

Abstract
Type 2 diabetes, which is mainly linked to obesity, is associated with increased incidence of liver cancer. We have previously found that in various models of obesity/diabetes, hyperinsulinemia maintains heightened hepatic expression of Cyclin D1, suggesting a plausible mechanism linking diabetes and liver cancer progression. Here we show that Cyclin D1 is greatly elevated in human livers with diabetes and is among the most significantly upregulated genes in obese/diabetic liver tumors. Liver-specific Cyclin D1 deficiency protected obese/diabetic mice against hepatic tumorigenesis, whereas lean/non-diabetic mice developed tumors irrespective of Cyclin D1 status. Cyclin D1 dependency positively correlated with liver cancer sensitivity to palbociclib, an FDA-approved CDK4 inhibitor, which was effective in treating orthotopic liver tumors under obese/diabetic conditions. The anti-diabetic drug metformin suppressed insulin-induced hepatic Cyclin D1 expression and protected against obese/diabetic hepatocarcinogenesis. These results indicate that the Cyclin D1-CDK4 complex represents a potential selective therapeutic vulnerability for liver tumors in obese/diabetic patients.
Introduction
Type 2 diabetes, particularly associated with obesity, is a current epidemic in the US and worldwide. Strong epidemiological data indicate that type 2 diabetes is tightly associated with increased incidence of different cancers, including liver malignancies (1). Furthermore, retrospective studies show that diabetes patients have worse prognosis than those without diabetes, suggesting the diabetic state might cause a more aggressive cancer phenotype and/or a poorer response to treatment. Insulin resistance associated with hyperinsulinemia might contribute to the increased risk of cancer in obesity/diabetes (2). Interestingly, diabetes patients treated with insulin or drugs that increase insulin secretion exhibit higher risks of cancer and worse outcomes compared to those treated with insulin-sensitizing agents such as metformin or thiazolidinediones (3). However, how the diabetic states and treatments contribute to a higher cancer incidence and/or worse patient survival is not completely understood.
D-type cyclins and cyclin-dependent kinases (CDK)-4 and 6 are critical regulators of the cell cycle G0-S transition necessary for cell division (4). We have previously identified that insulin fluctuations during feeding/fasting cycles modulates hepatic Cyclin D1 expression and CDK4 activity, leading to fine control of glucose metabolism through the GCN5-PGC1α axis. Notably, this signaling pathway remains constitutively hyperactive in diabetic and hyperinsulinemic states (5). Interestingly, the CDK/Rb pathway is associated with hepatocellular carcinoma (HCC), the most prevalent form of liver cancer. For example, germline mutations in Rb1 increase the susceptibility of malignancies, particularly osteosarcoma and HCC. In addition, alterations of the p16/Cyclin D1/Rb pathway are frequent in HCC. Furthermore, the risk for liver cancer is highest in individuals with diabetes (1). Thus, it is conceivable that obesity/diabetes and hyperinsulinemia increase the incidence of HCC through hepatic Cyclin D1/CDK4 hyperactivation.
In this study, we investigated whether obesity/diabetes-induced overexpression of Cyclin D1 in liver promote HCC. In dietary and genetic mouse models of diabetes, we found that hepatic Cyclin D1 was required for HCC initiation and progression. However, Cyclin D1 was dispensable for HCC formation under lean/non-diabetic conditions. The dependency on Cyclin D1 predicted the sensitivity of liver cancer to the CDK4 inhibitor palbociclib, which was able to effectively blunt the progression of experimental liver tumors in obese/diabetic conditions. Interestingly, the anti-diabetic drug metformin, functions as an insulin sensitizer, suppressed insulin-induced hepatic Cyclin D1 overexpression and protected against HCC in obese/diabetic mice. Our results indicate that Cyclin D1 represents a dependency and therapeutic vulnerability in HCC formed in obese/diabetic states.
