

Abstract
gem-Difluoroalkenes represent valuable synthetic handles for organofluorine chemistry; however, most reactions of this substructure proceed through reactive intermediates prone to eliminate a fluorine atom and generate monofluorinated products. Taking advantage of the distinct reactivity of gem-difluoroalkenes, we present a cobalt-catalyzed regioselective unsymmetrical dioxygenation of gem-difluoroalkenes using phenols and molecular oxygen which retains both fluorine atoms and provides β-phenoxy-β,β-difluorobenzyl alcohols. Mechanistic studies suggest that the reaction operates through a radical chain process initiated by Co(II)/O2/phenol and quenched by the Co-based catalyst. This mechanism enables the retention of both fluorine atoms, which contrasts most transition metal-catalyzed reactions of gem-difluoroalkenes that typically involve defluorination.
Graphical Abstract
Introduction
The introduction of fluorinated functional groups onto a small molecule typically perturbs physicochemical properties relevant to medicinal chemistry.1 For instance, the small size and high electronegativity of fluorine modulate the acidity and basicity of nearby functional groups,1b,2 which influences the solubility,3 lipophilicity,1 molecular conformation,4 and ligand-protein interactions relative to the non-fluorinated analogs.5 Further, positioning of the F-atoms at a metabolic soft spot can block cytochromes p450 mediated metabolic processes,6 thus likely increasing stability of the substructure relative to the parent non-fluorinated analogs.
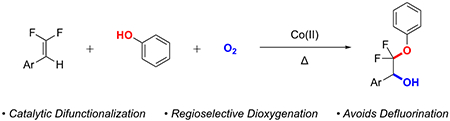
β,β-Difluoroalkenes represent a privileged functional group for accessing organo-fluorine compounds through convergent processes.7 This substructure displays unique reactivity relative to typical alkenes; specifically, the distinct electronic character of the termini enables differential functionalization of each carbon atom.7 Transition metal-free and transition-metal catalyzed reactions exploit this reactivity through selective addition of nucleophiles to the difluorinated position over the non-fluorinated position, to generate β-fluoroanionic7–8 or β-fluoroorganometallic9 intermediates, which readily undergo β-fluoride elimination to generate mono-fluorinated products (Figure 1a), even under biological conditions.10 Though this facile β-fluoride elimination under both basic and transition-metal catalyzed conditions enables access to monofluoroalkene-based products, the elimination reduces the synthetic utility of gem-difluoroalkenes towards accessing difluorinated products.
Figure 1:
Representative Reactions of gem-Difluoroalkenes.
To complement these studies, recently developed reactions have provided methods for functionalizing both the fluorinated and non-fluorinated carbon without eliminating fluoride via protonation of the anionic intermediate (Figure 1b),8a–c which does not allow difunctionalization processes. Fluorination-protonation has been achieved reactions in additional specialized cases, through intramolecular halo-lactonization with an iodonium intermediate to deliver related products that bear an additional iodine atom,11 or by exploiting highly-activated difluorinated precursors (e.g. acrylates12) using neat alcohol. In an additional example, allyl alcohol was added to an 18F labelled difluoroalkene in low yield (22 %) using KOH in MeCN,8d though the selectivity for formation of the difluorinated product versus the mono-fluorinated product was not reported. Alternatively, fluorination–functionalization sequences of gem-difluoroalkenes have recently been developed (Figure 1c),13 though by adding fluorine to the system, this strategy can only deliver CF3-based products. In an isolated example, a recently reported Pd-catalyzed carbonylation reaction of gem-difluoroalkenes enables access to difluorinated products (Figure 1d).14 In this case, the key Pd-alkyl intermediate apparently undergoes α-migratory insertion into the CO ligands faster than the β-fluoride elimination process. To complement these recently reported transformations, we present a cobalt-catalyzed unsymmetrical dioxygenation reaction of β,β-difluoroalkenes to generate β-phenoxy-β,β-difluorobenzyl alcohols (Figure 1e). Notably, the reaction proceeds through a radical intermediate that avoids β-fluoride elimination, and thus provides a distinct strategy for generating fluorinated substructures from readily accessible fluorinated synthons.7
Results and Discussion
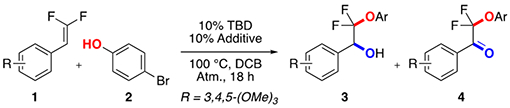
Following our previous work on the base-catalyzed hydrofunctionalization of gem-difluoroalkenes with thiophenols,8b we explored the nucleophilic addition of phenols to the difluoroalkenes.8c Under an atmosphere of O2 using 1,5,7-triazabicylco[4.4.0]dec-5-ene (TBD) as a base, we observed two intriguing side products bearing alcohol (3) or ketone (4) groups at the benzylic position in a nearly 1:1 ratio (Table 1, entry 1) with exclusive regioselectivity. In contrast to O2, use of MnO2, K2S2O8, NMO, and Oxone as oxidants provided a complex mixture of defluorinated products, and moreover, no yield of alcohol 3 (entries 2–5). Use of catalytic quantities of oxidizing metals, such as Fe(III) or Ag(I), failed to improve the selectivity between the alcohol and ketone products (entries 6–11). However, the use of a Co-based catalyst system provided the alcohol product in moderate yield and selectivity (entry 12). Further exploration revealed that Co salts reacted without TBD as an activating base (entries 13–17), and that use of Co(0), (I), and (IV) complexes decreased the reactivity and selectivity (entries 13, 14, 16), while the use of both Co(II) and (III) salts provided good yields (entries 12, 15). Attempts to reduce the loading of Co precatalyst introduced an induction period, while and attempts to reduce the load of phenol required long reaction times (48 h) to reach complete conversion, while also reducing the final yield of product. After thorough optimization, a simple system of Co(acac)2, 1,2-dichlorobenzene (DCB), O2, and 90–140 °C proved optimal (entry 17).
Table 1:
Optimization of Selective Dioxygenation of Difluoroalkenes [a]
 | |||||
|---|---|---|---|---|---|
| Entry | Additive | Atmosphere | Conv.[b] | 3[b] | 4[b] |
| 1 | – | O2 | 92 | 41 | 31 |
| 2 | MnO2 | Air | 100 | 0 | 0 |
| 3 | K2S2O8 | Air | 79 | 8 | 0 |
| 4 | NMO | Air | 33 | 0 | 0 |
| 5 | Oxone | Air | 63 | 0 | 0 |
| 6 | Pd(OAc)2 | O2 | 95 | 38 | 26 |
| 7 | FeCl3 | O2 | 96 | 38 | 23 |
| 8 | CuCl | O2 | 96 | 24 | 23 |
| 9 | AgNO3 | O2 | 95 | 32 | 27 |
| 10 | [Ir(cod)Cl]2 | O2 | 74 | 30 | 22 |
| 11 | RhCl3–H2O | O2 | 94 | 46 | 29 |
| 12 | Co(acac)2 | O2 | 94 | 74 | 13 |
| 13[c] | Co2(CO)8 | O2 | 96 | 8 | 13 |
| 14[c] | Co(PPh3)3Cl | O2 | 93 | 39 | 28 |
| 15[c] | Co(acac)3 | O2 | 95 | 65 | 6 |
| 16[c] | CoS2 | O2 | 87 | 5 | 8 |
| 17[c,d] | Co(acac)2 | O2 | 100 | 71[e] | 3 |
Standard conditions: 1 (1.0 equiv., 0.10 mmol), 2 (3 equiv., 0.30 mmol), DCB (0.25 M, 0.40 mL), TBD (10 mol%, 0.010 mmol), 100 °C, 18 h.
As determined by 19F NMR analysis of the crude reaction mixture using α,α,α-trifluorotoluene (TFT) as a standard (10 μL).
In the absence of TBD.
110 °C, 24 h.
isolated yield.
Under these conditions, a broad range of aromatic gem-difluoroalkenes were competent in the reaction, with most substrates providing >9:1 selectivity for the alcohol product over the ketone product (Table 2). Overall, the reaction yield was sensitive to the electronic character of the difluoroalkene, and adjustment to the reaction time and temperature provided synthetically useful yields of products. Highly-reactive electron-rich substrates thermally degraded at temperatures above 80 °C, and as such, reactions of these electron-rich substrates required lower reaction temperature and/or shorter reaction times to give the products in moderate to good yields (5a–f). More forcing conditions decomposed the starting materials, without providing the desired products. In contrast, electron-deficient substrates required more forcing conditions (120–140 °C), and yields were slightly reduced (5g–l). At temperatures above 140 °C, the substrate decomposed. Mono-ortho-substituted substrates reacted sluggishly, and despite elevated time and temperature full conversion was not achieved. However, the products were isolated in moderate yield, with the unreacted starting material recovered (5e, m). Bis-ortho-substituted substrates did not react (5n), and amine-containing substrates provided lower yields of product (5d).
Table 2:
Scope of β,β-Difluorostyrenes[a]
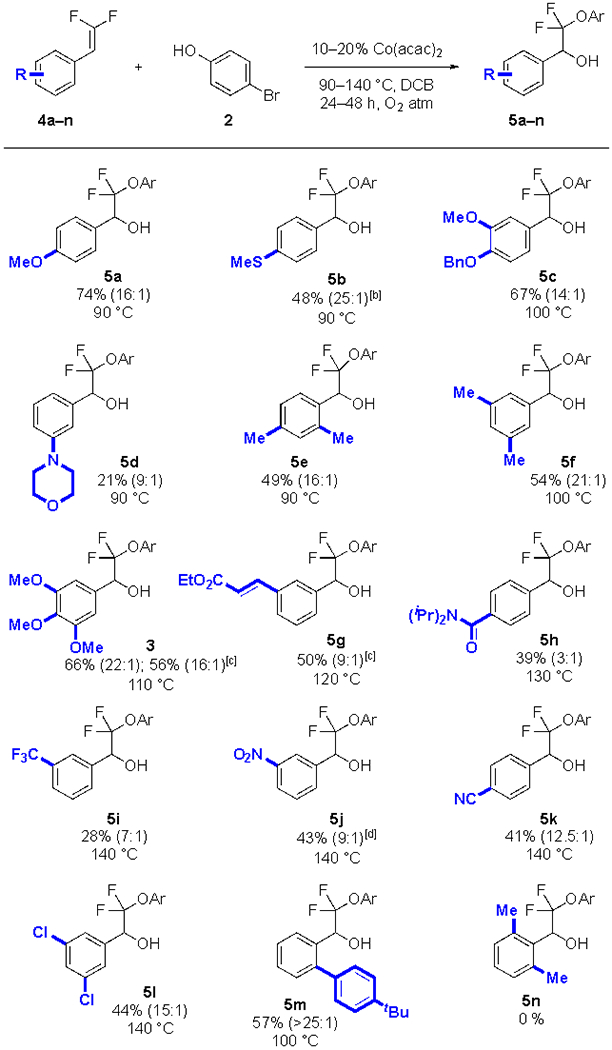 |
Standard conditions: 4a–n (1.0 equiv., 0.50 mmol), 2 (3.0 equiv., 1.5 mmol), DCB (0.25 M, 2.0 mL), Co(acac)2 (10 mol%, 0.050 mmol), temperature as indicated, for 24 h under an O2 atmosphere. The selectivity of alcohol:ketone was determined by 19F NMR analysis of the crude reaction mixture and is reported in parentheses. Yields of pure isolated product represent the average of 2 runs.
Co(acac)2 (20 mol%, 0.10 mmol).
1 (1.0 equiv., 1.50 mmol), 2 (3.0 equiv., 4.5 mmol), DCB (0.25 M, 6.0 mL), Co(acac)2 (10 mol%, 0.150 mmol)
48 h.
Many heterocycles were compatible with the reaction conditions (Table 3), and these substrates generally followed a similar reactivity pattern, in which electron-rich heterocycles performed better than electron deficient heterocycles under more mild conditions (7a–c vs. 7d). Heterocycles with an aliphatic amine (7c) or steric bulk at the ortho position (7e) reacted poorly. Unexpectedly, an ethylene-glycol acetal-protected aldehyde partially deprotected under the reaction conditions (7d), requiring reprotection on workup.
Table 3:
Scope of Heteroaryl β,β-Difluorostyrenes[a]
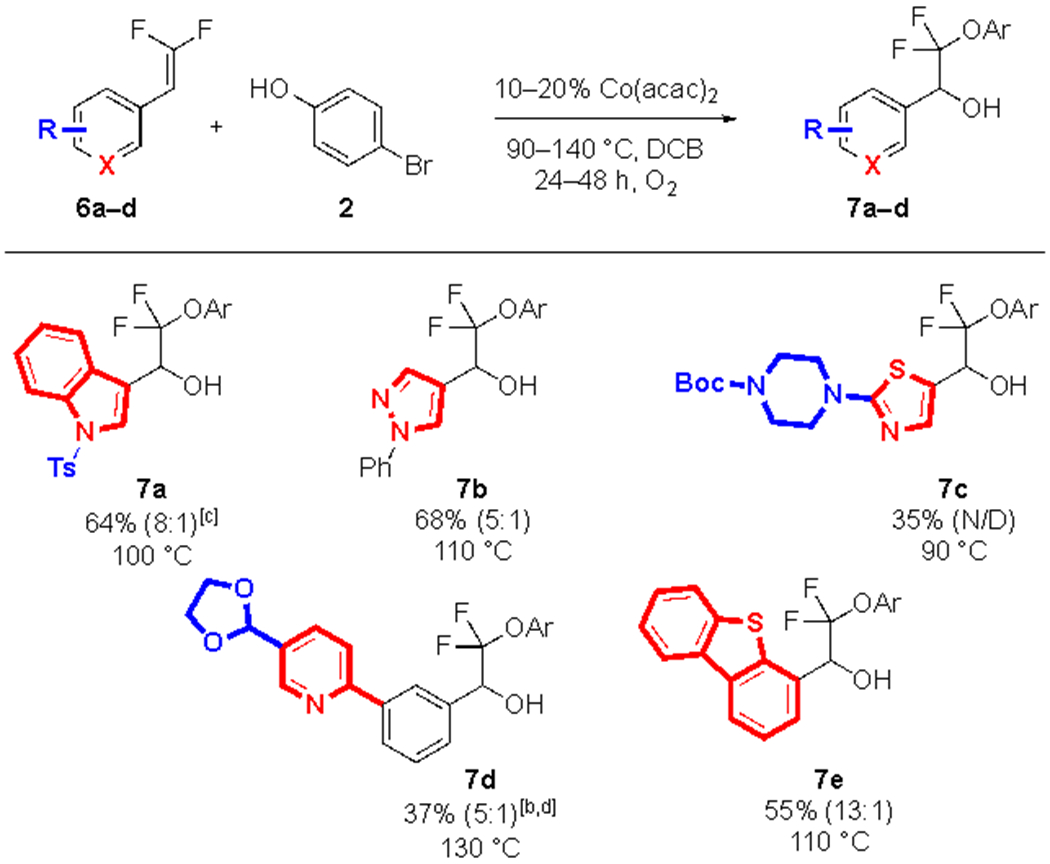 |
Standard conditions: 6a–e (1.0 equiv., 0.50 mmol), 2 (3.0 equiv., 1.5 mmol), DCB (0.25 M, 2.0 mL), Co(acac)2 (10 mol%, 0.050 mmol), temperature as indicated, for 24 h under an O2 atmosphere. The selectivity of alcohol:ketone was determined by 19F NMR analysis of the crude reaction mixture and is reported in parentheses. Yields of pure isolated product represent the average of 2 runs.
Co(acac)2 (20 mol%, 0.10 mmol).
36 h.
48 h, worked up with 4 N HCl/1,4-dioxane and ethylene glycol.
A range of phenolic coupling partners were successfully coupled to deliver difunctionalized products bearing many important functional groups (Table 4). Electron-deficient phenols reacted favourably (9a–f, 3), though the electronic character of the phenol did not greatly affect the overall yields of product (e.g. 9h–i). Similar to gem-difluoroalkene substrates, phenols bearing an ortho-substituent reacted sluggishly (9c,j–l), but still gave product in synthetically useful yields, with the remaining difluoroalkene recovered unreacted. At present, heterocyclic phenols are not competent substrates (e.g 9m, but also including substituted tetrazoles, thiadiazoles, and imidazoles); however, we remain optimistic that these substrates might become compatible with further adjustments to the catalyst system.
Table 4:
Scope of Phenol Nucleophiles[a]
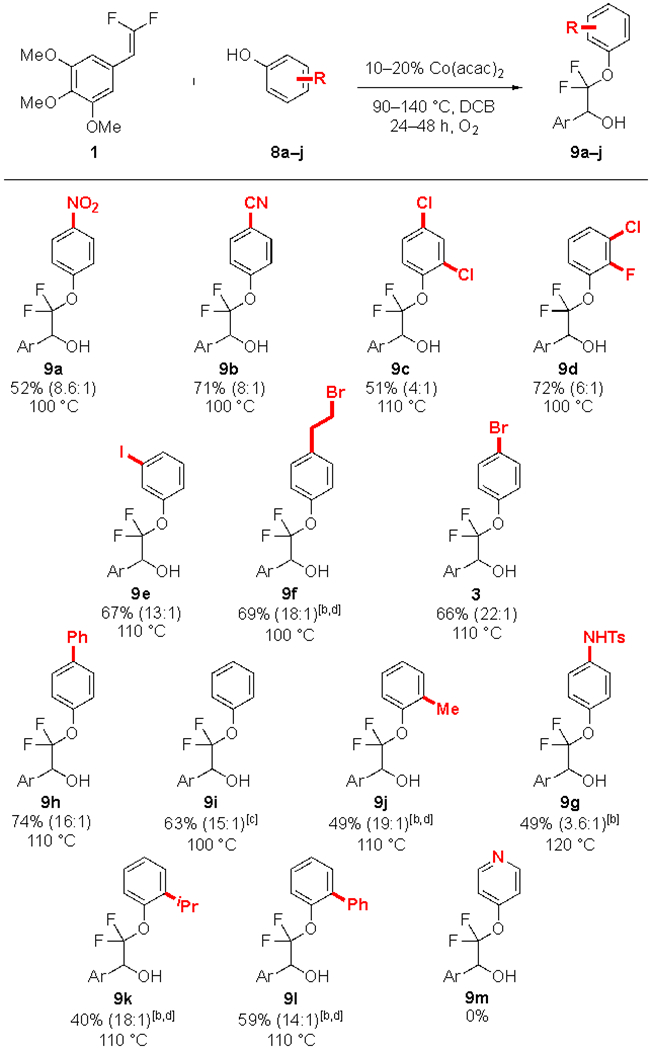 |
Standard conditions: 1 (1.0 equiv., 0.50 mmol), 8a–j (3.0 equiv., 1.5 mmol), DCB (0.25 M, 2.0 mL), Co(acac)2 (10 mol%, 0.050 mmol), temperature as indicated, for 24 h under an O2 atmosphere. The selectivity of alcohol:ketone was determined by 19F NMR analysis of the crude reaction mixture and is reported in parentheses. Yields of pure isolated product represent the average of 2 runs.
