

Abstract
Sulfide quinone oxidoreductase (SQOR) catalyzes the first step in sulfide clearance, coupling H2S oxidation to coenzyme Q reduction. Recent structures of human SQOR revealed a sulfur atom bridging the SQOR active site cysteines in a trisulfide configuration. Here, we assessed the importance of this cofactor using kinetic, crystallographic and computational modeling approaches. Cyanolysis of SQOR proceeds via formation of an intense charge transfer complex that subsequently decays to eliminate thiocyanate. We captured a disulfanyl-methanimido thioate intermediate in the SQOR crystal structure, revealing how cyanolysis leads to reversible loss of SQOR activity that is restored in the presence of sulfide. Computational modeling and MD simulations revealed an ~105-fold rate enhancement for nucleophilic addition of sulfide into the trisulfide versus a disulfide cofactor. The cysteine trisulfide in SQOR is thus critical for activity and provides a significant catalytic advantage over a cysteine disulfide.
Graphical Abstract
INTRODUCTION
Hydrogen sulfide (H2S) is a signaling molecule that exerts physiological effects in the cardiovascular, central nervous, and gastrointestinal systems.1–3 H2S is synthesized endogenously in mammals through the activities of cystathionine β-synthase4 and cystathionine γ-lyase,5 as well as 3-mercaptopyruvate sulfur transferase.6–7 Tissue concentrations ofH2S typically range from 10-80 nM.8–10 At higher concentrations, H2S can act as a respiratory poison that blocks the electron transport chain by inhibiting complex IV.11
Due to the dual effects of H2S, its levels must be strictly regulated. The accumulation of toxic concentrations of H2S is prevented by its oxidation to thiosulfate and sulfate via the mitochondrial sulfide oxidation pathway.12 The first and committed step in this pathway is catalyzed by sulfide quinone oxidoreductase (SQOR), an inner mitochondrial membrane-anchored flavoprotein, which is a member of the flavin disulfide reductase superfamily.13 SQOR couples H2S oxidation to coenzyme Q10 (CoQ10) reduction.12, 14–16 It transfers the oxidized sulfane sulfur to a small molecule acceptor, which is predicted to be glutathione (GSH) under physiological conditions.15–16 Inherited deficiency of SQOR presents as Leigh disease.17
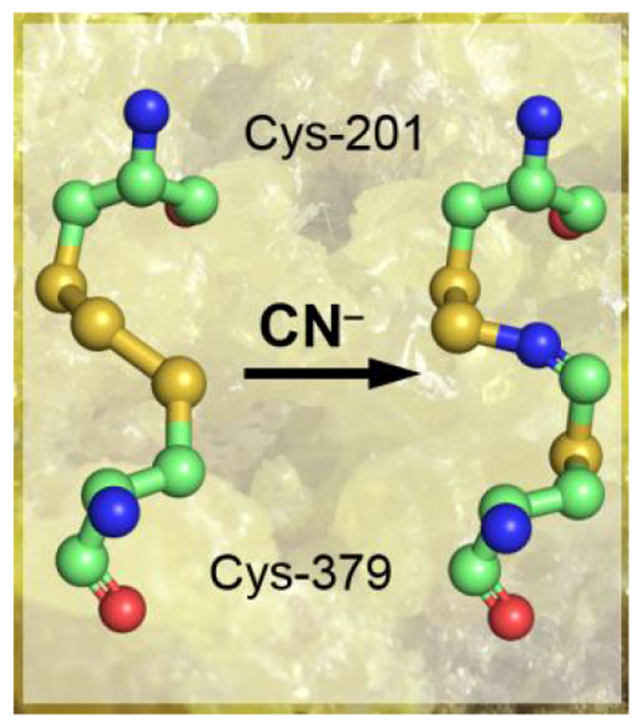
While the overall reaction catalyzed by SQORs are similar,18–20 the requirement of a small molecule acceptor by the human enzyme distinguishes it from most of the characterized bacterial homologs, which build long polysulfide chains and can release octasulfur rings as oxidation products.18–20 In contrast, the catalytic cycle of human SQOR resembles that of bacterial flavocytochrome c sulfide dehydrogenase, which couples the conversion of sulfide to hydrodisulfide with the reduction of cytochrome c.21 This catalytic mode is also seen in the Staphylococcus aureus bacterial SQOR, which is phylogenetically grouped with the human enzyme and utilizes coenzyme A as the acceptor.22 Unexpectedly, the crystal structures of human SQOR revealed the presence an additional sulfur bridging the active site cysteines in a trisulfide.23–24 The catalytic relevance of the trisulfide configuration is controversial, and it has been assigned as the inactive23 or active24 form of the enzyme. To our knowledge, a catalytically relevant cysteine trisulfide would be the first of its kind for a thiol-based redox active cofactor.
A postulated reaction mechanism for human SQOR that starts with the trisulfide as the resting form of the enzyme is shown in Figure 1. The reaction cycle proceeds via two half reactions. In the first half reaction, sulfide adds to the trisulfide at the solvent-accessible Cys-379 to form a 379Cys-SSH persulfide, which was previously captured in crystallo.24 The bridging sulfur is retained on 201Cys-SS− persulfide, which forms an unusually intense charge transfer (CT) complex with FAD that is centered at 675 nm.14, 16, 24–25 Sulfur transfer from 379Cys-SSH to a small molecule acceptor leads to regeneration of the active site trisulfide with the concomitant two-electron reduction of FAD. In the second half reaction, FADH2 transfers electrons to CoQ10, regenerating the resting enzyme and linking sulfide oxidation to mitochondrial energy metabolism by supplying reduced CoQ10 to Complex III in the electron transport chain.26
Figure 1.
