

Abstract
Myeloid cell leukemia sequence 1 (MCL-1) is an antiapoptotic protein that plays a key role in promoting cell survival in multiple myeloma (MM), acute myeloid leukemia (AML), and non-Hodgkin lymphoma (NHL). Overexpression of MCL-1 is associated with treatment resistance and poor prognosis; thus, MCL-1 inhibitors are rational therapeutic options for malignancies depending on MCL-1. Several MCL-1 inhibitors have entered clinical trials, including AZD5991, S64315, AMG 176, and AMG 397. A key area of investigation is whether MCL-1 inhibitors will complement the activity of BCL-2 inhibitors, such as venetoclax, and synergistically enhance anti-tumor efficacy when given in combination with other anti-cancer drugs. Another important question is whether a safe therapeutic window can be found for this new class of inhibitors. In summary, inhibition of MCL-1 shows potential as a treatment for hematologic malignancies and clinical evaluation of MCL-1 inhibitors is currently underway.
Keywords: MCL-1 inhibitor, multiple myeloma, acute myeloid leukemia, non-Hodgkin lymphoma, BH3-mimetic
1. Introduction
Programmed cell death, or apoptosis, is an evolutionarily conserved and tightly regulated process that plays a key role in the maintenance of cellular homeostasis throughout life, orchestrating the controlled elimination of aging, excessive, or early transformed cells, and the continued survival of vital cells during cell stress, differentiation, and development [1, 2]. Members of the B-cell lymphoma–2 (BCL-2) protein family, which share at least one BCL-2 homology (BH) domain, regulate apoptosis [3]. Apoptosis is prevented by antiapoptotic BCL-2 family proteins, including BCL-2, BCL-B, BCL-XL, BCL-W, myeloid cell leukemia sequence 1 (MCL-1), and A1 (also known as BFL1 in humans). These homologous BCL-2 family members inhibit activation of the proapoptotic multidomain proteins BAX and BAK. Prosurvival BCL-2 family proteins are antagonized by a group of proapoptotic BCL-2 homology 3 (BH3)-only proteins (eg, BIM, PUMA, BID, NOXA, and BAD) [1, 2, 4]. A variety of cell stress signals, including DNA damage induced by cytotoxic agents, may increase the expression and activity of BH3-only protein members [1]. If the surge in BH3-only protein activity exceeds the inhibitory capacity of prevailing prosurvival BCL-2 family members, the effector proteins BAX and BAK may be activated, resulting in outer mitochondrial membrane permeabilization and release of activators of the caspase cascade (Figure 1) [1, 2].
Figure 1. Overview of the role of MCL-1.
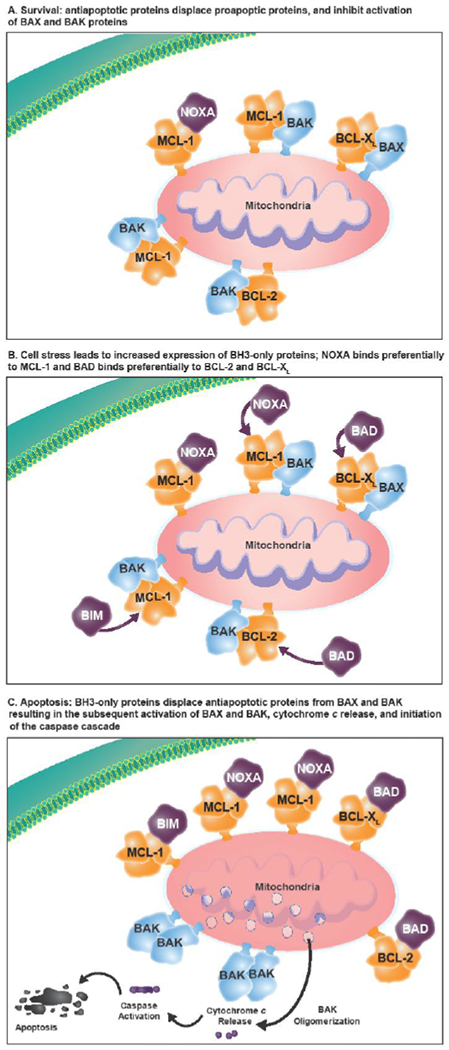
(A) The antiapoptotic proteins (eg, BCL-2, BCL-XL, BCL-W, MCL-1) are prosurvival proteins that bind the proapoptotic multidomain effectors BAK and BAX to prevent cell death, promoting cell survival. (B) A variety of cell stressors increase the expression of the proapoptotic sensors, including the BH3-only proteins (ie, BIM, BID, PUMA, NOXA, and BAD). (C) The BH3-only proteins subsequently displace or prevent the antiapoptotic proteins from binding to BAX and BAK, leading to cytochrome c release into the cytosol and activation of the caspase cascade, resulting in cell death. BCL-2=B-cell lymphoma–2; BH3=BCL-2 homology 3; MCL-1=myeloid cell leukemia sequence 1.
Biochemical studies have revealed important differences in the targeting specificity of the BH3-only proteins to their opposing prosurvival partners [4]. BIM binds to all known prosurvival proteins [5, 6], PUMA and BID bind nonselectively to BCL-2, BCL-XL, BCL-W, MCL-1, and A1, whereas BAD binding is restricted to BCL-2, BCL-XL, and BCL-W, and NOXA selectively binds to MCL-1 and A1 [5]. Similarly, BAX and BAK differ in their ability to associate with prosurvival proteins [4]. The balance and regulation of these diverse BCL-2 family member interactions can confer cell-type dependent differences in the sensitivity and resistance of different organs to cytotoxic insults [4].
MCL-1 plays an important role in the development of B cells, formation and maintenance of germinal-center B cells, and development and survival of existing plasma cells (PCs), naive T cells, and memory T cells [4, 7], whereas BCL-B appears to have an important role in B cell proliferation and plasmacyte differentiation [6]. In contrast, BCL-2 is critical for naive B cells and naive T cells, while erythroid progenitors and platelets are reliant on BCL-XL [4]. MCL-1 and BCL-XL also have important roles in the regulation of myelopoiesis and maintenance of mature myeloid cells, with MCL-1, and possibly BCL-XL, having a key role in allowing the healthy differentiation of myeloid precursors into granulocytes [1].
Data collected from murine knockout models have expanded the understanding of the role of the BCL-2 family of proteins throughout B-cell development. Loss of MCL-1 expression for as little as 2 days depleted B-cell subsets throughout multiple stages of development, whereas loss of BCL-XL expression for up to 4 days only affected immature B-cell maintenance [8]. In such studies, MCL-1 has been found to be essential for the survival of mature neutrophils, with mature neutrophil numbers reduced by 80%–90% in blood, spleen, and peritoneal exudates in knockout mice [9].
Murine knockout models have also expanded the understanding of the role of MCL-1 in multiple other tissue types. MCL-1 has been shown to be essential for cardiac homeostasis in adult murine models [10]; absence of MCL-1 led to loss of cardiomyocytes, increase in fibrosis, and rapid fatal cardiomyopathy [11]. Similarly, MCL-1 and BCL-XL have been found to work interdependently to maintain adult hepatic homeostasis, with both proteins required to prevent hepatic failure [12, 13]. An important preclinical observation is that in MCL-1 heterozygous mice with MCL-1 protein levels reduced by 30 to 50%, the reduction in MCL-1 levels had minimal effect on the general health of mice administered a range of cytotoxic drugs; effects were similar to those observed in similarly treated wild-type mice. These findings support the potential tolerability of MCL-1 inhibitors alone and in combination with conventional cytotoxic drugs [14].
2. Overview of the Role of MCL-1 in Cancer
In this review, we discuss the role of the antiapoptotic protein MCL-1 in hematologic cancers and review the current progress in the development and clinical evaluation of MCL-1 inhibitors. This is a narrative review; the PubMed database was searched for relevant studies describing the role of MCL-1 and MCL-1 inhibition in multiple myeloma (MM), acute myeloid leukemia (AML), and non-Hodgkin lymphoma (NHL); in addition, authors identified other relevant articles and data presented at congresses.
Overexpression of MCL-1 is a common aberration in solid tumors and hematologic cancers [15]. In cell line surveys of human hematologic cells, MCL-1 has been shown to play an important role in promoting cell survival in plasma cell myeloma [7, 16–18], AML [17, 19, 20], and lymphoma [21, 22] cell lines. Overexpression of MCL-1 has been implicated in resistance to radiotherapy [23], chemotherapy [24], and BH3-mimetics targeting BCL-2/BCL-XL [25, 26].
A number of growth factors (eg, vascular endothelial growth factor [VEGF], endothelial growth factor [EGF]), cytokines (eg, interleukin [IL]-3, IL-5, IL-6), and cytotoxic stimuli (eg, drugs, radiation) regulate MCL-1 transcription through cell-type dependent effects on signal transduction pathways such as the PI3K/Akt, JAK/STAT, p38/MAPK, and MEK/ERK pathways, with both antiapoptotic and proapoptotic stimuli involved [27]. In addition, it has been demonstrated that microRNAs (eg, miR-29b, miR-30, miR-137, and miR-197) downregulate expression of MCL-1, inhibiting cell growth and leading to apoptosis of MM or human hematopoietic cells in vivo [28–30].
To a greater extent than other members of the BCL-2 family of proteins, the transcriptional activity of MCL-1 does not directly correlate to MCL-1 protein levels which are also subject to posttranslational regulation in several ways. Posttranslational modification can occur, which may result in shortened forms of the protein [7, 31]. Proteasomal degradation via phosphorylation and polyubiquitination of the N-terminal domain of MCL-1 is mediated by kinases, such as JNK, Glycogen Synthase Kinase 3 (GSK-3) and ERK-1, and ubiquitin ligases, such as Mule, SCFβ-TrCP, SCFFbw7, APC/CCdc20, and Trim17 [32, 33]. Ubiquitination is reversible; Usp9x is a deubiquitinase that removes Lys 48-linked polyubiquitin chains that facilitate proteasomal degradation of MCL-1, thus promoting tumor survival [33, 34]. Non-proteasomal degradation via caspase-dependent cleavage at two sites within the N-terminus also disrupts the pro-apoptotic activity of MCL-1 [31, 33]. Cleavage of MCL-1 after Asp127 and Asp157 resulted in proteins lacking the first 127 and 157 amino acids, respectively, retaining the BH1 to BH3 domains associated with proapoptotic activity, losing the BH4 domain and the antiapoptotic activity associated with MCL-1 [35]. Of interest, the cleavage fragments of MCL-1 appear to have similar half-lives to parent protein and, in contrast with the cleavage fragments of other BCL-2 proteins, the shorter fragment (∆157-MCL-1) appears to continue to protect cells from apoptosis [36].
Available evidence indicates that MCL-1 is an important prosurvival protein, and that targeting MCL-1 may therefore be an effective approach to cancer treatment [37, 38]. As monotherapies, MCL-1 inhibitors may prove effective against cancers that depend on MCL-1 for survival. In combination, MCL-1 inhibition may overcome the effects of MCL-1 overexpression in mediating treatment resistance [15].