Co(acac)2 (20%, 0.10 mmol).
36 h.
48 h.
Mechanistic Overview:
Based on mechanistic studies and an analysis of previous literature, we propose a mechanism involving Co/O2-mediated generation of phenoxyl radical (PhO•, B) prior to engagement of the difluoroalkene (Figure 2). In this mechanism, Co plays key roles, as both an initiator and quencher of the catalytic sequence.
Figure 2:
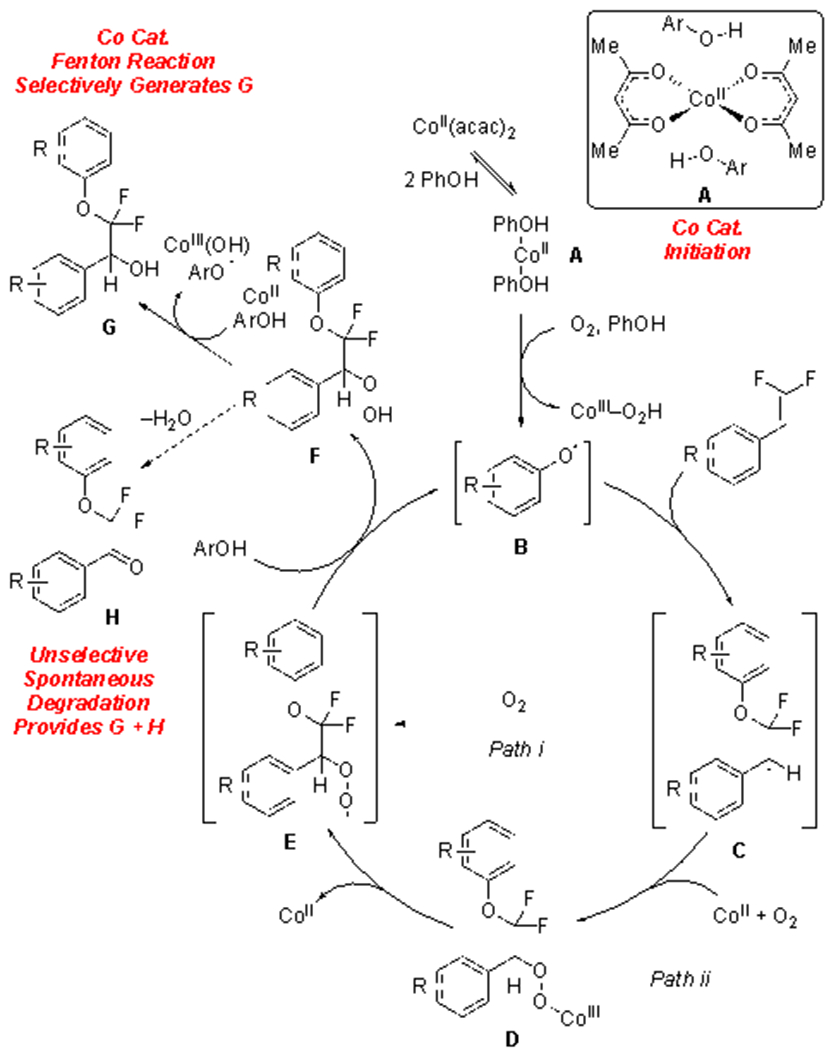
Proposed Co-Initiated Radical Chain Reaction
First, Co initiates a radical chain reaction with O2 and phenol (Figure 2) by reducing O2 to generate superoxide radical (O2•−),15 which subsequently abstracts H• from phenol to generate PhO• (B), Co(III), and a peroxide anion (HO2−).16 Reaction of PhO• (B) with the gem-difluoroalkene at the difluorinated position generates stabilized benzyl radical C. Notably, this reaction proceeds with regioselectivity comparable to that of the addition of S-based radicals to difluoroalkenes.8d,17 Subsequent reaction of radical C with O2 and CoII generates metalloperoxide D,15b,18 or with free O2 to generate peroxyl radical E.19 From D, fragmentation of the Co–O bond would generate peroxyl E,20 which can abstract H• from an appropriate donor,21 in this case presumably PhOH to regenerate PhO• (B) and to propagate the cycle, while also delivering hydroperoxide F. At this stage, Co-mediated fragmentation of F generates alcohol product G, consistent with previous cobalt-catalyzed Fenton decomposition reactions of hydroperoxides,20c,21 through an alkoxy radical intermediate.20c,22 In contrast, under Co-free conditions (Table 1, entry 1), hydroperoxide intermediate F might competitively eliminate H2O to generate phenone side product H;23 thus, by accelerating fragmentation of the O–O bond (F → G), the Co-based catalyst dictates the product distribution by diverting intermediate F.20c,21 This proposed mechanism explains the failure of other metals and oxidants to generate the alcohol-containing product, as metals and reagents that cannot effectively divert hydroperoxide F to alcohol G react with low selectivity (Table 1, entries 2–11).
Radical Intermediates:
Experimental support for radical intermediates includes the decreased yields of product using known radical traps. Specifically, the addition of butylated hydroxytoluene (BHT) and 1,4-benzoquinone inhibited the formation of desired product 5a without forming fluorinated adducts (Table S4). Further, conducting the reaction in the presence of BHT afforded a BHT-phenol adduct (observed by GC-MS), which combined with the lack of a fluorinated adducts (19F NMR). Moreover, under standard reaction conditions phenolic dimers and oligomers were observed by LCMS. Combined, these observations suggest that (1) radicals exist, and (2) the initial radical process initiates through the phenol, not the difluoroalkene.
Initiation Does Not Involve Alkene:
Proposed mechanisms initiating by the difluoroalkene reacting with either Co or O2 were discounted by a series of EPR experiments. Specifically, in stoichiometric experiments monitored by EPR at 10 K, the difluoroalkene did not react with Co(II) or Co(III) by ligation or oxidation (Figure 3A, B), as indicated by the X-band CW EPR spectra of Co(acac)2 in the presence of alkene 4a, which possessed spectral features that match those observed in the absence of the alkene (g tensor values = 6.9, 2.8, and 2.1; Figure 3A, 3B). Further, both of these spectra matched those measured at 4.2 K for both single crystal Co(acac)224 and polycrystalline Co(acac)2(H2O)2 diluted in a Mg lattice25 (g tensor values = 6.84, 2.74, and 1.88), again, confirming that the fluoroalkene does not directly react with Co species under our reaction conditions. Overall, the lack of reactivity of the fluoroalkene ruled out mechanisms involving initial epoxidation of the difluoroalkene, or involving electron transfer from the difluoroalkene to Co(III) to generate a difluoroalkene radical cation.26
Figure 3:
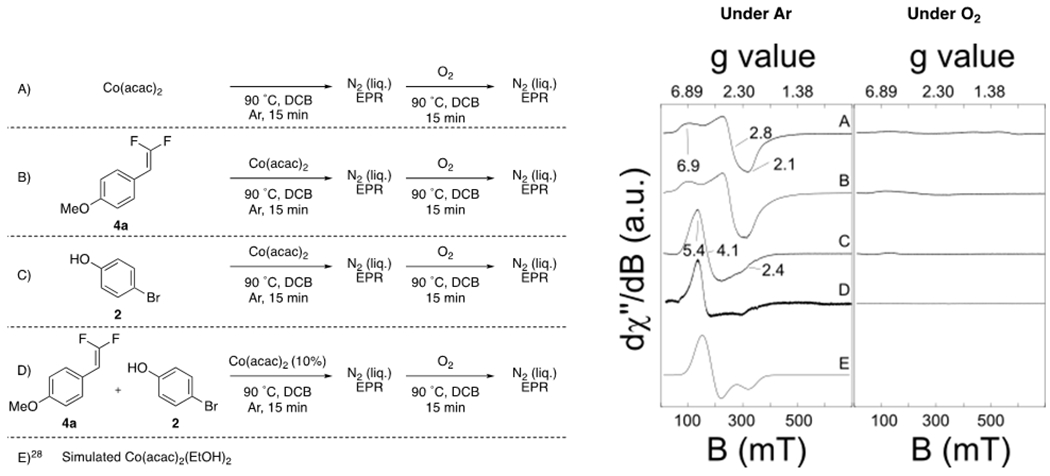
10 K EPR Analysis of Co Catalytic Center. Reaction Conditions: [A] Co(acac)2 (1.0 equiv., 0.10 mmol) was stirred in 0.40 mL of DCB at 90 °C in Ar for 15 min. A 100 μL sample was quenched in N2(l) (left). Then Ar was exchanged for O2 and reacted at 90 °C for 2 h. A 100 μL sample was quenched in N2(l) (right). [B] 4a (1.0 equiv., 0.10 mmol) was reacted with Co(acac)2 (1.0 equiv., 0.10 mmol) in 0.40 mL of DCB at 90 °C in Ar for 15 min. A 100 μL sample was quenched in N2(l) (left). Then Ar was exchanged for O2 and reacted at 90 °C for 2 h. A 100 μL sample was quenched in N2(l) (right). [C] 2 (3.0 equiv., 0.30 mmol) was stirred in the presence of Co(acac)2 (1.0 equiv., 0.10 mmol) in 0.40 mL of DCB at 90 °C in Ar for 15 min. A 100 μL sample was quenched in N2(l) (left). Then Ar was exchanged for O2 and reacted at 90 °C for 2 h. A 100 μL sample was quenched in N2(l) a (right). [D] 4a (1.0 equiv., 0.10 mmol) was reacted with 2 (3.0 equiv., 0.30 mmol) in the presence of Co(acac)2 (0.10 equiv., 0.010 mmol) in 0.40 mL of DCB at 90 °C in Ar for 15 min. A 100 μL sample was quenched in N2(l) (left). Then Ar was exchanged for O2 and reacted at 90 °C for 2 h. A 100 μL sample was quenched in N2(l) (right). [E] Calculated spectra using the EasySpin toolbox from Matlab27 for Co(acac)2(EtOH)2.28
Initiation of Reaction Involves PhOH, Co, and O2:
Evidence for early-stage generation of PhO• (Figure 2, A → B) derives from electron paramagnetic resonance (EPR) experiments using spin trapping reagents. Given the short lifetime of the expected reactive oxygen species and organic radicals, nitrone-based reagents were employed to form stable radical adducts with moderate half-lives and distinguishable EPR spectra for O- and C-centered radicals. Specifically, the reaction of Co(II), phenol, O2 and 5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide (BMPO) or 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) generated spin-trapped adducts with EPR spectra (Figure 4 and Figure S2)29 consistent with previous reports of Co(II) generating O-based radicals.28,30 The EPR spectrum of the phenoxyl radical–BMPO adduct possessed a 14N hyperfine coupling constant (1.4 mT, 39.2 MHz) and 1H hyperfine coupling constant (2.3 mT. 64.5 MHz) consistent with the 2,4-dichlorophenoxyacetic acid C-based radical trapped by DMPO (14N hyperfine coupling constant = 1.53 mT, 1H hyperfine coupling constant = 2.27 mT).31
Figure 4:
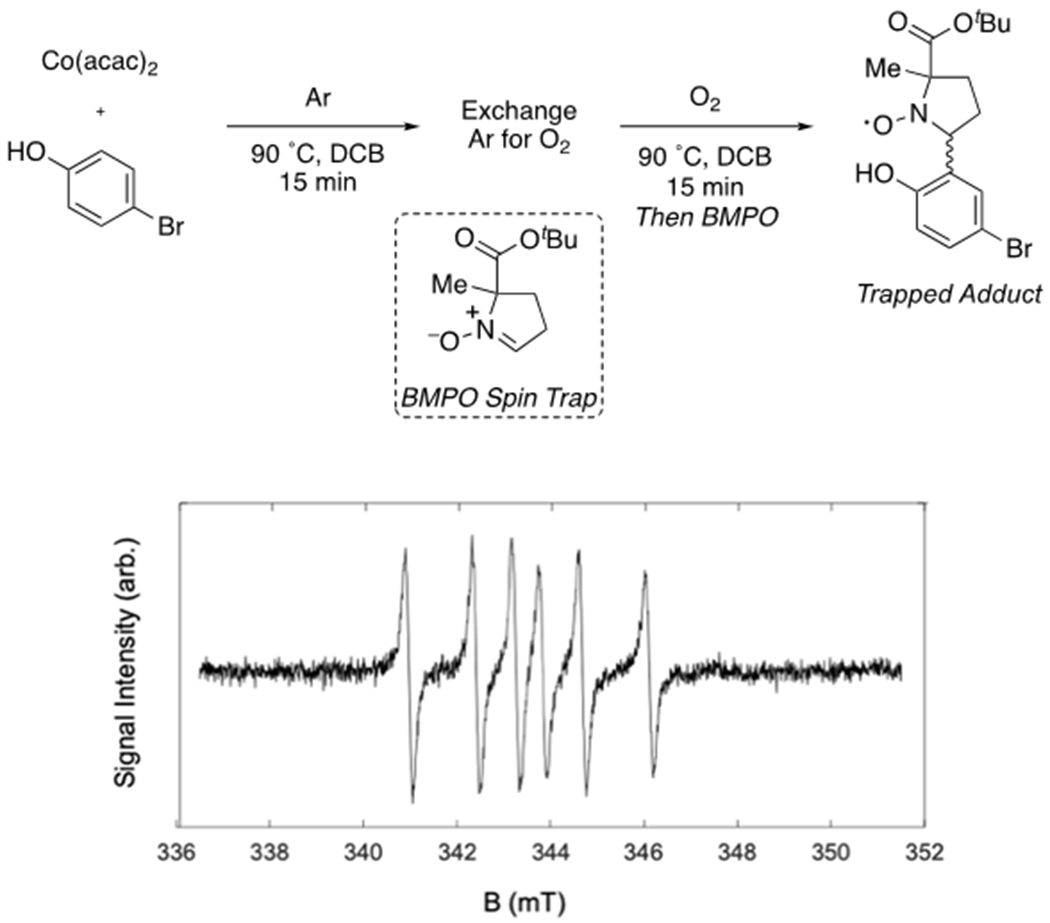
Room Temperature EPR Analysis of Radicals by Spin Trapping with BMPO. Reaction Conditions: Co(acac)2 (1.0 equiv., 0.10 mmol), 2 (3.0 equiv., 0.30 mmol) in 0.80 mL DCB, 90 °C, for 15 min under an Ar atmosphere, followed by the gas exchange of Ar for O2, heated at 90 °C for 15 min, followed by quenching with a solution of BMPO.
Roles of Co Catalyst:
Additional evidence supports Co(II) and O2 playing key roles in initiating the catalytic cycle. First, EPR studies under stoichiometric conditions [Co(II)/O2/PhOH] suggest the formation of initial Co(II) complex A bearing two phenolic ligands and two acetylacetonate ligands in an octahedral complex [Co(acac)2(PhOH)2]28 prior to activation of O2. This structural assignment was made by comparing the measured EPR g tensor values (Figure 3C,D g tensor = 5.8, 3.8, 2.5) to a known Co(acac)2(EtOH)2 complex, which in previous work, was assigned as an octahedral complex with axial alcohol ligands using a combination of density functional theory and EPR spectra (Figure 3E, g tensor = 5.8, 2.0).28 Notably, this same complex is observed as an early intermediate in catalytic reactions quenched by N2(l) and studied by EPR at 10 K, suggesting that complex A forms prior to activation of O2 (Figure 3C). This specific complex might serve as an initiator for generating superoxide [O2•−] and initiating a radical chain process.
Second, following formation of complex A, O2 oxidizes Co(II) to Co(III).15 This pre-catalytic oxidation of Co(II) was observed by 10 K EPR with or without either phenol or difluoroalkene (Figure 3, Left vs. Right Panels), and qualitatively by a color change from red [Co(II)] to green [Co(III)]. Previous reports indicate that this step concurrently generates a superoxide radical15–16 that might serve as the oxidant for the phenol. Supporting the role of O2 early in the reaction mechanism, in the absence of O2, no Co(II)- or Co(III)-catalyzed reaction of difluoroalkene 4a occurs (though electron-rich gem-difluoroalkenes thermally degrade upon extensive heating), presumably because PhO• cannot form (Table 5).
Table 5:
Reaction in the Absence of O2[a]
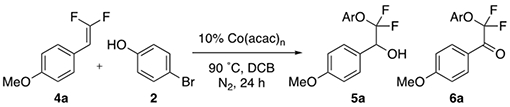 | |||
|---|---|---|---|
| Co | 4a Conv. | 5a Yield | 6a Yield |
| Co(acac)2 | 40% | 0% | 0% |
| Co(acac)3 | 40% | 0% | 0% |
Standard conditions: 4a (1.0 equiv., 0.1 mmol), 2 (3.0 equiv., 0.30 mmol), DCB (0.25 M, 0.40 mL), Co(acac)n (0.10 equiv., 0.010 mmol), 90 °C, for 24 h under an Ar atmosphere. The conversion of 4a and formation of 5a or 6a was determined by 19F NMR analysis standardized with 10 μL (0.080 mmol) of TFT.