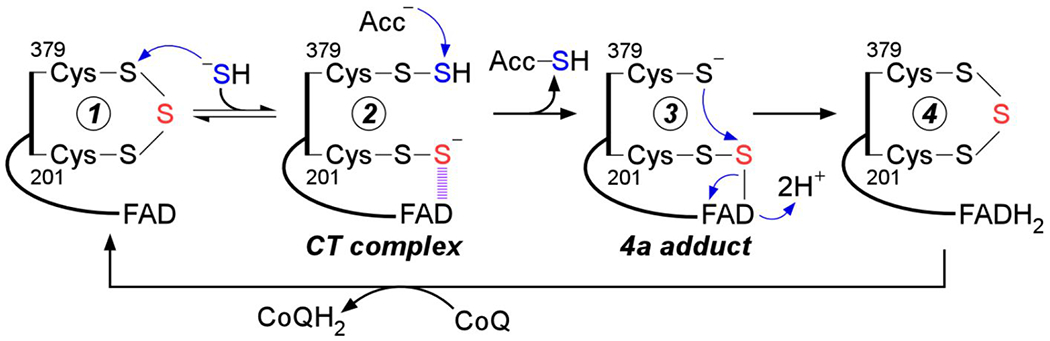
Postulated mechanism for sulfide oxidation catalyzed by human SQOR. Sulfide adds into the resting cysteine trisulfide (1) to generate a 379Cys-SSH persulfide and a 201Cys-SS− persulfide, with the latter participating in a CT complex with FAD (2). Sulfur transfer to a small molecule acceptor proceeds through a putative 4a adduct (3) to generate the reduced enzyme (4). Electron transfer from FADH2 to CoQ regenerates the resting enzyme. The oxidized sulfur and bridging sulfur in the cysteine trisulfide are labeled in blue and red, respectively.
In principle, an active site cysteine trisulfide provides several advantages over the conventional disulfide configuration seen in the mechanistically similar flavocytochrome c sulfide dehydrogenase.21 Sulfane sulfur species have increased electrophilic character versus their respective thiols,27 which would enhance the reactivity of the solvent-accessible Sγ of Cys-379 in SQOR towards nucleophilic addition by sulfide. Indeed, the rate of sulfide addition to the cysteine trisulfide of SQOR is estimated to be ~2 × 107-fold higher than the rate of sulfide addition to cysteine disulfide in solution (0.6 M−1s−1 at pH 7.4, 25 °C).28 The subsequent formation of persulfide rather than thiolate intermediate on Cys-201 would also enhance its reactivity for facilitating sulfur transfer and electron movement via the putative C4a adduct.
In this study, we report the spectral and kinetic characterization of cyanolysis-induced dismantling followed by sulfide-dependent rebuilding of the trisulfide cofactor. Cyanide treatment destabilized human SQOR and led to its inactivation with concomitant loss of the bridging sulfane sulfur. Addition of sulfide to inactive cyanide treated enzyme led to recovery of active SQOR, indicating that the oxidation state of the active site cysteines was preserved upon cyanide treatment. Crystallization of SQOR with cyanide led to the capture of a 379Cys N-(201Cys-disulfanyl)-methanimido thioate intermediate, providing insights into how the trisulfide can be rebuilt following cyanide treatment. Finally, computational modeling indicated that the trisulfide configuration provides a significant catalytic advantage over a disulfide in the SQOR reaction. Collectively, our study demonstrates that the cysteine trisulfide in SQOR is required for its catalytic activity, confers a catalytic edge over a disulfide, and contributes to its structural integrity.
EXPERIMENTAL SECTION
Materials—
The following reagents were purchased from Millipore Sigma: CoQ1, n-dodecyl-β-D-maltoside (DDM), potassium cyanide, sodium sulfide nonahydrate, and sodium sulfite. The phospholipid DHPC (1,2-diheptanoyl-sn-glycero-3-phosphocholine) was purchased from Avanti Polar Lipids (Alabaster, AL). DHLA was purchased from Cayman Chemical Company (Ann Arbor, MI).
Preparation of human SQOR–
Human SQOR was purified as detergent-solubilized recombinant enzyme as described previously.15 Human SQOR used for crystallization was purified in an identical procedure as described previously,15 except that DHPC (0.03% w/v) was substituted with DDM (0.05% w/v) as the solubilizing detergent.24
SQOR spectral analyses and activity assays–
The absorption spectra of SQOR were recorded on a temperature-controlled Shimadzu UV-2600 spectrophotometer in Buffer A (50 mM Tris, pH 8.0, containing 300 mMNaCl and 0.03% DHPC). The concentration of SQOR used in the spectral assays was estimated by the absorbance of the FAD cofactor, using an extinction coefficient of 11,500 M−1 cm−1 at 450 nm.14 SQOR activity was estimated by the rate of CoQ1 reduction (Δεox-red = 12,000 M−1cm−1) at 25 °C as described previously,15 using sulfite (800 μM) as the sulfur acceptor.