2.1. Role of MCL-1 in Multiple Myeloma
Approximately 40% of patients with multiple myeloma (MM) carry a gain or amplification of 1q21, the chromosome region containing the MCL-1 gene and the gene encoding for the IL−6 receptor (IL6R) [7]. Gain or amplification of 1q21 is associated with a significantly shorter progression-free survival and lower overall survival than are associated with normal 1q status [39]. MCL-1 expression can be upregulated by the paracrine effects of IL-6 expression on neighboring cells (ie, induction of JAK/STAT3 signaling and increase of MCL-1 and BCL-XL transcription) or it can be regulated independently of IL-6 by other signals from the bone marrow microenvironment (eg, through interferon-α, B-cell activating factor [BAFF], or a proliferation-inducing ligand [APRIL]) [7].
Observations from clinical studies in MM support the role of MCL-1 as an adverse prognostic disease marker. In 25 patients with newly diagnosed MM, event-free survival was significantly shorter in patients whose myeloma maintained higher levels of MCL-1 (ie, MCL-1 levels >2 standard deviations above the mean for normal PCs were associated with a shorter median overall survival of 12 months compared with not reached after 40 months of follow-up for the myeloma cohort with normal levels of MCL-1, P=0.002) [40]. Furthermore, MCL-1 gene expression appears to adaptively increase in relapsed disease [7, 41]. In one study assessing 60 consecutive myeloma samples (21 individual patients at diagnosis and 39 patients at relapse), cellular MCL-1 dependency, as assessed by exposing patient samples to a panel of BH3-mimetic drugs, was significantly higher in the relapse samples (69%) versus the diagnosis sample (33%; P=0.01) [41]. In contrast, the dependence on BCL-2 and BCL-XL were not significantly different between diagnosis and relapse [41]. Increased MCL-1 dependency was also found in some patients overexpressing cyclin D1 (CCND1) [41].
The importance of MCL-1 in the pathogenesis of MM has been inferred across a number of preclinical studies assessing the impact of MCL-1 expression on cell growth and differentiation. In vitro and in vivo studies have shown that MCL-1 is an important prosurvival factor in normal PCs; BCMA-mediated MCL-1 expression represents a PC survival pathway that is independent of Blimp-1–controlled PC differentiation, and loss of MCL-1 resulted in rapid loss of PCs [42]. Early studies using freshly isolated MM cells suggested that the expression of MCL-1 was required for survival; inhibition of MCL-1 induced rapid activation of apoptosis, even when there was continuous expression of other BCL-2 proteins [43].
Investigations using human cell lines confirm that the majority (17/25) of human myeloma cell lines tested were dependent on MCL-1 (≤25% viability) [16]. For example, inhibition of MCL-1 by the selective peptidyl ligand BIM2A resulted in cell death in 68% (17/25) of the human myeloma cell lines evaluated [16]. In contrast, only 20% of the overall population of cell lines were highly sensitive to BCL-2 inhibition (eg, venetoclax, concentration inhibitory to 50% of cells [IC50] <0.5 μM), with a nonoverlapping population comprising 20% of the cohort highly sensitive to BCL-XL inhibition (eg, A5463, IC50<0.5 μM) [16]. These experiments suggest MM cells are highly dependent on BCL-2 family proteins for survival [7], with MCL-1 being of importance in most cell lines (approximately 70%) [16].
In another study, depletion of MCL-1, but not of BCL-2 or BCL-XL, using antisense oligonucleotides, triggered a decrease in cellular viability of three different human myeloma cell lines, with apoptosis detected within 2 days and the effect being maximal at days 4 and 6 [44]. Depletion of MCL-1 resulted in apoptosis of MM cells even in the presence of IL-6 [44]. In MM cell lines, dexamethasone combined with MCL-1 inhibition enhanced apoptosis in cells in which MCL-1 inhibition already had a major apoptotic effect [44]. However, in cells in which MCL-1 inhibition alone was relatively ineffective, the combination with dexamethasone was synergistic and resulted in a large increase in apoptosis with MCL-1 depletion, sensitizing the myeloma cells to dexamethasone-induced apoptosis [44].
In summary, evidence from a number of preclinical studies shows that MCL-1 is a key antiapoptotic protein in MM, and inhibition of MCL-1 is a promising approach for the treatment of MM.
2.2. Role of MCL-1 in Acute Myeloid Leukemia
Human leukemia cells from newly diagnosed patients have been found to overexpress prosurvival BCL-2 family members [45, 46]. Of the BCL-2 family of proteins, MCL-1 was consistently high in nearly all bone marrow cell samples from newly diagnosed patients with AML [46]. High levels of antiapoptotic BCL-2 proteins, including MCL-1, were also found to be associated with relapse of AML [20].
Interest in targeting prosurvival proteins in AML has followed the approval by the US Food and Drug Administration of the BCL-2 inhibitor venetoclax in combination with DNA methyltransferase inhibitors or low-dose cytarabine for older patients with AML and those with comorbidities precluding the use of intensive chemotherapy [47–49]. Preclinical models have highlighted the important prosurvival role of MCL-1 in AML [20]. The development of MCL-1 inhibitors suitable for clinical development promoted preclinical screens to determine which cancers were most susceptible to apoptosis induced by these drugs [50, 51]. Hematologic cancers, including AML, were most sensitive, prompting the initiation of phase 1 studies for several inhibitors (AZD5991 [ClinicalTrials.gov NCT03218683], S64315 [NCT02979366, NCT03672695], AMG 176 [NCT03797261, NCT02675452], and AMG 397 [NCT03465540]) in this disease (discussed further in section 3.1) [52–57].
Overexpression of MCL-1 has been identified in chemotherapy-relapsed AML and as a major factor in the development of resistance to the dual BCL-2/BCL-XL inhibitor ABT-737 in AML cell lines [58, 59]. Several groups using different MCL-1 inhibitors have independently found that combined targeting of BCL-2 and MCL-1 has promise in AML, sparking intense interest to clinically develop this dual BH3-mimetic approach [17, 60–62].
2.3. Role of MCL-1 in Non-Hodgkin Lymphoma
The t(14;18) chromosomal translocation, which joins the IGH promoter with the BCL2 gene resulting in constitutive expression of the antiapoptotic BCL-2 protein, is present in approximately 80% of patients with follicular lymphomas [63] and 20%–30% of patients with diffuse large cell lymphomas [64, 65]. Additionally, overexpression of BCL-2 in NHL is more likely in patients with t(14;18)-positive disease than in those with t(14;18)-negative disease [63]. Moreover, increased levels of BCL-2 are associated with poorer prognosis in patients with NHL [22].
MCL-1 is widely expressed in malignant B cells, although the levels of MCL-1 in subtypes of NHL vary [66]. High-level expression of MCL-1 was found to be required for B-lymphoma cell survival [66]. MCL-1 expression was correlated with tumor grade and found to be predominant in high-grade versus low-grade lymphomas [67], which suggests an association with progressive disease [68].
Some patients with diffuse large B-cell lymphoma (DLBCL) have genetic alterations that may influence response to treatment. Approximately 10% of cell lines from DLBCL had MCL-1 mutations; approximately half of these were missense mutations, with the remainder being associated with copy number gain [64]. Additionally, rearrangements of the MYC oncogene in double-hit and triple-hit lymphomas (MYC and either BCL-2 or BCL-6; MYC and BCL-2 and BCL-6 rearrangements, respectively) occur in <10% of cases of B-cell lymphoma and are both associated with an aggressive clinical course and poor prognosis [64, 69, 70].
In NHL cell lines, antisense oligonucleotides specific to MCL-1 resulted in apoptosis associated with caspase-9 activation, indicating that MCL-1 prevents cytochrome c release [66]. Preclinical studies show that suppressing MCL-1 protein synthesis with homoharringtonine combined with the proteasome inhibitor bortezomib induced the BH3-only protein NOXA to disrupt the MCL-1 interaction with BAK, effectively reducing tumor growth and significantly increasing survival in murine double-hit models [71]. Therefore, a combined approach to downregulate MCL-1 and upregulate NOXA-mediated BAK activation may have a role in double-hit disease [71]. The MCL-1 inhibitor S64315 has been shown to prolong survival in models of aggressive lymphoma driven by the MYC oncogene [19]. Thus, there is strong preclinical rationale to investigate the therapeutic role of MCL-1 inhibitors in aggressive NHL.
3. MCL-1 Inhibitors
3.1. Direct MCL-1 Inhibitors
The search for a safe, effective, and selective MCL-1 inhibitor has proven challenging. The initial MCL-1 putative inhibitors identified were neither selective (eg, gossypol [72, 73] [under clinical evaluation for a range of solid and hematologic cancers] [74], apogossypolone [75], antimycin A [76, 77], obatoclax [78], and TW-37 [79, 80]) nor potent and did not cause cell death in a BAK/BAX-dependent manner [81, 82]. Historical difficulties in the development of effective MCL-1 inhibitors (eg, lack of specificity for MCL-1, poor pharmacokinetic profiles, limited cell membrane permeability) were reported [74]. In addition, the key binding site on MCL-1 is shallow and relatively inflexible compared with the binding site on BCL-2 and BCL-XL, making the development of MCL-1 inhibitors particularly difficult. Inhibitors that bind in the proximity of the P2 pocket, a binding region on MCL-1 that forms a large hydrophobic cavity in the presence of ligands, appear to have the most potential, as shown by nuclear magnetic resonance–based screening [15].
Selective MCL-1 inhibitors are in various stages of design and assessment, although only a few are undergoing clinical development (Table 1) [17, 19, 62, 83–86]. Given the physiologic role of MCL-1 in cardiac and hepatic tissues [10–13], pluripotent stem cells [87] and brain cells [88], it will be important to determine in any clinical development program whether a sufficiently wide therapeutic window exists to enable MCL-1 inhibitors to have a safe tolerability profile. A number of early MCL-1 inhibitors, including MIM1 (a polyphenol compound) [89], UMI-77 (a naphthol derivative, modified from UMI-59) [90], Roussi compound 2c (derived from meiogynin A) [91], compound 9 from Eutropics Pharmaceuticals (Cambridge, MA, USA) [51], and compound 12 from a Chinese research group [92], were created with the aim of finding selective and potent MCL-1 inhibitors (Table 2 [50, 51, 60, 89, 90, 92–99]). More recently, pyridoclax was found to disrupt the MCL-1–BIM interaction in living cells [96], as does VU661013 [60]. Similarly, a number of indole derivatives have been created and assessed for their apoptotic activity [74, 94, 95]. A-1210477 appeared to have some potential [100] and using BH3 profiling, A-1210477 was found to act in a mitochondria-dependent manner [82]; however, no in vivo activity for A-1210477 has been reported [101], and results of BH3 profiling suggest cell entry could prevent A-1210477 activity in vivo [82]. Structure-based design was also used to identify compound 42, an MCL-1 inhibitor that binds with picomolar affinity [102]. Compound 42 has been shown to have potent cellular activity, displacing BIM from MCL-1, and to cause tumor regression in murine models of MM and AML [102]. BIM SAHBA is a hydrocarbon-stapled peptide that targets the BCL-2 family of proteins and appears to preferentially displace BIM from MCL-1 versus BCL-2, as demonstrated in MCL-1 deficient mouse embryonic fibroblasts [103]. Results from preclinical studies suggest that a well-timed combination with a BH3 mimetic is likely to be required for clinical activity against disease such as DLBCL, where multiple oncoproteins are involved [103]. We are not aware of ongoing clinical development for any of the above-mentioned MCL-1 inhibitors.
