplamsa testing was completed through WiCell Core Service at University of Wisconsin.
Plasmids and transfection
pDONR223-AXL was a gift from William Hahn & David Root (Addgene plasmid #23945) and subcloned into the BamH1/EcoR1 restriction sites of the pcDNA6.0 expression vector (Life Technologies). Site-directed mutagenesis was performed to generate an AXL-Y821F mutant expression construct using a QuikChange II Site-Directed Mutagenesis Kit according to the manufacturer’s instructions. AXL-Y779F and AXL-Y866F mutants were synthesized by GenScript (Piscataway, NJ). The presence of 779F, 821F, and 866F mutations were confirmed by DNA sequencing. Transfection was performed using Lipofectamine3000 and Opti-MEMI (Life Technology) according to the manufacturer’s instructions. Blasticidin (3ug/mL) was used as the antibiotic for clonal selection.
siRNA transfection
Non-targeting control pool siRNA (Cat#D-001810) and SMARTpool siRNA targeting ABL1 (Cat#L-003100) were purchased from Dharmacon, Inc (Lafayette, CO) and utilized for transfection with Lipofectamine RNAiMAX (Life Technologies).
Cell proliferation assay and clonogenic assay
Cell proliferation and clonogenic assays using crystal violet were performed as described previously (24,25). All treatments were performed in triplicate.
Irradiation
Cells were irradiated with a Xstrahl X-ray System, Model RS225 (Xstrahl, UK) at a dose rate of 3.27 Gy/min at 30 cm FSD, tube voltage of 195 kV, current of 10 mA and filtration with 3 mm Al. Animals were irradiated with a Precision Xray XRAD 320 with 1 Gy/minute delivered at 320 kV/12.5 mA at 50 cm FSD with a beam hardening filter with half-value layer of 4 mm Cu. The delivered dose rate was confirmed by ionization chamber. Mice were shielded with custom-built lead jigs to limit radiation exposure to the rear quarter of the body.
Xenograft Flank Models
Female athymic nude mice (4–6 weeks old) were obtained from Envigo (Indianapolis, IN). Animal procedures and maintenance were conducted in accordance with institutional guidelines of University of Wisconsin. Patient derived xenografts (PDXs) or cell lines were inoculated by subcutaneous injection into the dorsal flank of each mouse and tumor volume was measured using a caliper. When tumors attained a volume of ~200mm3, mice were randomized into groups and treatment was initiated. Inhibitor or an equivalent volume of vehicle were administered by intraperitoneal injection (cetuximab/IgG) or oral gavage (R428, imatinib, vehicle). Tumors were collected within 3 hours of the last treatment for analysis of biochemical markers.
Immunoblot analysis
Whole-cell protein lysis, immunoprecipitation, and immunoblot analysis were performed as previously described (24). Antibodies were used according to the manufacturer’s instructions: AXL for immunoblot (Cell Signaling Technologies (CST) #8661), AXL for immunoprecipitation (Santa Cruz Biotechnology #166269), c-ABL (CST #2862), p-c-ABL-Y412 (Abcam #4717), EGFR (CST #4267), GAPDH (CST #5174), α-tubulin (MilliporeSigma #CP06).
Immunohistochemistry
Tumors were processed for immunohistochemistry as previously described (24). Ki67 (CST #9027, 1:400) antibody was used and bound antibodies were detected using the VECTASTAIN Universal Kit/HRP (Vector Laboratories) and 3,3’-diaminobenzidine substrates. Images are shown at a magnification of 20X and were quantified in three separate areas by counting the number of positive cells and creating an average and standard error that is presented in graphic form.
Reverse Phase Protein Array
Cells were plated on six-well plates and treated with 4Gy radiation the next day. Protein from 3 treated and 3 untreated (control) plates was isolated 4 hours or 24 hours after radiation treatment. Protein samples were submitted to the RPPA Core at MD Anderson for analysis.
Statistical analysis
Student’s t-test was used for comparisons between two groups. Differences were considered significant when P<.05. Due to the exploratory nature of this study, no multiple-testing corrections were applied to p-values.
To analyze the longitudinal tumor growth data (volume), linear mixed models were fit to the log-transformed data (beginning at treatment start date) using the ‘lme4’ package in R (V3.6.1). The fixed-effect model matrices were parameterized such that all main and interactive effects between treatment groups (drug1, drug2, vehicle and drug1+drug2) and time were estimated with the random-effect structure accommodated for inter-mouse variation via random intercepts. All models were estimated using the restricted maximum likelihood estimation (REML) criterion. Reported model p-values were estimated using Satterthwaite’s approximation as implemented in the ‘lmerTest’ package. Model assumptions were assessed using graphical analysis of model residuals and QQ plots about the estimated random effects (49–51).
For the two-drug experiments, a synergistic effect between the two treatments was assessed via inference about the three-way interaction between time, first drug, and second drug. Note that the HN30-AXL-WT cetuximab+imatinib experiment, the UW-SCC64 cetuximab+imatinib experiment, and the UW-SCC64 XRT+imatinib experiment were analyzed on the measured scale due to the presence of zero-volume, fully shrunk tumors (Figures 4A, 4C, and 6C).
Figure 4: c-ABL targeting overcomes cetuximab resistance.
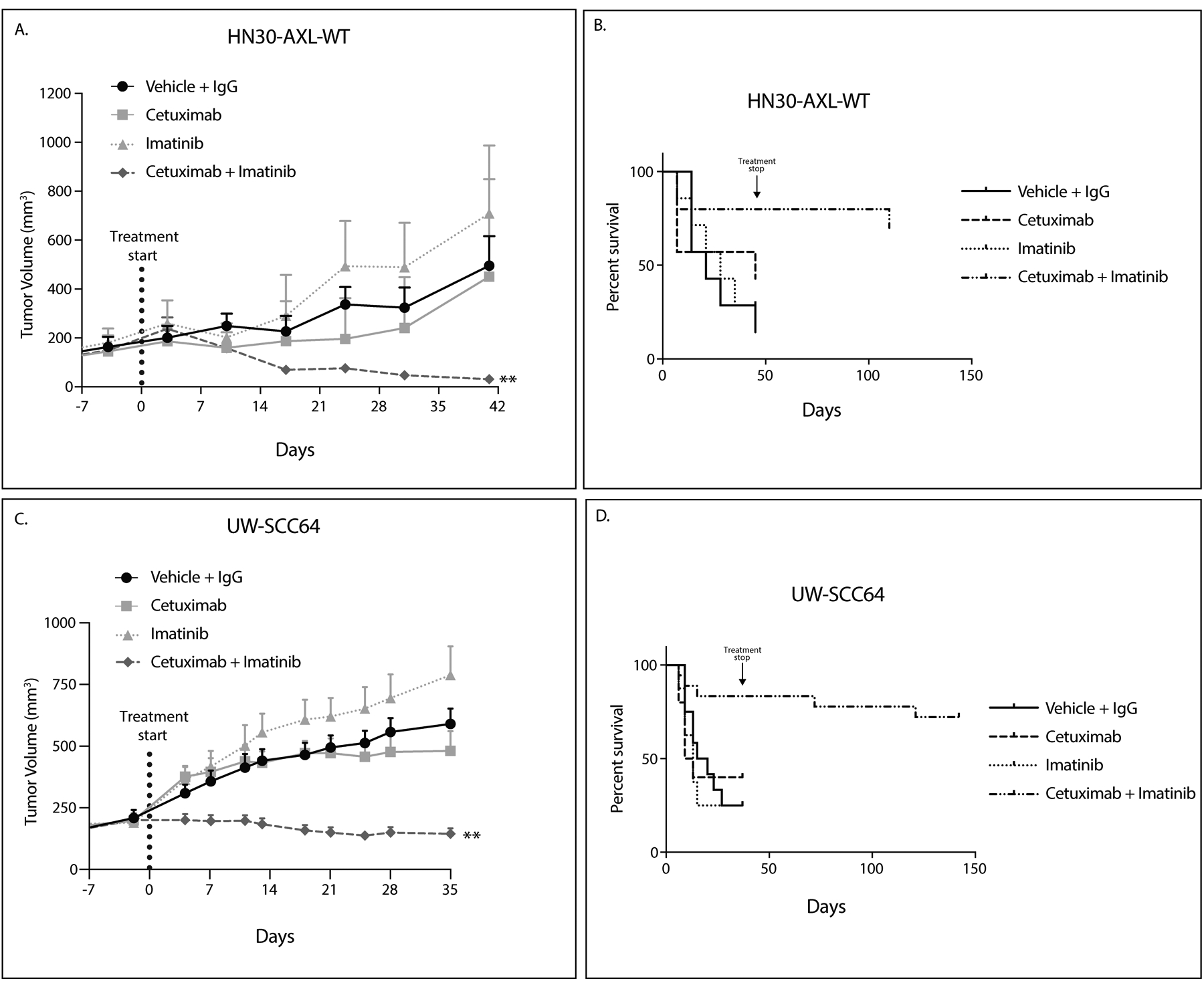
A,C: HN30-AXL-WT cells (A) or PDX UW-SCC64 (C) were implanted into flanks of nude mice and treated with vehicle (oral gavage, methylcellulose, twice daily) and human IgG (intraperitoneal, twice weekly) (HN30-AXL n=7, UW-SCC64 n=12), cetuximab (intraperitoneal, 0.2 mg/mouse, twice weekly) (HN30-AXL n=7, UW-SCC64 n=10), imatinib (oral gavage, 30mg/kg, once daily) (HN30-AXL n=7, UW-SCC64 n=8), or combination of cetuximab with imatinib (HN30-AXL n=10, UW-SCC64 n=18). Data was analyzed using linear mixed models. Volume data was untransformed given the presence of fully shrank tumors, resulting in volume readings of zero. **, P<.01 for combination compared to single treatments.
