

Abstract
Introduction.
16α-[18F]Fluoroestradiol (FES), a PET radiotracer for the estrogen receptor (ER) in breast cancer, was the first receptor-targeted PET radiotracer for oncology and is continuing to prove its value in clinical research, antiestrogen development, and breast cancer care. The story of its conception, design, evaluation and use in clinical studies parallels the evolution of the whole field of receptor-targeted radiotracers, one greatly influenced by the research and intellectual contributions of William C. Eckelman.
Methods and Results.
The development of methods for efficient production of fluorine-18, for conversion of [18F]fluoride ion into chemically reactive form, and for its rapid and efficient incorporation into suitable estrogen precursor molecules at high molar activity, were all methodological underpinnings required for the preparation of FES. FES binds to ER with very high affinity, and its in vivo uptake by ER-dependent target tissues in animal models was efficient and selective, findings that preceded its use for PET imaging in patients with breast cancer.
Advances in Knowledge and Implications for Patient Care.
Comparisons between ER levels measured by FES-PET imaging of breast tumors with tissue-specimen ER quantification by IHC and other methods show that imaging provided improved prediction of benefit from endocrine therapies. Serial imaging of ER by FES-PET, before and after dosing patients with antiestrogens, is used to determine the efficacious dose for established antiestrogens and to facilitate clinical development of new ER antagonists. Beyond FES imaging, PET-based hormone challenge tests, which evaluate the functional status of ER by monitoring rapid changes in tumor metabolic or transcriptional activity after a brief estrogen challenge, provide highly sensitive and selective predictions of whether or not there will be a favorable response to endocrine therapies. There is sufficient interest in the clinical applications of FES that FDA approval is being sought for its wider use in breast cancer.
Conclusions.
FES was the first PET probe for a receptor in cancer, and its development and clinical applications in breast cancer parallel the conceptual evolution of the whole field of receptor-binding radiotracers.
Keywords: receptor-targeting, radiopharmaceuticals, endocrine therapy, tamoxifen, hormone-challenge test, breast cancer
INTRODUCTION
For over a century, estrogens produced by the ovaries the were known to have remarkable stimulatory effects on the uterus and breast; in fact, as early as 1896, it was shown that oophorectomy (surgical removal of the ovaries) could be used to treat some breast cancers [1, 2]. Consequently, some sort of receptors for these hormones were presumed to be present in these target tissues. However, it was not until the early 1960’s, with the production of [3H]estradiol (Figure 1) and [3H]hexestrol at high molar activity, that specific, high affinity binding was detected in the uterus of rats and sheep [3, 4]. Shortly thereafter, the binding was shown to be due to a soluble protein [5], which was considered to be the long-sought-after estrogen receptor (ER). My early research at the time was focused on developing tritium-and radioiodine-labeled estrogens, some chemically or photochemically reactive, as molecular probes for characterizing the biophysical and biochemical properties of this ER [6, 7].
Figure 1.

Early estrogens radiolabeled at high molar activity. A. [3H]Estradiol labeled by radiotritiation of 6-dehydroestradiol. B. 3/5-[125I]Iodoestradiol radiolabeled by A-ring electrophilic radioiodination. C. 3-[125I]Iodo-meso-Hexestrol radiolabeled by electrophilic radioiodination, and separated from a product mixture of unlabeled, monoiodo, symmetrical and unsymmetrical diiodo, tritiodo, and tetraiodo products.
In the early 1970’s, I became aware of the possibility of using certain high molar activity estrogen analogs for imaging of ER in breast tumors by gamma scintigraphy, with the presumed benefit of locating ER-positive tumors and perhaps predicting whether these tumors might be responsive to endocrine therapies. At the time, endocrine therapy involved oophorectomy (sometimes even adrenalectomy or hypophysectomy), therapeutic operations that had begun in the 1890’s [1]); these invasive operations, however, provided beneficial responses in only about 35% of patients [8]. In the 1970’s, the antiestrogen tamoxifen, the first targeted agent in oncology, was beginning to replace surgical ovariectomy, but it did not improve the chance for clinical benefit [8]. Hence, the breast cancer field was ripe for the development of new diagnostic or predictive methods to improve the selection of patients who might benefit from endocrine therapies from those who might require cytotoxic chemotherapies.
My goal in this review is to chart an historical account of the progression of ideas and advances that led to the discovery by my laboratory in 1984 of 16α-[18F]fluoroestradiol (FES) [9, 10], the first PET imaging agent for a receptor target in cancer. I will cover these events and subsequent developments associated with the clinical uses of FES and related PET imaging paradigms to characterize ER activity in breast cancer that arose from a multi-decade collaboration between my laboratory at the University Illinois and numerous collaborators at Washington University. In doing so, I will attempt to present the major insights and advances within the context of the era in which they occurred; this is instructive, because the development of FES tracks closely with the technical and conceptual advances that were taking place in the field. Considering that this issue of Nuclear Medicine and Biology is to honor William C. Eckelman, I will at times connect these progressions to his many contributions as a leader, enabler, and promoter of the whole field of receptor-binding radiotracers; to him, I and all other participants in this field owe a great debt of gratitude.
It is important to preface the account that follows by noting that when we began our work on the development of an in vivo imaging agent for ER, the feasibility of actually accomplishing this was unclear: (a) Could receptor-targeted probes be synthesized rapidly and efficiently at the no-carrier-added level (i.e., without dilution with non-radioactive F-19 carrier) to achieve sufficiently high molar activities? (b) Would the receptor binding characteristics of these probes, together with the low levels of ER in tumors, enable enough activity to accumulate in the target and at sufficient contrast with non-target areas, so that an in vivo image based on receptor binding could actually be obtained? And finally, (c) could medically meaningful information be obtained from these images? The answers, fortunately, were yes in all cases, as detailed below.
INITIAL EFFORTS TO OBTAIN GAMMA-EMITTING ESTROGENS AS ER IMAGING AGENTS
Early attempts had been made to radiolabel the steroid, estradiol, with radioiodine, but substitution with this bulky halogen by electrophilic radioiodination occurred in the A-ring ortho to the phenol; this interfered with ER-ligand interaction and drastically lowered binding affinity (Figure 1). From my early work on ER probe development, I recognized that the very high affinity non-steroidal bisphenolic estrogen, hexestrol, was a better prospect for aromatic radioiodination, because placement of a single iodine on only one of the two phenols left the other one unsubstituted, allowing it to mimic the A-ring of estradiol, which was known to play a critical role for high affinity ER binding [6, 7]. Electrophilic iodination of hexestrol produced a mixture of five iodohexestrols, which could readily be separated, and binding measurements showed that the mono-iodohexestrol retained good ER binding affinity (Figure 1) [6, 7]; so, this compound seemed like a good prospect for in vivo imaging of ER.
When [125I]iodohexestrol was injected into immature female rats, however, we were surprised that very little activity accumulated in the uterus, the most ER-rich target tissue, while in the same model, [3H]estradiol showed very high and selective uterine uptake [7]. High activity from [125I]iodohexestrol was found throughout the body, a phenomenon that was puzzling, but we later realized was instructive (see below) [7]. Around this time and shortly thereafter, other researchers were working on ER imaging agents, using radioiodinated steroidal estrogens, with estradiol substituted at the 16α position [11] or with a 17α-(2-iodovinyl) group [12–17]. The 16α-[125I]iodoestradiol of Hochberg [11], in particular, seemed to have good ER binding affinities and greater promise for target-selective in vivo distribution than we had experienced with [125I]iodohexestrol.
A FORTUITOUS MEETING, BROMINE-77, AND CLARIFYING BINDING AFFINITY AND SELECTIVITY (BSI)
In late 1974, I had a fortuitous meeting with Michael Welch at Washington University Medical School in St. Louis, about a 3-hour drive from the University of Illinois at Urbana-Champaign for a discussion on in vivo imaging of breast cancers. Mike and I quickly recognized how mutually beneficial it would be to collaborate on this effort, with me providing expertise on receptor-targeted ligand design and synthesis, including adaptation to radiolabeling, and he on radioisotope production, radiolabeling, and animal experimentation, with a route to clinical imaging studies through colleagues at his institution. At the time, Mike was receiving periodic shipments of bromine-77 from Los Alamos National Laboratory that he was using to radiobrominate proteins; so, we decided to make some [77Br]bromoestrogens. My laboratory quickly produced a series of 16α-substituted bromoestrogens by electrophilic bromination [18], and we found that one of them bound to ER even better than estradiol (Figure 2) [18]. It proved relatively straightforward to accomplish electrophilic radiobromination at the tracer scale, producing 16α-[77Br]bromoestradiol [19], as well as some other analogs, in good yields and at very high molar activity [20, 21]. Biodistribution studies confirmed that these analogs were taken up efficiently and selectively by the uterus of immature female rats, in a fashion similar to that of [3H]estradiol [19]. (Immature animals were used because endogenous levels of estradiol are low.) There was also very good, selective uptake in an ERα-positive DMBA-induced mammary tumor in an adult rat [19].
Figure 2.

Bromo and iodoestrogens and their binding values: Binding Selectivity Index (BSI), Relative Binding Affinity (RBA), and Non-specific Binding (NSB). The symbols (---) and (+++) indicate relative specific uptake by the uterus in immature female rats.
Now, with a set of radiolabeled probes for ER binding in vivo, we began to look closely for relationships between their ligand binding characteristics and the efficiency and selectivity of their uptake by the uterus. It became apparent that their uptake characteristics were affected positively by the ER binding affinity but negatively by their non-specific binding. Accordingly, we developed an index that we termed the “binding selectivity index” (BSI), which was the ratio of their binding affinity for ER divided by their affinity for non-specific binding sites, the latter thought to be largely due to the binding of these lipophilic molecules by serum albumin as well as by their interaction with lipid components such as membranes [22, 23]. The receptor binding was easily determined by competitive radiometric binding assays with [3H]estradiol, and was expressed as a relative binding affinity (RBA) value, with E2 being assigned a value of 100. Their non-specific binding (NSB) could be measured by equilibrium dialysis, but NSB proved to be directly related to a measure of their lipophilicity, LogPo/w, which could be measured by octanol water partitioning [24] or by reversed phase HPLC [25]; even more conveniently, NSB proved to be proportional to the calculated cLogP values [23]. The NSB of E2 was assigned a value of 1, giving the BSI for E2 (BSI = RBA/NSB = 100/1) a value of 100.
BSI values that we obtained for a number of early compound are shown in Figure 2. More so than binding affinity (RBA) values alone, we found BSI values to be useful predictors of in vivo uptake efficiency and selectivity, and in particular to the retention of a tracer by the uterus as well as other tissues [26]. Comparison between 16α-[77Br]bromoestradiol (BE2) with 16α-[77Br]bromo-11β-methoxyestradiol (BME2) was especially instructive: The former compound had a BSI of 84 (130/1.5), rather similar to that for estradiol. The methoxy group in the latter was strategically placed at the 11β position because this substitution was known to improve the potency of estrogens in in vivo bioactivity assays, notably exemplified by the highly active estrogen, moxestrol [27]. Introduction of the polar 11β-methoxy group lowered ER binding affinity about 2.5 fold, but it reduced the non-specific binding 10-fold, resulting in an elevated BSI value of 390 (39/0.1). In the immature rat uterus, uterine BE2 activity had a half-life of 6 hours, whereas BME2 was retained with a half-life of 24 hours, although it could be rapidly washed out in 1 hour with a chase of unlabeled E2 [20]. Thus, target tissue engagement of different radiopharmaceuticals tracked more with their BSI value than their RBA value. This pharmacokinetic behavior, termed “target tissue trapping”, can be understood in terms of receptor rebinding [28], and with reference to a three-compartment model, it occurs when a dissociating ligand is rebound by the receptor at a much greater rate (k2 x [R]) than its exit from the extravascular compartment (k−1) [26]. It is likely that other ligands having very high BSI values will exhibit this type of target-tissue retention behavior.
Consideration of these specific and non-specific binding characteristics of ER radioligands enabled us to being to understand the poor biodistribution of [125I]iodohexestrol [7]. Despite its respectable binding affinity (RBA = 14), it had greatly elevated non-specific binding and was predicted to have a very modest BSI of only 1.4 (14/9.7) (Figure 2) [7]. We found, however, that the in vivo ER-specific binding of this compound was further compromised by a specific, high affinity, off-target binding. Because the o-iodophenol unit of iodohexestrol sufficiently resembles thyroxine, it was bound tightly by thyroxine-binding prealbumin, an abundant serum protein; by contrast, the steroid-based radioligands did not bind to this prealbumin [7]. Thus, to obtain favorable, target-selective biodistribution, it proved important to avoid interaction with other abundant specific binding proteins, in addition to avoiding excessive non-specific binding. The situation with another serum binding protein, sex-hormone binding globulin (SHBG), proved to be both very different and more complex, and will be discussed later [29, 30].
