

Abstract
Environmental DNA damaging agents continuously challenge the integrity of the genome through introducing a variety of DNA lesions. The DNA damage caused by environmental factors will lead to mutagenesis and subsequent carcinogenesis if they are not removed efficiently by repair pathways. Methods for detection of DNA damage and repair can be applied to identify, visualize and quantify the DNA damage formation and repair events, and they enable us to illustrate the molecular mechanisms of DNA damage formation, DNA repair pathways, mutagenesis and carcinogenesis. Ever since the discovery of double helix structure of DNA in 1953, a great number of methods have been developed to detect various types of DNA damage and repair. Rapid advances in sequencing technologies have facilitated the emergence of a variety of novel methods for detecting environmentally-induced DNA damage and repair at genome-wide scale during the last decade. In this review, we provide a historical overview of the development of various damage detection methods. We also highlight the current methodologies to detect DNA damage and repair, especially some next generation sequencing (NGS)-based methods.
Keywords: DNA damage, DNA repair, mutagenesis, next-generation sequencing, third-generation sequencing
INTRODUCTION
The natural environment is generally considered to play an important role in the origin and evolution of life on the earth. Nucleic acids, essential to all three domains of life (Archaea, Bacteria, and Eukarya), are constantly challenged by endogenous and exogenous sources of nucleic acid damaging agents. Endogenous reactive oxygen species (ROS) produced in cellular metabolism and nucleotide misincorporation during DNA replication can cause oxidation and mismatch of DNA bases (Lindahl and Barnes 2000). Environmental factors such as ultraviolet (UV) light, mycotoxins, polycyclic aromatic hydrocarbons (PAHs) and ionizing radiation can cause structural distortions and strand breaks of DNA (Hoeijmakers 2001). UV exposure results in the formation of cyclobutane pyrimidine dimer (CPD) and (6–4) pyrimidine-pyrimidone photoproduct [(6–4)PP] between two adjacent pyrimidines. Aflatoxin B1 (AFB1), known as the most toxic mycotoxin, forms AFB1-DNA adduct after bioactivation in the cell. Benzo[a]pyrene is the most common PAH and induces the benzo[a]pyrene diol epoxide (BPDE)-DNA adduct after being bioactivated. X ray irradiation causes deleterious single and/or double strand breaks in DNA. To cope with these challenges, organisms have evolved a variety of DNA repair mechanisms as well as global DNA damage responses. There are mainly five common repair pathways that recognize and remove different types of DNA damage: direct reversal repair, nucleotide excision repair, base excision repair, mismatch repair and double strand break repair (Fig. 1). Failure to repair DNA damage efficiently by the aforementioned repair mechanisms can cause mutagenesis and eventually lead to cancers.
Fig. 1.
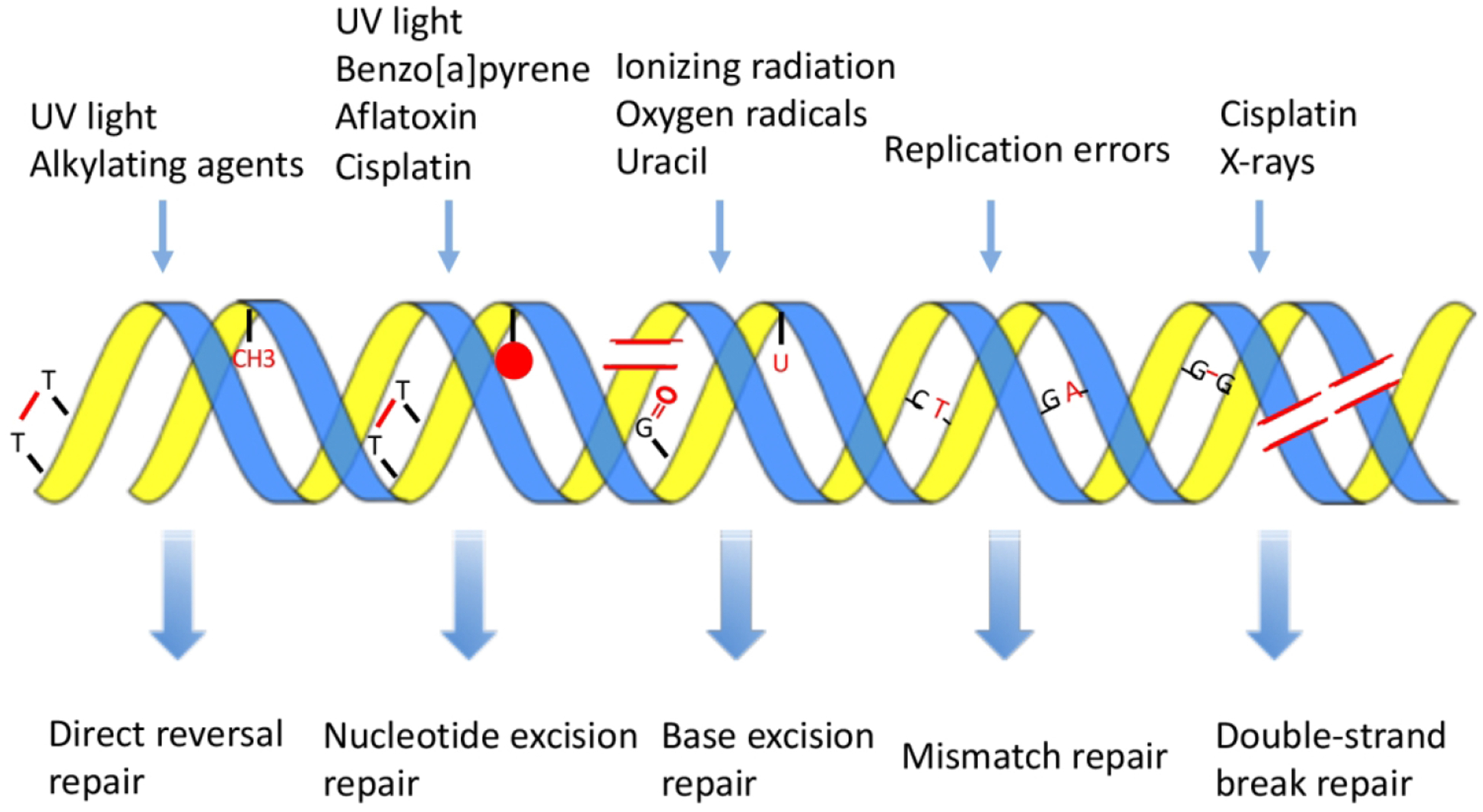
Schematic of various DNA damaging agents-induced DNA damage and the corresponding repair pathways. The top panel shows the different types of DNA damaging agents. The various DNA lesions caused by DNA damaging agents are shown in red in the middle panel. The lower panel shows the corresponding repair pathways that are responsible for the removal of those DNA lesions listed in the middle panel. This figure is redrawn based on reference (Hoeijmakers 2001).
To better understand the mechanisms of mutagenesis and carcinogenesis, it is critical to know the exact location of DNA damage and its repair efficiency across the whole genome. Prior to the advent of high-throughput sequencing technologies, a number of strategies were developed to detect environmentally-induced DNA damage and repair in various organisms. However, none of these strategies are applicable to detect DNA damage and repair at genome-wide scale with single-nucleotide resolution. Over the last decade a variety of high-throughput sequencing-based methods have been devised to detect genome-wide DNA damage and repair at single-nucleotide resolution (Sloan et al. 2018; Salk and Kennedy 2019). In this review, we focus on current methodologies for detecting environmentally-induced DNA damage and repair, especially the recently developed high-throughput sequencing-based methods.
HISTORICAL OVERVIEW OF METHODOLOGIES FOR DETECTING ENVIRONMENTALLY-INDUCED DNA DAMAGE AND REPAIR
There is no doubt that new technology inventions assist the advancement of science, and advances in science, in turn, facilitate the emergence of novel technologies. Since the discovery of the double helix structure of DNA through X-ray diffraction in 1953 (Watson and Crick 1953; Wilkins et al. 1953), researchers tended to answer biological questions at the molecular level. Although studies on biological responses to UV and ionizing radiation can be traced back to the 1930s and 1940s, the discovery of photolyase in 1958 marked the beginning of DNA repair field (Rupert et al. 1958; Rupert 1960; Sancar 2017). The isolation and identification of the UV-induced CPD by paper chromatography was regarded as a key breakthrough in 1960s (Varghese and Wang 1967). Meanwhile, radioactive labeling-based methods such as repair replication (Pettijohn and Hanawalt 1964) and unscheduled DNA synthesis (UDS) assay (Rasmussen and Painter 1964) were used to detect nucleotide excision repair in bacterial and mammalian cells. The fluorescence-based methods such as acridine orange staining (Gruzdev and Kishchenko 1978) and halo assay in which propidium iodide was used (Roti Roti and Wright 1987) were developed to detect DNA strand breaks and DNA loops in 1970s and 1980s. In 1977, the bulky DNA adduct caused by mycotoxin AFB1 was identified through high-performance liquid chromatography-mass spectrometry (HPLC-MS) (Essigmann et al. 1977). The Sanger sequencing (Sanger and Coulson 1975), the first generation of sequencing technology, and the recombinant DNA technology were invented (Jackson et al. 1972) and repair-related genes were then cloned and sequenced (Sancar and Rupert 1978a; Sancar and Rupp 1979; Sancar et al. 1980). Methods for identification of repair proteins encoded by cloned repair genes on plasmids were devised (‘maxicell’) and they were used for expression and purification of repair-related proteins (Sancar and Rupert 1978b; Sancar et al. 1979). These methods enabled the identification and purification of UvrA, UvrB, and UvrC proteins of Escherichia coli and the development of the in vitro excision assay in 1980s (Sancar et al. 1981a; Sancar et al. 1981b; Sancar et al. 1981c; Sancar and Rupp 1983). The excision assay, which detects the excised oligonucleotides carrying the damage, was the method that led to the discovery of the molecular mechanism of nucleotide excision repair in prokaryotes and eukaryotes (Sancar and Rupp 1983; Huang et al. 1992; Guzder et al. 1995). The single cell gel electrophoresis assay (comet assay) was also invented in 1980s to detect DNA strand breaks for single cell (Ostling and Johanson 1984; Freeman et al. 1986). The immunoassay-based methods such as radioimmunoassays (RIA) (Mitchell and Clarkson 1981), enzyme-linked immunosorbent assay (ELISA) (Leipold et al. 1983) and immunoslot blot (Wani et al. 1987) were invented and employed to quantify UV-induced DNA damage in 1980s . With the advent of polymerase chain reaction (PCR) (Saiki et al. 1985), the PCR-based methods such as quantitative PCR (qPCR) (Govan et al. 1990; Kalinowski et al. 1992) and ligation-mediated PCR (LMPCR) (Pfeifer et al. 1991) were developed and widely used for mapping DNA damage at nucleotide resolution in 1990s. The TUNEL (TdT-mediated dUTP-biotin nick end labeling) assay was used to in situ label DNA breaks for studying apoptosis in 1992 (Gavrieli et al. 1992). In 2010s, a method that combines the immunoprecipitation and microarray was reported for mapping UV-induced DNA damage at genome-wide scale in yeast (Teng et al. 2011) and at chromosome scale in humans (Zavala et al. 2014).
Over the last five years, a number of next generation sequencing (NGS)-based methods have emerged for detecting various types of DNA damage and repair across the entire genome at nucleotide resolution (Sloan et al. 2018). The emergence of these NGS-based methods revolutionized the DNA repair field by providing researchers the precise locations of DNA damage and repair at genome-wide level. With the development of the third-generation sequencing technologies (also called long-read sequencing), such as Pacific Biosciences Single Molecule Real-Time (SMRT) sequencing and Oxford Nanopore sequencing, a third-generation sequencing-based method, RADAR-seq, was recently developed for detecting the ribonucleotide incorporation and UV-induced DNA damage (Zatopek et al. 2019).
GEL ELECTROPHORESIS-BASED METHODS
DNA fragments in an electric field will migrate to the positively charged anode because of the negatively charged phosphate backbone of DNA. Agarose gel electrophoresis is a simple and efficient way for separating DNA fragments with a range from 50 bp to 500, 000 bp. Thus, the density of DNA strand breaks and other types of DNA damage which can be converted to strand breaks by chemical or enzymatic reaction can be analyzed by agarose gel electrophoresis under denaturing conditions (McMaster and Carmichael 1977). The comet assay is another gel electrophoresis-based method to detect strand breaks in individual cells on a microscope slide (Ostling and Johanson 1984; Freeman et al. 1986). The cells embedded in agarose gel are lysed to form nucleoids and the migration pattern of single nucleoid resembles comet observed by fluorescence microscope. The intensity of the comet tail depends on the frequency of strand breaks. This method won popularity after it was developed because of its simplicity and sensitivity. However, it can only be used for detecting the overall DNA damage and repair lacking the information on individual genes. The combination of comet and fluorescent in situ hybridization (FISH) method (Comet-FISH) allows observation of specific genes within the DNA comet (McKelvey-Martin et al. 1998; Spivak et al. 2009).
