

Abstract
RNA molecules have a variety of cellular functions that can drive disease pathologies. They are without a doubt one of the most intriguing yet controversial small-molecule drug targets. The ability to widely target RNA with small molecules could be revolutionary, once the right tools, assays, and targets are selected, thereby defining which biomolecules are targetable and what constitutes drug-like small molecules. Indeed, approaches developed over the past 5–10 years have changed the face of small molecule–RNA targeting by addressing historic concerns regarding affinity, selectivity, and structural dynamics. Presently, selective RNA–protein complex stabilizing drugs such as branaplam and risdiplam are in clinical trials for the modulation of SMN2 splicing, compounds identified from phenotypic screens with serendipitous outcomes. Fully developing RNA as a druggable target will require a target engagement-driven approach, and evolving chemical collections will be important for the industrial development of this class of target. In this review we discuss target-directed approaches that can be used to identify RNA-binding compounds and the chemical knowledge we have today of small-molecule RNA binders.
Introduction
The central dogma of molecular biology states that DNA is transcribed to RNA, which is then translated to protein. This dogma implies two notions: (1) only proteins are capable of the complex processes that enable a cell to function, and (2) nucleic acids, especially RNA, serve only as passive carriers of genetic information for protein synthesis. However, the recent Encyclopedia of DNA Elements (ENCODE) project determined that only ~1.5% of the human genome codes for proteins, yet more than 50% is transcribed from DNA to RNA.1 This finding suggests that RNAs have a more significant role to play in cellular function than previously believed (Fig. 1A). Furthermore, the advancement of next-generation sequencing and the field of epigenetics have attributed a myriad of functions to both coding and noncoding segments of RNA, including its indispensable role in the regulation of gene expression.2,3 Finally, at least 67% of the 20,000 proteins encoded in the human genome have intrinsically disordered regions that render them undruggable by conventional protein-focused methods,4,5 suggesting that they are only targetable at the transcriptional level. These discoveries have therefore poised RNA as a potentially critical player in the future of drug discovery.
Figure 1.
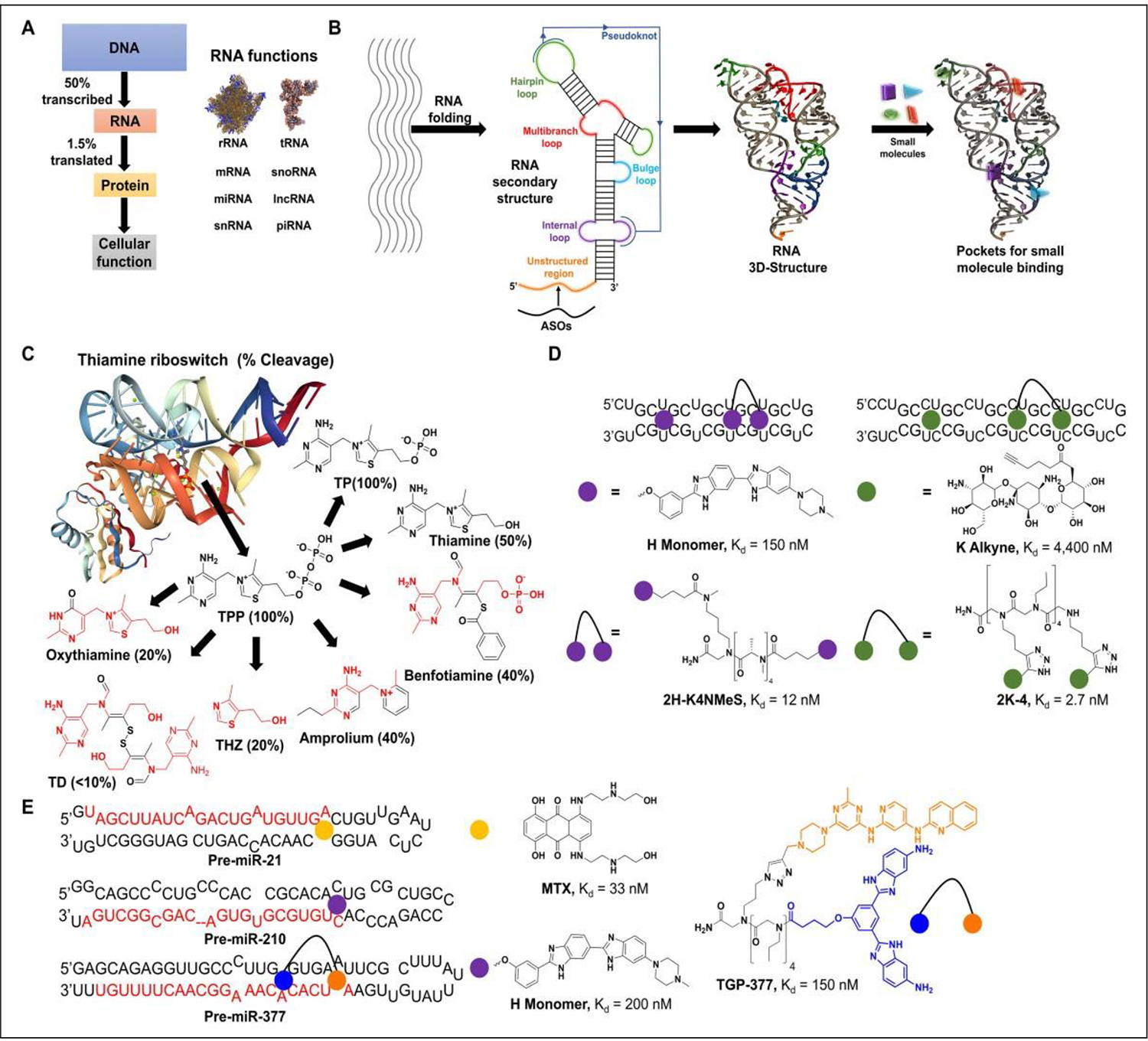
Schemes of key advances in RNA–small molecule binding. (A) Central dogma of molecular biology previously only showed RNA as a carrier of information, but yet >50% of the genome is transcribed and only 1.5% codes for protein, suggesting that RNA has alternate functions. (B) These coding and noncoding RNAs can adopt 3D folds that are both devoid and rich in structure that enable different targeting modalities such as ASOs to unstructured regions and small molecules to structured regions. (C) One example to highlight the relationship of RNA structure and function and how small molecules can perturb this system is the thiamine riboswitch that binds TPP and its derivatives. By mimicking the natural target ligand TPP, ligands with varying activity toward binding the riboswitch can be generated. (D) Other RNAs rich in structure that can be targeted by small molecules include r(CUG)exp and r(CCUG)exp in DM1 and DM2 by bis-benzimidazole derivatives and aminoglycosides like kanamycin with high affinity. (E) miRs are also rich in a structure that can be targeted for bioactive interactions, such as miR-21 by mitoxantrone, miR-210 by a bis-benzimidazole, and miR-377 by TGP-377, with low nanomolar affinities to their targets.
Initial efforts to drug RNA utilized antisense oligonucleotides (ASOs) that target RNA by Watson–Crick base pairing. This method has achieved moderate success in FDA-approved treatments6 but contains several limitations including complications with efficacy, delivery, and off-target effects.7 Its most notable drawback is its dependence on targeting unstructured regions of RNA, which is nevertheless intrinsically structured (Fig. 1B), limiting the amenability of ASOs to target RNA8,9 and necessitating new strategies. The pioneering discoveries of Zaug and Cech10 and Baer and Altman11 in the study of group I introns and ribonuclease P, respectively, revealed the intimate relationship between RNA structure and function. This work suggests that targeting an RNA’s structured regions may be a tractable means of drugging it. The 3D folds of RNA—including helical regions, hairpin loops, bulges, multibranched loops, and internal loops, as well as longer-range interactions like pseudoknots12 — create potential pockets for the binding of small-molecule drugs (Fig. 1B).
This was exemplified in the discovery of aminoglycoside binders to the bacterial ribosome’s 3D fold, specifically the ribosomal A site.13–15 Additional examples of small molecules that bind to RNA’s 3D structure and modulate its function accordingly arose from the discovery of riboswitches, including the thiamine riboswitch (Fig. 1C).16 Studies on riboswitches indicated that mimicking the native ligand can indeed enable the drugging of RNA by small molecules.17,18 Since the publication of these seminal works, more than 200 small-molecule binders to RNA have been identified19,20 to target a wide variety of RNAs, from repeat expansions that cause myotonic dystrophy types 1 (DM1; r[CUG]) and 2 (DM2; r[CCUG]) (Fig. 1D)21,22 to micro- RNAs (miRs) in cancer (Fig. 1E).23,24 RNA’s evolving biological role and disease relevance necessitates new ligands to target its structure, as well as new methods of doing so.
Many reviews are available that focus on individual methods used to identify RNA-binding small molecules. However, a comprehensive review summarizing all of the tools available for this purpose has yet to emerge. Thus, the focus of this review is to provide a broad overview of the methodologies available to identify RNA-binding small molecules and cite examples of these methods being used to identify ligands of potential therapeutic benefit. The methods described herein are divided into three categories: (1) label-based methods, such as fluorescent intercalator displacement (FID), Fö rster resonance energy transfer (FRET), differential scanning fluorimetry (DSF), micro-scale thermophoresis (MST), and surface plasmon resonance (SPR); (2) label-free methods, including small-molecule microarrays (SMMs), mass spectrometry (MS)-based approaches, and nuclear magnetic resonance (NMR) spectroscopy; and (3) predictive approaches like virtual screening. These methods will be described in the context of their individual merits for assessing small-molecule RNA-binding capacity and additional potential for high-throughput screening (HTS) where applicable.
Label-Based Methods for Identifying an RNA Binder
Small-molecule binding to RNA can be assessed by a number of methods, most of which involve labeling the RNA at its terminus with a fluorescent probe such as 5-carboxyfluorescein (5-FAM), cyanine dyes (Cy5, Cy3), rhodamine, and Alexa Fluor dyes (Fig. 2A), or internally with modified nucleotides. This fluorescence is then used to monitor physical changes that occur during binding, such as (1) changes in emission resulting from the label stacking / unstacking with the helix upon ligand binding;25,26 (2) alterations in molecular rotation by fluorescence polarization (FP);27 (3) displacement of RNA-binding proteins or beacons by FRET;28 (4) changes in molecular movement in a thermal gradient, that is, thermophoresis by MST;29 and (5) alterations in an RNA’s thermal stability by DSF.30 Other methods discussed herein include FID and SPR, both of which have seen extensive use in screening campaigns to identify RNA-binding matter. These methods will be discussed in the context of RNA small-molecule binding.
Figure 2.
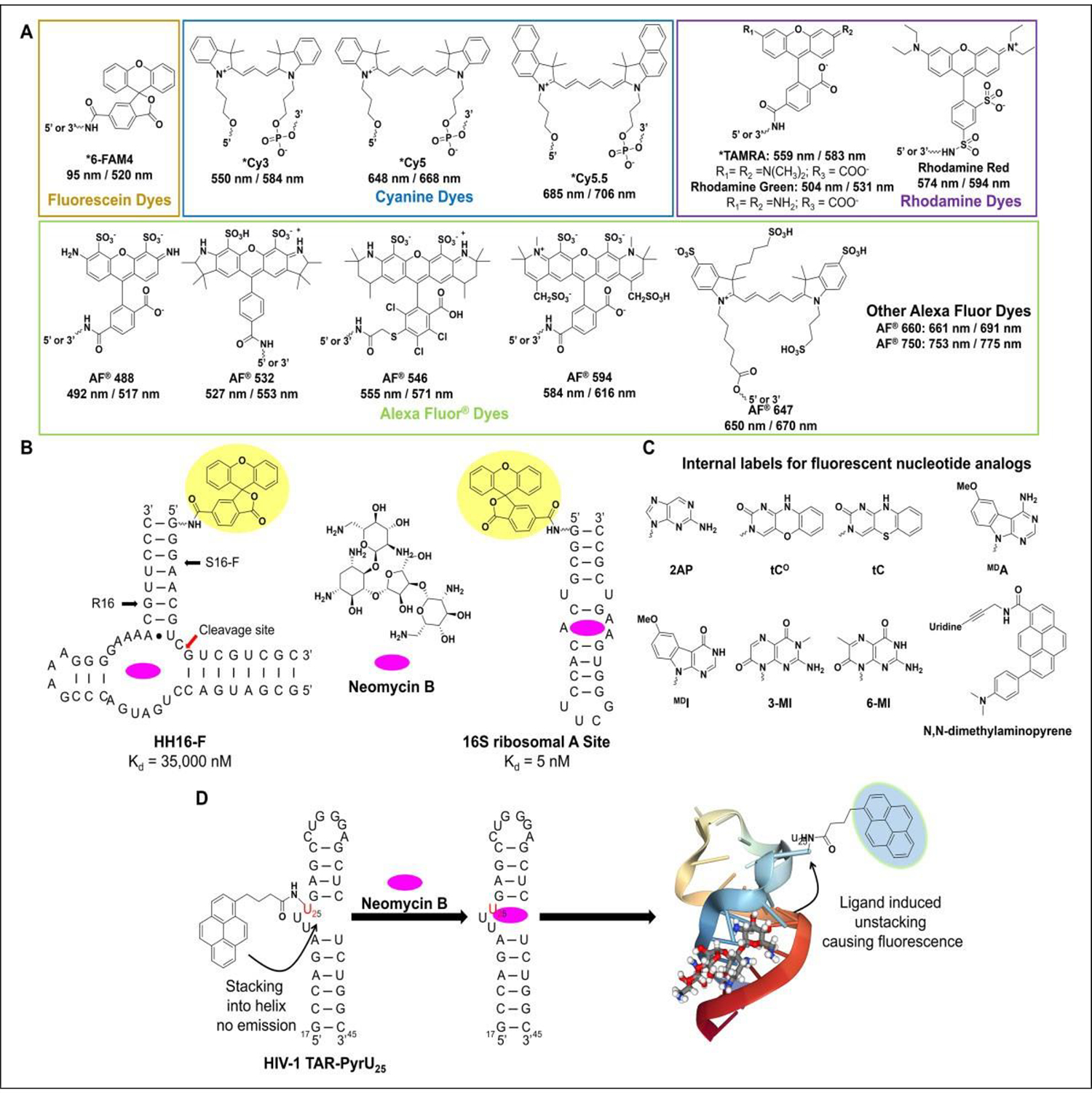
Common probes used for label-based methods to assess RNA binding. (A) Common probes used for 5´ or 3´ end labeling of oligonucleotides. These include fluorescein dyes (6-FAM); cyanine dyes such as Cy3, Cy5, and Cy5.5; rhodamine dyes such as TAMRA; and their derivates, the Alexa Fluor group of dyes. These are all common labels used for the study of RNA–small molecule binding and RNA structural dynamics. (B) HH16-F and the 16S ribosomal A site were labeled with 6-FAM and studied for neomycin B’s binding to the multibranch loop and A site, respectively. (C) Examples of fluorescent nucleoside analogs that can be used to internally label RNA. (D) Blount and Tor applied the incorporation of pyrene U to study the binding of neomycin B to HIV-1-TAR RNA. Labeling of U25 results in quenched emission with ligand binding displacing the pyrene and restoring fluorescence.
Direct Binding with End-Labeled RNAs
The fluorescence of labeled RNAs can be directly monitored to measure the binding interaction of small molecules to the RNA. This is due to differential stacking of the label with the helix, which can be sensitive to perturbations in the RNA’s structure that occur upon ligand binding. Numerous examples have demonstrated the application of this method to assess a small molecule’s RNA binding, notably the work of Llano-Sotelo and Chow. This group conjugated the S16 component of the hammerhead (HH) ribozyme with fluorescein on the 5´ terminus and, upon annealing with the R16 component, generated the HH16-F construct that could detect binding of neomycin B with a Kd of 35 mM (Fig. 2B).25 Using a similar strategy, the same group studied the binding of neomycin B to the 16S bacterial ribosomal A site, affording a Kd of 5 nM. This result is consistent with Kd values obtained by other means, such as SPR (19 nM).31 Despite their appealing simplicity, these methods have several limitations. Conjugation of the fluorophore to the RNA can perturb the RNA’s native structure, typically necessitating long linkers. Changes in the fluorophore’s photo-physical properties due to stacking with terminal bases, the magnitude of which is affected by linker length, can also dramatically change assay sensitivity. Signal intensity is also highly dependent on whether the tag is internal or at the termini.32 Furthermore, an RNA-binding event entails both local and global changes to the RNA structure33–39 that may not be detected with a terminally conjugated label.
Fluorescent Nucleotide Analogs
Internally labeled nucleobase analogs can serve to combat the limitations outlined above, as a number of strategies have been employed to increase their fluorescence properties. These methods include using isosteres, such as 2-aminopurine (2AP), that are intrinsically fluorescent; synthesizing base modified variants where the p system is extended; or using extended nucleobase analogs, where known fluorophores are conjugated onto the parent base via a rigid or flexible linker (Fig. 2C). Numerous reports are available for the application of 2AP and the study of small-molecule binding to RNA, and as a starting point we refer the reader to the recent review by McGovern-Gooch and Baird for further reading.40 Base-modified variants include methoxybenzodeazaadenine (MDA), methoxybenzodeazainosine (MDI), and tricyclic cytidine (tC). Okamoto et al. used the MDA and MDI modification and its analogs to create bases capable of discriminating between single-nucleotide polymorphisms (SNPs),41 whereas Lin et al. used tC fluorescence to study the discrimination of base pairs in DNA/RNA hybrid duplexes and demonstrated enhanced affinity with the modified base.42 Füchtbauer et al. synthesized and incorporated these tC analogs into RNA and showed that A-form geometry was maintained.43 Other analogs include pteridines such as 3- and 6-methylisoxanthopterin (3-MI and 6-MI) used to study RNA conformational dynamics.26,44 Studying extended analog variants, Okamoto et al. used pyrene conjugated to uridine (PyrU) to show the same discrimination between base pairing.45 Since these reports, extensive efforts have been made to synthesize new fluorescent nucleobase analogs in an attempt to expand the current repertoire of labels and improve the study of RNA small-molecule binding. We refer the reader to Tanpure et al.46 for a detailed summarization of these efforts.