Materials and Methods
Animal Procedures
All animal studies were performed according to protocols approved by Beth Israel Deaconess Medical Center’s Animal Care and Use Committee. All mice were housed with regular chow (12%-fat, 23%-protein, 65%-carbohydrates) or high-fat diet (HFD, 59%-fat, 15%-protein, 26%-carbohydrates; LabDiet) under a 12 hr light/12 hr dark cycle at 22°C. Liver-specific Cyclin D1 KO mice (LKO) were generated by crossing the Ccnd1flox/flox alleles with the albumin promoter-driven cre mice. In Figure 2 HCC model, DEN (25 mg/kg) was injected intraperitoneally (i.p.) into 14-day-old mice. After 4 weeks, mice were subjected to either chow- or HFD-feeding until sacrificed (6). In Figure 4 HCC model, 5-week-old male Leprdb mice from Jackson laboratories were injected with DEN (i.p., 80 mg/kg), followed by 50 mg/kg metformin treated through drinking water starting at 9 weeks of age, until sacrificed. The concentration of metformin in water was calculated basing on average daily water intake 20 mL and adjusted to body weight of mice over time. Glycemia was measured by tail bleed with a glucometer (OneTouch). Mice were examined for tumors at 36-week of age, and tumor load was quantified by counting the macro-tumor on surface of each liver lobe and presented as “average tumor no per animal”. For the orthotopic liver tumor model, Dt81Hepa1-6 murine hepatoma cells were injected intrasplenically in 0.2 mL PBS containing 0.2% albumin (7) into 11-week-old male congenic C57B/L6 or Leprdb/db mice. After ten days recovery/incubation, mice were treated daily with either vehicle or palbociclib (Selleck Chemical) via oral gavage (5) at indicated dose for six days. Tumor progression was analyzed by weighing the tumor-bearing livers and subtracting the average weight of five age-and-sex matched non-tumor Leprdb/db livers, and then further quantified by areas of tumor nodules in liver sections under microscope, followed by normalization to the whole area of the tissue captured. Tumor and non-tumor tissue was collected, rapidly frozen or fixed for biochemical, histological and immunochemical analyses.
Figure 2. Cyclin D1 deficiency protects mice from carcinogen-induced HCC formation in the context of obesity/type 2 diabetes.
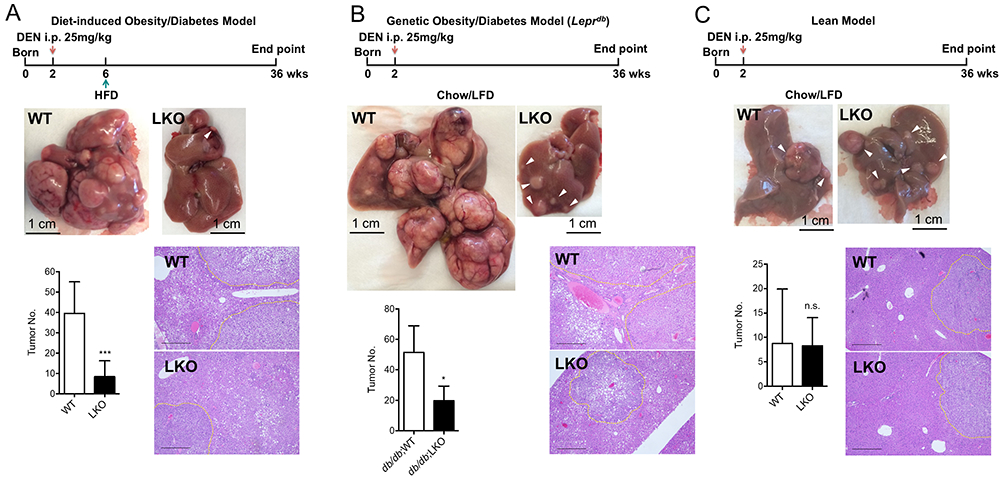
A. Deletion of Cyclin D1 in hepatocytes protects mice from liver cancer under diet-induced obese/type 2 diabetic conditions. In the diet-induced obesity/diabetes model, DEN-injected mice were subjected to HFD for the whole span of the experiments. Representative liver phenotype at the endpoint, tumor burden and liver histology between Cyclin D1 WT and liver-specific KO (LKO) mice were shown (WT, n=12; LKO, n=19). B. Deletion of Cyclin D1 in hepatocytes protects mice from liver cancer under genetic obese/type 2 diabetic conditions. The Leprdb alleles were introduced to genetically induce obesity/diabetes in DEN-injected mice. Representative liver phenotype at the endpoint, tumor burden and liver histology between Cyclin D1 WT and LKO mice were shown (WT, n=5; LKO, n=7). C. Hepatic Cyclin D1 is dispensable for liver cancer formation in lean condition. Representative liver phenotype at the endpoint, tumor burden and liver histology between Cyclin D1 WT and LKO mice kept on chow/LFD were shown (WT, n=17; LKO, n=11). Tumor burden was presented as average surface tumor number per mouse. Dash lines outlined tumor nodules in each H&E image. Scale bar represents 500 μm.
Figure 4. Metformin reduces Cyclin D1 expression and prevented HCC initiation in obese/diabetic mice.
