

Abstract
Genome-wide association studies (GWAS) have identified thousands of germline susceptibility loci associated with risk for cancer as well as a wide range of other traits and diseases. An interest of many investigators is identifying traits or diseases that share common susceptibility loci. We developed LDtrait (https://ldlink.nci.nih.gov/?tab=ldtrait) as an open access web tool for finding germline variation associated with multiple traits. LDtrait searches the NHGRI-EBI GWAS Catalog to identify susceptibility loci in linkage disequilibrium (LD) with a user-provided list of query variants. Options allow for modifying LD thresholds, calculating LD from a diverse set of reference populations, and downloading annotated variant lists. Results from example query searches highlight the utility of LDtrait in uncovering cross-trait associations for cancer risk and other traits. LDtrait accelerates etiologic understanding of cancer genetics by rapidly identifying genetic similarities with other traits or diseases.
Keywords: linkage disequilibrium, genome-wide association study, genetic susceptibility loci, LDtrait, LDlink, phenotype
Introduction
Understanding the contribution of germline genetic variation to the development of cancer has been an important aim of genetic research since releasing the first draft of the human genome(1). While genome-wide association studies (GWAS) have made considerable progress identifying germline variants associated with a vast range of studied cancers(2), the proportion of genetic variation explained by discovered susceptibility loci for most traits is low3. This supports the need for additional genetic association studies for uncovering additional relationships between genetic variants and cancer. As the list of identified genetic susceptibility loci continues to grow, it is increasingly important to better understand the biologic mechanisms by which these susceptibility variants confer risk. Improved understanding of mechanisms and pathways involved in carcinogenesis can be helpful in identifying individuals most likely to benefit from early interventions or focused treatments(3,4). However, most functional studies are resource intensive requiring large time and financial commitment to disentangle functional impacts of germline variants in a susceptibility locus.
A key complexity to mapping cancer-associated loci is linkage disequilibrium (LD). LD is the non-random distribution of allele frequencies at nearby loci in a defined population. Variants in high LD will have correlations between allele frequencies that are often inherited together and underlying haplotypes will not be observed at expected frequencies(5). LD facilitates GWAS by allowing for substantially fewer genomic variants to be directly genotyped, as tagging variants can be used to represent large numbers of correlated variants. However, LD complicates interpretation of GWAS susceptibility loci as dozens to hundreds of variants at a susceptibility locus have similar associations with cancer, but likely only a small subset of these have any functional effect related to the cancer of interest. Additionally, some loci are pleiotropic, impacting multiple traits or diseases, and may offer novel insights into carcinogenesis based on associations with other traits.
We developed LDtrait to help identify shared genetic architecture of cancer with various traits and diseases. LDtrait is an open source, publicly available web tool for searching for LD between a user defined list of input variants with a database of published GWAS susceptibility loci (https://www.ebi.ac.uk/gwas/home)(2). LDtrait harnesses population-specific LD metrics to identify variants in the input list and GWAS Catalog that are in high LD to generate evidence for pleiotropic effects of a locus that is evident across many traits or diseases. These cross-trait associations expand knowledge on cancer genotype-phenotype relationships and may provide useful clues for nominating important genes or pathways for functional investigation. LDtrait is part of the LDlink (https://ldlink.nci.nih.gov/)(6) suite of web-based modules that query LD based on 1000 Genomes Project population genetic data.
Materials & Methods
LDtrait (https://ldlink.nci.nih.gov/?tab=ldtrait) uses data from the NHGRI-EBI GWAS Catalog(2) to identify associations of a variant in linkage disequilibrium with other traits or diseases. To ensure LDtrait remains up to date, data from the GWAS Catalog is downloaded nightly from the EBI download site (https://www.ebi.ac.uk/gwas/api/search/downloads/alternative). Each entry is then parsed into JSON format and inserted into a non-relational, NoSQL MongoDB database (https://www.mongodb.com/). During the importing process each GWAS Catalog entry’s variant RefSeq (RS) number is queried in LDlink’s dbSNP MongoDB database (dbSNP build 151) to convert GWAS Catalog chromosomal coordinates (GRCh38) to LDlink chromosomal coordinates (GRCh37). If the variant’s RS number is not found, the entry from the GWAS Catalog is piped to an output file (https://ldlink.nci.nih.gov/tmp/ldtrait_error_snps.json) with an error message explaining why it failed to be inserted. A compound index is created on the GRCh37 chromosome and position which speeds up query performance when retrieving GWAS Catalog variants within a genomic window of interest. A timestamp of successful nightly downloads is also displayed on the website in the LDtrait module (e.g., “GWAS Catalog last updated on 3/25/2020, 01:09 PM (GMT-0400).”).