Third, EPR studies (10 K) of stoichiometric reactions of Co(II) or Co(III) and phenol under an inert atmosphere (Ar) suggest that Co(II)/O2/PhOH play key roles early in the catalytic cycle. In these reactions, no changes to the Co center were observed (Figure 3D) suggesting that O2 also participates in activating the phenol15a to generate phenoxyl radical B prior to reaction with the gem-difluoroalkene to generate benzyl radical C.
Finally, in control reactions both with and without Co, alcohol product G did not convert to ketone side product H, which effectively rules out a Co-mediated over-oxidation of alcohol G to deliver ketone H.
Combined, this data suggests that Co(II)/O2/PhOH react to generate PhO• (B), 15–16 prior to reaction with the gem-difluoroalkene to generate benzyl radical C. Reaction of C with Co(II)/O2 generates peroxide D,15b,18 followed by Co-mediated fragmentation of the Co–O bond to generate radical E and regenerate the Co(II) catalyst.20a,20b Intermediate E abstracts H• from phenol to generate hydroperoxide F,20c,21 which regenerates PhO• (B) to propagate the reaction. Finally, Co-mediated decomposition of hydroperoxide F generates product G.20c,21 Overall, by exploiting radical intermediates, this cobalt-catalyzed reaction retains both fluorine atoms, which contrasts other transition-metal-catalyzed reactions of gem-difluoroalkenes7a,9,32 that involve β-anionic or β-fluoro-organometallic intermediates that typically eliminate F− and deliver monofluorinated products.
Conclusion
In conclusion, the use of a Co-based catalyst system in an O2 environment enables the selective unsymmetrical dioxygenation of gem-difluoroalkenes in a process that avoids β-fluoride elimination. The reaction selectively generates a difunctionalized product containing a benzyl alcohol and an α,α-difluoroalkylether, thus rapidly and convergently generating compounds containing substructures that should be useful for medicinal chemistry and chemical biology. Mechanistic investigation implicates a radical mechanism that avoids discrete β-anionic or β-fluoro-organometallic intermediates, which overcomes the historical problem of β-fluoride elimination. More generally, this radical-based strategy provides a template for developing new organometallic transformations that will deliver useful fluorinated substructures while avoiding defluorination processes. Though the current system only functions on styrene systems, ongoing work aims to develop related reactions using aliphatic substrates.
EXPERIMENTAL PROCEDURES
General Considerations:
Unless otherwise noted, reactions were performed using oven-dried glassware, and heating was performed in a pre-heated oil bath. Selective dioxygenation reactions of phenols and difluorostyrenes were performed in 20 mL borosilicate glass scintillation vials sealed with a PTFE-lined screw-top cap. All other reactions were performed in round-bottom flasks sealed with rubber septa. Stainless-steel syringes were used to transfer air- and moisture-sensitive liquid reagents. Reactions were monitored by either 19F NMR with an internal standard of α,α,α–trifluorotoluene or by thin-layer chromatography (TLC) on UNIPLATE Silica Gel HLF plates, visualized by quenching of fluorescence. Column chromatography was conducted using a Teledyne Isco CombiFlash Rf 200 system utilizing gradient elution. Isolated yields reported in the manuscript represent an average of at least 2 independent runs of final compound deemed to be at least 95% pure by NMR. Yields reported in the supporting information refer to a single experiment.
Unless otherwise noted, reagents were purchased from commercial sources and used as received. Cobalt(II) 2,4-pentanedionate [Co(acac)2] was purchased from Alfa Aesar. 1,2–Dichlorobenzene (DCB, anhydrous, 99+%) and N-methylpyrrolidine (NMP, anhydrous) were purchased from Sigma Aldrich. Solvents, including dimethylformamide (DMF), toluene (PhMe), dichloromethane (DCM), methanol (MeOH), acetonitrile (MeCN), and tetrahydrofuran (THF) were used directly from a solvent purification system, in which solvent was dried by passage through two columns of activated alumina under argon. Other chemical abbreviations utilized in this document include: α,α,α-trifluorotoluene (TFT), sodium sulfate (Na2SO4), magnesium sulfate (MgSO4), ethyl acetate (EtOAc), diethyl ether (Et2O), ammonium chloride (NH4Cl), nbutyl lithium (nBuLi), sodium hydroxide (NaOH), room temperature (R.T.), tbutyl carbonate anhydride (Boc2O), potassium carbonate (K2CO3), 1,5,7–triazabicyclo[4.4.0]dec-5-ene (TBD), and hydrochloric acid (HCl).
Proton nuclear magnetic resonance (1H NMR) and fluorine nuclear magnetic resonance (19F NMR) were taken on a Bruker AVIIIHD 400 AVANCE spectrometer (400 and 376 MHz respectively). Proton and carbon nuclear magnetic resonance (13C NMR{1H}) were taken on a Bruker AVIII 500 Avance spectrometer with a CPDUL cryoprobe (500 and 126 MHz respectively). Chemical shifts (δ) for protons are reported in parts per million (ppm) downfield from tetramethylsilane, and are referenced to the proton resonance of residual solvent in the NMR solvent (CHCl3: δ = 7.26 ppm; DMSO: δ = 2.50 ppm). Chemical shifts (δ) for carbon are reported in ppm downfield from tetramethylsilane, and are referenced to the carbon resonance of the solvent residual peak (CDCl3: δ = 77.2 ppm; DMSO: δ = 39.52 ppm). Chemical shifts for fluorine are reported uncorrected in ppm upfield from trichlorofluoromethane (0 ppm). NMR data are represented as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet), coupling constant in Hertz (Hz), integration. Electron paramagnetic resonance (EPR) were taken on a Bruker EMXplus EPR spectrometer with an Oxford cryostat. High-resolution mass determinations were obtained either by electrospray ionization (ESI) on a Waters LCT Premier™ mass spectrometer or by atmospheric-pressure chemical ionization (APCI-hexane/PhMe) on a Waters Q-Tof Premier™, for which sample plus near mass internal exact mass standard were dissolved in hexane, and hexane or PhMe/hexane were used as ionization solvent. Infrared spectra were measured on a Perkin Elmer Spectrum Two Fourier Transform Infrared Spectrometer by drying samples on a diamond ATR Sample base plate. Uncorrected melting points were measured on a Thomas Hoover Capillary Melting Point apparatus.
General Procedure for the Preparation of Difluoroalkenes (A-1):
An oven-dried 3-neck round-bottomed flask equipped with a magnetic stirbar was charged with 1 equivalent of aryl aldehyde and 1.2 or 1.5 equivalents of triphenylphosphine. The system was sealed with three PFTE septa, and subsequently evacuated and backfilled with N2 three times. Dry NMP was added via syringe transfer (PTFE syringe with oven-dried stainless-steel needle), and the system was immersed in a preheated 100 °C oil bath. Once no solid reagents remained (approximately 2 min of heating), 1.5 or 1.8 equivalents of potassium bromodifluoroacetate were added portionwise over 0.5 h, with the rate of addition controlling the evolution of CO2 gas. Once all of the potassium bromodifluoroacetate was added, the solution was allowed to stir for 0.5–1 h. Upon completion, the reaction was cooled to R.T. and then quenched with H2O. Subsequently, Et2O was added to the reaction, and the mixture was washed with H2O (five times), and the aqueous layer was back-extracted with Et2O (two times). The combined organic layers were dried over Na2SO4 and concentrated. The crude material was dry-packed onto silica gel and then eluted through a plug of silica gel with EtOAc:hexanes (1:1) to remove triphenylphosphine oxide. Subsequently, the mother liquor was concentrated and subjected to flash chromatography using EtOAc and hexanes.
General Procedure for the Preparation of Difluoroalkenes (A-2):
An oven-dried 3-neck round-bottomed flask equipped with a magnetic stirbar was charged with 1 equivalent of aryl aldehyde and 1.2 or 1.5 equivalents of triphenylphosphine. The system was sealed with three PFTE septa, and subsequently evacuated and backfilled with N2 three times. Dry NMP was added via syringe transfer (PTFE syringe with oven-dried stainless-steel needle), and the system was immersed in a preheated 100 °C oil bath. Once no solid reagents remained (approximately 2 min of heating), 1.5 or 1.8 equivalents of potassium bromodifluoroacetate were added portionwise over 0.5 h, with the rate of addition controlling the evolution of CO2 gas. Once all of the potassium bromodifluoroacetate was added, the solution was allowed to stir for 0.5–1 h. Upon completion, the reaction was cooled to R.T. and then quenched with H2O. Subsequently, Et2O was added to the reaction, and the mixture was washed with H2O (five times), and the aqueous layer was back-extracted with Et2O (two times). The combined organic layers were dried over Na2SO4 and concentrated. The crude material was dry-packed onto silica gel and then eluted through a plug of silica gel with EtOAc:hexanes (1:1) to remove triphenylphosphine oxide. Subsequently, H2O2 (30% in H2O) was added to the mother liquor and allowed to react for 30 min to oxidize the residual triphenylphosphine. The reaction was washed with H2O (three times), dried over Na2SO4, concentrated, and subjected to flash chromatography using EtOAc and hexanes.
General Procedure for the Selective Unsymmetric Dioxygenation of Difluoroalkenes with Phenols (B):
Note: Reactions performed under an atmosphere of O2 are a fire hazard. Reactions should be performed in a fume hood, separated from sources of ignition and flammable solvents. On large scale, reactions should be quenched with a reductive solution to reduce the formation of organic peroxides.
An oven-dried 20 mL scintillation vial, equipped with a magnetic stirbar, was charged with difluoroalkene (0.50 mmol), phenol (1.50 mmol), and Co(acac)2 (0.050–0.20 mmol). The system was purged with O2 gas for 1 min before anhydrous DCB (2.0 mL) was added to the system under a stream of O2 gas. The system was sealed with a PTFE-lined screw-top cap and stirred for 1 min at R.T. Subsequently, the vial was placed into a pre-heated reaction block and stirred vigorously at 90–140 °C for 24–48 h. The vial was cooled to R.T., and 50 μL of TFT was added via microsyringe. The solution was diluted with approximately 1 mL of DCM and then stirred at R.T. for 10 min to allow adequate mixing. After mixing, an aliquot was removed from the vial and passed through a pad of silica gel into an NMR tube using acetone as eluent to remove Co(acac)2, after which the reaction was analyzed by 19F NMR for completion and selectivity. After 19F NMR analysis, the aliquot was sampled for TLC analysis (visualized with 10% phosphomolybdic acid in EtOH) then returned to the vial. Aqueous base (sat. NaOH or Na2CO3) was added to the solution and stirred for 30 min, and then extracted with DCM (four times). The combined organic layers were washed with brine, dried over Na2SO4, concentrated in vacuo, and then purified by flash chromatography using EtOAc and hexanes.
Preparation and Characterization of Substrates
5-(2,2-difluorovinyl)-1,2,3-trimethoxybenzene (1), 1-(2,2-difluorovinyl)-4-methoxybenzene (4a), (4-(2,2-difluorovinyl)phenyl)(methyl)sulfane (4b), 1-(benzyloxy)-4-(2,2-difluorovinyl)-2-methoxybenzene (4c), 4-(3-(2,2-difluorovinyl)phenyl)morpholine (4d), ethyl (E)-3-(3-formylphenyl)acrylate (4g-1), ethyl (E)-3-(3-(2,2-difluorovinyl)phenyl)acrylate (4g), 1-(2,2-difluorovinyl)-3-nitrobenzene (4j), 1,3-dichloro-5-(2,2-difluorovinyl)benzene (4l), 1-tosyl-1H-indole-3-carbaldehyde (6a-1), 1-phenyl-1H-pyrazole-4-carbaldehyde (6b-1), 2-(piperazin-1-yl)thiazole (6c-3), tert-butyl 4-(4-formylthiazol-2-yl)piperazine-1-carboxylate (6c-1), tert-butyl 4-(thiazol-2-yl)piperazine-1-carboxylate (6c-2), 2-(3-(2,2-difluorovinyl)phenyl)-5-(1,3-dioxolan-2-yl)pyridine (6d) 2-bromo-5-(1,3-dioxolan-2-yl)pyridine (6d-2), and 3-(5-(1,3-dioxolan-2-yl)pyridin-2-yl)benzaldehyde (6d-1) were prepared according to a previous literature report.8b 4-formyl-N,N-diisopropylbenzamide (4h-1)34 and N-(4-hydroxyphenyl)-4-methylbenzenesulfonamide (8g)35 were prepared according to revious reports.
1-(2,2-difluorovinyl)-2,4-dimethylbenzene (4e)
Following General Procedure A-2, 2,4-dimethylbenzaldehyde (3.20 mL, 22.0 mmol) was reacted with PPh3 (8.84 g, 33.0 mmol) and BrCF2CO2K (8.76 g, 40.0 mmol). Following workup, the product was purified by flash chromatography using a gradient of 0–10% EtOAc in hexanes, furnishing 1.57 g of desired product 4e (41% yield) as a clear oil; 1H NMR (400 MHz, CDCl3) δ 7.31 (dd, J = 8.44, 2.00 Hz, 1 H), 7.02 (dd, J = 4.24, 2.31 Hz, 2 H), 5.34 (dd, J = 25.61, 3.94 Hz, 1 H), 2.32 (s, 3 H), 2.27 (s, 3 H); 13C NMR{1H} (126 MHz, CDCl3) δ 156.2 (dd, J = 295.18, 288.05 Hz), 137.2, 135.8 (dd, J = 4.84, 1.67 Hz), 131.1, 128.1 (dd, J = 7.88, 1.99 Hz), 127.0, 126.0 (dd, J = 6.89, 4.94 Hz), 79.3 (dd, J = 28.66, 14.94 Hz), 21.2, 20.0; 19F NMR (376 MHz, CDCl3) δ −84.76 (dd, J = 33.14, 4.06 Hz, 1 F), −85.53 (ddd, J = 33.09, 25.53, 1.83 Hz, 1F); IR (film) 2923, 1726, 1616, 1569, 1505, 1453, 1379, 1345, 1281, 1250, 1235, 1180, 1111, 1074, 1037, 948, 917, 876, 836, 818, 765, 750, 721, 615, 581, 549, 534 cm−1; HRMS (TAPCI) m/z: [M+]+ Calcd for C10H10F2 168.0751; found 168.0745, 3.6 ppm.
1-(2,2-difluorovinyl)-3,5-dimethylbenzene (4f)
Following General Procedure A-2, 3,5-dimethylbenzaldehyde (2.10 mL, 15.0 mmol) was reacted with PPh3 (6.23 g, 22.5 mmol) and BrCF2CO2K (6.05 g, 27.0 mmol). Following workup, the product was purified by flash chromatography using a gradient of 0–10% EtOAc in hexanes, furnishing 1.163 g of desired product 4f (44% yield) as a clear oil; 1H NMR (400 MHz, CDCl3) δ 6.97 (bs, 2 H), 6.90 (bs, 1 H), 5.21 (dd, J = 26.41, 4.03 Hz, 1H), 2.32 (s, 6 H); 13C NMR{1H} (126 MHz, CDCl3) δ 156.3 (dd, J = 298.23, 287.44 Hz), 138.3, 130.3 (t, J = 6.70 Hz), 128.9 (t, J = 2.15 Hz), 125.6 (dd, J = 6.56, 3.67 Hz), 82.3 (dd, J = 28.88, 13.63 Hz), 21.4; 19F NMR (376 MHz, CDCl3) δ −82.39 (dd, J = 32.54, 26.54 Hz, 1 F), −84.62 (dd, J = 32.49, 3.99 Hz, 1F); IR (film) 3019, 2921, 2868, 1726, 1605, 1448, 1379, 1350, 1297, 1198, 1160, 1038, 965, 892, 851, 814, 765, 750, 715, 690, 583, 539, 515 cm−1; HRMS (TAPCI) m/z: [M+]+ Calcd for C10H10F2 168.0751; found 168.0744, 4.2 ppm.
4-(2,2-difluorovinyl)-N,N-diisopropylbenzamide (4h)
Following General Procedure A-2, compound 4h-1 (0.823, 3.60 mmol) was reacted with PPh3 (1.50 g, 5.30 mmol) and BrCF2CO2K (1.42 g, 6.50 mmol) in NMP (2.0 mL, 2 M). Following workup, the product was purified by flash chromatography using a gradient of 0–30% EtOAc in hexanes, furnishing 0.655 g of desired product 4h (69% yield) as a colorless solid (MP = 43–44 °C); 1H NMR (500 MHz, DMSO-D6, 25 °C) δ 7.41 (d, J = 8.09 Hz, 2 H), 7.29 (d, J = 8.21 Hz, 2 H), 5.85 (dd, J = 28.05, 4.06 Hz, 1 H), 3.61 (bs, 2.04, 2 H), 1.38–1.15 (m, 12 H); 1H NMR (500 MHz, DMSO-D6, 60 °C) δ 7.41 (d, J = 7.92 Hz, 2 H), 7.28 (d, J = 7.89 Hz, 2 H), 5.79 (dd, J = 27.83, 4.01 Hz, 1 H), 3.64 (hept, J = 6.41 Hz, 2 H), 1.28 (bs, 12 H); 13C NMR{1H} (126 MHz, DMSO-D6, 60 °C) δ 169.8, 156.1 (dd, J = 297.83, 286.17 Hz), 138.2 (t, J = 2.29 Hz), 130.5 (dd, J = 7.75, 5.77 Hz), 128.1 (dd, J = 6.61, 3.79 Hz), 126.3, 82.3 (dd, J = 29.45, 11.75 Hz), 20.9; 19F NMR (376 MHz, DMSO-D6, 25 °C) δ −82.16 (dd, J = 32.14, 28.07 Hz, 1 F), −84.02 (dd, J = 32.19, 4.05 Hz, 1 F); IR (film) 3434, 2252, 2126, 1729, 1660, 1345, 1276, 1052, 1024, 1005, 822, 760, 623 cm−1; HRMS (ESI+) m/z: [M+H]+ Calcd for C15H20F2NO 268.1513; found 268.1500, 1.3 mmu.