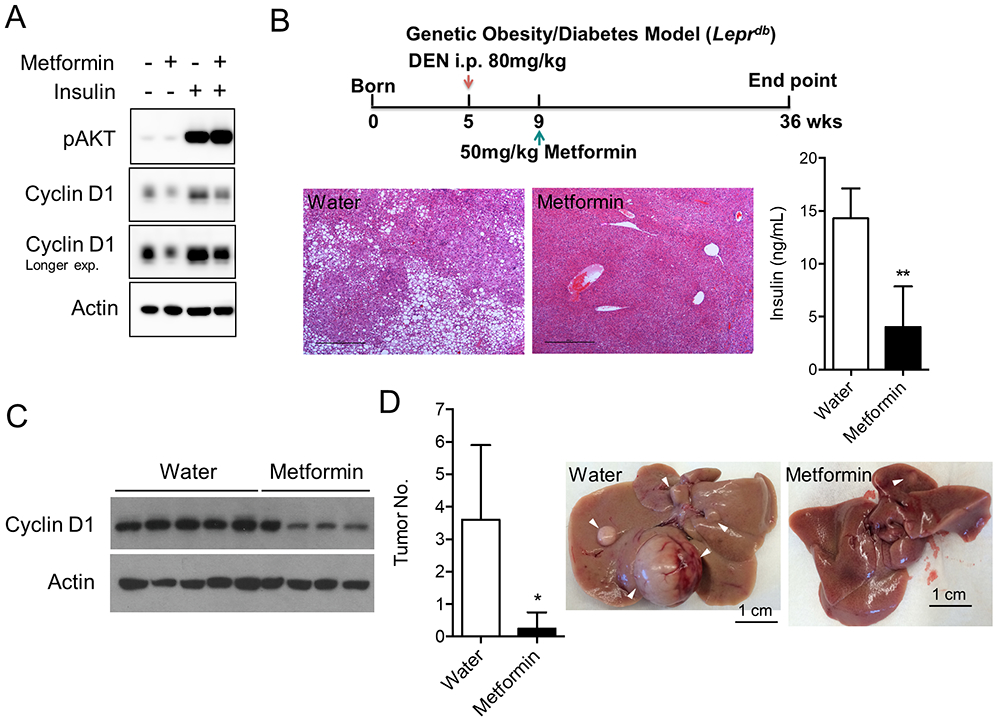
A. Metformin prevents insulin-induced Cyclin D1 protein levels in hepatocytes. Overnight serum starved AML12 murine hepatocytes were treated with control, metformin (10 mM), insulin (200 nM) or both for 4 h, followed by immunoblotting to detect Cyclin D1 expression. B. Design and metabolic phenotypes of the metformin treatment experiment. Metformin was given at 50 mg/kg for 27 weeks. Endpoint blood insulin level and liver histology of the db/db mice were shown. Scale bar represents 500 μm. C. Metformin treatment prevents the induction of Cyclin D1 in type 2 diabetic liver as detected by immunoblotting. D. Metformin treatment protects mice from DEN-induced liver cancer in diabetic mice (water, n=5; metformin, n=4).
Tissue Culture
AML12 murine hepatocyte cell line was obtained from ATCC, and maintained in DMEM/F12 (Sigma-Aldrich) with 10% FBS, 10 μg/ml insulin, 5.5 μg/ml transferrin, 5 ng/ml selenium, 40 ng/ml dexamethasone, 100 U/ml penicillin, and 100 mg/ml streptomycin. Dt81Hepa1-6 murine hepatoma cell line obtaining from Dr. Marc Bilodeau Laboratory was derived in vivo from the parental Hepa1-6 line (7), and maintained in DMEM (Sigma-Aldrich) with 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin. Their authentication was confirmed by in-house PCR of their respective marker genes: mouse albumin and human TGFα for AML12, mouse albumin and alpha-fetoprotein for Dt81Hepa1-6, upon their arrival in our laboratory. Mycoplasma contamination was tested with the PCR Mycoplasma Detection Kit (Lonza) once every two months. Both cells were cultured in a humidified incubator at 37°C with 5% CO2, and used for less than 10 passages upon thawing from liquid nitrogen.
Immunoblotting, Quantitative real time-PCR and RNA sequencing
Tissues were snap frozen using liquid nitrogen before gridding into powder on dry ice. For immunoblotting, pulverized tissues were lysed in RIPA buffer and blotted with the following antibodies: Cyclin D1 (Cell Signaling), actin (Cell Signaling), pJNK (Cell Signaling), Lamin B2 (Abcam), pSTAT3 (Cell Signaling), histone H3 (Abcam), and pAKT (Cell Signaling). Total RNA was isolated with Trizol (Invitrogen) by Direct-zol RNA MiniPrep kit (Zymo Research), and qPCR was carried out using SYBR Green PCR Master Mix (Bio-Rad). Experimental Ct values were normalized to 36B4 or actin, and relative mRNA expression was calculated using the DDCt method. RNASeq Library was prepared with NEBNext Ultra RNA kit, followed by sequencing with Single Read 75bp High Throughput Flow Cell at the DFCI Center for Cancer Computational Biology.