B, D: Data for studies in panels A and C are presented regarding overall survival. Survival was evaluated using tumor doubling time with “death” occurring when a tumor reached double its original volume. For HN30-AXL-WT, 5 of 10 tumors were still undetectable at 69 days after treatment was stopped. For UW-SCC64, 12 of 18 tumors were still not visible 107 days after treatment was stopped.
Figure 6: c-ABL plays a role in radiation resistance.
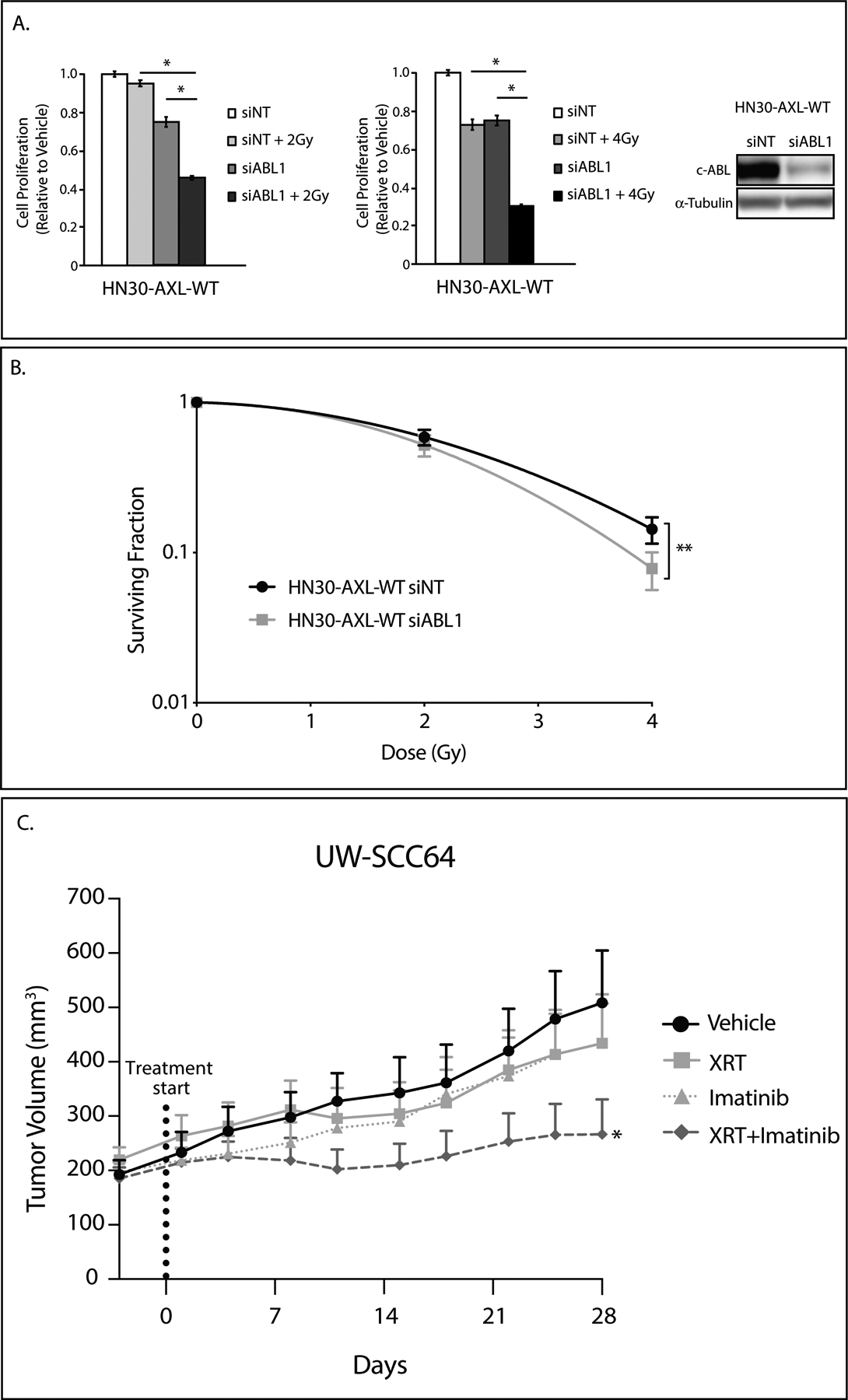
A: HN30-AXL-WT cells were transfected with siABL1 or nontargeting siRNA (siNT). 24 hours later, cells were re-plated at a lower confluency. 24 hours later, cells were treated with 2Gy or 4Gy of radiation and after 72 hours of treatment, relative colony numbers were determined by crystal violet assay. siNT and siABL1 treatment data are the same for both graphs. Mean values and SEs were derived from replicates within one experiment. Statistical analyses are representative of three independent experiments. *, P<.05. siABL1 knockdown of c-ABL was evaluated by immunoblot analysis of cell lysates with α-tubulin as loading control.
B: Radiosensitivity of HN30-AXL-WT transfected with siABL1 or siNT was evaluated by clonogenic survival analysis. Cells were transfected and 24 hours later re-plated at 200 cells per well. 24 hours later, cells were treated with radiation (0Gy, 2Gy, or 4Gy). 14 days later colonies were analyzed. Mean values, SEs, and statistical analyses were calculated from replicates within four experiments. **, P<.01.
C: PDX UW-SCC64 was implanted into flanks of nude mice and treated with vehicle (oral gavage, methylcellulose, once daily) (n=11), XRT (3Gy twice weekly) (n=10), imatinib (oral gavage, 30mg/kg, once daily) (n=11), or combination of radiation with imatinib (n=18) for 28 days. Data was analyzed using linear mixed models where volume was untransformed given the presence of fully shrank tumors, resulting in volume readings of zero. *, P<.05 for combination compared to single treatments.
In addition to this synergistic analysis, the fractional product method was used to evaluate therapeutic interactions between inhibitors in the two-drug in vivo experiments (26)(27). This method utilizes average final tumor volumes with each individual agent to calculate the expected (E) effect for an additive interaction as a product of the individual responses. The observed (O) effect is the final average tumor volume following combination treatment. The ratio of observed to expected (O:E) values was calculated and used to indicate synergy (O:E<1), additivity (O:E=1), or antagonism (O:E>1).