1-(2,2-difluorovinyl)-3-(trifluoromethyl)benzene (4i)
Following General Procedure A-1, 3-trifluoromethylbenzaldehyde (2.7 mL, 20mmol) was reacted with PPh3 (6.3 g, 24 mmol) and BrCF2CO2K (6.2 g, 30 mmol) in NMP (10 mL, 2 M). Following workup, the product was purified by flash chromatography using a gradient of 0–50% Et2O in pentane, furnishing 2.070 g of desired product 4i (50% yield) as a colorless oil; 1H NMR matched the previously reported spectrum.33
4-(2,2-difluorovinyl)benzonitrile (4k)
Following General Procedure A-1, 4-formylbenzonitrile (6.55 g, 50 mmol) was reacted with PPh3 (15.7 g, 60 mmol) and BrCF2CO2K (15.1 g, 75 mmol) in NMP (25 mL, 2 M). Following workup, the product was purified by flash chromatography using a gradient of 0–50% Et2O in pentane, furnishing 6.10 g of desired product 4k (74% yield) as a colorless solid; 1H NMR matched the previously reported spectrum.8b
4′-(tert-butyl)-2-(2,2-difluorovinyl)-1,1′-biphenyl (4m)
Following General Procedure A-2, compound 4m–1 (5.00 g, 0.021 mmol) was reacted with PPh3 (6.63 g, 0.025 mmol) and BrCF2CO2K (6.76 g, 0.031 mmol) in NMP (10.5 mL, 2 M). Following workup, the product was purified by flash chromatography using a gradient of 0–10% EtOAc in hexanes, furnishing 3.44 g of desired product 4m (60% yield) as a clear oil; 1H NMR matched that of the previously reported spectrum.8b
2-(2,2-difluorovinyl)-1,3-dimethylbenzene (4n)
Following General Procedure A-2, 2,6-dimethylbenzaldehyde (2.2 mL, 15.0 mmol) was reacted with PPh3 (5.91 g, 22.5 mmol) and BrCF2CO2K (6.17 g, 27.0 mmol). Following workup, the product was purified by flash chromatography using a gradient of 0–10% EtOAc in hexanes, furnishing 0.763 g of desired product 4n (28% yield) as a clear oil; 1H NMR (400 MHz, CDCl3) δ 7.14 (dd, J = 8.57, 6.35 Hz, 1 H), 7.07 (d, J = 7.53 Hz, 2 H), 5.23 (dd, J = 27.50, 2.26 Hz, 1 H), 2.29 (s, 6 H); 13C NMR{1H} (126 MHz, CDCl3) δ 154.9 (dd, J = 291.73, 288.36 Hz), 137.5 (dd, J = 2.57, 1.37 Hz), 127.8, 127.6, 78.1 (dd, J = 27.32, 20.62 Hz), 20.5 (d, J = 2.42 Hz); 19F NMR (376 MHz, CDCl3) δ −83.38 (dd, J = 32.45, 26.96 Hz, 1 F), −87.16 (dd, J = 33.11, 2.37 Hz, 1 F); IR (film) 3024, 2956, 2923, 2330, 1736, 1586, 1468, 1445, 1380, 1329, 1276, 1254, 1222, 1166, 1096, 1032, 932, 850, 802, 768, 746, 698, 599, 537 cm−1; HRMS (TAPCI) m/z: [M+]+ Calcd for C10H10F2 168.0751; found 168.0741, 1.0 mmu.
3-(2,2-difluorovinyl)-1-tosyl-1H-indole (6a)
Following General Procedure A-1, compound 6a-1 (9.09 g, 0.030 mmol) was reacted with PPh3 (9.58 g, 0.036 mmol) and BrCF2CO2K (9.81 g, 0.045 mmol) in NMP (15 mL, 2.0 M). Following workup, the product was purified by flash chromatography using a gradient of 0–50% EtOAc in hexanes, furnishing 4.80 g of desired product 6a (47% yield) as a tan solid; 1H NMR matched previously reported data.8b
4-(2,2-difluorovinyl)-1-phenyl-1H-pyrazole (6b)
Following General Procedure A-1, compound 6b-1 (1.50 g, 0.00870 mol) was reacted with PPh3 (2.84 g, 0.0100 mol) and BrCF2CO2K (3.22 g, 0.0130 mol) in NMP (4.3 mL, 2.0 M). Following workup, the product was purified by flash chromatography using a gradient of 0–50% EtOAc in hexanes, furnishing 1.31 g of desired product 6b (72% yield) as a colorless solid; 1H NMR matched previously reported data.8b
tert-butyl 4-(4-(2,2-difluorovinyl)thiazol-2-yl)piperazine-1-carboxylate (6c)
Following General Procedure A-1, compound 6c–1 (8.01 g, 0.0270 mol) was reacted with PPh3 (8.47 g, 0.0320 mol) and BrCF2CO2K (8.60 g, 0.0400 mol) in NMP (14 mL, 2.0 M). Following workup, the product was purified by flash chromatography using a gradient of 0–70% EtOAc in hexanes, furnishing 2.56 g of desired product 6c (29% yield) as a tan solid; 1H NMR matched previously reported data.8b
4-(2,2-difluorovinyl)dibenzo[b,d]thiophene (8c)
Following General Procedure A-2, compound dibenzo[b,d]thiophene-4-carbaldehyde8b (5.03 g, 0.0240 mol) was reacted with PPh3 (7.44 g, 0.0280 mol) and BrCF2CO2K (7.57 g, 0.0350 mol) in NMP (12 mL, 2.0 M). Following workup, the product was purified by flash chromatography using a gradient of 0–10% EtOAc in hexanes, furnishing 2.73 g of desired product 8c (47% yield) as a colorless solid; 1H NMR matched previously reported data.8b
N-(4-hydroxyphenyl)-4-methylbenzenesulfonamide (8g)
Prepared according to reference 2. 4-Aminophenol (1.50 g, 14.0 mmol) was dissolved in 50 mL DCM, and the resulting solution cooled to 0 °C under vigorous stirring. Pyridine (5.1 mL, 63 mmol) was added dropwise, and the resulting solution was stirred for 15 min. A solution of tosyl chloride (2.94 g, 15.4 mmol) in DCM (0.010 L) was added dropwise at 0 °C. The solution was warmed to R.T. and stirred overnight. 3 N HCl (50 mL) was added to quench the reaction, and the mixture was extracted DCM (three times, 15 mL each time). The organic layers were combined and washed with 3 N HCl (20 mL). The combined organic layers were dried over Na2SO4, dried in vacuo, and purified by flash chromatography (30–50% EtOAc in Hexanes) to provide 3.05 g (83% yield) of desired product 8g as a pale yellow solid; 1H NMR matched the previously reported spectrum.35
Experimental Procedures and Characterization of Compounds in Table 2:
2-(4-bromophenoxy)-2,2-difluoro-1-(3,4,5-trimethoxyphenyl)ethan-1-ol (3)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 110 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 10–35% EtOAc in hexanes, furnishing 0.148 g (71% yield) of desired product 3 as a yellow solid (MP = 93–95 °C); 1H NMR (400 MHz, CDCl3) δ 7.45–7.41 (m, 2 H), 7.03–6.99 (m, 2 H), 6.74 (s, 2 H), 5.00 (td, J = 7.24, 3.49 Hz, 1 H), 3.86 (s, 6H), 3.85 (s, 3 H), 3.14 (d, J = 3.74 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 153.1, 149.1 (t, J = 2.05 Hz), 138.4 (d, J = 2.06 Hz), 132.6, 131.0, 123.6, 122.4 (t, J = 273.70 Hz), 119.0, 105.0, 74.2 (t, J = 31.70 Hz), 61.0, 56.3; 19F NMR (376 MHz, CDCl3) δ −81.65 (dd, J = 141.05, 6.98 Hz, 1 F), −82.16 (dd, J = 140.99, 7.23 Hz, 1 F); IR (film) 3450, 2939, 1595, 1508, 1485, 1464, 1422, 1326, 1253, 1129, 1068, 1011, 829, 750, 710 cm−1; HRMS (ESI+) m/z: [M+]+ Calcd for C17H17BrF2O5 418.0227; found 418.0212, 3.6 ppm.
Experimental Procedure for Model Reaction (1.5 mmol)
2-(4-bromophenoxy)-2,2-difluoro-1-(3,4,5-trimethoxyphenyl)ethan-1-ol (3)
Following General Procedure B, 0.345 g (1.50 mmol) of compound 1 was reacted with 0.780 g (4.50 mmol) of 4-bromophenol in the presence of 0.039 g (0.15 mmol) of Co(acac)2 at 110 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 0–35% EtOAc in hexanes, furnishing 0.387 g (62% yield) of desired product 3 as a yellow solid.
2-(4-bromophenoxy)-2,2-difluoro-1-(4-methoxyphenyl)ethan-1-ol (5a)
Following General Procedure B, 0.085 g (0.50 mmol) of compound 4a was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 90 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 10–35% EtOAc in hexanes, furnishing 0.109 g (77% yield) of desired product 5a as a pale yellow solid (MP = 51–53 °C); 1H NMR (400 MHz, CDCl3) δ 7.47 (d, J = 8.64 Hz, 2 H), 7.44–7.41 (m, 2 H), 7.01 (d, J = 8.91 Hz, 2 H), 6.96–6.92 (m, 2H), 5.04 (td, J = 7.18, 4.29 Hz, 1 H), 3.83 (s, 3 H), 2.57 (d, J = 4.27 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 160.3, 149.2 (t, J = 2.40 Hz), 132.6, 129.1, 127.4, 123.6, 122.6 (t, J = 272.76 Hz), 119.0, 113.9, 74.1 (t, J = 31.82 Hz), 55.4; 19F NMR (376 MHz, CDCl3) δ −82.39 (d, J = 7.21 Hz, 2 F); IR (film) 3424, 2957, 2911, 2838, 1891, 1613, 1586, 1515, 1485, 1465, 1442, 1399, 1346, 1305, 1246, 1197, 1177, 1144, 1117, 1065, 1032, 1012, 939, 827, 800, 756, 745, 716, 691, 636, 593, 535, 493 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C15H13BrF2O3Cl 392.9705; found 392.9709, 1.0 ppm.
2-(4-bromophenoxy)-2,2-difluoro-1-(4-(methylthio)phenyl)ethan-1-ol (5b)
Following General Procedure B, 0.093 g (0.50 mmol) of compound 4b was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.026 g (0.100 mmol) of Co(acac)2 at 90 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 10–35% EtOAc in hexanes, furnishing 0.099 g (53% yield) of desired product 5b as a yellow solid (MP = 70–72 °C); 1H NMR (400 MHz, CDCl3) δ 7.45 (t, J = 8.70 Hz, 4 H), 7.28 (d, J = 8.49 Hz, 2 H), 7.01 (d, J = 8.40 Hz, 2 H), 5.04 (t, J = 7.06 Hz, 1 H), 2.83 (bs, 1 H), 2.50 (s, 3 H); 13C NMR{1H} (126 MHz, CDCl3) δ 149.0, 139.9, 132.6, 131.9, 128.3, 126.2, 123.6, 122.4 (t, J = 272.73 Hz), 119.0, 74.1 (t, J = 32.16 Hz), 15.6; 19F NMR (376 MHz, CDCl3) δ −82.27 (dd, J = 19.13, 7.12 Hz, 2 F); IR (film) 3397, 2921, 2051, 1892, 1728, 1601, 1484, 1436, 1405, 1346, 1251, 1210, 1195, 1146, 1092, 1066, 1012, 968, 941, 846, 818, 796, 758, 744, 685, 644, 539, 493 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C15H13BrF2O2SCl 408.9476; found 408.9482, 1.5 ppm.
1-(4-(benzyloxy)-3-methoxyphenyl)-2-(4-bromophenoxy)-2,2-difluoroethan-1-ol (5c)
Following General Procedure B, 0.131 g (0.500 mmol) of compound 4c was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 100 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 10–35% EtOAc in hexanes, furnishing 0.150 g (68% yield) of desired product 5c as an light orange solid (MP = 87–88 °C); 1H NMR (400 MHz, CDCl3) δ 7.44 (td, J = 7.21, 6.81, 1.94 Hz, 4 H), 7.41–7.34 (m, 2 H), 7.34–7.28 (m, 1 H), 7.11 (d, J = 1.93 Hz, 1 H), 7.06–6.96 (m, 3 H), 6.89 (d, J = 8.29 Hz, 1 H), 5.17 (s, 2 H), 5.00 (t, J = 7.12 Hz, 1 H), 3.90 (s, 3H), 2.83 (bs, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 149.6, 149.1 (d, J = 2.75 Hz), 148.8, 137.0, 132.5, 128.7, 128.3, 128.0, 127.4, 123.6, 122.5 (t, J = 272.85 Hz), 120.6, 119.0, 113.4 (d, J = 1.62 Hz), 111.3, 74.1 (t, J = 31.44 Hz), 71.0, 56.2; 19F NMR (376 MHz, CDCl3) δ −81.89 (dd, J = 141.33, 7.27 Hz, 1 F), −82.28 (dd, J = 141.33, 7.27 Hz, 1 F); IR (film) 3458, 3033, 2917, 2849, 1735, 1607, 1594, 1514, 1484, 1464, 1454, 1421, 1382, 1337, 1252, 1202, 1138, 1065, 1033, 1012, 914, 844, 827, 800, 738, 696, 648, 551, 494 cm−1; HRMS (ESI+) m/z: [M+K]+ Calcd for C22H19BrF2O4K 503.0072; found 503.0078, 1.2 ppm.
2-(4-bromophenoxy)-2,2-difluoro-1-(3-morpholinophenyl)ethan-1-ol (5d)
Following General Procedure B, 0.113 g (0.500 mmol) of compound 4d was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 90 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 15–50% EtOAc in hexanes, furnishing 0.044 g (21% yield) of desired product 5d as an orange oil; 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 8.86 Hz, 2 H), 7.31 (t, J = 7.92 Hz, 1 H), 7.10 (t, J = 2.00 Hz, 1 H), 7.04 (d, J = 7.93 Hz, 1 H), 7.01 (d, J = 8.62 Hz, 2 H), 6.94 (ddd, J = 8.26, 2.55, 0.96, 1 H), 5.04 (t, J = 7.24 Hz, 1 H), 3.88–3.85 (m, 4 H), 3.20–3.17 (m, 4 H), 2.72 (d, J = 3.57 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 151.5, 149.1 (d, J = 2.26 Hz), 136.3, 132.6, 129.3, 123.6, 122.5 (t, J = 272.72 Hz), 119.5, 119.0, 116.4, 115.1, 74.7 (t, J = 31.27 Hz), 67.0, 49.4; 19F NMR (376 MHz, CDCl3) δ −81.94 (t, J = 8.00 Hz, 2 F); IR (film) 3377, 2965, 2857, 1727, 1604, 1584, 1485, 1448, 1380, 1343, 1304, 1243, 1202, 1145, 1115, 1067, 1012, 997, 978, 962, 933, 888, 827, 785, 756, 737, 698, 644, 529, 494 cm−1; HRMS (ESI+) m/z: [M+H]+ Calcd for C18H19BrF2NO3 414.0516; found 414.0521, 1.2 ppm.
2-(4-bromophenoxy)-1-(2,4-dimethylphenyl)-2,2-difluoroethan-1-ol (5e)
Following General Procedure B, 0.084 g (0.50 mmol) of compound 4e was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 90 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 0–20% EtOAc in hexanes, furnishing 0.089 g (50% yield) of desired product 5e as a pale oil; 1H NMR (400 MHz, CDCl3) δ 7.58 (d, J = 7.96 Hz, 1 H), 7.43 (d, J = 8.90 Hz, 2 H), 7.11 (d, J = 8.24 Hz, 1 H), 7.04 (d, J = 2.23 Hz, 2 H), 7.02 (s, 1 H), 5.35 (td, J = 7.18, 3.79 Hz, 1 H), 2.85 (d, J = 4.35 Hz, 1 H), 2.40 (s, 3 H), 2.35 (s, 3 H); 13C NMR{1H} (126 MHz, CDCl3) δ 149.1 (t, J = 2.25 Hz), 138.7, 136.7, 132.5, 131.3, 130.9, 127.3 (t, J = 1.73 Hz), 127.1, 123.5, 123.0 (t, J = 273.10 Hz), 118.8, 70.3 (t, J = 31.67 Hz), 21.2, 19.6; 19F NMR (376 MHz, CDCl3) δ −81.41 (dd, J = 140.67, 7.53 Hz, 1 F), −81.85 (dd, J = 140.45, 7.12 Hz, 1 F); IR (film) 3381, 2923, 1616, 1583, 1484, 1249, 1196, 1142, 1065, 1012, 826, 809, 760, 748, 720, 691, 494 cm−1; HRMS (ESI+) m/z: [M+Cl]− Calcd for C16H15BrF2O2Cl 390.9912; found 390.9920, 2.0 ppm.