Serum and Liver Biochemistry Measurements
Serum samples were gathered from blood collected by cardiac puncture. Insulin was measured with Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem). Liver triglycerides were detected with kit from Sigma according to manufacturer’s instruction.
Histology
Rodent tissue samples were fixed in 10% buffered formalin overnight and stored in 70% ethanol prior to paraffin embedding, sectioning and hematoxylin/eosin staining by Rodent Histopathology Core, Harvard Medical School (HMS). Study involving human specimens was conducted on a protocol approved by the Partners Institutional Review Board, and followed ethical guidelines in accordance with Declaration of Helsinki, with written informed consent obtained from patients. Cyclin D1 IHC was performed on the de-ID slides by the Specialized Histopathology Core at HMS. For each sample, a pathologist blindly scored both extent and intensity basing on the following criteria: extent: 0 represents negative, 1 represents <5%, 2 is 5 to 25%, 3 is 25 to 50% and 4 is more than 50%; intensity: negative is scored 0, week is 1, moderate is 2 and strong is 3. Finally, the extent score and intensity score of the same sample were added up to get the final quantification.
Correlation analysis
Correlation between Cyclin D1 and CDK4/6 inhibitor palbociclib was analyzed based on DepMap portal (https://depmap.org/portal). Dependency on CCND1 gene was determined by either CRISPR gene editing from the Broad’s Project Achilles (8) DepMap Public 19Q3 dataset or RNAi from the combined Broad, Novartis, Marcotte datasets. Palbociclib sensitivity was presented as log2 fold changes obtained from Broad’s PRISM Repurposing Primary Screen(9), or IC50 or AUC sensitivity score from Sanger’s Genomics of Drug Sensitivity in Cancer (GDSC) dataset (10-13). Only human liver cancer cell lines were included in the analyses, with correlation computed by Pearson correlation coefficients.
Statistics
All statistics were performed with GraphPad Prism. In general, for two experimental comparisons, a two-tailed unpaired Student’s t-test was used; for multiple comparisons, one-way ANOVA was applied, if not otherwise indicated. When cells were used for experiments, three replicates per treatment were chosen as an initial sample size. All n values defined in the legends refer to biological replicates unless otherwise indicated. Statistical significance is represented by asterisks corresponding to *P < 0.05, **P < 0.01, and ***P < 0.001.
Results
Cyclin D1 is aberrantly upregulated in liver and HCC with type 2 diabetes
We have previously identified that changes in hepatic Cyclin D1 expression and CDK4 activity modulate gluconeogenesis (5). Here, we have extended these observations to obesity/diabetes induced by long-term high-fat diet (HFD) feeding, finding that it strongly increased hepatic Cyclin D1 expression at both protein (Fig. 1A) and mRNA levels (Fig. S1A). Similar effects (Fig. 1A, Fig. S1A) were also observed in Leptin receptor mutant obese mice (Leprdb, referred to as db/db thereafter), which develop spontaneous diabetes with severe hyperinsulinemia. Consistently, Cyclin D1 is significantly higher in human liver specimens from patients with diabetes when compared to healthy liver (Fig. 1B). Next, we compared gene expression profiles between liver tumors formed in lean or obese/diabetic conditions. Of interest, only a short list of genes was significantly upregulated in HCC with obesity/diabetes (Fig. S1B), and the most significantly upregulated one was Cyclin D1, which was further validated at both mRNA (Fig. 1C) and protein levels (Fig. 1D, Fig. S1C). Collectively, these data show that the cell cycle regulator Cyclin D1 is upregulated in both liver and HCC with obesity/diabetes, suggesting this oncoprotein might promote liver cancer under this metabolic condition.
Figure 1. Cyclin D1 is aberrantly upregulated in type 2 diabetic liver and HCC.