Generally speaking, the methods referenced above are applied to the study of RNA conformational dynamics, but in theory they can be applied to the study of small-molecule binding to RNA as well. This is exemplified by Blount and Tor’s work with identifying binders to HIV-1 TAR using pyrene U-labeled TAR RNA (Fig. 2D).47 Compared with end-labeled RNAs, these internally labeled constructs are more sensitive to changes in the local and global structure of the RNA upon ligand binding, affording greater insight into both the binding event and the binding-related changes to the RNA’s structure.
Fluorescence Polarization
FP is the selective excitation of a fluorophore using plane polarized light. The selection arises from the orientation of the molecules and is such that only a fraction of them will have their dipole in a parallel orientation relative to the excitation polarization. Depending on the species that the fluorophore is conjugated to—that is, a small molecule, nucleic acid, or protein—the species will tumble at different rates, a result of Brownian motion relative to the lifetime of the fluorophore, randomizing its orientation and with small species tumbling faster than larger species. Thus, when a photon is emitted, it will be unpolarized, and the slower-tumbling larger species will retain some of the original polarization (Fig. 3A). In addition, when two species bind together, they influence each other’s molecular rotation and cause retention of polarization upon binding. To maximize the observed change in polarization, the species that has the largest change in mobility upon binding should be labeled. Other factors to consider include the fluorescence lifetime of the fluorophore, solvent viscosity, and solvent composition, as they have significant effects on molecular motion and changes in fluorescence quantum yield during the binding event. A more detailed discussion of these effects is reviewed by Jameson and Ross.48
Figure 3.
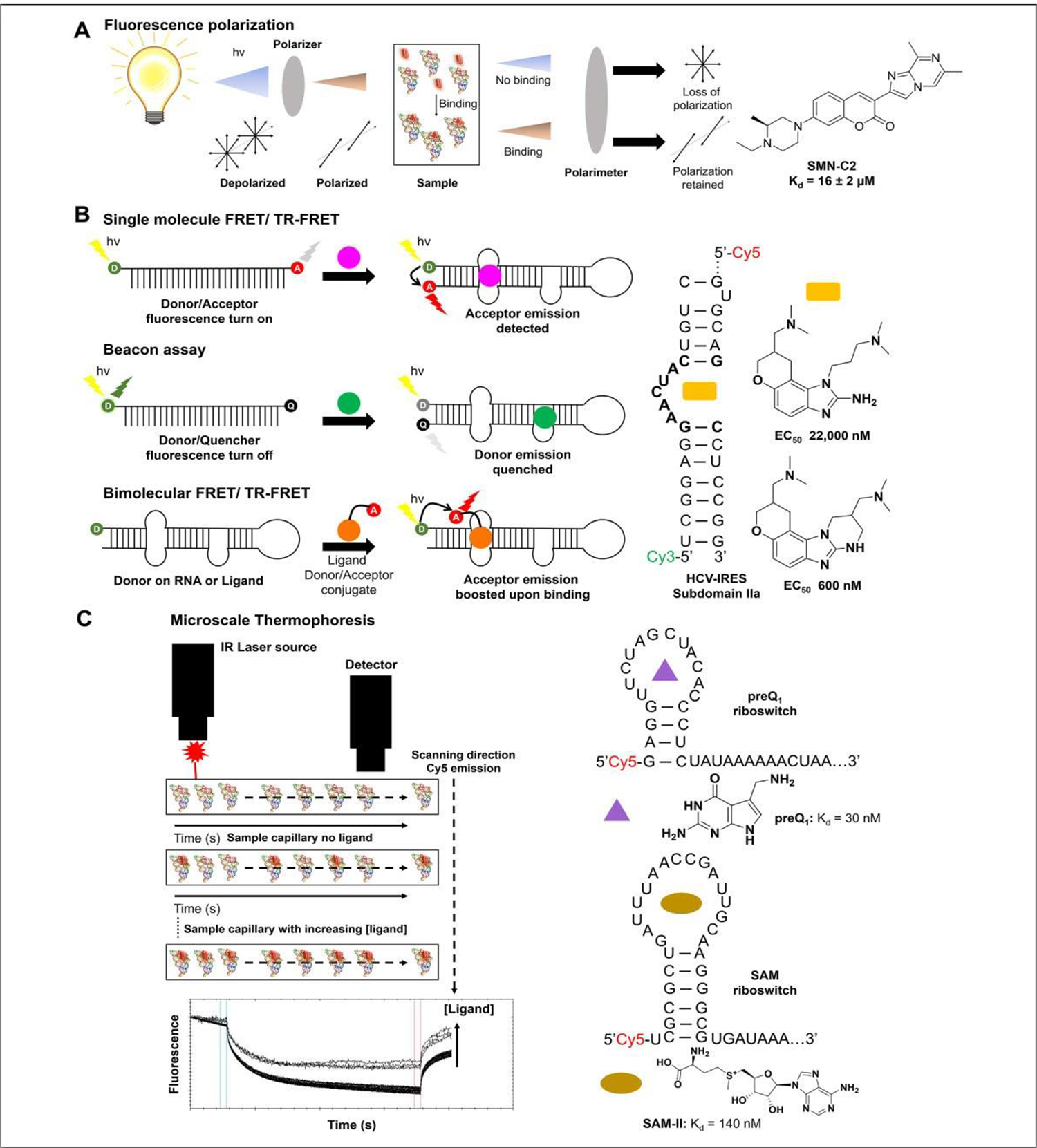
Schematics of label-based assays with examples. (A) FP assays use plane polarized light to measure the molecular rotation of a fluorescently labeled sample. If the fluorescent binder is not bound to the dye, then its rapid molecular rotation results in depolarization. If the molecule does bind, then polarization is retained, and that change is measured using a polarimeter. Compound SMN-C2 was identified to bind exon 7 of the SMN2 mRNA via FP with a Kd of 16 + 2 mM. (B) FRET can be used in multiple modes to measure the binding of small molecules to RNA. In smFRET, the donor–acceptor pair is attached to the same molecule, allowing one to measure ligand stabilization of an RNA upon binding. If the ligand and RNA are labeled separately, then disruption of the complex can be measured to identify an inhibitor. Two compounds were identified to bind the HCV-IRES with EC50 values of 22,000 and 600 nM, respectively, which are structurally similar. (C) MST measures the change in thermophoretic movement of a dye-labeled RNA in a temperature gradient before and after ligand binding. The preQ1 and SAM-II ligands’ affinity was measured to their respective riboswitches by MST, affording Kd values of 30 and 140 nM, respectively, which are comparable to those previously reported.
The FP assay has seen widespread use in the study of protein–nucleic acid and nucleic acid–small molecule binding analyses. Here, we focus on the latter application. In a pioneering study, PTC Therapeutics and F. Hoffmann-La Roche identified three novel splicing modifiers, SMN-C1, SMN-C2, and SMN-C3, that correct improper splicing of survival of motor neuron 2 (SMN2) in spinal muscular atrophy (SMA) patients.49 This prompted a study of the molecules’ mode of action, initiated by J. Wang and colleagues, who demonstrated by the synthesis of an SMN- C2-Phe-coumaine (SMN-C2-Phe-Cu) analog that SMN-C2 binds an AGGAAG stretch in exon 7 of SMN2 with a Kd of 16 + 2 mM, which was confirmed by cleavage analysis in vitro (Fig. 3A).50 Aside from splicing sites, FP assays have been extensively applied in the study of RNA and DNA aptamer binding to their cognate ligands. Y. Wang and colleagues used FP assays to study the binding of tobramycin to a J6RNA aptamer known to bind aminoglycosides, revealing a Kd of 0.77 nM.51 These studies resulted in clinical trials of risdiplam, which is awaiting a decision by the FDA.52 In parallel, studies by Novartis identified the com- pound LMI070 (branaplam) as a splicing modulator of SMN2.53,54 It is important to note that the mode of action for these ligands is their ability to stabilize the interaction between the SMN2 transcript and the subsequent splicing factors that enable proper processing and thus functioning as a molecular glue.
FRET-Based Approaches for RNA Binding
FRET and beacon assays are widely used methods for studying small-molecule binding to RNA in vitro and in vivo. This method uses a donor fluorochrome and an acceptor fluorochrome to form a donor–acceptor pair that is then conjugated to the biomolecule(s) of interest. The donor’s emission spectrum must sufficiently overlap with the excitation of the acceptor and be in sufficient proximity (1–10 nm) to allow FRET.55 This process is the nonradiative transfer of energy between two fluorochromes in close proximity due to the coupling of their dipole moments, the efficiency of which is affected by many factors, the most important being the selection of the FRET pair. The following factors should also be considered for optimal assay performance: (1) the fluorescence lifetimes of the donor and the acceptor, (2) photobleaching of the fluorochromes, (3) the quantum yields of the donor and acceptor, and (4) bleed-through of donor emission into the acceptor emission channel. Emphasis is placed on points 1, 3, and 4 as these predominantly affect the assay’s sensitivity. FRET assays can be used to study both intra- and intermolecular binding events, where the donor–acceptor pair is conjugated to one species in the former case or two different species in the latter case (Fig. 3B). This gives rise to different types of FRET-based assays to assess ligand binding to nucleic acids, including single-molecule FRET (smFRET), FRET melting (FRET-melt), time-resolved FRET (TR-FRET), and beacon assays.
Single-Molecule FRET
smFRET studies the changes of a biomolecule that is simultaneously labeled with both the donor and acceptor fluorochromes and has seen extensive use in identifying RNA-binding small molecules. Hermann and colleagues used a Cy3/Cy5 FRET pair conjugated to a mimic of the hepatitis C virus internal ribosome entry site (HCV-IRES) subdomain IIa riboswitch, aiming to identify novel benzimidazole modulators of the riboswitch’s conformation to inhibit viral translation. In doing so, two structurally similar derivatives were identified, exhibiting EC50 values of 22,00035,56 and 600 nM,57 respectively, in FRET binding assays (Fig. 3B).35,57,58 Further, Benz et al. utilized a 5´- FAM/3´-TAMRA smFRET pair on telomeric DNA sequences, which revealed stabilizers of G-quadruplexes (G-quad), while thermal melting of the hits ranked them by their relative degree of stabilization via a change in melting temperature (ΔTm). This approach yielded four DNA-selective G-quad stabilizers from a library of 97 compounds, with IC50 values ranging from 13.4 to 3.0 mM and ΔTm values ranging from 28 to 14 °C.28 Using the same technique, Rahman et al. screened >2300 compounds and identified 13 novel G-quad stabilizers exhibiting similar changes in ΔTm.59 However, in the aforementioned studies, bulk FRET was measured via emission of the acceptor fluorochrome, with no delay after donor excitation. This method has the potential to reduce assay sensitivity due to donor emission bleed-through into the acceptor emission channel, potentially leading to false negatives.
Time-Resolved FRET
To overcome the shortcomings of bulk FRET measurements, TR-FRET can be employed. TR-FRET combines time-resolved fluorescence with FRET and hinges on the use of a short-lifetime donor with a long-lifetime acceptor, coupled with a delay in measurement of acceptor emission. This delay ensures sufficient decay of donor emission and background fluorescence, maximizing signal-to-noise ratios and assay sensitivity. Utilizing this approach, the intermolecular interaction of r(CUG) repeats that bind to muscleblind-like protein 1 (MBNL1) was studied using a biotinylated RNA with a streptavidin-XL665 tag and an MBNL1-His6/Anti-His-Tb as the donor–acceptor pair.60 This RNA protein complex is responsible for DM1- associated splicing defects, and it is believed that disruption via small-molecule binding to the r(CUG)exp can provide lead therapeutics. Parkesh et al. identified 17 compounds from a computationally optimized library of 40 small molecules to inhibit MBNL1 binding by >85%, with IC50 values ranging from 50 to 1000 mM.60,61 Rzuczek et al. further examined this model, identifying 28 r(CUG)exp RNA binders from a library of 320 small molecules (IC50 30–130 mM), with 5 of 28 being bioactive in cellulis.62 Using multivalent approaches to rationally design RNA-binding ligands, bioactivity and potency were improved 1000-fold to low nanomolar levels.21
Beacon Assays
Beacon assays also use a FRET-based sensor to study the presence of a specific nucleic acid in a PCR, or the binding of ligands to a specific sequence. Initially discovered by Tyagi and Kramer in their effort to study the origin and movement of mRNAs63 in cells, the beacon probe is usually 25–35 nucleotides long and conjugated with a donor fluorophore on the 5´ end and a quencher fluorophore on the 3´ end, forming a donor–quencher (D/Q) pair. The first and last 5–7 nucleotides are complementary, forming a hairpin that brings the D/Q pair into close proximity and quenches the donor’s fluorescence. The single-stranded apical loop then targets the DNA or RNA of interest via Watson–Crick base pairs. Upon hybridization with the target DNA/RNA, the D/Q pair is separated, restoring the donor’s fluorescence. Factors to consider when designing a beacon probe include the size and sequence of the targeting loop for efficient hybridization to the target, the size of the stem for hairpin formation, and the selection of an adequate D/Q pair to maximize the signal-to-noise ratio. The interplay of these properties on assay design is summarized in greater detail by Zheng et al.64
Beacon assays have been applied successfully to identify RNA-binding small molecules to a variety of targets like miRs and viruses. miRs are 22- to 23-nucleotide short non-coding RNAs that bind to the 3´ UTR of a target mRNA via Watson–Crick base pairing, downregulating its translation.65 Bose et al. employed a DNA beacon (5´-FAM/3´-BHQ1) targeting the guide strand of miR-27a. The group identified six aminoglycosides that inhibited pre-miR-27a processing in vitro and further demonstrated that these aminoglycosides were bioactive in MCF-7 cells, displaying an IC50 of 20 mM.66 Unlike the two-component beacon used by Bose et al., Bell et al. used a single-molecule beacon to study compounds capable of inhibiting binding of the Gag protein to stem loop 3 (SL3) in the human immunodeficiency virus 1 (HIV-1) Ψ packaging domain. This afforded NSC260594 from the Library of Pharmacologically Active Compounds (LOPAC) collection of 1280 compounds, establishing it as the best hit out of 78 primary hits with an IC50 of 4.5 mM.67 Interestingly, further study of this compound by selective 2´-hydroxyl acylation analyzed by primer extension (SHAPE) demonstrated that NSC260594 induced global changes in the packaging domain that limit incorporation of the viral guide RNA into virions.68
The Arenz group also employed a single-molecule beacon to study the inhibition of Dicer processing of pre-let-7 by dodecapeptides. As a proof of concept, the aminoglycoside kanamycin was shown to bind to and inhibit processing by Dicer, as indicated by a 40% reduction in emission from the D/Q probe at 100 μM. Analysis of the three dodecapep- tides S117 (Ac-NH-SSIYALEPDQKG-CONH2), S417 (Ac-NH-RYNIKKEFNEFG-CONH2), and S186 (Ac-NH-AKPYSQRRKTSG-CONH2) demonstrated 10%–20% inhibition for S117 and S417 and 85% for S186, with dose- dependent reductions in fluorescence observed for all RNA constructs tested.69 The Duca group used a similar beacon approach to identify inhibitors of miR-372. Using neomycin–nucleobase and unnatural nucleobase conjugates, they showed that the conjugates designated as 3e and 3f were able to achieve ~100% inhibition of in vitro Dicer processing and bound to the pre-miR with Kd values of 16 ± 8.8 and 14 ± 7.6 μM.70 Staedel and colleagues used this approach to screen a library of 640 polyamines identifying a spermine–amidine conjugate that bound with a Kd of 150 nM and inhibited Dicer processing with an IC50 of 1.1 μM. The bioactivity of this ligand, measured by cell growth inhibition, was further confirmed in gastric cancer cells.71
Microscale Thermophoresis
MST is the measure of molecular movement in a microscopic temperature gradient created by heating the sample via infrared radiation (Fig. 3C). The rate and extent of thermophoretic movement is sensitive to a variety of factors, including size, charge, molecular rotation, structural conformation, and hydration shell, all of which are perturbed upon ligand binding. First described by Ludwig and colleagues in 1856, thermophoresis has since been extensively applied in the study of protein–ligand binding interactions.72 MST provides a robust means of assessing molecular binding events and has multiple advantages, such as requiring small sample volumes (as little as 10 μL) and using only picomoles of biomolecule per sample, in comparison with isothermal titration calorimetry (ITC) and SPR, which require an order of magnitude or more material. MST can also be used with virtually any biomolecule that can be conjugated with an appropriate dye, such as Cy5. Furthermore, due to its method of measuring binding, it is highly sensitive to changes in charge, solvation, conformation, and size upon binding to obtain a Kd. The user is nevertheless advised to consider aggregation of planar aromatic ligands and direct interactions with the cyanine label, as these could provide misleading results in the fluorescence and thermophoresis measurements. The use of crowding agents like bovine serum albumin (BSA) or mild detergents like 0.01% (v/v) Tween-20 can help to mitigate these effects. A comprehensive review of the method’s capabilities is discussed by Jerabek-Willemsen et al.73,74
Recent studies have applied MST to study the binding of small molecules to RNA. One of the first to apply this method to RNA was Gaffarogullari et al. in 2013, studying the catalytic activity of the Diels–Alderase ribozyme that catalyzes Diels–Alder cycloaddition reactions. Using this method, the group not only obtained the Kd of each substrate to the ribozyme but also determined a cooperativity effect of 2.6-fold between the diene and dienophile for binding to the active site. They also found that entropy drove the energy penalty observed for dissociation of the diene.75 In addition, Moon et al. showed that MST could be applied to the binding of Rev to Rev response element (RRE) RNA (Kd = 4.8 nM), as well as to the inhibition of that interaction by competing off REV with neomycin (Ki = 2300 nM). The group also determined the binding constants of the prequeuosine1 riboswitch from two different organisms to cognate ligands PreQ1 (Kd = 26 nM) and SAMII (Kd = 140 nM), on par with affinities previously reported in the literature (Fig. 3C).29 Disney et al. has also employed this method extensively, assessing the binding of ligands to noncoding RNAs such as miR-96 (Kd = 40 nM),76,77 miR-21 (Kd = 24 nM),76 miR-210 (Kd = 200 nM),78 and r(GC)4 base pairs (Kd = 12.5 nM).79 MST provides a robust binding method capable of studying any RNA of interest and, compared with other fluorescence methods, it is highly sensitive. However, it has limited throughput due to instrument design, making it ideal as a secondary validation system for leads obtained initially by HTS.