THE BEGINNING OF A NEW FIELD AND THE INFLUENCE OF WILLIAM C ECKELMAN
As we were working to define the ligand binding characteristics needed for effective ER-targeted imaging, others in the field were grappling with these same issues. Bill Eckelman, who himself was working on ER imaging in breast cancer [12], took in 1980 an early transformative step in support of the field by convening a small conference entitled “Receptor-Binding Radiotracers,” the report of which appeared in two volumes with the same title, published by the CRC press in 1982. My contribution at the conference became a 33-page chapter entitled “In Vivo and In Vitro Steroid Receptor Assays in the Design of Estrogen Pharmaceuticals” [23]. In it, I covered the issues outlined above, namely, the elements of ligand binding and receptor characteristics that need to be considered carefully and quantitatively in the design of receptor-targeted radiotracers. The conference and volume covered a multitude of diverse topics critical to the success and utility of receptor-targeted imaging.
This meeting and the books that emerged from it also marked the beginning of a continuing series of contributions through which Bill Eckelman provided wise counsel to those practicing in the field of receptor-binding radiotracer research as it was developing. Many of these took the form of reviews of new concepts and technologies [31–34] and observations on the need to refine probe design [35–46], items that were often published as short commentaries in Nuclear Medicine and Biology, the journal for which he served as Editor-in-Chief for more than 30 years, beginning in 1985. Bill was also instrumental in building the infrastructure that supported the field through his efforts in creating the Society of Radiopharmaceutical Sciences (SRS) and by helping with the organization of many of the biennial international meetings of SRS, which have served as an important focal point for both new and established investigators to meet and exchange findings and ideas. The 24th of these meetings is scheduled for the summer of 2021, in Nantes, France.
ATTEMPTS TO IMAGE MAMMARY TUMORS IN ANIMALS AND BREAST TUMORS IN WOMEN WITH Br-77 E2. A SWITCH TO F-18 WITH PROSPECTS OF PET IMAGING OF ER
We were encouraged by the relative ease with which we could prepare these bromine-77 labeled estrogens, by their high and understandable ER binding affinity and selectivity, and by their excellent target-selective uptake efficacy and selectivity in biodistribution studies. Our experience in using them for imaging ER-positive tumor targets in vivo, however, proved disappointing. With 2D gamma imaging, we could see in rats some accumulation of [77Br]BE2 activity in DMBA-induced mammary tumors above a relatively high background level of activity [18, 19]. Imaging of breast tumors in humans with [77Br]BE2, however, was poor, as extensive scattering from other sites, notably liver, further compromised the weak images of tumors [47].
These challenges of in vivo ER-targeted imaging with bromine-77 radiolabeled estrogens prompted us to switch isotopes to fluorine-18, which also allowed us to benefit from the rapid development of PET imaging at Washington University. The fluorine atom is small, not much larger than a hydrogen, but it is polar and induces a large dipole moment in a C-F bond. Nevertheless, estriol, which also has a polar hydroxyl group at the 16α position, is a very active and high affinity estrogen; so, this looked like a logical site for fluorine-18 substitution.
There were a number of challenges that needed to be overcome to accomplish this: (a) In contrast to bromine, introduction of fluorine by electrophilic routes is very challenging, particularly at the tracer level in high molar activity. (b) Nucleophilic radiofluorination can be difficult, because hydration by even small amounts of water markedly compromises the inherent high nucleophilicity of fluorine ion, and the adequate drying and solubilizing of fluorine ion is not straightforward. (c) Finally, cyclotron production of fluorine-18, as initially done in gas targets by the 20Ne(d,α)18F reaction, gave high yields, but efficient extraction of the activity from the target in a chemically reactive form (e.g., as H[18F]F) proved to be quite cumbersome [48]. These were not problems unique to us, as a number of other sites worldwide were being challenged by the same issues in making fluorine-18 a practical radionuclide for labeling PET radiotracers. However, within a relatively short period of time, we and others were obtaining large quantities of [18F]fluoride ion from H[18O]O water targets by the 18O(p,n)18F reaction. By drying it quickly by azeotropic distillation with acetonitrile in the presence of tetrabutyl ammonium salts, or other lipophilic cations, we could produce [18F]fluorine in an organic soluble, chemically reactive form [9, 10, 49].
An additional, unexpected challenge we encountered was a major dilution of the molar activity of [18F]fluoride ion present in target water after extraction from the target, which initially greatly reduced the molar activity of FES. The source of the non-radioactive fluoride carrier was traced to Teflon (polyperfluoroethylene!) tubing that released fluoride ion either due to degradation from the radiation field during cyclotron target bombardment or during passage of high levels of radioactive material through the tubing. After we used polyethylene tubing to replace all of the Teflon lines used during isotope production and transfers during the radiochemical syntheses, we obtained FES product at the very high molar activities required for in vivo imaging of low abundance targets such as ER [10]. A more extensive study of fluoride ion release from Teflon lines during radiochemical syntheses has recently been published [50].
PREPARATION OF VARIOUS [18F]ESTROGENS, AND THE BIRTH OF FES
Relatively quickly, we prepared a number of fluorine-labeled steroidal and non-steroidal estrogens [9, 51–53], but we soon focused on the 16 epimers of fluoroestradiol. These could be prepared by [18F]fluoride ion displacement in THF, which proceeded with inversion of stereochemistry of the corresponding epimeric 16 trifloxy estrones; a second triflate served to protect the 3-phenol (Figure 3). The 17-ketone was then reduced with LiAlH4 in ether at −78 °C, this solvent and the low temperature giving the 17β-epimeric alcohol with good selectivity; the 3-O-triflate was then removed upon warming to RT [9]. Of the two 16-fluoroestradiols, the 16α epimer bound to ER about twice as well as the 16β isomer, with calculated BSI values of 114 (80/0.7) for 16α and 43 (30/0.7) for 16β [54]. Hence, for further evaluation, we focused on 16α-[18F]fluoroestradiol, which we named, FES (Figure 4).
Figure 3.

Synthesis of the two 16-[18F]fluoroestradiols. FES (the 16α-epimer, bottom right) was carried forward for PET imaging studies.
Figure 4.

FES, 16α-[18F]Fluoroestradiol, the featured compound.
With appropriate precautions for preserving the high molar activity of [18F]fluoride ion (see above), we routinely obtained FES at molar activities in excess of 1000 Ci/mmol (37 GBq/μmol), which was sufficient for in vivo PET imaging. As an aside, it is worth noting that since our original publication of this synthesis of FES [10], a number of alternative routes have been reported; some of these avoided stereochemical issues at C-17 [55–57] or were developed to be more adaptable to automated synthesis systems [58–60], which were not available at the time of our original work.
Molar activities were typically determined by HPLC analysis relative to a set of mass standards; however, we realized that any unlabeled, receptor-binding material that might be collected together with the radiolabeled sample but not evident as a discrete peak (e.g., by continuous leakage from a column that had previously been used for preparative work with large amounts of steroidal compounds), could have the net effect of reducing molar activities by competing for binding to ER. Thus, after full decay of a sample of FES, we would occasionally determine unlabeled mass in terms of its ability to bind to estrogen receptor in competitive binding assays with a [3H]estradiol tracer. From these, we could calculate an “effective molar activity” (termed, at the time, “effective specific activity”) [61]. In a good preparations of FES, the molar activity and effective molar activity were the same.
In initial screening biodistribution studies in immature female rats, FES showed high uptake in the uterus and ovary that was nearly completely blocked by prior administration of a high, blocking dose of unlabeled E2 (Figure 5). Uptake in other, non-target organs varied, but was essentially unaffected by a blocking dose of E2; uptake in bone, which is an indication of metabolic defluorination, was minimal. Other F-18 labeled estrogens that we prepared around the same time showed no particular advantage over FES [10]; so, FES became the focus of our clinical development efforts done in collaboration with Washington University colleagues, which are discussed below. (More recently, versions of FES in which strategically placed substituents, including unlabeled fluorine, appear to enhance binding characteristics and reduce metabolic clearance, are reported to provide somewhat better images than those obtained with FES [62].)
Figure 5.

Tissue biodistribution of FES in immature rats alone or blocked with unlabeled estradiol. From reference 10.
EFFORTS TO IMPROVE FES AND CONSIDERATION OF SPECIFIC STEROID BINDING PROTEINS IN BLOOD
Despite the favorable in vivo behavior of FES and while work on its clinical use was unfolding, we continued to explore structural alterations of this probe through which its ER binding affinity might be increased and its non-specific binding and metabolic inactivation reduced, with the aim of obtaining higher target tissue uptake and reduced levels of off-target activity. We believed that these improvements might prove critical for the ultimate utility in the more challenging task of imaging of ER-positive breast tumors in humans, where ER levels are present at much lower levels than in the rat uterus. None of the fluorine-18 labeled non-steroidal estrogens we explored stood out in terms of improved binding characteristics or target uptake efficacy and selectivity in biodistribution studies [51–53]; so, we kept our focus on steroidal systems, eventually preparing and evaluating in vitro and in animals more than 20 analogs of FES (some are shown in Figure 6).
Figure 6.

FES analogs with 11β and 17α substituents, and 16β-epimers. Some had elevated relative binding affinity (RBA) values (and lowered non-specific binding (NSB), giving very high binding selectivity index (BSI) values – not shown). In immature rats, βFMOX had the highest uterine %ID/g, nearly 3x that of FES, but gave very poor distribution in humans, presumed due to low SHBG binding (see text).
With steroidal estrogens, it was known that a 17α-ethynyl group would block hepatic oxidation of the 17β-alcohol to the lower affinity estrone, an 11β-ethyl group would raise binding affinity (at the cost of increased lipophilicity), and an 11β-methoxy group would preserve good binding but reduce lipophilicity, with a potential benefit in reduced non-specific binding. We did obtain the expected effect of these alterations in terms of ER binding affinity and lipophilicity, and some compounds had markedly higher BSI values than FES, and also showed significantly improved uterine uptake efficacy [23, 25, 63–65]. Because of our experience with the spurious binding of [125I]iodohexestrol by a specific binding protein in serum (thyroxine-binding prealbumin) [23], we also measured the binding of these FES analogs to two serum steroid-binding proteins [29, 66]. We were not alone in having an interest in the how such steroid-binding proteins in blood might be altering the uptake and distribution of FES, in particular whether binding by high levels of such serum proteins might sequester FES in the circulation, reducing its access to ER-mediated uptake by breast tumors [30, 67].
Steroid binding transport proteins in blood are well known, and although it is somewhat controversial, there are thoughts that in humans, sex hormone binding globulin (SHBG, also known as SBP) might not only assist in the transport of lipophilic estrogens and androgens, shielding them from metabolism, but might also facilitate their uptake into target tissues through interactions of the steroid-bound SHBG with cell surface receptors [68, 69]. On the other hand, high affinity binding of estrogens by SHBG might have the opposite effect by sequestering them and hindering their target cell uptake, as was suggested in some studies of FES in women [30, 67]. SHBG levels in women is particularly high during pregnancy, but is still found at very significant levels during adulthood. While SHBG is not found in rodent species most frequently used in biodistribution studies (rats and mice), these rodents contain another estrogen-binding serum protein, alpha fetoprotein (AFP), which is at very high levels in neonates but is still present at significant levels in the immature rats used for our biodistribution studies. Curiously, AFP in humans does not bind steroids, but its binding in rodent blood might be facilitating steroid transport. We began to routinely assay FES and our other FES analogs for their binding to SHBG and AFP by competitive radiometric binding assays [25]. Again, with E2 as the binding standard (100%), FES had an affinity of 9.5% for SHBG and of 101% for AFP [25]. Notably, the other analogs having high ER binding affinities and low predicted non-specific binding due to substitutions at 11β and/or 17α, however, often had drastically lower affinities for both of these blood binding proteins [25].
Some years later, in the early 1990’s when FES was already being used successfully in humans to image ER in breast tumors, we compared FES with 11β-methoxy-17α-ethynyl-16β-[18F]fluoroestradiol (βFMOX). βFMOX had a reasonably good BSI of 116 (78/0.67), but was particularly notable in having a three-fold higher uptake than FES in rat uterus (Figure 6); it also had very low affinity for both AFP and SHBG [65]. On the basis of these intriguing features, we advanced it to a small trial in breast cancer patients, but we were astounded by the remarkably unpromising results: βFMOX activity hardly redistributed from the site of injection [70]! Our only explanation is that the very low binding affinity of βFMOX for SHBG (0.037%) [65] accounted for its very poor distribution properties in humans. By contrast, FES, with its significant SHBG binding affinity 9.5% that of estradiol, had excellent distribution and target uptake behavior in humans. Thus, significant binding of these [18F]-labeled estrogens to SHBG appears to be necessary for their efficient transport to target sites in humans.