Table 1.
Overview of Compounds With Activity Against MCL-1 Currently Undergoing Clinical Evaluation
| Clinical Trials* |
|||||||
|---|---|---|---|---|---|---|---|
| Compound | Category/Binding Site | In Vitro Potency | In Vitro or In Vivo Properties | Identifier | Study Phase | Treatment Regimen/Route of Administration | Tumor Type |
| Currently undergoing clinical evaluation | |||||||
| AZD5991 [17, 86] | MCL-1 inhibition via BAK-dependent mechanism/ ligand-binding pocket |
KI: 200 pM IC50: 0.72 nM |
100% tumor regression in mouse models after single dose; synergistic in vivo efficacy with SOC regimens | NCT03218683 | 1 | Dose finding, administration every 21 days for 9 cycles/IV | Relapsed or refractory hematologic malignancies |
| S63845 [19] | Inhibition of MCL-1/BAK interaction/BH3-binding groove |
KI: <1.2 nM Kd: 0.19 nM |
25 mg/kg well tolerated and highly effective against mouse tumors | – | – | – | – |
| S64315 (MIK665) [83] | – | – | – | NCT02992483 | 1 | Part 1: dose finding/IV Part 2: expansion study of RDE/IV |
Relapsed or refractory lymphoma or MM |
| NCT02979366 | 1 | Starting dose: 50 mg once weekly/IV | AML, MDS | ||||
| NCT03672695 | 1 | Recommended phase 2 dose finding study in combination with venetoclax/S64315, IV; venetoclax, oral | AML | ||||
| AMG 176 [62, 84] | Selective MCL-1 inhibition/BH3-binding groove | KI: <1 nM | Rapid and robust induction of apoptosis in tumor xenografts after a single dose | NCT02675452 | 1 | Part 1: dose finding/IV Part 2: combination regimens/IV |
Relapsed or refractory MM or AML |
| NCT03797261 | 1 | Dose finding: various combinations of AMG 176 and venetoclax/AMG176, IV; venetoclax, oral | Relapsed or refractory hematologic malignancies | ||||
| AMG 397 [85] | Selective MCL-1 inhibition/– | – | – | NCT03465540 | 1 | Dose finding/oral | Relapsed or refractory MM, AML, NHL |
AML=acute myeloid leukemia; BH3=BCL-2 homology 3; IC50=concentration inhibitory to 50% of cells; IV=intravenous; MCL-1=myeloid cell leukemia sequence 1; MDS=myelodysplastic syndrome; MM=multiple myeloma; NHL=non-Hodgkin lymphoma; RDE=recommended dose for expansion; SOC=standard of care.
Clinical trial data available from www.ClinicalTrials.gov.
Table 2.
Overview of Compounds With Activity Against MCL-1 Not Currently Undergoing Clinical Evaluation
| Compound | Category/Binding Site | In Vitro Potency | In Vitro or In Vivo Properties |
|---|---|---|---|
| Clinical evaluation not yet under way | |||
| VU661013 [60] | MCL-1 inhibition/ destabilizes BIM/MCL-1 association |
KI: 97 pM | Growth inhibition in AML cell lines in vitro; dose-dependent decrease in tumor burden in murine models |
| Compound 42 [102] | Displaces BIM from MCL-1 | Ki: 70–300 pM | Tumor regression in murine models |
| BIM SAHBA [103] | Preferentially displaces BIM from MCL-1 | EC50: 2–18 μM | – |
| Clinical evaluation does not appear to be progressing | |||
| Fesik compound 53 [95] | Inhibition of MCL-1/BH3 peptide binding | KI: 0.055 μM | – |
| A-1210477 [50, 94] | Inhibition of MCL-1/disrupts BIM binding | KI: 0.0004–0.0005 μM | – |
| Zhang compound 12 [92] | Inhibition of MCL-1/BH3 peptide binding |
KI: 0.48 μM IC50: 2.2μM |
Inhibits MCL-1-dependent cells in vitro |
| MIM1 [89] | Inhibition of MCL-1/BAK interaction | IC50: 4.2 μM | Induces apoptosis in leukemia cells |
| UMI-77 [90] | Inhibition of MCL-1/BAK interaction | KI: 0.49 μM | Daily administration (5 d/wk for 2 weeks) significantly inhibited tumor growth in BxPC-3 xenograft mouse model |
| Roussi compound 2c [98] | Inhibition of MCL-1 activity | KI: 0.46 μM | – |
| Cardone compound 9 [51] | Inhibition of MCL-1/BH3 peptide binding | IC50: 0.31 μM | High activity against a panel of human-derived cancer cell lines |
| Compound 34 [97] | Inhibition of MCL-1/disrupts BIM binding | IC50: 6.1 μM | – |
| Chai compound 7 [93] | Inhibition of MCL-1/BH3 peptide binding | KI: 8 μM | – |
| Pyridoclax [96] | Inhibition of MCL-1/disrupts BIM binding | – | Sensitizes ovarian cancer cells to BCL-XL inhibition |
| Liu & Wang A1 [99] | Inhibition of MCL-1/BH3 peptide binding | KI: 0.18 μM | – |
AML=acute myeloid leukemia; BH3=BCL-2 homology 3; EC50=half-maximal concentration of drug; IC50=concentration inhibitory to 50% of cells; MCL-1=myeloid cell leukemia sequence 1.
AZD5991 is a macrocyclic molecule, selective for MCL-1 (Ki=200 pM), with highly potent activity (IC50=0.72 nM) [17] reported to act via a mitochondria-dependent manner, being highly MCL-1–specific at the cellular level [82]. AZD5991 binds directly to MCL-1 at the ligand-binding pocket and induces caspase-3/7 activation and cell death via a BAK-dependent mechanism and reduces MCL-1 levels in AZD5991-sensitive but not AZD5991-resistant cells. Taken together, these results suggest that activation of caspases by AZD5991 subsequently reduces MCL-1 levels [17]. AZD5991 has a dose-dependent antitumor effect, resulting in 52% tumor growth inhibition 10 days after administration of 10 mg/kg through to complete tumor regression after 100 mg/kg in MM models in mice. The activity of AZD5991 against MM and AML subcutaneous tumors in murine models was enhanced when AZD5991 was administered in combination with bortezomib (proteasome inhibitor) or venetoclax (BCL-2 inhibitor), respectively [17]. Activity against leukemic cells in the bone marrow of mice has also been reported [17]. Efficacious doses were associated with minimal bodyweight changes in mice during the study periods, leading to the conclusion that monotherapy and combination treatments were well tolerated in preclinical studies [17]. However, given that AZD5991 has weaker binding to mouse MCL-1 than human MCL-1, safety results from first-in-human studies will be important to assess. A phase 1 dose-finding study of AZD5991, administered intravenously every 21 days for 9 cycles in patients with relapsed or refractory hematologic malignancies, is ongoing [52].
S63845 is a selective inhibitor of MCL-1, exhibiting activity in panels of MM, AML, lymphoma, and leukemia cell lines, as well as primary AML patient samples [19]. S63845 binds with high affinity (KD=0.19 nM) and specificity to the BH3-binding groove of MCL-1, activating the BAX/BAK–dependent apoptotic pathway [19]. The MM cell lines most sensitive to S63845 had detectable levels of MCL-1 but barely detectable levels of BCL-XL; similarly, the sensitivity of AML cell lines to S63845 inversely correlated with BCL-XL mRNA levels [19]. Interestingly, t(4;14) MM cell lines were sensitive to S63845, suggesting that MCL-1 inhibition may be effective in MM disease associated with a poor prognosis and refractory to standard chemotherapy [19]. S63845 has synergistic activity in vitro against AML cell lines when combined with daunorubicin or hypomethylating agents, such as decitabine [61, 104]. S63845 also has potent activity against primary human AML cells in combination with BCL-2 co-targeting in both in vitro and in vivo models [61]. Interestingly, this activity was more potent against leukemic rather than normal hematopoietic progenitors [61]. It is likely that S63845 initiates apoptosis through the disruption of the interaction between MCL-1 and BAK and/or the release of endogenous BH3-only proteins, such as BIM, after binding to MCL-1 [105]. In murine models, S63845 (25 mg/kg) caused only a minor reduction in some leukocyte subsets and caused no changes in the major organs or skeletal muscle, indicating that S63845 was well tolerated at efficacious doses in preclinical studies [19]. It was suggested that the intermittent periods of MCL-1 inhibition resulting from drug treatment account for the tolerability of S63845 at effective doses in contrast to the serious impact on multiple cell types observed following irreversible loss of MCL-1 after gene knockout [19].
S64315 belongs to the same series of compounds as S63845. Minimal data on S64315 have been released to date; however, it is currently undergoing clinical evaluation [83]. Phase 1 dose-finding studies are under way for S64315 (ie, MIK665) using a 2-part study design to assess a preliminary maximum tolerated dose (via dose escalation), and then further assessing tolerability and preliminary antitumor activity in MM (NCT02992483) and AML (NCT02979366) expansion cohorts [55, 106]. Another study is planned to assess S64315 in combination with venetoclax in patients with AML (NCT03672695) [56].
AMG 176 is a potent and selective MCL-1 inhibitor that binds to the BH3-binding groove of MCL-1; the binding affinity of AMG 176 and its related analog AM-8621 for human MCL-1 is in the picomolar range [62, 84], with AMG 176 shown to cause cytochrome c release (ie, apoptosis) only from MCL-1 dependent mitochondria [82]. The AM-8621 analog, a tool MCL-1 inhibitor used to characterize the mechanism of action of AMG 176 in vitro, was found to disrupt the interaction between MCL-1 and BAK (IC50=43 nM) and between MCL-1 and BIM, and to have on-target MCL-1–mediated activity [62]. Cells with high BCL-XL expression were most likely to be resistant to AM-8621, and those with high BAK expression were most likely to be sensitive to AM-8621 [62]. MM cells were most likely to be sensitive to AM-8621, whereas AML and diffuse large B-cell lymphoma cell lines were more heterogeneous, with sensitivity to either AM-8621, venetoclax, or both [62]. Given its superior pharmacokinetic properties compared with AM-8621, AMG 176 underwent further clinical development [62]. A single oral dose of AMG 176 resulted in rapid and robust induction of apoptosis in MM xenografts, as measured by activated BAK, cleaved caspase 3, and cleaved poly (ADP-ribose) polymerase (PARP) [62]. Similarly, twice-weekly administration of AMG 176 (30 or 60 mg/kg) resulted in a dose-dependent reduction in tumor burden in an orthotopic model of AML in mice [62].