Differential Scanning Fluorimetry
DSF has seen widespread use in the identification of ligands binding to proteins.80,81 Recently, this method has been adapted to include RNA–ligand binding interactions, initially utilized by Silvers et al. studying RNA stability in the presence of Mg+ and F− for the fluoride riboswitch.30 The method uses an RNA-binding dye, like RiboGreen or SYBR Green, that discriminates between structured and unstructured RNA (single- vs double-stranded) by binding and emitting fluorescence. As the temperature of the sample increases, the RNA begins to unfold, causing a decrease in fluorescence along with a signal reduction due to thermally induced dye diffusion. This experiment affords a Tm, the temperature at which 50% of the RNA is unfolded and is a direct measure of the RNA’s thermal stability under these conditions. The ligand can then be added, after which the Tm is again measured to determine the gain or loss in RNA stability as a result of binding. This method has many advantages over normal fluorescence and calorimetric approaches, such as its insensitivity to pipetting errors, its small sample volumes, and its amenability to HTS campaigns as a result of the previous two points.
One example of this approach is the study of HIV transactivating response element (TAR)-binding ligands like acepromazine and its analogs by Sztuba-Solinska et al.82 Initially identified from SMMs, DSF studies identified two compounds that exhibited 4–8 °C increases in Tm, with a thienopyridine being most active. Direct binding by 2AP assays (see McGovern-Gooch and Baird for more detail40) afforded a Kd of 2.4 ± 1.1 μM. More recently, Baird and colleagues utilized the differential scanning FRET method to study the effect of Mg2+ ions on the conformation landscape of the c-di-GMP aptamer, identifying that maximal binding-induced conformation changes occur at near-physiological Mg2+ concentrations.83 Demonstrating the high-throughput nature of this assay, Matarlo et al. identified natural product inhibitors of pre-miR-21 biogenesis. By screening a library of >3800 pure natural products, 10 compounds were identified as hits to bind to pre-miR-21 and stabilize its structure, with minimal cytotoxicity in HCT-116 cells. The compound bPGN was identified as having a Kd of 400 nM and a demonstrated knockdown of miR-21, as well as an upregulation of programmed cell death protein 4 (PDCD4) and phosphatase and tensin homolog (PTEN), both downstream targets of miR-21.84 This method is relatively nascent in its application to the study of RNA-binding small molecules but has significant potential to provide high-throughput analysis of ligand effects on RNA stability, otherwise unattainable by conventional UV melting.
Fluorescent Intercalator Displacement
FID assays are a commonly used method for identifying novel RNA-binding matter. Initially used to identify ligands that bound DNA using intercalators such as ethidium bromide (EtBr)85,86 and thiazole orange (TO),87,88 the method has since been applied to the study of RNA–ligand interactions, employing a wider array of indicators. The assay functions by using a fluorescent indicator of known affinity to the RNA of interest and then precomplexing them together, enhancing or quenching the indicator’s emission. A library of small molecules can be subsequently screened against the complex to identify potential RNA-binding ligands, because as unknown ligands bind to the RNA, the indicator’s emission is altered (quenched/enhanced). This change can assess the binding capacity of each ligand to the RNA and then rank them accordingly. Common indicators include EtBr, TO89 and its derivatives TO-Pro-190,91 and TO-Pro-3, X2S,92 2AP,93,94 and SYBR dyes.95
FID assays using small molecules have been employed with great success in the identification of novel RNA binders. Zhang et al. identified mitoxantrone and sanguinarine as binders to the RRE of HIV-1 RNA, using X2S as their indicator with a hit rate of 0.2%.92 Similarly, Tran and Disney identified eight RNA binders (hit rate, 19%) from a library of small molecules focused on RNA-binding chemotypes such as benzimidazoles and pentamidines, with Kd values ranging from 4 to 160 μM.91 Incorporating a more high-throughput orientation, Haniff et al. used TO-Pro-1 bound to r(AAUU), r(AUAU), r(GGCC), and r(GCGC) base pairs to study whether small molecules can selectively bind to these base pairs, as well as their ability to discriminate between the orientation of the pairs in tandem (i.e., AU/AU or alternating AU/UA). This work identified 28 novel RNA-binding ligands from a library of 3200 compounds (hit rate, 0.8%). This library yielded AU and GC pair-selective base pair binders with Kd values ranging from 0.04 to 3 μM.79
An alternative to RNA-binding small-molecule indicators is the use of RNA-binding protein indicators. RNA-binding proteins such as the trans-activator of transcription (Tat) and Rev protein of HIV-1 have been extensively studied for their capacity to bind TAR and RRE RNA, respectively.96 To increase the generalizability of this approach, Gobel, Crothers, and colleagues identified that the highly basic and positively charged regions of the Tat protein, specifically residues 49–57 (RKKRRQRRR),97–99 contributed to its RNA-binding properties. This was successfully applied as a minimized fragment to identify RNA–small molecule interactions by Matsumoto et al.,100 Patwardhan et al.,101,102 and Weeks and Crothers.98 Similarly, Luedtke and Tor85,103 found that residues 34–50 (TRQARRNRRRRQRERQR), which contribute to Rev’s RNA-binding capacity, could be used in HTS to identify selective disrupters of the RRE–Rev interaction.85,103 In these and other studies, peptide fragments were labeled with 5-FAM and tetramethylrhodamine (TAMRA) on the N- and C- terminus, respectively, for Tat100 to create a FRET pair as the readout. In the case of Rev, the peptide was labeled with 5-FAM only, with subsequent binding being measured by FP. Such a strategy was also applied by Wang et al. in their study of the binding of aminoglycosides to 16S ribosomal RNA.104 These two methods provide unique advantages and disadvantages to the study of RNA–small molecule interactions and are discussed in depth by Wicks et al.105
Surface Plasmon Resonance
SPR, like many of the methods described herein, has seen widespread application in the study of protein binding. SPR occurs when polarized light hits a metal surface at the interface of materials with different refractive indices. At the appropriate angle of incidence, surface electrons of the metal become excited and oscillate, absorbing some light while reflecting the rest. If one of the surface compositions is changed, its refractive index also changes, thereby shifting the resonance angle required to absorb light.106
Win and colleagues identified codeine binding aptamers by affinity selection using Sepharose beads conjugated with codeine. Lead aptamers were then assessed for their binding affinity to codeine using SPR. This was completed by chemical modification of a CM5 sensor chip to display codeine, affording two aptamers with Kd values of 2 and 4 μM, respectively.107 Conversely, Hendrix et al. used biotinylated RNAs to bind to the surface of a CM5 sensor chip, revealing that neomycin B bound to three sites in domain II of RRE with submicromolar affinities.108 The advantage of using the latter biotinylated method over the former chemical modification is found in the simplicity and ease by which the surface can be conjugated.
One alternative to using chip-based SPR methods is bio-layer interferometry (BLI), functioning on the same principles as SPR, but instead of measuring the shift in resonance angle of resonating plasmons, it measures the interference patterns of reflected light. Generally, a BLI sensor consists of two reflective surfaces, one inside the sensor that does not interact with the analyte, and one outside the sensor that does. The internal surface serves as a reference, while the external surface measures the change in refractive index as a result of analyte binding. This method has seen many applications in the study of nucleic acid binding, including recent work by Disney et al. that has applied this method extensively to the study of r(CGG)exp, r(CUG)exp, and r(G4C2)exp expansions. The group identified high-affinity binders such as 2HE-5NME (Kd = 50 nM),109 3K-4 (Kd = 5 nM), and a hydroxy ellipticine derivative dubbed 4 (Kd = 260 nM)110 to r(CGG)exp, r(CUG)exp, and r(G4C2)exp, respectively, producing bioactive ligands. The advantage of this method over fluorescence-based methods such as FRET and FP is that it does not require the species of interest to be labeled and utilizes relatively small amounts of material, making it amenable for HTS applications. Since the experiment is conducted over time, assessment of both kinetic and steady-state binding is permitted, providing a selection of leads not simply based on Kd but also on kon and koff. In addition to generating a wealth of information to guide drug-based lead identification, the increasing capability of instrumentation enabling 384-well formats is enhancing HTS capabilities.
Label-Free Methods for Identifying RNA Binders
Small-Molecule Microarrays
SMMs offer unique advantages over plate-based assays for the identification of RNA-binding ligands. SMMs are highly miniaturized, therefore using less material; they have an excellent signal-to-noise ratio due to their small sample volumes; and they can exponentially increase the throughput with which RNA-binding ligands can be identified. This method involves the printing of small molecules onto a glass surface, either covalently111 or noncovalently,112 using a robotic printer. Once adhered, a labeled RNA is hybridized to the glass surface and then washed to remove unbound species. Hits are identified by accumulation of the label onto the points at which compounds were printed (Fig. 4). These hits are then taken for secondary validation by alternative binding assays such as those mentioned above. Disney et al. and Schneekloth et al. have both done extensive work in the use of SMMs to identify small molecules binding to RNA. Two-dimensional combinatorial screening (2DCS), a method created by the Disney lab, utilizes glass slides coated with an agarose matrix used to bind small molecules to the surface. A γ-32 P-labeled RNA library is then hybridized with the array in the presence of excess competitor oligonucleotides and washed, followed by excision of bound RNA and subsequent sequencing (Fig. 4A). This library versus library screening enables the assessment of millions of binding interactions simultaneously. Furthermore, this approach not only affords the identification of novel RNA-binding compounds, but also deconvolutes the sequence specificity of its binding to RNA in a target-agnostic fashion, culminating in the Disney group’s Inforna database of small molecule–RNA interactions (Fig. 4A).19,113 Many leads have arisen in response to a variety of RNA targets since the establishment of this approach, yielding numerous bioactive small-molecule interactions with RNA (SMIRNAs). In seminal works by Velagapudi et al.,113,114 inhibitors of miR-96 and miR-10b were identified, both with micromolar in cellulis activity. Inhibitors of repeat-associated defects in DM1 have been identified with nanomolar affinity and bioactivity21 in preclinical models. Multiple inhibitors to miRs have also been developed in response to miR-96,115 miR-515,116 miR-21,76 miR-210,117 and the miR-17–92112 cluster with low micromolar to nanomolar Kd values (Fig. 4B).
Figure 4.
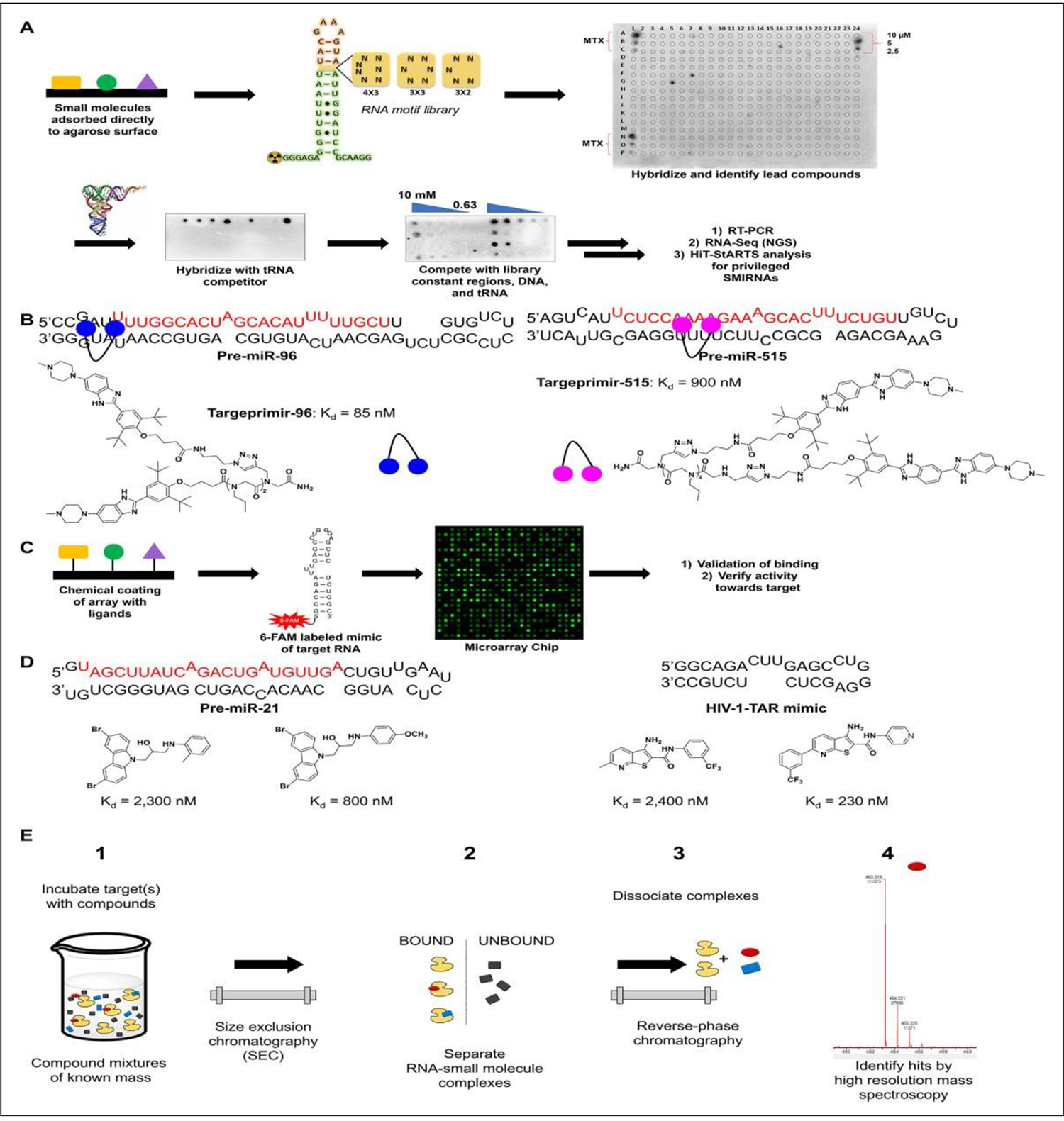
Schematic of SMM methodologies to identify RNA binders. (A) Schematic of 2DCS. Small molecules can be directly absorbed onto or site-specifically conjugated to the surface of agarose-coated microarrays, which are then hybridized with a radiolabeled RNA library. Hits are subjected to competitive screening with tRNA, and then tRNA, DNA, and oligonucleotides mimicking the constant regions to all library members (green and red sequences). Hits are then excised and sequenced by next-generation sequencing and deconvoluted by HiT-StARTS analysis in a target agnostic manner, identifying privileged RNA–small molecule interactions that inform drug design. (B) Inforna identified small molecules that bind the Drosha sites of pre-miR-96 and pre-miR-515. These small molecules were then dimerized to yield Targaprimir-96 and Targaprimir-515 with Kd values of 85 and 900 nM, respectively.112,116 (C) Schematic of SMM methodology developed by Schneekloth et al. This method uses a dye-labeled mimic of the RNA of interest and then subjects it to binding on an array of covalently linked molecules. Hits are identified by fluorescence. (D) Dye-labeled SMMs identified hits to pre-miR-21’s Dicer site with Kd values of 2300 and 800 nM. Hits were also identified to HIV TAR RNA with Kd values of 2400 and 230 nM. (E) ALIS (Automated Ligand Identification System) process developed by Merck. This method involves equilibration of the RNA target with a mixture of compounds (1), SEC purifying the RNA–small molecule complexes (2), reverse-phase HPLC dissociating the RNA–small molecule complex (3), and MS that identifies small-molecule hits (4). Image modified from Rizvi and Nickbarg.133
In contrast to the Disney group’s 2DCS library versus library screening, other increasingly targeted approaches using SMMs have also been employed. Such methods as those developed by Schneekloth et al.118 use dye-labeled mimics of the target RNA to scan their arrays for novel binders. These arrays are constructed by covalently displaying the ligands on the surface of the array, which differs from the current 2DCS methods, which require no such linkage (Fig. 4C). Using this method, Sztuba-Solinska et al. identified binders to the TAR hairpin with a Kd of 2.4 μM,82 while Connelly et al. identified nanomolar inhibitors of miR-21 (Fig. 4D).119 In both of these studies, diversity was achieved via the chemical space, identifying numerous hits to a single target. Indeed, this approach can rapidly identify new ligands, but prior knowledge of the RNA structure is required to assess potential binding sites, and rigorous validation by functional assays is also necessary. Both methodologies mentioned above provide robust methods to identify new RNA-binding small molecules, the former in a broad context and the latter in a more targeted fashion. However, these assays do not provide any information on functional activity or mechanism, and thus require rigorous secondary confirmation to establish binding affinity, activity, and mechanism.