Our experience βFMOX dissuaded us from continuing efforts to enhance the properties of FES through alterations that disabled their binding to steroid binding proteins. It did, however, encourage us to consider the varied roles that serum binding proteins might play in the in vivo distribution of receptor-binding radiotracers. It remains an issue that is not fully resolved and was encountered again in other studies we conducted on PET probes for the androgen receptor in prostate cancer [71, 72].
CLINICAL DEVELOPMENT OF FES – EARLY STUDIES AND THE FIRST PET IMAGE OF A RECEPTOR IN HUMAN BREAST CANCER
Following our first report in 1984 of the favorable biodistribution characteristics of FES in immature female rats [10], we imaged a series of DMBA-induced mammary tumors in adult female rats. We examined as a function of time, metabolism and tumor uptake of FES, as well as blood volume and blood flow in tumors and possible correlations between uptake and ER levels in tumors and in the uterus, finding that the relationships among these factors were complex [73]. We also performed a dose-response study of FES in immature female rats [74]; increasing doses of FES were obtained by adding progressively larger quantities of unlabeled FES. An interesting finding from this study was that uptake in the very ER-rich uterus, and to a lesser extent the ovaries, required a rather high mass dose to saturate. By contrast, other tissues not normally thought to be ER-containing, such as muscle and esophagus, appeared to have some levels of ER, sufficient to give low but measurable levels of specific uptake of FES activity. Interestingly, these sparse sites were saturated by addition of even small amounts of unlabeled FES. Concerns that the uptake of FES in very ER-rich target tissues might be “flow limited” and not clearly proportional to ER levels were moderated by the fact the ER levels in breast tumors were known to be considerably lower than those in rat uterus [23].
Our first PET images of ER-positive breast tumors in women were published in 1988 in Radiology [75]. One picture, presented in advance of the publication at the 1987 national meeting of the Society of Nuclear Medicine, was highlighted as “Image of the Year” (Figure 7) [76]. As it was the first PET image of a receptor in cancer, it represented a milestone in the development of receptor-targeted radiotracers for PET imaging. This exciting in vivo observation of ER in breast tumors by FES-PET prompted us to consider deeply in what ways FES-PET imaging might prove valuable in improving the management of breast cancer.
Figure 7.

FES-PET of a breast tumor. Image of the Year at the 1987 National Meeting of the Society of Nuclear Medicine. From reference 76.
STRATEGIES FOR PREDICTING A FAVORABLE RESPONSE TO ENDOCRINE THERAPIES IN BREAST CANCER – EX VIVO ASSAYS OF ESTROGEN RECEPTOR
Although it was long known that about 35% of all breast cancers would respond favorably to endocrine therapies [8], prior to the discovery of ER, predicting in advance which cancers would respond was not possible. Starting in the early 1970’s, it became possible to evaluate the level of ER in the breast tumor fine-needle aspirates or excision biopsy. This was initially done by radioligand binding assays with [3H]estradiol, first analyzing binding by sucrose density gradient ultracentrifugation and then more conveniently by charcoal-dextran ligand adsorption assays. Both of these assays were technically demanding, requiring considerable biochemical expertise, and the ligand-adsorption assays typically quantified both E2 binding capacity and affinity.
Starting in 1985 and continuing unto the 1990’s, the cumbersome radioligand binding assay began to be replaced by immunological-based assays using monoclonal antibodies for ER for colorometric enzyme-linked immunosorbent assays (ELISA) or microscopic immunohistochemical (IHC) analyses, both of which could be more conveniently (and reliably) performed by clinical pathology departments [77]. Quantification of ER levels, however, became more indirect, and particularly with IHC assays, various scoring metrics needed to be developed to express ER positivity in terms of ER colorimetric staining intensity and distribution across a field of cancer cells [78]. Although they were more convenient, the results of these ex vivo assays of ER level remained imperfect predictors of clinical benefit from endocrine therapies for a number of reasons (see below), with predictive values being similar to the older radiometric methods. About 70% of breast cancers were considered to be ER-positive by these assays, which meant that overall the chance that a woman with ER-positive disease would respond favorably to endocrine therapy (which at the time meant treatment with tamoxifen) was only increased to 50%. The chance for a favorable response in IHC ER-negative disease, however, was low [79, 80].
Before considering in what ways FES-PET imaging of breast cancer might be used to improve management of breast cancer, in particular the prediction of a favorable response to endocrine therapies vs. the need for radiation or chemotherapy, it is important to distinguish between primary and metastatic disease and note the impact of other methods for characterizing the tumor. Primary breast tumors with no evidence of spread (Stage 1) are surgically excised (sometimes after neoadjuvant endocrine therapy, i.e., prior to surgery). If the tumor is ER-positive (by IHC), patients are typically treated with adjuvant endocrine therapy for 5–10 years to prevent recurrence. Recurrences in ER-positive disease do occur, often after many years [81, 82], and some women with ER-positive cancers elect to have chemotherapy, as it might be more effective than endocrine therapies, even though it might not be needed [83]. Recently, a major clinical trial (TAILORx) demonstrated that a 21-gene profile used to determine a risk of recurrence score (Oncotype-Dx) can effectively identify those patients with ER-positive and node-negative disease who will do well on endocrine therapies alone [84]. However, for more advanced ER-positive breast cancers with node involvement, FES-PET might add value in predicting response to endocrine therapies alone in these breast cancers.
With recurrent and metastatic ER-positive breast cancers, the situation is very different, and it may even be difficult to ascertain ER positivity at all of the metastatic sites. Nevertheless, endocrine therapies often remain effective, with a response rate of ca. 25–50%, depending on the extent of prior therapies; the prediction of success from continued endocrine therapies, however, is difficult [85, 86]. Hence, it is in the context of advanced breast cancer that the value of FES-PET is most likely to add predictive benefit.
EXPLORING THE CLINICAL UTILITY OF FES-PET IMAGING
Clinical assays for ER levels in breast tumors all involve ex vivo analysis of ER that evolved from ones directly measuring the ligand-binding function of ER with [3H]estradiol to detection now of an antigen in the ER with antibodies. By contrast, FES-PET imaging provides a non-invasive, in vivo measurement of the ligand binding function of ER in situ (recently reviewed [79, 87]. Multiple tumor sites throughout the body can be evaluated without patient discomfort, even sites that would be challenging to reach by needle biopsy (e.g., mediastinal nodes and brain lesions) and sites where tissue processing can affect the ER assay (e.g., bone lesions) [88]. Thus, inter-tumoral heterogeneity can be detected by FES-PET [89, 90], as can changes in ER level in the metastases compared with the primary tumor, which might have been obtained at an earlier time and are found to be different in a substantial number of cases [87, 91]. Although the signal from the PET image of the tumor is averaged over the voxel size inherent to the imaging device, variations in ER levels might, in principle, be detectable in a large lesion. More significantly, sampling errors in ER IHC analyses from percutaneous needle biopsies due to intra-tumoral heterogeneity in ER levels could be avoided [92], as could spurious differences arising from gene expression changes due to a wound response [93] that might be encountered in repeat biopsies.
Beginning in 1988 together with colleagues at Washington University, we published a series of clinical studies on FES-PET: We showed that the specific uptake for FES (% injected dose/gram) in a series of primary breast tumors correlated quantitatively with the level of ER determined subsequently on tumor biopsies by radioligand binding assays [75]. Subsequently, we found that FES tumor uptake in recurrent, metastatic disease also correlated with radiometric ER determinations in the primary tumor, though somewhat less well [94], with differences similar to that between ER assays on repeat biopsies and between the primary vs. metastatic lesions. We also found no correlation between tumor uptake by FES-PET and [18F]fluorodeoxyglucose (FDG)-PET [95], and we found that FES-PET was predictive of benefit from endocrine therapy [96]. There have been and continue to be many studies by other groups evaluating the potential of FES-PET alone or in combination of FDG-PET for delineating more accurately the prognosis of breast cancers and prediction of the success of endocrine vs. chemotherapies in both the neoadjuvant stage [97] and in advanced breast cancers. The results show the added value of FES-PET over that from ER IHC assays and from FDG-PET measurements alone [98], and in appraising patients that were otherwise difficult to evaluate [79, 98–102].
There are currently a number of active clinical trials (some multi-center) evaluating the predictive value of FES-PET, often together with FDG-PET or other targeted imaging agents, or its ability to identify metastases and intra-patient heterogeneity (NCT01957332), (NCT02398773), (NCT03726931), and (EUDRACT 2013–000-287–29). Suggestions for best practices in the clinical use of FES-PET [103] and simulation of the cost effectiveness of FES-PET vs. conventional work-up of patients with metastatic breast cancer have been reported by the Groningen group [104]. Recently, Zionexa, a French radiopharmaceutical company, has obtained approval for the clinical use of FES in France; they have also filed an NDA for FES (EstroTep®) with the US FDA. If FDA approval is successful, FES could soon become commercially available through an agreement between Zionexa and PETNET; this would greatly facilitate wider use of FES.
It is also of note that FES has become the standard in vivo probe for pharmacodynamic target occupancy studies used to determine appropriate dosing of currently used ER antagonists dosing (antiestrogens such as tamoxifen and Fulvestrant) required for full antagonist efficacy [105, 106]. This has even been extended to patient-specific dose refinement [107]. FES-PET is also used in dose-ranging studies to guide clinical trials of several new antiestrogens in patients with wild type ER [108] and will likely also be useful in tumors having constitutively active mutant forms of ER in their tumors and metastases [109]. In animal models, FES-PET was used to monitor increases in ER levels in tumors responding to inhibition of PIK3CA, where it served an indicator of the return to endocrine therapy responsiveness [110]; such a use of FES-PET might prove of value in other situations where growth factor pathways in breast cancers are being targeted.
BEYOND THE BINDING FUNCTION OF ER: USING PET IMAGING TO EVALUATE THE ACTIVITY OF ER THROUGH HORMONE CHALLENGE TESTS
In clinical management of ER-positive breast cancer, which represents about 70% of disease, it is known that only about half of patients with these tumors will benefit from endocrine therapies. While prediction is improved by the direct assessment of the binding function of ER in vivo and in situ using FES-PET [94–96], a significant number of breast tumors that have ER do not respond to endocrine therapies. This presents the challenge of discriminating between “functional” and “non-functional” ER in tumors in terms of the capacity of the ER regulatory signal transduction axis to mediate the antitumor effects of an appropriate endocrine therapy.
Over the past several years, breast cancer researchers have been addressing this issue in multiple ways, using increasingly available and comprehensive “-omics” approaches, developing databases of genomic, cistromic, transcriptomic, and proteomic analyses on biopsy samples from many breast tumors, and more recently extending to liquid biopsies of circulating tumor cells and tumor DNA in metastatic breast cancer. Various “signatures” extracted from these troves of information are then used to define certain molecular pathological subtypes that can be correlated with disease prognosis and prediction of therapy response. This classification does include the level of ER mRNA or protein, but beyond this, the signatures are thought to encompass features that will indicate whether or not ER retains its function as a mediator of endocrine therapy.
We have noted above the utility of the Oncotype Dx assay of the expression levels of 21 ER-associated genes in predicting the risk of recurrence in primary/early stage breast cancers in patients receiving endocrine therapy alone. As yet, however, there are no –omics assays that are robustly predictive of benefit from endocrine-only therapy in advanced/metastatic disease. In this regard, PET imaging presents an interesting alternative approach of directly accessing whether ER in tumors remains functional through the use of “hormone challenge tests”. Challenge test paradigms, such as the glucose tolerance test for diabetes and the dexamethasone suppression test for adrenal gland function, are widely used in endocrinology to determine on a short time scale the function of various organ systems, and this experimental paradigm can be applied to PET imaging as well.