To date, MCL-1 inhibitors under development have a reduced affinity for murine MCL-1. Therefore, a human MCL-1 knock-in mouse model has been developed, replacing the murine MCL-1 gene with its human ortholog, to enable a better understanding of the pharmacodynamics and tolerability of MCL-1 inhibition in vivo [62, 107]. In a human MCL-1 knock-in mouse model, intravenous administration of S63845 was tolerated to a maximum dose of 12.5 mg/kg; regression of Eμ-Myc lymphoma was observed in 60% of mice administered S63845 12.5 mg/kg [107]. Similarly, oral administration of AMG 176 (30 and 60 mg/kg) was tolerated with no evidence of overt systemic toxicity, as was the combination of AMG 176 and venetoclax [62]. At doses that were well tolerated, AMG 176 treatment resulted in a dose-dependent decrease in levels of B cells, monocytes, neutrophils, eosinophils, basophils, and reticulocytes in blood and bone marrow; these changes in biochemistry could be useful pharmacodynamic endpoints for assessing treatment [62]. A phase 1 dose-finding study for AMG 176 in patients with relapsed or refractory MM or AML (NCT02675452) has been initiated [53]. In addition, a phase 1 dose-finding study of AMG 176 in combination with venetoclax in patients with relapsed or refractory AML, NHL, or DLBCL has also commenced [54]. Although AMG 176 was administered orally in preclinical studies, in clinical trials currently under way, AMG 176 is being evaluated as an intravenous infusion. Intravenous administration of MCL-1 inhibitors may allow for more precise pharmacokinetic profiles to be achieved with respect to drug exposure compared with oral administration. Oral administration, however, is more convenient for patients [101]
AMG 397, an oral small-molecule inhibitor of MCL-1, is the only oral MCL-1 inhibitor to reach the clinic thus far [101]. Preclinical data in the literature are sparse; however, clinical evaluation is under way [57]. It will be important to assess the impact of the route of administration on the efficacy and safety of MCL-1 inhibitors in clinical practice [101]. The phase 1 dose-finding studies involving AMG 176 (NCT02675452) [53] and AMG 397 (NCT03465540) [57] in patients with MM, NHL, or AML are currently on clinical hold to evaluate a safety signal for cardiac toxicity [108]. The dose-finding combination trial of AMG 176 and venetoclax (NCT03797261) is also currently suspended based on this safety signal [54].
In summary, several direct MCL-1 inhibitors have been found to be well tolerated at efficacious doses in preclinical studies. Clinical studies are ongoing to establish the benefit-risk profile of MCL-1 inhibitors in patients with hematologic malignancies. At the time of writing, no clinical data have been reported for any MCL-1 inhibitor in peer-reviewed publications.
3.2. Indirect MCL-1 Inhibitors
In addition to compounds that cause apoptosis through MCL-1 inhibition, there is a range of compounds that cause apoptosis at least in part through a reduction in MCL-1 cellular levels by reducing expression of MCL-1 or by increasing posttranslational degradation. Therefore, in addition to MCL-1 inhibition, interruption of key factors in the regulation of MCL-1 may offer potential therapeutic targets for cancer treatment.
Indirect MCL-1 inhibitors include the following [109–120]: RS-F3, isolated from the marine sponge Subarea clavata [109]; maritoclax, a natural product of marinopyrrole A [110, 111]; WP1130, a partially selective Usp9x deubiquitinase inhibitor [112]; selinexor, an XPO1-selective inhibitor [113]; necrostatin-1, a potential inhibitor of necroptosis [114]; TM-233, a novel analog of 1’-acetoxychavicol acetate [115]; spautin-1, a novel autophagy inhibitor [116]; PIK-75, a kinase inhibitor [117]; cyclin-dependent kinase 9 (CDK9) inhibitors, which inhibit the transcription of MCL-1 [111]; ABC294640, a sphingosine kinase 2 inhibitor [118]; YM155, which prevents the regulation of MCL-1 expression via IL-6 stimulation [119]; and asiatic acid, which attenuates expression of BCL-2 proteins and MCL-1 in human leukemia cell lines [120].
Among these indirect MCL-1 inhibitors, CDK9 inhibitors have most recently entered the clinic. CDK9, one of a family of 13 protein kinases, forms the catalytic core of the positive transcription elongation factor, which is an enzyme critical for stimulating transcription of key development and stimulus responsive genes [121]. Nonselective CDK9 inhibitors are associated with significant toxicity associated with off-target activity [121]. As a transcriptional activator, CDK9 is necessary for the expression of MCL-1, thereby reducing intracellular MCL-1 levels, which has a very short half-life. Alvocidib (flavopiridol), dinaciclib, and AZD4573 are CDK9 inhibitors undergoing clinical evaluation [111, 122, 123]. Alvocidib, which inhibits the phosphorylation of the carboxyl-terminal domain of RNA polymerase II, is associated with a reduction in the transcription of antiapoptotic genes and, consequently, a reduction in antiapoptotic protein, leading to apoptosis of primary chronic lymphocytic leukemia cells [124]. Alvocidib causes a rapid downregulation of MCL-1 in both MCL-1-dependent (SKBR3) and MCL-1-independent (HCC-1806) breast cancer cell lines; apoptosis occurs more rapidly in MCL-1-dependent cells [125]. Alvocidib also inhibits the transcription of BCL-2, although the decline in BCL-2 levels is less rapid than the decline in MCL-1 levels [124]. A newer generation CDK9 inhibitor, dinaciclib, causes a time-dependent loss of MCL-1, BIM, and NOXA protein, and a more modest loss of BAK protein, leading to PARP cleavage and cellular destruction [111], with lower concentrations (up to 10 nM) that are potentially more achievable clinically, associated with inhibition of CDK2 kinase activity [126]. Dinaciclib has demonstrated efficacy in the treatment of hematologic malignancies [127–129]. AZD4573, the newest CDK9 inhibitor to enter clinical trials, is a selective CDK9 inhibitor being evaluated in a first-in-human study in patients with hematologic malignancies [130].
3.3. Combination Therapy With MCL-1 Inhibitors
Given that antiapoptotic proteins appear to be able, at least in part, to compensate for one another, combination therapy with BCL-2 inhibitors could provide a synergistic response [22, 41] and overcome drug resistance observed with BCL-2 inhibition alone [17], and appears to be important in disease such as DLBCL, where a number of oncoproteins are involved [103].
Of the BCL-2 inhibitor combinations, combinations with venetoclax appear the most promising (Table 1) and help mitigate the evolution of resistance to venetoclax monotherapy [131, 132]. Early evidence suggested that venetoclax was least effective against cell lines with high MCL-1 and BCL-XL levels [133]. Downregulation of MCL-1 with PI3K/AKT/mTOR inhibitors had no effect alone, but in combination with venetoclax sensitized previously resistant cell lines to venetoclax, without having any effect on BCL-XL levels [133]. Subsequently, a variety of complex and heterogeneous mechanisms have been reported to cause venetoclax resistance in leukemia and lymphoma cell lines [132]. However, evidence points to MCL-1 being one mediator of venetoclax resistance, in model systems [131] and in some patient samples [134]. Significant upregulation of MCL-1 was observed in venetoclax-resistant HBL2 and MAVER1 mantle cell lymphoma cell lines; furthermore, overexpression of MCL-1 was associated with decreased venetoclax sensitivity [131]. More recently, it has been demonstrated that acquired increases in MCL-1 protein are associated with secondary resistance to venetoclax in some patients with chronic lymphocytic leukemia, including where MCL-1 gene amplification is present in subclones that become dominant at disease progression [134]. Resistance due to increases in MCL-1 and BCL-XL was reversed by concurrent exposure to MCL-1 and BCL-XL inhibitors, respectively [132]. There was evidence that increases in MCL-1 levels were variably the result of either reduction in MCL-1 degradation or gene amplification [132]. For example, FLT3-ITD or PTPN11 mutations, which enhance the expression of MCL-1 and BCL-XL, have also been associated with resistance to venetoclax [135, 136]. Decreases in proapoptotic proteins also resulted in venetoclax resistance, including reductions in NOXA, BAX, and TP53 levels [131, 132, 137]; BAX deficiency may represent innate rather than acquired resistance [132]. Venetoclax can also inhibit PTEN expression and upregulate the AKT pathway activation, leading to venetoclax resistance in B-cell lymphoma [138].
The combination of AZD5991 and the BCL-2 selective inhibitor venetoclax resulted in a sharp reduction in MCL-1 levels, but other members of the BCL-2 family were unaffected [17]. Furthermore, tumor regression was observed after combination therapy in mice models, suggesting combination therapy overcomes the resistance observed with monotherapy [17].
AML cell lines [139, 140] and primary AML cells [61, 140] were more sensitive to the combination of the MCL-1 inhibition (eg, with the selective inhibitor S63845 or doxycycline) and BCL-2–specific inhibitors (eg, venetoclax) than inhibition of either protein alone [139]. Similarly, the combination of VU661013, a MCL-1 inhibitor that destabilizes BIM/MCL-1, and venetoclax was shown to be effective in venetoclax-resistant AML cells ex vivo [60]. Venetoclax was subsequently approved in November 2018 for the treatment of AML in combination regimens with azacitadine or decitabine, or low-dose cytarabine [141]. Azacitidine has been associated with MCL-1 downregulation, which may explain the synergistic outcomes reported with the combination of azacitidine or decitabine and venetoclax [47, 142].
A-1210477 and venetoclax showed synergy in vitro against human AML cell lines, and it was suggested that the synergistic effect resulted from the combination therapy disrupting the binding of BIM to both MCL-1 and BCL-2 [143]. Synergistic effects were also observed in combination with venetoclax against AML progenitor cells [100] and in combination with ABT-263 against melanoma cells [144].
In multiple AML cell lines, synergistic activity has been seen when combining AM-8621, an analog of AMG 176, with cytarabine, decitabine, or doxorubicin [62]. The combination of AM-8621 and venetoclax displayed greater synergy in AML cell lines compared with the other combinations [62]; similar inhibition was observed with the combination of AMG 176 and venetoclax in AML [62]. In human MCL-1 knock-in mouse models, the combination of AMG 176 (30 mg/kg, twice weekly) and venetoclax (50 mg/kg, daily) resulted in significant decreases in peripheral blood B cells and monocytes compared with venetoclax monotherapy, which resulted in significant reductions in B cells only [62]. As discussed previously, dose-finding studies are under way to assess the safety and preliminary efficacy of venetoclax in combination with MCL-1 inhibitors S64315 (NCT03672695) [56] and AMG 176 (NCT03797261) [54] in hematologic cancers.
The combination of MEK and MCL-1 inhibition also appears to be a promising therapeutic strategy. In murine models using solid tumor cell lines, the combination of MCL-1 (AMG 176 or AM-4907) and MEK inhibition (trametinib) led to greater tumor regression than trametinib alone or the trametinib/navitoclax combination [145]. Prior inhibition of BCL-XL with navitoclax led to cells becoming extremely sensitive to combination MEK and MCL-1 inhibition, but not to MEK inhibition alone [145]. In contrast, prior inhibition of MCL-1 did not enhance sensitivity to BCL-XL inhibition [145].