MS-Based Approaches
Historic MS-based approaches for identifying small molecules that bind to nucleic acid focused on direct MS detection of nucleic acid–small molecule complexes.120–126 These include approaches such as MASS (multitarget affinity/specificity screening)127 and DOLCE-MS (detection of oligonucleotide ligand complexes by ESI-MS).128 Limitations due to buffer restraints (MS interferers, i.e., K+ and Mg+), RNA size, and the need for high mass resolution and accurate mass detection hinder the use of these approaches for general large-scale screening for small molecules targeting RNA. Indirect affinity selection–mass spectrometry (AS-MS) methods have been developed for screening large multiplexed compound sets under a variety of conditions.126,129–131 Recently, Merck (Kenilworth, NJ) applied their AS-MS method, referred to as the Automated Ligand Detection System (ALIS), to screen compounds against numerous RNA targets, an approach validated with ncRNA riboswitches, their ligands, and a large unbiased small-molecule library.132–134 Here, RNA is incubated with mass-encoded small-molecule mixtures, small molecule–RNA complexes are purified by size exclusion chromatography (SEC), and the complexes dissociate through reverse-phase chromatography. Bound ligands are detected by high-resolution MS (Fig. 4E). This approach has been recently used to screen ~50,000 compounds against 42 distinct RNAs, which include untranslated regions (UTRs) long noncoding (lnc) RNAs, repeat elements, and small nucleolar (sno) RNAs.133 Selectivity across RNA and protein targets was assessed and numerous (n =545) selective RNA binders (bind only the single indicated RNA target) were identified. Furthermore, cheminformatics hit evaluation provided insight into the physicochemical properties and chemical substructures that direct general and specific RNA binding. Remarkably, many of the RNA targeting hits can be classified as classic drug-like molecules.133 Beyond hit identification, ALIS has applications in affinity ranking, Kd determination, and competition investigations.133 The use of denaturants such as urea can also confirm structure requirements of ligand binding.133 Advantages of the AS-MS approach include its applicability to general screening, minimal assay development, no target tagging, and limited susceptibility to artifacts.133 Sample purification via chromatography prior to MS means that a variety of native assay buffers can be used as the small molecule will be purified prior to detection, and therefore common MS interferers are not a problem. In addition, the ALIS approach by Merck has additional advantages as it is easy to automate, requires lower sample amounts, has enhanced sensitivity and resolution, emits compound precharacterization requirements and is less prone to false positives/irreproducible results due to column degradation.133 Although no RNA length limitations have been reported, questions still exist around RNA folding and whether longer RNAs may unfold or aggregate under such conditions. An additional challenge with the AS-MS method is the requirement to emit compounds classified as “nonflyers,” which can represent a significant proportion of a standard drug discovery screening collection and the poor recovery of compounds with weak binding affinities.
Dynamic Combinatorial Chemistry
An inherent limitation of the methods described above is the use of “fixed” ligand libraries generated by traditional combinatorial chemistry approaches. This culminates into a screen where the target is unable to influence the ligand pool and select the most optimal binder. One solution to this problem is the use of dynamic combinatorial libraries (DCLs) generated through dynamic combinatorial chemistry (DCC). This concept hinges on the use of “soluble” ligand binding fragments that can undergo reversible chemistry to form more complex binders and whose reaction equilibrium shifts in response to the target’s interaction with those complex species. In other words, upon binding, the ligand drives the reaction equilibrium to make more of the high-affinity species and depletes low-affinity species from the pool.135,136
Since its first mention in the late 1990s, this methodology has been successfully applied to identify RNA-binding ligands by the Marchán and Miller groups. For example, the Marchán group, using the stem loop at the tau exon-10-intron 10 junction, used a DCL consisting of acridine, neamine, and azaquinolone derivatives with varying spacer lengths for the sulfide reactive group and two aromatic tripeptides (H-Cys-Tyr-Arg-NH2 = TyrP and [H-Cys-Trp-Arg-NH2][S-S] = TrpP). Through use of biotinylated RNA, the selection was carried out, isolating bound ligands by streptavidin pulldown. Analysis of the ligand pools found amplification of the Acr1-Nea and Acr2-Nea2 ligands, which displayed EC50 values of 5.9 and 2.1 mM, respectively, and was confirmed by optical melting analysis to stabilize the tau hairpin.137
The Miller group has conducted extensive analyses of RNA binders deploying DCC to generate RNA-directed DCLs. For example, due to the biases of DCLs to produce dimers as a result of the entropic penalty for forming trimeric or higher-order species, the Miller group developed resin-bound dynamic combinatorial chemistry (RBDCC), whereby phase separation of the individual units enables display of the full library complexity, or the use of bifunctional resin partners that can form ternary resin-bound dynamic combinatorial libraries (RBDCLs).138,139 Gareiss and colleagues applied these methodologies to identify lead molecules capable of displacing MBNL1 from r(CUG) repeats. To do this, an RBDCL with 11,325 members was constructed from 150 unique resin-bound cysteine-containing building blocks. This library was incubated with Cy3-labeled r(CUG)10 and bound beads were picked by fluorescence and deconvoluted to identify the ligand, which resulted in four unique ligands that bound with Kd values ranging from 41 to 1.9 μM.140 Ofori and colleagues then used ligand 4 from Gareiss et al. and derivatized it with benzo[g]quinoline to produce an eight-compound library of ligands that were screened for binding to r(CUG) repeats by SPR and fluorescence titration. Two stereoisomeric derivatives exhibited Kd values of 40 and 70 nM, respectively, and were further tested in cellulo and in vivo, resulting in modest correction of MBNL1-mediated splicing defects in Clcn1 and Atp2a1 in HASLR mice.141
Rolling Circle Amplification
Rolling circle amplification (RCA) has seen extensive use in biosensing applications of DNA and RNA. The method uses a circular DNA template that has a hybridization site for the analyte of interest. Upon hybridization, isothermal primer extension from the analyte is carried out, creating a concatemer, or repeated DNA sequences in series, that amplifies the detectable signal of the analyte for readout. While this method is highly sensitive, it has had few uses to study the ability of small-molecule ligands to inhibit RNA biology. The Arenz group utilized this methodology to study the inhibition of the RNA-induced silencing complex cleavage of miR-122 in vitro by aminoglycosides such as kanamycin A/B and neomycin. Here, using Sybr Gold, they measured the amount of concatemer produced upon cleavage by recombinant “minimal RISC,” kanamycin A/B, and neomycin B and found that concatemer production was reduced by ~20%, ~40%, and ~70%, respectively, indicating an inhibition of pre-miR-122 cleavage by the minimal RISC.142
Fragment-Based Approaches by NMR Spectroscopy and X-Ray Crystallography
The development of HTS technologies has led to many drug leads.143 However, screening of large compound libraries has yielded either few hits or false positives against challenging targets.143 An alternative strategy, fragment-based drug discovery (FBDD), consists of screening smaller sets of fragments for chemical starting points.143 Fragments, defined as having less than 20 nonhydrogen atoms, allow for more efficient sampling of the chemical space than drug-like molecules.143 Small fragments may bind to a greater number of sites than larger, more complicated molecules, allowing them to bind to targets such as RNAs that lack well-defined binding pockets.143–145 While fragments are typically weak binders, lead compounds may be developed by growing, merging, or linking fragment hits together.144,146
NMR is a sensitive technique suitable for finding weak binders such as those with single-digit millimolar Kd values.146,147 NMR methods for identifying fragments consist of both target- and ligand-based screens. Target-based screens monitor changes in the spectra of an unlabeled or isotopically labeled target upon addition of a ligand.144,148,149 These methods may identify binding sites and determine binding affinities.144,150 However, assignment of the target resonances may be complicated by spectral overlap.144,149 Titration measurements are also slower than other biophysical methods or biochemical assays, and target-based methods require greater amounts of biomolecule target than ligand-based methods.149
Ligand-based methods monitor changes in the spectra of a ligand upon addition of a target.144,148,151 Screens can be carried out with cocktails of fragments and do not require assignment of the target resonances, nor large amounts of the target.152 Methods used for ligand-based screens include saturation transfer difference (STD), water–ligand observation with gradient spectroscopy (WaterLOGSY), and T2 relaxation-edited techniques.144,147,148,151
In an STD experiment, resonances of the target are selectively irradiated by a radiofrequency field.148 The saturation is transferred to bound ligands, and then to free ligands in fast exchange with bound ligands. This is where signals are detected.148,151 A separate spectrum is then taken with the irradiation frequency applied away from the target or ligand spectral range, and the spectra are subtracted.148,153 Only resonances of ligands that bind to the protein or other targets remain. Ligand sites that are not close to the target and give consequently weaker signals may be identified by STD and used as fragment growing or linking sites.144 Stronger STD signals are displayed for protons in close proximity to the target experiment.144,147,148,151 In a WaterLOGSY experiment, the water signal is irradiated, and magnetization is transferred from bound water to bound ligand.148,153 Bound ligands display signals of the same sign as target resonances.144,147,148,154
T2 relaxation-edited experiments rely on the difference in T2 relaxation rates between small-molecule ligands and macromolecule targets.148,150,155–157 Larger molecules have longer rotational correlation times (or slower tumbling) due to enhanced spin–spin interactions, leading to shorter T2 relaxation times and broader lines.150,155,157 In a T2-edited spectrum, binding of a ligand to a macromolecule leads to broadening of the ligand resonances because the ligand adopts the shorter relaxation time of the macromolecule.150,157 The T2 relaxation time may be measured using a Carr–Purcell–Meiboom–Gill (CPMG) pulse to follow the signal decay as a function of the relaxation delay.157 These methods are used to identify fragments that bind to a target, but with certain limitations.
Ligand-based methods may be used with mixtures of ligands and do not provide information about ligand binding sites.144,147 STD and WaterLOGSY methods require small amounts of unlabeled biomolecule target in the micromolar range,147,151,153 but they are often run with excess ligand over the target, a condition that may lead to a pH change in solution if the ligands contain acidic or basic moieties.149 Furthermore, the high concentrations of compounds necessary for these methods may make it difficult to distinguish site-specific binding from nonspecific binding or aggregation.144,149 Compound-induced partial unfolding or precipitation of the target may also give the appearance of binding or nonbinding, respectively.144,149 Finally, ligand-based methods become challenging with high-affinity binders (Kd < 0.1 μM), which give weaker signals.144,148
Structures of target–fragment complexes such as those mentioned previously may be further elucidated by NMR or x-ray crystallography.144 NMR has been traditionally used to determine structures and dynamics of biomolecules in solution.144 However, defining full NMR solution structures is a time-intensive process limited to small- to medium-sized biomolecules and requires a complete assignment of backbone resonances.144 X-ray crystallography is commonly used to rapidly generate high-resolution structures of ligand–target complexes, but it does not provide information about affinity, and crystal contacts may block ligand binding sites.144,149 Additionally, x-ray crystallography usually provides a single structure, without the dynamics observed for ligand–target complexes in solution. Further, overly flexible biomolecules may not be amendable to crystallization.144,149
Lee et al. employed a target-based NMR screen to identify compounds that bound to the influenza A virus (IAV) promoter (Fig. 5 left).158 The IAV promoter contains a conserved panhandle structure that is involved in transcription and replication.159 The group screened a library of 4279 fragments in groups of 20 for fragments inducing conformational changes in the IAV promoter RNA, resulting in line broadening or in chemical shift changes in imino proton resonances in 1D NMR spectra.158 This screen yielded seven compounds, among which DPQ was determined by a 1D NMR titration experiment to bind with the highest affinity (Kd 50.5 ± 9 μM).158 NMR revealed that a structure of DPQ in complex with the IAV panhandle bound to the promoter RNA’s internal loop, near the (A-A)-U motif, and induced conformational changes in base pairs close to the internal loop to create a binding pocket.158 Furthermore, in cell-based assays with Madin–Darby canine kidney (MDCK) cells infected with H1N1 and H3N2 strains of IAV and influenza B virus, DPQ inhibited viral replication of each with EC50 values of 71.6 ± 28.8 (H1N1), 275.5 ± 97.6 (H3N2), and 113.7 ± 8.9 mM (IBV), respectively.158 Cell viability assays also showed that DPQ was nontoxic at concentrations up to 500 μM.158 This work indicates that an NMR-based fragment screen can be applied to the identification of bioactive scaffolds against influenza, and potentially other disease-inducing RNAs.
Figure 5.
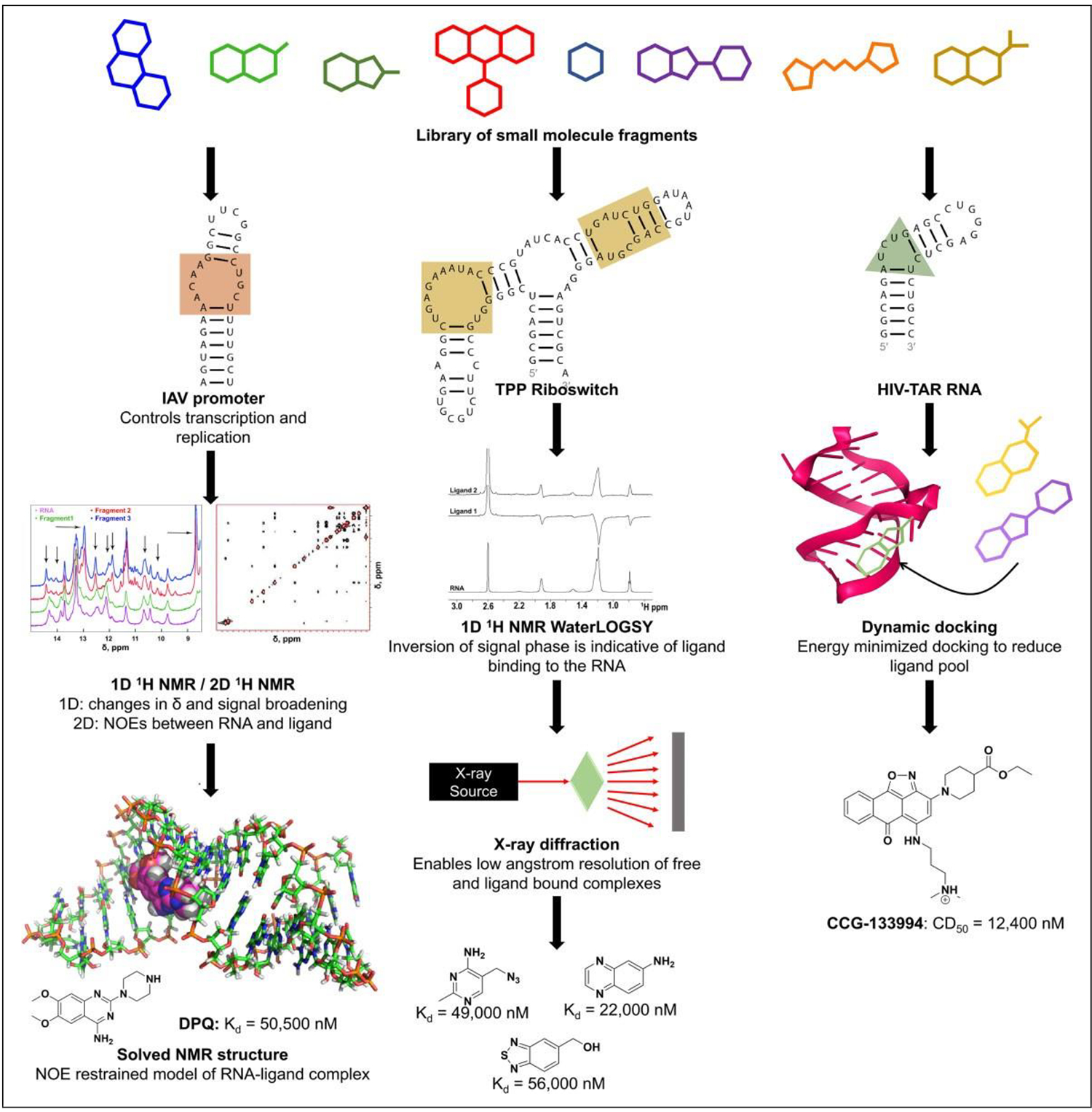
NMR, x-ray, and computational methods to screen for compounds that bind RNA. Left: Lee et al. used NMR spectroscopy to screen for ligands that bind to influenza A RNA promoter and determine the structure of one of the hits, DPQ, in complex with the RNA.158 Center: Cressina et al. screened a library of 1300 compounds for those that bind to the E. coli TPP riboswitch and used WaterLOGSY and T2 relaxation-edited NMR experiments to confirm binding of 20 hits.152 Warner et al. determined x-ray structures of four of the hits in complex with the riboswitch, the highest-affinity binder among which is shown.162 Right: Al-Hashimi’s group used virtual screening to identify ligands that bind to HIV-1 TAR RNA.167,178 The highest-affinity binder identified from EBVS is shown with its competitive dose to displace 50% of the Tat peptide value or CD50.178
Garavís et al. identified fragments from a fragment library of 355 fluorinated compounds that bound to telomeric repeat-containing RNA (TERRA) using 19F NMR.160 These lncRNAs, transcribed from telomeres, contain an average of 34 r(UUAGGG) repeats that fold in vivo into G-quadruplexes.160 TERRA promotes heterochromatin formation at telomeres, protecting telomere ends from the DNA damage response, and its expression is elevated in various human cancer cells.160,161 The group acquired 1D 19F NMR spectra with and without a CPMG T2 filter on cocktail samples containing eight fragments each. Upon addition of the target molecule to compounds, signals of binders broadened while signals of nonbinders did not change. The group ultimately identified 20 compounds from the NMR screen and validated the hits using 1H NMR STD experiments.160 All seven hits thermally stabilized TERRA2, as determined by circular dichroism (CD) melting experiments.160 Taken together, this showed that fragment-based screening can be applied to identify ligands targeting RNA G-quadruplexes.160
On certain occasions, a combination of fragment-based methods may elucidate different aspects of ligand–RNA binding. For example, one screen examined fragments binding to the thiamine pyrophosphate (TPP) riboswitch and interfering with the riboswitch mechanism, modulating downstream protein synthesis.152,162 The Escherichia coli TPP riboswitch contains a 78-nucleotide aptamer domain that binds to TPP and causes structural changes in a 90-nucleotide expression domain, sequestering the ribosome binding site and leading to translational attenuation.152 Cressina et al. screened a library of 1300 fragments by competition equilibrium dialysis and obtained 20 hits.152 The group confirmed binding of these 20 hits by Water-LOGSY and T2 relaxation-edited NMR experiments. Warner et al. solved crystal structures of four of the fragments in complex with the TPP riboswitch using small-angle x-ray scattering (SAXS) experiments (Fig. 5 center).162 These fragments bind to the aminopyrimidine binding site of the riboswitch in a mode similar to the aminopyrimidine moiety of TPP, and form stacking interactions with G42 and A43 as well as hydrogen bonds with nucleotides in J3/2 and P2. Further characterization of TPP that had bound to one of the fragments using SAXS and SHAPE revealed G72’s importance in the partial folding of the riboswitch, which achieves complete folding in the presence of TPP. Taken together, these methods identified and determined structures of TPP riboswitch in complex with fragment inhibitors.