Despite of the simplicity and sensitivity of the conventional slide-based comet assay, it has high sample-to-sample variation and is labor-intensive for genotoxicity screening. The recently developed CometChip technology overcomes these limitations by using micro-patterned vertical casting cassettes and a macrowell former to create a 96-well agarose gel. Each of the 96 wells has about 500 microwells and only one single cell can be loaded into each microwell. After electrophoresis, the DNA will be stained with SYBR Gold and image data acquired will be analyzed by the Comet Analysis Software (Wood et al. 2010; Ge et al. 2014; Sykora et al. 2018). This method is quite useful for high throughput genotoxicity testing and measurement of DNA damage and repair (Ngo et al. 2019). Like the conventional comet assay, it can detect the overall level of strand breaks but cannot provide information on the locations of DNA damage and repair in the genome.
RADIOACTIVE LABELING-BASED METHODS
Radioactive isotopes such as 32P and 3H have been widely used in studies of biological and medical sciences. In DNA repair field, the repair replication (Pettijohn and Hanawalt 1964) and UDS assay (Rasmussen and Painter 1964) were first used to measure nucleotide excision repair in bacterial and mammalian cells respectively. The [14C]thymine, [14C]5-bromouracil, C3H3-thymine and [3H]5- bromouracil were used in the repair replication method and the [3H]thymine was included in the UDS assy. The 32P-postlabeling assay, which was devised in 1981, is a general way to measure the DNA damage level and repair rate (Randerath et al. 1981). Because of the high sensitivity of the radioactive labeling method, it gained wide applications and was combined with other techniques (e.g. immunoassay and PCR) to develop novel methods for detection of DNA damage and repair both in vivo and in vitro. However, the radioactive labeling-based methods are generally labor-intensive and require stringent radioactive safety standards.
Detection of DNA damage and repair in specific genes
Methods such as comet assay, immunoslot blot and UDS are designed to measure the total levels of DNA damage and repair either in cell populations or individual cells. For studying repair in specific genes, radioactive hybridization probe, restriction enzymes, PCR amplification and radioactive end labeling are generally used to isolate and label the specific genes of interest. The Southern blot assay uses restriction enzymes to cut off specific DNA fragments, creates strand breaks at DNA lesion sites within the DNA fragments by repair enzymes, and measures the frequency of DNA lesions by hybridization with radioactive probe after alkaline gel electrophoresis (Bohr et al. 1985; Thomas et al. 1988). Although the Southern blot has high sensitivity and wide applications for various DNA lesions, it only measures the total frequency of DNA damage in specific genes and cannot achieve nucleotide resolution.
A number of radioactive labeling-based methods were then developed to detect DNA damage and repair in specific genes with single-nucleotide resolution. In these methods, cleavage at DNA damage sites by repair enzymes (e.g. T4 Endo V digests at CPD damage sites) or chemical treatment [e.g. piperidine cleaves at (6–4) PP damage sites] or blockage of DNA polymerase at damage site during the primer extension was applied to detect the DNA damage and repair. They are generally divided into two categories based on their distinct strategies: The first category, which includes LMPCR (Pfeifer et al. 1991), single-strand ligation PCR (sslig-PCR) (Grimaldi et al. 1994) and primer extension (Wellinger and Thoma 1996; Wellinger and Thoma 1997), uses PCR or multiple times of primer extension to amplify the damage signal . The second category including indirect end-labeling (Smerdon and Thoma 1990), oligonucleotide-facilitated end-labeling (Kunala and Brash 1992) and oligonucleotide/streptavidin magnetic bead-facilitated end-labeling (Li and Waters 1996; Li et al. 2000) utilizes radioactive end-labeling of the unamplified DNA fragments carrying information about damage positions . The methods in the first category have higher sensitivity and can be used in mammalian cells that have large genome size. However, the efficiency variations in ligation and PCR amplification steps may cause bias.Although the methods in the second category have lower sensitivity, they exclude potential bias produced in the first category of methods because they do not amplify the damage signal with PCR. And they have been used in organisms with lower genome size such as E.coli and yeast (Li and Waters 1997; Li et al. 2014a; Li et al. 2014b).
Despite of the single-nucleotide resolution, these methods can only measure DNA damage and repair in a short region (<500 bp) due to the limited resolvability of DNA fragment on sequencing gels. Moreover, methods that depend on the cleavage at damage sites by repair enzyme or chemical reaction are not applicable for other types of non-cleavable DNA lesions such as benzo[a]pyrene and aflatoxin-induced DNA damage.
Excision repair assays
Methods that detect various types of DNA damage and repair can be used for identifying DNA damage, measuring DNA damage level and repair rate, and localizing DNA damage sites at nucleotide resolution. Whereas, it is the combination of maxicell method, incision assay and excision assay that led to the discovery of the molecular mechanism of nucleotide excision repair (Sancar 2016). In addition, a repair patch assay was developed to determine the size and sequence of the newly synthesized repair patch after excision repair in 1990 (Sibghat-Ullah et al. 1990; Huang et al. 1992; Reardon et al. 1997).
The incision assay (also known as endonuclease-sensitive site assay) was first developed to measure the damage-dependent incisions of DNA by repair enzymes such as T4 Endo V or chemical reactions by using alkaline sucrose gradient (Seeberg et al. 1976), nitrocellulose filter binding (Seeberg and Steinum 1982) or agarose gel electrophoresis (Sancar and Rupp 1983). To detect the damage-dependent incisions at nucleotide resolution, it was further optimized by using the 5’ or the 3’ end-radiolabeled DNA fragments carrying damage at specific sites and the denaturing polyacrylamide gel to visualize the exact incision sites (Sancar and Rupp 1983). By using this method, the molecular mechanism of nucleotide excision repair in E.coli was uncovered in 1983. It was shown that the UvrABC excision nucleases remove pyrimidine dimers by cutting the 8th phosphodiester bond 5’ to the lesion and the 4th or 5th phosphodiester bond 3’ to the lesion (Sancar and Rupp 1983). One decade later, the molecular mechanism of bacterial transcription-coupled repair, one subpathway of nucleotide excision repair, was discovered by using the incision assay in a reconstituted strand-specific repair system and the mysterious mfd− phenotype (Witkin 1966) was then clarified (Selby and Sancar 1993).
The excision assay is the method that detects the excised damage-carrying oligonucleotides (nominal 30-mer) by dual incisions during the nucleotide excision repair. This powerful method for studying the mechanism of the excision nucleases is classified into two categories: in vitro and in vivo. For in vitro excision assay, the purified excision nucleases or cell-free extract, cofactors such as ATP, and the lesion-containing DNA substrate are used to reconstitute the nucleotide excision repair. The excision assay and the incision assay only differ in the radiolabel position in the DNA substrate. In the in vitro excision assay, the DNA substrate can be radiolabeled in the vicinity of the lesion before the repair reaction and the excised oligonucleotide carrying the radiolabel is resolved on a polyacrylamide gel (Van Houten et al. 1986; Huang et al. 1992). If the substrate is not radiolabeled, the excised oligonucleotide released in the repair reaction can be radiolabeled by terminal deoxynucleotidyl transferase before being resolved on a sequencing gel (Guzder et al. 1995) or detected by Southern hybridization after being separated on a sequencing gel (Moggs et al. 1996). The molecular mechanism of human nucleotide excision repair was uncovered by using an in vitro excision assay system consisting of human cell-free extract and a plasmid carrying thymine dimers or a thymine-psoralen monoadduct adjacent to 32P label (Huang et al. 1992; Svoboda et al. 1993). Similarly, using the in vitro excision assay consisting of cell-free extracts from mouse brain and liver, the circadian oscillation of nucleotide excision repair activity and XPA protein level was identified (Kang et al. 2009; Kang et al. 2010). The in vivo excision assay was developed based on the in vitro finding that the excised oligonucleotide released from DNA is tightly bound to the TFIIH, a 10-subunit protein complex involved in transcription initiation and nucleotide excision repair (Mu et al. 1995; Mu et al. 1996; Kemp et al. 2012). In the in vivo excision assay, the excised oligonucleotide can be isolated by either DNA damage-specific immunoprecipitation after Hirt extraction (Hirt 1967) or TFIIH immunoprecipitation after gentle cell lysis. The isolated oligonucleotides are then 3ʹ or 5’ end-radiolabeled and resolved on a sequencing gel (Hu et al. 2013). Alternatively, the captured oligonucleotides can be 3’ end-labeled with biotin and detected by using horseradish peroxidase (HRP)-conjugated streptavidin and chemiluminescent reagents (Choi et al. 2014). An integrated method that combines the in vivo excision assay, immunoblot blot and Western blot was devised to detect the excised oligonucleotides, the rate of nucleotide excision repair and DNA damage response signaling in parallel from the same population of cells (Choi et al. 2015; Song et al. 2017). As the DNA fragmentation occurs during apoptosis, the in vivo excision assay is also applicable to simultaneously detect the apoptotic DNA fragments and the excised oligonucleotides (Baek et al. 2018).
Based on the phosphorothioate sequencing method (Gish and Eckstein 1988) in which phosphorothioate bonds are hydrolyzed preferentially by iodoethanol, the repair patch assay (Sibghat-Ullah et al. 1990) was devised to identify the size and sequence of the repair patch synthesized after excision repair. During the nucleotide excision repair, the gap, generated after the excision of the damage-carrying DNA fragment, is filled in by DNA polymerase and then ligated by DNA ligase (Kemp 2019). To determine whether the gap is enlarged or not before new strand synthesis, it is required to know the exact size and sequence of the newly synthesized repair patch. In the repair patch assay, the repair synthesis is carried out in the presence of one dNTP(αS) and three dNTPs (four reactions in which either A, G, T, C are in the form of dNTP(αS) and the other three nucleotides in the form of canonical dNTPs). Following repair synthesis reaction, DNA fragments carrying the repair patch are terminally labeled with 32P, and incubated in the presence of iodoethanol. The cleavage at the position of dNTP(αS) gives rise to a sequence ladder exactly matching the repair gap. Thus, this method reveals the exact boundaries of the incision sites and the repair patch. This nucleotide resolution method has been applied to determine the repair patches in prokaryotic and eukaryotic cells (Sibghat-Ullah et al. 1990; Huang et al. 1992; Reardon et al. 1997).
FLUORESCENCE-BASED METHODS
In combination with fluorescence microscopy and flow cytometry, the fluorescence-based methods for detection of DNA damage and repair are widely used because of the high sensitivity and specificity, the wide range of fluorophores for selection and the ease of use. In general, there are mainly four categories for the fluorescence-based methods: fluorescent-dye staining, affinity-based binding assay, enzyme-mediated fluorescent labeling and host cell reactivation assay. In addition, a number of luminescent oligonucleotide-based methods for detecting the activities of DNA repair enzymes are available and will not be discussed here (Leung et al. 2013; Wilson and Kool 2018). There are some limitations of the fluorescence-based methods: they cannot provide any genomic sequence information and photobleaching caused by photochemical destruction of fluorophores is a concern in the quantitative analysis.
Fluorescent-dye staining
The fluorescent-dye staining is a straightforward method for visualizing DNA. The DNA intercalating fluorescence dyes such as acridine orange and propidium iodide were first used to detect DNA strand breaks (Gruzdev and Kishchenko 1978) and DNA loops (Roti Roti and Wright 1987). Three other fluorescence dyes, ethidium bromide, SYBR Green I, and GelRed are now commonly used in the Comet assay. This method is easy to use but it has relatively low sensitivity and requires pure DNA for staining. To increase the sensitivity and specificity, a strategy that uses direct covalent modification of DNA damage with fluorescent molecules such as near-infrared probe (Condie et al. 2015) and modified alkoxyamine (Wei et al. 2015) was developed to detect and quantify the abasic sites .
Affinity-based binding assay
In the affinity-based binding assay, DNA damage and repair are visualized by using the binding affinity of DNA damage specific antibodies or fluorophore-conjugated DNA repair proteins to DNA lesions. Antibodies against various DNA lesions such as CPD, (6–4)PP, BPDE-DNA adduct, AFB1-DNA adduct and 8-Oxoguanine have already been used to detect DNA damage and repair both in vivo and in vitro. Meanwhile, antibodies against DNA damage biomarkers such as γ-H2AX which is a biomarker for DNA double-strand breaks are also utilized to detect DNA damage and repair (Pilch et al. 2003; Bonner et al. 2008). Immunoassay-based methods such as RIA (Mitchell and Clarkson 1981), ELISA (Leipold et al. 1983), immunoslot blot (Wani et al. 1987) and microplate-formatted cell-based immunoassay for NER of UV photoproducts (M-CINUP) (Nishinaga et al. 2012) use the same strategy to detect and quantify various types of DNA damage . These methods heavily rely on the specificity of the primary antibody and require a secondary fluorophore-conjugated antibody to visualize the signal.
A more direct approach was developed to detect DNA damage and repair by using fluorophore-conjugated DNA repair proteins that bind to DNA lesions directly. For example, the bacteriophage Mu Gam protein, which recognizes double strand breaks, was fused to GFP to monitor the formation and repair of double strand breaks in E. coli and mammalian cells (Shee et al. 2013).The truncated p53-binding protein 1 (53BP1) that accumulate at double strand break sites was fused to Apple fluorescent protein to localize sites of double strand breaks by in vivo imaging (Yang et al. 2015). In another study, the DNA damage binding protein 2 (DDB2) was fused to FLAG-HA tag, hybridized onto fibroblasts treated with UV irradiation, and revealed by anti-HA immunofluorescence (Dreze et al. 2014). Although the DDB2 proteo-probe is quite useful for monitoring DNA damage and repair, it requires the secondary antibody for visualizing the signal.