Stopped flow spectroscopy–
All stopped flow experiments were conducted at 4 °C on a SF-DX2 double mixing stopped-flow system from Hi-Tech Scientific, equipped with a photodiode array detector (300-700 nm range). The concentrations reported in the figure legends for stopped flow experiments are before 1:1 (v/v) mixing.
Detection of sulfane sulfur in SQOR–
SQOR was assayed for sulfane sulfur using the cold cyanolysis method as described previously.24 The data for mol of sulfane sulfur per mol SQOR monomer are presented as the mean ± SD of three independent preparations of SQOR.
Thermal denaturation assays–
The thermal stabilities of untreated SQOR, and cyanide pre-treated SQOR before and after sulfide treatment, were assessed using 300 μL of enzyme (5 μM) in a quartz cuvette, housed in a temperature-controlled Shimadzu UV-2600 spectrophotometer. SQOR was allowed to equilibrate at 20 °C for 2 min before initiating the assay by increasing the temperature by 1 °C min−1. Thermal denaturation was monitored by the increase in absorbance at 600 nm. The midpoint of transition at 600 nm toward the maximum absorbance, defined as Tagg,29 was used as the basis of comparison for thermal stabilities of SQOR species.
Crystallization of SQOR-CoQ1 with cyanide–
SQOR-CoQ1 crystals were grown at 20 °C by the hanging drop vapor diffusion method using solubilized human SQOR (17.4 mg mL−1) in 50 mM Tris-HCl pH 8.0, containing NaCl (300 mM) and n-dodecyl β-D-maltoside (0.05% w/v) supplemented with CoQ1 (5 mM, in 100% DMSO). The SQOR solution was then mixed 1:1 (v/v) with the reservoir solution composed of 200 mM ammonium tartrate dibasic, pH 6.6, and PEG 3350 (20% w/v), yielding a final CoQ1 concentration of 2.5 mM. The resulting SQOR-CoQ1 crystals were soaked with potassium cyanide (1.25 mM) for 40 min, followed by cryoprotection in the aforementioned reservoir solution supplemented with glycerol (35% v/v) before freezing in liquid nitrogen.
X-ray data collection and structure determination–
Diffraction data for SQOR-CoQ1 + cyanide crystals were collected at the LS-CAT beamline 21-ID-D (Advanced Photon Source, Argonne National Laboratory) at 1.12723 Å wavelength. The diffraction images were processed using HKL2000.30 The molecular replacement solution for SQOR-CoQ1 + cyanide was determined using SQOR-CoQ1 (PDB ID: 60IB) as a search model. The final structures were completed using alternate cycles of manual fitting in Coot31 and refinement in REFMAC5.32 The stereochemical quality of the final models was assessed using MolProbity.33
MD simulations of SQOR-
The crystal structure of human SQOR-CoQ1 + sulfide in the trisulfide state complexed with FAD (PDB: 6016, monomer A, 2.56 Ǻ resolution) was used as a starting point.24 CoQ1 was manually docked by superimposing another structure of SQOR-CoQ1 (PDB: 60IB, monomer A, 2.03 Å resolution). Two systems were simulated: SQOR in a Cys-201-Cys-379 disulfide state (SQOR-SS) and in the trisulfide state (SQOR-SSS). Protonation and tautomers of titratable residues, and missing hydrogen atoms, were added with the ProToss utility.34 Systems were solvated with a periodic truncated octahedral box of TIP3P water extended up to 12 Å around solute, then neutralized with six Cl− ions with the leap utility of AmberToolsl7.35 Both were then minimized, heated to 310 K (500 ps, NVT), and equilibrated at 1 atm (1 ns, NPT) prior to conducting the simulations (600 ns, NPT). Minimization and simulation was carried out using the pmemd.cuda module of AMBER 16.35 For describing the protein, the AMBER ff14SB force field was used for standard residues, whereas the gaffioxce field was used for FAD and CoQ1 ligands, along with RESP charges.36 The central sulfur atom of the trisulfide moiety was treated as a separate residue with zero charge. All parameters for describing the trisulfide moiety were present in the ff14SB force field, except for the S–S–S bonds, which were taken from the gaff force field. An 8.0 Å cutoff was used for treating direct non-bonding interactions, and long-range interactions were treated with the Particle Mesh Ewald (PME) method.37 For MD simulations, temperature and pressure (in NPT simulations) were controlled by means of the Langevin thermostat38 and the Monte Carlo barostat,39 respectively. Distances involving hydrogen atoms were constrained with SHAKE40 and a time integration step of 2 femtoseconds was used. Harmonic restraints of 10 kcal mol−1 Å−2 were applied to a water-bridged hydrogen bond observed in SQOR crystal structures24 between the protonated amino group of the Lys-207 side chain and the N5 atom in FAD. Trajectory processing and analysis was done with the cpptraj module of AmberToolsl7.35 Convergence of simulations was monitored following the Cα-RMSD (see Figure S1). In order to extract representative structures from the MD 600 ns trajectories, clustering analysis (5 clusters) was performed for each system, using a hierarchical-agglomerative algorithm with cpptraj.35
Reactivity descriptors from QM/MM calcidations on SQOR–
Descriptors of intrinsic reactivity derived from the electronic structure of Cys-379 and Cys-201 were calculated at the QM/MM level in the framework of a conceptual DFT41 at the M06-2X-D3/6-31+G(d,p) level of theory42–44 combined with a classical description of the enzyme using the aforementioned force fields. Global softness (S) and electrophilic Fukui function condensed to the Sγ atom were calculated according to equations (1) and (2):
| (1) |
where IP and EA respectively represent the ionization potential and electron affinity of the system of interest, determined using the vertical ΔSC] (self-consistent field) approximation,45 and
| (2) |
where qSγ(N) and qSγ(N+1) represent the atomic charge on the Sγ atoms of Cys-379/Cys-201, calculated using a Natural Population Analysis,46 both in the system of reference bearing N electrons and after addition of one extra electron. The atomic electrophilic softness (, calculated as S times ) is a local descriptor that can be used to compare Sγ intrinsic reactivity in Cys-379 and Cys-201 across SQOR-SS and SQOR-SSS. The electronic structure of each macromolecular system was thus obtained through single-point calculations performed on representative structures extracted from MD simulations using the additive QM/MM scheme implemented in AMBER1635 interfaced with Gaussian 09 Rev. D.0147 with a QM region comprising 379Cys-CH2-S(S)S-CH2-201Cys.