Predictive Approaches by Virtual Screening
The high cost of HTS in time and resources has led to an increased use of virtual screening to identify new scaffolds against therapeutic targets.163,164 Virtual screening samples the chemical space and removes inactive compounds before experimental testing using in vitro assays.163 One strategy employed by virtual screening is the docking of large virtual libraries of compounds to the 3D structure of a target.163,164 Molecular docking estimates the binding mode of a ligand in a receptor as well as its binding affinity.164 Several algorithms and scoring functions for protein–ligand docking have been adapted for RNA docking. However, challenges with modeling the charged structure and conformational flexibility of RNA targets limit their success, especially considering that RNA can undergo large conformational changes upon ligand binding.165–167 In addition, relatively few experimental RNA–ligand structures are available to develop scoring functions calculating the free energy of ligand binding.165,166 Docking algorithms developed to address these issues are discussed in detail below.
Molecular Recognition with a Driven dynamics OptimizeR (MORDOR) is a program developed for induced-fit docking of ligands to RNA.165 In this methodology, the ligand and receptor undergo conformational changes to yield an optimal fit.165,168 MORDOR uses molecular simulations to model ligand and RNA flexibility during docking and calculates a score using the total energy of the complex, which includes a solvation term.165 This method was used to screen for compounds against the human telomerase RNA, and the binding of a subset was subsequently confirmed by NMR STD experiments.169
Daldrop et al. applied DOCK 3.5.54 to screen for compounds binding to a guanine riboswitch (GRA).170 The group adapted a scoring function to RNA–ligand binding by using RNA-specific parameters to calculate van der Waals and electrostatic energies.170 Crystal structures were determined for three of the ligands in complex with GRA that had been identified by docking.162 Lang et al. reported an updated version of DOCK, DOCK 6, that added AMBER generalized Born and Poisson–Boltzmann implicit solvent models to physics-based scoring functions.166
The docking of 70 RNA–ligand complexes using these solvent models, in combination with explicit water molecules and counter-ions, improved the success rate of reproducing experimentally determined structures.166 Furthermore, DrugScoreRNA and LigandRNA are two knowledge-based, stand-alone functions for scoring ligand–RNA complexes that derive potentials from experimentally determined 3D structures of ligand–RNA complexes.171,172 While DrugScoreRNA uses distance-dependent potentials to score RNA–ligand complexes, LigandRNA uses distance- and angle-dependent potentials.171,172 When tested on a set of 42 complexes, a combination of LigandRNA and DOCK 6 correctly identified ligand poses in 47.6% of cases compared with 35.7% for LigandRNA or DOCK 6 alone and 31.0% for DrugScoreRNA.172
In addition to the above, Morley and Afshar developed RiboDOCK for docking ligands to RNA.173 The scoring function of the program includes both attractive and repulsive potentials. The attractive potentials account for parallel p-p aromatic stacking (Sarom) and interactions between a positively charged ligand carbon and RNA acceptor (SposC-acc), such as a guanidinium carbon and RNA carbonyl oxygen. The repulsive interactions account for donor–donor (Sdon–don) and acceptor–acceptor (Sacc–acc) repulsion. This program was then used to identify compounds that target the bacterial ribosomal A site.173,174 An updated program based on RiboDOCK, rDock, was later released.175,176 Terms that were added to the rDock scoring function include a van der Waals potential and attractive and repulsive potentials, in addition to external restraint terms such as cavity and pharmacophore restraints.175,176 When tested with a set of 56 RNA–ligand complexes, rDock predicted at least one correct pose in 98% of cases.175
An alternative approach to virtual screening for ligands against RNAs combines NMR spectroscopy with computational methods to generate ensembles of conformations of an RNA. Molecular dynamics (MD) force fields used to model the conformational space of RNAs have had limited accuracy. The Al-Hashimi group carried out an 80 ns MD simulation on NMR structures of HIV-1 TAR RNA and then selected 20 conformers (dubbed TARNMR-MD) that agreed with experimentally determined NMR residual dipolar couplings (RDCs) for elongated TAR RNA.167,177
RDCs report on the angle between internuclear bond vectors in a biomolecule and an external magnetic field.178,179 The group applied virtual screening of 51,000 small molecules to each of the 20 conformers using the ICM docking program (Molsoft) and experimentally validated six of them for binding to TAR and inhibiting its interaction with Tat.167
Al-Hashimi’s group later generated an ensemble of 20 TAR structures using four sets of RDCs and 8.2 μs MD simulation, and then carried ensemble-based virtual screening (EBVS) using ICM to identify TAR binders.178,180 They assembled three small-molecule libraries of hits and nonhits from experimental HTS for docking, and then assessed the performance of EBVS by analyzing the enrichment factor (EF) and area under the curve (AUC) of a receiver operating characteristic (ROC) curve.178 EF measures the number of true positives in a subset of compounds relative to random screening of the database.181 An ROC curve is a plot of the true-positive rate against the false-positive rate and measures the ability of docking to separate active compounds from inactive compounds, while AUC values range from 0.5 for random selection to 1.0 for perfect enrichment.182 EBVS of 2% of nonhits in a library consisting of 78 hits and 103,349 nonhits identified 42% of hits for an EF of 21 and AUC of 0.88. Similar results were obtained for the other two libraries. Hit compounds in cell-based assays were significantly enriched, with AUC values ranging from 0.91 to 0.94.178 Altogether, these studies demonstrate that EBVS may be used to identify bioactive compounds that bind to HIV-1 TAR RNA (Fig. 5 right).
Databases of Known RNA-Binding Ligands
An enormous amount of data describing ligand–RNA interactions is available, but unfortunately it is challenging to mine. To aid future RNA drug discovery attempts, it would be helpful to compile all protocols and experimental data in searchable databases. This knowledge can be used to identify scaffolds of interest for RNA-binding optimization or tools for screening approaches. Several freely available databases (Inforna,19 SMMRNA,183 R-BIND,184 G4LDB,185 and NALDB186) documenting a catalog of small molecule–RNA interactions exist, with insight ranging from basic selectivity and affinitymeasurements to detailed compound parameters and experimental insight. Inforna (https://disney.florida.scripps.edu/software/) is a database of experimentally determined, privileged RNA motif–small molecule interactions alongside interactions reported in the literature.19 The motifs, or structural elements, within an RNA target of interest are compared with the database of interactions, informing lead compounds for further investigation and a chemical similarity searching feature to expand those leads.19 SMMRNA and R-BIND (https://rbind.-chem.duke.edu/) were/are interactive databases that displayed multiple binding parameters, including molecular weight, hydrogen donor and acceptor count, XlogP, number of rotatable bonds, and number of aromatic rings,183,184 and SMMRNA also housed 2D and 3D structures for ~770 unique RNA-binding compounds.183 The G- quadruplex ligand database, G4LDB, was a second database that focused on reported G-quadruplex ligands.184 It contained >800 G-quadruplex ligands with ~4,000 activity records. The data set consisted of physical properties, 3D ligand structures and design, in vitro and/or in cell binding, and activity data.185 NALDB (http://bsbe.iiti.ac.in/bsbe/naldb/HOME.php) provides detailed information about small molecules targeting all nucleic acids.186 It contains more than 3500 ligand entries and, similar to the databases mentioned above, provides data sets consisting of ligand physical properties, 3D ligand structures, and activity data.186
RNA-Binding Scaffolds
While the interest in screening RNA structures to identify small molecules able to bind them/modulate their biology has initially remained essentially academic, this picture has drastically changed. There is an increased interest of the pharmaceutical industry in assessing the possibility of drugging RNA targets using corporate compound collections. This has not only triggered a change in the type of RNA target structures under investigation but also increased the chemical diversity reported to interact with RNA.
The 2008 comprehensive review by Thomas and Hergenrother187 constitutes an outstanding reference point to assess the evolution of chemical equity studied for its capacity to interact with RNA. By the time this review was published, there was still a clear focus to use reasonably characterized available RNA binders like aminoglycosides, oxazolidinone antibiotics, aromatic bases, and intercalating agents as starting point points to develop more specific binders. The late 1990s and early 2000s have witnessed the initiation of more thorough screening efforts to identify novel chemotypes with improved selectivity and toxicity profiles as well as better potential for further optimization toward therapeutic applications. Early attempts in screening large compound libraries demonstrated a low hit rate and yielded hits with challenging properties.188 Some interesting developments were achieved in identifying more suitable chemistry already by the mid-2000s, as exemplified by MS-based efforts by Ionis scientists,189 resulting in the optimization of a benzimidazole series of HCV-IRES IIa binders190 possessing low micromolar activity in an HCV replicon assay together with minimal cellular toxicity. Other hit-finding strategies like fragment-based approaches have also been applied early on to RNA targets, resulting in the confirmation of some privileged scaffolds as illustrated in an NMR screen to identify E. coli A site rRNA binders reported by AbbVie researchers.191 This study confirmed benzimidazoles and 2-amino-benzimidazoles as privileged scaffolds for RNA interactions but also highlighted 2-amino-quinoline and 2-amino-pyridine scaffolds as alternative leads with good potential for optimization.
The emergence of miRs as potential therapeutic targets and miR biogenesis inhibition as a potentially interesting mechanism of action for small molecules has triggered several developments in the screening approaches to identify binders for specific Dicer and Drosha sites on miR targets. The Disney group has successfully applied its combined 2DCS and cheminformatics platform to identify numerous inhibitors against primary (pri) and precursor (pre) miRs as well as binders of intronic and expanded repeats implicated in different neurological disorders. Although 2DCS was initially validated using aminoglycosides,192 this approach has enabled the identification of miR binders across a range of chemotypes. While a dimerization strategy to optimize the hits into cell-active and -selective miR biogenesis inhibitors has often been applied by the Disney group,193 several hits were demonstrated to be useful as monomeric binder, including hits based on 1,8-diamino-2,7-naphtyridine194 and again 2-substituted benzimidazole.117 A recent development of this technique has allowed the Disney group to study the potential of clinical compounds as well as compounds targeting specific protein classes like kinase inhibitors to also bind and potentially modulate RNA.112,195 In particular, three topoisomerase inhibitors (doxorubicin, epirubicin, and mitoxantrone) bound RNA, with an optimal motif identified in oncogenic miR-21, the levels of which are downregulated upon compound treatment.181 While a broader outcome still needs to be published, these studies shed an interesting light on a potential secondary pharmacology, centered on RNA modulation, that compounds developed against protein targets might possess and that, to this day, has been difficult to study and exploit and thus has been largely overlooked.
SMMs have also been successfully used by the Schneekloth group to identify compounds that modulate Dicer processing of pre-miR-21 in vitro,119 but they more recently broadened their approach to lncRNA Malat-1, identifying binders that specifically recognize the ENE triplex sub-structure that could yield interesting tools to study Malat-1 function.196 RNA binders identified by Schneekloth and coworkers have again underlined benzimidazoles as a privileged scaffold, but also evidenced other useful chemotypes like N-substituted carbazoles.
Beyond microarrays, efforts toward identifying oncogenic miR-21 biogenesis inhibitors have been reported by the Garner and O’Keefe groups using their catalytic enzyme-linked click chemistry assay197 and a DSF-based screen,84 respectively. Interestingly, both of these groups used natural products/natural product extracts in their compound sets and identified scaffolds that were further characterized, including tetracyclines derivatives and butylcycloheptyl prodiginine. The Al-Hashimi and Hargrove groups have also investigated natural product scaffolds using virtual screening of a dynamic ensemble of HIV-TAR, identifying amiloride derivatives as an interesting set of binders.17
While some attempts to identify RNA-specific pharmacophores and to define an RNA binder’s chemical space have been disclosed,20,76,198 these have been based on a relatively limited data set, and there is still a scarcity of data when it comes to systematic screening of a large and diverse compound set with properties in line for subsequent medicinal chemistry optimization. One such attempt has been recently reported by Merck scientists using ALIS,127 described above. More recently, Merck’s diversity set of ~50,000 members and a set of functionally annotated compounds (~5100 compounds) were studied for binding an array of 42 RNA targets, thus probing interactions between drug-like molecules and biologically important RNA structures.133,199 The binders identified were largely biased toward G quadruplexes (1097 of ~1420 compounds, or 77%), and only 119 were selective for one of the 42 RNAs screened. Although the data generated are somewhat limited in scope and predictive power for RNA in general because of the bias for G-quadruplexes, these studies did identify features enriched in selective and general RNA binders as well as differences between RNA and protein binders. Further, the physicochemical spaces occupied by the RNA and protein binders overlap and are within a drug-like space. Interestingly, based on the hits identified and using a combination of machine learning and nearest-neighbor selection based on chemical and biological similarity, an RNA-focused library (~3700 compounds) was assembled. This library was subsequently screened against a subset of 32 of the initial RNA targets, yielding a hit rate of 0.32% (G quadruplexes excluded), a 32-fold improvement compared with the initial diversity set. Collectively, these studies suggest that using quality RNA binders as seeds, chemoinformatic approaches could facilitate the discovery of novel relevant RNA ligands. It will be interesting to see how these compounds advance to in cellulis activity. In complementary studies, an FID-based screen of the LOPAC library was conducted by Tran and Disney to study its potential for RNA binders, identifying eight hits displaying alkyl pyridinium, indole, 2-phenyl benzimidazole, and 2-phenyl indole scaffolds. Identification of their privileged RNA-binding space identified AU-rich hairpin loops as their preferred binding partners with Kd values ranging from 4 to 160 μM.91 Similarly, through use of 30,000 small molecules from The Scripps Research Institute’s (TSRI) and National Cancer Institute’s (NCI) collections, they were computationally analyzed for potential to bind RNA, yielding a 1987-compound set that was both chemically diverse and drug-like. From these screens, 239 novel RNA binders were identified. Analysis of the hits identified 13 privileged scaffolds that include phenyl imidazolines, 2-aminopyrimides, 2-indoles, benzimidazoles, and anilines as RNA binders, with all hits falling within the drug-like space upon comparison of their physiochemical properties to FDA-approved drugs. Further investigation identified a privileged RNA-binding partner in the SL1, SL2, and SL3 stems of the 30 UTR of HCV that, upon binding, inhibited viral replication in an HCV replicon assay by a mechanism not previously shown before.76
While affinity selection will yield binders that will require thorough follow-up characterization in order to validate the potential functional activity of the identified hits, the wealth of information generated in that way will certainly be very informative to refine our views of both the chemical and target space of RNA binders and help expand the chemical diversity useful for RNA modulation. At a stage where our understanding of how to rationally design and optimize small molecules with the potential to selectively modulate RNA function is still in its infancy, additional similar efforts will be useful to help medicinal chemists fully realize the potential of RNA as both a primary and a secondary pharmacologic target.