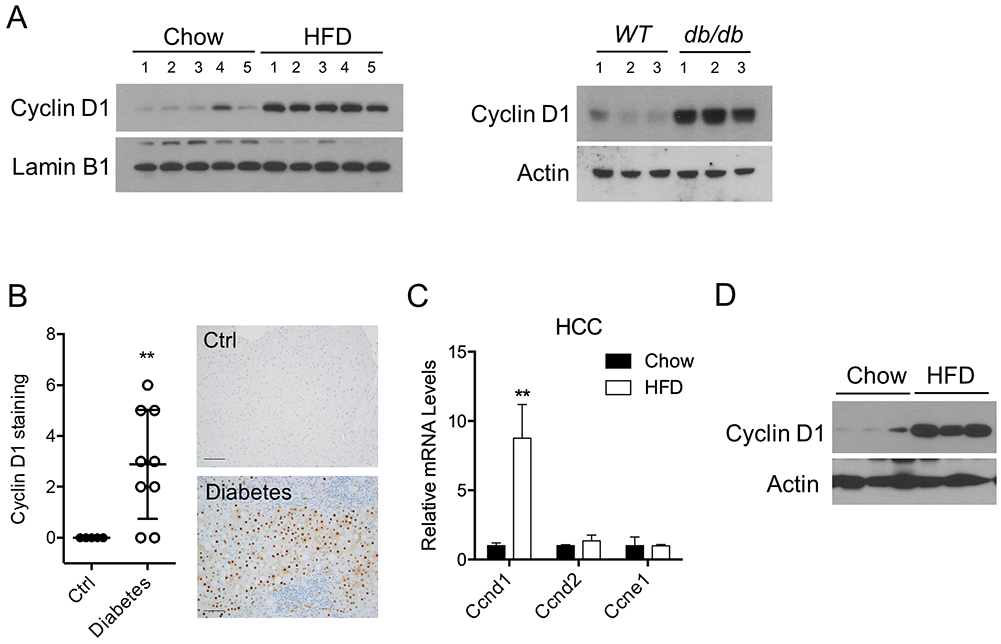
A. Cyclin D1 protein expression is significantly increased in liver of dietary and genetic obese/type 2 diabetic mice. Mice were kept with either chow/low-fat diets (Chow) or high-fat diets (HFD) for nine months. Leprdb mice, referred as db/db for short, were used as a genetic model of obesity/diabetes. B. Cyclin D1 expression is elevated in clinical liver specimens from individuals with diabetes, as determined by immunohistochemistry (IHC). Physiologically normal liver samples were used as control. Representative images were shown for the staining pattern and intensity. Scale bar represents 100 μm. C-D. Expression levels of Cyclin D1 are significantly enhanced in obese/diabetic HCC, as determined by qPCR (C) and immunoblotting (D). Liver cancers were derived in mice administered with chemical carcinogen diethylnitrosomine (DEN) at postnatal day 14, followed by either chow or HFD feeding for 30 weeks.
Cyclin D1 deficiency protects mice from carcinogen-induced HCC formation and development only in the context of type 2 diabetes
Based on our results that Cyclin D1 levels are strongly increased in obese/diabetic liver and tumors, we hypothesized that Cyclin D1 overexpression might mediate the liver cancer-promoting effects of obesity/diabetes. To test this, we employed a murine HCC model induced by the hepatic carcinogen diethylnitrosamine (DEN) (6) in combination with dietary and genetic models of diabetes, in which wild-type (WT) and liver-specific Cyclin D1 KO (LKO) mice were fed with HFD or crossed to db/db background. Strikingly, while obese/diabetic mice developed large amounts of HCC as expected, Cyclin D1 deficiency strongly protected against liver tumors (Fig. 2A, B). To the contrary, within the same time frame, lean non-diabetic mice, regardless of the Cyclin D1 status, developed HCC (Fig. 2C), albeit smaller in both size and quantity when compared to their obese/diabetic counterparts (6), indicating that Cyclin D1 is selectively necessary for HCC in obese/diabetic conditions.
Interestingly, Cyclin D1 deletion in hepatocytes did not substantially affect the induction nor progression of diabetes, as reflected by comparable body weight gain (Fig. S2A), hepatic lipid accumulation (Fig. S2B-D), as well as blood glucose and insulin levels (Fig. S2C, D) between WT and LKO mice over the course of 9-month HFD feeding. Similar observations were found in the DEN-treated mice (Fig. S3A-D), ruling out the possibility that loss of Cyclin D1 prevented tumor formation through reversing the diabetic phenotypes. Similarly, hepatic Cyclin D1 had marginal effect on the inflammatory responses to obesity/diabetes (Fig. S3E-F), suggesting Cyclin D1 deletion protected against HCC independently of hepatic inflammation. Together, these results indicate that chronic elevated levels of Cyclin D1 in obese/diabetic mice are necessary for carcinogen-induced HCC development. In contrast, the normal levels of Cyclin D1 protein that fluctuate during fed/fasting cycles in lean mice do not contribute to liver cancer.