Enzyme-mediated fluorescent labeling
The enzyme-mediated fluorescent labeling strategy uses various DNA repair enzymes to process the DNA damage and/or incorporate fluorescently-modified nucleotides at damage sites for visualization. In the TUNEL assay, the terminal deoxynucleotidyl transferase was used to label 3’- hydroxyl termini of DNA double strand breaks with biotin-dUTP or fluorescently-modified nucleotide in single cells for detecting DNA double strand breaks during apoptosis (Gavrieli et al. 1992; Loo 2002). This method is limited to detect strand breaks with 3’-hydroxyl termini only. In another study, the pyrimidine dimer glycosylase (PDG), fluorescently-labeled dNTP and DNA polymerase I were used to visualize single strand breaks and UV-induced DNA damage (Lee et al. 2013). Then, this method was extended to detect 8-Oxoguanine, CPD, (6–4)PP, uracil and abasic site by adding an enzymatic repair cocktail containing formamidopyrimidine DNA glycosylase (FPG), Endo VIII, pyrimidine dimer glycosylase (PDG), uracil-DNA glycosylase (UDG) and Endo IV (Zirkin et al. 2014). These enzymes recognize and process their DNA damage substrates, and the DNA polymerase and ligase perform the incorporation of the fluorescently-labeled nucleotides and ligation respectively. This method is quite useful because the enzyme cocktail can be tailored for visualizing specific type of DNA damage (Kang et al. 2016; Lee et al. 2016). However, this approach is applied only on extracted DNA in vitro.
An optimized cell-based Repair Assisted Damage Detection (RADD) method was developed to detect a broad spectrum of DNA damage by using the same labeling strategy as the aforementioned TUNEL assays within the cell (Holton et al. 2018). This technique uses CSK (a hypotonic buffer) treatment and mild permeabilization before treating with DNA damage processing mix (UDG, FPG, T4PDG, Endo IV, Endo VIII) and DNA gap filling mix (Klenow DNA polymerase, biotin-dUTP). It can be applied in various types of cells from different species without requirement of DNA extraction. More recently, a multi-color fluorescent labeling assay was devised to simultaneously detect oxidative damage and photoproducts caused by UV irradiation (Torchinsky et al. 2019). In this assay, the extracted DNA carrying oxidative damage and photoproducts are labeled two times consecutively. Each damage type is treated with specific repair enzymes and labeled with a distinct fluorescent nucleotide. After labeling, the DNA will be stained and stretched on a glass slide for imaging and quantification. This useful methodology requires only 50 ng of DNA sample for efficient labeling and can be adapted to detect other types of DNA damage.
Host cell reactivation assay
Host cell reactivation (HCR) assay measures the repair capacity of host cells to repair the in vitro DNA carrying damage. Generally, damaged viruses or plasmids are delivered into host cells and the replication reactivation of the damaged viruses or plasmids indicates the host repair capacity (Aaronson and Lytle 1970; Zavadova 1971). This assay has certain advantages over direct DNA-damaging treatment because the physiology of the cell will not be disturbed by the treatment of DNA-damaging agent. With the development of real time qPCR, a method termed oligonucleotide retrieval assay (ORA) was developed to evaluate nucleotide excision repair capacity of host cells transfected with synthetic biotinylated oligonucleoti des carrying CPD damage (Shen et al. 2014). This method uses the similar strategy with HCR to quantify the repair capacity of host cells. After repair incubation, the biotinylated oligonucleotides are retrieved by streptavidin beads and subjected to qPCR, in which damaged oligonucleotides are not as efficiently amplified as repaired ones, for quantification of the repaired oligonucleotides.
The green fluorescent protein (GFP) from the jellyfish, Aequorea victoria, and its derivatives have been utilized as a tool to track proteins since the cloning and expression of GFP in E. coli and Caenorhabditis elegans (Prasher et al. 1992; Chalfie et al. 1994). A fluorescence-based multiplex flow-cytometric HCR assay (FM-HCR) was developed to simultaneously measure the repair capacity in up to four repair pathways by using plasmid reporters expressing different fluorescent proteins (Nagel et al. 2014b). The repair pathways that can be measured by FM-HCR include nucleotide excision repair, base excision repair, mismatch repair, homologous recombination repair, nonhomologous end joining, and methylguanine methyltransferase repair. The fluorescent plasmid reporters carrying DNA lesions to be removed by different repair pathways are transfected into cells and the different fluorescent signals will be collected by a flow cytometry after repair incubation. The 96-well microplate flow-cytometric sample processing employed in FM-HCR enables the rapid and high-throughput analysis of repair capacity in multiple repair pathways. Meanwhile, an NGS-based HCR assay (HCR-Seq) was devised to measure transcriptional bypass of DNA lesions by deep sequencing of mRNA including the plasmid reporter transcripts. The FM-HCR and HCR-Seq are quite useful for comprehensive analysis of repair capacity in basic research and clinical applications (Nagel et al. 2014a; Nagel et al. 2017).
NEXT-GENERATION SEQUENCING-BASED METHODS
Determination of the genome-wide distribution of environmentally-induced DNA damage and the heterogeneous repair kinetics at nucleotide resolution is critical for characterizing genotoxicity and linking specific environmental carcinogen exposure to mutagenesis and subsequent carcinogenesis. Although a number of radioactive labeling-based methods were developed to detect DNA damage and repair at single-nucleotide resolution, they are limited to small genomic regions. A wide range of mass spectrometry-based DNA adductomic methods are extremely useful for simultaneous identification and quantification of multiple DNA adducts, but these methods cannot provide genome sequence information (Kanaly et al. 2006; Balbo et al. 2014; Hemeryck et al. 2016; Chang et al. 2018).
The advent of NGS technology has transformed the genome-related research by providing high-throughput, low cost and high accuracy DNA sequence data. In the last five years, a growing number of NGS-based DNA adductomic methods have been developed to detect, characterize and quantify various types of environmentally-induced DNA damage and repair at genome-wide level (Hu and Adar 2017; Panahi et al. 2018; Sloan et al. 2018; Salk and Kennedy 2019). As covalent modifications of DNA bases caused by exposures to environmental carcinogens hinder the PCR amplification step during the NGS library preparation procedure, the standard NGS method must be specifically modified for measuring DNA damage and repair. In the burgeoning field of NGS-based DNA adductomics, there are mainly three strategies to circumvent the obstacle. The first strategy uses immunoprecipitation or biotin-streptavidin capture to enrich DNA fragments carrying DNA damage. The DNA damage can be directly reversed or bypassed by translesion DNA polymerase in one round of primer extension before the PCR amplification step. The second strategy enzymatically or chemically creates a nick at the damage site and ligates the sequencing adaptor for NGS library preparation. The third one takes advantages of the DNA damage immunoprecipitation and the stoppage of high-fidelity DNA polymerase before the lesion during primer extension to enrich the DNA damage and localize the damage position.
Besides the three aforementioned strategies, there is another strategy using in situ end-labeling to detect double strand breaks (Vitelli et al. 2017). A number of methods developed to detect double strand breaks are based on this strategy: BLESS (Breaks Labeling, Enrichment on Streptavidin and next-generation Sequencing) (Crosetto et al. 2013), END-seq (Canela et al. 2016), BLISS (Breaks Labeling In Situ and Sequencing) (Yan et al. 2017), i-BLESS (immobilized-BLESS) (Biernacka et al. 2018). There are also two methods available for detection of single strand breaks: SSB-seq (Baranello et al. 2014) and SSiNGLe (single-strand break mapping at nucleotide genome level) (Cao et al. 2019). These methods are not discussed in detail in this review due to the space limitation.
DNA damage enrichment and damage reversal or bypass-based strategy
This strategy uses immunoprecipitation or biotin-streptavidin capture to enrich DNA fragments carrying DNA damage. For those DNA damage that can block PCR amplification, they are directly reversed or bypassed by translesion DNA polymerase in one round of primer extension before the PCR amplification of the NGS library. This strategy includes single-strand DNA-associated protein immunoprecipitation followed by sequencing (SPI-seq) (Zhou et al. 2013), 8-oxo-7,8-dihydroguanine sequencing (OG-Seq) (Ding et al. 2017), OxiDIP-Seq (Amente et al. 2019), Damaged DNA Immunoprecipitation and next generation sequencing (DDIP-seq) (Alhegaili et al. 2019), eXcision Repair sequencing (XR-seq) (Hu et al. 2015) and translesion eXcision Repair sequencing (tXR-seq) (Li et al. 2017). The SPI-seq was designed to map double strand breaks across the fission yeast genome. Similar to ChIP-seq, SPI-seq detects the enrichment of Rad52, which binds to the single-strand DNA at double strand break sites, to determine the hotspots of double strand breaks. OG-seq and OxiDIP-Seq have been applied to measure oxidative damage. As shown in Figure 2, the cell lysis and genomic DNA extraction are performed after treatment with DNA damaging agent. In OG-Seq (Fig. 2A), the OG is chemically labeled with biotin and the streptavidin magnetic beads are used to enrich the double stranded DNA carrying OG damage. And the strands complementary to the biotinylated ones will be released and sequenced (Ding et al. 2017). Instead of using chemical labeling, the OxiDIP-Seq directly uses an anti-8-Oxoguanine antibody to enrich the DNA fragments containing oxidative damage (Amente et al. 2019). In 2017, a novel experimental method which is similar to DDIP-seq (Fig. 2B) was used to study the effect of genome architecture on CPD susceptibility in human lung fibroblast. In this method, the sonicated DNA fragments were subjected to CPD lesion immunoprecipitation and photolyases were used to remove the CPDs and (6–4)PPs before the NGS library preparation and sequencing.
Fig. 2.
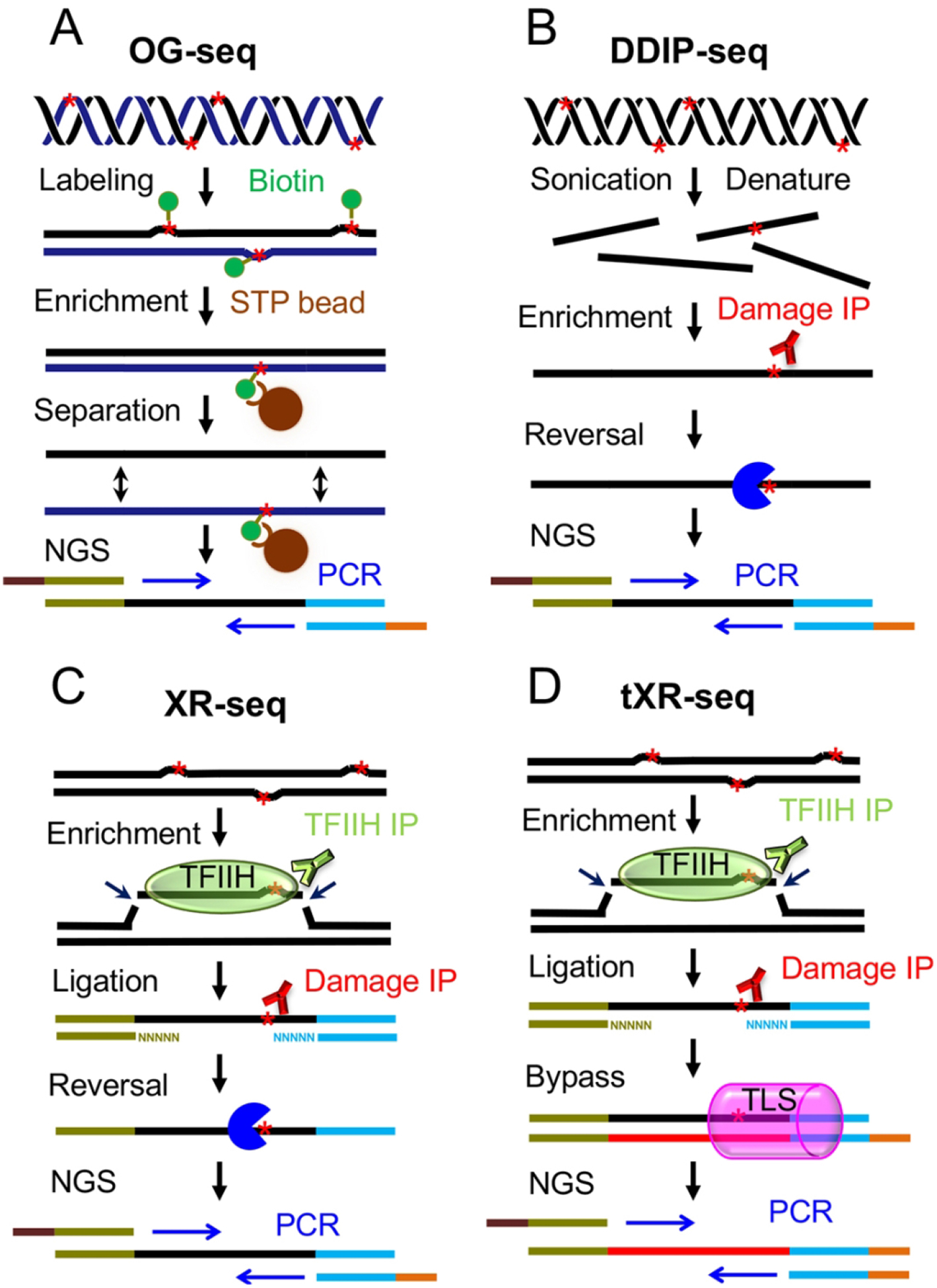
Schematic representation of four methods using the DNA damage enrichment and damage reversal or bypass-based strategy for detection of DNA damage and repair. (A) OG-seq for detection of 8-Oxoguanine (Ding et al. 2017). After fragmentation of genomic DNA containing oxidative damage, 8-Oxoguanines indicated by red stars are chemically labeled with biotin and enriched by streptavidin (STP) beads. Then, the double strands of DNA fragments are separated, and the strand (black) complementary to the damage containing strand (dark blue) will be amplified by PCR and sequenced by NGS. (B) Overview of DDIP-seq method (Alhegaili et al. 2019). The genomic DNA carrying CPDs represented by red stars is sonicated and denatured, and DNA strands containing damage are enriched by CPD immunoprecipitation. The CPDs are removed by repair enzymes (the dark blue pie), and then the strands are subjected to standard NGS library preparation procedure for sequencing. (C) XR-seq method for mapping repair of UV damage (Hu et al. 2015). The excised oligonucleotides carrying the UV damage indicated by red star are released in complex with TFIIH and enriched by TFIIH immunoprecipitation. After adapter ligation, the excised oligonucleotides are further purified by UV damage immunoprecipitation. The UV damage in the excised oligonucleotides are reversed by photolyases, and the excised oligonucleotides are amplified by PCR followed by NGS. Besides UV damage, the XR-seq can also be applied to detect repair of cisplatin-adducts which are reversed by sodium cyanide (Hu et al. 2016). (D) tXR-seq method for mapping repair of CPD and BPDE-DNA adducts (Li et al. 2017). The steps of DNA damage enrichment and ligation are similar to XR-seq. Instead of using damage reversal strategy, tXR-seq makes use of translesion DNA synthesis (TLS) polymerase (indicated by pink can) to bypass the DNA damage during one cycle of primer extension. The primer extension products are then amplified and subjected to NGS. This method can be applied to essentially all types of DNA damage that are removed by nucleotide excision repair.