With the increasing use of tamoxifen as endocrine therapy in the 1970’s and 80’s, an interesting phenomenon was observed in the clinic: When some women with ER-positive breast cancers began taking tamoxifen, they experienced a period of increased pain from their disease 1–2 weeks after the start of therapy, but thereafter they clearly responded to the antagonistic effect of tamoxifen; this phenomenon was termed a “tamoxifen flare” [111]. Although considered an antiestrogen, tamoxifen is not a full antagonist and is better termed a mixed agonist/antagonist. In fact, because it has variable levels of agonist and antagonist activity in different target tissues, it is classified as a selective estrogen receptor modulator (SERM) [112]. While tamoxifen is an antagonist in the breast, at low concentrations tamoxifen actually stimulates breast cancer cell proliferation [113], although it becomes inhibitory when it eventually reaches therapeutic levels in blood, which for a slow-clearing drug like tamoxifen takes some time [114]. Tamoxifen flare was attributed to the transient agonist effect from this mixed agonist/antagonist when, early during the course of therapy, circulating levels are low and thus stimulatory [115]. Clinically evident tamoxifen flare, however, has a very limited predictive value, because it is quite rare, with many responders failing to experience a transient worsening of symptoms, and some patients experiencing increased pain due to the progression of non-responsive disease.
We conceived of performing a “tamoxifen challenge test” to establish by longitudinal PET imaging of tumor glycolytic activity using FDG-PET whether a breast tumor that responded to an ER agonist-challenge by an increase in metabolic activity—a “metabolic flare”—would also respond to endocrine therapy; this idea was first discussed publically at a National Cancer Institute workshop in the mid 1990’s [116]. We thought that such a PET-based in vivo challenge test would determine more specifically whether the ER in individual breast tumors in advanced disease was still functional. We also imagined that it might improve the predictions of endocrine therapy benefit than those based on ex vivo assays of ER by IHC. We considered that it might even be more predictive than the in situ measurement of ER by FES-PET, because FES-PET assesses only the binding activity of ER, not its functional activities, such as regulating tumor cell metabolism or proliferation.
Hormone-Challenge Tests of Metabolic Flare.
Our initial studies enrolled patients with advanced disease, most of whom were treatment-naïve; by clinical assays all were ER-positive (and most also PR positive) and thus candidates for tamoxifen therapy, with an expected response rate of ca. 50% [117, 118]. Our plan, illustrated in Figure 8, was to combine the determination of ER levels by FES-PET, at baseline before initiation of tamoxifen therapy and then after 7–10 days of tamoxifen, with a parallel assessment of tumor metabolism, i.e., glucose uptake by FDG-PET. The baseline FES-PET signal would be a measure of ER level, and the expected decrease between baseline and treatment would indicate the degree to which tamoxifen was occupying ER and blocking its action on metabolism (although full blockage of ER would likely have required an even longer period of tamoxifen treatment than 7–10 days used for the challenge). A change between the two FDG-PET images would indicate an endocrine perturbation of tumor metabolism, and because the second image was timed to coincide with the time of peak tamoxifen clinical flare, we hoped that in some patients we might see an upward change representative of a “metabolic flare”, which would be an indication that ER was functional. The critical issue was whether a metabolic flare would prove to be a more robust predictor of tamoxifen therapy response than was ER IHC or clinical flare, or even by monitoring ER by FES-PET. After the four images, the patients were followed clinically to ascertain whether they were responding to continued tamoxifen treatment by oncologists who were blinded to the imaging results. As expected for the 40 ER-positive patients in this study, we obtained a clinical response rate of almost exactly 50%, with 21 responders and 19 non-responders.
Figure 8.

Four PET Imaging Protocol for a Tamoxifen Challenge Test to Predict Endocrine Responsiveness. Baseline FES-PET is an in vivo measure of ER binding in the tumor, and the difference between baseline and stimulated FES-PET represents the extent of ER occupancy by tamoxifen and its metabolites. The difference between baseline and stimulated FDG-PET represents the extent of tumor metabolic flare.
The predictive values of FES-PET and FDG-PET were high (Figure 9): With a standard uptake value (SUV) cutoff of 2 for baseline FES-PET, the positive-predictive value (PPV) was 79% and the negative predictive value (NPV) was 88%. As all patients were ER positive by IHC, this ex vivo assay had no predictive value in this patient population. Considerable blockade of the FES-PET image was seen in the second image, but in some cases it was incomplete, because the 7–10 day treatment period selected for maximum tamoxifen flare was likely insufficient for the full therapeutic effect of tamoxifen to be reached. Intriguingly, the individual FDG-PET values were not predictive, but the difference in FDG-PET values before and after tamoxifen was very predictive; with a cutoff of a 10% increase or greater in FDG-PET SUV values, the PPV was 91% and the NPV 94%. Overall, in this patient population, the predictive values from the longitudinal imaging with FDG-PET, a test of ER functional status, was remarkably high. Significantly, as well, the initial FES-PET value, representing ER level through ER in situ ligand binding, also afforded much greater predictive values than the standard clinical measurement of ER by IHC, which for these ER-positive patients was only 50%. This predictive value of FES-PET in advanced breast cancer has been seen in other studies [79] and is being further evaluated in a multicenter clinical trial, ECOG-ACRIN trial EAI142, NCT02398773.
Figure 9.

7–10 Day Tamoxifen Challenge Test to Predict Endocrine Responsiveness. Predictive values of FES-PET SUV (left) and % change in FDG-PET SUV (right) after 7–10 days on tamoxifen for benefit from continued tamoxifen therapy in women with treatment-naïve advanced breast cancer. From reference 118.
More recently, because of improved outcomes found in several clinical trials [119], estrogen deprivation therapy with aromatase inhibitors has largely replaced antiestrogen therapy with tamoxifen in many ER-positive breast cancers. This change, however, obviated detection of tumor metabolic flare as an early response to endocrine therapy, because aromatase inhibitors reduce endogenous estradiol levels and therefore do not cause any transient agonist stimulation. In addition, although it worked, the tamoxifen metabolic flare relied on the transient character of this SERM as an agonist, which is both incomplete and dose dependent. Furthermore, because it took 7–10 days for the change in metabolism to develop maximally with tamoxifen, the overall challenge test was relatively slow, making it inconvenient for patients who needed to return for the second image(s) after a considerable delay.
Based on these considerations, we redesigned the challenge test to use a short-term, 1-day stimulus with a high dose of the full agonist estradiol, after which patients would be transitioned to salvage endocrine therapy with either aromatase inhibitors or the full ER antagonist, fulvestrant. The only other change was the elimination of the second FES-PET image, which provided no useful information beyond that from the baseline image. The patient cohort in this study included 51 patients with advanced breast cancer, all of whom were ER positive (based either on prior analysis of the primary tumor or of concurrent metastases) and most PR positive. The majority of them had extensive prior chemo- and/or radiation therapy, as well as endocrine therapy, including tamoxifen [120]. As expected for this patient group, a response rate of 30% was found, and cutoff values very similar to those in our first study [117, 118] were used: FES-PET SUV of 2 and an increase in FDG-PET SUV of 12% or greater.
Our findings support the premise that with disease progression in breast cancer, the ER, even though still present, becomes progressively disconnected from the regulation of breast cancer cell metabolism, proliferation and progression (Figure 10). Thus, we found that FES-PET images of ER level in these heavily pre-treated patients had lost much of its predictive value for benefit from endocrine therapy alone, with PPV and NPV values declining to 43% and 74%, respectively; it was also not predictive of overall survival. By contrast, the increase in metabolic activity, now determined more acutely (and conveniently) after only a 1-day treatment with the full agonist estradiol, remained robustly predictive of endocrine therapy benefit, with PPV and NPV values of 100% and 94%, respectively, very comparable to those found in the tamoxifen challenge study on less heavily pretreated patients; it was also predictive of overall survival [120]. The receiver operating characteristic (ROC) plot from this study had an area under the curve (AOC) value close to 90% [120]. In a related study, different doses of estradiol were used in the FDG-PET challenge, and a lower dose of estradiol was found to be effective in eliciting a robust response [121].
Figure 10.

1-Day Estradiol Challenge Test to Predict Endocrine Responsiveness. Predictive values of FES-PET SUV (left) and % change in FDG-PET SUV (right) after a 1-day challenge with estradiol for benefit from salvage endocrine therapy in women with advanced breast cancer who had extensive prior endocrine, radiation, or chemotherapies. From reference 120.
Hormone-Challenge Test for ER-Regulated Gene Expression: the Progesterone Receptor.
Breast cancer biopsies are routinely assayed for levels of both ER and progesterone receptor (PR). Because it is an estrogen-regulated gene [122–124], PR was considered long ago to be a biomarker for ER having functional capacity for mediating benefit from endocrine therapy [125, 126]. Considerable disagreement has developed about the predictive value of PR measurements [127–131], however, and the most recent recommendation from the American Society of Clinical Oncologists (ASCO 2020) is that in early breast cancer, assay of PR should be considered optional [132]. Though not widely discussed, it is possible that estimates of the predictive value of PR assays in breast cancer are being confounded by the low levels of estradiol in postmenopausal women with breast cancer (insufficient for activating a PR response through ER), by the heterogeneity in PR expression levels throughout tumors (sampling errors from fine needle biopsy) [128, 133, 134], by mutations in the PR gene [135], or by hyperactivity of growth factor signaling pathways [136]. Balancing these uncertainties against the success of our 1-day estradiol challenge paradigm in predicting benefit from endocrine therapy through an increase in tumor metabolism by FDG-PET, we were curious whether we might find a change in PR levels, monitored by PET, after a similar 1-day estradiol challenge that might be more meaningful than single PR measurements by IHC. Such a challenge test would also be assessing the regulatory function of ER at the level of transcription, and this might be more robust than assessing ER function through changes in tumor glycolytic activity.
To undertake this gene expression-based challenge test, we used FFNP (Furanyl Fluoro-Nor-Progesterone, Figure 11), a PET imaging agent we developed for PR that showed efficient and selective uptake by the uterus of estrogen-primed immature rats [137]. We had already used it to image PR in breast cancer patients [138], and it is the subject of a current clinical trial (NCT03212170). It should be mentioned that the development of FFNP was the product of considerable efforts to obtain a ligand with high binding affinity for PR binding, low non-specific binding, and robust resistance to the rapid metabolism of many progestins that takes place in humans [139], but surprisingly not in rodents [137].
Figure 11.
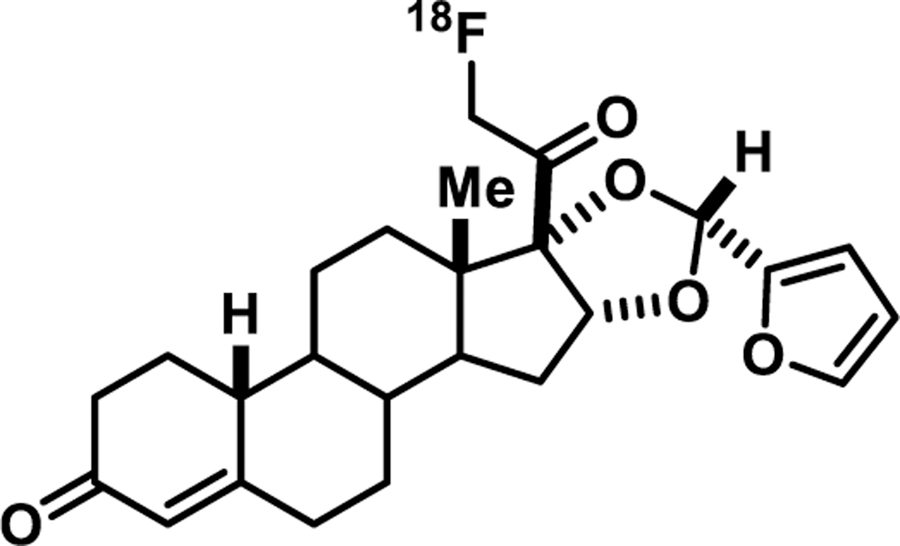
FFNP, 21-[18F]Fluoro-Furanyl-19-Nor-Progesterone
In preliminary proof-of-principle studies with two ER-positive syngeneic murine mammary cancer models, one that was estrogen responsive (SSM3) and one that was not (SSM2), we showed by both longitudinal Western blot analyses and FFNP-PET imaging, that PR levels decreased with estradiol deprivation and increased upon a 1-day estradiol stimulation in the ER-positive responsive SSM3 but not the ER-positive non-responsive SSM2 tumor system [140, 141]. In a clinical study just being completed (NCT00968409), an increase in PR has been monitored after a 1-day estradiol challenge in 43 women with ER-positive advanced breast cancer; these results will be reported shortly. Further clinical work with FFNP will be facilitated by an expedited radiosynthetic method recently reported [142].
Other Possible Hormone Challenge Tests, Other Modes for Testing, and Comparison with Monitoring Disease Response to Therapy.