DFT-PCM modeling of reaction mechanisms and barriers for sulfide nucleophilic attack-
The mechanism of the reaction of the sulfide anion, manually docked and oriented as guided by our previous models of similar reactions,48 was characterized at the M06-2X-D3/6-31+G(d,p)-PCM level of theory, previously validated by us to model reactions of the sulfide anion in a similar system.48 A simplified representation of the catalytic disulfide/trisulfide and FAD at the active site of SQOR was used including CH3SSSCH3/CH3SSCH3 and the flavin. The structures of each reactant complex, transition and product complex were fully optimized and verified by the inspection of the eigenvalues of the Hessian matrix at the same level. Thermochemical corrections at 298 K and 1 atm were calculated under usual approximations of statistical thermodynamics (rigid rotor, harmonic frequencies) as implemented in Gaussian 09 Rev. D0147 The effects exerted by the bulk protein on the active site structure along the reaction and reaction barrier were introduced using the IEF-PCM continuum model49 with a dielectric constant ε = 10.125. The reactive systems were placed in a molecular shaped cavity constructed using Bondi’s radii50 and including non-electrostatic (cavitation, repulsion and dispersion) contributions. In order to connect transition states with reactant complexes and product complexes, we calculated the IRC reaction path51 using the HPC algorithm.52
RESULTS
Formation and decay of the cyanide-induced CT complex in SQOR–
Mixing SQOR with cyanide led to the formation of an intense CT complex characterized by an absorbance maximum at 695 nm and a shift in the FAD peak from 450 nm to 420 nm, with isosbestic points at 430 nm and 505 nm (Figure 2A). These spectral features are similar to the CT complexes seen previously with other nucleophiles.25, 53 From the dependence of the rate of CT complex formation on the concentration of cyanide, the following parameters were obtained: kon = 10,500 ± 120 M−1 s−1, koff = 3.7 ± 0.6 s−1, and KD(app) = 348 ± 50 μM at 4 °C (Figure 2B,C). The CT complex is an intermediate in the catalytic cycle of SQOR, and sulfide addition to the CT intermediates formed by alternative nucleophiles leads to their decay with the concomitant reduction of FAD.25, 53 Similarly, addition of sulfide immediately following cyanide-induced CT complex formation led to its decay with concurrent reduction of FAD (Figure 2D). This result indicates that the CT complex formed in the presence of cyanide can participate in the first half reaction leading to FADH2 formation.
Figure 2.
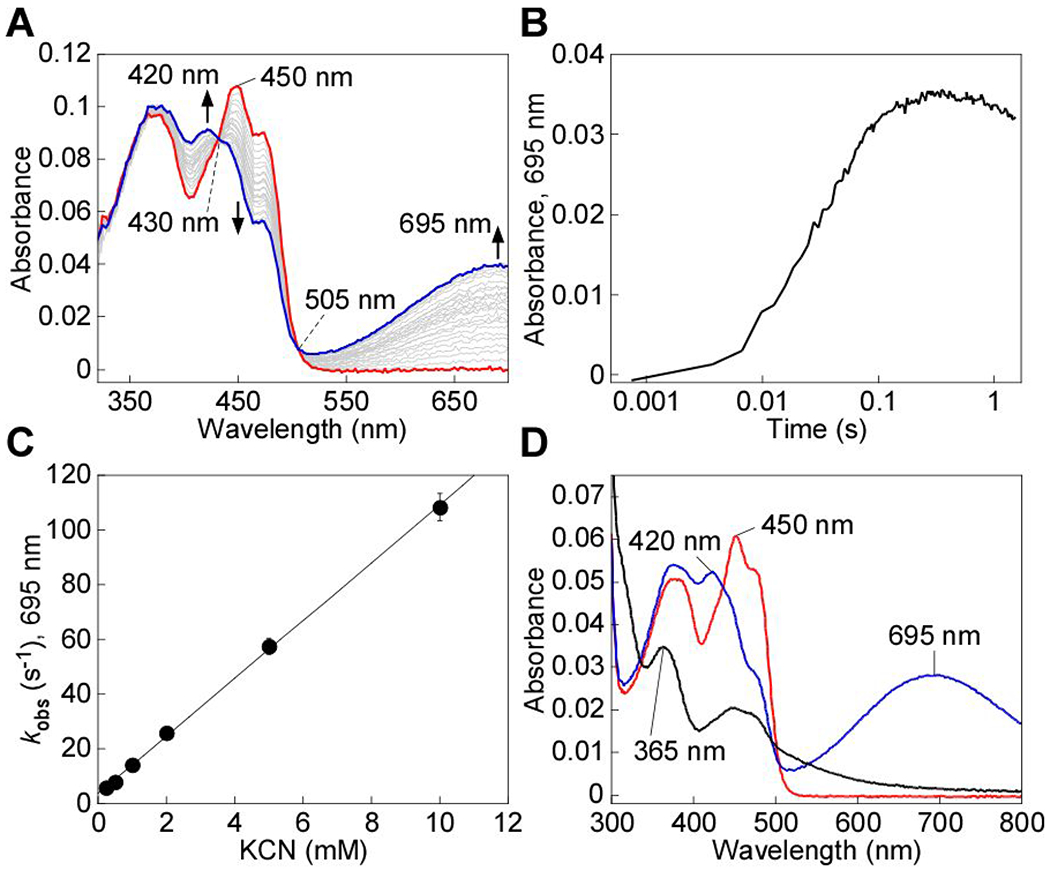