It was reported that regions of lamina-associated heterochromatin are more susceptible to UV damage than the regions of euchromatin (Garcia-Nieto et al. 2017). The DDIP-seq method uses DNA damage immunoprecipitation to enrich sonicated DNA fragments carrying solar-simulated radiation-induced damage and PreCR Repair Mix to remove the CPDs and (6–4)PPs before PCR amplification (Alhegaili et al. 2019). Like ChIP-seq, these methods can detect DNA damage across the whole genome, but they have relatively low resolution ranging from 100 to 1000 bp.
The XR-seq (Fig. 2C) was developed to measure the nucleotide excision repair of UV- and cisplatin-caused DNA damage at single-nucleotide resolution following the studies on the fate of the excised oligonucleotide released during nucleotide excision repair (Mu et al. 1996; Kemp et al. 2012; Hu et al. 2013; Hu et al. 2015; Hu et al. 2016). It was found that the excised oligonucleotide was released in complex with TFIIH and XPG which incises the damage containing strand at the 3’ side of the lesion. In the XR-seq, mammalian cells are lysed gently after treatment with UV or cisplatin and the excised oligonucleotides are isolated by TFIIH or XPG immunoprecipitation. For some organisms such as E. coli (Adebali et al. 2017a), yeast (Li et al. 2018), Arabidopsis thaliana (Oztas et al. 2018) and Drosophila melanogaster (Deger et al. 2019), anti-DNA damage antibody can be used to capture the excised oligonucleotides when there are no anti-TFIIH or anti-XPG antibodies available. Then, the isolated oligonucleotides are ligated with adapters and further purified by specific DNA damage immunoprecipitation. The UV-induced DNA damage and cisplatin-DNA adducts on the adapter-ligated excised oligonucleotides are reversed by photolyases and sodium cyanide respectively before the PCR amplification (Hu et al. 2015; Hu et al. 2016). After sequencing, the reads from XR-seq are aligned to human genome and the damage sites can be identified based on the dual incision mode of human nucleotide excision repair.
Although the XR-seq is quite useful for studying the effects of a wide range of factors on nucleotide excision repair in E. coli and humans (Adar et al. 2016; Adebali et al. 2017a; Adebali et al. 2017b; Hu et al. 2017a; Hu et al. 2017b; Chiou et al. 2018), it is limited by its requirement of damage reversal by repair enzymes or chemical reactions because not all types of DNA damage on the excised oligonucleotides can be reversed enzymatically or chemically. To overcome this shortcoming, the tXR-seq (Fig. 2D) was devised and applied to map the repair of UV- and benzo[a]pyrene-caused DNA damage (Li et al. 2017). The tXR-seq and XR-seq share the same DNA damage enrichment procedure. They differ at the step before the PCR amplification. The tXR-seq uses the human translesion synthesis polymerases η and κ to bypass the CPD and BPDE-DNA damage respectively during the one-cycle primer extension. Then the primer extension products are amplified by PCR to create a library for sequencing and subsequent bioinformatic analysis. As tXR-seq does not require the DNA damage reversal by repair enzymes or chemical reactions and there are plenty of translesion synthesis polymerases commercially available for bypassing various types of DNA damage, it can be applied to map virtually all DNA lesions that are removed by nucleotide excision repair.
The key advantage of XR-seq and tXR-seq is that the repair is directly and purely measured by isolating all the repair products (the excised oligonucleotides) other than subtracting one large percentage of damage at later time point from another one at early time point. They have been applied in a wide range of organisms to study nucleotide excision repair-related cellular processes such as transcription, histone modifications, mutagenesis, circadian rhythm oscillation (Adar et al. 2016; Hu et al. 2017a; Oztas et al. 2018; Yang et al. 2018; Hu et al. 2019; Yang et al. 2019a; Yang et al. 2019b; Yimit et al. 2019). With advances in the detection of excised oligonucleotides from human skin epidermis after UVB exposure (Choi et al. 2019), the XR-seq and the tXR-seq have great potential to be used in clinical applications. For both XR-seq and tXR-seq, the drawback is that the real rate of repair cannot be measured because the excised oligonucleotides are continuously produced and degraded by nucleases during the nucleotide excision repair. These methods can only capture a snapshot of the ongoing repair of DNA damage at one time point. The specificity of DNA damage antibody is a variable that needs to be taken into account when interpreting results from methods using DNA damage immunoprecipitation. It is not an issue if the right antibody is chosen based on the experimental design.
Nick creation and ligation-based strategy
The nick creation and ligation-based strategy uses nicking endonucleases or chemical reactions to create a nick at damage sites before or after fragmentation of genomic DNA, and ligates the adapter to the 5’ or 3’ end at the nick site by using ligases before PCR amplification and subsequent sequencing. Based on this strategy, a good number of methods have been developed to detect various types of DNA damage such as UV damage, alkylation damage, oxidative damage, ribonucleotide incorporation and cisplatin-DNA adducts in different organisms: Excision-seq (Bryan et al. 2014), Ribose-seq (Koh et al. 2015), hydrolytic end sequencing (HydEn-seq) (Clausen et al. 2015), polymerase usage sequencing (Pu-seq) (Daigaku et al. 2015), Lesion-Adjoining Fragment Sequencing (LAF-Seq) (Li et al. 2015), embeded ribonucleotide sequencing (emRriboSeq) (Ding et al. 2015), endonuclease sequencing (EndoSeq) (Ding et al. 2015), CPD-seq (Mao et al. 2016), N-methylpurine-sequencing (NMP-seq) (Mao et al. 2017), Click-Code-Seq (Wu et al. 2018), and adductSeq (Premi et al. 2019).
The Excision-seq (Fig. 3A) uses E.coli UDG and T4 Endo IV to create a nick at the uracil site in the DNA, and utilizes S. pombe UVDE, which cleaves 5’ to CPD and (6–4)PP, and photolyases to create ligatable 5’ ends at CPD or (6–4)PP sites (Bryan et al. 2014). In Excision-seq, the fragmentation of genomic DNA depends on the repair enzyme digestion. Although this method can reach high resolution, it requires high level of DNA damage for library construction, which limits its wide application. For example, the UV dose used in this method needs to be around 10,000 J/m2 which is much higher than routinely used dose.
Fig. 3.
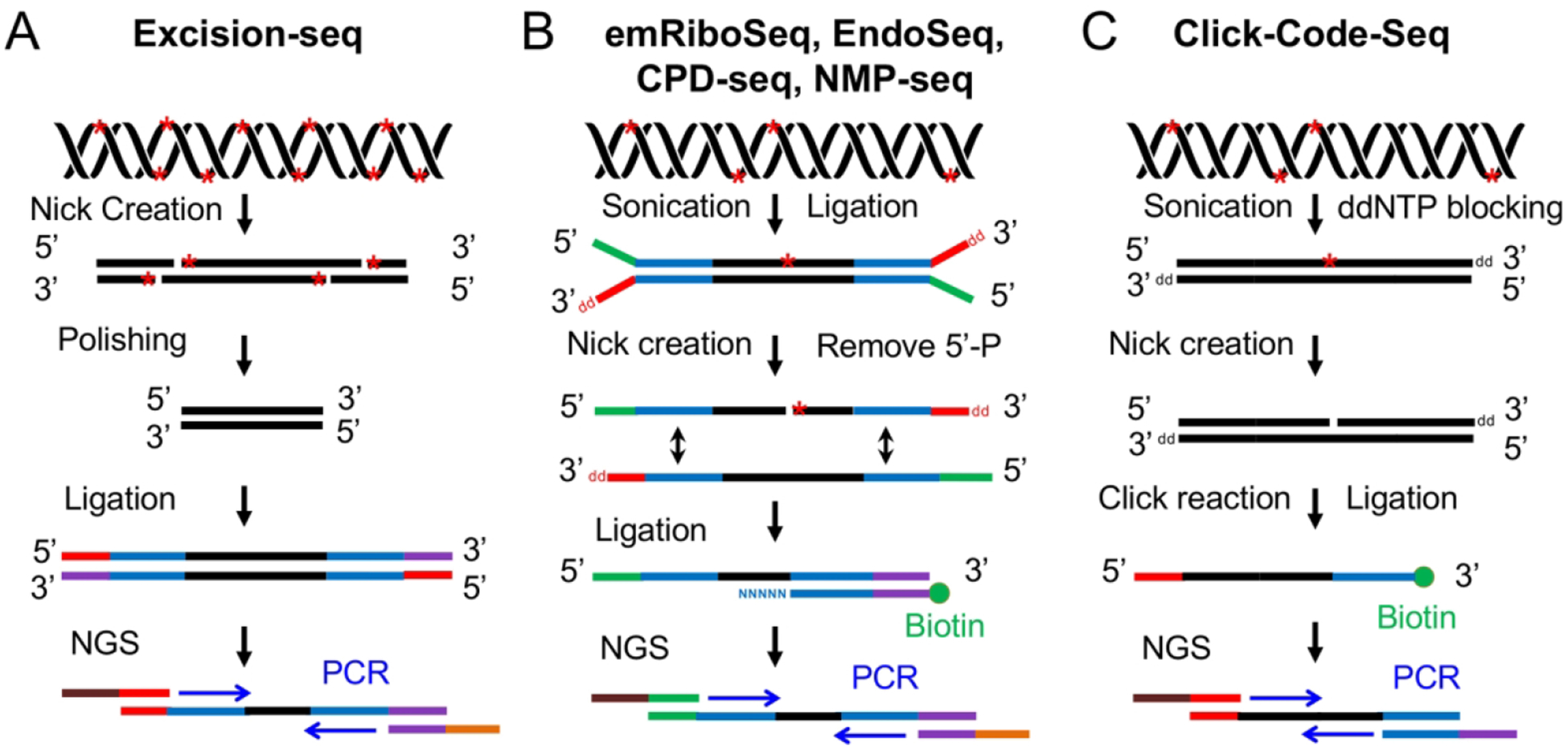
Schematic of six NGS-based methods using nick creation and ligation-based strategy for detection of DNA damage. (A) Excision-seq method for detection of uracil and UV damage indicated by red stars in DNA (Bryan et al. 2014). The UDG and Endo IV are used to digest uracil, and UVDE and photolyases are applied to nick at sites of UV damage. After polishing, adapter ligation and PCR amplification, the library is subjected to NGS. (B) Overview of experimental workflow for emRiboSeq, EndoSeq, CPD-seq, NMP-seq (Ding et al. 2015; Mao et al. 2016; Mao et al. 2017). After sonication and ligation, the DNA damage (red stars) are recognized and digested by specific repair enzymes to create a nick 5’ to the lesion. Then, the 5’-P group is removed and the second adapter is ligated to the DNA fragments containing 3’-OH group. Following ligation, the ligation products are amplified by PCR and subjected to NGS. (C) Click-Code-Seq method for detection of oxidative damage (Wu et al. 2018). After sonication and ddNTP blocking, one nucleotide gap is created by FPG and APE1 repair enzymes. Then, a prop-dGTP is incorporated to fill the gap and a 5’-azido-modified code sequence is ligated to the 3’ end by click reaction followed by the 5’ end adaptor ligation. The ligation products are amplified and sequenced by NGS.