2-(4-bromophenoxy)-1-(3,5-dimethylphenyl)-2,2-difluoroethan-1-ol (5f)
Following General Procedure B, 0.084 g (0.50 mmol) of compound 4f was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 100 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 0–15% EtOAc in hexanes, furnishing 0.099 g (44% yield) of desired product 5f as a tan solid (MP = 79–81 °C); 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 8.88 Hz, 2 H), 7.16 (bs, 2 H), 7.04 (bs, 2 H), 7.02 (bs, 1 H), 5.00 (t, J = 7.25 Hz, 1 H), 2.74 (bs, 1 H), 2.36 (s, 6 H); 13C NMR{1H} (126 MHz, CDCl3) δ 149.2, 138.1, 135.2, 132.5, 130.9, 125.6 (d, J = 1.49 Hz), 123.6, 122.5 (t, J = 272.07 Hz), 118.9, 74.6 (t, J = 31.54 Hz), 21.5; 19F NMR (376 MHz, CDCl3) δ −81.59 (dd, J = 140.92, 7.09 Hz, 1 F), −82.16 (dd, J = 140.89, 7.38 Hz, 1 F); IR (film) 3395, 3011, 2919, 2051, 1891, 1760, 1609, 1583, 1484, 1399, 1379, 1345, 1251, 1199, 1143, 1114, 1066, 1012, 953, 938, 905, 886, 828, 803, 786, 762, 744, 716, 699, 686, 645, 561, 536, 493 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C16H15BrF2O2Cl 390.9912; found 390.9921, 2.3 ppm.
ethyl (E)-3-(3-(2-(4-bromophenoxy)-2,2-difluoro-1-hydroxyethyl)phenyl)acrylate (5g)
Following General Procedure B, 0.119 g (0.500 mmol) of compound 4g was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 120 °C for 48 h. After workup with sat. Na2CO3 (aq.), the product was purified by flash chromatography using a gradient of 5–35% EtOAc in hexanes, furnishing 0.109 g (51% yield) of desired product 5g as an orange oil; 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 16.16 Hz, 2 H), 7.56 (d, J = 7.42 Hz, 1 H), 7.52 (dt, J = 7.88, 1.47 Hz, 1 H), 7.42–7.38 (m, 3 H), 6.98 (m, 2 H), 6.45 (d, J = 16.02 Hz, 1 H), 5.10 (t J = 7.05 Hz, 1 H), 4.25 (q, J = 7.11 Hz, 2 H), 3.53 (bs, 1 H), 1.32 (t, J = 7.13 Hz, 3 H); 13C NMR{1H} (126 MHz, CDCl3) δ 167.2, 148.9 (t, J = 2.32 Hz), 144.4, 136.3, 134.5, 132.5, 129.7, 128.9, 128.6, 127.5, 122.3 (t, J = 273.04 Hz), 119.0, 118.7, 73.8 (t, J = 31.46 Hz), 60.8, 14.4; 19F NMR (376 MHz, CDCl3) δ −81.74 (dd, J = 140.69, 6.93 Hz, 1 F), −82.25 (dd, J = 140.78, 7.25 Hz,1 F); IR (film) 3418, 2982, 2051, 1891, 1693, 1584, 1484, 1438, 1397, 1368, 1308, 1252, 1225, 1188, 1148, 1113, 1098, 1066, 1012, 983, 863, 843, 825, 794, 757, 734, 696, 651, 581, 558, 493, 465 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C19H17BrF2O4Cl 460.9967; found 460.9999, 6.9 ppm.
4-(2-(4-bromophenoxy)-2,2-difluoro-1-hydroxyethyl)-N,N-diisopropylbenzamide (5h)
Following General Procedure B, 0.134 g (0.500 mmol) of compound 4h was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 130 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 15–50% EtOAc in hexanes, furnishing 0.094 g (41% yield) of desired product 5h as a colorless solid (MP = 182–183 °C); 1H NMR (500 MHz, DMSO-D6, 60 °C) δ 7.57 (dd, J = 8.45, 3.06 Hz, 4 H), 7.29 (d, J = 7.74 Hz, 2 H), 7.08 (d, J = 8.34 Hz, 2 H), 6.51 (d, J = 5.64 Hz, 1 H), 5.10 (q, J = 7.08 Hz, 1 H), 3.66–3.63 (m, 2 H), 1.28 (bs, 12 H); 13C NMR{1H} (126 MHz, DMSO-D6, 60 °C) δ 169.3, 148.8 (d, J = 2.31 Hz), 138.7, 137.2, 132.3, 127.7, 124.7, 123.2, 122.6 (t, J = 272.21 Hz), 117.7, 72.0 (t, J = 31.34 Hz), 54.5, 20.2; 19F NMR (376 MHz, CDCl3) δ −81.80 (dd, J = 140.26, 7.03 Hz, 1 F), −82.22 (dd, J = 140.26, 6.48 Hz, 1 F); IR (film) 3250, 2974, 2935, 1602, 1515, 1483, 1457, 1407, 1381, 1372, 1349, 1275, 1252, 1209, 1195, 1161, 1141, 1082, 1064, 1038, 1012, 919, 883, 854, 808, 765, 750, 681, 631, 610, 577, 548, 527, 497 cm−1; HRMS (ESI+) m/z: [M+Na]+ Calcd for C21H24BrF2NO3Na 478.0805; found 478.0813, 1.7 ppm.
2-(4-bromophenoxy)-2,2-difluoro-1-(3-(trifluoromethyl)phenyl)ethan-1-ol (5i)
Following General Procedure B, 0.104 g (0.500 mmol) of compound 4i was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 140 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 0–50% EtOAc in hexanes, furnishing 0.057 g (28% yield) of desired product 5i as a colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.82 (s, 1 H), 7.73 (d, J = 7.8 Hz, 1 H), 7.65 (d, J = 7.8 Hz, 1 H), 7.52 (t, J = 7.8 Hz, 1 H), 7.45 – 7.37 (m, 2 H), 7.06 – 6.91 (m, 2 H), 5.15 (td, J = 7.0, 3.6 Hz, 1 H), 2.81 (d, J = 4.0 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 148.72, 135.96, 132.55, 131.08, 131.36 – 130.33 (q, J = 32.49 Hz), 128.82, 125.89 (q, J = 4.0 Hz), 124.63 (q, J = 4.2 Hz), 123.97 (d, J = 272.9 Hz), 123.36, 124.32 – 119.63 (t, J = 273.01 Hz), 119.13, 74.39 – 72.98 (t, J = 31.2 Hz); 19F NMR (376 MHz, CDCl3) δ −82.05 (dd, J = 140.9, 6.7 Hz, 1 F), −82.53 (dd, J = 141.3, 7.0 Hz, 1 F); IR (film) 3414, 1584, 1485, 1327, 1250, 1161, 1122, 1064, 1012, 828, 794, 751, 737, 701, 669, 491 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C15H10BrF5O2Cl 430.9478; found 430.9504, 2.6 ppm.
2-(4-bromophenoxy)-2,2-difluoro-1-(3-nitrophenyl)ethan-1-ol (5j)
Following General Procedure B, 0.093 g (0.50 mmol) of compound 4j was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 140 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 10–35% EtOAc in hexanes, furnishing 0.084 g (45% yield) of desired product 5j as an orange oil; 1H NMR (400 MHz, CDCl3) δ 8.46 (t, J = 1.89 Hz, 1 H), 8.27 (ddd, J = 6.22, 2.30, 1.09 Hz, 1 H), 7.91 (d, J = 7.82 Hz, 1 H), 7.61 (t, J = 7.99 Hz, 1 H), 7.45 (d, J = 8.88 Hz, 2 H), 7.00 (d, J = 9.03 Hz, 2 H), 5.23 (td, J = 6.90, 3.91 Hz, 1 H), 2.91 (d, J = 3.95 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 148.7 (t, J = 2.07 Hz), 148.4, 137.1, 133.9, 132.7, 129.5, 124.1, 123.5, 123.0, 122.0 (t, J = 273.01 Hz), 119.4, 73.5 (t, J = 31.89 Hz); 19F NMR (376 MHz, CDCl3) δ −82.02 (dd, J = 140.93, 6.87 Hz, 1 F), −82.56 (dd, J = 140.85, 6.99 Hz, 1 F). IR (film) 3469, 3094, 2919, 2052, 1890, 1619, 1584, 1529, 1484, 1444, 1400, 1351, 1276, 1251, 1195, 1151, 1115, 1066, 1012, 935, 909, 883, 843, 827, 808, 764, 750, 728, 699, 688, 647, 546, 492 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C14H10BrF2NO4Cl 407.9450; found 407.9453, 0.7 ppm.
4-(2-(4-bromophenoxy)-2,2-difluoro-1-hydroxyethyl)benzonitrile (5k)
Following General Procedure B, 0.083 g (0.50 mmol) of compound 4k was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 140 °C for 24 h. After workup with sat. Na2CO3 (aq.), the product was purified by flash chromatography using a gradient of 0–50% EtOAc in hexanes, furnishing 0.073 g (41% yield) of desired product 5k as a colorless solid (MP = 128–130 °C); 1H NMR (400 MHz, CDCl3) δ 7.69 – 7.57 (m, 4 H), 7.41 – 7.32 (m, 2 H), 6.91 (d, J = 9.0 Hz, 1 H), 5.10 (td, J = 6.9, 3.5 Hz, 2 H), 2.91 (d, J = 4.0 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 148.6, 140.1, 132.6, 132.1, 128.5, 123.4, 121.9 (t, J = 237.2 Hz) 119.3, 118.5, 112.9, 73.6 (t, J = 31.9 Hz); 19F NMR (376 MHz, CDCl3) δ −81.75 (dd, J = 140.7, 6.9 Hz, 1 F), −82.32 (dd, J = 140.7, 7.0 Hz, 1 F); IR (film) 3399, 2908, 2239, 1611, 1580, 1485, 1251, 1152, 1065, 1011, 848, 825, 804, 763, 578, 551, 494 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C15H10BrF2NO2Cl 387.9557; found 387.9583, 2.6 ppm.
2-(4-bromophenoxy)-1-(3,5-dichlorophenyl)-2,2-difluoroethan-1-ol (5l)
Following General Procedure B, 0.104 g (0.500 mmol) of compound 4l was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 140 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 0–50% EtOAc in hexanes, furnishing 0.087 g (44% yield) of desired product 5l as a colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.48 (dd, J = 5.4, 3.5 Hz, 4 H), 7.42 (t, J = 1.9 Hz, 1 H), 7.12 – 6.96 (m, 2 H), 5.07 (td, J = 6.8, 3.7 Hz, 1 H), 2.85 (d, J = 4.0 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 148.6, 138.2, 135.0, 132.6, 129.2, 126.3, 123.4, 121.8 (t, J = 272.7), 119.3, 74.2 (t, J = 32.4 Hz); 19F NMR (376 MHz, CDCl3) δ −81.76 (dd, J = 140.8, 6.8 Hz, 1 F), −82.32 (dd, J = 140.9, 6.9 Hz, 1 F); IR (film) 3400, 3083, 1592, 1572, 1484, 1435, 1206, 1150, 1065, 1011, 795, 739, 674, 491 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C14H9BrCl3F2O2 430.8825; found 430.8837, 1.2 ppm.
2-(4-bromophenoxy)-1-(4′-(tert-butyl)-[1,1′-biphenyl]-2-yl)-2,2-difluoroethan-1-ol (5m)
Following General Procedure B, 0.136 g (0.500 mmol) of compound 4m was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.026 g (0.100 mmol) of Co(acac)2 at 100 °C for 48 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 5–25% EtOAc in hexanes, furnishing 0.139 g (60% yield) of desired product 5m as an orange oil; 1H NMR (400 MHz, CDCl3) δ 7.81 (t, J = 1.66 Hz, 1 H), 7.66 (dt, J = 7.39, 1.71 Hz, 1 H), 7.60 (d, J = 8.45 Hz, 2 H), 7.52 (d, J = 2.18 Hz, 2 H), 7.52–7.49 (m, 2 H), 7.45 (d, J = 8.86 Hz, 2 H), 7.04 (d, J = 8.77 Hz, 2 H), 5.16 (t, J = 7.11 Hz, 1 H), 3.00 (bs, 1 H), 1.40 (s, 9 H); 13C NMR{1H} (126 MHz, CDCl3) δ 150.7, 149.1, 141.3, 137.9, 135.8, 132.5, 128.8, 127.8, 126.9, 126.5, 126.4, 125.9, 123.6, 122.5 (t, J = 272.36 Hz), 119.0, 74.5 (t, J = 31.35 Hz), 34.7, 31.5; 19F NMR (376 MHz, CDCl3) δ −81.63 (dd, J = 140.63, 7.08 Hz, 1 F), −82.03 (dd, J = 140.63, 7.16 Hz, 1 H); IR (film) 3401, 3065, 2962, 2904, 2867, 1580, 1483, 1399, 1363, 1252, 1209, 1140, 1115, 1067, 1012, 954, 906, 881, 839, 825, 766, 739, 705, 675, 645, 632, 585, 545, 522, 492 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C24H23BrClF2O2 495.0538; found 495.0516, 4.4 ppm.
Experimental Procedures and Characterization of Products in Table 3:
2-(4-bromophenoxy)-2,2-difluoro-1-(1-tosyl-1H-indol-3-yl)ethan-1-ol (7a)
Following General Procedure B, 0.167 g (0.500 mmol) of compound 6a was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 100 °C for 36 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 15–40% EtOAc in hexanes, furnishing 0.162 g (62% yield) of desired product 7a as an orange solid (MP = 53–55 °C); 1H NMR (400 MHz, CDCl3) δ 7.99 (dt, J = 8.49, 0.88 Hz), 1 H), 7.77 (d, J = 8.53 Hz, 3 H), 7.73 (d, J = 7.75 Hz, 1 H), 7.45–7.41 (m, 2 H), 7.34 (ddd, J = 8.38, 7.16, 1.28 Hz, 1 H), 7.28–7.24 (m, 1 H), 7.21 (d, J = 7.65 Hz, 2 H), 6.98 (dd, J = 8.76, 1.07 Hz, 2 H), 5.35 (td, J = 6.73, 4.53 Hz, 1 H), 2.68 (d, J = 4.95 Hz, 1 H), 2.34 (s, 3 H); 13C NMR{1H} (126 MHz, CDCl3) δ 149.0 (d, J = 1.96 Hz), 145.4, 135.2, 132.6, 130.1, 129.1, 127.0, 125.8 (d, J = 2.00 Hz), 125.2, 123.6, 123.5, 122.5 (t, J = 272.36 Hz), 120.9 (d, J = 1.82 Hz), 119.2, 117.0 (d, J = 1.66 Hz), 113.8, 69.1 (t, J = 33.67 Hz), 21.7; 19F NMR (376 MHz, CDCl3) δ −81.47 (dd, J = 140.26, 6.78 Hz, 1 F), −82.05 (dd, J = 140.24, 7.22 Hz, 1 F); IR (film) 3509, 3113, 2924, 2052, 1913, 1596, 1566, 1485, 1447, 1340, 1368, 1278, 1255, 1189, 1172, 1122, 1084, 1066, 1012, 972, 907, 834, 811, 764, 744, 733, 703, 678, 657, 599, 571, 537, 492 cm−1; HRMS (ESI−) m/z: [M–H]− Calcd for C23H17BrF2NO4S 520.0030; found 520.0041, 2.1 ppm.
2-(4-bromophenoxy)-2,2-difluoro-1-(1-phenyl-1H-pyrazol-4-yl)ethan-1-ol (7b)
Following General Procedure B, 0.103 g (0.500 mmol) of compound 6b was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 110 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 15–40% EtOAc in hexanes, furnishing 0.140 g (71% yield) of desired product 7b as a yellow solid (MP = 70–72 °C); 1H NMR (400 MHz, CDCl3) δ 8.04 (s, 1 H), 7.82 (s, 1 H), 7.65 (d, J = 7.51 Hz, 2 H), 7.43 (dd, J = 8.90, 7.22 Hz, 4 H), 7.29 (t, J = 7.45 Hz, 1 H), 7.04 (d, J = 8.57 Hz, 2 H), 5.16 (td, J = 6.91, 3.91 Hz, 1 H), 3.59 (d, J = 5.46 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 149.0, 140.1 (d, J = 1.95 Hz), 139.8, 132.6, 129.6, 127.0, 126.6 (d, J = 1.86 Hz), 123.6, 122.5 (t, J = 271.91 Hz), 119.4, 119.1, 118.9 (d, J = 1.87 Hz), 67.7 (t, J = 33.26 Hz); 19F NMR (376 MHz, CDCl3) δ −82.60 (dd, J = 140.85, 6.21 Hz, 1 F), −83.07 (dd, J = 141.08, 6.84 Hz, 1 F); IR (film) 3279, 2923, 1680, 1600, 1572, 1504, 1485, 1405, 1257, 1209, 1148, 1114, 1067, 1043, 1012, 955, 904, 826, 804, 756, 690, 492 cm−1; HRMS (ESI+) m/z: [M+H]+ Calcd for C17H14BrF2N2O2 395.0207; found 395.0220, 3.3 ppm.
tert-butyl 4-(5-(2-(4-bromophenoxy)-2,2-difluoro-1-hydroxyethyl)thiazol-2-yl)piperazine-1-carboxylate (7c)
Following General Procedure B, 0.166 g (0.500 mmol) of compound 6c was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 90 °C for 24 h. After workup with sat. Na2CO3 (aq.), the product was purified by flash chromatography using a gradient of 15–60% EtOAc in hexanes, furnishing 0.095 g (36% yield) of desired product 7c as a brown solid (MP = 60–61 °C); 1H NMR (400 MHz, CDCl3) δ 7.46 (d, J = 8.69 Hz, 2 H), 7.23 (d, J = 0.68 Hz, 1 H), 7.06 (d, J = 8.62 Hz, 2 H), 5.19 (t, J = 6.29 Hz, 1 H), 3.54 (dd, J = 6.43, 3.50 Hz, 4 H), 3.47 (td, J = 5.10, 1.76 Hz, 4 H), 1.47 (s, 9 H); 13C NMR{1H} (126 MHz, CDCl3) δ 173.0, 154.7, 148.9, 140.2, 132.7, 123.6, 122.0 (t, J = 271.72 Hz), 120.5, 119.2, 80.5, 69.8 (t, J = 34.07 Hz), 48.2, 28.5; 19F NMR (376 MHz, CDCl3) δ −81.66 (dd, J = 140.43, 5.78 Hz, 1 F), −82.45 (dd, J = 140.58, 6.84 Hz, 1 F); IR (film) 3333, 2977, 2928, 2862, 2249, 2103, 1690, 1584, 1514, 1484, 1454, 1420, 1366, 1285, 1250, 1234, 1202, 1162, 1134, 1065, 1012, 997, 970, 905, 860, 843, 829, 805, 771, 757, 731, 692, 646, 632, 552, 493, 463 cm−1; HRMS (ESI+) m/z: [M+H]+ Calcd for C20H25BrF2N3O4S 520.0717; found 520.0735, 3.5 ppm.