AM-8621 has shown synergistic inhibitory activity with dexamethasone in MM cell lines [62]. In addition, proteasome inhibitors have been shown to induce the BH3-only protein NOXA, resulting in indirect inhibition of MCL-1 [18]; thus, combination therapy between proteasome inhibitors and MCL-1 inhibitors may be synergistic. For example, the combination of the proteasome inhibitor carfilzomib and TG02, a multikinase inhibitor that targets JAK2 and CDK9, increased NOXA levels and decreased MCL-1 protein levels in MM cell lines, leading investigators to conclude that further clinical evaluation of the combination is warranted [146]. In mouse models, the combination of AMG 176 with the proteasome inhibitor carfilzomib achieved significantly greater inhibition of tumor burden than either agent alone in MM [62].
Dual combination of systemic MCL-1 and BCL-XL inhibitors do not appear to be under clinical evaluation for hematologic cancers. This is likely a consequence of liver toxicity that is likely to occur if these two pro-survival proteins are systemically co-targeted [13]. For this strategy to be successful, the targeted delivery of at least one of the BH3-mimetics to the tumor of interest (eg, antibody-directed conjugates) will likely be required to limit exposure of non-tumor tissues to toxicity associated with dual BCL-XL/MCL-1 targeting. Furthermore, the dose-dependent thrombocytopenia associated with BCL-XL inhibition may limit the use of MCL-1/BCL-XL combination [133, 147].
4. Conclusions and Future Considerations
Members of the BCL-2 protein family have important functions in the regulation of apoptosis and interact through complex pathways. Of these proteins, the antiapoptotic protein MCL-1 appears to have a critical role in promoting the survival of hematologic cancer cell lines. In particular, MCL-1 has a prosurvival role for MM, AML, and NHL cell lines and primary cells.
Advances in the identification of selective inhibitors of MCL-1 offer the potential for a targeted treatment option in patients with hematologic malignancies. Preclinical evidence supports MCL-1 inhibition as a promising therapeutic strategy, and a number of MCL-1 inhibitors have been identified (Figure 2). The selective MCL-1 inhibitors AZD5991, S64315, AMG 176, and AMG 397 are currently being evaluated in phase 1 dose-finding studies in MM, AML, NHL, and other hematologic malignancies. Preclinical evidence also suggests that combining MCL-1 inhibitors with proteasome or BCL-2 inhibitors may be more effective at least in some instances, and phase 1 studies are under way with both AMG 176 and S64315 in combination with venetoclax.
Figure 2. Treatment summary of MCL-1 inhibitors.
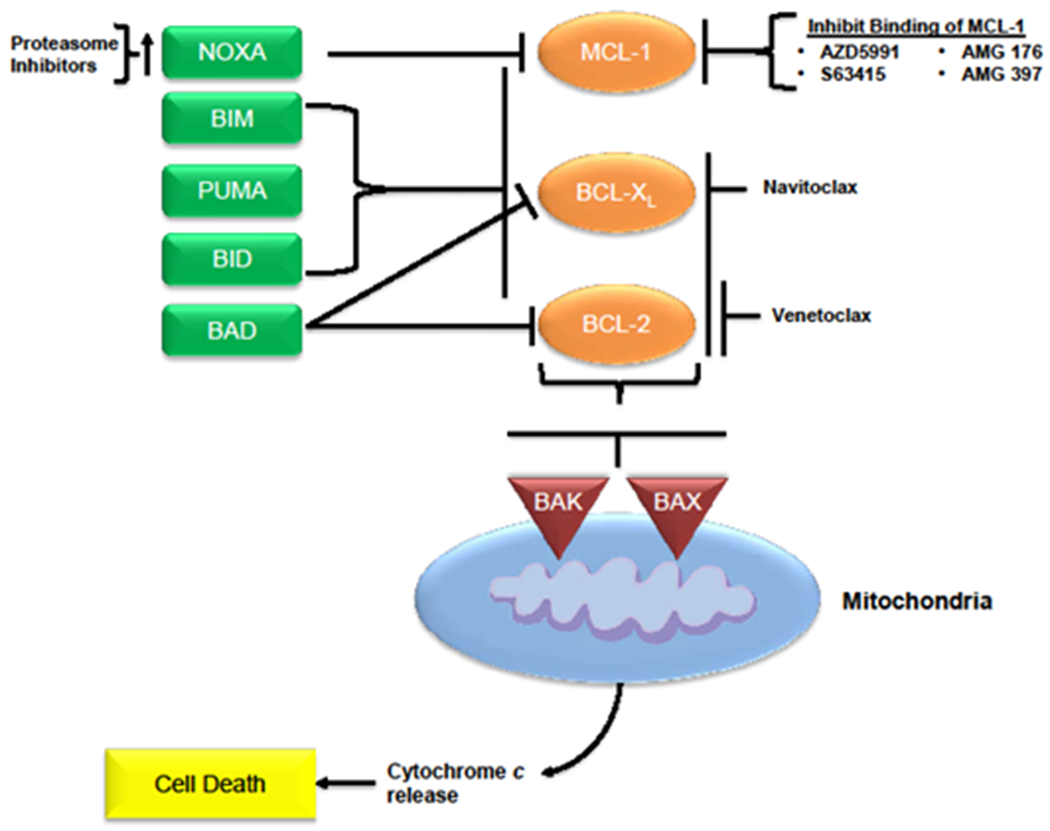
Description of the role of MCL-1 inhibitors in the promotion of apoptosis of malignant cells. Compounds that inhibit the binding or transcription of MCL-1 lead to an increase in activity of the proapoptotic multidomain effectors BAK and BAX, leading to cell death. Combinations of agents that inhibit MCL-1 and BCL-2 or BCL-XL have the potential to result in greater activation of the proapoptotic proteins and enhance cell death, as do combinations of agents that inhibit MCL-1 and induce the BH3-only proteins (eg, NOXA). BCL-2=B-cell lymphoma–2; BH3=BCL-2 homology 3; CDK9=cyclin-dependent kinase 9; MCL-1=myeloid cell leukemia sequence 1.
In addition to assessing the tolerability and efficacy of MCL-1 inhibitors in ongoing clinical trials, further clinical evaluation will be needed to understand the optimal role of MCL-1 inhibitors in treatment of hematologic malignancies, including how best to incorporate MCL-1 inhibitors into treatment algorithms. The benefit-risk profile of MCL-1 inhibitors as monotherapy and in various combination regimens will need to be explored and fully understood given recent safety concerns. Finally, to optimize treatment outcomes, it will be important to identify those patients most likely to benefit from MCL-1 inhibition.
In summary, MCL-1 inhibition is a potentially important strategy under investigation for the treatment of hematologic malignancies, including MM, AML, and NHL.
5. Practice Points
Overexpression of MCL-1 is an adverse prognostic marker in hematologic malignancies such as MM, AML, and NHL and is associated with treatment resistance.
Therapy targeting MCL-1 could offer a novel treatment approach for patients relapsing on current treatment options.
MCL-1 inhibitors could potentially synergize with other classes of drugs or standard of care therapies when given in combination regimens.
6. Research Agenda
Phase 1 dose-finding studies in MM, AML, and NHL are currently underway for a number of selective MCL-1 inhibitors, and should define single agent safety and preliminary efficacy.
A major clinical objective will be to determine whether a safe therapeutic window can be found for this new class of inhibitors, as preclinical gene knockout studies (ie permanent complete inhibition) highlight MCL-1 as having physiologic roles in maintenance of cardiac and hepatic tissues.
Defining the efficacy and tolerability of MCL-1 inhibitors in combination with other classes of drugs (including BCL-2 inhibitors, proteasome inhibitors, and MEK inhibitors) in hematologic malignancies will be a high priority.
Understanding whether predictive drug profiling has clinical utility in the management of hematologic malignancies requiring combination therapy is also a priority.
Acknowledgments
The authors thank Jesse Potash (Amgen Inc., Thousand Oaks, CA, USA) and Lee Hohaia and Meghan Johnson (Complete Healthcare Communications, LLC, North Wales, PA, USA), whose work was funded by Amgen Inc., for medical writing assistance in the preparation of this manuscript. This study was supported in part by the UC Davis Paul Calabresi Career Development Award for Clinical Oncology as funded by the National Cancer Institute/National Institutes of Health through grant #5K12-CA138464.
Role of the Funding Source
This work was funded by Amgen Inc. Several of the authors are employed by Amgen Inc., and had a role in writing this manuscript and in the decision to submit the manuscript for publication.
Conflict of Interest
AHW reports receiving honoraria from Novartis, Astellas, Pfizer, Macrogenics, Abbvie, Genentech, Servier, Celgene, Amgen, Astra Zeneca, and Janssen; reports receiving research funding from Novartis, Celgene, Abbvie, Servier, Astra Zeneca, and Amgen; and is a former employee of the Walter and Eliza Hall Institute and receives a fraction of its royalty stream related to venetoclax. AWR reports past research funding from AbbVie, Janssen, and Servier and is an employee of the Walter and Eliza Hall Institute and receives a fraction of its royalty stream related to venetoclax. ASpencer reports receiving consulting fees from Celgene, Janssen, Secura Bio, Specialised Therapeutics Australia, AbbVie, Servier, Haemalogix, and Sanofi; speakers bureaus for Celgene, Janssen, and Takeda; grant/research support from Celgene, Janssen, Amgen, Takeda, Servier, and Haemalogix; and honoraria from Celgene, Janssen, Amgen, Takeda, Secura Bio, Specialised Therapeutics Australia, AbbVie, Servier, Haemalogix, and Sanofi. ASR reports research funding from Amgen and consulting fees from Amgen, Celgene, and Karyopharm. DS reports speakers bureaus for Amgen, Celgene, and Takeda; advisory boards for Amgen, Celgene, Takeda, Karyopharm, Sanofi, Merck, and Celularity; and research funding from Celgene. RBW reports receiving honoraria from Agios, Amphivena, Aptevo, Argenx, Astellas, BioLineRx, BiVictrix, Boehringer Ingelheim, Covagen, Emergent Biosolutions, Jazz, Kite, Pfizer, and Seattle Genetics; reports receiving research funding from Agios, Aptevo, Arog, Amgen, BioLineRx, Jazz, Macrogenics, Pfizer, Seattle Genetics, Selvita, and Stemline; and has a financial interest in Amphivena. SC, PH, ZM, KM, and PKM are employed by and own stock in Amgen. AStein reports speakers bureaus for Amgen, Celgene, and Stemline; and receiving consulting fees from Amgen.