It is worth noting that hormone challenge tests of endocrine-therapy responsiveness in breast cancers could be generalized by using longitudinal PET imaging to monitor the effect of a hormonal perturbation by estradiol on a variety of tumor cell characteristics other than glucose uptake and PR gene up-regulation; the general scheme is illustrated in Figure 12. Using other established imaging probes, changes in the level of hypoxia [143], angiogenesis, DNA synthesis, cell proliferation, lipid metabolism, protein synthesis, as well as other physiological and biochemical characteristics of tumors could be monitored. The value of any such hormone-receptor mediated challenge test would depend on its robustness, that is, the magnitude of signal change after hormone stimulation and especially the predictive value of the change for subsequent tumor response to endocrine therapy.
Figure 12.
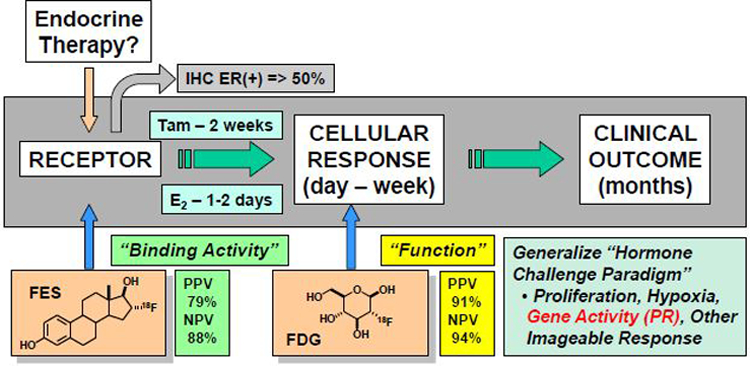
General scheme for hormone-challenge tests in advanced ER-positive breast cancer. At baseline, the presence of ER is quantified by FES-PET and glucose uptake by FDG-PET. After hormone stimulation (7–10 days tamoxifen or 1 day estradiol), glucose uptake is again quantified by FDG-PET. An increase in FDG-PET SUV of >12% predicts benefit from continued endocrine therapy with very high sensitivity and specificity, even in highly pretreated patients. Such tests could potentially also be based on the response to a hormone challenge monitored with PET probes for other physiological, biochemical, or transcriptional responses regulated by estrogen action through an active ER.
The hormone challenge we have used recently tests whether the ER signaling axis remains intact in advanced breast cancer by following an increase in tumor metabolism or gene expression after a brief positive stimulus to the tumor by the ER agonist, estradiol. In principle, however, it should be possible to obtain useful information on ER function by monitoring a decrease in the same parameters to a negative challenge, by withdrawal of estrogens (see, for example, [140, 141]). Because most breast cancers are found in postmenopausal women, when endogenous estradiol levels are already very low, the signal decrease from a further decrease in estradiol would likely be relatively small.
Because various types of longitudinal imaging are typically used to make an assessment of whether a therapy, endocrine or otherwise, is effective, one should compare such tests of therapy response with the hormone-challenge tests for therapy responsiveness (i.e., predicting response) that we have been describing. The major differences are: (a) the predictive power of the effect (can be very high for responsiveness), (b) the time interval required to make the assessment (potentially very short for responsiveness, longer for response), and (c) the potential detriment to the patient from an ineffective therapy during the time period required to assess response. The last of these may be particularly concerning, because disease progression that occurs during the more extended time interval required to establish “lack of response” to a particular therapy (sometimes up to several months) might make treatment by an alternative therapy overall less effective than had that therapy been selected earlier after a much shorter-term assay to predict therapy responsiveness.
CONCLUSION
The first portion of this review traced the design, development, validation, and clinical uses of FES as a PET probe for ER levels in breast cancer, showing how each stage of the evolution of this PET imaging agent rode upon a series of waves that represented technical and conceptual advances taking place in the field of radiopharmaceutical sciences. These include selection of a ligand design having appropriate specific and non-specific binding and pharmacokinetic properties, production of suitable radionuclide precursors at appropriate molar activities and in chemically reactive forms for radiolabeling, the actual radiochemical synthesis of the imaging probe, and the validation of its uptake specificity and efficiency in animal models before demonstration of utility for non-invasive, in vivo quantification of ER levels measured by receptor binding activity in situ in cancer patients. Applications to improve prediction of individual breast cancer responsiveness to endocrine therapies and to facilitate dose selection and new ER antagonist drug development then followed. The early contributions of Bill Eckelman in support of the field of receptor-binding radiotracers provided an encouraging and facilitating influence on this phase of my work.
By focusing on the development of FES and its use in breast cancer, I have not described the use of FES-PET in other medical situations (endometrial cancer [144], ovarian cancer [145]), or its suggested value in predicting response of breast cancer patients to the addition of docetaxel to endocrine therapy [146]. I have also not covered work by other groups using radioiodinated estrogens in clinical breast cancer studies [147, 148]. There are also a number of PET imaging agents to assess the level of other regulatory factors, such as the growth factor receptor HER2/neu, that when elevated in some breast cancers is an important therapy target [149].
The second portion of this review was entitled “Beyond Binding Function of ER”, and can be considered a response to challenges that led us to devise strategies to assess not just the presence of ER as a potential target for endocrine therapies by FES-PET, but to assess whether ER was still functional through its response to an acute, brief endocrine challenge. The results from the studies that we have conducted in women with breast cancer appear to have remarkably accurate predictive value, and the expanded use of such endocrine challenge tests to improve the treatment of breast and potentially other hormone-regulated cancers deserves further investigation. We hope that William Eckelman is pleased that we have worked to advance his expressed interests in having in vivo imaging address significant medical issues, obtain meaningful results [43, 150, 151], and advance personalized medicine [152–155].
Acknowledgements
I am grateful to the many students and postdocs in my lab who have contributed to the development of our ER and PR-targeted PET imaging agents. Although there are too many to name, the early contributions at Illinois by Steven Senderoff, Scott Landvatter, Dale Kiesewetter, Henry VanBrocklin, Kathryn Carlson, and Martin Pomper stand out. At Washington University Medical School, the support of Mike Welch, as well as his friendship and that of his family, were immensely beneficial; critical as well was the help of many of his coworkers and colleagues, for early chemistry and preclinical animal work, again noting only a few, Karen McElvany, Carla, Mathias, Tim Tewson and Mike Kilbourn, and for clinical and experimental imaging, Barry Siegel, Farrokh Dehdashti, Mark Mintun, Andrea McGuire, Joanne Mortimer, Ruby Chan, and Amy Fowler. Research support from the National Institutes of Health, the Department of Energy, the American Cancer Society, and the Breast Cancer Research Foundation are gratefully acknowledged. In preparing this report, I am also thankful for helpful comments from Farrokh Dehdashti, Barry Siegel, Hannah Linden, Amy Fowler, Marty Pomper, Kathryn Carlson, and Benita Katzenellenbogen.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- [1].Beatson GT. Meeting IX.—May 20, 1896: On the Treatment of Inoperable Cases of Carcinoma of the Mamma: Suggestions for a New Method of Treatment, with Illustrative Cases. Transactions. Medico-Chirurgical Society of Edinburgh 1896;15:153. [PMC free article] [PubMed] [Google Scholar]
- [2].Clarke MJ. Ovarian ablation in breast cancer, 1896 to 1998: milestones along hierarchy of evidence from case report to Cochrane review. BMJ 1998;317:1246–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jensen E and Jacobson H. Recent Progr. Hormone Res 1962;18:387. [Google Scholar]
- [4].Glascock RF and Hoekstra WG. Selective accumulation of tritium-labelled hexoestrol by the reproductive organs of immature female goats and sheep. Biochem. J 1959;72:673–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Toft D and Gorski J. A receptor molecule for estrogens: isolation from the rat uterus and preliminary characterization. Proc. Natl. Acad. Sci. U. S. A 1966;55:1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Katzenellenbogen JA and Hsiung HM. Iodohexestrols. I. Synthesis and photoreactivity of iodinated hexestrol derivatives. Biochemistry 1975;14:1736–41. [DOI] [PubMed] [Google Scholar]
- [7].Katzenellenbogen JA, Hsiung HM, Carlson KE, McGuire WL, Kraay RJ, and Katzenellenbogen BS. Iodohexestrols. II. Characterization of the binding and estrogenic activity of iodinated hexestrol derivatives, in vitro and in vivo. Biochemistry 1975;14:1742–50. [DOI] [PubMed] [Google Scholar]
- [8].Kaufmann M A review of endocrine options for the treatment of advanced breast cancer. Oncology 1997;54:2–5. [DOI] [PubMed] [Google Scholar]
- [9].Kiesewetter DO, Katzenellenbogen JA, Kilbourn MR, and Welch MJ. Synthesis of 16-fluoroestrogens by unusually facile fluoride ion displacement reactions: prospects for the preparation of fluorine-18 labeled estrogens. J. Org. Chem 1984;49:4900–5. [Google Scholar]
- [10].Kiesewetter DO, Kilbourn MR, Landvatter SW, Heiman DF, Katzenellenbogen JA, and Welch MJ. Preparation of four fluorine-18-labeled estrogens and their selective uptakes in target tissues of immature rats. J. Nucl. Med 1984;25:1212–21. [PubMed] [Google Scholar]
- [11].Hochberg RB and Rosner W. Interaction of 16 alpha-[125I] iodo-estradiol with estrogen receptor and other steroid-binding proteins. Proc. Natl. Acad. Sci. U. S. A 1980;77:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McManaway M, Jagoda E, Eckelman W, Larson S, Francis B, Gibson R, et al. Binding characteristics and biological activity of 17α-[125I] iodovinyl-11β-methoxyestradiol, an estrogen receptor-binding radiopharmaceutical, in human breast cancer cells (MCF-7). Cancer Res 1986;46:2386–9. [PubMed] [Google Scholar]
- [13].Kabalka GW, Gooch EE, Hsu HC, Washburn LC, Sun TT, and Hayes RL. Rapid and mild syntheses of radioiodinated estrogen derivatives via organoborane technology. Applications of Nuclear and Radiochemistry: Elsevier; 1982, p. 197–203.