1-(3-(5-(1,3-dioxolan-2-yl)pyridin-2-yl)phenyl)-2-(4-bromophenoxy)-2,2-difluoroethan-1-ol (7d)
Following General Procedure B, 0.145 g (0.500 mmol) of compound 6d was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 90 °C for 24 h. The reaction was cooled to R.T. and a solution of 4 N HCl in 1,4–dioxane (2.0 mL) and ethylene glycol (1.0 mL) were added. The solution was stirred for 2 h at 130°C temp. The reaction was cooled to R.T. and a solution of 4 N HCl in 1,4–dioxane (2.0 mL) and ethylene glycol (1.0 mL) were added. The solution was stirred for 2 h at 130 °C. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 15–40% EtOAc in hexanes, furnishing 0.095 g (40% yield) of desired product 7d as a brown solid (MP = 85–87 °C); 1H NMR (400 MHz, CDCl3) δ 8.76 (d, J = 2.14 Hz,1 H), 8.09 (t, J = 1.72 Hz, 1 H), 7.92 (dt, J = 7.85, 1.47 Hz, 1 H), 7.86 (dd, J = 8.19, 2.21 Hz, 1 H), 7.72 (dd, J = 8.05, 0.85 Hz, 1 H), 7.59 (d, J = 7.70 Hz, 1 H), 7.46 (t, J = 7.74 Hz, 1 H), 7.39 (d, J = 8.69 Hz, 2 H), 6.98 (d, J = 8.71 Hz, 2 H), 5.89 (s, 1 H), 5.09 (t, J = 7.10 Hz, 1 H), 4.36 (bs, 1 H), 4.15–4.04 (m, 4 H); 13C NMR{1H} (126 MHz, CDCl3) δ 158.0, 149.2, 148.2, 139.0, 136.5, 135.5, 132.5, 132.4, 128.9, 128.6, 127.8, 126.8, 123.6, 122.6 (t, J = 273.01 Hz), 120.8, 118.9, 102.0, 74.1 (t, J = 31.69 Hz), 65.6; 19F NMR (376 MHz, CDCl3) δ −81.75 (t, J = 5.71 Hz, 2 F); IR (film) 3054, 2890, 1726, 1602, 1570, 1485, 1413, 1357, 1264, 1252, 1202, 1145, 1067, 1027, 1012, 983, 942, 908, 841, 796, 735, 703, 650, 579, 494 cm−1; HRMS (ESI+) m/z: [M+H]+ Calcd for C22H19BrF2NO4 478.0466; found 478.0448, 3.8 ppm.
2-(4-bromophenoxy)-1-(dibenzo[b,d]thiophen-4-yl)-2,2-difluoroethan-1-ol (7e)
Following General Procedure B, 0.123 g (0.500 mmol) of compound 6e was reacted with 0.260 g (1.50 mmol) of 4-bromophenol in the presence of 0.026 g (0.100 mmol) of Co(acac)2 at 110 °C for 48 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 5–20% EtOAc in hexanes, furnishing 0.125 g (57% yield) of desired product 7e as a pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 8.16 (ddd, J = 7.05, 3.87, 1.88 Hz, 2 H), 7.86–7.84 (m, 1 H), 7.72 (d, J = 7.45 Hz, 1 H), 7.53–7.46 (m, 3 H), 7.40–7.38 (m, 2 H), 7.00 (d, J = 8.59 Hz, 2 H), 5.45 (td, J = 7.07, 2.82 Hz, 1 H), 3.20 (d, J = 3.92 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 149.0 (d, J = 3.01 Hz), 139.5, 139.2, 136.4, 135.3, 132.5, 129.9, 127.1, 126.2, 124.7, 124.6, 123.4, 122.7, 122.3, 121.7, 118.9, 73.7 (t, J = 32.23 Hz); 19F NMR (376 MHz, CDCl3) δ −81.01 (dd, J = 139.12, 6.75 Hz, 1 F), −81.76 (dd, J = 139.29, 7.33 Hz, 1 F); IR (film) 3412, 3064, 2922, 1888, 1762, 1583, 1550, 1525, 1484, 1444, 1401, 1342, 1276, 1250, 1196, 1147, 1111, 1099, 1066, 1038, 1021, 1012, 938, 904, 827, 793, 750, 706, 688, 646, 627, 577, 556, 492 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C20H13BrF2O2SCl 468.9476; found 468.9471, 1.1 ppm.
Experimental Procedures and Characterization of Compounds in Table 4:
2,2-difluoro-2-(4-nitrophenoxy)-1-(3,4,5-trimethoxyphenyl)ethan-1-ol (9a)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted with 0.209 g (1.50 mmol) of 4-nitrophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 100 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 15–40% EtOAc in hexanes, furnishing 0.107 g (56% yield) of desired product 9a as an yellow solid (MP = 133–135 °C); 1H NMR (400 MHz, CDCl3) δ 8.23 (d, J = 9.19 Hz, 2 H), 7.30 (d, J = 9.27 Hz, 2 H), 6.77 (s, 2 H), 5.07 (td, J = 7.19, 4.02 Hz, 1 H), 3.89 (s, 6 H), 3.87 (s, 3 H), 2.73 (d, J = 4.03 Hz); 13C NMR{1H} (126 MHz, CDCl3) δ 155.0 (d, J = 1.57 Hz), 153.4, 145.2, 138.9 (d, J = 1.59 Hz), 130.3, 125.5, 122.6 (t, J = 275.02 Hz), 121.5 (d, J = 1.64 Hz), 105.0, 74.4 (t, J = 31.34 Hz), 61.0, 56.4; 19F NMR (376 MHz, CDCl3) δ −82.02 (dd, J = 139.86, 7.08 Hz, 1 F), −82.44 (dd, J = 139.86, 7.43 Hz, 1 F); IR (film) 3460, 2925, 1594, 1524, 1492, 1463, 1423, 1348, 1326, 1254, 1129, 1004, 856, 749, 707 cm−1; HRMS (ESI−) m/z: [M+Na]− Calcd for C17H17F2NO7Na 408.0871; found 408.0874, 0.7 ppm.
4-(1,1-difluoro-2-hydroxy-2-(3,4,5-trimethoxyphenyl)ethoxy)benzonitrile (9b)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted with 0.179 g (1.50 mmol) of 4-hydroxybenzonitrile in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 100 °C for 24 h. After workup with sat. Na2CO3 (aq.), the product was purified by flash chromatography using a gradient of 10–40% EtOAc in hexanes, furnishing 0.138 g (82% yield) of desired product 9b as a pale yellow solid (MP = 39–42 °C); 1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 8.77 Hz, 2 H), 7.26 (d, J = 8.80 Hz, 2 H), 6.77 (s, 2 H), 5.06 (td, J = 7.22, 4.04 Hz, 1 H), 3.89 (s, 6 H), 3.87 (s, 3 H), 2.67 (d, J = 4.05 Hz); 13C NMR{1H} (126 MHz, CDCl3) δ 153.5, 153.3, 138.8, 133.9, 130.4, 122.6 (t, J = 274.84 Hz), 122.0, 118.2, 109.6, 105.0, 74.5 (t, J = 31.07 Hz), 61.0, 56.4; 19F NMR (376 MHz, CDCl3) δ −82.13 (d, J = 3.66 Hz, 1 F), −82.15 (d, J = 3.53 Hz, 1 F); IR (film) 3456, 2938, 2841, 2231, 1594, 1503, 1462, 1422, 1326, 1298, 1252, 1236, 1126, 1074, 1004, 922, 843, 809, 790, 768, 733, 702, 661, 640, 548, 465 cm−1; HRMS (ESI+) m/z: [M+K]+ Calcd for C18H17BrF2NO5K 404.0712; found 404.0717, 1.2 ppm.
2-(2,4-dichlorophenoxy)-2,2-difluoro-1-(3,4,5-trimethoxyphenyl)ethan-1-ol (9c)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted with 0.245 g (1.50 mmol) of 2,4-dichlorophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 110 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 10–40% EtOAc in hexanes, furnishing 0.095 g (47% yield) of desired product 9c as a yellow oil; 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 2.43 Hz, 1 H), 7.28–7.25 (m ,1 H), 7.21 (dd, J = 8.81, 2.42 Hz, 1 H), 6.79 (s, 2 H), 5.09 (t, J = 7.27 Hz, 1 H), 3.88 (s, 6 H), 3.86 (s, 3 H), 2.89 (bs, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 153.1, 144.7 (d, J = 1.88 Hz), 138.6, 131.6, 130.3, 130.2, 128.1, 127.8, 123.8 (t, J = 1.84 Hz), 122.6 (t, J = 275.45 Hz), 105.0, 74.4 (t, J = 30.98 Hz), 60.9, 56.2; 19F NMR (376 MHz, CDCl3) δ −81.59 (dd, J = 138.37, 6.85 Hz, 1 F), −82.67 (dd, J = 138.48, 7.65 Hz, 1 F); IR (film) 3444, 3081, 2940, 2839, 2251, 1594, 1508, 1475, 1463, 1422, 1384, 1325, 1261, 1235, 1185, 1125, 1096, 1075, 1002, 910, 868, 841, 812, 791, 770, 734, 687, 663, 632, 568, 530 cm−1; HRMS (ESI+) m/z: [M+K]+ Calcd for C17H16Cl2F2O5K 446.9980; found 446.9998, 4.0 ppm.
2-(3-chloro-2-fluorophenoxy)-2,2-difluoro-1-(3,4,5-trimethoxyphenyl)ethan-1-ol (9d)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted with 0.156 mL (0.220 g, 1.50 mmol) of 3-chloro-4-fluorophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 100 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 10–40% EtOAc in hexanes, furnishing 0.132 g (67% yield) of desired product 9d as a pale solid (MP = 115–117 °C); 1H NMR (400 MHz, CDCl3) δ 7.23 (ddd, J = 8.08, 6.36, 1.61 Hz, 1 H), 7.18 (ddd, J = 8.52, 6.62, 1.30 Hz, 1 H), 7.01 (td, J = 8.26, 1.86 Hz, 1 H), 6.77 (s, 2 H), 5.07 (t, J = 7.23 Hz, 1 H), 3.85 (s, 6 H), 3.84 (s, 3 H), 3.42 (bs, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 153.1, 138.3, 130.8, 127.7, 124.0 (d, J = 5.69 Hz), 122.6 (t, J = 276.48 Hz), 122.5, 122.4 (d, J = 15.56 Hz), 105.0, 74.2 (t, J = 31.07 Hz), 60.9, 56.2; 19F NMR (376 MHz, CDCl3) δ −81.90 (dt, J = 138.37, 6.47 Hz, 1 F), −82.46 (dt, J = 138.62, 6.16 Hz, 1 F), −130.23 (q, J = 5.94 Hz, 1 F); IR (film) 3461, 2942, 2841, 2105, 1596, 1510, 1481, 1460, 1421, 1326, 1273, 1230, 1186, 1124, 1098, 1074, 1002, 941, 912, 850, 819, 793, 762, 748, 737, 719, 698, 663, 623, 591, 573, 528, 467 cm−1; HRMS (ESI+) m/z: [M+H]+ Calcd for C17H17ClF3O5 393.0717; found 393.0734, 1.7 mmu.
2,2-difluoro-2-(3-iodophenoxy)-1-(3,4,5-trimethoxyphenyl)ethan-1-ol (9e)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted in the dark with 0.330 g (1.50 mmol) of 3-iodophenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 110 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 10–35% EtOAc in hexanes, furnishing 0.156 g (67% yield) of desired product 9e as a pale solid (MP = 123–126 °C); 1H NMR (400 MHz, CDCl3) δ 7.56 (dt, J = 7.82, 1.28 Hz, 1 H), 7.51 (t, J = 1.90 Hz, 1 H), 7.13 (ddd, J = 8.34, 2.23, 1.03 Hz, 1 H), 7.06 (t, J = 8.00 Hz, 1 H), 6.77 (s, 2 H), 5.02 (td, J = 7.14, 3.90 Hz, 1 H), 3.89 (s, 6 H), 3.87 (s, 3 H), 2.67 (d, J = 3.26 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 153.3, 150.3 (t, J = 2.14 Hz), 135.1, 131.0, 130.9, 130.6, 122.5 (t J = 272.99 Hz), 121.2, 105.0, 93.7, 74.4 (t, J = 31.16 Hz), 61.0, 56.4; 19F NMR (376 MHz, CDCl3) δ −81.89 (ddd, J = 141.02, 7.26, 7.02 Hz, 2 F); IR (film) 3448, 2936, 1580, 1508, 1500, 1466, 1422, 1336, 1326, 1238, 1129, 997, 845, 758, 706 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C17H17F2IO5Cl 500.9777; found 500.9782, 1.0 ppm.
2-(4-(2-bromoethyl)phenoxy)-2,2-difluoro-1-(3,4,5-trimethoxyphenyl)ethan-1-ol (9f)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted with 0.302 g (1.50 mmol) of 4(2-bromoethyl)phenol in the presence of 0.026 g (0.10 mmol) of Co(acac)2 at 100 °C for 48 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 10–45% EtOAc in hexanes, furnishing 0.162 g (69% yield) of desired product 9f as a red oil; 1H NMR (400 MHz, CDCl3) δ 7.16 (d, J = 8.56 Hz, 2 H), 7.08 (d, J = 8.21 Hz, 2 H), 6.77 (s, 2 H), 5.02 (td, J = 7.24, 3.11 Hz, 1 H), 3.86 (s, 6 H), 3.85 (s, 3 H), 3.54 (t, J = 7.49 Hz, 2 H), 3.12 (t, J = 7.47 Hz, 2 H); 13C NMR{1H} (126 MHz, CDCl3) δ 153.2, 148.9 (t, J = 2.37 Hz), 138.5 (d, J = 1.63 Hz), 136.6, 130.9, 129.8, 122.5 (t, J = 271.38 Hz), 121.9, 105.0, 74.4 (t, J = 31.77 Hz), 61.0, 56.3, 38.7, 32.9; 19F NMR (376 MHz, CDCl3) δ −81.56 (dd, J = 141.41, 7.45 Hz, 1 F), −81.99 (dd, J = 141.40, 7.81 Hz, 1 F); IR (film) 3446, 2939, 2839, 2250, 1758, 1593, 1507, 1462, 1421, 1325, 1235, 1200, 1125, 1064, 1019, 1002, 910, 831, 809, 764, 751, 731, 697, 646, 551, 531 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C19H21BrF2O5Cl 481.0229; found 481.0247, 3.7 ppm.
N-(4-(1,1-difluoro-2-hydroxy-2-(3,4,5-trimethoxyphenyl)ethoxy)phenyl)-4-methylbenzenesulfonamide (9g)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted with 0.395 g (1.50 mmol) of N-(4-hydroxyphenyl)-4-methylbenzenesulfonamide in the presence of 0.026 g (0.10 mmol) of Co(acac)2 at 120 °C for 24 h. The product was purified without workup by flash chromatography using a gradient of 20–60% EtOAc in hexanes, furnishing 0.134 g (53% yield) of desired product 9g as an orange solid (MP = 72–75 °C); 1H NMR (400 MHz, CDCl3) δ 7.61 (d, J = 8.27 Hz, 2 H), 7.19 (d, J = 7.78 Hz, 2 H), 7.16 (bs, 1 H), 7.02–6.96 (m, 4 H), 6.74 (s, 2 H), 4.98 (t, J = 6.94 Hz, 1 H), 3.84 (s, 9 H), 3.10 (bs, 1 H), 2.35 (s, 3 H); 13C NMR {1H} (126 MHz, CDCl3) δ 153.1, 147.4 (d, J = 1.85 Hz), 144.2, 138.4, 135.9, 134.2, 131.0, 129.8, 127.3, 123.0, 122.6, 122.4 (t, J = 271.93 Hz), 105.0, 74.4 (t, J = 31.91 Hz), 61.0, 56.3, 21.6; 19F NMR (376 MHz, CDCl3) δ −84.19 (dd, J = 141.15, 5.92 Hz, 1 F), −84.73 (dd, J = 141.11, 7.18 Hz, 1 F); IR (film) 3468, 3247, 2941, 2840, 2253, 1595, 1505, 1462, 1423, 1398, 1326, 1299, 1275, 1253, 1234, 1201, 1186, 1153, 1126, 1090, 1068, 1018, 1001, 909, 845, 814, 798, 765, 729, 706, 694, 663, 582, 565, 547, 511 cm−1; HRMS (ESI+) m/z: [M+Na]+ Calcd for C24H25F2NO7SNa 532.1218; found 532.1227, 1.7 ppm.