Cyanide-induced CT complex formation in SQOR. A, SQOR (10 μM, red line) in 100 mM potassium phosphate, pH 7.4 containing 0.03% DHPC, was mixed 1:1 (v/v) with KCN (4 mM) and monitored over 1.5 s at 4 °C for the formation of the cyanide-induced CT complex at 695 mn (blue line). B, Representative stopped flow kinetic trace for the reaction in (A) monitored at 695 mn. C, Dependence of the kobs at 4 °C for cyanide-induced CT complex formation on cyanide concentration. The data are representative of two independent experiments, with each data point obtained in triplicate. D, SQOR (5 μM, red line) was treated with KCN (5 mM) to form the CT complex (blue line), immediately followed by the addition of Na2S (200 μM) and incubated for 5 min at 20 °C, which led to CT complex decay and FAD reduction (black line). The data are representative of three independent experiments.
Decay of the cyanide-induced CT complex and cyanolysis of the cysteine trisulfide–
Extended incubation of the cyanide-induced CT complex in the presence of excess cyanide led to its slow decay (Figure 3A). A kobs of 0.15 ± 0.02 min−1 at 20 °C was observed for the decay of the CT complex in the presence of 5-10 mM KCN (Figure 3B). The FAD spectrum following CT decay was slightly altered from that in native SQOR. Thus, a blue shift in the absorbance maximum from 450 nm to 447 nm and a narrowing of the 380 nm absorption peak were seen (Figure 3A). The altered spectral features were observed even after the enzyme was desalted to remove excess cyanide, suggesting a change in the flavin electronic environment. Cyanolysis yields ~1 mol of sulfane sulfur per mol SQOR monomer,24 consistent with the presence of the trisulfide in the enzyme as isolated. As expected, cyanolysis of cyanide pre-treated and desalted enzyme yielded no detectable sulfane sulfur (0 ± 0.1 mol sulfane sulfur per mol SQOR monomer). We also assessed the effects of sulfite treatment, which forms a strong CT complex when added to native SQOR.24 However, sulfite did not elicit spectral changes in cyanide pre-treated and desalted SQOR (Figure 3C).
Figure 3.
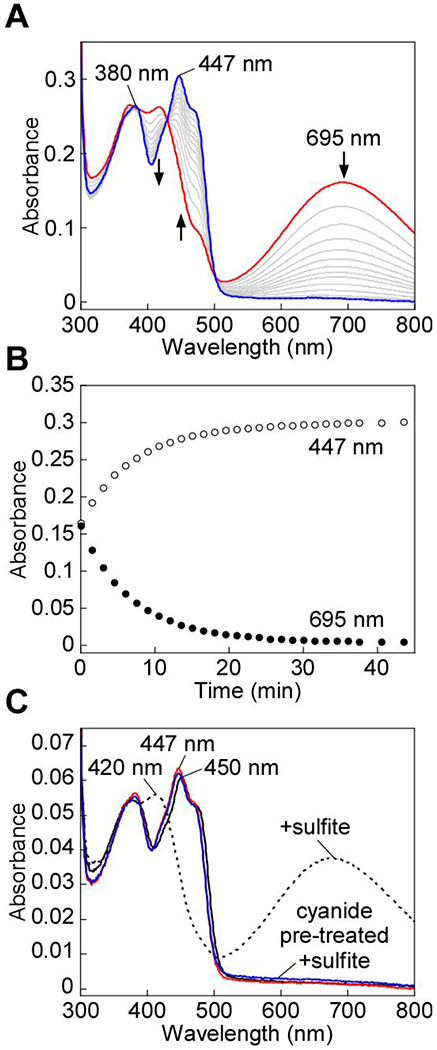
Cyanide-induced CT complex decay in SQOR. A, SQOR (25 μM) in Buffer A was treated with KCN (10 mM) to form the CT complex (red line), which was monitored over 43 min at 20 °C for the complete decay of the CT complex (blue line). B, Kinetic traces for the decay of the cyanide-induced CT complex in (A), monitored at 450 nm (open circles) and 695 nm (closed circles). C, SQOR (5 μM. solid black line) was treated with sodium sulfite (5 mM) and incubated for 1 min at 20 °C to form the CT complex (dashed black line). In tandem, SQOR (5 μM) pre-treated with KCN (10 mM) and desalted (solid red line) was then treated with sodium sulfite (5 mM). CT complex formation was not observed after incubation for 1 min at 20 °C (solid blue line). The data are representative of three independent experiments.