References
- [1].Kollek M, Muller A, Egle A, Erlacher M. Bcl-2 proteins in development, health, and disease of the hematopoietic system. FEBS J 2016;283(15):2779–810. [DOI] [PubMed] [Google Scholar]
- [2].Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol 2014;15(1):49–63. [DOI] [PubMed] [Google Scholar]
- [3].Zhuang J, Brady HJ. Emerging role of Mcl-1 in actively counteracting BH3-only proteins in apoptosis. Cell Death Differ 2006;13(8):1263–7. [DOI] [PubMed] [Google Scholar]
- [4].Carrington EM, Zhan Y, Brady JL, Zhang JG, Sutherland RM, Anstee NS, et al. Anti-apoptotic proteins BCL-2, MCL-1 and A1 summate collectively to maintain survival of immune cell populations both in vitro and in vivo. Cell Death Differ 2017;24(5):878–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 2005;17(3):393–403. [DOI] [PubMed] [Google Scholar]
- [6].Hamouda MA, Jacquel A, Robert G, Puissant A, Richez V, Cassel R, et al. BCL-B (BCL2L10) is overexpressed in patients suffering from multiple myeloma (MM) and drives an MM-like disease in transgenic mice. J Exp Med 2016;213(9):1705–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Slomp A, Peperzak V. Role and Regulation of Pro-survival BCL-2 Proteins in Multiple Myeloma. Front Oncol 2018;8:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vikstrom IB, Slomp A, Carrington EM, Moesbergen LM, Chang C, Kelly GL, et al. MCL-1 is required throughout B-cell development and its loss sensitizes specific B-cell subsets to inhibition of BCL-2 or BCL-XL. Cell Death Dis 2016;7(8):e2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dzhagalov I, St John A, He YW. The antiapoptotic protein Mcl-1 is essential for the survival of neutrophils but not macrophages. Blood 2007;109(4):1620–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev 2013;27(12):1365–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev 2013;27(12):1351–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cazanave SC, Gores GJ. The liver’s dance with death: two Bcl-2 guardian proteins from the abyss. Hepatology 2009;50(4):1009–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hikita H, Takehara T, Shimizu S, Kodama T, Li W, Miyagi T, et al. Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology 2009;50(4):1217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Brinkmann K, Grabow S, Hyland CD, Teh CE, Alexander WS, Herold MJ, et al. The combination of reduced MCL-1 and standard chemotherapeutics is tolerable in mice. Cell Death Differ 2017;24(12):2032–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Belmar J, Fesik SW. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther 2015;145:76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gong JN, Khong T, Segal D, Yao Y, Riffkin CD, Garnier JM, et al. Hierarchy for targeting prosurvival BCL2 family proteins in multiple myeloma: pivotal role of MCL1. Blood 2016;128(14):1834–44. [DOI] [PubMed] [Google Scholar]
- [17].Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun 2018;9(1):5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Touzeau C, Maciag P, Amiot M, Moreau P. Targeting Bcl-2 for the treatment of multiple myeloma. Leukemia 2018;32(9):1899–907. [DOI] [PubMed] [Google Scholar]
- [19].Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016;538(7626):477–82. [DOI] [PubMed] [Google Scholar]
- [20].Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev 2012;26(2):120–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kelly GL, Grabow S, Glaser SP, Fitzsimmons L, Aubrey BJ, Okamoto T, et al. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev 2014;28(1):58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Phillips DC, Xiao Y, Lam LT, Litvinovich E, Roberts-Rapp L, Souers AJ, et al. Loss in MCL-1 function sensitizes non-Hodgkin’s lymphoma cell lines to the BCL-2-selective inhibitor venetoclax (ABT-199). Blood Cancer J 2015;5:e368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Trivigno D, Essmann F, Huber SM, Rudner J. Deubiquitinase USP9x confers radioresistance through stabilization of Mcl-1. Neoplasia 2012;14(10):893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Michels J, Obrist F, Vitale I, Lissa D, Garcia P, Behnam-Motlagh P, et al. MCL-1 dependency of cisplatin-resistant cancer cells. Biochem Pharmacol 2014;92(1):55–61. [DOI] [PubMed] [Google Scholar]
- [25].Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011;471(7336):110–4. [DOI] [PubMed] [Google Scholar]
- [26].Williams MM, Lee L, Hicks DJ, Joly MM, Elion D, Rahman B, et al. Key Survival Factor, Mcl-1, Correlates with Sensitivity to Combined Bcl-2/Bcl-xL Blockade. Mol Cancer Res 2017;15(3):259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Le Gouill S, Podar K, Harousseau JL, Anderson KC. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle 2004;3(10):1259–62. [DOI] [PubMed] [Google Scholar]
- [28].Zhang YK, Wang H, Leng Y, Li ZL, Yang YF, Xiao FJ, et al. Overexpression of microRNA-29b induces apoptosis of multiple myeloma cells through down regulating Mcl-1. Biochem Biophys Res Commun 2011;414(1):233–9. [DOI] [PubMed] [Google Scholar]
- [29].Li XH, Ha CT, Xiao M. MicroRNA-30 inhibits antiapoptotic factor Mcl-1 in mouse and human hematopoietic cells after radiation exposure. Apoptosis 2016;21(6):708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yang Y, Li F, Saha MN, Abdi J, Qiu L, Chang H. miR-137 and miR-197 Induce Apoptosis and Suppress Tumorigenicity by Targeting MCL-1 in Multiple Myeloma. Clin Cancer Res 2015;21(10):2399–411. [DOI] [PubMed] [Google Scholar]
- [31].Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett 2010;584(14):2981–9. [DOI] [PubMed] [Google Scholar]
- [32].Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol 2007;27(11):4006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mojsa B, Lassot I, Desagher S. Mcl-1 ubiquitination: unique regulation of an essential survival protein. Cells 2014;3(2):418–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010;463(7277):103–7. [DOI] [PubMed] [Google Scholar]
- [35].Herrant M, Jacquel A, Marchetti S, Belhacene N, Colosetti P, Luciano F, et al. Cleavage of Mcl-1 by caspases impaired its ability to counteract Bim-induced apoptosis. Oncogene 2004;23(47):7863–73. [DOI] [PubMed] [Google Scholar]
- [36].Clohessy JG, Zhuang J, Brady HJ. Characterisation of Mcl-1 cleavage during apoptosis of haematopoietic cells. Br J Haematol 2004;125(5):655–65. [DOI] [PubMed] [Google Scholar]
- [37].Grabow S, Delbridge AR, Aubrey BJ, Vandenberg CJ, Strasser A. Loss of a Single Mcl-1 Allele Inhibits MYC-Driven Lymphomagenesis by Sensitizing Pro-B Cells to Apoptosis. Cell Rep 2016;14(10):2337–47. [DOI] [PubMed] [Google Scholar]
- [38].Quinn BA, Dash R, Azab B, Sarkar S, Das SK, Kumar S, et al. Targeting Mcl-1 for the therapy of cancer. Expert Opin Investig Drugs 2011;20(10):1397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shah V, Sherborne AL, Walker BA, Johnson DC, Boyle EM, Ellis S, et al. Prediction of outcome in newly diagnosed myeloma: a meta-analysis of the molecular profiles of 1905 trial patients. Leukemia 2018;32(1):102–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wuilleme-Toumi S, Robillard N, Gomez P, Moreau P, Le Gouill S, Avet-Loiseau H, et al. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005;19:1248–52. [DOI] [PubMed] [Google Scholar]
- [41].Gomez-Bougie P, Maiga S, Tessoulin B, Bourcier J, Bonnet A, Rodriguez MS, et al. BH3-mimetic toolkit guides the respective use of BCL2 and MCL1 BH3-mimetics in myeloma treatment. Blood 2018;132(25):2656–69. [DOI] [PubMed] [Google Scholar]
- [42].Peperzak V, Vikstrom I, Walker J, Glaser SP, LePage M, Coquery CM, et al. Mcl-1 is essential for the survival of plasma cells. Nat Immunol 2013;14(3):290–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhang B, Gojo I, Fenton RG. Myeloid cell factor-1 is a critical survival factor for multiple myeloma. Blood 2002;99(6):1885–93. [DOI] [PubMed] [Google Scholar]
- [44].Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, et al. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood 2002;100(1):194–9. [DOI] [PubMed] [Google Scholar]
- [45].Zhang Z, Liu Y, Song T, Xue Z, Shen X, Liang F, et al. An antiapoptotic Bcl-2 family protein index predicts the response of leukaemic cells to the pan-Bcl-2 inhibitor S1. Br J Cancer 2013;108(9):1870–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Xiang Z, Luo H, Payton JE, Cain J, Ley TJ, Opferman JT, et al. Mcl1 haploinsufficiency protects mice from Myc-induced acute myeloid leukemia. J Clin Invest 2010;120(6):2109–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019;133(1):7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wei AH, Strickland SA Jr., Hou JZ, Fiedler W, Lin TL, Walter RB, et al. Venetoclax Combined With Low-Dose Cytarabine for Previously Untreated Patients With Acute Myeloid Leukemia: Results From a Phase Ib/II Study. J Clin Oncol 2019;37(15):1277–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].DiNardo CD, Rausch CR, Benton C, Kadia T, Jain N, Pemmaraju N, et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am J Hematol 2018;93(3):401–7. [DOI] [PubMed] [Google Scholar]
- [50].Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis 2015;6:e1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Richard DJ, Lena R, Bannister T, Blake N, Pierceall WE, Carlson NE, et al. Hydroxyquinoline-derived compounds and analoguing of selective Mcl-1 inhibitors using a functional biomarker. Bioorg Med Chem 2013;21(21):6642–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Study of AZD5991 in Relapsed or Refractory Haematologic Malignancies. Available at: https://clinicaltrials.gov/ct2/show/NCT03218683 Accessed December 4, 2019.
- [53].AMG 176 First in Human Trial in Subjects With Relapsed or Refractory Multiple Myeloma and Subjects With Relapsed or Refractory Acute Myeloid Leukemia. Available at: https://clinicaltrials.gov/ct2/show/NCT02675452 Accessed December 4, 2019.
- [54].A Study of Venetoclax and AMG 176 in Patients With Relapsed/Refractory Hematologic Malignancies. Available at: https://clinicaltrials.gov/ct2/show/NCT03797261 Accessed December 4, 2019.
- [55].Phase I Study of S64315 Administred Intravenously in Patients With Acute Myeloid Leukaemia or Myelodysplastic Syndrome. Available at: https://clinicaltrials.gov/ct2/show/NCT02979366 Accessed December 4, 2019.
- [56].Phase I Dose Escalation Study of Intravenously Administered S64315 in Combination With Orally Administered Venetoclax in Patients With Acute Myeloid Leukaemia. Available at: https://clinicaltrials.gov/ct2/show/NCT03672695 Accessed December 4, 2019.
- [57].Safety, Tolerability, Pharmacokinetics and Efficacy of AMG 397 in Subjects With Multiple Myeloma, NHL, and AML. Available at: https://clinicaltrials.gov/ct2/show/NCT03465540 Accessed December 4, 2019.