To query LDtrait, a list of variants is needed as input. Variant RS numbers or genomic coordinates (e.g., chr22:25875265, GRCh37) can either be entered one per line in the text entry box or uploaded as a file that contains one variant per line. A maximum of 50 variants are permitted for each LDtrait query. All input variants must match a bi-allelic variant in dbSNP build 151. A reference population must also be selected from the drop down menu to calculate LD metrics. At least one 1000 Genomes Project(7) sub-population is required, but more than one may be selected. Additionally, the user must select whether R2 or D′ is their LD metric of preference by using the R2 or D′ toggle as well as the respective threshold for returning results.
When a query is submitted LDtrait checks that all input variant details are valid, identifies the genomic location of each input variant and then defines a user-specified window around each query variant. These genomic regions are then queried and variants matching these coordinates are pulled from the GWAS Catalog database and tested for pairwise LD with the query variants using custom, previously-published Python scripts(6). LDtrait returns a list of queried variants in LD with a GWAS-associated variant reported in the GWAS Catalog. Each variant in the list is a clickable link that brings up a detailed table showing disease or trait-associated variants that are in LD with the query variant. The returned details table includes information on GWAS trait, genomic position, alleles, R2, D’, effect size (95% confidence interval) and P-value. External links lead to the variant pair in LDpair as well as to the corresponding entry in the GWAS Catalog. A download link is also available to download an annotated variant list of associated traits in tab-delimited format that lists all query variant RS numbers and their respective GWAS Catalog results.
Results
To demonstrate the utility of LDtrait for identifying pleiotropic relationships relevant for cancer risk, LDtrait was queried to investigate the associations of several variants known to be associated with different GWAS traits and diseases. A total of three variants were selected from the literature(8–10), the CEU 1000 Genomes Project(7) population was chosen as the reference population, and R2 was selected as the LD metric with the default threshold of 0.1 (Figure 1). The total LDtrait query took approximately 5 seconds.
Figure 1.
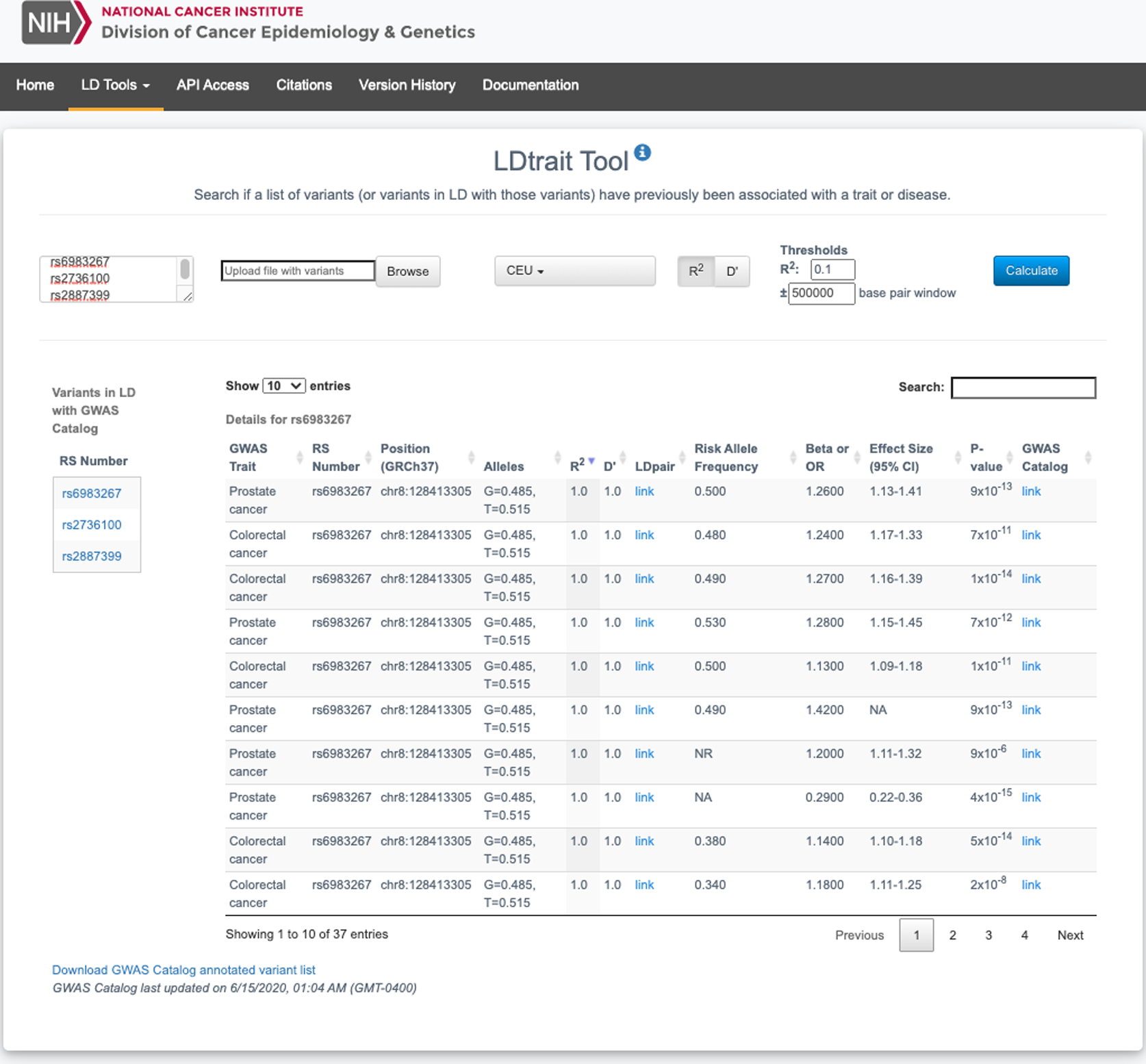
Screenshot demonstrating example LDtrait query and detailed table output.
The first queried variant was rs6983267(8), which is one of the first reported cancer susceptibility variants at the 8q24 locus. Our query returned 37 GWAS Catalog entries for variants in LD with rs6983267 (Supplemental Table 1). A top disease association returned by the query was prostate cancer(8,11), which is a good positive control as rs6983267 lies around the known prostate cancer 8q24 region. The other cancer association returned by the query was for colorectal cancer(12,13), suggesting pleiotropic cancer effects for this locus.