For mapping ribonucleotide incorporation, the Ribose-seq, HydEn-seq and Pu-seq make use of alkaline hydrolysis to cleave at embedded ribonucleotides, and emRiboSeq (Fig. 3B) uses RNase H2 to make the cleavage. Although the alkaline treatment is simple and straightforward, it may cause background by hydrolysis at abasic sites. The replacement of RNase H2 with one specific nicking endonuclease in emRiboSeq is defined as EndoSeq (Fig. 3B). The EndoSeq can be applied to detect UV damage and apurinic/apyrimidinic (AP) sites, and it requires the generation of 3’-OH group after enzymatic digestion for subsequent biotinylated second adapter ligation.
The N-methylpurines (NMPs) caused by environmental methylating agents are removed by base excision repair. The alkyladenine glycosylase (AAG) recognizes and excises the NMPs generating AP sites in humans. The AP sites are then cleaved by AP endonuclease 1 (APE1) before the repair synthesis and ligation during base excision repair (Bauer et al. 2015). Both LAF-Seq and NMP-seq (Fig. 3B) make use of AAG and APE1 to create nicks at NMPs and ligate adapters to the 3’ ends for the following PCR amplification and sequencing. The two methods are useful for mapping NMPs at single-nucleotide resolution. The LAF-Seq can only detect NMPs at gene size level, whereas the NMP-seq can be used for detection of NMPs at genome-wide scale.
The CPD-seq (Fig. 3B) was devised to determine the CPD damage sites across the whole genome with single-nucleotide resolution in yeast (Mao et al. 2016). The T4 Endo V and APE1 are used to create a nick upstream the CPD sites and generate the ligatable 3’-OH group. This useful method has been applied to investigate the effects of transcription factor binding and nucleosome structure on CPD damage formation and repair as well as mutagenesis (Mao et al. 2016; Brown et al. 2018; Mao et al. 2018). The DNA strand breakage generated during DNA extraction and purification and the inefficient adaptor ligation can introduce the background signal which can be seen in the no UV treatment group (Mao et al. 2016). When the CPD damage level is relatively low, it might be challenging to detect the damage by using the CPD-seq method. The Damage-seq method makes use of DNA damage immunoprecipitation and stoppage of DNA polymerase before the damage site to detect a variety of DNA lesions with high sensitivity and single-nucleotide resolution (Hu et al. 2016; Hu et al. 2017a). Following the development of CPD-seq and Damage-seq, another method termed adductSeq was recently devised to detect the CPD hyperhotspots in humans (Premi et al. 2019). The adductSeq can also examine CPDs across the entire genome at single-nucleotide resolution by using T4 Endo V and photolyases to generate a nick and ligatable 5’ end at a CPD site. Semirandom primers are used to create a double-stranded end at the lesion site for subsequent adapter ligation and PCR amplification. To reduce the background, USER enzyme and shrimp alkaline phosphatase (rSAP) are used for blocking nonspecific ligation to adapters. Meanwhile, an elegant statistical method termed freqSeq was developed to identify the CPD hyperhotspots. The freqSeq uses ratio-based analysis to identify the highly recurrent CPDs based on the results from adductSeq. Interestingly, instead of using the endonuclease to create a nick at damage site, another method named Ad-Seq was developed to map DNA-adducts using the 5’ to 3’ exonuclease activity of two exonucleases (Harismendy and Howell 2018). The exonucleases Lambda and Rec-Jf continuously digest both double strand and single strand DNA from 5’ end until they encounter the DNA-adduct. The digestion resistant DNA-adducts are then subjected to 3’ C tailing and HG9 primer extension followed by adapter ligation and PCR amplification. This method has been used for mapping UV damage and cisplatin-DNA adducts, however, it also needs high level of DNA damage for enrichment of DNA adducts due to the background issue.
For single-nucleotide mapping of oxidative damage, the Click-Code-Seq (Fig. 3C) uses FPG and APE1 to remove the 8-oxoGuanine, creating one nucleotide gap. The gap is then filled with a synthetic O-3ʹ-propargyl modified nucleotide (prop-dGTP) by using a DNA polymerase. The 3ʹ-alkynyl modified DNA is ligated to a 5ʹ-azido-modified code sequence through a copper(I)-catalyzed click reaction. In this way, the 8-oxoGuanine sites are labeled with a biocompatible code sequence which is suitable for sequencing. After the 5’ end adaptor ligation, the ligation products are subjected to PCR amplification and NGS (Wu et al. 2018). This smart way of labeling 8-oxoGuanine sites enables the nucleotide-resolution mapping of oxidative damage at genome-wide scale, surpassing the OG-seq method.
Despite of the wide applications of the nick creation and ligation-based methods, the specificity and sensitivity of the strategy mainly depend on the nicking endonucleases applied in the specific application. Most importantly, the measurement of oxidative DNA damage in cellular DNA is quite challenging because of the low level of oxidative formation and the occurrence of artefactual DNA oxidation during DNA extraction (Cadet et al. 2011).
DNA damage enrichment and primer extension-based strategy
The DNA damage enrichment and primer extension-based strategy applies damage-specific immunoprecipitation and the stoppage of DNA polymerase before the damage during primer extension to determine the damage position. Two NGS-based damage-detecting methods have applied this strategy to map DNA damage formation and repair: one is the aforementioned Damage-seq (Hu et al. 2016), the other is cisplatin-seq (Shu et al. 2016).
In the Damage-seq (Fig. 4A), the damage-specific antibody is used to enrich the damage-containing DNA fragments ligated to the first adapter and a biotinylated primer is extended by the Q5 DNA polymerase which will stop before the damage site. The primer extension products are then ligated to the second adapter and amplified by PCR for sequencing. To increase the sensitivity of this method for detection of low levels of damage, a high-sensitivity damage sequencing (HS-Damage-seq) method was devised by adding a subtractive hybridization step in the original Damage-seq procedure to remove the undamaged strands before PCR amplification (Hu et al. 2017a). This HS-Damage-seq can even detect the remaining CPD damage after 48 hours of repair in human fibroblast cells treated with 10 J/m2 UVC. It has been applied to detect UV damage and cisplatin-DNA adducts (Hu et al. 2017a; Yimit et al. 2019). The Damage-seq can be adapted to detect essentially all types of DNA damage that can block the DNA polymerase if there is an available damage-specific antibody. The cisplatin-seq (Fig. 4B) makes use of the high mobility group box 1 protein (HMGB1), a nuclear protein that preferentially binds to the cisplatin-DNA adducts, to enrich the double-stranded DNA fragments carrying the cisplatin-DNA adducts. Then, the following steps including the primer extension, the ligation of the second adapter and the PCR amplification are similar with Damage-seq (Shu et al. 2016).
Fig. 4.
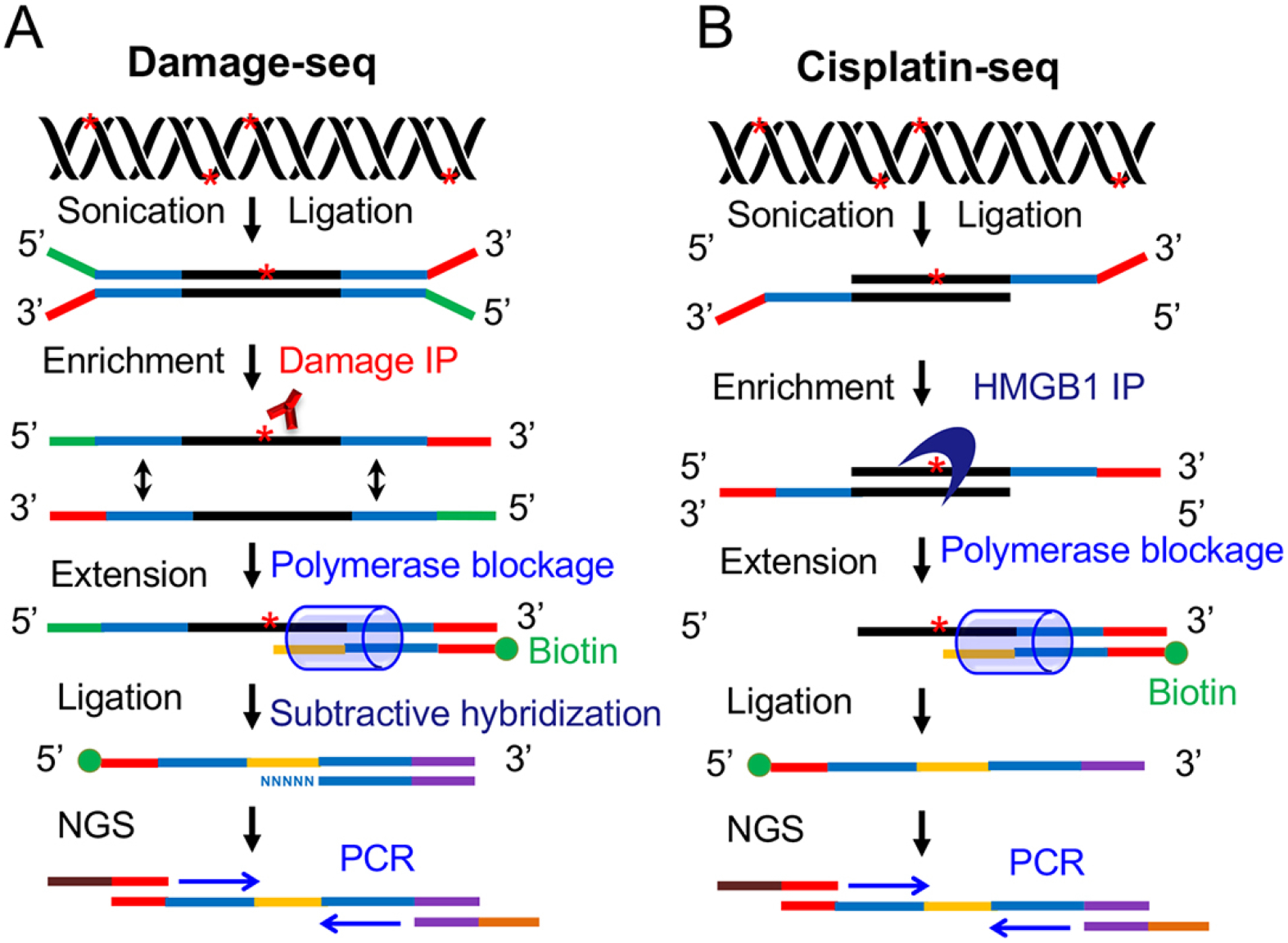
Schematic of two methods using DNA damage enrichment and primer extension-based strategy for detection of DNA damage. (A) Damage-seq method for detection of any type of damage that can block the DNA polymerase (Hu et al. 2016; Hu et al. 2017a). After sonication, ligation and denaturation, the DNA strands carrying damage are enriched by damage immunoprecipitation followed by one cycle of primer extension in which the high-fidelity DNA polymerase stops before the lesion. Following a subtractive hybridization step, the products of primer extension are then amplified by PCR for subsequent NGS. (B) Cisplatin-seq method for detection of cisplatin-DNA adducts (Shu et al. 2016). Following sonication and ligation, the DNA fragments containing cisplatin-DNA adducts are enriched by HMGB1 domain A immunoprecipitation followed by primer extension step which is similar to Damage-seq. Then, the primer extension products are ligated to a second adapter and amplified by PCR for the following NGS.
In contrast to the cisplatin-seq which can only be used for detection of cisplatin-DNA adducts, the Damage-seq is a general method and can be adapted to map various types of DNA damage under the condition that there are available damage-specific antibodies. The DNA damage enrichment step greatly reduces the level of background signal and therefore increases the sensitivity of this strategy. At the same time, the specificity of this strategy virtually depends on the specificity of the antibody or protein used.
THIRD-GENERATION SEQUENCING-BASED METHODS
The third-generation sequencing technologies (also known as long-read sequencing) including the Single Molecular Real-Time sequencing (SMRT) by Pacific Biosciences (PacBio) and Nanopore sequencing by Oxford detect the nucleotide sequences at single molecular level (Branton et al. 2008; Eid et al. 2009). The SMRT sequencing reads the nucleotide sequences by real-time imaging of the incorporation of differently fluorescent-labeled nucleotides by polymerase anchored in a small well (70 nm diameter and 100 nm depth). The Nanopore sequencing detects the nucleotide sequences by recording the ionic current shift when negatively charged DNA strand is forced by electrophoresis to pass through the biological nanopore formed within a phospholipid bilayer. The third-generation sequencing can produce long reads (more than 10 kb for SMRT) and does not require PCR amplification. In contrast, the short-read NGS-based methods for detection of DNA damage and repair generally need DNA damage enrichment and PCR amplification for high throughput sequencing. Thus, the NGS-based methods measure the relative amount of DNA damage and repair across the whole genome rather than the absolute amount.