Sulfide-mediated regeneration of the active site trisulfide–
We next assessed the impact of cyanide treatment on SQOR activity under steady state turnover conditions. Surprisingly, the specific activity of SQOR in the standard assay was similar for the cyanide pre-treated (369 ± 30 μmol min−1 mg−1) and native (360 ± 10 μmol min−1 mg−1) enzymes (Figure S1). This result suggested that cyanide treated enzyme can be reactivated by rebuilding the trisulfide following cyanolysis of SQOR.
We therefore monitored the rate at which the trisulfide is rebuilt, using formation of the sulfide-induced CT complex as a measure of the active enzyme (Figure 4A).14, 25 For this, the kinetics of CT complex formation was assessed following rapid mixing of sulfide with cyanide pre-treated SQOR. Compared to native SQOR (kohs = 19.4 ± 2 s−1), CT complex formation was ~12-fold slower with cyanide pre-treated SQOR (1.6 ± 0.2 s−1) (Figure 4B). The lag in the absorbance increase at 675 nm indicated that trisulfide rebuilding limits the rate of CT complex formation in cyanide pre-treated SQOR. Consistent with this postulate, incubation of cyanide pre-treated SQOR with sulfide for 1 h at 4 °C led to FAD reduction (Figure 4C), signaling reformation of the active trisulfide-containing SQOR under these conditions. The presence of excess sulfide, which serves as both the sulfur donor and acceptor, led to FADH2 accumulation in the absence of CoQ1.14, 25 The 447 nm FAD absorption peak observed in cyanide pre-treated SQOR (Figure 2A) shifted to 450 nm following incubation with sulfide (Figure 4D), indicating recovery of the native FAD microenvironment. Furthermore, addition of sulfite to regenerated SQOR resulted in the formation of a robust CT complex, indicating the presence of a trisulfide in the active site (Figure 4D). Regeneration of the trisulfide was confirmed by cold cyanolysis, which detected 1.1 ± 0.3 mol of sulfane sulfur per mol SQOR monomer.
Figure 4.
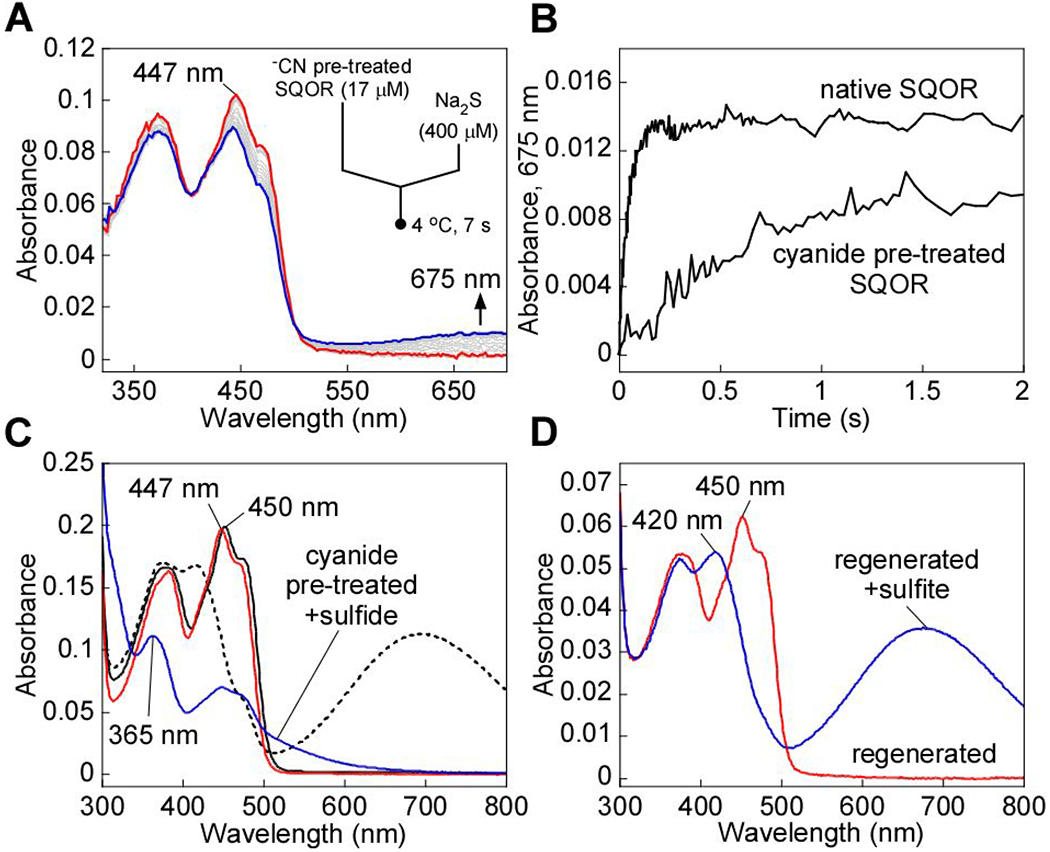
Regeneration of cyanide pre-treated SQOR by sulfide. A, Cyanide pre-treated SQOR (17 μM, red line) in Buffer A was rapidly mixed 1:1 (v/v) with Na2S (400 μM) and monitored over a period of 7 s at 4 °C for fonnation of the sulfide-induced CT complex (blue line). B, Comparison of the kinetic traces at 675 mn for cyanide pre-treated SQOR, as shown in A, versus native SQOR mixed with Na2S (400 μM) under the same conditions. C, SQOR (17 μM, solid black line) in Buffer A was treated with KCN (10 mM) to fonn the CT complex (dashed black line) and monitored over 40 min at 20 °C for the complete decay of the CT complex and desalted to remove excess cyanide (red line). Cyanide pre-treated SQOR was then incubated with Na2S (300 μM) for 1 h at 4 °C, which led to FAD reduction (blue trace). D, Cyanide pre-treated SQOR, pre-incubated with sulfide under the same conditions as (A) and desalted (5 μM, red line), was treated with sulfite (5 mM) and incubated for 1 min to fonn the sulfite-induced CT complex (blue line). The data are representative of three independent experiments.