- [14].Hanson RN, Seitz DE, and Botarro JC. E-17-[125 I] Iodovinylestradiol: An estrogen-receptor-seeking radiopharmaceutical. J. Nucl. Med 1982;23:436. [PubMed] [Google Scholar]
- [15].DeSombre ER, Mease RC, Sanghavl J, Singh T, Seevers RH, and Hughes A. Estrogen receptor binding affinity and uterotrophic activity of triphenylhaloethylenes. J. Steroid Biochem 1988;29:583–90. [DOI] [PubMed] [Google Scholar]
- [16].Ali H, Rousseau J, and Van Lier J. Synthesis of (17α, 20E/Z) iodovinyl testosterone and 19-nortestosterone derivatives as potential radioligands for androgen and progesterone receptors. J. Steroid Biochem. Mol. Biol 1994;49:15–29. [DOI] [PubMed] [Google Scholar]
- [17].Rijks LJ, Bakker PJ, van Tienhoven G, Noorduyn LA, Boer GJ, Rietbroek RC, et al. Imaging of estrogen receptors in primary and metastatic breast cancer patients with iodine-123-labeled Z-MIVE. J. Clin. Oncol 1997;15:2536–45. [DOI] [PubMed] [Google Scholar]
- [18].Heiman DF, Senderoff SG, Katzenellenbogen JA, and Neeley RJ. Estrogen receptor-based imaging agents. 1. Synthesis and receptor binding affinity of some aromatic and D-ring halogenated estrogens. J. Med. Chem 1980;23:994–1002. [DOI] [PubMed] [Google Scholar]
- [19].Katzenellenbogen JA, Senderoff SG, McElvany KD, O’Brien H, and Welch MJ. 16α-[77Br] bromoestradiol-17β: a high specific-activity, gamma-emitting tracer with uptake in rat uterus and induced mammary tumors. J. Nucl. Med 1981;22:42–7. [PubMed] [Google Scholar]
- [20].Katzenellenbogen JA, McElvany KD, Senderoff SG, Carlson KE, Landvatter SW, and Welch MJ. 16 alpha-[77Br]bromo-11 beta-methoxyestradiol-17 beta: a gamma-emitting estrogen imaging agent with high uptake and retention by target organs. J. Nucl. Med 1982;23:411–9. [PubMed] [Google Scholar]
- [21].Senderoff SG, McElvany KD, Carlson KE, Heiman DF, Katzenellenbogen JA, and Welch MJ. Methodology for the synthesis and specific activity determination of 16α-[77Br]-Bromoestradiol-17 β and 16α-[77Br]-11 β-methoxyestradiol-17 β, Two estrogen receptor-binding radiopharmaceuticals. Int. J. Appl. Radiat. Isot 1982;33:545–51. [DOI] [PubMed] [Google Scholar]
- [22].Katzenellenbogen J The development of gamma-emitting hormone analogs as imaging agents for receptor-positive tumors. Prog. Clin. Biol. Res 1981;75:313–27. [PubMed] [Google Scholar]
- [23].Katzenellenbogen J, Heiman D, Carlson K, and Lloyd J. In vivo and in vitro steroid receptor assays in the design of estrogen radiopharmaceuticals In: Eckelman WC editor. Receptor-binding radiotracers I Boca Raton, FL: CRC Press; 1982, p. 93–126. [Google Scholar]
- [24].Kochanny MJ, VanBrocklin HF, Kym PR, Carlson KE, O’Neil JP, Bonasera TA, et al. Fluorine-18 labeled progestin ketals: synthesis and target tissue uptake selectivity of potential imaging agents for receptor-positive breast tumors. J. Med. Chem 1993;36:1120–7. [DOI] [PubMed] [Google Scholar]
- [25].Pomper MG, VanBrocklin H, Thieme AM, Thomas RD, Kiesewetter DO, Carlson KE, et al. 11. beta.-methoxy-, 11. beta.-ethyl, and 17. alpha.-ethynyl-substituted 16. alpha.-fluoroestradiols: receptor-based imaging agents with enhanced uptake efficiency and selectivity. J. Med. Chem 1990;33:3143–55. [DOI] [PubMed] [Google Scholar]
- [26].Katzenellenbogen JA. Estrogen and progestin radiopharmaceuticals for imaging breast cancer In: Pavlik EJ editor. Estrogens, progestins, and their antagonists: Springer; 1997, p. 197–242. [PubMed] [Google Scholar]
- [27].Raynaud JP, Martin PM, Bouton MM, and Ojasoo T. 11beta-Methoxy-17-ethynyl-1,3,5(10)-estratriene-3,17beta-diol (moxestrol), a tag for estrogen receptor binding sites in human tissues. Cancer Res 1978;38:3044–50. [PubMed] [Google Scholar]
- [28].Frost J Pharmacokinetic Aspects of the In Vivo, Noninvasive Study of Neuroreceptors in Man In: Eckelman WC editor. Receptor-binding radiotracers II Boca raton, FL: CRC Press; 1982, p. 25–39. [Google Scholar]
- [29].McElvany KD, Carlson KE, Katzenellenbogen JA, and Welch MJ. Factors affecting the target site uptake selectivity of estrogen radiopharmaceuticals: serum binding and endogenous estrogens. J. Steroid Biochem 1983;18:635–41. [DOI] [PubMed] [Google Scholar]
- [30].Peterson LM, Kurland BF, Link JM, Schubert EK, Stekhova S, Linden HM, et al. Factors influencing the uptake of 18F-fluoroestradiol in patients with estrogen receptor positive breast cancer. Nucl. Med. Biol 2011;38:969–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Channing MA, Huang BX, and Eckelman WC. Analysis of residual solvents in 2-[(18)F]FDG by GC. Nucl. Med. Biol 2001;28:469–71. [DOI] [PubMed] [Google Scholar]
- [32].Ma Y, Kiesewetter D, Lang L, and Eckelman WC. Application of LC-MS to the analysis of new radiopharmaceuticals. Mol. Imaging Biol 2003;5:397–403. [DOI] [PubMed] [Google Scholar]
- [33].Aloj L, Carson RE, Lang L, Herscovitch P, and Eckelman WC. Measurement of transferrin receptor kinetics in the baboon liver using dynamic positron emission tomography imaging and [18F] holo-transferrin. Hepatology 1997;25:986–90. [DOI] [PubMed] [Google Scholar]
- [34].Eckelman WC. The use of PET and knockout mice in the drug discovery process. Drug Discovery Today 2003;8:404–10. [DOI] [PubMed] [Google Scholar]
- [35].Eckelman WC and Gibson RE. The design of site-directed radiopharmaceuticals for use in drug discovery. Nuclear imaging in drug discovery, development, and approval: Springer; 1993, p. 113–34.
- [36].Eckelman WC. Accelerating drug discovery and development through in vivo imaging. Nucl. Med. Biol 2002;29:777–82. [DOI] [PubMed] [Google Scholar]
- [37].Eckelman WC. The application of receptor theory to receptor-binding and enzyme-binding oncologic radiopharmaceuticals. Nucl. Med. Biol 1994;21:759–69. [DOI] [PubMed] [Google Scholar]
- [38].Eckelman W The use of in vitro models to predict the distribution of receptor binding radiotracers in vivo. Nucl. Med. Biol 1989;16:233–45. [DOI] [PubMed] [Google Scholar]
- [39].Eckelman WC, Kilbourn MR, and Mathis CA. Radiopharmaceutical space. Nucl. Med. Biol 2006;33:829. [DOI] [PubMed] [Google Scholar]
- [40].Eckelman WC and Mathis CA. Targeting proteins in vivo: in vitro guidelines. Nucl. Med. Biol 2006;2:161–4. [DOI] [PubMed] [Google Scholar]
- [41].Eckelman WC and Mathis CA. Molecular targets. Nucl. Med. Biol 2006;33:1. [DOI] [PubMed] [Google Scholar]
- [42].Eckelman WC, Kilbourn MR, and Mathis CA. Specific to nonspecific binding in radiopharmaceutical studies: it’s not so simple as it seems! Nucl. Med. Biol 2009;36:235–7. [DOI] [PubMed] [Google Scholar]
- [43].Eckelman WC. Targeted molecular imaging: target significance and probe validation. JACC Cardiovasc. Imaging 2012;5:616–8. [DOI] [PubMed] [Google Scholar]
- [44].Eckelman WC, Lau C-Y, and Neumann RD. Perspective, the one most responsive to change. Nucl. Med. Biol 2014;4:297–8. [DOI] [PubMed] [Google Scholar]
- [45].Eckelman WC, Frank JA, and Brechbiel M. Theory and practice of imaging saturable binding sites. Invest. Radiol 2002;37:101–6. [DOI] [PubMed] [Google Scholar]
- [46].Eckelman WC. Sensitivity of new radiopharmaceuticals. Nucl. Med. Biol 1998;25:169–73. [DOI] [PubMed] [Google Scholar]
- [47].McElvany KD, Katzenellenbogen JA, Shafer KE, Siegel BA, Senderoff SG, and Welch MJ. 16α-[77Br] bromoestradiol: dosimetry and preliminary clinical studies. J. Nucl. Med. 1982;23:425–30. [PubMed] [Google Scholar]
- [48].Tewson TJ, Welch MJ, and Raichle ME. [18F]-labeled 3-deoxy-3-fluoro-D-glucose: synthesis and preliminary biodistribution data. J. Nucl. Med 1978;19:1339–45. [PubMed] [Google Scholar]
- [49].Welch MJ, Kilbourn MR, Mathias CJ, Mintun MA, and Raichle ME. Comparison in animal models of 18F-spiroperidol and 18F-haloperidol: potential agents for imaging the dopamine receptor. Life Sci 1983;33:1687–93. [DOI] [PubMed] [Google Scholar]
- [50].Berridge M, Apana S, and Hersh J. Teflon radiolysis as the major source of carrier in fluorine-18. J. Labelled Compd. Radiopharm 2009;52:543–8. [Google Scholar]
- [51].Bergmann KE, Landvatter SW, Rocque PG, Carlson KE, Welch MJ, and Katzenellenbogen JA. Oxohexestrol derivatives labeled with fluorine-18. Synthesis, receptor binding and in vivo distribution of two non-steroidal estrogens as potential breast tumor imaging agents. Nucl. Med. Biol 1994;21:25–39. [DOI] [PubMed] [Google Scholar]
- [52].Landvatter SW and Katzenellenbogen JA. Nonsteroidal estrogens: synthesis and estrogen receptor binding affinity of derivatives of (3R*, 4S*)-3, 4-bis (4-hydroxyphenyl) hexane (hexestrol) and (2R*, 3S*)-2, 3-bis (4-hydroxyphenyl) pentane (norhexestrol) functionalized on the side chain. J. Med. Chem 1982;25:1300–7. [DOI] [PubMed] [Google Scholar]
- [53].Landvatter SW, Kiesewetter DO, Kilbourn MR, Katzenellenbogen JA, and Welch MJ. (2R∗, 3S∗)-1-[18fluoro-2, 3-bis (4-hydroxyphenyl) pentane ([18F] fluoronorhexestrol), A positron-emitting estrogen that shows highly-selective, receptor-mediated uptake by target tissues in vivo. Life Sci 1983;33:1933–8. [DOI] [PubMed] [Google Scholar]
- [54].Katzenellenbogen JA. The pharmacology of steroid radiopharmaceuticals: specific and non-specific binding and uptake selectivity In: Nunn AD editor. Radiopharmaceuticals: Chemistry and Pharmacology: M Dekker; 1992, p. 297–331. [Google Scholar]
- [55].Lim JL, Zheng L, Berridge MS, and Tewson TJ. The use of 3-methoxymethyl-16 beta, 17 beta-epiestriol-O-cyclic sulfone as the precursor in the synthesis of F-18 16 alpha-fluoroestradiol. Nucl. Med. Biol 1996;23:911–5. [DOI] [PubMed] [Google Scholar]
- [56].Kil HS, Cho HY, Lee SJ, Oh SJ, and Chi DY. Alternative synthesis for the preparation of 16alpha-[(18) F]fluoroestradiol. J Labelled Comp Radiopharm 2013;56:619–26. [DOI] [PubMed] [Google Scholar]
- [57].Zhou D, Lin M, Yasui N, Al-Qahtani MH, Dence CS, Schwarz S, et al. Optimization of the preparation of fluorine-18-labeled steroid receptor ligands 16alpha-[18F] fluoroestradiol (FES),[18F] fluoro furanyl norprogesterone (FFNP), and 16beta-[18F] fluoro-5alpha-dihydrotestosterone (FDHT) as radiopharmaceuticals. J. Labelled Compd. Radiopharm 2014;57:371–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Fedorova O, Nikolaeva V, and Krasikova R. Automated SPE-based synthesis of 16alpha-[(18)F]fluoroestradiol without HPLC purification step. Appl. Radiat. Isot 2018;141:57–63. [DOI] [PubMed] [Google Scholar]
- [59].Shi J, Afari G, and Bhattacharyya S. Rapid synthesis of [18F]fluoroestradiol: remarkable advantage of microwaving over conventional heating. J Labelled Comp Radiopharm 2014;57:730–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Liang S, Lan X, Zhang Y, Xu X, and Li B. Fully automatic synthesis of [(1)(8)F]FES for reporter gene hERL expression imaging. Nucl. Med. Commun 2012;33:29–33. [DOI] [PubMed] [Google Scholar]
- [61].Brodack JW, Kilbourn MR, Welch MJ, and Katzenellenbogen JA. NCA 16α-[18F] fluoroestradiol-17β: the effect of reaction vessel on fluorine-18 resolubilization, product yield, and effective specific activity. Int. J. Rad. Appl. Instrum. A 1986;37:217–21. [DOI] [PubMed] [Google Scholar]
- [62].Paquette M, Phoenix S, Ouellet R, Langlois R, van Lier JE, Turcotte EE, et al. Assessment of the novel estrogen receptor PET tracer 4-fluoro-11beta-methoxy-16alpha-[(18)F]fluoroestradiol (4FMFES) by PET imaging in a breast cancer murine model. Mol. Imaging Biol 2013;15:625–32. [DOI] [PubMed] [Google Scholar]
- [63].VanBrocklin HF, Liu A, Welch MJ, O’Neil JP, and Katzenellenbogen JA. The synthesis of 7α-methyl-substituted estrogens labeled with fluorine-18: potential breast tumor imaging agents. Steroids 1994;59:34–45. [DOI] [PubMed] [Google Scholar]