Theoretically, the third-generation sequencing can directly detect some types of damaged base modifications. Indeed, the SMRT sequencing and Nanopore sequencing have been shown to directly detect oxidative damage, interstrand crosslink or UV damage in a few studies (Clark et al. 2011; Feng et al. 2013; Schadt et al. 2013; An et al. 2015; Zhang et al. 2015). A SMRT sequencing-based method termed RAre DAmage and Repair sequencing (RADAR-seq) was devised to detect the ribonucleotide incorporation and CPD damage in Thermococcus kodakarensis and E. coli (Zatopek et al. 2019). Because the 6mA and 4mC can be distinctively detected by SMRT sequencing, the RADAR-seq makes use of Bst FL DNA polymerase and Taq DNA ligase to replace the DNA damage with a patch of DNA bases containing 6mA and 4mC after nick creation at damage sites by repair enzymes. This promising method can be applied to detect rare and different types of DNA damage simultaneously at genome-wide level, and study cellular processes such as DNA repair and replication.
As the RADAR-seq uses only 6mA and 4mC to localize the damage site, it cannot achieve single-nucleotide resolution. The overlapping of two patches of DNA bases can challenge the identification of damage sites as well. The RADAR-seq can now only be applied in organisms with small genome size because of the low throughput of SMRT sequencing technology.
CONCLUSIONS AND PERSPECTIVES
Understanding the dynamic environmentally-induced DNA damage formation and repair events in the cell and how these events relate to mutagenesis and subsequent carcinogenesis requires the methodologies for detecting DNA damage and repair. The development of NGS technology has enabled us to study DNA damage and repair at genome-wide level and with single-nucleotide resolution. However, current NGS-based methodologies still have limitations such as short read-length, high input DNA requirement and PCR amplification bias. The breakthroughs in DNA sequencing technology will advance scientific research in the field of DNA damage and repair. For example, the improvements to the high error rates in the third-generation sequencing technology will expand its application in detecting cancerous mutations and different types of DNA damage. Besides, the emerging single-molecule super-resolution imaging technique allows us to directly visualize individual repair proteins and the events of DNA damage and repair in single cells. The further development of this imaging technique will help us to study the heterogeneity of DNA damage and repair in single cells.
The recent findings from the Pan Cancer Analysis of Whole Genomes (PCAWG) project have enabled us to understand cancer’s complexity on an unprecedented scale (Campbell 2020). In the near future, it is intriguing to explore how different cellular processes such as DNA damage formation and repair, DNA damage response, circadian clock oscillation, replication, transcription, histone modifications, chromosome looping, mutagenesis and carcinogenesis interplay with each other. We are entering an era of big data in which collaborations among research scientists, computer scientists, physician scientists are needed.
ACKNOWLEDGMENTS
We would like to thank Dr. Jinchuan Hu for his insightful comments on this manuscript. We apologize to authors whose work was not cited due to the space and topic limitations. This work was supported by the National Institute of Environmental Health Sciences K99ES030015 (to W.L.) and ES027255 (to A. S.), and National Institute of General Medical Sciences GM118102 (to A. S.).
REFERENCES
- Aaronson SA, Lytle CD. 1970. Decreased host cell reactivation of irradiated SV40 virus in xeroderma pigmentosum. Nature 228(5269):359–361. [DOI] [PubMed] [Google Scholar]
- Adar S, Hu J, Lieb JD, Sancar A. 2016. Genome-wide kinetics of DNA excision repair in relation to chromatin state and mutagenesis. Proc Natl Acad Sci U S A 113(15):E2124–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adebali O, Chiou YY, Hu J, Sancar A, Selby CP. 2017a. Genome-wide transcription-coupled repair in Escherichia coli is mediated by the Mfd translocase. Proc Natl Acad Sci U S A 114(11):E2116–E2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adebali O, Sancar A, Selby CP. 2017b. Mfd translocase is necessary and sufficient for transcription-coupled repair in Escherichia coli. J Biol Chem 292(45):18386–18391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhegaili AS, Ji Y, Sylvius N, Blades MJ, Karbaschi M, Tempest HG, Jones GDD, Cooke MS. 2019. Genome-Wide Adductomics Analysis Reveals Heterogeneity in the Induction and Loss of Cyclobutane Thymine Dimers across Both the Nuclear and Mitochondrial Genomes. Int J Mol Sci 20(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amente S, Di Palo G, Scala G, Castrignano T, Gorini F, Cocozza S, Moresano A, Pucci P, Ma B, Stepanov I, Lania L, Pelicci PG, Dellino GI, Majello B. 2019. Genome-wide mapping of 8-oxo-7,8-dihydro-2’-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Res 47(1):221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An N, Fleming AM, White HS, Burrows CJ. 2015. Nanopore detection of 8-oxoguanine in the human telomere repeat sequence. ACS Nano 9(4):4296–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S, Han S, Kang D, Kemp MG, Choi JH. 2018. Simultaneous detection of nucleotide excision repair events and apoptosis-induced DNA fragmentation in genotoxin-treated cells. Sci Rep 8(1):2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbo S, Turesky RJ, Villalta PW. 2014. DNA adductomics. Chem Res Toxicol 27(3):356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranello L, Kouzine F, Wojtowicz D, Cui K, Przytycka TM, Zhao K, Levens D. 2014. DNA break mapping reveals topoisomerase II activity genome-wide. Int J Mol Sci 15(7):13111–13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer NC, Corbett AH, Doetsch PW. 2015. The current state of eukaryotic DNA base damage and repair. Nucleic Acids Res 43(21):10083–10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernacka A, Zhu Y, Skrzypczak M, Forey R, Pardo B, Grzelak M, Nde J, Mitra A, Kudlicki A, Crosetto N, Pasero P, Rowicka M, Ginalski K. 2018. i-BLESS is an ultra-sensitive method for detection of DNA double-strand breaks. Commun Biol 1:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohr VA, Smith CA, Okumoto DS, Hanawalt PC. 1985. DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 40(2):359–369. [DOI] [PubMed] [Google Scholar]
- Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. 2008. GammaH2AX and cancer. Nat Rev Cancer 8(12):957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, Butler T, Di Ventra M, Garaj S, Hibbs A, Huang X, Jovanovich SB, Krstic PS, Lindsay S, Ling XS, Mastrangelo CH, Meller A, Oliver JS, Pershin YV, Ramsey JM, Riehn R, Soni GV, Tabard-Cossa V, Wanunu M, Wiggin M, Schloss JA. 2008. The potential and challenges of nanopore sequencing. Nat Biotechnol 26(10):1146–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AJ, Mao P, Smerdon MJ, Wyrick JJ, Roberts SA. 2018. Nucleosome positions establish an extended mutation signature in melanoma. PLoS Genet 14(11):e1007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan DS, Ransom M, Adane B, York K, Hesselberth JR. 2014. High resolution mapping of modified DNA nucleobases using excision repair enzymes. Genome Res 24(9):1534–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J, Douki T, Ravanat JL. 2011. Measurement of oxidatively generated base damage in cellular DNA. Mutat Res 711(1–2):3–12. [DOI] [PubMed] [Google Scholar]
- Campbell PJ, Getz G, Korbel JO et al. . 2020. Pan-cancer analysis of whole genomes. Nature 578(7793):82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela A, Sridharan S, Sciascia N, Tubbs A, Meltzer P, Sleckman BP, Nussenzweig A. 2016. DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Mol Cell 63(5):898–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Salazar-Garcia L, Gao F, Wahlestedt T, Wu CL, Han X, Cai Y, Xu D, Wang F, Tang L, Ricciardi N, Cai D, Wang H, Chin MPS, Timmons JA, Wahlestedt C, Kapranov P. 2019. Novel approach reveals genomic landscapes of single-strand DNA breaks with nucleotide resolution in human cells. Nat Commun 10(1):5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC. 1994. Green fluorescent protein as a marker for gene expression. Science 263(5148):802–805. [DOI] [PubMed] [Google Scholar]
- Chang YJ, Cooke MS, Hu CW, Chao MR. 2018. Novel approach to integrated DNA adductomics for the assessment of in vitro and in vivo environmental exposures. Arch Toxicol 92(8):2665–2680. [DOI] [PubMed] [Google Scholar]
- Chiou YY, Hu J, Sancar A, Selby CP. 2018. RNA polymerase II is released from the DNA template during transcription-coupled repair in mammalian cells. J Biol Chem 293(7):2476–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Gaddameedhi S, Kim SY, Hu J, Kemp MG, Sancar A. 2014. Highly specific and sensitive method for measuring nucleotide excision repair kinetics of ultraviolet photoproducts in human cells. Nucleic Acids Res 42(4):e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Han S, Kemp MG. 2019. Detection of the small oligonucleotide products of nucleotide excision repair in UVB-irradiated human skin. DNA Repair (Amst) 86:102766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Kim SY, Kim SK, Kemp MG, Sancar A. 2015. An Integrated Approach for Analysis of the DNA Damage Response in Mammalian Cells: NUCLEOTIDE EXCISION REPAIR, DNA DAMAGE CHECKPOINT, AND APOPTOSIS. J Biol Chem 290(48):28812–28821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark TA, Spittle KE, Turner SW, Korlach J. 2011. Direct detection and sequencing of damaged DNA bases. Genome Integr 2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen AR, Lujan SA, Burkholder AB, Orebaugh CD, Williams JS, Clausen MF, Malc EP, Mieczkowski PA, Fargo DC, Smith DJ, Kunkel TA. 2015. Tracking replication enzymology in vivo by genome-wide mapping of ribonucleotide incorporation. Nat Struct Mol Biol 22(3):185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condie AG, Yan Y, Gerson SL, Wang Y. 2015. A Fluorescent Probe to Measure DNA Damage and Repair. PLoS One 10(8):e0131330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosetto N, Mitra A, Silva MJ, Bienko M, Dojer N, Wang Q, Karaca E, Chiarle R, Skrzypczak M, Ginalski K, Pasero P, Rowicka M, Dikic I. 2013. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Methods 10(4):361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigaku Y, Keszthelyi A, Muller CA, Miyabe I, Brooks T, Retkute R, Hubank M, Nieduszynski CA, Carr AM. 2015. A global profile of replicative polymerase usage. Nat Struct Mol Biol 22(3):192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deger N, Yang Y, Lindsey-Boltz LA, Sancar A, Selby CP. 2019. Drosophila, which lacks canonical transcription-coupled repair proteins, performs transcription-coupled repair. J Biol Chem 294(48):18092–18098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Taylor MS, Jackson AP, Reijns MA. 2015. Genome-wide mapping of embedded ribonucleotides and other noncanonical nucleotides using emRiboSeq and EndoSeq. Nat Protoc 10(9):1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Fleming AM, Burrows CJ. 2017. Sequencing the Mouse Genome for the Oxidatively Modified Base 8-Oxo-7,8-dihydroguanine by OG-Seq. J Am Chem Soc 139(7):2569–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreze M, Calkins AS, Galicza J, Echelman DJ, Schnorenberg MR, Fell GL, Iwai S, Fisher DE, Szuts D, Iglehart JD, Lazaro JB. 2014. Monitoring repair of UV-induced 6–4-photoproducts with a purified DDB2 protein complex. PLoS One 9(1):e85896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323(5910):133–138. [DOI] [PubMed] [Google Scholar]
- Essigmann JM, Croy RG, Nadzan AM, Busby WF Jr., Reinhold VN, Buchi G, Wogan GN. 1977. Structural identification of the major DNA adduct formed by aflatoxin B1 in vitro. Proc Natl Acad Sci U S A 74(5):1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Fang G, Korlach J, Clark T, Luong K, Zhang X, Wong W, Schadt E. 2013. Detecting DNA modifications from SMRT sequencing data by modeling sequence context dependence of polymerase kinetic. PLoS Comput Biol 9(3):e1002935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman SE, Blackett AD, Monteleone DC, Setlow RB, Sutherland BM, Sutherland JC. 1986. Quantitation of radiation-, chemical-, or enzyme-induced single strand breaks in nonradioactive DNA by alkaline gel electrophoresis: application to pyrimidine dimers. Anal Biochem 158(1):119–129. [DOI] [PubMed] [Google Scholar]
- Garcia-Nieto PE, Schwartz EK, King DA, Paulsen J, Collas P, Herrera RE, Morrison AJ. 2017. Carcinogen susceptibility is regulated by genome architecture and predicts cancer mutagenesis. EMBO J 36(19):2829–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrieli Y, Sherman Y, Ben-Sasson SA. 1992. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119(3):493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Prasongtanakij S, Wood DK, Weingeist DM, Fessler J, Navasummrit P, Ruchirawat M, Engelward BP. 2014. CometChip: a high-throughput 96-well platform for measuring DNA damage in microarrayed human cells. J Vis Exp(92):e50607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gish G, Eckstein F. 1988. DNA and Rna Sequence Determination Based on Phosphorothioate Chemistry. Science 240(4858):1520–1522. [DOI] [PubMed] [Google Scholar]