Conclusion
Pharmaceutical drug discovery over the decades has been predominantly directed toward several targets - enzymes, G-protein-coupled receptors, and proteases.200,201 Expanding this targetable space is a noted priority among academics and companies and is led by omics/profiling initiatives.202–204 The continued development of new molecular entities such as ASOs, mRNAs, and small activating (sa) RNAs will breathe new life into numerous previously written off “undruggable” protein targets.205–209 RNA (translatable and nontranslatable) displays an enormity of function with roles correlating or even driving disease.210,211 Prior to 2000, attempts made to develop small molecules that target RNA were wrought with selectivity and affinity challenges.187 Regardless, RNA targeting small molecules demonstrated some success with various classes of marketed antimicrobials, alongside that of riboswitches that utilize endogenous ligands for modulation.187 Interest in RNA–small molecule targeting over the last 5–10 years has erupted, with numerous biotech and pharmaceutical companies investing resources in the development of RNA targeting platforms (https://cen.acs.org/articles/95/i47/RNA-drug-hunters.html), a consequence of the development of novel methods and modalities that have broken down historical barriers that rendered this field dormant and controversial (https://www.the-scientist.com/lab-tools/drug-discovery-techniques-open-the-door-to-rna-targeted-drugs-65903). These companies are utilizing several enabling technologies to drive RNA targeting drug discovery, with a good proportion involving target engagement-driven projects that could piggyback on nonclassical protein drug discovery. The selection of correct hit identification approaches is key to the target-directed identification of compounds that have favorable properties for selective RNA targeting. The use of phenotypic assays as a primary screen with follow-up protein target deconvolution is common practice (especially within industry).212,213 Beyond bacterial riboswitch and spliceswitch screens, deconvolution of RNA targets is less common.193 A recent report that found RNA rather than protein to be the primary target for an established cancer drug indeed demonstrates the importance of RNA target deconvolution for understanding drug mode of action.195
Here, we have discussed the different approaches for the target-directed screening of small molecules against RNA targets, many of which can also be applied to other targets such as proteins. Due to the dynamics of RNA, difficulty in associating function–structure in vitro, and noted selectivity challenges with compounds displaying certain properties (e.g., intercalators), there is a preference to use unlabeled screening approaches that can assess target selectivity, affinity, and/or function in a single well. For binding approaches using labeled RNA, selectivity can easily be incorporated in the primary screen by using unlabeled competitor RNA. For unlabeled approaches, this is more difficult and instead cost-effective follow-up assays are required to reduce primary hit collections. Secondary assays preferably assess functional responses in vitro or in cell-based assays such as monitoring RNA–protein binding (e.g., FP,27 electromobility shift assays [EMSAs],214 alpha screen,215 and cat-ELCCA216), RNA levels and structure (e.g., sequencing, RT-qPCR, SHAPE,217 and rG4-seq218), direct function (e.g., reporter assays219 and processing assays [splicing, Dicer, or Drosha]), and phenotypic outputs (e.g., differentiation and proliferation). Most of these assays are costly and low throughput and require RNA labeling or further on-target cellular follow-up. Improving small-molecule libraries by biasing against promiscuous RNA binders, furthering our understanding of RNA-binding scaffolds and RNA–small molecule interactions, and developing HTS approaches that can cost-effectively monitor target engagement and selectivity in parallel will help rapidly produce high-quality RNA-binding hits for lead optimization, moving the “modern” RNA targeting field beyond tool development and validation.
Acknowledgments
We also would like to acknowledge the many works that could not be summarized in this review and their contribution to this evolving field.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding for these efforts has been provided by the taxpayers of the United States of America in the form of grants from the National Institutes of Health (R01 GM97455, DP1 NS096898, P01 NS09914, and R33 NS096032 to M.D.D.) and by AstraZeneca.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Clamp M; Fry B; Kamal M; et al. Distinguishing Protein-Coding and Noncoding Genes in the Human Genome. Proc. Natl. Acad. Sci. U.S.A 2007, 104, 19428–19433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wei JW; Huang K; Yang C; et al. Non-Coding RNAs as Regulators in Epigenetics (Review). Oncol. Rep 2017, 37, 3. [DOI] [PubMed] [Google Scholar]
- 3.Wurm AA; Pina C Long Non-Coding RNAs as Functional and Structural Chromatin Modulators in Acute Myeloid Leukemia. Front. Oncol 2019, 9, 899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunker AK; Romero P; Obradovic Z; et al. Intrinsic Protein Disorder in Complete Genomes. Genome Informatics 2000, 11, 161–171. [PubMed] [Google Scholar]
- 5.Oldfield CJ; Cheng Y; Cortese MS; et al. Comparing and Combining Predictors of Mostly Disordered Proteins. Biochemistry 2005, 44, 1989–2000. [DOI] [PubMed] [Google Scholar]
- 6.Stein CA; Castanotto D FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther 2017, 25, 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rinaldi C; Wood MJA Antisense Oligonucleotides: The Next Frontier for Treatment of Neurological Disorders. Nat. Rev. Neurol 2017, 14, 9. [DOI] [PubMed] [Google Scholar]
- 8.Eckardt S; Romby P; Sczakiel G Implications of RNA Structure on the Annealing of a Potent Antisense RNA Directed against the Human Immunodeficiency Virus Type 1. Biochemistry 1997, 36, 12711–12721. [DOI] [PubMed] [Google Scholar]
- 9.Stull RA; Taylor LA; Szoka FC Jr. Predicting Antisense Oligonucleotide Inhibitory Efficacy: A Computational Approach Using Histograms and Thermodynamic Indices. Nucleic Acids Res 1992, 20, 3501–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaug AJ; Cech TR The Intervening Sequence RNA of Tetrahymena Is an Enzyme. Science 1986, 231, 470–475. [DOI] [PubMed] [Google Scholar]
- 11.Baer M; Altman S A Catalytic RNA and Its Gene from Salmonella typhimurium. Science 1985, 228, 999–1002. [DOI] [PubMed] [Google Scholar]
- 12.Mathews DH; Disney MD; Childs JL; et al. Incorporating Chemical Modification Constraints into a Dynamic Programming Algorithm for Prediction of RNA Secondary Structure. Proc. Natl. Acad. Sci. U.S.A 2004, 101, 7287–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moazed D; Noller HF Interaction of Antibiotics with Functional Sites in 16S Ribosomal RNA. Nature 1987, 327, 389–394. [DOI] [PubMed] [Google Scholar]
- 14.Stern S; Wilson RC; Noller HF Localization of the Binding Site for Protein S4 on 16 S Ribosomal RNA by Chemical and Enzymatic Probing and Primer Extension. J. Mol. Biol 1986, 192, 101–110. [DOI] [PubMed] [Google Scholar]
- 15.Inoue T; Cech TR Secondary Structure of the Circular Form of the Tetrahymena rRNA Intervening Sequence: A Technique for RNA Structure Analysis Using Chemical Probes and Reverse Transcriptase. Proc. Natl. Acad. Sci. U. S.A 1985, 82, 648–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winkler W; Nahvi A; Breaker RR Thiamine Derivatives Bind Messenger RNAs Directly to Regulate Bacterial Gene Expression. Nature 2002, 419, 952–956. [DOI] [PubMed] [Google Scholar]
- 17.Blount KF; Wang JX; Lim J; et al. Antibacterial Lysine Analogs That Target Lysine Riboswitches. Nat. Chem. Biol 2007, 3, 44–49. [DOI] [PubMed] [Google Scholar]
- 18.Pedrolli DB; Matern A; Wang J; et al. A Highly Specialized Flavin Mononucleotide Riboswitch Responds Differently to Similar Ligands and Confers Roseoflavin Resistance to Streptomyces davawensis. Nucleic Acids Res 2012, 40, 8662–8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Disney MD; Winkelsas AM; Velagapudi SP; et al. Inforna 2.0: A Platform for the Sequence-Based Design of small Molecules Targeting Structured RNAs. ACS Chem. Biol 2016, 11, 1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morgan BS; Forte JE; Culver RN; et al. Discovery of Key Physicochemical, Structural, and Spatial Properties of RNA-Targeted Bioactive Ligands. Angew. Chem. Int. Ed. Engl 2017, 56, 13498–13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rzuczek SG; Colgan LA; Nakai Y; et al. Precise Small-Molecule Recognition of a Toxic CUG RNA Repeat Expansion. Nat. Chem. Biol 2016, 13, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Childs-Disney JL; Yildirim I; Park H; et al. Structure of the Myotonic Dystrophy Type 2 RNA and Designed Small Molecules That Reduce Toxicity. ACS Chem. Biol 2014, 9, 538–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim K; Chadalapaka G; Lee SO; et al. Identification of Oncogenic MicroRNA-17–92/ZBTB4/Specificity Protein Axis in Breast Cancer. Oncogene 2011, 31, 1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monroig P. d. C.; Chen L; Zhang S; et al. Small Molecule Compounds Targeting miRNAs for Cancer Therapy. Adv. Drug. Deliv. Rev 2015, 81, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Llano-Sotelo B; Chow CS RNA-Aminoglycoside Antibiotic Interactions: Fluorescence Detection of Binding and Conformational Change. Bioorg. Med. Chem. Lett 1999, 9, 213–216. [DOI] [PubMed] [Google Scholar]
- 26.Parsons J; Hermann T Conformational Flexibility of Ribosomal Decoding-Site RNA Monitored by Fluorescent Pteridine Base Analogues. Tetrahedron 2007, 63, 3548–3552. [Google Scholar]
- 27.Hafner M; Vianini E; Albertoni B; et al. Displacement of Protein-Bound Aptamers with Small Molecules Screened by Fluorescence Polarization. Nat. Protoc 2008, 3, 579. [DOI] [PubMed] [Google Scholar]
- 28.Benz A; Singh V; Mayer TU; et al. Identification of Novel Quadruplex Ligands from Small Molecule Libraries by FRET-Based High-Throughput Screening. Chembiochem 2011, 12, 1422–1426. [DOI] [PubMed] [Google Scholar]
- 29.Moon MH; Hilimire TA; Sanders AM; et al. Measuring RNA–Ligand Interactions with Microscale Thermophoresis. Biochemistry 2018, 57, 4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silvers R; Keller H; Schwalbe H; et al. Differential Scanning Fluorimetry for Monitoring RNA Stability. Chembiochem 2015, 16, 1109–1114. [DOI] [PubMed] [Google Scholar]
- 31.Llano-Sotelo B; Azucena EF Jr., Kotra LP; et al. Aminoglycosides Modified by Resistance Enzymes Display Diminished Binding to the Bacterial Ribosomal Aminoacyl-tRNA Site. Chem. Biol 2002, 9, 455–463. [DOI] [PubMed] [Google Scholar]
- 32.Nazarenko I; Pires R; Lowe B; et al. Effect of Primary and Secondary Structure of Oligodeoxyribonucleotides on the Fluorescent Properties of Conjugated Dyes. Nucleic Acids Res 2002, 30, 2089–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hall KB RNA in Motion. Curr. Opin. Chem. Biol 2008, 12, 612–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Al-Hashimi HM; Walter NG RNA Dynamics: It Is about Time. Curr. Opin. Struct. Biol 2008, 18, 321–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boerneke MA; Hermann T Conformational Flexibility of Viral RNA Switches Studied by FRET. Methods 2015, 91, 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hermann T; Patel DJ RNA Bulges as Architectural and Recognition Motifs. Structure 2000, 8, R47–R54. [DOI] [PubMed] [Google Scholar]
- 37.Leulliot N; Varani G Current Topics in RNA-Protein Recognition: Control of Specificity and Biological Function through Induced Fit and Conformational Capture. Biochemistry 2001, 40, 7947–7956. [DOI] [PubMed] [Google Scholar]
- 38.Mandal M; Breaker RR Gene Regulation by Riboswitches. Nat. Rev. Mol. Cell. Biol 2004, 5, 451–463. [DOI] [PubMed] [Google Scholar]
- 39.Shajani Z; Deka P; Varani G Decoding RNA Motional Codes. Trends Biochem. Sci 2006, 31, 421–424. [DOI] [PubMed] [Google Scholar]
- 40.McGovern-Gooch KR; Baird NJ Fluorescence-Based Investigations of RNA-Small Molecule Interactions. Methods 2019, 167, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okamoto A; Saito Y; Saito I Design of Base-Discriminating Fluorescent Nucleosides. J. Photochem. Photobiol. C 2005, 6, 108–122. [Google Scholar]
- 42.Lin K-Y; Jones RJ; Matteucci M Tricyclic 20-Deoxycytidine Analogs: Syntheses and Incorporation into Oligodeoxynucleotides Which Have Enhanced Binding to Complementary RNA. J. Am. Chem. Soc 1995, 117, 3873–3874. [Google Scholar]
- 43.Füchtbauer AF; Preus S; Börjesson K; et al. Fluorescent RNA Cytosine Analogue—An Internal Probe for Detailed Structure and Dynamics Investigations. Sci. Rep 2017, 7, 2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi X; Herschlag D Fluorescence Polarization Anisotropy to Measure RNA Dynamics In Methods in Enzymology; Academic Press: Amsterdam, 2009; pp 287–302. [DOI] [PubMed] [Google Scholar]
- 45.Okamoto A; Tainaka K; Nishiza K-I; et al. Monitoring DNA Structures by Dual Fluorescence of Pyrene Derivatives. J. Am. Chem. Soc 2005, 127, 13128–13129. [DOI] [PubMed] [Google Scholar]
- 46.Tanpure AA; Pawar MG; Srivatsan SG Fluorescent Nucleoside Analogs: Probes for Investigating Nucleic Acid Structure and Function. Isr. J. Chem 2013, 53, 366–378. [Google Scholar]
- 47.Blount KF; Tor Y Using Pyrene-Labeled HIV-1 TAR to Measure RNA–Small Molecule Binding. Nucleic Acids Res 2003, 31, 5490–5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jameson DM; Ross JA Fluorescence Polarization/Anisotropy in Diagnostics and Imaging. Chem. Rev 2010, 110, 2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naryshkin NA; Weetall M; Dakka A; et al. SMN2 Splicing Modifiers Improve Motor Function and Longevity in Mice with Spinal Muscular Atrophy. Science 2014, 345, 688–693. [DOI] [PubMed] [Google Scholar]
- 50.Wang J; Schultz PG; Johnson KA Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc. Natl. Acad. Sci. U.S.A 2018, 115, E4604–E4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y; Killian J; Hamasaki K; et al. RNA Molecules That Specifically and Stoichiometrically Bind Aminoglycoside Antibiotics with High Affinities. Biochemistry 1996, 35, 12338–12346. [DOI] [PubMed] [Google Scholar]
- 52.Poirier A; Weetall M; Heinig K; et al. Risdiplam Distributes and Increases SMN Protein in Both the Central Nervous System and Peripheral Organs. Pharmacol. Res. Perspect 2018, 6, e00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheung AK; Hurley B; Kerrigan R; et al. Discovery of Small Molecule Splicing Modulators of Survival Motor Neuron-2 (SMN2) for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem 2018, 61, 11021–11036. [DOI] [PubMed] [Google Scholar]
- 54.Palacino J; Swalley SE; Song C; et al. SMN2 Splice Modulators Enhance U1–Pre-mRNA Association and Rescue SMA Mice. Nat. Chem. Bio 2015, 11, 511–517. [DOI] [PubMed] [Google Scholar]
- 55.Lakowicz JR Principles of Fluorescence Spectroscopy, 2nd Ed.; Kluwer Academic/Plenum, New York, 1999. [Google Scholar]
- 56.Boerneke MA; Dibrov SM; Gu J; et al. Functional Conservation Despite Structural Divergence in Ligand-Responsive RNA Switches. Proc. Natl. Acad. Sci. U.S.A 2014, 111, 15952–15957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dibrov SM; Ding K; Brunn ND; et al. Structure of a Hepatitis C Virus RNA Domain in Complex with a Translation Inhibitor Reveals a Binding Mode Reminiscent of Riboswitches. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 5223–5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parsons J; Castaldi MP; Dutta S; et al. Conformational inhibition of the Hepatitis C Virus Internal Ribosome Entry Site RNA. Nat. Chem. Biol 2009, 5, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rahman KM; Tizkova K; Reszka AP; et al. Identification of Novel Telomeric G-Quadruplex-Targeting Chemical Scaffolds through Screening of Three NCI Libraries. Bioorg. Med. Chem. Lett 2012, 22, 3006–3010. [DOI] [PubMed] [Google Scholar]