Results
AXL targeting overcomes cetuximab resistance
Previously, we identified that AXL plays a critical role in mediating resistance to cetuximab therapy in vitro using established HNSCC cell lines (4). Here, we advanced these findings using established PDXs with known cetuximab responses: UW-SCC1 (1A) and UW-SCC64 (1B) as previously published (28) (clinical characteristics in Supplemental Table S1). We implanted these two PDXs into the flanks of nude mice and treated with vehicle and IgG, cetuximab (0.2 mg/mouse, twice weekly), R428 (25mg/kg, twice daily), or the combination (n=9–14 tumors/treatment group) (Figure 1A and 1B). In both studies, there was noticeable growth delay of tumors receiving combination therapy indicating that blockade of AXL signaling could resensitize tumors to cetuximab treatment. In regard to UW-SCC1, the model fit to the data suggested evidence of a synergistic effect between R428 and cetuximab (P<.0001). While there was no evidence of a synergistic effect between the two drugs in UW-SCC64 PDX (P=.67), the slope of the tumor-growth curve for combination treatment was still significantly less than that of the cetuximab-only group (P<.001) and analysis by the fractional product method indicated that the combination therapy worked better together than either drug alone (Supplemental Figure S6). Tumors from these studies were stained for Ki67 expression—a nuclear marker of proliferation (Figure 1C and 1D) and evaluated for the presence of apoptotic bodies (Supplemental Figure S2). Quantification of Ki67 expression revealed that tumors treated with the combination therapy demonstrated reduced Ki67 positive staining as compared to cetuximab or R428 therapies alone while the presence of apoptotic bodies increased after combination therapy. This data supported additional studies showing that Ki67 can be used as a surrogate biomarker for cetuximab response in head and neck xenografts (Supplemental Figure S1). These results indicate the advantageous effect of R428 on cetuximab-resistant tumors and that Ki67 could be a marker of response of resensitization to cetuximab from AXL targeting.
Figure 1: AXL targeting overcomes cetuximab resistance.
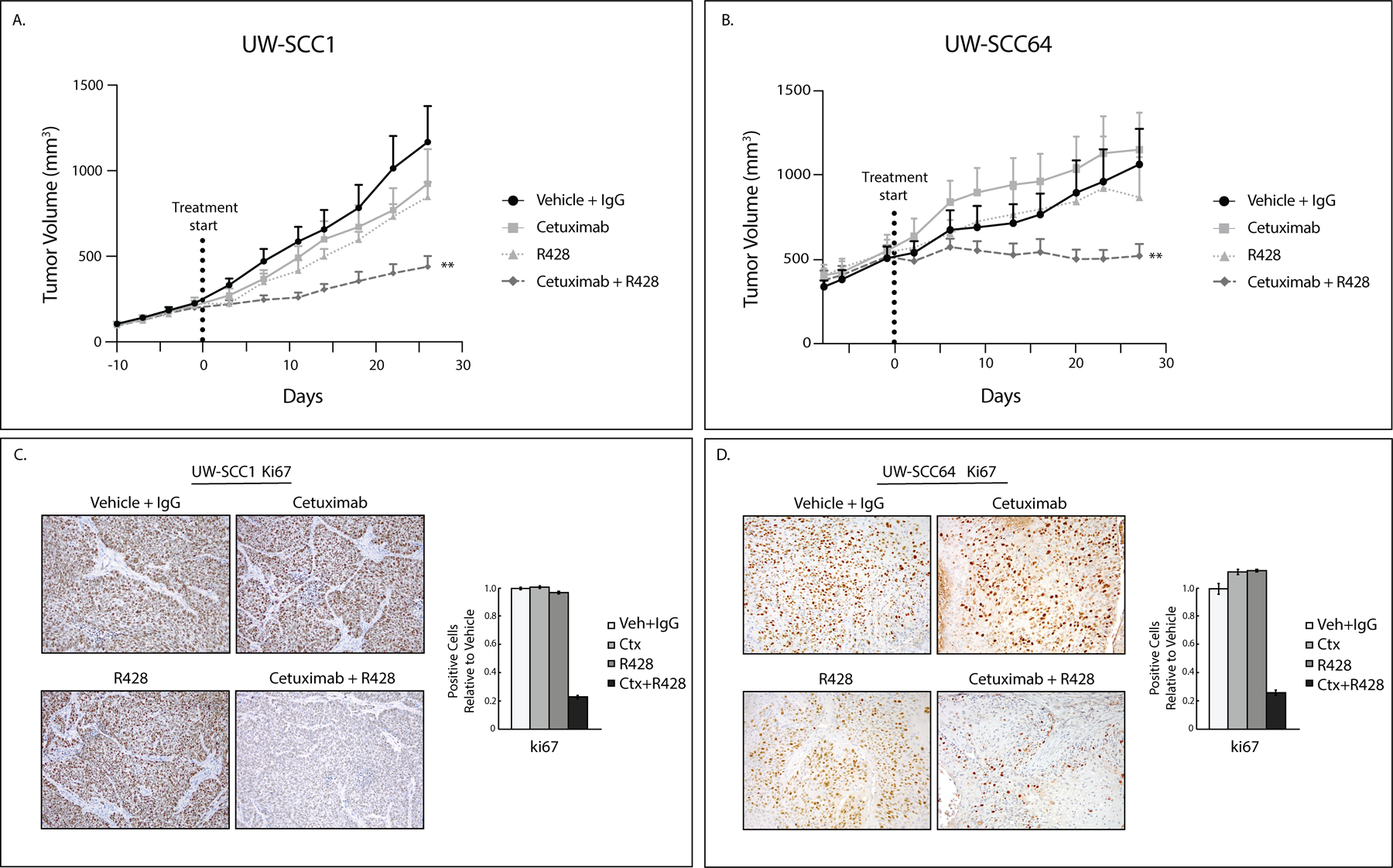
A, B: UWSCC PDXs (1 and 64) were implanted into flanks of nude mice and treated with vehicle (oral gavage, methylcellulose, twice daily) and human IgG (intraperitoneal, twice weekly) (UW-SCC1 n=9, UW-SCC64 n=13), cetuximab (intraperitoneal, 0.2 mg/mouse, twice weekly) (UW-SCC1 n=9, UW-SCC64 n=11), R428 (oral gavage, 25mg/kg, twice daily) (UW-SCC1 n=10, UW-SCC64 n=10), or combination of cetuximab with R428 for 28 days (UW-SCC1 n=9, UW-SCC64 n=14). Data was analyzed using linear mixed models where volume was modeled on natural-log scale. **, P<.01 for UW-SCC1 combination compared to single treatments and UW-SCC64 combination compared to cetuximab only.
C, D: Tumors from studies shown in 1A and 1B were stained for Ki67 using IHC. Image quantification shown.
Y821 of AXL is critical for mediating resistance to cetuximab
To determine how AXL mediates cetuximab resistance, we mutated three critical signaling tyrosines Y779, Y821, and Y866 to phenylalanine (29). These mutants, along with AXL wildtype (WT), were stably overexpressed in the cetuximab-sensitive cell line HN30 and tested for response to cetuximab in vitro (Figure 2A). HN30-Vector was sensitive to cetuximab treatment whereas the HN30-AXL-WT cells were resistant. Furthermore, the HN30-AXL-Y779F cells and HN30-AXL-Y866F cells were both resistant to cetuximab with a less than a 20% decrease in cell number; however, the HN30-AXL-Y821F cells (clone 2 and clone 4) were sensitive to cetuximab treatment with a greater than 30% response. P-values were calculated using treatment response as compared via student’s t-test to the AXL-WT overexpressing cell line. To verify that the AXL-Y821F mutation inhibited AXL activity, HN30-AXL-Y821F along with HN30-AXL-WT cells were treated with the combination of cetuximab and R428. Although treatment with R428 resensitized HN30-AXL-WT cells to cetuximab therapy, R428 did not enhance the growth inhibitory effects of cetuximab in cells bearing the Y821 mutation (Supplemental Figure S3). To confirm the sensitivity of Y821 mutants to cetuximab, we implanted HN30-Vector, HN30-AXL-WT, and HN30-AXL-Y821F-C2 cells into flanks of nude mice and treated with IgG or cetuximab (0.2 mg/mouse) twice weekly (n=13–16 tumors/treatment group) (Figure 2B). The growth curve for cetuximab-treated tumors within HN30-Vector and HN30-AXL-Y821F experiments were significantly delayed in growth as compared to that of IgG group (P<.0001 for both) indicating sensitivity. No significant difference in growth curves was found between cetuximab and IgG for HN30-AXL-WT tumors (P=.11) indicating resistance. These results demonstrate that Y821 of AXL could regulate a signaling pathway that is critical for mediating resistance to cetuximab treatment.
Figure 2: Y821 of AXL is critical for resistance to cetuximab.