- [64].VanBrocklin HF, Pomper MG, Carlson KE, Welch MJ, and Katzenellenbogen JA. Preparation and evaluation of 17-ethynyl-substituted 16 alpha-[18F]fluoroestradiols: selective receptor-based PET imaging agents. Int. J. Rad. Appl. Instrum. B 1992;19:363–74. [DOI] [PubMed] [Google Scholar]
- [65].VanBrocklin HF, Rocque PA, Lee HV, Carlson KE, Katzenellenbogen JA, and Welch MJ. 16β-[18F] fluoromoxestrol: a potent, metabolically stable positron emission tomography imaging agent for estrogen receptor positive human breast tumors. Life Sci 1993;53:811–9. [DOI] [PubMed] [Google Scholar]
- [66].VanBrocklin HF, Brodack JW, Mathias CJ, Welch MJ, Katzenellenbogen JA, Keenan JF, et al. Binding of 16 alpha-[18F]fluoro-17 beta-estradiol to alphafetoprotein in Sprague-Dawley female rats affects blood levels. Int. J. Rad. Appl. Instrum. B 1990;17:769–73. [DOI] [PubMed] [Google Scholar]
- [67].Tewson TJ, Mankoff DA, Peterson LM, Woo I, and Petra P. Interactions of 16alpha-[18F]-fluoroestradiol (FES) with sex steroid binding protein (SBP). Nucl. Med. Biol 1999;26:905–13. [DOI] [PubMed] [Google Scholar]
- [68].Noé G, Cheng YC, Dabiké M, and Croxatto HB. Tissue uptake of human sex hormone-binding globulin and its influence on ligand kinetics in the adult female rat. Biol. Reprod 1992;47:970–6. [DOI] [PubMed] [Google Scholar]
- [69].Hammes A, Andreassen TK, Spoelgen R, Raila J, Hubner N, Schulz H, et al. Role of endocytosis in cellular uptake of sex steroids. Cell 2005;122:751–62. [DOI] [PubMed] [Google Scholar]
- [70].Jonson SD, Bonasera TA, Dehdashti F, Cristel ME, Katzenellenbogen JA, and Welch MJ. Comparative breast tumor imaging and comparative in vitro metabolism of 16alpha-[18F]fluoroestradiol-17beta and 16beta-[18F]fluoromoxestrol in isolated hepatocytes. Nucl. Med. Biol 1999;26:123–30. [DOI] [PubMed] [Google Scholar]
- [71].Downer JB, Jones LA, Engelbach JA, Lich LL, Mao W, Carlson KE, et al. Comparison of animal models for the evaluation of radiolabeled androgens. Nucl. Med. Biol 2001;28:613–26. [DOI] [PubMed] [Google Scholar]
- [72].Parent EE, Carlson KE, and Katzenellenbogen JA. Synthesis of 7α-(fluoromethyl) dihydrotestosterone and 7α-(fluoromethyl) nortestosterone, structurally paired androgens designed to probe the role of sex hormone binding globulin in imaging androgen receptors in prostate tumors by positron emission tomography. J. Org. Chem 2007;72:5546–54. [DOI] [PubMed] [Google Scholar]
- [73].Mathias CJ, Welch MJ, Katzenellenbogen JA, Brodack JW, Kilbourn MR, Carlson KE, et al. Characterization of the uptake of 16 alpha-([18F]fluoro)-17 beta-estradiol in DMBA-induced mammary tumors. Int. J. Rad. Appl. Instrum. B 1987;14:15–25. [DOI] [PubMed] [Google Scholar]
- [74].Katzenellenbogen JA, Mathias CJ, VanBrocklin HF, Brodack JW, and Welch MJ. Titration of the in vivo uptake of 16 alpha-[18F]fluoroestradiol by target tissues in the rat: competition by tamoxifen, and implications for quantitating estrogen receptors in vivo and the use of animal models in receptor-binding radiopharmaceutical development. Nucl. Med. Biol 1993;20:735–45. [DOI] [PubMed] [Google Scholar]
- [75].Mintun M, Welch M, Siegel B, Mathias C, Brodack J, McGuire A, et al. Breast cancer: PET imaging of estrogen receptors. Radiology 1988;169:45–8. [DOI] [PubMed] [Google Scholar]
- [76].Wagner HN Jr. SNM Highlights as History: 1987. J. Nucl. Med 2004;45:N27. [Google Scholar]
- [77].King WJ, DeSombre ER, Jensen EV, and Greene GL. Comparison of immunocytochemical and steroid-binding assays for estrogen receptor in human breast tumors. Cancer Res 1985;45:293–304. [PubMed] [Google Scholar]
- [78].Harvey JM, Clark GM, Osborne CK, and Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J. Clin. Oncol 1999;17:1474–81. [DOI] [PubMed] [Google Scholar]
- [79].Kumar M, Salem K, Tevaarwerk AJ, Strigel RM, and Fowler AM. Recent Advances in Imaging Steroid Hormone Receptors in Breast Cancer. J. Nucl. Med 2019:jnumed. 119.228858. [DOI] [PMC free article] [PubMed]
- [80].van Kruchten M, de Vries EG, Brown M, de Vries EF, Glaudemans AW, Dierckx RA, et al. PET imaging of oestrogen receptors in patients with breast cancer. Lancet Oncol 2013;14:e465–e75. [DOI] [PubMed] [Google Scholar]
- [81].Pan H, Gray R, Braybrooke J, Davies C, Taylor C, McGale P, et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med 2017;377:1836–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Waks AG and Winer EP. Breast Cancer Treatment: A Review. JAMA 2019;321:288–300. [DOI] [PubMed] [Google Scholar]
- [83].Fisher B, Costantino J, Redmond C, Poisson R, Bowman D, Couture J, et al. A randomized clinical trial evaluating tamoxifen in the treatment of patients with node-negative breast cancer who have estrogen-receptor–positive tumors. N. Engl. J. Med 1989;320:479–84. [DOI] [PubMed] [Google Scholar]
- [84].Sparano JA, Gray RJ, Makower DF, Albain KS, Saphner TJ, Badve SS, et al. Clinical Outcomes in Early Breast Cancer With a High 21-Gene Recurrence Score of 26 to 100 Assigned to Adjuvant Chemotherapy Plus Endocrine Therapy: A Secondary Analysis of the TAILORx Randomized Clinical Trial. JAMA Oncol 2019. [DOI] [PMC free article] [PubMed]
- [85].Mouridsen H, Palshof T, Patterson J, and Battersby L. Tamoxifen in advanced breast cancer. Cancer Treat. Rev 1978;5:131–41. [DOI] [PubMed] [Google Scholar]
- [86].Wittliff JL. Steroid-hormone receptors in breast cancer. Cancer 1984;53:630–43. [DOI] [PubMed] [Google Scholar]
- [87].Fowler AM, Clark AS, Katzenellenbogen JA, Linden HM, and Dehdashti F. Imaging Diagnostic and Therapeutic Targets-Steroid Receptors in Breast Cancer. J. Nucl. Med 2016;57:75S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Gertych A, Mohan S, Maclary S, Mohanty S, Wawrowsky K, Mirocha J, et al. Effects of tissue decalcification on the quantification of breast cancer biomarkers by digital image analysis. Diagn. Pathol 2014;9:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kurland BF, Peterson LM, Lee JH, Linden HM, Schubert EK, Dunnwald LK, et al. Between-patient and within-patient (site-to-site) variability in estrogen receptor binding, measured in vivo by 18F-fluoroestradiol PET. J. Nucl. Med 2011;52:1541–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Nienhuis HH, van Kruchten M, Elias SG, Glaudemans A, de Vries EFJ, Bongaerts AHH, et al. (18)F-Fluoroestradiol Tumor Uptake Is Heterogeneous and Influenced by Site of Metastasis in Breast Cancer Patients. J. Nucl. Med 2018;59:1212–8. [DOI] [PubMed] [Google Scholar]
- [91].Amir E, Miller N, Geddie W, Freedman O, Kassam F, Simmons C, et al. Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer. J. Clin. Oncol 2012;30:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Sass PA, Dąbrowski M, Charzyńska A, and Sachadyn P. Transcriptomic responses to wounding: meta-analysis of gene expression microarray data. BMC Genomics 2017;18:850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].McGuire AH, Dehdashti F, Siegel BA, Lyss AP, Brodack JW, Mathias CJ, et al. Positron tomographic assessment of 16 alpha-[18F] fluoro-17 beta-estradiol uptake in metastatic breast carcinoma. J. Nucl. Med 1991;32:1526–31. [PubMed] [Google Scholar]
- [95].Dehdashti F, Mortimer JE, Siegel BA, Griffeth LK, Bonasera TJ, Fusselman MJ, et al. Positron tomographic assessment of estrogen receptors in breast cancer: comparison with FDG-PET and in vitro receptor assays. J. Nucl. Med 1995;36:1766–74. [PubMed] [Google Scholar]
- [96].Mortimer JE, Dehdashti F, Siegel BA, Katzenellenbogen JA, Fracasso P, and Welch MJ. Positron emission tomography with 2-[18F]Fluoro-2-deoxy-D-glucose and 16alpha-[18F]fluoro-17beta-estradiol in breast cancer: correlation with estrogen receptor status and response to systemic therapy. Clin. Cancer Res 1996;2:933–9. [PubMed] [Google Scholar]
- [97].Park JH, Kang MJ, Ahn J-H, Kim JE, Jung KH, Gong G, et al. Phase II trial of neoadjuvant letrozole and lapatinib in Asian postmenopausal women with estrogen receptor (ER) and human epidermal growth factor receptor 2 (HER2)-positive breast cancer [Neo-ALL-IN]: highlighting the TILs, ER expressional change after neoadjuvant treatment, and FES-PET as potential significant biomarkers. Cancer Chemother. Pharmacol 2016;78:685–95. [DOI] [PubMed] [Google Scholar]
- [98].Kurland BF, Peterson LM, Lee JH, Schubert EK, Currin ER, Link JM, et al. Estrogen Receptor Binding (18F-FES PET) and Glycolytic Activity (18F-FDG PET) Predict Progression-Free Survival on Endocrine Therapy in Patients with ER+ Breast Cancer. Clin. Cancer Res 2017;23:407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].van Kruchten M, Glaudemans AW, de Vries EF, Beets-Tan RG, Schroder CP, Dierckx RA, et al. PET imaging of estrogen receptors as a diagnostic tool for breast cancer patients presenting with a clinical dilemma. J. Nucl. Med 2012;53:182–90. [DOI] [PubMed] [Google Scholar]
- [100].Linden HM, Peterson LM, and Fowler AM. Clinical Potential of Estrogen and Progesterone Receptor Imaging. PET Clin 2018;13:415–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Liao GJ, Clark AS, Schubert EK, and Mankoff DA. 18F-Fluoroestradiol PET: Current Status and Potential Future Clinical Applications. J. Nucl. Med 2016;57:1269–75. [DOI] [PubMed] [Google Scholar]
- [102].Evangelista L, Guarneri V, and Conte PF. 18F-Fluoroestradiol Positron Emission Tomography in Breast Cancer Patients: Systematic Review of the Literature & Meta-Analysis. Curr Radiopharm 2016;9:244–57. [DOI] [PubMed] [Google Scholar]
- [103].Venema CM, Apollonio G, Hospers GA, Schröder CP, Dierckx RA, de Vries EF, et al. Recommendations and technical aspects of 16α-[18F] fluoro-17β-estradiol PET to image the estrogen receptor in vivo: the Groningen experience. Clin. Nucl. Med 2016;41:844–51. [DOI] [PubMed] [Google Scholar]
- [104].Koleva-Kolarova R, Greuter M, Van Kruchten M, Vermeulen K, Feenstra T, Buskens E, et al. The value of PET/CT with FES or FDG tracers in metastatic breast cancer: a computer simulation study in ER-positive patients. Br. J. Cancer 2015;112:1617–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Linden HM, Kurland BF, Peterson LM, Schubert EK, Gralow JR, Specht JM, et al. Fluoroestradiol positron emission tomography reveals differences in pharmacodynamics of aromatase inhibitors, tamoxifen, and fulvestrant in patients with metastatic breast cancer. Clin. Cancer Res 2011;17:4799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].van Kruchten M, de Vries EG, Glaudemans AW, van Lanschot MC, van Faassen M, Kema IP, et al. Measuring residual estrogen receptor availability during fulvestrant therapy in patients with metastatic breast cancer. Cancer Discov 2015;5:72–81. [DOI] [PubMed] [Google Scholar]
- [107].Heidari P, Deng F, Esfahani SA, Leece AK, Shoup TM, Vasdev N, et al. Pharmacodynamic imaging guides dosing of a selective estrogen receptor degrader. Clin. Cancer Res 2015;21:1340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Wang Y, Ayres KL, Goldman DA, Dickler MN, Bardia A, Mayer IA, et al. 18F-fluoroestradiol PET/CT measurement of estrogen receptor suppression during a phase I trial of the novel estrogen receptor-targeted therapeutic GDC-0810: using an imaging biomarker to guide drug dosage in subsequent trials. Clin. Cancer Res 2017;23:3053–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kumar M, Salem K, Michel C, Jeffery JJ, Yan Y, and Fowler AM. (18)F-Fluoroestradiol PET Imaging of Activating Estrogen Receptor-alpha Mutations in Breast Cancer. J. Nucl. Med 2019;60:1247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Bosch A, Li Z, Bergamaschi A, Ellis H, Toska E, Prat A, et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor–positive breast cancer. Sci. Transl. Med 2015;7:283ra51–ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Plotkin D, Lechner JJ, Jung WE, and Rosen PJ. Tamoxifen flare in advanced breast cancer. JAMA 1978;240:2644–6. [PubMed] [Google Scholar]