The next variant we queried was rs2736100(9), a germline variant on chromosome 5 near telomerase reverse transcriptase (TERT), to identify additional reported GWAS associations in LD at this TERT locus. TERT is a vital component for making the enzyme telomerase, which maintains telomere length at chromosomal ends and likely has pleiotropic relationships with cancer and other traits. Our LDtrait query for rs2736100 found 145 GWAS traits and diseases with associated variants in LD with rs2736100 (Supplemental Table 2). Unsurprisingly, a top hit for rs2736100 included telomere length(14), but we also identified pleiotropic associations with red blood cell counts(15) and several tumor types (e.g., glioma(16), lung(17), and testicular(18)).
Our final queried variant was rs2887399(10). This germline variant is associated with mosaic chromosome Y loss in men and is on chromosome 14 near T Cell Leukemia/Lymphoma 1A (TCL1A). Our query returned 8 GWAS traits associated with variants in LD with rs2887399 (Supplemental Table 3). As expected, the main trait found to be associated with rs2887399 was mosaic loss of chromosome Y(19,20). Another associated trait included neutrophil count(21) which is interesting in light of recent papers on mosaic Y loss indicating changes in blood cell counts, particularly neutrophil counts(20,22).
Discussion
LDtrait is a quick and easy-to-use open access tool for identifying pleiotropic effects of cancer susceptibility variants. By quickly calculating LD structure and aggregating results from the GWAS Catalog, LDtrait can aid cancer researchers in accelerating etiologic understanding of discovered cancer susceptibility loci by identifying other traits or diseases with similar genetic associations. As demonstrated by our example queries, LDtrait provides expected associations from other association studies on the same tumor as well as identifies interesting pleiotropic relationships with different traits or diseases. A few publicly available tools are similar to LDtrait in terms of functionality like DistiLD by Pallejà et al.(23) also allows for identifying potential GWAS studies associated with germline variants; however, there are several distinct differences between LDtrait and DistiLD. DistiLD for example codes diseases in ICD-10, and can query their database by SNPs, genes, or diseases. In comparison, LDtrait uses the more comprehensive 1000 Genomes Project reference for LD calculations and provides additional control over the reference population used to calculate LD or the R2/D’ threshold. LDtrait also has a more comprehensive graphical user interface rather than returning a basic text description of associations. For example, for the LDtrait query on rs6983267 as shown in the previous paragraph, DistiLD returned the following result: “Identification of Genetic Susceptibility Loci for Colorectal Tumors in a Genome-wide Meta-analysis.(PMID:23266556)”. This output does not include crucial information such as p value, risk allele or effect size that is included in standard LDtrait output. We also looked into GCTA-LD. While GCTA-LD calculates LD, it does not offer functionality for querying published associations between disease traits and single nucleotide variants. We have included LDtrait as a new module within the publicly available LDlink(6) suite of LD tools and anticipate this new analysis tool to be of great utility for identifying novel pleiotropic associations relevant for cancer and other diseases.