Cyanolysis of the bridging sulfur decreases SQOR protein stability
We consistently observed that cyanide treatment led to an increased tendency for SQOR to aggregate at temperatures above 20 °C, indicating that loss of the bridging sulfur in the active site trisulfide leads to protein instability. We therefore investigated the thermal stability of SQOR with and without cyanide pretreatment. Native SQOR exhibited a Tagg of 66 ± 1 °C, compared to 36.5 ± 0.5 °C for cyanide pre-treated enzyme (Figure 5). Incubation of cyanide pre-treated SQOR in the presence of excess sulfide restored stability to that the native state (Tagg of 65.5 ±0.5 °C). Thus, the decrease in thermal stability of SQOR upon loss of the bridging sulfur was largely reversed upon regeneration of the cysteine trisulfide.
Figure 5.
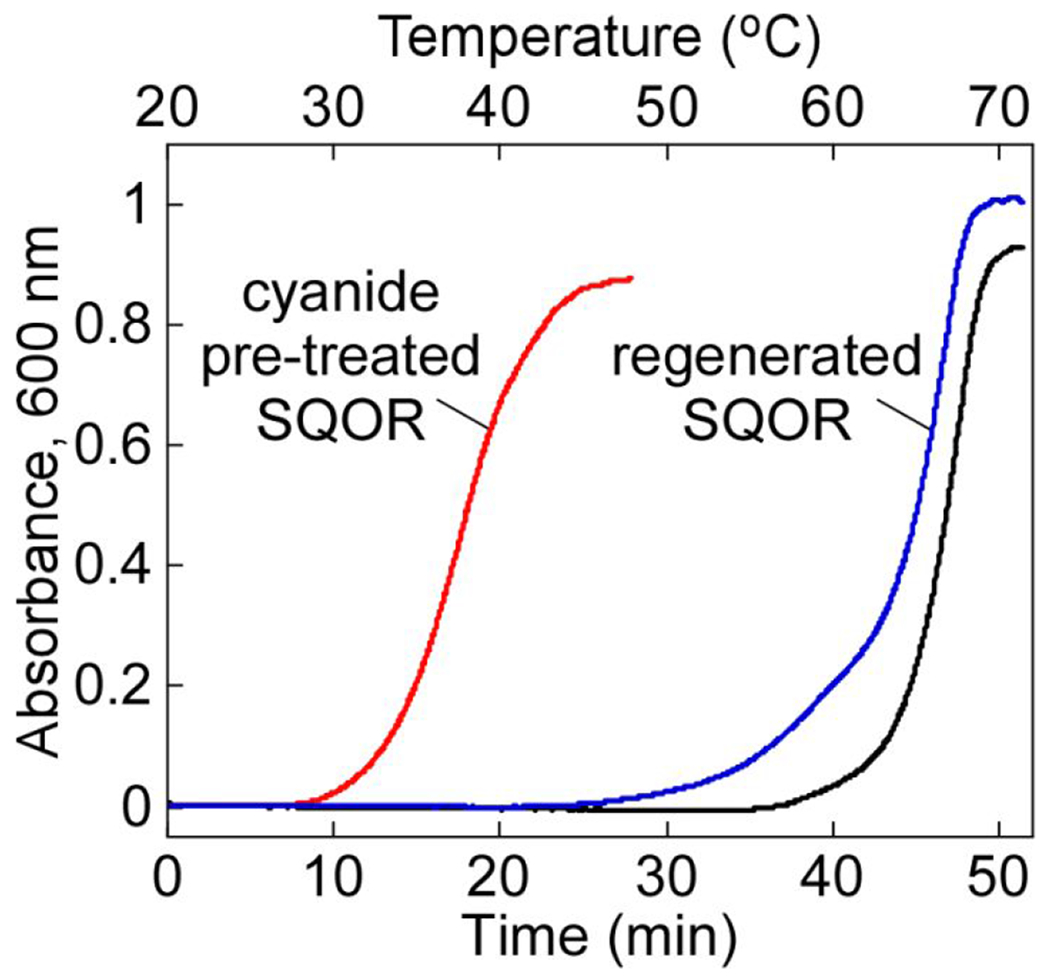
Effect of bridging sulfur extraction on SQOR protein stability. SQOR (20 μM) in Buffer A was pre-treated with KCN (10 mM) for 45 min at 20 °C and desalted, followed by incubation with Na2S (300 μM) for 1 h at 4 °C and a second desalting. A final SQOR concentration of 5 μM was used for the thermal denaturation assays. The stability of native SQOR (black line) versus cyanide pre-treated SQOR before (red line) and after (blue line) incubation with sulfide was monitored by the increase in absorbance at 600 nm. The data are representative of three independent experiments.