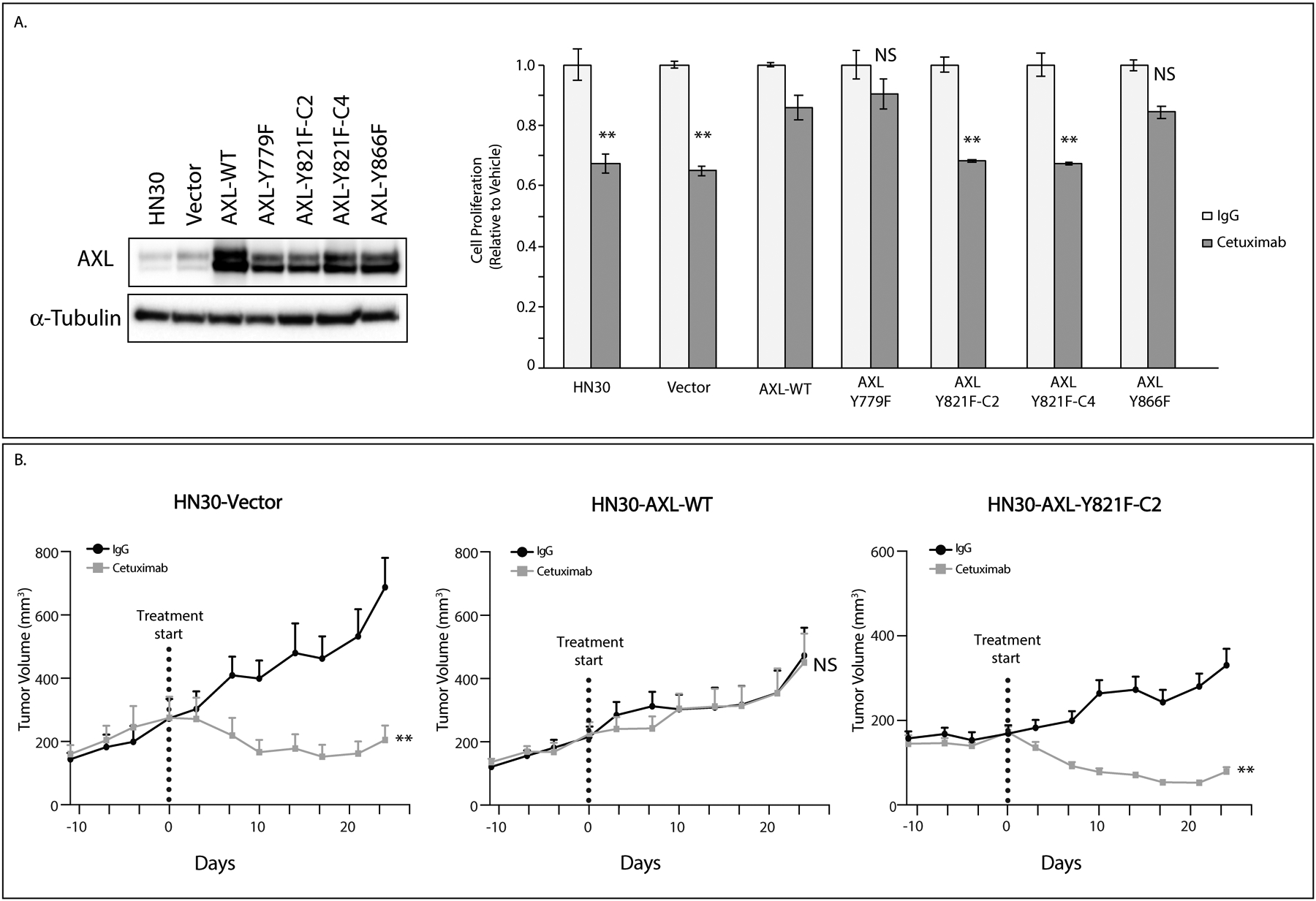
A: pcDNA6.0-Vector control, pcDNA6.0-AXL-WT, and pcDNA6.0-AXL mutants (Y779F, Y821F, and Y866F) were stably overexpressed in HN30 cell line. Cells were treated with IgG or cetuximab (100nM) for 72 hours and relative cell numbers were determined by crystal violet assay. Mean values and SEs were derived from replicates within one experiment. Statistical analyses compare cetuximab response to AXL-WT cells and are representative of four independent experiments. **, P<.01. Endogenous AXL expression was determined by immunoblot analysis of cell lysates with α-tubulin as loading control.
B: HN30-Vector, HN30-AXL-WT, and HN30-AXL-Y821F-C2 cells were injected into flanks of nude mice and treated with IgG or cetuximab (0.2mg/mouse) twice weekly for 28 days. Data was analyzed using linear mixed models where volume was modeled on natural-log scale. **, P<.01; NS, not significant. Tumor numbers: HN30-Vect: IgG n=13, Ctx n=14. HN30-AXL-WT: IgG n=14, Ctx n=14. HN30-AXL-Y821F-C2: IgG n=16, Ctx n=15.
c-ABL plays a role in cetuximab resistance via tyrosine 821 of AXL
To explore signaling pathways that AXL uses to mediate cetuximab resistance, we performed Reverse Phase Protein Array (RPPA) analysis which used 304 antibodies to detect endogenous protein level changes between HN30-Vector, HN30-AXL-WT, and HN30-AXL-Y821F(clone 2) cells. From normalized values provided, protein expression of c-ABL kinase was higher in HN30-AXL-WT cells while lower in the HN30-AXL-Y821F mutant cells (Figure 3A). To validate this finding, we evaluated endogenous levels of c-ABL kinase and phosphorylated c-ABL in the HN30-AXL-WT and mutant cell lines via immunoblot (Figure 3B). There was less total and activated c-ABL in the HN30-AXL-Y821F cells as compared to HN30-AXL-WT, HN30-AXL-Y779F, and HN30-AXL-Y866F cells. This correlation suggests that Y821 of AXL signaling may lead to regulation of c-ABL expression. Since HN30-AXL-Y821F cells were also sensitive to cetuximab treatment (Figure 2A), we hypothesized that c-ABL may be playing a role in cetuximab resistance. To test this hypothesis, we knocked down ABL with siRNA transfection and treated cells with cetuximab (Figure 3C). Loss of c-ABL expression resensitized to cetuximab. Pharmacological inhibition of c-ABL signaling with the ABL kinase inhibitor PPY-A in cetuximab-resistant HN30-AXL-WT cells was also sufficient to resensitize cells to cetuximab treatment (Figure 3D). Furthermore, we observed elevated expression levels of c-ABL in a model of HN30 cells with acquired resistance to cetuximab and resensitization to cetuximab upon ABL inhibition via siRNA or PPY-A treatment (Supplemental Figure S4). To confirm that AXL is signaling through c-ABL, HN30-AXL-WT cells were transfected with siABL1 and treated with cetuximab and R428 (Supplemental Figure S5). Treatment with R428 did not provide an additional decrease in cell proliferation to siABL1 cells treated with cetuximab (siABL1+Ctx vs siABL1+Ctx+R428). There was a moderate additional decrease in proliferation when c-ABL was knocked down in cells treated with the combination of R428 and cetuximab (R428+Ctx vs R428+Ctx+siABL1). These data indicate that genetic modulation of c-ABL downstream of AXL is advantageous. Collectively, these results indicate that AXL is signaling through Y821 to c-ABL kinase to promote cetuximab resistance.
Figure 3: c-ABL plays a role in cetuximab resistance.