- Govan HL 3rd, Valles-Ayoub Y, Braun J. 1990. Fine-mapping of DNA damage and repair in specific genomic segments. Nucleic Acids Res 18(13):3823–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi KA, McAdam SR, Souhami RL, Hartley JA. 1994. DNA damage by anti-cancer agents resolved at the nucleotide level of a single copy gene: evidence for a novel binding site for cisplatin in cells. Nucleic Acids Res 22(12):2311–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruzdev AD, Kishchenko GP. 1978. Fluorescence polarization of stretched polytene chromosomes stained with acridine orange. Biophys Struct Mech 4(2):97–110. [DOI] [PubMed] [Google Scholar]
- Guzder SN, Habraken Y, Sung P, Prakash L, Prakash S. 1995. Reconstitution of yeast nucleotide excision repair with purified Rad proteins, replication protein A, and transcription factor TFIIH. J Biol Chem 270(22):12973–12976. [DOI] [PubMed] [Google Scholar]
- Harismendy O, Howell SB. 2018. Ad-Seq, a genome-wide DNA-adduct profiling assay. bioRxiv. [Google Scholar]
- Hemeryck LY, Moore SA, Vanhaecke L. 2016. Mass Spectrometric Mapping of the DNA Adductome as a Means to Study Genotoxin Exposure, Metabolism, and Effect. Anal Chem 88(15):7436–7446. [DOI] [PubMed] [Google Scholar]
- Hirt B 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26(2):365–369. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH. 2001. Genome maintenance mechanisms for preventing cancer. Nature 411(6835):366–374. [DOI] [PubMed] [Google Scholar]
- Holton NW, Ebenstein Y, Gassman NR. 2018. Broad spectrum detection of DNA damage by Repair Assisted Damage Detection (RADD). DNA Repair (Amst) 66–67:42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Adar S. 2017. The Cartography of UV-induced DNA Damage Formation and DNA Repair. Photochem Photobiol 93(1):199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Adar S, Selby CP, Lieb JD, Sancar A. 2015. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev 29(9):948–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Adebali O, Adar S, Sancar A. 2017a. Dynamic maps of UV damage formation and repair for the human genome. Proc Natl Acad Sci U S A 114(26):6758–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Choi JH, Gaddameedhi S, Kemp MG, Reardon JT, Sancar A. 2013. Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J Biol Chem 288(29):20918–20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Li W, Adebali O, Yang Y, Oztas O, Selby CP, Sancar A. 2019. Genome-wide mapping of nucleotide excision repair with XR-seq. Nat Protoc 14(1):248–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Lieb JD, Sancar A, Adar S. 2016. Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc Natl Acad Sci U S A 113(41):11507–11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Selby CP, Adar S, Adebali O, Sancar A. 2017b. Molecular mechanisms and genomic maps of DNA excision repair in Escherichia coli and humans. J Biol Chem 292(38):15588–15597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JC, Svoboda DL, Reardon JT, Sancar A. 1992. Human nucleotide excision nuclease removes thymine dimers from DNA by incising the 22nd phosphodiester bond 5’ and the 6th phosphodiester bond 3’ to the photodimer. Proc Natl Acad Sci U S A 89(8):3664–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, Symons RH, Berg P. 1972. Biochemical method for inserting new genetic information into DNA of Simian Virus 40: circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proc Natl Acad Sci U S A 69(10):2904–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowski DP, Illenye S, Van Houten B. 1992. Analysis of DNA damage and repair in murine leukemia L1210 cells using a quantitative polymerase chain reaction assay. Nucleic Acids Res 20(13):3485–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaly RA, Hanaoka T, Sugimura H, Toda H, Matsui S, Matsuda T. 2006. Development of the adductome approach to detect DNA damage in humans. Antioxid Redox Signal 8(5–6):993–1001. [DOI] [PubMed] [Google Scholar]
- Kang TH, Lindsey-Boltz LA, Reardon JT, Sancar A. 2010. Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc Natl Acad Sci U S A 107(11):4890–4895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TH, Reardon JT, Kemp M, Sancar A. 2009. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc Natl Acad Sci U S A 106(8):2864–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Lee J, Kim J, Oh Y, Kim D, Lee J, Lim S, Jo K. 2016. Analysis of alcohol-induced DNA damage in Escherichia coli by visualizing single genomic DNA molecules. Analyst 141(14):4326–4331. [DOI] [PubMed] [Google Scholar]
- Kemp MG. 2019. Damage removal and gap filling in nucleotide excision repair. Enzymes 45:59–97. [DOI] [PubMed] [Google Scholar]
- Kemp MG, Reardon JT, Lindsey-Boltz LA, Sancar A. 2012. Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J Biol Chem 287(27):22889–22899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh KD, Balachander S, Hesselberth JR, Storici F. 2015. Ribose-seq: global mapping of ribonucleotides embedded in genomic DNA. Nat Methods 12(3):251–257, 253 p following 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunala S, Brash DE. 1992. Excision repair at individual bases of the Escherichia coli lacI gene: relation to mutation hot spots and transcription coupling activity. Proc Natl Acad Sci U S A 89(22):11031–11035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kim Y, Lim S, Jo K. 2016. Single-molecule visualization of ROS-induced DNA damage in large DNA molecules. Analyst 141(3):847–852. [DOI] [PubMed] [Google Scholar]
- Lee J, Park HS, Lim S, Jo K. 2013. Visualization of UV-induced damage on single DNA molecules. Chem Commun (Camb) 49(42):4740–4742. [DOI] [PubMed] [Google Scholar]
- Leipold B, Remy W, Adelmann-Grill B. 1983. Measurement of ultraviolet light-induced photolesions in mammalian DNA by microELISA. J Immunol Methods 60(1–2):69–76. [DOI] [PubMed] [Google Scholar]
- Leung CH, Zhong HJ, Lu LH, Chan DSH, Ma DL. 2013. Luminescent and colorimetric strategies for the label-free DNA-based detection of enzyme activity. Briefings in Functional Genomics 12(6):525–535. [DOI] [PubMed] [Google Scholar]
- Li M, Ko T, Li S. 2015. High-resolution Digital Mapping of N-Methylpurines in Human Cells Reveals Modulation of Their Induction and Repair by Nearest-neighbor Nucleotides. J Biol Chem 290(38):23148–23161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Waters R. 1996. Nucleotide level detection of cyclobutane pyrimidine dimers using oligonucleotides and magnetic beads to facilitate labelling of DNA fragments incised at the dimers and chemical sequencing reference ladders. Carcinogenesis 17(8):1549–1552. [DOI] [PubMed] [Google Scholar]
- Li S, Waters R. 1997. Induction and repair of cyclobutane pyrimidine dimers in the Escherichia coli tRNA gene tyrT: Fis protein affects dimer induction in the control region and suppresses preferential repair in the coding region of the transcribed strand, except in a short region near the transcription start site. J Mol Biol 271(1):31–46. [DOI] [PubMed] [Google Scholar]
- Li S, Waters R, Smerdon MJ. 2000. Low- and high-resolution mapping of DNA damage at specific sites. Methods 22(2):170–179. [DOI] [PubMed] [Google Scholar]
- Li W, Adebali O, Yang Y, Selby CP, Sancar A. 2018. Single-nucleotide resolution dynamic repair maps of UV damage in Saccharomyces cerevisiae genome. Proc Natl Acad Sci U S A 115(15):E3408–E3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Giles C, Li S. 2014a. Insights into how Spt5 functions in transcription elongation and repressing transcription coupled DNA repair. Nucleic Acids Res 42(11):7069–7083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Hu J, Adebali O, Adar S, Yang Y, Chiou YY, Sancar A. 2017. Human genome-wide repair map of DNA damage caused by the cigarette smoke carcinogen benzo[a]pyrene. Proc Natl Acad Sci U S A 114(26):6752–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Selvam K, Ko T, Li S. 2014b. Transcription bypass of DNA lesions enhances cell survival but attenuates transcription coupled DNA repair. Nucleic Acids Res 42(21):13242–13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T, Barnes DE. 2000. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol 65:127–133. [DOI] [PubMed] [Google Scholar]
- Loo DT. 2002. TUNEL assay. An overview of techniques. Methods Mol Biol 203:21–30. [DOI] [PubMed] [Google Scholar]
- Mao P, Brown AJ, Esaki S, Lockwood S, Poon GMK, Smerdon MJ, Roberts SA, Wyrick JJ. 2018. ETS transcription factors induce a unique UV damage signature that drives recurrent mutagenesis in melanoma. Nat Commun 9(1):2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Brown AJ, Malc EP, Mieczkowski PA, Smerdon MJ, Roberts SA, Wyrick JJ. 2017. Genome-wide maps of alkylation damage, repair, and mutagenesis in yeast reveal mechanisms of mutational heterogeneity. Genome Res 27(10):1674–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao P, Smerdon MJ, Roberts SA, Wyrick JJ. 2016. Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc Natl Acad Sci U S A 113(32):9057–9062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKelvey-Martin VJ, Ho ET, McKeown SR, Johnston SR, McCarthy PJ, Rajab NF, Downes CS. 1998. Emerging applications of the single cell gel electrophoresis (Comet) assay. I. Management of invasive transitional cell human bladder carcinoma. II. Fluorescent in situ hybridization Comets for the identification of damaged and repaired DNA sequences in individual cells. Mutagenesis 13(1):1–8. [DOI] [PubMed] [Google Scholar]
- McMaster GK, Carmichael GG. 1977. Analysis of single- and double-stranded nucleic acids on polyacrylamide and agarose gels by using glyoxal and acridine orange. Proc Natl Acad Sci U S A 74(11):4835–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DL, Clarkson JM. 1981. The development of a radioimmunoassay for the detection of photoproducts in mammalian cell DNA. Biochim Biophys Acta 655(1):54–60. [DOI] [PubMed] [Google Scholar]
- Moggs JG, Yarema KJ, Essigmann JM, Wood RD. 1996. Analysis of incision sites produced by human cell extracts and purified proteins during nucleotide excision repair of a 1,3-intrastrand d(GpTpG)-cisplatin adduct. J Biol Chem 271(12):7177–7186. [DOI] [PubMed] [Google Scholar]
- Mu D, Hsu DS, Sancar A. 1996. Reaction mechanism of human DNA repair excision nuclease. J Biol Chem 271(14):8285–8294. [DOI] [PubMed] [Google Scholar]
- Mu D, Park CH, Matsunaga T, Hsu DS, Reardon JT, Sancar A. 1995. Reconstitution of human DNA repair excision nuclease in a highly defined system. J Biol Chem 270(6):2415–2418. [DOI] [PubMed] [Google Scholar]
- Nagel ZD, Chaim IA, Samson LD. 2014a. Inter-individual variation in DNA repair capacity: a need for multi-pathway functional assays to promote translational DNA repair research. DNA Repair (Amst) 19:199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel ZD, Kitange GJ, Gupta SK, Joughin BA, Chaim IA, Mazzucato P, Lauffenburger DA, Sarkaria JN, Samson LD. 2017. DNA Repair Capacity in Multiple Pathways Predicts Chemoresistance in Glioblastoma Multiforme. Cancer Res 77(1):198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel ZD, Margulies CM, Chaim IA, McRee SK, Mazzucato P, Ahmad A, Abo RP, Butty VL, Forget AL, Samson LD. 2014b. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc Natl Acad Sci U S A 111(18):E1823–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo LP, Owiti NA, Swartz C, Winters J, Su Y, Ge J, Xiong A, Han J, Recio L, Samson LD, Engelward BP. 2019. Sensitive CometChip assay for screening potentially carcinogenic DNA adducts by trapping DNA repair intermediates. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishinaga M, Kurata R, Onishi K, Kuriyama K, Wakasugi M, Matsunaga T. 2012. Establishment of a microplate-formatted cell-based immunoassay for rapid analysis of nucleotide excision repair ability in human primary cells. Photochem Photobiol 88(2):356–362. [DOI] [PubMed] [Google Scholar]
- Ostling O, Johanson KJ. 1984. Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem Biophys Res Commun 123(1):291–298. [DOI] [PubMed] [Google Scholar]
- Oztas O, Selby CP, Sancar A, Adebali O. 2018. Genome-wide excision repair in Arabidopsis is coupled to transcription and reflects circadian gene expression patterns. Nature Communications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panahi Y, Fattahi A, Zarei F, Ghasemzadeh N, Mohammadpoor A, Abroon S, Nojadeh JN, Khojastefard M, Akbarzadeh A, Ghasemnejad T. 2018. Next-generation sequencing approaches for the study of genome and epigenome toxicity induced by sulfur mustard. Arch Toxicol 92(12):3443–3457. [DOI] [PubMed] [Google Scholar]
- Pettijohn D, Hanawalt P. 1964. Evidence for Repair-Replication of Ultraviolet Damaged DNA in Bacteria. J Mol Biol 9:395–410. [DOI] [PubMed] [Google Scholar]