- 60.Chen CZ; Sobczak K; Hoskins J; et al. Two High-Throughput Screening Assays for Aberrant RNA-Protein Interactions in Myotonic Dystrophy Type 1. Anal. Bioanal. Chem 2012, 402, 1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parkesh R; Childs-Disney JL; Nakamori M; et al. Design of a Bioactive Small Molecule That Targets the Myotonic Dystrophy Type 1 RNA via an RNA Motif-Ligand Database and Chemical Similarity Searching. J. Am. Chem. Soc 2012, 134, 4731–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rzuczek SG; Southern MR; Disney MD Studying a Drug-Like, RNA-Focused Small Molecule Library Identifies Compounds That Inhibit RNA Toxicity in Myotonic Dystrophy. ACS Chem. Biol 2015, 10, 2706–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tyagi S; Kramer FR Molecular Beacons: Probes That Fluoresce upon Hybridization. Nat. Biotechnol 1996, 14, 303–308. [DOI] [PubMed] [Google Scholar]
- 64.Zheng J; Yang R; Shi M; et al. Rationally Designed Molecular Beacons for Bioanalytical and Biomedical Applications. Chem. Soc. Rev 2015, 44, 3036–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ha M; Kim VN Regulation of MicroRNA Biogenesis. Nat. Rev. Mol. Cell. Biol 2014, 15, 509. [DOI] [PubMed] [Google Scholar]
- 66.Bose D; Jayaraj GG; Kumar S; et al. A Molecular-Beacon-Based Screen for Small Molecule Inhibitors of miRNA Maturation. ACS Chem. Biol 2013, 8, 930–938. [DOI] [PubMed] [Google Scholar]
- 67.Bell NM; L’Hernault A; Murat P; et al. Targeting RNA-Protein Interactions within the Human Immunodeficiency Virus Type 1 Lifecycle. Biochemistry 2013, 52, 9269–9274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ingemarsdotter CK; Zeng J; Long Z; et al. An RNA-Binding Compound That Stabilizes the HIV-1 gRNA Packaging Signal Structure and Specifically Blocks HIV-1 RNA encapsidation. Retrovirology 2018, 15, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Davies BP; Arenz C A Homogenous Assay for Micro RNA Maturation. Angew. Chem. Int. Ed 2006, 45, 5550–5552. [DOI] [PubMed] [Google Scholar]
- 70.Vo DD; Staedel C; Zehnacker L; et al. Targeting the Production of Oncogenic MicroRNAs with Multimodal Synthetic Small Molecules. ACS Chem. Bio 2014, 9, 711–721. [DOI] [PubMed] [Google Scholar]
- 71.Staedel C; Tran TPA; Giraud J; et al. Modulation of Oncogenic miRNA Biogenesis Using Functionalized Polyamines. Sci. Rep 2018, 8, 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Seidel SAI; Wienken CJ; Geissler S; et al. Label-Free Microscale Thermophoresis Discriminates Sites and Affinity of Protein–Ligand Binding. Angew. Chem. Int. Ed. Engl 2012, 51, 10656–10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jerabek-Willemsen M; André T; Wanner R; et al. MicroScale Thermophoresis: Interaction Analysis and Beyond. J. Mol. Struct 2014, 1077, 101–113. [Google Scholar]
- 74.Jerabek-Willemsen M; Wienken CJ; Braun D; et al. Molecular Interaction Studies Using Microscale Thermophoresis. Assay Drug Dev. Technol 2011, 9, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gaffarogullari EC; Krause A; Balbo J; et al. Microscale Thermophoresis Provides Insights into Mechanism and Thermodynamics of Ribozyme Catalysis. RNA Biol 2013, 10, 1815–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Childs-Disney JL; Tran T; Vummidi BR; et al. A Massively Parallel Selection of Small Molecule-RNA Motif Binding Partners Informs Design of an Antiviral from Sequence. Chem 2018, 4, 2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li Y; Disney MD Precise Small Molecule Degradation of a Noncoding RNA Identifies Cellular Binding Sites and Modulates an Oncogenic Phenotype. ACS Chem. Biol 2018, 13, 3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Costales MG; Suresh B; Vishnu K; et al. Targeted Degradation of a Hypoxia-Associated Non-Coding RNA Enhances the Selectivity of a Small Molecule Interacting with RNA. Cell Chem. Biol 2019, 26, 1180–1186.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haniff HS; Graves A; Disney MD Selective Small Molecule Recognition of RNA Base Pairs. ACS Combi. Sci 2018, 20, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DeSantis K; Reed A; Rahhal R; et al. Use of Differential Scanning Fluorimetry as a High-Throughput Assay to Identify Nuclear Receptor Ligands. Nucl. Recept. Signal 2012, 10, nrs.10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vivoli M; Novak HR; Littlechild JA; et al. Determination of Protein-Ligand Interactions Using Differential Scanning Fluorimetry. JoVE 2014, e51809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sztuba-Solinska J; Shenoy SR; Gareiss P; et al. Identification of Biologically Active, HIV TAR RNA-Binding Small Molecules Using Small Molecule Microarrays. J. Am. Chem. Soc 2014, 136, 8402–8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baird NJ; Inglese J; Ferré-D’Amaré AR Rapid RNA–Ligand Interaction Analysis through High-Information Content Conformational and Stability Landscapes. Nat. Commun 2015, 6, 8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Matarlo JS; Krumpe LRH; Heinz WF; et al. The Natural Product Butylcycloheptyl Prodiginine Binds Pre- miR-21, Inhibits Dicer-Mediated Processing of pre-miR-21, and Blocks Cellular Proliferation. Cell Chem. Biol 2019, 26, 1133–1142.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Luedtke NW; Tor Y Fluorescence-Based Methods for Evaluating the RNA Affinity and Specificity of HIV-1 Rev-RRE Inhibitors. Biopolymers 2003, 70, 103–119. [DOI] [PubMed] [Google Scholar]
- 86.Sau SP; Kumar P; Sharma PK; et al. Fluorescent Intercalator Displacement Replacement (FIDR) Assay: Determination of Relative Thermodynamic and Kinetic Parameters in Triplex Formation—A Case Study Using Triplex-Forming LNAs. Nucleic Acids Res 2012, 40, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Boger DL; Tse WC Thiazole Orange as the Fluorescent Intercalator in a High Resolution FID Assay for Determining DNA Binding Affinity and Sequence Selectivity of Small Molecules. Bioorg. Med. Chem 2001, 9, 2511–2518. [DOI] [PubMed] [Google Scholar]
- 88.Lee LG; Chen C-H; Chiu LA Thiazole Orange: A New Dye for Reticulocyte Analysis. Cytometry 1986, 7, 508–517. [DOI] [PubMed] [Google Scholar]
- 89.Krishnamurthy M; Schirle NT; Beal PA Screening Helix-Threading Peptides for RNA Binding Using a Thiazole Orange Displacement Assay. Bioorg. Med. Chem 2008, 16, 8914–8921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Asare-Okai PN; Chow CS A Modified Fluorescent Intercalator Displacement Assay for RNA Ligand Discovery. Anal. Biochem 2011, 408, 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tran T; Disney MD Identifying the Preferred RNA Motifs and Chemotypes That Interact by Probing Millions of Combinations. Nat. Commun 2012, 3, 1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang J; Umemoto S; Nakatani K Fluorescent Indicator Displacement Assay for Ligand-RNA Interactions. J. Am. Chem. Soc 2010, 132, 3660–3661. [DOI] [PubMed] [Google Scholar]
- 93.Bradrick TD; Marino JP Ligand-Induced Changes in 2-Aminopurine Fluorescence as a Probe for Small Molecule Binding to HIV-1 TAR RNA. RNA 2004, 10, 1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chao PW; Chow CS Monitoring AminoglycosideInduced Conformational Changes in 16S rRNA through Acrylamide Quenching. Bioorg. Med. Chem 2007, 15, 3825–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sarpong K; Datta B Nucleic-Acid-Binding Chromophores as Efficient Indicators of Aptamer-Target Interactions. J. Nucleic Acids 2012, 2012, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Karn J; Dingwall C; Finch JT; et al. RNA Binding by the Tat and Rev Proteins of HIV-1. Biochimie 1991, 73, 9–16. [DOI] [PubMed] [Google Scholar]
- 97.Weeks K; Ampe C; Schultz S; et al. Fragments of the HIV-1 Tat Protein Specifically Bind TAR RNA. Dis. Markers 1990, 249, 1281–1285. [DOI] [PubMed] [Google Scholar]
- 98.Weeks KM; Crothers DM RNA Recognition by Tat-Derived Peptides: Interaction in the Major Groove? Cell 1991, 66, 577–588. [DOI] [PubMed] [Google Scholar]
- 99.Ludwig V; Krebs A; Stoll M; et al. Tripeptides from Synthetic Amino Acids Block the Tat–TAR Association and Slow Down HIV Spread in Cell Cultures. Chembiochem 2007, 8, 1850–1856. [DOI] [PubMed] [Google Scholar]
- 100.Matsumoto C; Hamasaki K; Mihara H; et al. A High-Throughput Screening Utilizing Intramolecular Fluorescence Resonance Energy Transfer for the Discovery of the Molecules That Bind HIV-1 TAR RNA Specifically. Bioorg. Med. Chem. Lett 2000, 10, 1857–1861. [DOI] [PubMed] [Google Scholar]
- 101.Patwardhan NN; Cai Z; Newson CN; et al. Fluorescent Peptide Displacement as a General Assay for Screening Small Molecule Libraries against RNA. Org. Biomol. Chem 2019, 17, 1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Patwardhan NN; Ganser LR; Kapral GJ; et al. Amiloride as a New RNA-Binding Scaffold with Activity against HIV-1 TAR. MedChemComm 2017, 8, 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Luedtke NW; Tor Y A Novel Solid-Phase Assembly for Identifying Potent and Selective RNA Ligands. Angew. Chem. Int. Ed. Engl 2000, 112, 1858–1860. [DOI] [PubMed] [Google Scholar]
- 104.Wang Y; Hamasaki K; Rando RR Specificity of Aminoglycoside Binding to RNA Constructs Derived from the 16s rRNA Decoding Region and the HIV-RRE Activator Region. Biochemistry 1997, 36, 768–779. [DOI] [PubMed] [Google Scholar]
- 105.Wicks SL; Hargrove AE Fluorescent Indicator Displacement Assays to Identify and Characterize Small Molecule Interactions with RNA. Methods 2019, 167, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hoa XD; Kirk AG; Tabrizian M Towards Integrated and Sensitive Surface Plasmon Resonance Biosensors: A Review of Recent Progress. Biosens. Bioelectron 2007, 23, 151–160. [DOI] [PubMed] [Google Scholar]
- 107.Win MN; Klein JS; Smolke CD Codeine-Binding RNA Aptamers and Rapid Determination of Their Binding Constants Using a Direct Coupling Surface Plasmon Resonance Assay. Nucleic Acids Res 2006, 34, 5670–5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hendrix M; Priestley ES; Joyce GF; et al. Direct Observation of Aminoglycoside-RNA Interactions by Surface Plasmon Resonance. J. Am. Chem. Soc 1997, 119, 3641–3648. [DOI] [PubMed] [Google Scholar]
- 109.Yang W-Y; He F; Strack RL; et al. Small Molecule Recognition and Tools to Study Modulation of r(CGG)exp in Fragile X-Associated Tremor Ataxia Syndrome. ACS Chem. Biol 2016, 11, 2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang Z-F; Ursu A; Childs-Disney JL; et al. The Hairpin Form of r(G4C2)exp in c9ALS/FTD Is Repeat-Associated Non-ATG Translated and a Target for Bioactive Small Molecules. Cell Chem. Biol 2019, 26, 179–190.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hergenrother PJ; Depew KM; Schreiber SL Small-Molecule Microarrays: Covalent Attachment and Screening of Alcohol-Containing Small Molecules on Glass Slides. J. Am. Chem. Soc 2000, 122, 7849–7850. [Google Scholar]
- 112.Velagapudi SP; Luo Y; Tran T; et al. Defining RNA-Small Molecule Affinity Landscapes Enables Design of a Small Molecule Inhibitor of an Oncogenic Noncoding RNA. ACS Cent. Sci 2017, 3, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Velagapudi SP; Gallo SM; Disney MD Sequence-Based Design of Bioactive Small Molecules That Target Precursor MicroRNAs. Nat. Chem. Biol 2014, 10, 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Velagapudi SP; Disney MD Two-Dimensional Combinatorial Screening Enables the Bottom-Up Design of a MicroRNA-10b Inhibitor. Chem. Commun 2014, 50, 3027–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Velagapudi SP; Cameron MD; Haga CL; et al. Design of a Small Molecule against an Oncogenic Noncoding RNA. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 5898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Costales MG; Hoch DG; Abegg D; et al. A Designed Small Molecule Inhibitor of a Non-Coding RNA Sensitizes HER2 Negative Cancers to Herceptin. J. Am. Chem. Soc 2019, 141, 2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Costales MG; Haga CL; Velagapudi SP; et al. Small Molecule Inhibition of MicroRNA-210 Reprograms an Oncogenic Hypoxic Circuit. J. Am. Chem. Soc 2017, 139, 3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Abulwerdi FA; Schneekloth JS Jr. Microarray-Based Technologies for the Discovery of Selective, RNA-Binding Molecules. Methods 2016, 103, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Connelly CM; Boer RE; Moon MH; et al. Discovery of Inhibitors of MicroRNA-21 Processing Using Small Molecule Microarrays. ACS Chem. Biol 2017, 12, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xavier KA; Eder PS; Giordano T RNA as a Drug Target: Methods for Biophysical Characterization and Screening. Trends Biotechnol 2000, 18, 349–356. [DOI] [PubMed] [Google Scholar]
- 121.Riccardi Sirtori F; Altomare A; Carini M; et al. MS Methods to Study Macromolecule-Ligand Interaction: Applications in Drug Discovery. Methods 2018, 144, 152. [DOI] [PubMed] [Google Scholar]
- 122.Feng WY Mass Spectrometry in Drug Discovery: A Current Review. Curr. Drug. Discov. Technol 2004, 1, 295–312. [DOI] [PubMed] [Google Scholar]
- 123.Thomas B; Akoulitchev AV Mass Spectrometry of RNA. Trends Biochem. Sci 2006, 31, 173–181. [DOI] [PubMed] [Google Scholar]
- 124.Hari Y; Nyakas A; Stucki SR; et al. Elucidation of Nucleic Acid-Drug Interactions by Tandem Mass Spectrometry. Chimia (Aarau) 2014, 68, 164–167. [DOI] [PubMed] [Google Scholar]
- 125.Scalabrin M; Palumbo M; Richter SN Highly Improved Electrospray Ionization-Mass Spectrometry Detection of G-Quadruplex-Folded Oligonucleotides and Their Complexes with Small Molecules. Anal. Chem 2017, 89, 8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Annis DA; Nickbarg E; Yang X; et al. Affinity Selection-Mass Spectrometry Screening Techniques for Small Molecule Drug Discovery. Curr. Opin. Chem. Biol 2007, 11, 518–526. [DOI] [PubMed] [Google Scholar]
- 127.Cummins LL; Chen S; Blyn LB; et al. Multitarget Affinity/Specificity Screening of Natural Products: Finding and Characterizing High-Affinity Ligands from Complex Mixtures by Using High-Performance Mass Spectrometry. J. Nat. Prod 2003, 66, 1186–1190. [DOI] [PubMed] [Google Scholar]
- 128.Greig MJ; Robinson JM Detection of Oligonucleotide-Ligand Complexes by ESI-MS (DOLCE-MS) as a Component of High Throughput Screening. J. Biomol. Screen 2000, 5, 441–454. [DOI] [PubMed] [Google Scholar]
- 129.Whitehurst CE; Annis DA Affinity Selection-Mass Spectrometry and Its Emerging Application to the High Throughput Screening of G Protein-Coupled Receptors. Comb. Chem. High Throughput Screen 2008, 11, 427–438. [DOI] [PubMed] [Google Scholar]
- 130.Walker SS; Degen D; Nickbarg E; et al. Affinity Selection-Mass Spectrometry Identifies a Novel Antibacterial RNA Polymerase Inhibitor. ACS Chem. Biol 2017, 12, 1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.O’Connell TN; Ramsay J; Rieth SF; et al. Solution-Based Indirect Affinity Selection Mass Spectrometry—A General Tool for High-Throughput Screening of Pharmaceutical Compound Libraries. Anal. Chem 2014, 86, 7413–7420. [DOI] [PubMed] [Google Scholar]
- 132.Rizvi NF; Howe JA; Nahvi A; et al. Discovery of Selective RNA-Binding Small Molecules by Affinity-Selection Mass Spectrometry. ACS Chem. Biol 2018, 13, 820. [DOI] [PubMed] [Google Scholar]
- 133.Rizvi NF; Nickbarg EB RNA-ALIS: Methodology for Screening Soluble RNAs as Small Molecule Targets Using ALIS Affinity-Selection Mass Spectrometry. Methods 2019, 167, 28. [DOI] [PubMed] [Google Scholar]
- 134.Flusberg DA; Rizvi NF; Kutilek V; et al. Identification of G-Quadruplex-Binding Inhibitors of MYC Expression through Affinity Selection-Mass Spectrometry. SLAS Discov 2019, 24, 142. [DOI] [PubMed] [Google Scholar]
- 135.Otto S; Furlan RLE; Sanders JKM Dynamic Combinatorial Chemistry. Drug Discov. Today 2002, 7, 117–125. [DOI] [PubMed] [Google Scholar]
- 136.McAnany JD; Miller BL Dynamic Combinatorial Chemistry as a Rapid Method for Discovering Sequence-Selective RNA-Binding Compounds In Methods in Enzymology; Hargrove AE, Ed.; Academic Press: Amsterdam, 2019; p 67. [DOI] [PubMed] [Google Scholar]
- 137.López-Seńın P; Gómez-Pinto I; Grandas A, et al. Identification of Ligands for the Tau Exon 10 Splicing Regulatory Element RNA by Using Dynamic Combinatorial Chemistry. Chem. Eur. J 2011, 17, 1946–1953. [DOI] [PubMed] [Google Scholar]