- [112].McDonnell DP. The molecular pharmacology of SERMs. Trends Endocrinol. Metab 1999;10:301–11. [DOI] [PubMed] [Google Scholar]
- [113].Katzenellenbogen BS, Kendra KL, Norman MJ, and Berthois Y. Proliferation, hormonal responsiveness, and estrogen receptor content of MCF-7 human breast cancer cells grown in the short-term and long-term absence of estrogens. Cancer Res 1987;47:4355–60. [PubMed] [Google Scholar]
- [114].Fabian C, Sternson L, El-Serafi M, Cain L, and Hearne E. Clinical pharmacology of tamoxifen in patients with breast cancer: correlation with clinical data. Cancer 1981;48:876–82. [DOI] [PubMed] [Google Scholar]
- [115].Reddel RR and Sutherland RL. Tamoxifen stimulation of human breast cancer cell proliferation in vitro: a possible model for tamoxifen tumour flare. Eur. J. Cancer Clin. Oncol 1984;20:1419–24. [DOI] [PubMed] [Google Scholar]
- [116].Katzenellenbogen JA, Coleman RE, Hawkins RA, Krohn KA, Larson SM, Mendelsohn J, et al. Tumor receptor imaging: proceedings of the National Cancer Institute workshop, review of current work, and prospective for further investigations. Clin. Cancer Res 1995;1:921–32. [PubMed] [Google Scholar]
- [117].Dehdashti F, Flanagan FL, Mortimer JE, Katzenellenbogen JA, Welch MJ, and Siegel BA. Positron emission tomographic assessment of “metabolic flare” to predict response of metastatic breast cancer to antiestrogen therapy. Eur. J. Nucl. Med 1999;26:51–6. [DOI] [PubMed] [Google Scholar]
- [118].Mortimer JE, Dehdashti F, Siegel BA, Trinkaus K, Katzenellenbogen JA, and Welch MJ. Metabolic flare: indicator of hormone responsiveness in advanced breast cancer. J. Clin. Oncol 2001;19:2797–803. [DOI] [PubMed] [Google Scholar]
- [119].Berry J Are all aromatase inhibitors the same? A review of controlled clinical trials in breast cancer. Clin. Ther 2005;27:1671–84. [DOI] [PubMed] [Google Scholar]
- [120].Dehdashti F, Mortimer JE, Trinkaus K, Naughton MJ, Ellis M, Katzenellenbogen JA, et al. PET-based estradiol challenge as a predictive biomarker of response to endocrine therapy in women with estrogen-receptor-positive breast cancer. Breast Cancer Res. Treat 2009;113:509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Ellis MJ, Gao F, Dehdashti F, Jeffe DB, Marcom PK, Carey LA, et al. Lower-dose vs high-dose oral estradiol therapy of hormone receptor–positive, aromatase inhibitor–resistant advanced breast cancer: A phase 2 randomized study. JAMA 2009;302:774–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Kraus WL and Katzenellenbogen BS. Regulation of progesterone receptor gene expression and growth in the rat uterus: modulation of estrogen actions by progesterone and sex steroid hormone antagonists. Endocrinology 1993;132:2371–9. [DOI] [PubMed] [Google Scholar]
- [123].Eckert RL and Katzenellenbogen BS. Effects of estrogens and antiestrogens on estrogen receptor dynamics and the induction of progesterone receptor in MCF-7 human breast cancer cells. Cancer Res 1982;42:139–44. [PubMed] [Google Scholar]
- [124].May FE, Johnson MD, Wiseman LR, Wakeling AE, Kastner P, and Westley BR. Regulation of progesterone receptor mRNA by oestradiol and antioestrogens in breast cancer cell lines. J. Steroid Biochem 1989;33:1035–41. [DOI] [PubMed] [Google Scholar]
- [125].Horwitz KB and McGuire WL. Predicting response to endocrine therapy in human breast cancer: a hypothesis. Science 1975;189:726–7. [DOI] [PubMed] [Google Scholar]
- [126].McGuire WL, Horwitz KB, Pearson OH, and Segaloff A. Current status of estrogen and progesterone receptors in breast cancer. Cancer 1977;39:2934–47. [DOI] [PubMed] [Google Scholar]
- [127].Boland M, Ryan É, Dunne E, Aherne T, Bhatt N, and Lowery A. Meta-analysis of the impact of progesterone receptor status on oncological outcomes in oestrogen receptor-positive breast cancer. Br. J. Surg 2019. [DOI] [PubMed]
- [128].Purdie C, Quinlan P, Jordan L, Ashfield A, Ogston S, Dewar J, et al. Progesterone receptor expression is an independent prognostic variable in early breast cancer: a population-based study. Br. J. Cancer 2014;110:565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Inda MA, Blok EJ, Kuppen PJ, Charehbili A, den Biezen-Timmermans EC, van Brussel A, et al. Estrogen Receptor pathway activity score to predict clinical response or resistance to neo-adjuvant endocrine therapy in primary breast cancer. Mol. Cancer Ther 2019. [DOI] [PubMed]
- [130].Wu J-R, Zhao Y, Zhou X-P, and Qin X. Estrogen receptor 1 and progesterone receptor are distinct biomarkers and prognostic factors in estrogen receptor-positive breast cancer: Evidence from a bioinformatic analysis. Biomed. Pharmacother 2020;121:109647. [DOI] [PubMed] [Google Scholar]
- [131].Li Y, Yang D, Yin X, Zhang X, Huang J, Wu Y, et al. Clinicopathological Characteristics and Breast Cancer–Specific Survival of Patients With Single Hormone Receptor–Positive Breast Cancer. JAMA Network Open 2020;3:e1918160-e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Allison KH, Hammond MEH, Dowsett M, McKernin SE, Carey LA, Fitzgibbons PL, et al. Estrogen and Progesterone Receptor Testing in Breast Cancer: American Society of Clinical Oncology/College of American Pathologists Guideline Update. Arch. Pathol. Lab. Med 2020. [DOI] [PubMed]
- [133].Lin Y, Hatem J, Wang J, Quinn A, Hicks D, and Tang P. Tissue microarray-based immunohistochemical study can significantly underestimate the expression of HER2 and progesterone receptor in ductal carcinoma in situ of the breast. Biotech. Histochem 2011;86:345–50. [DOI] [PubMed] [Google Scholar]
- [134].Allott EH, Geradts J, Sun X, Cohen SM, Zirpoli GR, Khoury T, et al. Intratumoral heterogeneity as a source of discordance in breast cancer biomarker classification. Breast Cancer Res 2016;18:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Fowler AM, Salem K, DeGrave M, Ong IM, Rassman S, Powers GL, et al. Progesterone Receptor Gene Variants in Metastatic Estrogen Receptor Positive Breast Cancer. Horm. Cancer 2020;11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Cui X, Schiff R, Arpino G, Osborne CK, and Lee AV. Biology of progesterone receptor loss in breast cancer and its implications for endocrine therapy. J. Clin. Oncol 2005;23:7721–35. [DOI] [PubMed] [Google Scholar]
- [137].Buckman BO, Bonasera TA, Kirschbaum KS, Welch MJ, and Katzenellenbogen JA. Fluorine-18-labeled progestin 16 alpha, 17 alpha-dioxolanes: development of high-affinity ligands for the progesterone receptor with high in vivo target site selectivity. J. Med. Chem 1995;38:328–37. [DOI] [PubMed] [Google Scholar]
- [138].Dehdashti F, Laforest R, Gao F, Aft RL, Dence CS, Zhou D, et al. Assessment of progesterone receptors in breast carcinoma by PET with 21–18F-fluoro-16alpha,17alpha-[(R)-(1’-alpha-furylmethylidene)dioxy]-19-norpregn-4-ene-3,20-dione. J. Nucl. Med 2012;53:363–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Verhagen A, Studeny M, Luurtsema G, Visser G, De Goeij C, Sluyser M, et al. Metabolism of a [18F] fluorine labeled progestin (21-[18F] fluoro-16α-ethyl-19-norprogesterone) in humans: a clue for future investigations. Nucl. Med. Biol 1994;21:941–52. [DOI] [PubMed] [Google Scholar]
- [140].Chan SR, Fowler AM, Allen JA, Zhou D, Dence CS, Sharp TL, et al. Longitudinal noninvasive imaging of progesterone receptor as a predictive biomarker of tumor responsiveness to estrogen deprivation therapy. Clin. Cancer Res 2015;21:1063–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Fowler AM, Chan SR, Sharp TL, Fettig NM, Zhou D, Dence CS, et al. Small-animal PET of steroid hormone receptors predicts tumor response to endocrine therapy using a preclinical model of breast cancer. J. Nucl. Med 2012;53:1119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Basuli F, Zhang X, Blackman B, White ME, Jagoda EM, Choyke PL, et al. Fluorine-18 Labeled Fluorofuranylnorprogesterone ([18F] FFNP) and Dihydrotestosterone ([18F] FDHT) Prepared by “Fluorination on Sep-Pak” Method. Molecules 2019;24:2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Cheng J, Lei L, Xu J, Sun Y, Zhang Y, Wang X, et al. 18F-fluoromisonidazole PET/CT: a potential tool for predicting primary endocrine therapy resistance in breast cancer. J. Nucl. Med 2013;54:333–40. [DOI] [PubMed] [Google Scholar]
- [144].Yamada S, Tsuyoshi H, Tsujikawa T, Okazawa H, and Yoshida Y. Predictive Value of 16alpha-[18F]-Fluoro-17beta-Estradiol PET as a Biomarker of Progestin Therapy Resistance in Patients With Atypical Endometrial Hyperplasia and Low-Grade Endometrial Cancer. Clin. Nucl. Med 2019;44:574–5. [DOI] [PubMed] [Google Scholar]
- [145].van Kruchten M, de Vries EF, Arts HJ, Jager NM, Bongaerts AH, Glaudemans AW, et al. Assessment of estrogen receptor expression in epithelial ovarian cancer patients using 16alpha-18F-fluoro-17beta-estradiol PET/CT. J. Nucl. Med 2015;56:50–5. [DOI] [PubMed] [Google Scholar]
- [146].Gong C, Yang Z, Sun Y, Zhang J, Zheng C, Wang L, et al. A preliminary study of 18 F-FES PET/CT in predicting metastatic breast cancer in patients receiving docetaxel or fulvestrant with docetaxel. Sci. Rep 2017;7:6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Bennink RJ, van Tienhoven G, Rijks LJ, Noorduyn AL, Janssen AG, and Sloof GW. In vivo prediction of response to antiestrogen treatment in estrogen receptor-positive breast cancer. J. Nucl. Med 2004;45:1–7. [PubMed] [Google Scholar]
- [148].Bennink RJ, Rijks LJ, van Tienhoven G, Noorduyn LA, Janssen AG, and Sloof GW. Estrogen receptor status in primary breast cancer: iodine 123-labeled cis-11beta-methoxy-17alpha-iodovinyl estradiol scintigraphy. Radiology 2001;220:774–9. [DOI] [PubMed] [Google Scholar]
- [149].Henry KE, Ulaner GA, and Lewis JS. Clinical Potential of Human Epidermal Growth Factor Receptor 2 and Human Epidermal Growth Factor Receptor 3 Imaging in Breast Cancer. PET Clinics 2018;13:423–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Anderson C Molecular Nuclear Medicine: The Challenge of Genomics and Proteomics to Clinical Practice: Springer Science & Business Media,2003. [Google Scholar]
- [151].Cannon DM, Klaver JK, Gandhi SK, Solorio G, Peck SA, Erickson K, et al. Genetic variation in cholinergic muscarinic-2 receptor gene modulates M2 receptor binding in vivo and accounts for reduced binding in bipolar disorder. Mol. Psychiatry 2011;16:407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Eckelman WC and Vera DR. From bench to bedside. Nucl. Med. Biol 2003;30:793–4. [DOI] [PubMed] [Google Scholar]
- [153].Kelloff GJ, Krohn KA, Larson SM, Weissleder R, Mankoff DA, Hoffman JM, et al. The progress and promise of molecular imaging probes in oncologic drug development. Clin. Cancer Res 2005;11:7967–85. [DOI] [PubMed] [Google Scholar]
- [154].Eckelman WC, Reba RC, and Kelloff GJ. Targeted imaging: an important biomarker for understanding disease progression in the era of personalized medicine. Drug Discovery Today 2008;13:748–59. [DOI] [PubMed] [Google Scholar]
- [155].Eckelman W Choosing a target for targeted radionuclide therapy using biomarkers to personalize treatment. J Diagn Imaging Ther 2014;1:103–9. [Google Scholar]