2-([1,1′-biphenyl]-4-yloxy)-2,2-difluoro-1-(3,4,5-trimethoxyphenyl)ethan-1-ol (9h)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted with 0.255 g (1.50 mmol) of 4-phenylphenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 110 °C for 24 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 5–40% EtOAc in hexanes, furnishing 0.157 g (75% yield) of desired product 9h as a pale yellow solid (MP = 54–56 °C); 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 8.54 Hz, 4 H), 7.43 (t, J = 7.50 Hz, 2 H), 7.37–7.33 (m, 1 H), 7.22 (dd, J = 8.65, 0.91 Hz, 2 H), 6.81 (s, 2 H), 5.06 (td, J = 7.09, 3.94 Hz, 1 H), 3.90 (s, 6 H), 3.88 (s, 3 H), 2.90 (d, J = 4.01 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 153.2, 149.4, 140.3, 139.1, 138.5, 131.0, 129.0, 128.2, 127.5, 127.2, 122.6 (t, J = 272.36 Hz), 122.0, 105.1, 74.6 (t, J = 31.88 Hz), 61.0, 56.3; 19F NMR (376 MHz, CDCl3) δ −81.48 (dd, J = 141.06, 6.88 Hz, 1 F), −81.99 (dd, J = 141.05, 7.20 Hz, 1 F); IR (film) 3443, 2939, 2838, 2251, 1903, 1594, 1509, 1486, 1462, 1421, 1325, 1289, 1235, 1184, 1125, 1064, 1008, 909, 842, 807, 758, 730, 698, 651, 551, 531, 500 cm−1; HRMS (ESI+) m/z: Calcd for C23H22F2O5Na [M+Na]+ 439.1333; found 439.1344, 2.5 ppm.
2,2-difluoro-2-phenoxy-1-(3,4,5-trimethoxyphenyl)ethan-1-ol (9i)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted with 0.141 g (1.50 mmol) of phenol in the presence of 0.013 g (0.050 mmol) of Co(acac)2 at 100 °C for 36 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 15–40% EtOAc in hexanes, furnishing 0.106 g (62% yield) of desired product 9i as an off-white solid (MP = 100–101 °C); 1H NMR (400 MHz, CDCl3) δ 7.32 (dd, J = 8.53, 7.23 Hz, 2 H), 7.20 (t, J = 7.42 Hz, 1 H), 7.14 (d, J = 7.18 Hz, 2 H), 6.78 (s, 2 H), 5.03 (ddd, J = 9.10, 6.73, 2.85 Hz, 1 H), 3.87 (s, 6 H), 3.86 (s, 3 H), 3.07 (d, J = 3.91 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 153.1, 150.0, 138.4, 131.1, 129.5, 125.8, 122.5 (t, J = 271.92 Hz), 121.7, 105.0, 74.5 (t, J = 31.90 Hz), 61.0, 58.2; 19F NMR (376 MHz, CDCl3) δ −81.49 (dd, J = 141.10, 6.77 Hz, 1 F), −81.97 (dd, J = 141.10, 7.31 Hz, 1 F); IR (film) 3442, 2940, 2839, 1771, 1592, 1508, 1491, 1462, 1422, 1325, 1291, 1235, 1194, 1125, 1078, 1062, 1026, 1003, 921, 898, 839, 787, 754, 732, 702, 690, 660, 558, 530, 485 cm−1; HRMS (ESI+) m/z: [M+H]+ Calcd for C17H19F2O5 341.1201; found 341.1195, 1.8 ppm.
2,2-difluoro-2-(o-tolyloxy)-1-(3,4,5-trimethoxyphenyl)ethan-1-ol (9j)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted with 0.16 mL (1.50 mmol) of o-cresol in the presence of 0.026 g (0.100 mmol) of Co(acac)2 at 110 °C for 48 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 5–25% EtOAc in hexanes, furnishing 0.089 g (50% yield) of desired product 9j as an orange oil; 1H NMR (400 MHz, CDCl3) δ 7.21–7.07 (m, 4 H), 6.80 (s, 2 H), 5.07 (dd, J = 7.79, 5.83 Hz, 1 H), 3.88 (s, 6 H), 3.86 (s, 3 H), 2.93 (bs, 1 H), 2.05 (s, 3 H); 13C NMR{1H} (126 MHz, CDCl3) δ 153.2, 148.4 (d, J = 2.00 Hz), 138.6, 131.30, 131.25, 131.18, 126.8, 125.9, 122.7 (t, J = 271.11 Hz), 122.0 (d, J = 1.66 Hz), 105.1, 74.7 (t, J = 31.83 Hz), 61.0, 56.3, 16.3; 19F NMR (376 MHz, CDCl3) δ −80.33 (dd, J = 141.28, 5.87 Hz, 1 F), −82.40 (dd, J = 141.25, 7.82 Hz, 1 F); IR (film) 3445, 2939, 2839, 1594, 1507, 1492, 1461, 1421, 1325, 1251, 1234, 1178, 1125, 1062, 1003, 922, 844, 819, 787, 745, 712, 694, 660, 559, 527 cm−1; HRMS (ESI+) m/z: [M+Na]+ Calcd for C18H20F2O5Na 377.1177; found 377.1179, 0.5 ppm.
2,2-difluoro-2-(2-isopropylphenoxy)-1-(3,4,5-trimethoxyphenyl)ethan-1-ol (9k)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted with 0.21 mL (0.204 g, 1.50 mmol) of 2-isopropylphenol in the presence of 0.026 g (0.10 mmol) of Co(acac)2 at 110 °C for 48 h. After workup 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 5–35% EtOAc in hexanes, furnishing 0.080 g (42% yield) of desired product 9k as a black solid; 1H NMR (400 MHz, CDCl3) δ 7.24 (dt, J = 7.74, 2.51 Hz, 2 H), 7.15 (ddd, J = 8.03, 5.34, 2.08 Hz, 2 H), 6.81 (s, 2 H), 5.09 (dt, J = 8.66, 4.45 Hz, 1 H), 3.87 (s, 6 H), 3.86 (s, 3 H), 2.82 (p, J = 6.92 Hz, 1 H), 2.74 (d, J = 3.95 Hz, 1 H), 1.03 (dd, J = 6.92, 1.01 Hz, 6 H); 13C NMR{1H} (126 MHz, CDCl3) δ 153.3, 147.1 (d, J = 2.01 Hz), 141.5, 138.6, 131.2 (d, J = 1.87 Hz), 126.7, 126.6, 126.2, 122.7 (dd, J = 271.38, 2.53 Hz), 121.8, 105.1, 74.9 (dd, J = 32.84, 30.16 Hz), 61.0, 56.3, 26.4, 23.1 (d, J = 16.51 Hz); 19F NMR (376 MHz, CDCl3) δ −79.34 (dd, J = 140.88, 4.75 Hz, 1 F), −83.16 (dd, J = 140.73, 8.56 Hz, 1 F); IR (film) 3452, 2964, 2840, 1595, 1508, 1488, 1461, 1422, 1385, 1363, 1325, 1275, 1250, 1234, 1179, 1126, 1084, 1060, 1033, 1004, 910, 836, 812, 785, 754, 732, 698, 661, 573, 530, 473 cm−1; HRMS (ESI−) m/z: [M+Cl]− Calcd for C20H24F2O5Cl 417.1280; found 417.1280, 0.0 ppm.
2-([1,1′-biphenyl]-2-yloxy)-2,2-difluoro-1-(3,4,5-trimethoxyphenyl)ethan-1-ol (9l)
Following General Procedure B, 0.115 g (0.500 mmol) of compound 1 was reacted with 0.255 g (1.50 mmol) of 2-phenylphenol in the presence of 0.026 g (0.10 mmol) of Co(acac)2 at 110 °C for 48 h. After workup with 1 N NaOH (aq.), the product was purified by flash chromatography using a gradient of 5–40% EtOAc in hexanes, furnishing 0.141 g (68% yield) of desired product 9l as a pale yellow solid (MP = 42 °C); 1H NMR (400 MHz, CDCl3) δ 7.42–7.31 (m, 8 H), 7.29 (dd, J = 7.31, 1.53 Hz, 1 H), 6.56 (s, 2 H), 4.82 (td, J = 7.11, 4.18 Hz, 1 H), 3.85 (s, 3 H), 3.78 (s, 6 H), 2.32 (d, J = 4.17 Hz, 1 H); 13C NMR{1H} (126 MHz, CDCl3) δ 153.0, 147.0, 138.3, 137.8, 135.2, 131.3, 130.8, 129.3, 129.1, 128.5, 128.3, 128.1, 127.4, 125.9, 125.4, 122.6 (t, J = 273.57 Hz), 121.9, 104.9, 74.5 (t, J = 31.33 Hz), 60.9, 56.1; 19F NMR (376 MHz, CDCl3) δ −80.71 (dd, J = 139.63, 7.17 Hz, 1 F), −81.67 (dd, J = 139.53, 7.09 Hz, 1 F); IR (film) 3454, 3059, 2940, 2838, 1595, 1506, 1479, 1463, 1422, 1325, 1264, 1236, 1189, 1127, 1070, 1009, 910, 838, 774, 736, 700, 661, 613, 566, 530, 474 cm−1; HRMS (ESI+) m/z: [M+K]+ Calcd for C23H22F2O5K 455.1072; found 455.1076, 0.9 ppm.
Supplementary Material
Acknowledgements
We thank the National Institutes of Health (R35 GM124661) and the Madison and Lila Self Graduate Fellowship (D.L.O) for supporting this work. NMR Instrumentation was provided by NIH Shared Instrumentation Grants S10OD016360 and S10RR024664, NSF Major Research Instrumentation Grants 9977422 and 0320648, and NIH Center Grant P20GM103418. Support for the EPR instrument was provided by NSF Chemical Instrumentation Grant # 0946883.
Footnotes
Supporting Information availability Statement
The Supporting Information is available free of charge on the ACS Publications website.
Reaction optimization, Copies of NMR spectra for synthesized compounds and procedures for mechanistic studies
The authors declare no competing financial interest.
REFERENCES
- (1).a) Bégué J-P; Bonnet-Delpon D Bioorganic and Medicinal Chemistry of Fluorine; Wiley-VCH:Weinheim, 2008; [Google Scholar]; b) Gillis EP.; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA. Applications of Fluorine in Medicinal Chemistry J. Med. Chem. 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- (2).Spahn V; Del Vecchio G; Labuz D; Rodriguez-Gaztelumendi A; Massaly N; Temp J; Durmaz V; Sabri P; Reidelbach M; Machelska H; Weber M; Stein C A nontoxic pain killer designed by modeling of pathological receptor conformations Science 2017, 355, 966–969. [DOI] [PubMed] [Google Scholar]
- (3).a) Huchet QA; Kuhn B; Wagner B; Kratochwil NA.; Fischer H; Kansy M; Zimmerli D; Carreira EM; Muller K. Fluorination Patterning: A Study of Structural Motifs That Impact Physicochemical Properties of Relevance to Drug Discovery J. Med. Chem. 2015, 58, 9041–9060; [DOI] [PubMed] [Google Scholar]; b) Huchet QA.; Trapp N; Kuhn B; Wagner B.; Fischer H.; Kratochwil NA.; Carreira EM.; Muller K. Partially fluorinated alkoxy groups - Conformational adaptors to changing environments J. Fluorine Chem. 2017, 198, 34–46. [Google Scholar]
- (4).a) Hunter L. The C-F bond as a conformational tool in organic and biological chemistry Beilstein J Org Chem 2010, 6, 38; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Thiehoff C.; Rey YP.; Gilmour R. The Fluorine Gauche Effect: A Brief History Isr. J. Chem. 2017, 57, 92–100. [Google Scholar]
- (5).a) Xing L; Keefer C; Brown ME Fluorine multipolar interaction: Toward elucidating its energetics in binding recognition J. Fluorine Chem. 2017, 198, 47–53; [Google Scholar]; b) Muller K; Faeh C.; Diederich F. Fluorine in pharmaceuticals: looking beyond intuition Science 2007, 317, 1881–1886; [DOI] [PubMed] [Google Scholar]; c) Olsen JA.; Banner DW; Seiler P; Obst Sander U.; D’Arcy A.; Stihle M; Muller K.; Diederich F. A fluorine scan of thrombin inhibitors to map the fluorophilicity/fluorophobicity of an enzyme active site: evidence for C-F…C=O interactions Angew. Chem. Int. Ed. Engl. 2003, 42, 2507–2511; [DOI] [PubMed] [Google Scholar]; d) Paulini R; Muller K; Diederich F. Orthogonal multipolar interactions in structural chemistry and biology Angew. Chem. Int. Ed. Engl. 2005, 44, 1788–1805. [DOI] [PubMed] [Google Scholar]
- (6).a) Martone RL; Zhou H; Atchison K; Comery T; Xu JZ; Huang X; Gong X; Jin M; Kreft A; Harrison B; Masyer SC; Aschmies S; Gonzales C; Zaleska MM; Riddell DR; Wagner E; Lu P.; Sun SC; Sonnenberg-Reines J; Oganesian A; Adkins K; Leach MW; Clarke DW.; Huryn D; Abou-Gharbia M; Magolda R; Bard J; Frick G; Raje S; Forlow SB.; Balliet C; Burczynski ME; Reinhart PH.; Wan HI; Pangalos MN.; Jacobsen JS. Begacestat (GSI-953): a novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein gamma-secretase for the treatment of Alzheimer’s disease J. Pharmacol. Exp. Ther 2009, 331, 598–608; [DOI] [PubMed] [Google Scholar]; b) Mayer SC; Kreft AF.; Harrison B.; Abou-Gharbia M.; Antane M.; Aschmies S; Atchison K.; Chlenov M; Cole DC.; Comery T.; Diamantidis G.; Ellingboe J; Fasn K.; Galante R.; Gonzales C.; Ho DM.; Hoke ME.; Hu Y.; Huryn D.; Jain U.; Jin M; Kremer K.; Kubrak D.; Lin M; Lu P.; Magolda R.; Martone R; Moore W.; Oganesian A.; Pangalos MN; Porte A; Reinhart P.; Resnick L.; Riddell DR.; Sonnenberg-Reines J.; Stock JR.; Sun SC.; Wagner E; Wang T; Woller K.; Xu Z; Zaleska MM.; Zeldis J; Zhang M.; Zhou H; Jacobsen JS. Discovery of begacestat, a Notch-1-sparing gamma-secretase inhibitor for the treatment of Alzheimer’s disease J. Med. Chem. 2008, 51, 7348–7351. [DOI] [PubMed] [Google Scholar]
- (7).a) Zhang X; Cao S Recent advances in the synthesis and CF functionalization of gem-difluoroalkenes Tetrahedron Lett. 2017, 58, 375–392; [Google Scholar]; b) Orsi DL; Altman RA Exploiting the unusual effects of fluorine in methodology Chem. Commun. 2017, 53, 7168–7181. [DOI] [PubMed] [Google Scholar]
- (8).a) Qiao Y; Si T; Yang MH; Altman RA Metal-free trifluoromethylation of aromatic and heteroaromatic aldehydes and ketones J. Org. Chem 2014, 79, 7122–7131; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Orsi DL; Easley BJ; Lick AM; Altman RA Base Catalysis Enables Access to alpha,alpha-Difluoroalkylthioethers Org. Lett 2017, 19, 1570–1573; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Orsi DL; Yadav MR; Altman RA Organocatalytic strategy for hydrophenolation of gem-difluoroalkenes Tetrahedron 2019, 75, 4325–4336; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Frost AB; Brambilla M; Exner RM; Tredwell M Synthesis and Derivatization of 1,1-[(18) F]Difluorinated Alkenes Angew. Chem. Int. Ed. Engl. 2019, 58, 472–476. [DOI] [PubMed] [Google Scholar]
- (9).a) Tian P; Feng C; Loh TP Rhodium-catalysed C(sp(2))-C(sp(2)) bond formation via C-H/C-F activation Nat Commun 2015, 6, 7472–7478; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xiong Y; Huang T; Ji X; Wu J; Cao S Nickel-catalyzed Suzuki-Miyaura type cross-coupling reactions of (2,2-difluorovinyl)benzene derivatives with arylboronic acids Org Biomol Chem 2015, 13, 7389–7392; [DOI] [PubMed] [Google Scholar]; c) Dai W; Shi H; Zhao X; Cao S Sterically Controlled Cu-Catalyzed or Transition-Metal-Free Cross-Coupling of gem-Difluoroalkenes with Tertiary, Secondary, and Primary Alkyl Grignard Reagents Org. Lett. 2016, 18, 4284–4287; [DOI] [PubMed] [Google Scholar]; d) Kong L; Zhou X; Li X Cobalt(III)-Catalyzed Regio- and Stereoselective alpha-Fluoroalkenylation of Arenes with gem-Difluorostyrenes Org. Lett. 2016, 18, 6320–6323; [DOI] [PubMed] [Google Scholar]; e) Thornbury RT; Toste FD Palladium-Catalyzed Defluorinative Coupling of 1-Aryl-2,2-Difluoroalkenes and Boronic Acids: Stereoselective Synthesis of Monofluorostilbenes Angew. Chem. Int. Ed. Engl. 2016, 55, 11629–11632; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Cai SH; Ye L; Wang DX; Wang YQ; Lai LJ; Zhu C; Feng C; Loh TP Manganese-catalyzed synthesis of monofluoroalkenes via C-H activation and C-F cleavage Chem. Commun. 2017, 53, 8731–8734. [DOI] [PubMed] [Google Scholar]
- (10).McCune CD; Beio ML; Sturdivant JM; de la Salud-Bea R; Darnell BM; Berkowitz DB Synthesis and Deployment of an Elusive Fluorovinyl Cation Equivalent: Access to Quaternary alpha-(1′-Fluoro)vinyl Amino Acids as Potential PLP Enzyme Inactivators J. Am. Chem. Soc. 2017, 139, 14077–14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Morikawa T; Kumadaki I; Shiro M Electrophilic Cyclization Reaction of Gem-Difluoroolefin Derivatives - Syntheses of 6,6-Difluorotetrahydro-2-Pyrones and 2,2-Difluorotetrahydropyran Via Halogen Induced Cyclization Chem. Pharm. Bull. 1985, 33, 5144–5146. [Google Scholar]