Cyclin D1 correlates with sensitivity to CDK4 inhibition in obese/diabetic liver cancer.
Cyclin D1 is a surrogate partner of CDK4 kinase, target of the FDA-approved abemaciclib, palbociclib and ribociclib (14). The selective requirement of Cyclin D1 prompted us to determine whether CDK4 inhibition would be effective for treating liver tumors with obesity/diabetes. We searched publicly available datasets and found that the dependency on Cyclin D1 positively correlates with sensitivity to the CDK4 inhibitor palbociclib across a variety of human liver cancer cell lines (Fig. 3A, Fig. S4A-D). We therefore examined the efficacy of palbociclib on liver cancer in vivo. Consistent with the genetic deletion and correlation data, pharmacological inhibition of CDK4 significantly compromised the progression of liver cancer in obese/diabetic mice (Fig. 3B). Considering the higher level of Cyclin D1 in obese/diabetic HCC (Fig. 1G), we predicted CDK4 inhibition would be more effective in this metabolic subset of liver cancer. To address this, we administered palbociclib to tumor-bearing lean and obese/diabetic mice. Despite the difference in basal tumor formation reflecting the tumor promoting effects of obesity/diabetes, liver cancer formed in the obese/diabetic background was more vulnerable to CDK4 inactivation (Fig. 3C). Taken together, these results indicate that Cyclin D1 dependency in liver cancer predicts susceptibility to CDK4 inhibition, especially in the context of obesity and diabetes.
Figure 3. Cyclin D1 correlates with sensitivity to CDK4 inhibition in obese/diabetic liver cancer.
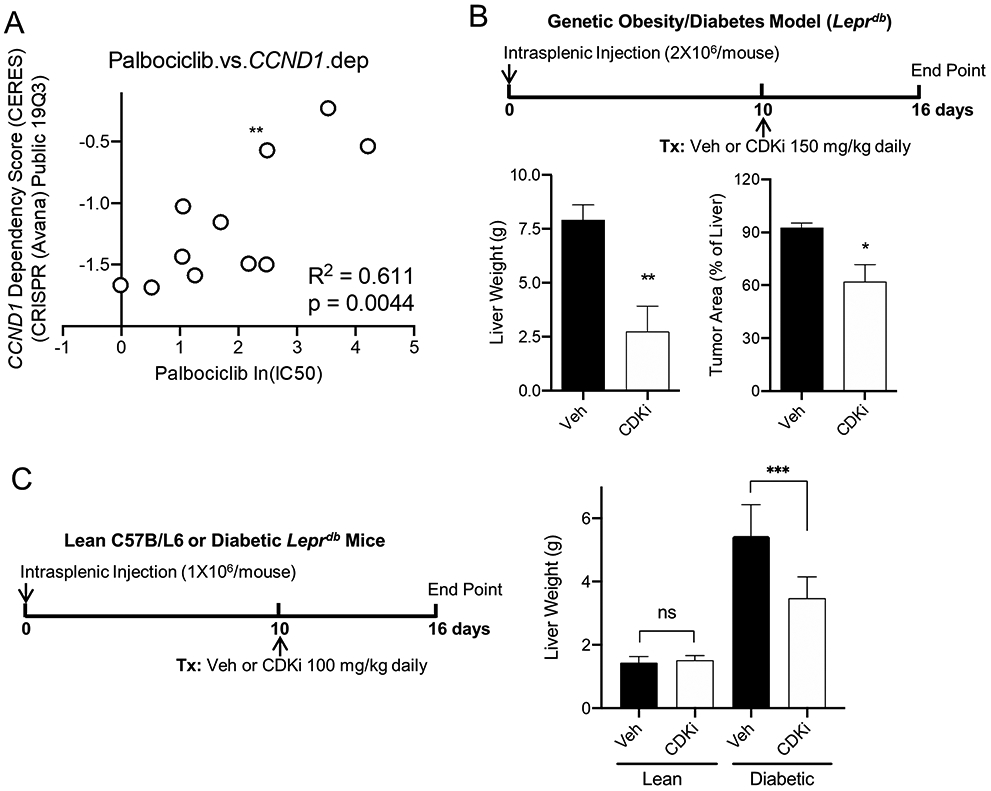
A. Dependency on CCND1 gene correlates positively with sensitivity to CDK4 inhibitor palbociclib across a panel of human liver cancer cell lines. Dependency on CCND1 gene was determined by CRISPR gene editing from the Broad’s Project Achilles DepMap Public 19Q3 dataset. Palbociclib sensitivity was presented as IC50 from Sanger’s Genomics of Drug Sensitivity in Cancer dataset. B. CDK4 inhibitor palbociclib is effective in suppressing the progression of obese/diabetic liver tumors (n=5). Diagram depicts the protocol for orthotopic liver cancer and treatment model. Murine hepatoma was established in the liver of db/db mice via intrasplenic injection. Upon tumor established at day 10 post injection, daily treatment was given via oral gavage for six days, followed by tumor assessment. Tumor loads were measured by subtracting the weight of normal liver from the weights of tumor-bearing livers, or by quantifying the microscopic cancerous areas of the H&E slides. C. Liver cancers developed in obese/diabetic mice are more vulnerable to CDK4 inhibitor (n=5). Upon hepatoma development in lean or obese/diabetic mice, treatments with either vehicle (Veh) or palbociclib were given at a relatively lower dose, followed by tumor assessment.