- [58].Kaufmann SH, Karp JE, Svingen PA, Krajewski S, Burke PJ, Gore SD, et al. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 1998;91(3):991–1000. [PubMed] [Google Scholar]
- [59].Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006;10(5):375–88. [DOI] [PubMed] [Google Scholar]
- [60].Ramsey HE, Fischer MA, Lee T, Gorska AE, Arrate MP, Fuller L, et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov 2018;8(12):1566–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Moujalled DM, Pomilio G, Ghiurau C, Ivey A, Salmon J, Rijal S, et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia 2019;33(4):905–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, et al. AMG 176, a Selective MCL1 Inhibitor, Is Effective in Hematologic Cancer Models Alone and in Combination with Established Therapies. Cancer Discov 2018;8(12):1582–97. [DOI] [PubMed] [Google Scholar]
- [63].Chang CM, Schroeder JC, Huang WY, Dunphy CH, Baric RS, Olshan AF, et al. Non-Hodgkin lymphoma (NHL) subtypes defined by common translocations: utility of fluorescence in situ hybridization (FISH) in a case-control study. Leuk Res 2010;34(2):190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017;171(2):481–94 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ennishi D, Mottok A, Ben-Neriah S, Shulha HP, Farinha P, Chan FC, et al. Genetic profiling of MYC and BCL2 in diffuse large B-cell lymphoma determines cell-of-origin-specific clinical impact. Blood 2017;129(20):2760–70. [DOI] [PubMed] [Google Scholar]
- [66].Michels J, O’Neill JW, Dallman CL, Mouzakiti A, Habens F, Brimmell M, et al. Mcl-1 is required for Akata6 B-lymphoma cell survival and is converted to a cell death molecule by efficient caspase-mediated cleavage. Oncogene 2004;23(28):4818–27. [DOI] [PubMed] [Google Scholar]
- [67].Cho-Vega JH, Rassidakis GZ, Admirand JH, Oyarzo M, Ramalingam P, Paraguya A, et al. MCL-1 expression in B-cell non-Hodgkin’s lymphomas. Hum Pathol 2004;35(9):1095–100. [DOI] [PubMed] [Google Scholar]
- [68].Kuramoto K, Sakai A, Shigemasa K, Takimoto Y, Asaoku H, Tsujimoto T, et al. High expression of MCL1 gene related to vascular endothelial growth factor is associated with poor outcome in non-Hodgkin’s lymphoma. Br J Haematol 2002;116(1):158–61. [DOI] [PubMed] [Google Scholar]
- [69].Friedberg JW. How I treat double-hit lymphoma. Blood 2017;130(5):590–6. [DOI] [PubMed] [Google Scholar]
- [70].Huang W, Medeiros LJ, Lin P, Wang W, Tang G, Khoury J, et al. MYC/BCL2/BCL6 triple hit lymphoma: a study of 40 patients with a comparison to MYC/BCL2 and MYC/BCL6 double hit lymphomas. Mod Pathol 2018;31(9):1470–8. [DOI] [PubMed] [Google Scholar]
- [71].Nguyen T, Parker R, Zhang Y, Hawkins E, Kmieciak M, Craun W, et al. Homoharringtonine interacts synergistically with bortezomib in NHL cells through MCL-1 and NOXA-dependent mechanisms. BMC Cancer 2018;18(1):1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem 2003;46(20):4259–64. [DOI] [PubMed] [Google Scholar]
- [73].Oliver CL, Bauer JA, Wolter KG, Ubell ML, Narayan A, O’Connell KM, et al. In vitro effects of the BH3 mimetic, (-)-gossypol, on head and neck squamous cell carcinoma cells. Clin Cancer Res 2004;10(22):7757–63. [DOI] [PubMed] [Google Scholar]
- [74].Wan Y, Dai N, Tang Z, Fang H. Small-molecule Mcl-1 inhibitors: Emerging anti-tumor agents. Eur J Med Chem 2018;146:471–82. [DOI] [PubMed] [Google Scholar]
- [75].Arnold AA, Aboukameel A, Chen J, Yang D, Wang S, Al-Katib A, et al. Preclinical studies of Apogossypolone: a new nonpeptidic pan small-molecule inhibitor of Bcl-2, Bcl-XL and Mcl-1 proteins in Follicular Small Cleaved Cell Lymphoma model. Mol Cancer 2008;7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Tzung SP, Kim KM, Basanez G, Giedt CD, Simon J, Zimmerberg J, et al. Antimycin A mimics a cell-death-inducing Bcl-2 homology domain 3. Nat Cell Biol 2001;3(2):183–91. [DOI] [PubMed] [Google Scholar]
- [77].Zhai D, Jin C, Satterthwait AC, Reed JC. Comparison of chemical inhibitors of antiapoptotic Bcl-2-family proteins. Cell Death Differ 2006;13(8):1419–21. [DOI] [PubMed] [Google Scholar]
- [78].Acoca S, Cui Q, Shore GC, Purisima EO. Molecular dynamics study of small molecule inhibitors of the Bcl-2 family. Proteins 2011;79(9):2624–36. [DOI] [PubMed] [Google Scholar]
- [79].Wang G, Nikolovska-Coleska Z, Yang CY, Wang R, Tang G, Guo J, et al. Structure-based design of potent small-molecule inhibitors of anti-apoptotic Bcl-2 proteins. J Med Chem 2006;49(21):6139–42. [DOI] [PubMed] [Google Scholar]
- [80].Mohammad RM, Goustin AS, Aboukameel A, Chen B, Banerjee S, Wang G, et al. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin Cancer Res 2007;13(7):2226–35. [DOI] [PubMed] [Google Scholar]
- [81].van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006;10(5):389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Villalobos-Ortiz M, Ryan J, Mashaka TN, Opferman JT, Letai A. BH3 profiling discriminates on-target small molecule BH3 mimetics from putative mimetics. Cell Death Differ 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Xiang W, Yang CY, Bai L. MCL-1 inhibition in cancer treatment. Onco Targets Ther 2018;11:7301–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Caenepeel S, Belmontes B, Sun J, Cajulis E, Coxon A, Moody G, et al. Preclinical evaluation of AMG 176, a novel, potent and selective Mcl-1 inhibitor with robust anti-tumor activity in Mcl-1 dependent cancer models. Presented at: Americal Association for Cancer Research, April 1–5, 2017, 2017; Washington, DC, USA. [Google Scholar]
- [85].Oncology Pipeline. Amgen Inc; Available at: https://www.amgenpipeline.com/pipeline/#oncology Accessed February 11, 2019. [Google Scholar]
- [86].Hird AW, Secrist JP, Adam A, Belmonte MA, Gangl E, Gibbons F, et al. AZD5991: A potent and selective macrocyclic inhibitor of Mcl-1 for treatment of hematologic cancers [abstract]. Presented at: American Association for Cancer Research Annual Meeting 2017, 2017 Apr 1–5, 2017; Washington, DC. [Google Scholar]
- [87].Rasmussen ML, Kline LA, Park KP, Ortolano NA, Romero-Morales AI, Anthony CC, et al. A Non-apoptotic Function of MCL-1 in Promoting Pluripotency and Modulating Mitochondrial Dynamics in Stem Cells. Stem Cell Reports 2018;10(3):684–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hasan SM, Sheen AD, Power AM, Langevin LM, Xiong J, Furlong M, et al. Mcl1 regulates the terminal mitosis of neural precursor cells in the mammalian brain through p27Kip1. Development 2013;140(15):3118–27. [DOI] [PubMed] [Google Scholar]
- [89].Cohen NA, Stewart ML, Gavathiotis E, Tepper JL, Bruekner SR, Koss B, et al. A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem Biol 2012;19(9):1175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady AS, et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther 2014;13(3):565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Beekman AM, Howell LA. Small-Molecule and Peptide Inhibitors of the Pro-Survival Protein Mcl-1. ChemMedChem 2016;11(8):802–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wang Z, Xu W, Song T, Guo Z, Liu L, Fan Y, et al. Fragment-Based Design, Synthesis, and Biological Evaluation of 1-Substituted-indole-2-carboxylic Acids as Selective Mcl-1 Inhibitors. Arch Pharm (Weinheim) 2017;350(1). [DOI] [PubMed] [Google Scholar]
- [93].Bernardo PH, Sivaraman T, Wan KF, Xu J, Krishnamoorthy J, Song CM, et al. Structural insights into the design of small molecule inhibitors that selectively antagonize Mcl-1. J Med Chem 2010;53(5):2314–8. [DOI] [PubMed] [Google Scholar]
- [94].Bruncko M, Wang L, Sheppard GS, Phillips DC, Tahir SK, Xue J, et al. Structure-guided design of a series of MCL-1 inhibitors with high affinity and selectivity. J Med Chem 2015;58(5):2180–94. [DOI] [PubMed] [Google Scholar]
- [95].Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM, et al. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem 2013;56(1):15–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Gloaguen C, Voisin-Chiret AS, Sopkova-de Oliveira Santos J, Fogha J, Gautier F, De Giorgi M, et al. First evidence that oligopyridines, alpha-helix foldamers, inhibit Mcl-1 and sensitize ovarian carcinoma cells to Bcl-xL-targeting strategies. J Med Chem 2015;58(4):1644–68. [DOI] [PubMed] [Google Scholar]
- [97].Li R, Cheng C, Balasis ME, Liu Y, Garner TP, Daniel KG, et al. Design, synthesis and evaluation of marinopyrrole derivatives as selective inhibitors of Mcl-1 binding to pro-apoptotic Bim and dual Mcl-1/Bcl-xL inhibitors. Eur J Med Chem 2015;90:315–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Desrat S, Remeur C, Geny C, Riviere G, Colas C, Dumontet V, et al. From meiogynin A to the synthesis of dual inhibitors of Bcl-xL and Mcl-1 anti-apoptotic proteins. Chem Commun (Camb) 2014;50(62):8593–6. [DOI] [PubMed] [Google Scholar]
- [99].Ding X, Li Y, Lv L, Zhou M, Han L, Zhang Z, et al. De novo design, synthesis and evaluation of benzylpiperazine derivatives as highly selective binders of Mcl-1. ChemMedChem 2013;8(12):1986–2014. [DOI] [PubMed] [Google Scholar]
- [100].Fiskus W, Cai T, DiNardo CD, Kornblau SM, Borthakur G, Kadia TM, et al. Superior efficacy of cotreatment with BET protein inhibitor and BCL2 or MCL1 inhibitor against AML blast progenitor cells. Blood Cancer J 2019;9(2):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Hird AW, Tron AE. Recent advances in the development of Mcl-1 inhibitors for cancer therapy. Pharmacol Ther 2019;198:59–67. [DOI] [PubMed] [Google Scholar]
- [102].Lee T, Christov PP, Shaw S, Tarr JC, Zhao B, Veerasamy N, et al. Discovery of Potent Myeloid Cell Leukemia-1 (Mcl-1) Inhibitors That Demonstrate in Vivo Activity in Mouse Xenograft Models of Human Cancer. J Med Chem 2019;62(8):3971–88. [DOI] [PubMed] [Google Scholar]
- [103].Hadji A, Schmitt GK, Schnorenberg MR, Roach L, Hickey CM, Leak LB, et al. Preferential targeting of MCL-1 by a hydrocarbon-stapled BIM BH3 peptide. Oncotarget 2019;10(58):6219–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Anstee NS, Bilardi RA, Ng AP, Xu Z, Robati M, Vandenberg CJ, et al. Impact of elevated anti-apoptotic MCL-1 and BCL-2 on the development and treatment of MLL-AF9 AML in mice. Cell Death Differ 2019;26(7):1316–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Merino D, Whittle JR, Vaillant F, Serrano A, Gong JN, Giner G, et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Sci Transl Med 2017;9(401). [DOI] [PubMed] [Google Scholar]