- 138.Gromova AV; Ciszewski JM; Miller BL Ternary Resin-Bound Dynamic Combinatorial Chemistry. Chem. Commun 2012, 48, 2131–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McNaughton BR; Miller BL Resin-Bound Dynamic Combinatorial Chemistry. Org. Lett 2006, 8, 1803–1806. [DOI] [PubMed] [Google Scholar]
- 140.Gareiss PC; Sobczak K; McNaughton BR; et al. Dynamic Combinatorial Selection of Molecules Capable of Inhibiting the (CUG) Repeat RNA-MBNL1 Interaction In Vitro: Discovery of Lead Compounds Targeting Myotonic Dystrophy (DM1). J. Am. Chem. Soc 2008, 130, 16254–16261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ofori LO; Hoskins J; Nakamori M; et al. From Dynamic Combinatorial ‘Hit’ to Lead: In Vitro and In Vivo Activity of Compounds Targeting the Pathogenic RNAs That Cause Myotonic Dystrophy. Nucleic Acids Res 2012, 40, 6380–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hesse M; Arenz C A Rapid and Versatile Assay for Ago2-Mediated Cleavage by Using Branched Rolling Circle Amplification. Chembiochem 2016, 17, 304. [DOI] [PubMed] [Google Scholar]
- 143.Macarron R; Banks MN; Bojanic D; et al. Impact of High-Throughput Screening in Biomedical Research. Nat. Rev. Drug Discov 2011, 10, 188–195. [DOI] [PubMed] [Google Scholar]
- 144.Harner MJ; Frank AO; Fesik SW Fragment-Based Drug Discovery Using NMR Spectroscopy. J. Biomol. NMR 2013, 56, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cross R The RNA Drug Hunters. C&EN 2017, 95, 16–18. [Google Scholar]
- 146.Erlanson DA; Fesik SW; Hubbard RE; et al. Twenty Years On: The Impact of Fragments on Drug Discovery. Nat. Rev. Drug. Discov 2016, 15, 605. [DOI] [PubMed] [Google Scholar]
- 147.Dalvit C; Pevarello P; Tato M; et al. Identification of Compounds with Binding Affinity to Proteins via Magnetization Transfer from Bulk Water. J. Biomol. NMR 2000, 18, 65–68. [DOI] [PubMed] [Google Scholar]
- 148.Becker W; Bhattiprolu KC; Gubensak N; et al. Investigating Protein-Ligand Interactions by Solution Nuclear Magnetic Resonance Spectroscopy. Chemphyschem 2018,19, 895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Davis BJ; Erlanson DA Learning from Our Mistakes: The ‘Unknown Knowns’ in Fragment Screening. Bioorg. Med. Chem. Lett 2013, 23, 2844–2852. [DOI] [PubMed] [Google Scholar]
- 150.Shortridge MD; Hage DS; Harbison GS; et al. Estimating Protein-Ligand Binding Affinity Using High-Throughput Screening by NMR. J. Comb. Chem 2008, 10, 948–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mayer M; Meyer B Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem. Int. Ed. Engl 1999, 38, 1784–1788. [DOI] [PubMed] [Google Scholar]
- 152.Cressina E; Chen L; Abell C; et al. Fragment Screening against the Thiamine Pyrophosphate Riboswitch thiM. Chem. Sci 2011, 2, 157–165. [Google Scholar]
- 153.Antanasijevic A; Ramirez B; Caffrey M Comparison of the Sensitivities of WaterLOGSY and Saturation Transfer Difference NMR Experiments. J. Biomol. NMR 2014, 60, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Huang R; Bonnichon A; Claridge TD; et al. Protein-Ligand Binding Affinity Determination by the waterLOGSY Method: An Optimised Approach Considering Ligand Rebinding. Sci. Rep 2017, 7, 43727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Foster MP; McElroy CA; Amero CD Solution NMR of Large Molecules and Assemblies. Biochemistry 2007, 46, 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hajduk PJ; Olejniczak ET; Fesik SW One-Dimensional Relaxation-and Diffusion-Edited NMR Methods for Screening Compounds That Bind to Macromolecules. J. Am. Chem. Soc 1997, 119, 12257–12261. [Google Scholar]
- 157.Calabrese DR; Connelly CM; Schneekloth JS Ligand-Observed NMR Techniques to Probe RNA-Small Molecule Interactions In Methods in Enzymology; Hargrove AE, Ed.; Academic Press: Amsterdam, 2019; p 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lee M-K; Bottini A; Kim M; et al. A Novel Small-Molecule Binds to the Influenza A Virus RNA Promoter and Inhibits Viral Replication. Chem. Commun 2014, 50, 368–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu G; Park HS; Pyo HM; et al. Influenza A Virus Panhandle Structure Is Directly Involved in RIG-I Activation and Interferon Induction. J. Virol 2015, 89, 6067–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Garav´ıs M; Lopez-Mendez B; Somoza A; et al. Discovery of Selective Ligands for Telomeric RNA GQuadruplexes (TERRA) through 19F-NMR Based Fragment Screening. ACS Chem. Biol 2014, 9, 1559–1566. [DOI] [PubMed] [Google Scholar]
- 161.Deng Z; Wang Z; Xiang C; et al. Formation of Telomeric Repeat-Containing RNA (TERRA) Foci in Highly Proliferating Mouse Cerebellar Neuronal Progenitors and Medulloblastoma. J. Cell. Sci 2012, 125, 4383–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Warner KD; Homan P; Weeks KM; et al. Validating Fragment-Based Drug Discovery for Biological RNAs: Lead Fragments Bind and Remodel the TPP Riboswitch Specifically. Chem. Biol 2014, 21, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ma D-L; Chan DS-H; Leung C-H Molecular Docking for Virtual Screening of Natural Product Databases. Chem. Sci 2011, 2, 1656–1665. [Google Scholar]
- 164.Daldrop P; Brenk R Structure-Based Virtual Screening for the Identification of RNA-Binding Ligands. Methods Mol. Biol 2014, 1103, 127–139. [DOI] [PubMed] [Google Scholar]
- 165.Guilbert C; James TL Docking to RNA via Root-Mean-Square-Deviation-Driven Energy Minimization with Flexible Ligands and Flexible Targets. J. Chem. Inf. Model 2008, 48, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lang PT; Brozell SR; Mukherjee S; et al. DOCK 6: Combining Techniques to Model RNA-Small Molecule Complexes. RNA 2009, 15, 1219–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stelzer AC; Frank AT; Kratz JD; et al. Discovery of Selective Bioactive Small Molecules by Targeting an RNA Dynamic Ensemble. Nat. Chem. Biol 2011, 7, 553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wei BQ; Weaver LH; Ferrari AM; et al. Testing a Flexible-Receptor Docking Algorithm in a Model Binding Site. J. Mol. Biol 2004, 337, 1161–1182. [DOI] [PubMed] [Google Scholar]
- 169.Pinto IG; Guilbert C; Ulyanov NB; et al. Discovery of Ligands for a Novel Target, the Human Telomerase RNA, Based on Flexible-Target Virtual Screening and NMR. J. Med. Chem 2008, 51, 7205–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Daldrop P; Reyes FE; Robinson DA; et al. Novel Ligands for a Purine Riboswitch Discovered by RNA Ligand Docking. Chem. Biol 2011, 18, 324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Pfeffer P; Gohlke H DrugScoreRNA—Knowledge-Based Scoring Function to Predict RNA-Ligand Interactions. J. Chem. Inf. Model 2007, 47, 1868–1876. [DOI] [PubMed] [Google Scholar]
- 172.Philips A; Milanowska K; Lach G; et al. LigandRNA: Computational Predictor of RNA-Ligand Interactions. RNA 2013, 19, 1605–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Morley SD; Afshar M Validation of an Empirical RNA Ligand Scoring Function for Fast Flexible Docking Using Ribodock. J. Comput. Aided Mol. Des 2004, 18, 189–208. [DOI] [PubMed] [Google Scholar]
- 174.Foloppe N; Chen IJ; Davis B; et al. A Structure-Based Strategy to Identify New Molecular Scaffolds Targeting the Bacterial Ribosomal A-Site. Bioorg. Med. Chem 2004, 12, 935–947. [DOI] [PubMed] [Google Scholar]
- 175.Ruiz-Carmona S; Alvarez-Garcia D; Foloppe N; et al. rDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol 2014, 10, e1003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Vernalis Research R&D Team. rDock, 2013.1; Vernalis Research: Cambridge, UK, 2015. [Google Scholar]
- 177.Frank AT; Stelzer AC; Al-Hashimi HM; et al. Constructing RNA Dynamical Ensembles by Combining MD and Motionally Decoupled NMR RDCs: New Insights into RNA Dynamics and Adaptive Ligand Recognition. Nucleic Acids Res 2009, 37, 3670–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ganser LR; Lee J; Rangadurai A; et al. High-Performance Virtual Screening by Targeting a High-Resolution RNA Dynamic Ensemble. Nat. Struct. Mol. Biol 2018, 25, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Borkar AN; Bardaro MF Jr., Camilloni C; et al. Structure of a Low-Population Binding Intermediate in Protein-RNA Recognition. Proc. Natl. Acad. Sci. U.S.A 2016, 113, 7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Salmon L; Bascom G; Andricioaei I; et al. A General Method for Constructing Atomic-Resolution RNA Ensembles Using NMR Residual Dipolar Couplings: The Basis for Interhelical Motions Revealed. J. Am. Chem. Soc 2013, 135, 5457–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Li H; Zhang H; Zheng M; et al. An Effective Docking Strategy for Virtual Screening Based on Multi-Objective Optimization Algorithm. BMC Bioinformatics 2009, 10, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Triballeau N; Acher F; Brabet I; et al. Virtual Screening Workflow Development Guided by the “Receiver Operating Characteristic” Curve Approach. Application to High-Throughput Docking on Metabotropic Glutamate Receptor Subtype 4. J. Med. Chem 2005, 48, 2534–2547. [DOI] [PubMed] [Google Scholar]
- 183.Mehta A; Sonam S; Gouri I; et al. SMMRNA: A Database of Small Molecule Modulators of RNA. Nucleic Acids Res 2014, 42, D132–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Morgan BS; Sanaba BG; Donlic A; et al. R-BIND: An Interactive Database for Exploring and Developing RNA Targeted Chemical Probes. ACS Chem. Biol 2019, 14, 2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Li Q; Xiang JF; Yang QF; et al. G4LDB: A Database for Discovering and Studying G-Quadruplex Ligands. Nucleic Acids Res 2013, 41, D1115–D1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kumar Mishra S; Kumar A NALDB: Nucleic Acid Ligand Database for Small Molecules Targeting Nucleic Acid. Database (Oxford) 2016, 2016, baw002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Thomas JR; Hergenrother PJ Targeting RNA with Small Molecules. Chem. Rev 2008, 108, 1171–1224. [DOI] [PubMed] [Google Scholar]
- 188.Mei HY; Cui M; Lemrow SM; et al. Discovery of Selective, Small-Molecule Inhibitors of RNA Complexes—II. Self-Splicing Group I Intron Ribozyme. Bioorg. Med. Chem 1997, 5, 1185–1195. [DOI] [PubMed] [Google Scholar]
- 189.Swayze EE; Jefferson EA; Sannes-Lowery KA; et al. SAR by MS: A Ligand Based Technique for Drug Lead Discovery against Structured RNA Targets. J. Med. Chem 2002, 45, 3816–3819. [DOI] [PubMed] [Google Scholar]
- 190.Seth PP; Miyaji A; Jefferson EA; et al. SAR by MS: Discovery of a New Class of RNA-Binding Small Molecules for the Hepatitis C Virus: Internal Ribosome Entry Site IIA Subdomain. J. Med. Chem 2005, 48, 7099–7102. [DOI] [PubMed] [Google Scholar]
- 191.Yu L; Oost TK; Schkeryantz JM; et al. Discovery of Aminoglycoside Mimetics by NMR-Based Screening of Escherichia coli A-Site RNA. J. Am. Chem. Soc 2003,125, 4444–4450. [DOI] [PubMed] [Google Scholar]
- 192.Childs-Disney JL; Wu M; Pushechnikov A; et al. A Small Molecule Microarray Platform to Select RNA Internal Loop-Ligand Interactions. ACS Chem. Biol 2007, 2, 745–754. [DOI] [PubMed] [Google Scholar]
- 193.Disney MD Targeting RNA with Small Molecules to Capture Opportunities at the Intersection of Chemistry, Biology, and Medicine. J. Am. Chem. Soc 2019, 141, 6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Haga CL; Velagapudi SP; Strivelli JR; et al. Small Molecule Inhibition of miR-544 Biogenesis Disrupts Adaptive Responses to Hypoxia by Modulating ATM-mTOR Signaling. ACS Chem. Biol 2015, 10, 2267–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Velagapudi SP; Costales MG; Vummidi BR; et al. Approved Anti-Cancer Drugs Target Oncogenic Non-Coding RNAs. Cell. Chem. Biol 2018, 25, 1086–1094.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Abulwerdi FA; Xu W; Ageeli AA; et al. Selective Small-Molecule Targeting of a Triple Helix Encoded by the Long Noncoding RNA, MALAT1. ACS Chem. Biol 2019,14, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lorenz DA; Vander Roest S; Larsen MJ; et al. Development and Implementation of an HTS-Compatible Assay for the Discovery of Selective Small-Molecule Ligands for Pre-microRNAs. SLAS Discov 2018, 23, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Angelbello AJ; Gonzalez AL; Rzuczek SG; et al. Development of Pharmacophore Models for Small Molecules Targeting RNA: Application to the RNA Repeat Expansion in Myotonic Dystrophy Type 1. Bioorg. Med. Chem. Lett 2016, 26, 5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Rizvi NF; Santa Maria JP; Nahvi A; et al. Targeting RNA with Small Molecules: Identification of Selective, RNA-Binding Small Molecules Occupying Drug-Like Chemical Space. SLAS Discov 2020, 25, 384–396. [DOI] [PubMed] [Google Scholar]
- 200.Santos R; Ursu O; Gaulton A; et al. A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug. Discov 2017, 16, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rask-Andersen M; Almen MS; Schioth HB Trends in the Exploitation of Novel Drug Targets. Nat. Rev. Drug Discov 2011, 10, 579–590. [DOI] [PubMed] [Google Scholar]
- 202.Scudellari M Protein-Slaying Drugs Could Be the Next Blockbuster Therapies. Nature 2019, 567, 298. [DOI] [PubMed] [Google Scholar]
- 203.Sztuba-Solinska J; Chavez-Calvillo G; Cline SE Unveiling the Druggable RNA Targets and Small Molecule Therapeutics. Bioorg. Med. Chem 2019, 27, 2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Makley LN; Gestwicki JE Expanding the Number of ‘Druggable’ Targets: Non-Enzymes and Protein-Protein Interactions. Chem. Biol. Drug. Des 2013, 81, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sahin U; Kariko K; Tureci O mRNA-Based Therapeutics—Developing a New Class of Drugs. Nat. Rev. Drug Discov 2014, 13, 759–780. [DOI] [PubMed] [Google Scholar]
- 206.Crooke ST; Witztum JL; Bennett CF; et al. RNA Targeted Therapeutics. Cell Metab 2018, 27, 714. [DOI] [PubMed] [Google Scholar]
- 207.Yoon S; Rossi JJ Therapeutic Potential of Small Activating RNAs (saRNAs) in Human Cancers. Curr. Pharm. Biotechnol 2018, 19, 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Setten RL; Lightfoot HL; Habib NA; et al. Development of MTL-CEBPA: Small Activating RNA Drug for Hepatocellular Carcinoma. Curr. Pharm. Biotechnol 2018,19, 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lieberman J Tapping the RNA World for Therapeutics. Nat. Struct. Mol. Biol 2018, 25, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zhang X; Hong R; Chen W; et al. The Role of Long Noncoding RNA in Major Human Disease. Bioorg. Chem 2019, 92, 103214. [DOI] [PubMed] [Google Scholar]
- 211.Kharel P; Balaratnam S; Beals N; et al. The Role of RNA G-Quadruplexes in Human Diseases and Therapeutic Strategies. Wiley Interdiscip. Rev. RNA 2020, 11, e1568. [DOI] [PubMed] [Google Scholar]
- 212.Kubota K; Funabashi M; Ogura Y Target Deconvolution from Phenotype-Based Drug Discovery by Using Chemical Proteomics Approaches. Biochim. Biophys. Acta. Proteins Proteom 2019, 1867, 22. [DOI] [PubMed] [Google Scholar]
- 213.Wagner BK The Resurgence of Phenotypic Screening in Drug Discovery and Development. Expert Opin. Drug Discov 2016, 11, 121. [DOI] [PubMed] [Google Scholar]
- 214.Hellman LM; Fried MG Electrophoretic Mobility Shift Assay (EMSA) for Detecting Protein-Nucleic Acid Interactions. Nat. Protoc 2007, 2, 1849–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Pedram Fatemi R; Salah-Uddin S; Modarresi F; et al. Screening for Small-Molecule Modulators of Long Noncoding RNA-Protein Interactions Using AlphaScreen. J. Biomol. Screen 2015, 20, 1132–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lorenz DA; Kaur T; Kerk SA; et al. Expansion of cat-ELCCA for the Discovery of Small Molecule Inhibitors of the pre-let-7-Lin28 RNA-Protein Interaction. ACS Med. Chem. Lett 2018, 9, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Martin S; Blankenship C; Rausch JW; et al. Using SHAPE-MaP to Probe Small Molecule-RNA Interactions. Methods 2019, 167, 105. [DOI] [PubMed] [Google Scholar]
- 218.Kwok CK; Marsico G; Sahakyan AB; et al. rG4-seq Reveals Widespread Formation of G-Quadruplex Structures in the Human Transcriptome. Nat. Methods 2016,13, 841. [DOI] [PubMed] [Google Scholar]
- 219.Halder K; Benzler M; Hartig JS Reporter Assays for Studying Quadruplex Nucleic Acids. Methods 2012, 57, 115–121. [DOI] [PubMed] [Google Scholar]