The anti-diabetic drug metformin reduced Cyclin D1 expression and prevented HCC initiation in obese/diabetic mice
That obesity/diabetes-associated hyperinsulinemia sustains Cyclin D1 expression contributing to liver cancer suggests anti-diabetic drugs that function as insulin sensitizers and decrease hyperinsulinemia, such as metformin, might reduce liver cancer risk. Importantly, metformin treatment in hepatocytes blocked Cyclin D1 induction by high insulin (Fig. 4A). It has been reported that DEN, given at the adult stage even at higher doses, has minimal effect on driving liver cancer, unless in combination with a second boost such as phenobarbital or obesity/diabetes (10). Accordingly, we decided to use the carcinogen at a later age in obese/diabetic mice, followed by continuous treatment with metformin (Fig. 4B). In addition to reduced blood insulin level and hepatic fat deposition (Fig. 4B), metformin largely diminished Cyclin D1 expression in the liver (Fig. 4C). Consistent with liver Cyclin D1 deficiency (Fig 2), metformin treated mice with reduced Cyclin D1 levels were protected against liver tumor (Fig 4D). Taken together, these results indicate that the insulin sensitizing anti-diabetic drug metformin lowers Cyclin D1 expression in obese/diabetic liver and impedes subsequent hepatocarcinogenesis.
Discussion
Despite obesity/diabetes has been considered an important risk factor for HCC, the underlying mechanisms remains elusive. Obesity/diabetes-associated parameters, including hyperinsulinemia, hyperglycemia, hyperlipidemia, insulin resistance, inflammation and gut microbiota are likely to play a role; however, little attention has been given to tumor intrinsic dependencies that occur specifically in obesity/diabetes but not in tumors formed in lean/non-diabetic patients. HCC is the most common primary liver cancer whose incidence and prognosis are prominently affected by obesity/diabetes. A number of studies, including large-scale GWAS and whole-exome sequencing, have revealed a collection of genes frequently altered in HCC. Among these identified genes, Cyclin D1 is of particular interest because Cyclin D1 levels have been reported to be upregulated in certain HCC cohorts (15,16), while no change or even reduced in others (17,18). However, the possible association of these cohorts to obesity/diabetes is unknown. Our studies indicate that obesity/diabetes represents a potential disease factor that establishes elevated Cyclin D1 expression in these HCC subsets. We also show the specific Cyclin D1 dependency of HCC formation and progression under obesity/diabetes conditions, providing potential basis for patient stratification and therapy.
The specific requirement of Cyclin D1 for HCC in obesity/diabetes suggests that these HCC subsets might be selectively vulnerable to pharmacological inhibition of CDK4/6, surrogate partners of Cyclin D1. CDK4/6 inhibitors such as palbociclib have been demonstrated to restrict tumor growth in preclinical models of HCC, however have been mainly associated with RB1 mutation status (14). Notably, our studies show that, dependency on Cyclin D1 expression predicts palbociclib sensitivity, especially in obesity/diabetes. Additionally, obese/diabetic liver is characterized by depletion of certain T lymphocyte subpopulation (19), echoing the fact that Cyclin D1-CDK4 can stabilize PD-L1 to create an immunosuppressive microenvironment (20); accordingly, targeting CDK4 in the diabetic setting can directly suppress cancer cell proliferation, while simultaneously might compromise the PD-L1-mediated immunosuppression, providing extra therapeutic benefits by unleashing the antitumor immunity. Therefore, further testing the efficacy of CDK4/6 inhibition in combination with immunotherapeutic agents might offer an additional therapeutic option for HCC patients with diabetes.