- [106].Phase I Study of MIK665, a Mcl-1 Inhibitor, in Patients With Refractory or Relapsed Lymphoma or Multiple Myeloma. Available at: https://clinicaltrials.gov/ct2/show/NCT02992483 Accessed December 4, 2019. [Google Scholar]
- [107].Brennan MS, Chang C, Tai L, Lessene G, Strasser A, Dewson G, et al. Humanized Mcl-1 mice enable accurate preclinical evaluation of MCL-1 inhibitors destined for clinical use. Blood 2018;132(15):1573–83. [DOI] [PubMed] [Google Scholar]
- [108].Amgen. Amgen highlights new data from Kyprolis (carfilzomib) and oncology pipeline at IMW 2019. Amgen; Available at: https://www.amgen.com/media/news-releases/2019/09/amgen-highlights-new-data-from-kyprolis-carfilzomib-and-oncology-pipeline-at-imw-2019/ Accessed December 4, 2019. [Google Scholar]
- [109].Florean C, Kim KR, Schnekenburger M, Kim HJ, Moriou C, Debitus C, et al. Synergistic AML Cell Death Induction by Marine Cytotoxin (+)-1(R), 6(S), 1’(R), 6’(S), 11(R), 17(S)-Fistularin-3 and Bcl-2 Inhibitor Venetoclax. Mar Drugs 2018;16(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Doi K, Liu Q, Gowda K, Barth BM, Claxton D, Amin S, et al. Maritoclax induces apoptosis in acute myeloid leukemia cells with elevated Mcl-1 expression. Cancer Biol Ther 2014;15(8):1077–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Varadarajan S, Poornima P, Milani M, Gowda K, Amin S, Wang HG, et al. Maritoclax and dinaciclib inhibit MCL-1 activity and induce apoptosis in both a MCL-1-dependent and -independent manner. Oncotarget 2015;6(14):12668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Peterson LF, Sun H, Liu Y, Potu H, Kandarpa M, Ermann M, et al. Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood 2015;125(23):3588–97. [DOI] [PubMed] [Google Scholar]
- [113].Luedtke DA, Su Y, Liu S, Edwards H, Wang Y, Lin H, et al. Inhibition of XPO1 enhances cell death induced by ABT-199 in acute myeloid leukaemia via Mcl-1. J Cell Mol Med 2018;22(12):6099–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Jie H, He Y, Huang X, Zhou Q, Han Y, Li X, et al. Necrostatin-1 enhances the resolution of inflammation by specifically inducing neutrophil apoptosis. Oncotarget 2016;7(15):19367–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Sagawa M, Tabayashi T, Kimura Y, Tomikawa T, Nemoto-Anan T, Watanabe R, et al. TM-233, a novel analog of 1’-acetoxychavicol acetate, induces cell death in myeloma cells by inhibiting both JAK/STAT and proteasome activities. Cancer Sci 2015;106(4):438–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Shao S, Li S, Qin Y, Wang X, Yang Y, Bai H, et al. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int J Oncol 2014;44(5):1661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Thomas D, Powell JA, Vergez F, Segal DH, Nguyen NY, Baker A, et al. Targeting acute myeloid leukemia by dual inhibition of PI3K signaling and Cdk9-mediated Mcl-1 transcription. Blood 2013;122(5):738–48. [DOI] [PubMed] [Google Scholar]
- [118].Venkata JK, An N, Stuart R, Costa LJ, Cai H, Coker W, et al. Inhibition of sphingosine kinase 2 downregulates the expression of c-Myc and Mcl-1 and induces apoptosis in multiple myeloma. Blood 2014;124(12):1915–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Wagner V, Hose D, Seckinger A, Weiz L, Meissner T, Reme T, et al. Preclinical efficacy of sepantronium bromide (YM155) in multiple myeloma is conferred by down regulation of Mcl-1. Oncotarget 2014;5(21):10237–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Wu Q, Lv T, Chen Y, Wen L, Zhang J, Jiang X, et al. Apoptosis of HL-60 human leukemia cells induced by Asiatic acid through modulation of B-cell lymphoma 2 family proteins and the mitogen-activated protein kinase signaling pathway. Mol Med Rep 2015;12(1):1429–34. [DOI] [PubMed] [Google Scholar]
- [121].Krystof V, Baumli S, Furst R. Perspective of cyclin-dependent kinase 9 (CDK9) as a drug target. Curr Pharm Des 2012;18(20):2883–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Zeidner JF, Karp JE. Clinical activity of alvocidib (flavopiridol) in acute myeloid leukemia. Leuk Res 2015;39(12):1312–8. [DOI] [PubMed] [Google Scholar]
- [123].Lyle L, Daver N. Current and emerging therapies for patients with acute myeloid leukemia: a focus on MCL-1 and the CDK9 pathway. Am J Manag Care 2018;24(16 Suppl):S356–S65. [PubMed] [Google Scholar]
- [124].Chen R, Keating MJ, Gandhi V, Plunkett W. Transcription inhibition by flavopiridol: mechanism of chronic lymphocytic leukemia cell death. Blood 2005;106(7):2513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Xiao Y, Nimmer P, Sheppard GS, Bruncko M, Hessler P, Lu X, et al. MCL-1 Is a Key Determinant of Breast Cancer Cell Survival: Validation of MCL-1 Dependency Utilizing a Highly Selective Small Molecule Inhibitor. Mol Cancer Ther 2015;14(8):1837–47. [DOI] [PubMed] [Google Scholar]
- [126].Choudhary GS, Tat TT, Misra S, Hill BT, Smith MR, Almasan A, et al. Cyclin E/Cdk2-dependent phosphorylation of Mcl-1 determines its stability and cellular sensitivity to BH3 mimetics. Oncotarget 2015;6(19):16912–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Flynn J, Jones J, Johnson AJ, Andritsos L, Maddocks K, Jaglowski S, et al. Dinaciclib is a novel cyclin-dependent kinase inhibitor with significant clinical activity in relapsed and refractory chronic lymphocytic leukemia. Leukemia 2015;29(7):1524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Gojo I, Sadowska M, Walker A, Feldman EJ, Iyer SP, Baer MR, et al. Clinical and laboratory studies of the novel cyclin-dependent kinase inhibitor dinaciclib (SCH 727965) in acute leukemias. Cancer Chemother Pharmacol 2013;72(4):897–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Kumar SK, LaPlant B, Chng WJ, Zonder J, Callander N, Fonseca R, et al. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 2015;125(3):443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Study to Assess Safety, Tolerability, Pharmacokinetics and Antitumor Activity of AZD4573 in Relapsed/Refractory Haematological Malignancies. Available at: https://clinicaltrials.gov/ct2/show/NCT03263637?term=AZD4573&rank=1 Accessed December 4, 2019.
- [131].Prukova D, Andera L, Nahacka Z, Karolova J, Svaton M, Klanova M, et al. Co-targeting of BCL2 with venetoclax and MCL1 with S63845 is synthetically lethal in vivo in relapsed mantle cell lymphoma. Clin Cancer Res 2019. [DOI] [PubMed] [Google Scholar]
- [132].Tahir SK, Smith ML, Hessler P, Rapp LR, Idler KB, Park CH, et al. Potential mechanisms of resistance to venetoclax and strategies to circumvent it. BMC Cancer 2017;17(1):399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Choudhary GS, Al-Harbi S, Mazumder S, Hill BT, Smith MR, Bodo J, et al. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis 2015;6:e1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Guieze R, Liu VM, Rosebrock D, Jourdain AA, Hernandez-Sanchez M, Martinez Zurita A, et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019;36(4):369–84 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Chyla B, Daver N, Doyle K, McKeegan E, Huang X, Ruvolo V, et al. Genetic Biomarkers Of Sensitivity and Resistance to Venetoclax Monotherapy in Patients With Relapsed Acute Myeloid Leukemia. Am J Hematol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov 2016;6(10):1106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Nechiporuk T, Kurtz SE, Nikolova O, Liu T, Jones CL, D’Alessandro A, et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov 2019;9(7):910–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Pham LV, Huang S, Zhang H, Zhang J, Bell T, Zhou S, et al. Strategic Therapeutic Targeting to Overcome Venetoclax Resistance in Aggressive B-cell Lymphomas. Clin Cancer Res 2018;24(16):3967–80. [DOI] [PubMed] [Google Scholar]
- [139].Greaves G, Milani M, Butterworth M, Carter RJ, Byrne DP, Eyers PA, et al. BH3-only proteins are dispensable for apoptosis induced by pharmacological inhibition of both MCL-1 and BCL-XL. Cell Death Differ 2019;26(6):1037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Teh TC, Nguyen NY, Moujalled DM, Segal D, Pomilio G, Rijal S, et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia 2018;32(2):303–12. [DOI] [PubMed] [Google Scholar]
- [141].VENCLEXTA (Venetoclax). Full Prescribing Information, AbbVie Inc, North Chicago, IL, 2018. [Google Scholar]
- [142].Bose P, Gandhi V, Konopleva M. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma 2017;58(9):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Luedtke DA, Niu X, Pan Y, Zhao J, Liu S, Edwards H, et al. Inhibition of Mcl-1 enhances cell death induced by the Bcl-2-selective inhibitor ABT-199 in acute myeloid leukemia cells. Signal Transduct Target Ther 2017;2:17012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Mukherjee N, Strosnider A, Vagher B, Lambert KA, Slaven S, Robinson WA, et al. BH3 mimetics induce apoptosis independent of DRP-1 in melanoma. Cell Death Dis 2018;9(9):907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Nangia V, Siddiqui FM, Caenepeel S, Timonina D, Bilton SJ, Phan N, et al. Exploiting MCL1 Dependency with Combination MEK + MCL1 Inhibitors Leads to Induction of Apoptosis and Tumor Regression in KRAS-Mutant Non-Small Cell Lung Cancer. Cancer Discov 2018;8(12):1598–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Ponder KG, Matulis SM, Hitosugi S, Gupta VA, Sharp C, Burrows F, et al. Dual inhibition of Mcl-1 by the combination of carfilzomib and TG02 in multiple myeloma. Cancer Biol Ther 2016;17(7):769–77.27246906 [Google Scholar]
- [147].Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol 2012;30(5):488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Touzeau C, Ryan J, Guerriero J, Moreau P, Chonghaile TN, Le Gouill S, et al. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 2016;30(3):761–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Del Gaizo Moore V, Letai A. BH3 profiling--measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer Lett 2013;332(2):202–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Montero J, Letai A. Dynamic BH3 profiling-poking cancer cells with a stick. Mol Cell Oncol 2016;3(3):e1040144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Lehmberg TZ, Valentin R, Fernandes SM, Brown JR, Davids MS. Dynamic BH3 Profiling Predicts Patient Response and MRD Status in Chronic Lymphocytic Leukemia (CLL) Patients Undergoing Frontline Treatment with Kinase Inhibitor Plus FCR (KI + FCR). Presented at: American Society of Hematology, December 1–4, 2018, 2108; San Diego, CA, USA. [Google Scholar]