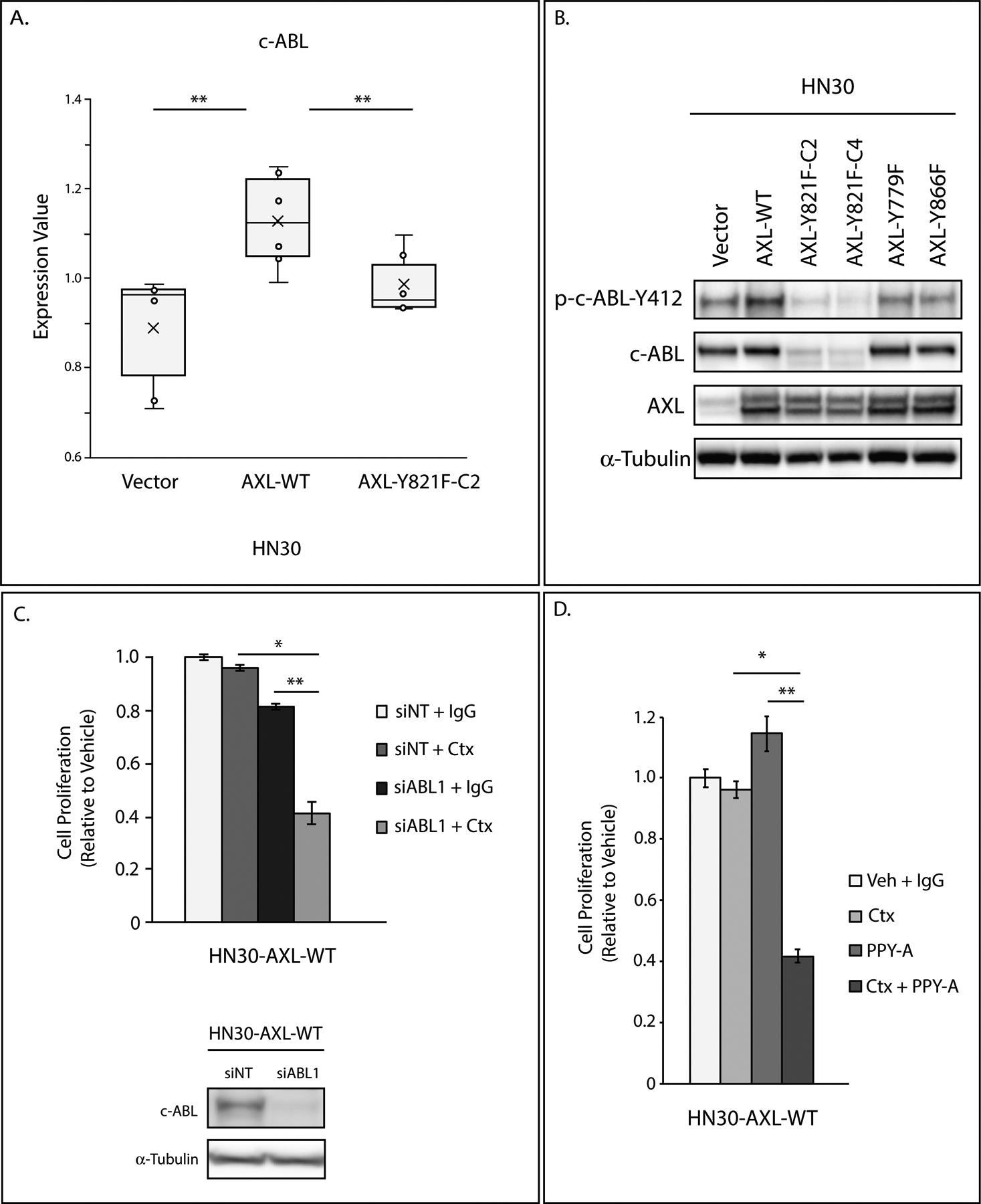
A: Lysate of HN30-Vector, HN30-AXL-WT, and HN30-AXL-Y821F(clone 2) cells were analyzed via RPPA. Expression values of c-ABL are presented in box plot format. Mean, SEs, and student’s t-test were calculated based on 3 replicates from two experiments. **, P<.01.
B: Endogenous phospho-c-ABL-Y412, c-ABL, and AXL expression were determined by immunoblot analysis of cell lysates from HN30-Vector, HN30-AXL-WT, and HN30-AXL mutants (Y779F, Y821F, Y866F) with α-tubulin as loading control.
C: HN30-AXL-WT cells were transfected with siABL1 or nontargeting siRNA (siNT). 24 hours later, cells were treated with IgG or cetuximab (100nM), and after 72 hours of treatment, relative cell numbers were determined by crystal violet assay. Mean values and SEs were derived from replicates within one experiment. Statistical analyses are representative of three independent experiments. *, P<.05; **, P<.01. siABL1 knockdown of c-ABL was evaluated by immunoblot analysis of cell lysates with α-tubulin as loading control.
D: HN30-AXL-WT cells were treated with DMSO and IgG, cetuximab (100nM), PPY-A (500nM), or combination of cetuximab and PPY-A for 72 hours and relative cell numbers were determined by crystal violet assay. Mean values and SEs were derived from replicates within one experiment. Statistical analyses are representative of five independent experiments. *, P<.05; **, P<.01.
c-ABL targeting overcomes cetuximab resistance, leading to tumor regression and limiting tumor recurrence
Because inhibition of c-ABL was able to resensitize to cetuximab in vitro, we hypothesized that c-ABL inhibition could enhance cetuximab sensitivity in vivo and may represent a novel therapeutic approach in patients who are refractory to cetuximab therapy or to increase the response to therapy.
To investigate this, we first used the genetically modified cetuximab resistant HN30-AXL-WT cells. These were implanted into flanks of nude mice and treated with vehicle and IgG, cetuximab (0.2 mg/mouse, twice weekly), ABL inhibitor imatinib (30mg/kg, once daily), or combination (n=7–10 tumors/treatment group) (Figure 4A). A synergistic effect of cetuximab and imatinib was seen in HN30-AXL-WT tumors (P<.001). After 41 days of treatment for HN30-AXL-WT, visible tumors from each treatment group were collected while mice from combination therapy group that did not have visible tumors were kept for observation. 5 of 10 HN30-AXL-WT tumors were still undetectable at 69 days after treatment was stopped. To better visualize the tumor regression of this study, a Kaplan-Meier curve was generated using doubling time of tumor volumes as criteria for “death” (Figure 4B). This curve shows that only 3 of 10 HN30-AXL-WT tumors doubled in volume in entirety of the study. This data suggests that not only can inhibition of c-ABL overcome cetuximab resistance, but it can lead to complete tumor regression with limited recurrence.
To test this in a more clinically relevant model, we used PDX UW-SCC64. This was implanted into flanks of nude mice and treated with vehicle and IgG, cetuximab (0.2 mg/mouse, twice weekly), ABL inhibitor imatinib (30mg/kg, once daily), or combination (n=8–18 tumors/treatment group) (Figure 4C). Again, a synergistic effect between cetuximab and imatinib (P<.0001). After 35 days of treatment for UW-SCC64, visible tumors from each treatment group were collected while mice from combination therapy group that did not have visible tumors were kept for observation. For UW-SCC64 tumors, 12 of 18 tumors were still not visible 107 days after treatment was stopped. The Kaplan-Meier curve generated using doubling time of tumor volumes as criteria for “death” (Figure 4D) shows that only 5 of 18 UW-SCC64 tumors doubled in volume in entirety of the study. In both studies, there was a dramatic delay in growth of tumors receiving combination therapy and several tumors regressed and remained completely undetectable in both models tested. This remarkable vanishing of 50–67% of tumors after receiving combination treatment strongly reinforces the idea that inhibition of c-ABL kinase signaling can resensitize tumors to cetuximab treatment.
Tyrosine 821 of AXL is critical for mediating resistance to radiation
Previous studies in our laboratory and others demonstrated the role of AXL in promoting resistance to radiation therapy (6,7). Based on our studies of AXL receptor signaling and the critical function of Y821 in resistance to cetuximab (Figure 2), we hypothesized that Y821 signaling might also play a role in resistance to radiation therapy. Therefore, we used the genetically modified HN30 cell lines—Vector, AXL-WT, Y821F—and tested their response to radiation treatment via clonogenic survival analysis (Figure 5A). The survival curves generated from these experiments indicated that HN30-Vector and HN30-AXL-Y821F cells were sensitive to radiation while HN30-AXL-WT cells were not as responsive. This data suggests that Y821 of AXL is critical in mediating resistance to radiation therapy.
Figure 5: AXL targeting overcomes resistance to radiation therapy.