An important clinical implication of these studies is the choice of therapy for patients with diabetes. Based on the fact that hyperinsulinemia is a major risk factor linking obesity/diabetes and liver cancer (2,3), classes of anti-diabetic therapies that function as insulin sensitizers and decrease hyperinsulinemia would be likely to reduce liver cancer. Along these lines we show here that the insulin sensitizing anti-diabetic drug metformin corrects hyperinsulinemia, suppresses hepatic Cyclin D1 overexpression and protects against liver cancer in obese/diabetic mice. Metformin-mediated Cyclin D1 reduction is independent of the canonical AKT-GSK3β pathway, suggests metformin has direct anti-tumorigenic effects via suppressing Cyclin D1 accumulation, underscoring others’ reports using metformin to treat cancers either as a single agent or in combination with chemotherapy (3). Accordingly, metformin will have a double hit protecting obese/diabetic liver cancer by correcting diabetes, and by blocking insulin-induced hepatic Cyclin D1 levels.
In conclusion, obesity/diabetes causes hepatic Cyclin D1 overexpression that selectively promotes liver cancer in the context of these metabolic diseases. Thus, the druggable Cyclin D1-CDK4 kinase complex represents a tangible vulnerability that has therapeutic implications in obese/diabetic liver cancer patients.
Supplementary Material
Significance.
Obesity/diabetes-associated liver tumors are specifically vulnerable to Cyclin D1 deficiency and CDK4 inhibition, suggesting that the obese/diabetic environment confers cancer-selective dependencies that can be therapeutically exploited.
Acknowledgements
K.S. was partially funded by a post-doctoral fellowship from the Charles A. King Program. C.D.J.T. was partially funded by a post-doctoral fellowship from the American Diabetes Association (ADA, 1-16-PDF-111). Marc Bilodeau holds the Novartis/Canadian Liver Foundation Hepatology Research Chair at the Université de Montréal. P.S. was funded by NIH (R01 CA202634). P.P. was funded by the ADA (7-12-MN-68), DFCI Claudia Adams Barr Award, and NIH (R01 CA181217, NIDDK DK089883 and DK081418).
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- 1.Shikata K, Ninomiya T, Kiyohara Y. Diabetes mellitus and cancer risk: review of the epidemiological evidence. Cancer science 2013;104:9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Satija A, Spiegelman D, Giovannucci E, Hu FB. Type 2 diabetes and risk of cancer. BMJ 2015;350:g7707. [DOI] [PubMed] [Google Scholar]
- 3.Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and cancer: a consensus report. Diabetes care 2010;33:1674–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 2011;11:558–72 [DOI] [PubMed] [Google Scholar]
- 5.Lee Y, Dominy JE, Choi YJ, Jurczak M, Tolliday N, Camporez JP, et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature 2014;510:547–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010;140:197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lacoste B, Raymond VA, Cassim S, Lapierre P, Bilodeau M. Highly tumorigenic hepatocellular carcinoma cell line with cancer stem cell-like properties. PLoS One 2017;12:e0171215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a Cancer Dependency Map. Cell 2017;170:564–76 e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu C, Mannan AM, Yvone GM, Ross KN, Zhang YL, Marton MA, et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat Biotechnol 2016;34:419–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2013;41:D955–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. Addendum: The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2019;565:E5–E6 [DOI] [PubMed] [Google Scholar]
- 13.Cancer Cell Line Encyclopedia C, Genomics of Drug Sensitivity in Cancer C. Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015;528:84–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bollard J, Miguela V, Ruiz de Galarreta M, Venkatesh A, Bian CB, Roberto MP, et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2017;66:1286–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishida N, Fukuda Y, Komeda T, Kita R, Sando T, Furukawa M, et al. Amplification and overexpression of the cyclin D1 gene in aggressive human hepatocellular carcinoma. Cancer Res 1994;54:3107–10 [PubMed] [Google Scholar]
- 16.Joo M, Kang YK, Kim MR, Lee HK, Jang JJ. Cyclin D1 overexpression in hepatocellular carcinoma. Liver 2001;21:89–95 [DOI] [PubMed] [Google Scholar]
- 17.Peng SY, Chou SP, Hsu HC. Association of downregulation of cyclin D1 and of overexpression of cyclin E with p53 mutation, high tumor grade and poor prognosis in hepatocellular carcinoma. Journal of hepatology 1998;29:281–9 [DOI] [PubMed] [Google Scholar]
- 18.Jung YJ, Lee KH, Choi DW, Han CJ, Jeong SH, Kim KC, et al. Reciprocal expressions of cyclin E and cyclin D1 in hepatocellular carcinoma. Cancer letters 2001;168:57–63 [DOI] [PubMed] [Google Scholar]
- 19.Ma C, Kesarwala AH, Eggert T, Medina-Echeverz J, Kleiner DE, Jin P, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016;531:253–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018;553:91–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.