- Pfeifer GP, Drouin R, Riggs AD, Holmquist GP. 1991. In vivo mapping of a DNA adduct at nucleotide resolution: detection of pyrimidine (6–4) pyrimidone photoproducts by ligation-mediated polymerase chain reaction. Proc Natl Acad Sci U S A 88(4):1374–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilch DR, Sedelnikova OA, Redon C, Celeste A, Nussenzweig A, Bonner WM. 2003. Characteristics of gamma-H2AX foci at DNA double-strand breaks sites. Biochem Cell Biol 81(3):123–129. [DOI] [PubMed] [Google Scholar]
- Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ. 1992. Primary structure of the Aequorea victoria green-fluorescent protein. Gene 111(2):229–233. [DOI] [PubMed] [Google Scholar]
- Premi S, Han L, Mehta S, Knight J, Zhao D, Palmatier MA, Kornacker K, Brash DE. 2019. Genomic sites hypersensitive to ultraviolet radiation. Proc Natl Acad Sci U S A 116(48):24196–24205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randerath K, Reddy MV, Gupta RC. 1981. 32P-labeling test for DNA damage. Proc Natl Acad Sci U S A 78(10):6126–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen RE, Painter RB. 1964. Evidence for Repair of Ultra-Violet Damaged Deoxyribonucleic Acid in Cultured Mammalian Cells. Nature 203:1360–1362. [DOI] [PubMed] [Google Scholar]
- Reardon JT, Thompson LH, Sancar A. 1997. Rodent UV-sensitive mutant cell lines in complementation groups 6–10 have normal general excision repair activity. Nucleic Acids Res 25(5):1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roti Roti JL, Wright WD. 1987. Visualization of DNA loops in nucleoids from HeLa cells: assays for DNA damage and repair. Cytometry 8(5):461–467. [DOI] [PubMed] [Google Scholar]
- Rupert CS. 1960. Photoreactivation of transforming DNA by an enzyme from bakers’ yeast. J Gen Physiol 43:573–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupert CS, Goodgal SH, Herriott RM. 1958. Photoreactivation in vitro of ultraviolet-inactivated Hemophilus influenzae transforming factor. J Gen Physiol 41(3):451–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N. 1985. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230(4732):1350–1354. [DOI] [PubMed] [Google Scholar]
- Salk JJ, Kennedy SR. 2019. Next-Generation Genotoxicology: Using Modern Sequencing Technologies to Assess Somatic Mutagenesis and Cancer Risk. Environ Mol Mutagen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A. 2016. Mechanisms of DNA Repair by Photolyase and Excision Nuclease (Nobel Lecture). Angew Chem Int Ed Engl 55(30):8502–8527. [DOI] [PubMed] [Google Scholar]
- Sancar A 2017. Claud S. Rupert (1919–2017): The Father of DNA Repair. Photochem Photobiol 93(4):1133–1134. [DOI] [PubMed] [Google Scholar]
- Sancar A, Clarke ND, Griswold J, Kennedy WJ, Rupp WD. 1981a. Identification of the uvrB gene product. J Mol Biol 148(1):63–76. [DOI] [PubMed] [Google Scholar]
- Sancar A, Hack AM, Rupp WD. 1979. Simple method for identification of plasmid-coded proteins. J Bacteriol 137(1):692–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A, Kacinski BM, Mott DL, Rupp WD. 1981b. Identification of the uvrC gene product. Proc Natl Acad Sci U S A 78(9):5450–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A, Rupert CS. 1978a. Cloning of the phr gene and amplification of photolyase in Escherichia coli. Gene 4(4):295–308. [DOI] [PubMed] [Google Scholar]
- Sancar A, Rupert CS. 1978b. Determination of plasmid molecular weights from ultraviolet sensitivities. Nature 272(5652):471–472. [DOI] [PubMed] [Google Scholar]
- Sancar A, Rupp WD. 1979. Cloning of uvrA, lexC and ssb genes of Escherichia coli. Biochem Biophys Res Commun 90(1):123–129. [DOI] [PubMed] [Google Scholar]
- Sancar A, Rupp WD. 1983. A novel repair enzyme: UVRABC excision nuclease of Escherichia coli cuts a DNA strand on both sides of the damaged region. Cell 33(1):249–260. [DOI] [PubMed] [Google Scholar]
- Sancar A, Stachelek C, Konigsberg W, Rupp WD. 1980. Sequences of the recA gene and protein. Proc Natl Acad Sci U S A 77(5):2611–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A, Wharton RP, Seltzer S, Kacinski BM, Clarke ND, Rupp WD. 1981c. Identification of the uvrA gene product. J Mol Biol 148(1):45–62. [DOI] [PubMed] [Google Scholar]
- Sanger F, Coulson AR. 1975. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol 94(3):441–448. [DOI] [PubMed] [Google Scholar]
- Schadt EE, Banerjee O, Fang G, Feng Z, Wong WH, Zhang X, Kislyuk A, Clark TA, Luong K, Keren-Paz A, Chess A, Kumar V, Chen-Plotkin A, Sondheimer N, Korlach J, Kasarskis A. 2013. Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases. Genome Res 23(1):129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeberg E, Nissen-Meyer J, Strike P. 1976. Incision of ultraviolet-irradiated DNA by extracts of E. coli requires three different gene products. Nature 263(5577):524–526. [DOI] [PubMed] [Google Scholar]
- Seeberg E, Steinum AL. 1982. Purification and properties of the uvrA protein from Escherichia coli. Proc Natl Acad Sci U S A 79(4):988–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby CP, Sancar A. 1993. Molecular mechanism of transcription-repair coupling. Science 260(5104):53–58. [DOI] [PubMed] [Google Scholar]
- Shee C, Cox BD, Gu F, Luengas EM, Joshi MC, Chiu LY, Magnan D, Halliday JA, Frisch RL, Gibson JL, Nehring RB, Do HG, Hernandez M, Li L, Herman C, Hastings PJ, Bates D, Harris RS, Miller KM, Rosenberg SM. 2013. Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. Elife 2:e01222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JC, Fox EJ, Ahn EH, Loeb LA. 2014. A rapid assay for measuring nucleotide excision repair by oligonucleotide retrieval. Sci Rep 4:4894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu X, Xiong X, Song J, He C, Yi C. 2016. Base-Resolution Analysis of Cisplatin-DNA Adducts at the Genome Scale. Angew Chem Int Ed Engl 55(46):14246–14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibghat-Ullah, Sancar A, Hearst JE. 1990. The repair patch of E. coli (A)BC excinuclease. Nucleic Acids Res 18(17):5051–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Broz AK, Sharbrough J, Wu Z. 2018. Detecting Rare Mutations and DNA Damage with Sequencing-Based Methods. Trends Biotechnol 36(7):729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smerdon MJ, Thoma F. 1990. Site-specific DNA repair at the nucleosome level in a yeast minichromosome. Cell 61(4):675–684. [DOI] [PubMed] [Google Scholar]
- Song J, Kemp MG, Choi JH. 2017. Detection of the Excised, Damage-containing Oligonucleotide Products of Nucleotide Excision Repair in Human Cells. Photochem Photobiol 93(1):192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak G, Cox RA, Hanawalt PC. 2009. New applications of the Comet assay: Comet-FISH and transcription-coupled DNA repair. Mutat Res 681(1):44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda DL, Taylor JS, Hearst JE, Sancar A. 1993. DNA repair by eukaryotic nucleotide excision nuclease. Removal of thymine dimer and psoralen monoadduct by HeLa cell-free extract and of thymine dimer by Xenopus laevis oocytes. J Biol Chem 268(3):1931–1936. [PubMed] [Google Scholar]
- Sykora P, Witt KL, Revanna P, Smith-Roe SL, Dismukes J, Lloyd DG, Engelward BP, Sobol RW. 2018. Next generation high throughput DNA damage detection platform for genotoxic compound screening. Sci Rep 8(1):2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Bennett M, Evans KE, Zhuang-Jackson H, Higgs A, Reed SH, Waters R. 2011. A novel method for the genome-wide high resolution analysis of DNA damage. Nucleic Acids Res 39(2):e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DC, Morton AG, Bohr VA, Sancar A. 1988. General method for quantifying base adducts in specific mammalian genes. Proc Natl Acad Sci U S A 85(11):3723–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchinsky D, Michaeli Y, Gassman NR, Ebenstein Y. 2019. Simultaneous detection of multiple DNA damage types by multi-colour fluorescent labelling. Chem Commun (Camb) 55(76):11414–11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houten B, Gamper H, Hearst JE, Sancar A. 1986. Construction of DNA substrates modified with psoralen at a unique site and study of the action mechanism of ABC excinuclease on these uniformly modified substrates. J Biol Chem 261(30):14135–14141. [PubMed] [Google Scholar]
- Varghese AJ, Wang SY. 1967. Cis-syn thymine homodimer from ultra-violet irradiated calf thymus DNA. Nature 213(5079):909–910. [DOI] [PubMed] [Google Scholar]
- Vitelli V, Galbiati A, Iannelli F, Pessina F, Sharma S, d’Adda di Fagagna F. 2017. Recent Advancements in DNA Damage-Transcription Crosstalk and High-Resolution Mapping of DNA Breaks. Annu Rev Genomics Hum Genet 18:87–113. [DOI] [PubMed] [Google Scholar]
- Wani AA, D’Ambrosio SM, Alvi NK. 1987. Quantitation of pyrimidine dimers by immunoslot blot following sublethal UV-irradiation of human cells. Photochem Photobiol 46(4):477–482. [DOI] [PubMed] [Google Scholar]
- Watson JD, Crick FH. 1953. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 171(4356):737–738. [DOI] [PubMed] [Google Scholar]
- Wei S, Shalhout S, Ahn YH, Bhagwat AS. 2015. A versatile new tool to quantify abasic sites in DNA and inhibit base excision repair. DNA Repair (Amst) 27:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellinger RE, Thoma F. 1996. Taq DNA polymerase blockage at pyrimidine dimers. Nucleic Acids Res 24(8):1578–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellinger RE, Thoma F. 1997. Nucleosome structure and positioning modulate nucleotide excision repair in the non-transcribed strand of an active gene. EMBO J 16(16):5046–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins MH, Stokes AR, Wilson HR. 1953. Molecular structure of deoxypentose nucleic acids. Nature 171(4356):738–740. [DOI] [PubMed] [Google Scholar]
- Wilson DL, Kool ET. 2018. Fluorescent Probes of DNA Repair. ACS Chem Biol 13(7):1721–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin EM. 1966. Radiation-induced mutations and their repair. Science 152(3727):1345–1353. [DOI] [PubMed] [Google Scholar]
- Wood DK, Weingeist DM, Bhatia SN, Engelward BP. 2010. Single cell trapping and DNA damage analysis using microwell arrays. Proc Natl Acad Sci U S A 107(22):10008–10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, McKeague M, Sturla SJ. 2018. Nucleotide-Resolution Genome-Wide Mapping of Oxidative DNA Damage by Click-Code-Seq. J Am Chem Soc 140(31):9783–9787. [DOI] [PubMed] [Google Scholar]
- Yan WX, Mirzazadeh R, Garnerone S, Scott D, Schneider MW, Kallas T, Custodio J, Wernersson E, Li Y, Gao L, Federova Y, Zetsche B, Zhang F, Bienko M, Crosetto N. 2017. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat Commun 8:15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang KS, Kohler RH, Landon M, Giedt R, Weissleder R. 2015. Single cell resolution in vivo imaging of DNA damage following PARP inhibition. Sci Rep 5:10129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Adebali O, Wu G, Selby CP, Chiou YY, Rashid N, Hu J, Hogenesch JB, Sancar A. 2018. Cisplatin-DNA adduct repair of transcribed genes is controlled by two circadian programs in mouse tissues. Proc Natl Acad Sci U S A 115(21):E4777–E4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Hu J, Selby CP, Li W, Yimit A, Jiang Y, Sancar A. 2019a. Single-nucleotide resolution analysis of nucleotide excision repair of ribosomal DNA in humans and mice. J Biol Chem 294(1):210–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Liu Z, Selby CP, Sancar A. 2019b. Long-term, genome-wide kinetic analysis of the effect of the circadian clock and transcription on the repair of cisplatin-DNA adducts in the mouse liver. J Biol Chem 294(32):11960–11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimit A, Adebali O, Sancar A, Jiang Y. 2019. Differential damage and repair of DNA-adducts induced by anti-cancer drug cisplatin across mouse organs. Nat Commun 10(1):309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatopek KM, Potapov V, Maduzia LL, Alpaslan E, Chen L, Evans TC Jr., Ong JL, Ettwiller LM, Gardner AF. 2019. RADAR-seq: A RAre DAmage and Repair sequencing method for detecting DNA damage on a genome-wide scale. DNA Repair (Amst) 80:36–44. [DOI] [PubMed] [Google Scholar]
- Zavadova Z 1971. Host-cell repair of vaccinia virus and of double stranded RNA of encephalomyocarditis virus. Nat New Biol 233(38):123. [PubMed] [Google Scholar]
- Zavala AG, Morris RT, Wyrick JJ, Smerdon MJ. 2014. High-resolution characterization of CPD hotspot formation in human fibroblasts. Nucleic Acids Res 42(2):893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Price NE, Fang X, Yang Z, Gu LQ, Gates KS. 2015. Characterization of Interstrand DNA-DNA Cross-Links Using the alpha-Hemolysin Protein Nanopore. ACS Nano 9(12):11812–11819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZX, Zhang MJ, Peng X, Takayama Y, Xu XY, Huang LZ, Du LL. 2013. Mapping genomic hotspots of DNA damage by a single-strand-DNA-compatible and strand-specific ChIP-seq method. Genome Res 23(4):705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirkin S, Fishman S, Sharim H, Michaeli Y, Don J, Ebenstein Y. 2014. Lighting up individual DNA damage sites by in vitro repair synthesis. J Am Chem Soc 136(21):7771–7776. [DOI] [PubMed] [Google Scholar]