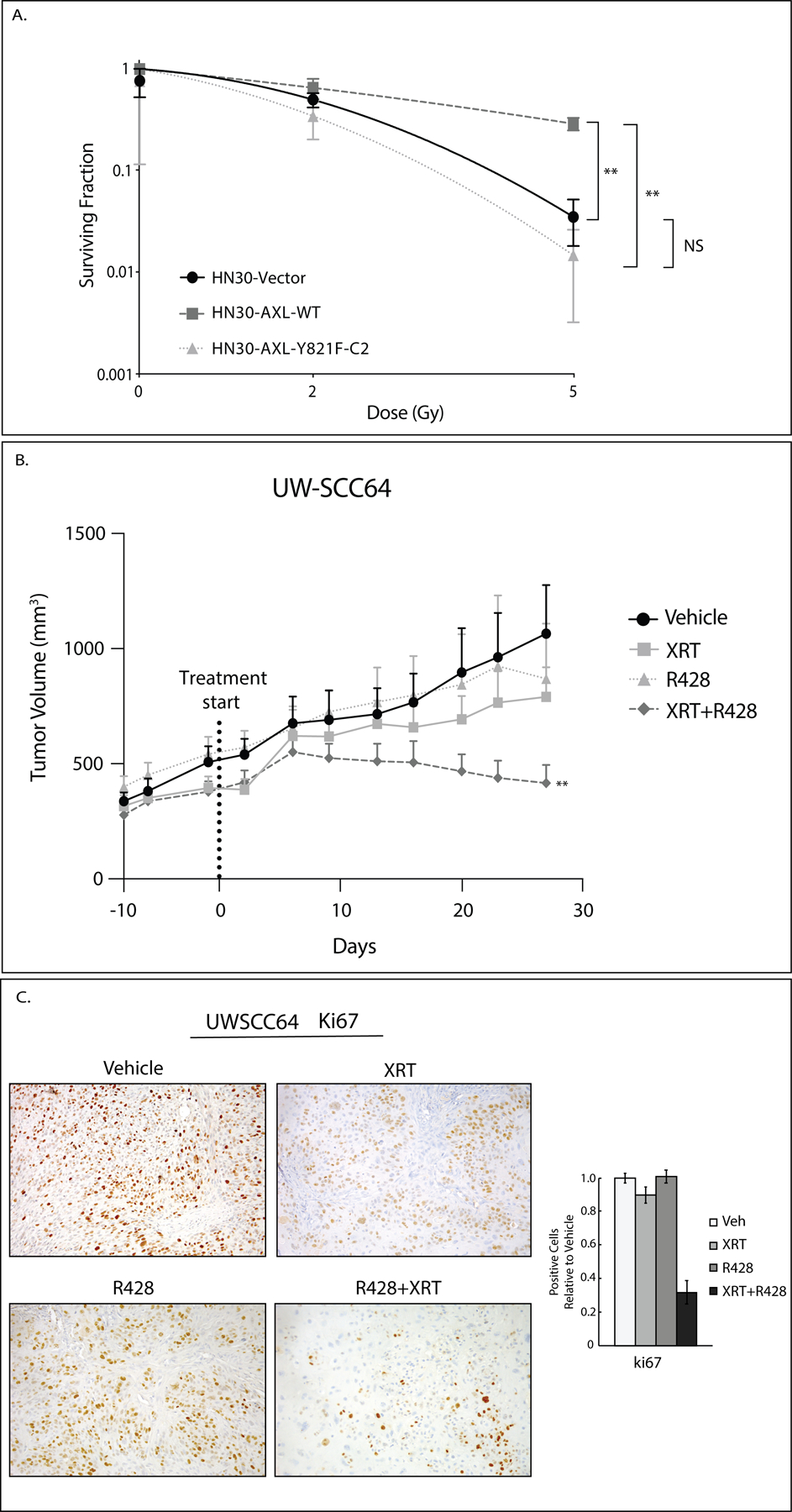
A: Radiosensitivity of HN30 cell lines (Vector, AXL-WT, AXL-Y821F-C2) was evaluated by clonogenic survival analysis. Cells were plated and 24 hours later treated with radiation (0Gy, 2Gy, 5Gy). Mean values, SEs, and statistical analyses were derived from replicates within the experiment. **, P<.01; NS, not significant.
B: PDX UW-SCC64 was implanted into flanks of nude mice and treated with vehicle (oral gavage, methylcellulose, twice daily) (n=13), XRT (3Gy twice weekly) (n=10), R428 (oral gavage, 25mg/kg, twice daily) (n=10), or combination of radiation with R428 (n=11) for 28 days. The vehicle and R428 arms are the same as shown in Figure 1B. Data was analyzed using linear mixed models where volume was modeled on natural-log scale. **, P<.01 for combination compared to single treatments.
C: Tumors from study shown in 5B were stained for Ki67 using IHC. Image quantification is shown.
AXL targeting overcomes resistance to radiation therapy
To determine if Y821 of AXL mediates radiation resistance, we hypothesized that inhibition of AXL signaling should resensitize tumors to radiation treatment. To test this, PDX UW-SCC64 was implanted into flanks of nude mice and treated with vehicle, radiation (3Gy twice weekly), AXL inhibitor R428 (25mg/kg, twice daily), or combination of radiation with R428 (n=10–13 tumors/treatment group) (Figure 5B). A synergistic effect between R428 and radiation was seen (P<.01) indicating that blockade of AXL caused a resensitization of tumors to radiation treatment. Tumors from these studies were stained for Ki67 expression (Figure 5C). Quantification of the expression reveals that the number of cells positive for Ki67 expression did not change after radiation or R428 therapy alone but is reduced after combination therapy suggesting that Ki67 may be a biomarker of response of resensitization to radiation from AXL targeting.
c-ABL plays a role in radiation resistance
Since HN30-AXL-Y821F cells were sensitive to radiation treatment (Figure 5A) and Y821 of AXL signaling regulates c-ABL expression (Figure 3A and 3B), we hypothesized that c-ABL may be playing a role in radiation resistance. To investigate, we targeted ABL signaling by siRNA in HN30-AXL-WT cells and treated with radiation (0Gy, 2Gy, 4Gy). We measured radiation response via cell proliferation assay (Figure 6A) and clonogenic survival analysis (Figure 6B). Together these results showed that HN30-AXL-WT cells were more responsive to radiation treatment after ABL knockdown with siRNA. To expand this finding, PDX UW-SCC64 was injected into flanks of nude mice and treated with vehicle, radiation (3Gy twice weekly), ABL inhibitor imatinib (30mg/kg, once daily), or combination of radiation with imatinib (n=10–18 tumors/treatment group) (Figure 6C). The model fit to the data provides evidence of a synergistic effect in regard to longitudinal tumor growth (P=.028) between imatinib and radiation indicating that blockade of c-ABL caused a resensitization of tumors to radiation therapy. In summary, these results suggest Y821 of AXL is signaling to c-ABL kinase to promote radiation resistance and that blockade of c-ABL signaling can overcome such resistance.
Discussion
In the current report, we identified tyrosine 821 of the AXL receptor tyrosine kinase as a critical mediator of resistance to cetuximab and radiation therapy by activating the c-ABL signaling cascade. Most notably, the reported findings suggest that utilizing imatinib, a c-ABL kinase inhibitor, in combination with cetuximab leads to profound tumor regression and impacts tumor recurrence in models of HNSCC. Together, this work suggests that repurposing this combination of FDA approved molecular therapeutics in HNSCC patients that become refractory to cetuximab and/or radiation therapy may have a profound impact on survival.
Previous studies about the structure of AXL identified three critical tyrosines Y779, Y821, and Y866 as the most prominent tyrosines in AXL signaling. Early AXL studies revealed tyrosine 821’s role in binding of p85 subunit of PI-3K and Grb2 (30) whereas Braunger et al also found that Y779 and Y866 were critical nodes for signaling with Y821 acting as a docking site for multiple effectors including PLCγ, p85 proteins, Grb2, c-src, and lck (29,31). The mutagenesis studies presented herein indicate that Y821, not Y779 nor Y866 is critical for mediating resistance to both cetuximab and radiation treatment. Collectively, our finding that AXL is signaling through Y821 to c-ABL warrants further studies delineating the signaling cascade and the role of Y821 in mediating other mechanisms of resistance.
AXL has demonstrated a role in resistance to both cetuximab and radiation therapy (4–7,19,32–34), and thus AXL has emerged as a major therapeutic target in many cancer types including HNSCC. However, how AXL imparts resistance is poorly understood. Here we report that c-ABL may be a critical signaling axis leading to resistance. Supporting this finding are several studies performed by other groups showing imatinib combined with radiation treatment was very effective at delaying tumor and cell growth in different types of cancer including desmoid tumors, glioblastoma, breast cancer, and bladder cancer through various mechanisms (35–41). Our study corroborates these findings by showing that the combination of c-ABL inhibition, via genetic or pharmacological approaches, was able to overcome resistance to radiation in HNSCC PDX model systems. Furthermore, as supported by others, the addition of imatinib to conventional radiotherapy could have a significant clinical impact (42).
c-ABL has also been shown to mediate resistance to molecular-targeted therapies including EGFR-targeted treatments like lapatinib and cetuximab. Lapatinib when combined with imatinib demonstrated a significant therapeutic benefit in drug-resistant breast cancer (43,44). Another study by Murray et al demonstrated the ability of c-ABL to modulate tumor sensitivity to antibody-dependent cell cytotoxicity (ADCC) (45). The authors showed that ABL1 knockdown and imatinib treatment enhanced ADCC and reduced cell proliferation while c-ABL overexpression decreased ADCC sensitivity. These results indicate that resistance to molecular targeting agents such as lapatinib and cetuximab may share mechanisms of resistance through the activation of c-ABL. The data presented in this report suggests that c-ABL signaling, emanating from AXL Y821, is a critical pathway for resistance to cetuximab, and the combination of imatinib and cetuximab had a dramatic impact on the growth of cetuximab-resistant tumors. It will be important to further evaluate if c-ABL is a critical signal for the mechanisms of action of cetuximab to strengthen the idea that this targeting strategy should be investigated clinically. Overall, these previous studies of c-ABL suggest that c-ABL is critical in mediating resistance to various therapeutic modalities including radiation and cetuximab. This supports the finding reported here that tumors overexpressing AXL may be more prone to resistance due to the regulation of c-ABL by AXL-Y821.