- (12).Paleta O; Svoboda J; Dědek V Haloacrylic acids XX. Dimerisation of trifluoroacrylates. Kinetically and thermodynamically controlled reactions J. Fluorine Chem. 1983, 23, 171–191. [Google Scholar]
- (13).a) Gao B; Zhao Y.; Hu J AgF-mediated fluorinative cross-coupling of two olefins: facile access to alpha-CF3 alkenes and beta-CF3 ketones Angew. Chem. Int. Ed. Engl. 2015, 54, 638–642; [DOI] [PubMed] [Google Scholar]; b) Gao B; Zhao Y; Ni C; Hu J AgF-mediated fluorinative homocoupling of gem-difluoroalkenes Org. Lett. 2014, 16, 102–105; [DOI] [PubMed] [Google Scholar]; c) Zhang B; Zhang X; Hao J; Yang C Palladium-Catalyzed Direct Approach to α-Trifluoromethyl Alcohols by Selective Hydroxylfluorination of gem-Difluoroalkenes Eur. J. Org. Chem 2018, 2018, 5007–5015; [Google Scholar]; d) Tang HJ; Lin LZ; Feng C; Loh TP Palladium-Catalyzed Fluoroarylation of gem-Difluoroalkenes Angew. Chem. Int. Ed. Engl. 2017, 56, 9872–9876; [DOI] [PubMed] [Google Scholar]; e) Tian P; Wang CQ; Cai SH; Song S; Ye L; Feng C; Loh TP F(-) Nucleophilic-Addition-Induced Allylic Alkylation J. Am. Chem. Soc. 2016, 138, 15869–15872; [DOI] [PubMed] [Google Scholar]; f) Liu H; Ge L; Wang DX; Chen N; Feng C Photoredox-Coupled F-Nucleophilic Addition: Allylation of gem-Difluoroalkenes Angew Chem Int Ed Engl 2019, 58, 3918–3922; [DOI] [PubMed] [Google Scholar]; g) Hu J; Yang Y; Lou Z; Ni C; Hu J Fluoro-Hydroxylation of gem-Difluoroalkenes: Synthesis of 18 O-labeled α-CF3 Alcohols Chin. J. Chem . 2018, 36, 1202–1208. [Google Scholar]
- (14).Liu J; Yang J; Ferretti F; Jackstell R; Beller M Pd-Catalyzed Selective Carbonylation of gem-Difluoroalkenes: A Practical Synthesis of Difluoromethylated Esters Angew. Chem. Int. Ed. Engl. 2019, 58, 4690–4694. [DOI] [PubMed] [Google Scholar]
- (15).a) Wang YH; Goldsmith ZK; Schneider PE; Anson CW; Gerken JB; Ghosh S; Hammes-Schiffer S; Stahl SS Kinetic and Mechanistic Characterization of Low-Overpotential, H2O2-Selective Reduction of O2 Catalyzed by N2O2-Ligated Cobalt Complexes J. Am. Chem. Soc. 2018, 140, 10890–10899; [DOI] [PubMed] [Google Scholar]; b) Ma X; Herzon SB Synthesis of Ketones and Esters from Heteroatom-Functionalized Alkenes by Cobalt-Mediated Hydrogen Atom Transfer J. Org. Chem. 2016, 81, 8673–8695. [DOI] [PubMed] [Google Scholar]
- (16).a) Zhang H; Guo LH; Wang D; Zhao L; Wan B Light-induced efficient molecular oxygen activation on a Cu(II)-grafted TiO2/graphene photocatalyst for phenol degradation ACS Appl Mater Interfaces 2015, 7, 1816–1823; [DOI] [PubMed] [Google Scholar]; b) Zabik NL; Anwar S; Ziu I; Martic-Milne S Electrochemical reactivity of bulky-phenols with superoxide anion radical Electrochim. Acta 2019, 296, 174–180; [Google Scholar]; c) Rene A; Abasq ML; Hauchard D; Hapiot P How do phenolic compounds react toward superoxide ion? A simple electrochemical method for evaluating antioxidant capacity Anal. Chem. 2010, 82, 8703–8710; [DOI] [PubMed] [Google Scholar]; d) Peng Z; Chen Y; Bruce PG; Xu Y Direct Detection of the Superoxide Anion as a Stable Intermediate in the Electroreduction of Oxygen in a Non-Aqueous Electrolyte Containing Phenol as a Proton Source Angew. Chem. Int. Ed. Engl. 2015, 54, 8165–8168; [DOI] [PubMed] [Google Scholar]; e) Ma D; Zhong J; Li J; Burda C; Duan R Preparation and photocatalytic performance of MWCNTs/BiOCl: Evidence for the superoxide radical participation in the degradation mechanism of phenol Appl. Surf. Sci. 2019, 480, 395–403; [Google Scholar]; f) Chen Y; Zhang X; Feng S Contribution of the Excited Triplet State of Humic Acid and Superoxide Radical Anion to Generation and Elimination of Phenoxyl Radical Environ. Sci. Technol. 2018, 52, 8283–8291; [DOI] [PubMed] [Google Scholar]; g) Aboul-Enein HY; Kruk I; Kladna A; Lichszteld K; Michalska T Scavenging effects of phenolic compounds on reactive oxygen species Biopolymers 2007, 86, 222–230. [DOI] [PubMed] [Google Scholar]
- (17).Suda M Radical-Addition Reactions on 1,1-Difluoro-1-Olefins Tetrahedron Lett. 1981, 22, 2395–2396. [Google Scholar]
- (18).Watanabe E; Kaiho A; Kusama H; Iwasawa N Cobalt-salen complex-catalyzed oxidative generation of alkyl radicals from aldehydes for the preparation of hydroperoxides J. Am. Chem. Soc. 2013, 135, 11744–11747. [DOI] [PubMed] [Google Scholar]
- (19).a) Dobras G; Orlińska B Aerobic oxidation of alkylaromatic hydrocarbons to hydroperoxides catalysed by N-hydroxyimides in ionic liquids as solvents Applied Catalysis A: General 2018, 561, 59–67; [Google Scholar]; b) Asano Y; Nagasawa Y; Yamaguchi E; Itoh A Aerobic Photooxidative Synthesis of beta-Alkoxy Monohydroperoxides Using an Organo Photoredox Catalyst Controlled by a Base Chem Asian J 2018, 13, 409–412; [DOI] [PubMed] [Google Scholar]; c) Santamaria J; Jroundi R; Rigaudy J Electron transfer activation. Hydroperoxide intermediates in a novel and selective procedure for benzylic oxidations Tetrahedron Lett. 1989, 30, 4677–4680. [Google Scholar]
- (20).a) Spier E; Neuenschwander U; Hermans I Insights into the cobalt(II)-catalyzed decomposition of peroxide Angew. Chem. Int. Ed. Engl. 2013, 52, 1581–1585; [DOI] [PubMed] [Google Scholar]; b) Wu J-M; Kunikawa S; Tokuyasu T; Masuyama A; Nojima M; Kim H-S; Wataya Y Co-catalyzed autoxidation of alkene in the presence of silane. The effect of the structure of silanes on the efficiency of the reaction and on the product distribution Tetrahedron 2005, 61, 9961–9968; [Google Scholar]; c) Nishinaga A; Yamato H; Abe T; Maruyama K; Matsuura T Asymmetric induction in oxygenation of styrene catalyzed by cobalt schiff base complex Tetrahedron Lett. 1988, 29, 6309–6312. [Google Scholar]
- (21).a) Lončarević D; Krstić J; Dostanić J; Manojlović D; Čupić Ž; Jovanović DM Cyclohexane oxidation and cyclohexyl hydroperoxide decomposition by poly(4-vinylpyridine-co-divinylbenzene) supported cobalt and chromium complexes Chem. Eng. J. 2010, 157, 181–188; [Google Scholar]; b) Talsi EP; Chinakov VD; Babenko VP; Sidelnikov VN; Zamaraev KI Role of alkylperoxo complexes of cobalt (III) in catalytic oxidation of cyclohexane by cumene hydroperoxide in the presence of bis (acetylacetonato) cobalt (II) J. Mol. Catal. 1993, 81, 215–233. [Google Scholar]
- (22).a) Olson AS; Jameson AJ; Kyasa SK; Evans BW; Dussault PH Reductive Cleavage of Organic Peroxides by Iron Salts and Thiols ACS Omega 2018, 3, 14054–14063; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Guan H; Sun S; Mao Y; Chen L; Lu R.; Huang J; Liu L Iron(II)-Catalyzed Site-Selective Functionalization of Unactivated C(sp(3) )-H Bonds Guided by Alkoxyl Radicals Angew. Chem. Int. Ed. Engl. 2018, 57, 11413–11417; [DOI] [PubMed] [Google Scholar]; c) Chevallier E; Jolibois RD; Meunier N; Carlier P; Monod A “Fenton-like” reactions of methylhydroperoxide and ethylhydroperoxide with Fe2+ in liquid aerosols under tropospheric conditions Atmos. Environ. 2004, 38, 921–933; [Google Scholar]; d) Su YL; De Angelis L; Tram L; Yu Y; Doyle MP Catalytic Oxidative Cleavage Reactions of Arylalkenes by tert-Butyl Hydroperoxide - A Mechanistic Assessment J. Org. Chem. 2020, 85, 3728–3741; [DOI] [PubMed] [Google Scholar]; e) Laine DF; Cheng IF Analysis of hydrogen peroxide and an organic hydroperoxide via the electrocatalytic Fenton reaction Microchem. J. 2009, 91, 78–81. [Google Scholar]
- (23).Zhang F; Du P; Chen J; Wang H; Luo Q; Wan X Co-catalyzed synthesis of 1,4-dicarbonyl compounds using TBHP oxidant Org. Lett. 2014, 16, 1932–1935. [DOI] [PubMed] [Google Scholar]
- (24).Bencini A; Benelli C; Gatteschi D; Zanchini C ESR spectra of some adducts of cobalt(II) acetylacetonate Inorg. Chim. Acta 1980, 40, X115–116. [Google Scholar]
- (25).Bencini A; Benelli C; Gatteschi D; Zanchini C ESR spectra of low-symmetry high-spin cobalt(II) complexes. 6. 6-Methylquinoline, pyridine, and water adducts of cobalt(II) acetylacetonate Inorg. Chem. 1980, 19, 3027–3030. [Google Scholar]
- (26).a) Morimoto T; Hirano M; Echigoya K; Sato T Oxidation by cobalt(III) acetate. Part 10. Effects of ring substituents on the product distributions in the oxidation of β-methylstyrenes by cobalt(III) acetate in acetic acid J Chem Soc Perk T 2 1986, 1205–1209; [Google Scholar]; b) Hirano M; Morimoto T Oxidation by cobalt(III) acetate. Part 6. A novel synthesis of the glycol monoacetates from aromatic olefins in wet acetic acid J Chem Soc Perk T 2 1984, 1033–1036; [Google Scholar]; c) Hirano M; Kitamura E; Morimoto T Oxidation by cobalt(III) acetate. Part 2. Oxidation of styrene in acetic acid J Chem Soc Perk T 2 1980, 4, 569–573. [Google Scholar]
- (27).Stoll S; Schweiger A EasySpin, a comprehensive software package for spectral simulation and analysis in EPR J Magn Reson 2006, 178, 42–55. [DOI] [PubMed] [Google Scholar]
- (28).Pietrzyk P; Srebro M; Radon M; Sojka Z; Michalak A Spin ground state and magnetic properties of cobalt(II): relativistic DFT calculations guided by EPR measurements of bis(2,4-acetylacetonate)cobalt(II)-based complexes J. Phys. Chem. A 2011, 115, 2316–2324. [DOI] [PubMed] [Google Scholar]
- (29).Sokolova R; Tarabek J; Papouskova B; Kocabova J; Fiedler J; Vacek J; Marhol P; Vavrikova E; Kren V Oxidation of the Flavonolignan Silybin. In situ EPR Evidence of the Spin-Trapped Silybin Radical Electrochim. Acta 2016, 205, 118–123. [Google Scholar]
- (30).a) Kuppusamy R; Muralirajan K; Cheng C-H Cobalt(III)-Catalyzed [5 + 1] Annulation for 2H-Chromenes Synthesis via Vinylic C–H Activation and Intramolecular Nucleophilic Addition Acs Catal 2016, 6, 3909–3913 [Google Scholar]; b) Vinck E; Murphy DM; Fallis IA; Strevens RR; Van Doorslaer S Formation of a cobalt(III)-phenoxyl radical complex by acetic acid promoted aerobic oxidation of a Co(II)salen complex Inorg. Chem. 2010, 49, 2083–2092. [DOI] [PubMed] [Google Scholar]
- (31).Teixeira MC; Telo JP; Duarte NF; Sa-Correia I The herbicide 2,4-dichlorophenoxyacetic acid induces the generation of free-radicals and associated oxidative stress responses in yeast Biochem. Biophys. Res. Commun. 2004, 324, 1101–1107. [DOI] [PubMed] [Google Scholar]
- (32).a) Yokota M; Fujita D; Ichikawa J. Activation of 1,1-difluoro-1-alkenes with a transition-metal complex: palladium(II)-catalyzed Friedel-Crafts-type cyclization of 4,4-(difluorohomoallyl)arenes Org. Lett. 2007, 9, 4639–4642 [DOI] [PubMed] [Google Scholar]; b) Yu L; Tang ML.; Si CM; Meng Z.; Liang Y; Han J; Sun X. Zinc-Mediated Decarboxylative Alkylation of Gem-difluoroalkenes Org. Lett. 2018, 20, 4579–4583 [DOI] [PubMed] [Google Scholar]; c) Yang L; Ji WW; Lin E; Li JL; Fan WX.; Li Q; Wang H. Synthesis of Alkylated Monofluoroalkenes via Fe-Catalyzed Defluorinative Cross-Coupling of Donor Alkenes with gem-Difluoroalkenes Org. Lett. 2018, 20, 1924–1927 [DOI] [PubMed] [Google Scholar]; d) Tan DH; Lin E; Ji WW; Zeng YF.; Fan WX; Li QJ; Gao H.; Wang HG. Copper-Catalyzed Stereoselective Defluorinative Borylation and Silylation of gem-Difluoroalkenes Adv. Synth. Catal. 2018, 360, 1032–1037 [Google Scholar]; e) Zhang J; Dai W; Liu Q; Cao S. Cu-Catalyzed Stereoselective Borylation of gem-Difluoroalkenes with B2pin2 Org. Lett 2017, 19, 3283–3286 [DOI] [PubMed] [Google Scholar]; f) Zell D; Dhawa U; Muller V; Bursch M; Grimme S; Ackermann L C-F/C-H Functionalization by Manganese(I) Catalysis: Expedient (Per)Fluoro-Allylations and Alkenylations Acs Catal 2017, 7, 4209–4213; [Google Scholar]; g) Sakaguchi H; Uetake Y.; Ohashi M.; Niwa T; Ogoshi S; Hosoya T. Copper-Catalyzed Regioselective Monodefluoroborylation of Polyfluoroalkenes en Route to Diverse Fluoroalkenes J. Am. Chem. Soc 2017, 139, 12855–12862 [DOI] [PubMed] [Google Scholar]; h) Lu X; Wang Y.; Zhang B; Pi JJ; Wang XX; Gong TJ.; Xiao B; Fu Y. Nickel-Catalyzed Defluorinative Reductive Cross-Coupling of gem-Difluoroalkenes with Unactivated Secondary and Tertiary Alkyl Halides J. Am. Chem. Soc. 2017, 139, 12632–12637 [DOI] [PubMed] [Google Scholar]; i) Li J; Lefebvre Q; Yang H.; Zhao Y; Fu H Visible light photocatalytic decarboxylative monofluoroalkenylation of alpha-amino acids with gem-difluoroalkenes Chem. Commun 2017, 53, 10299–10302 [DOI] [PubMed] [Google Scholar]; j) Kong L.; Liu B.; Zhou X; Wang F; Li X. Rhodium(iii)-catalyzed regio- and stereoselective benzylic alpha-fluoroalkenylation with gem-difluorostyrenes Chem. Commun 2017, 53, 10326–10329 [DOI] [PubMed] [Google Scholar]; k) Watabe Y.; Kanazawa K.; Fujita T; Ichikawa J. Nickel-Catalyzed Hydroalkenylation of Alkynes through C-F Bond Activation: Synthesis of 2-Fluoro-1,3-dienes Synthesis-Stuttgart 2017, 49, 3569–3575 [Google Scholar]; l) Xie J; Yu J; Rudolph M; Rominger F; Hashmi AS. Monofluoroalkenylation of Dimethylamino Compounds through Radical-Radical Cross-Coupling Angew. Chem. Int. Ed. Engl. 2016, 55, 9416–9421. [DOI] [PubMed] [Google Scholar]
- (33).Li Q; Lin J-H; Deng Z-Y; Zheng J; Cai J; Xiao J-C Wittig gem-difluoroolefination of aldehydes with difluoromethyltriphenylphosphonium bromide J. Fluorine Chem 2014, 163, 38–41. [Google Scholar]
- (34).Xiao P; Tang Z; Wang K; Chen H; Guo Q; Chu Y; Gao L; Song Z Chemoselective Reduction of Sterically Demanding N,N-Diisopropylamides to Aldehydes J. Org. Chem. 2018, 83, 1687–1700. [DOI] [PubMed] [Google Scholar]
- (35).Chen K; Liu S; Wang D; Hao W-J; Zhou P; Tu S-J; Jiang B Silver/Scandium-Cocatalyzed Bicyclization of β-Alkynyl Ketones Leading to Benzo[c]xanthenes and Naphtho[1,2-b]benzofurans J. Org. Chem 2017, 82, 11524–11530. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.