Supplementary Material
Significance:
The new GWAS search tool LDtrait will expedite discovery of shared genetic components underlying seemingly unrelated diseases and may offer novel insights into cancer research.
Acknowledgements
Support for LDtrait comes from the Division of Cancer Epidemiology and Genetics 2019 Informatic Tool Challenge as well as the Intramural Research Program of the National Cancer Institute.
Abbreviatoins
- GWAS
genome-wide association study
- LD
linkage disequilibrium
- RS
RefSeq
Footnotes
Application URL
Conflict of Interest Statement
The authors declare no relevant conflicts of interest
References
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. [DOI] [PubMed] [Google Scholar]
- 2.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet. 2017;101:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freedman ML, Monteiro ANA, Gayther SA, Coetzee GA, Risch A, Plass C, et al. Principles for the post-GWAS functional characterization of cancer risk loci. Nat Genet. 2011;43:513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ardlie KG, Kruglyak L, Seielstad M. Patterns of linkage disequilibrium in the human genome. Nat Rev Genet. 2002;3:299–309. [DOI] [PubMed] [Google Scholar]
- 6.Machiela MJ, Chanock SJ. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinforma Oxf Engl. 2015;31:3555–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, et al. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–9. [DOI] [PubMed] [Google Scholar]
- 9.Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. 2013;45:422–7, 427e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou W, Machiela MJ, Freedman ND, Rothman N, Malats N, Dagnall C, et al. Mosaic loss of chromosome Y is associated with common variation near TCL1A. Nat Genet. 2016;48:563–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–5. [DOI] [PubMed] [Google Scholar]
- 12.Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007;39:984–8. [DOI] [PubMed] [Google Scholar]
- 13.Tanskanen T, van den Berg L, Välimäki N, Aavikko M, Ness-Jensen E, Hveem K, et al. Genome-wide association study and meta-analysis in Northern European populations replicate multiple colorectal cancer risk loci. Int J Cancer. 2018;142:540–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Cao L, Li Z, Zhou D, Liu W, Shen Q, et al. A genome-wide association study identifies a locus on TERT for mean telomere length in Han Chinese. PloS One. 2014;9:e85043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kamatani Y, Matsuda K, Okada Y, Kubo M, Hosono N, Daigo Y, et al. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat Genet. 2010;42:210–5. [DOI] [PubMed] [Google Scholar]
- 16.Walsh KM, Codd V, Smirnov IV, Rice T, Decker PA, Hansen HM, et al. Variants near TERT and TERC influencing telomere length are associated with high-grade glioma risk. Nat Genet. 2014;46:731–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Z, Wu C, Shi Y, Guo H, Zhao X, Yin Z, et al. A genome-wide association study identifies two new lung cancer susceptibility loci at 13q12.12 and 22q12.2 in Han Chinese. Nat Genet. 2011;43:792–6. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, McGlynn KA, Rajpert-De Meyts E, Bishop DT, Chung CC, Dalgaard MD, et al. Meta-analysis of five genome-wide association studies identifies multiple new loci associated with testicular germ cell tumor. Nat Genet. 2017;49:1141–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wright DJ, Day FR, Kerrison ND, Zink F, Cardona A, Sulem P, et al. Genetic variants associated with mosaic Y chromosome loss highlight cell cycle genes and overlap with cancer susceptibility. Nat Genet. 2017;49:674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Terao C, Momozawa Y, Ishigaki K, Kawakami E, Akiyama M, Loh P-R, et al. GWAS of mosaic loss of chromosome Y highlights genetic effects on blood cell differentiation. Nat Commun. 2019;10:4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramsuran V, Kulkarni H, He W, Mlisana K, Wright EJ, Werner L, et al. Duffy-null-associated low neutrophil counts influence HIV-1 susceptibility in high-risk South African black women. Clin Infect Dis Off Publ Infect Dis Soc Am. 2011;52:1248–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin S-H, Loftfield E, Sampson JN, Zhou W, Yeager M, Freedman ND, et al. Mosaic chromosome Y loss is associated with alterations in blood cell counts in UK Biobank men. Sci Rep. 2020;10:3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pallejà A, Horn H, Eliasson S, Jensen LJ. DistiLD Database: diseases and traits in linkage disequilibrium blocks. Nucleic Acids Res. 2012;40:D1036–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.