In the current study, we identified the interaction of AXL and c-ABL signaling in the context of cetuximab and radiation resistance. Our findings were consistent with a previous study by Hong et al showing that c-ABL regulation by AXL was able to promote cisplatin resistance in esophageal cancer (46). In this study, the authors showed that AXL associates with c-ABL protein and sequesters it in the cytosol thus preventing translocation of c-ABL to the nucleus in response to DNA damage. AXL’s ability to prevent DNA-damage-induced apoptosis by blocking nuclear translocation of c-ABL and consequently promote cisplatin resistance supports our finding that AXL could be signaling through c-ABL in order to mediate cetuximab or radiation resistance and prevent cell death. However, our data differs from that reported by Hong et al in that Y821 appeared to regulate total levels of c-ABL expression. Future work will be centered on the mechanism of how c-ABL mediates resistance and whether it is through mechanisms of c-ABL nuclear shuttling or activating transcription factors important for other pathways. One potential pathway could be the regulation of neuregulin, a HER3 ligand. HER3 and neuregulin have been strongly implicated in resistance to cetuximab therapy (24,47–49). In one specific instance, it was reported that AXL regulated neuregulin expression which activated HER3 and subsequent dimerization with EGFR resulted in subcellular functions and cetuximab resistance (50). Together, it is possible to imagine that the Y821/c-ABL pathway may be critical in regulating cetuximab resistance by regulating neuregulin and the HER3 pathway. We are currently investigating along these lines.
In summary, the work presented herein identifies a unique targeting approach for HNSCC patients that become refractory to cetuximab or radiation therapy. We identified a novel signaling pathway that emanates from Y821 of the AXL receptor tyrosine kinase and activates c-ABL kinase. We demonstrate that inhibition of c-ABL in combination with cetuximab has marked anti-tumor activity. These results are currently being translated into clinical trials testing the combination of cetuximab and imatinib in HNSCC patients to fully investigate this new and novel therapeutic strategy in the HNSCC armamentarium.
Supplementary Material
Translational Relevance.
Head and neck squamous cell carcinoma (HNSCC) is a devastating disease that greatly impacts quality of life because of the location of the involved tissues. Although therapeutic mainstays include radiation and targeted therapies like cetuximab, there is either a limited response to these treatment approaches or resistance develops over time. These consequences emphasize a critical need for development of strategies that work better and are safer for patients. Using preclinical models, we demonstrate that blockade of the receptor tyrosine kinase AXL is able to resensitize tumors to cetuximab and radiation treatment. Furthermore, inhibition of downstream signaling from AXL to c-ABL kinase is even more effective at overcoming resistance. Combination treatment using imatinib and cetuximab exhibited substantial growth inhibition and survival benefit and provides strong evidence for clinical testing of these targeted therapies in resistant disease.
Acknowledgements:
The authors thank the University of Wisconsin Translational Research Initiatives in Pathology laboratory (TRIP) supported by the UW Department of Pathology and the Office of the Director-NIH (S10OD023526) for use of its facilities and services as well as the Functional Proteomics RPPA Core facility which is supported by MD Anderson Cancer Center Support Grant # 5 P30 CA016672-40. The authors would also like to thank Prashanth J Prabakaran and Adam D Swick for their technical assistance.
Financial Support: Research reported in this publication was supported, in part, by the Wisconsin Head & Neck Cancer SPORE (DLW, RJK, and JYB, P50 DE026787), NIH grant from Cellular and Molecular Pathology Graduate Training Program (NKM, 5 T32 GM81061-7), a pilot grant from the UW Institute for Clinical and Translational Research (ICTR) and the UW Carbone Cancer Center (DLW and RJK, ICTR-UWCH RD 12), and the University of Wisconsin Carbone Cancer Center Support Grant (P30 CA014520). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68(6):394–424 doi 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med 2006;354(6):567–78 doi 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 3.Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008;359(11):1116–27 doi 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 4.Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Luthar N, et al. AXL mediates resistance to cetuximab therapy. Cancer Res 2014;74(18):5152–64 doi 10.1158/0008-5472.CAN-14-0294. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 5.Skinner HD, Giri U, Yang LP, Kumar M, Liu Y, Story MD, et al. Integrative Analysis Identifies a Novel AXL-PI3 Kinase-PD-L1 Signaling Axis Associated with Radiation Resistance in Head and Neck Cancer. Clin Cancer Res 2017;23(11):2713–22 doi 10.1158/1078-0432.CCR-16-2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brand TM, Iida M, Stein AP, Corrigan KL, Braverman CM, Coan JP, et al. AXL Is a Logical Molecular Target in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res 2015;21(11):2601–12 doi 10.1158/1078-0432.CCR-14-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aguilera TA, Rafat M, Castellini L, Shehade H, Kariolis MS, Hui AB, et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat Commun 2016;7:13898 doi 10.1038/ncomms13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong CC, Lay JD, Huang JS, Cheng AL, Tang JL, Lin MT, et al. Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett 2008;268(2):314–24 doi 10.1016/j.canlet.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 9.Kurokawa M, Ise N, Omi K, Goishi K, Higashiyama S. Cisplatin influences acquisition of resistance to molecular-targeted agents through epithelial-mesenchymal transition-like changes. Cancer Sci 2013;104(7):904–11 doi 10.1111/cas.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin JZ, Wang ZJ, De W, Zheng M, Xu WZ, Wu HF, et al. Targeting AXL overcomes resistance to docetaxel therapy in advanced prostate cancer. Oncotarget 2017;8(25):41064–77 doi 10.18632/oncotarget.17026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dufies M, Jacquel A, Belhacene N, Robert G, Cluzeau T, Luciano F, et al. Mechanisms of AXL overexpression and function in Imatinib-resistant chronic myeloid leukemia cells. Oncotarget 2011;2(11):874–85 doi 10.18632/oncotarget.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meyer AS, Miller MA, Gertler FB, Lauffenburger DA. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative breast cancer cells. Sci Signal 2013;6(287):ra66 doi 10.1126/scisignal.2004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balaji K, Vijayaraghavan S, Diao L, Tong P, Fan Y, Carey JP, et al. AXL Inhibition Suppresses the DNA Damage Response and Sensitizes Cells to PARP Inhibition in Multiple Cancers. Mol Cancer Res 2017;15(1):45–58 doi 10.1158/1541-7786.MCR-16-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van der Mijn JC, Broxterman HJ, Knol JC, Piersma SR, De Haas RR, Dekker H, et al. Sunitinib activates Axl signaling in renal cell cancer. Int J Cancer 2016;138(12):3002–10 doi 10.1002/ijc.30022. [DOI] [PubMed] [Google Scholar]
- 15.Zhou L, Liu XD, Sun M, Zhang X, German P, Bai S, et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016;35(21):2687–97 doi 10.1038/onc.2015.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet 2012;44(8):852–60 doi 10.1038/ng.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bae SY, Hong JY, Lee HJ, Park HJ, Lee SK. Targeting the degradation of AXL receptor tyrosine kinase to overcome resistance in gefitinib-resistant non-small cell lung cancer. Oncotarget 2015;6(12):10146–60 doi 10.18632/oncotarget.3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 2013;19(1):279–90 doi 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elkabets M, Pazarentzos E, Juric D, Sheng Q, Pelossof RA, Brook S, et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015;27(4):533–46 doi 10.1016/j.ccell.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res 2009;69(17):6871–8 doi 10.1158/0008-5472.CAN-08-4490. [DOI] [PubMed] [Google Scholar]
- 21.Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun 2014;5:5712 doi 10.1038/ncomms6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo G, Gong K, Ali S, Ali N, Shallwani S, Hatanpaa KJ, et al. A TNF-JNK-Axl-ERK signaling axis mediates primary resistance to EGFR inhibition in glioblastoma. Nat Neurosci 2017;20(8):1074–84 doi 10.1038/nn.4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadahiro H, Kang KD, Gibson JT, Minata M, Yu H, Shi J, et al. Activation of the Receptor Tyrosine Kinase AXL Regulates the Immune Microenvironment in Glioblastoma. Cancer Res 2018;78(11):3002–13 doi 10.1158/0008-5472.CAN-17-2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Iida M, Brand TM, Starr MM, Huppert EJ, Luthar N, Bahrar H, et al. Overcoming acquired resistance to cetuximab by dual targeting HER family receptors with antibody-based therapy. Mol Cancer 2014;13:242 doi 10.1186/1476-4598-13-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006;1(5):2315–9 doi 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 26.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010;70(2):440–6 doi 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 27.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984;22:27–55. [DOI] [PubMed] [Google Scholar]
- 28.Swick AD, Prabakaran PJ, Miller MC, Javaid AM, Fisher MM, Sampene E, et al. Cotargeting mTORC and EGFR Signaling as a Therapeutic Strategy in HNSCC. Mol Cancer Ther 2017;16(7):1257–68 doi 10.1158/1535-7163.MCT-17-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braunger J, Schleithoff L, Schulz AS, Kessler H, Lammers R, Ullrich A, et al. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene 1997;14(22):2619–31 doi 10.1038/sj.onc.1201123. [DOI] [PubMed] [Google Scholar]
- 30.Fridell YW, Jin Y, Quilliam LA, Burchert A, McCloskey P, Spizz G, et al. Differential activation of the Ras/extracellular-signal-regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol Cell Biol 1996;16(1):135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O’Bryan JP, Frye RA, Cogswell PC, Neubauer A, Kitch B, Prokop C, et al. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol 1991;11(10):5016–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leonard B, Brand TM, O’Keefe RA, Lee ED, Zeng Y, Kemmer JD, et al. BET Inhibition Overcomes Receptor Tyrosine Kinase-Mediated Cetuximab Resistance in HNSCC. Cancer Res 2018;78(15):4331–43 doi 10.1158/0008-5472.CAN-18-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu S, Dai H, Li T, Tang Y, Fu W, Yuan Q, et al. Broad RTK-targeted therapy overcomes molecular heterogeneity-driven resistance to cetuximab via vectored immunoprophylaxis in colorectal cancer. Cancer Lett 2016;382(1):32–43 doi 10.1016/j.canlet.2016.08.022. [DOI] [PubMed] [Google Scholar]
- 34.Yang S, Zhang X, Qu H, Qu B, Yin X, Zhao H. Cabozantinib induces PUMA-dependent apoptosis in colon cancer cells via AKT/GSK-3beta/NF-kappaB signaling pathway. Cancer Gene Ther 2019. doi 10.1038/s41417-019-0098-6. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Moding EJ, Million L, Avedian R, Ghanouni P, Kunder C, Ganjoo KN. Concurrent Imatinib and Radiation Therapy for Unresectable and Symptomatic Desmoid Tumors. Sarcoma 2017;2017:2316839 doi 10.1155/2017/2316839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weigel MT, Dahmke L, Schem C, Bauerschlag DO, Weber K, Niehoff P, et al. In vitro effects of imatinib mesylate on radiosensitivity and chemosensitivity of breast cancer cells. BMC Cancer 2010;10:412 doi 10.1186/1471-2407-10-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yerushalmi R, Nordenberg J, Beery E, Uziel O, Lahav M, Luria D, et al. Combined antiproliferative activity of imatinib mesylate (STI-571) with radiation or cisplatin in vitro. Exp Oncol 2007;29(2):126–31. [PubMed] [Google Scholar]
- 38.Oertel S, Krempien R, Lindel K, Zabel A, Milker-Zabel S, Bischof M, et al. Human glioblastoma and carcinoma xenograft tumors treated by combined radiation and imatinib (Gleevec). Strahlenther Onkol 2006;182(7):400–7 doi 10.1007/s00066-006-1445-8. [DOI] [PubMed] [Google Scholar]
- 39.Quick QA, Gewirtz DA. Enhancement of radiation sensitivity, delay of proliferative recovery after radiation and abrogation of MAPK (p44/42) signaling by imatinib in glioblastoma cells. Int J Oncol 2006;29(2):407–12. [PubMed] [Google Scholar]
- 40.Holdhoff M, Kreuzer KA, Appelt C, Scholz R, Na IK, Hildebrandt B, et al. Imatinib mesylate radiosensitizes human glioblastoma cells through inhibition of platelet-derived growth factor receptor. Blood Cells Mol Dis 2005;34(2):181–5 doi 10.1016/j.bcmd.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 41.Qiao B, Kerr M, Groselj B, Teo MT, Knowles MA, Bristow RG, et al. Imatinib radiosensitizes bladder cancer by targeting homologous recombination. Cancer Res 2013;73(5):1611–20 doi 10.1158/0008-5472.CAN-12-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koppenhagen P, Dickreuter E, Cordes N. Head and neck cancer cell radiosensitization upon dual targeting of c-Abl and beta1-integrin. Radiother Oncol 2017;124(3):370–8 doi 10.1016/j.radonc.2017.05.011. [DOI] [PubMed] [Google Scholar]
- 43.Lo YH, Ho PC, Zhao H, Wang SC. Inhibition of c-ABL sensitizes breast cancer cells to the dual ErbB receptor tyrosine kinase inhibitor lapatinib (GW572016). Anticancer Res 2011;31(3):789–95. [PubMed] [Google Scholar]
- 44.Liu L, Shen W, Zhu Z, Lin J, Fang Q, Ruan Y, et al. Combined inhibition of EGFR and c-ABL suppresses the growth of fulvestrant-resistant breast cancer cells through miR-375-autophagy axis. Biochem Biophys Res Commun 2018;498(3):559–65 doi 10.1016/j.bbrc.2018.03.019. [DOI] [PubMed] [Google Scholar]
- 45.Murray JC, Aldeghaither D, Wang S, Nasto RE, Jablonski SA, Tang Y, et al. c-Abl modulates tumor cell sensitivity to antibody-dependent cellular cytotoxicity. Cancer Immunol Res 2014;2(12):1186–98 doi 10.1158/2326-6066.CIR-14-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hong J, Peng D, Chen Z, Sehdev V, Belkhiri A. ABL regulation by AXL promotes cisplatin resistance in esophageal cancer. Cancer Res 2013;73(1):331–40 doi 10.1158/0008-5472.CAN-12-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene 2008;27(28):3944–56 doi 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baro M, Lopez Sambrooks C, Burtness BA, Lemmon MA, Contessa JN. Neuregulin Signaling Is a Mechanism of Therapeutic Resistance in Head and Neck Squamous Cell Carcinoma. Mol Cancer Ther 2019. doi 10.1158/1535-7163.MCT-19-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang D, Qian G, Zhang H, Magliocca KR, Nannapaneni S, Amin AR, et al. HER3 Targeting Sensitizes HNSCC to Cetuximab by Reducing HER3 Activity and HER2/HER3 Dimerization: Evidence from Cell Line and Patient-Derived Xenograft Models. Clin Cancer Res 2017;23(3):677–86 doi 10.1158/1078-0432.CCR-16-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brand TM, Iida M, Corrigan KL, Braverman CM, Coan JP, Flanigan BG, et al. The receptor tyrosine kinase AXL mediates nuclear translocation of the epidermal growth factor receptor. Sci Signal 2017;10(460) doi 10.1126/scisignal.aag1064. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Bates Douglas, Maechler Martin, Bolker Ben, Walker Steve (2015). Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software, 67(1), 1–48. doi: 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- 52.R Core Team (2019). R: A language and environment for statistical computing R Foundation for Statistical Computing, Vienna, Austria: URL https://www.R-project.org/. [Google Scholar]
- 53.Kuznetsova A, Brockhoff PB, Christensen RHB (2017). “lmerTest Package: Tests in Linear Mixed Effects Models.” _Journal of Statistical Software_, *82*(13), 1–26. doi: 10.18637/jss.v082.i13 (URL: 10.18637/jss.v082.i13). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.