Dithiol-mediated reduction of FAD in SQOR–
As an alternative to cyanolysis, we attempted to extract the bridging sulfur from the SQOR trisulfide using DTT, which, in principle, can reduce the trisulfide to generate free thiols on Cys-379 and Cys-201. Unexpectedly, treatment with DTT led to bleaching of the yellow color associated with SQOR (Figure 6A), while cold cyanolysis revealed that the trisulfide was intact (1.0 ± 0.2 mol of sulfane sulfur per mol SQOR monomer). Given the known substrate promiscuity of SQOR,25, 53 we postulate that DTT adds to the resting trisulfide, forming a mixed disulfide and a CT complex. In the second step, an intramolecular displacement by the second thiol in the 379Cys-S-S-DTT adduct leads to elimination of oxidized DTT, reduction of FAD, and regeneration of the trisulfide (Figure 6B).
Figure 6.
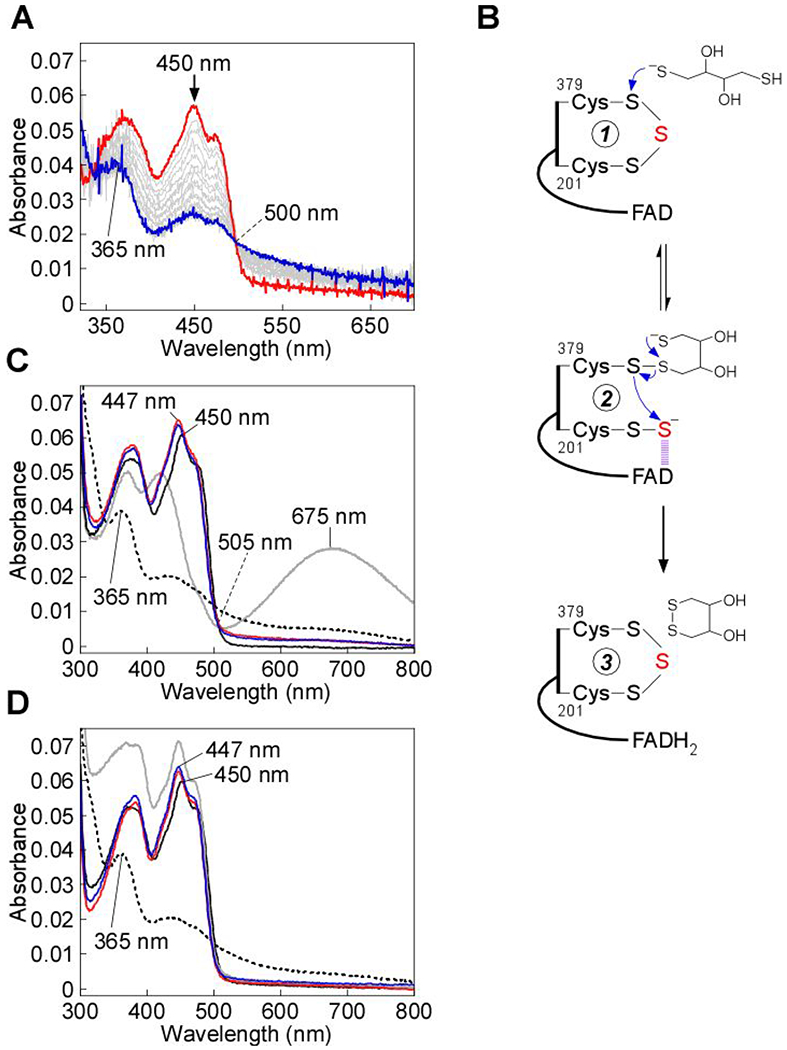
Dithiol-mediated reduction of FAD in SQOR. A, SQOR (10 μM) in Buffer A was rapidly mixed 1:1 (v/v) with DTT (400 μM) and FAD reduction was monitored over 7 s at 4 °C. B, Proposed mechanism for the addition of DTT into the SQOR cysteine trisulfide, leading to FAD reduction. DTT adds into the cysteine trisulfide (1) at the solvent-accessible Cys-379 to generate a mixed disulfide and 201Cys-SS− (2). An intramolecular thiol-disulfide exchange then regenerates the SQOR cysteine trisulfide, with electrons moving into FAD (3). C, SQOR (5 μM) in Buffer A (solid black line), was treated with DTT (200 μM), leading to FAD reduction (dashed black line), or β-mercaptoethanol (200 μM), leading to stable CT complex formation (solid gray line). FAD reduction was not observed in cyanide pre-treated SQOR (5 μM, solid red line) upon treatment with DTT (200 μM, solid blue line). D, SQOR (5 μM) under the same conditions as in (C) (solid black line), was treated with DHLA (200 μM), leading to FAD reduction (dashed black line), followed by re-oxidation by addition of CoQ1 (180 μM, solid gray line). FAD reduction was not observed in cyanide pre-treated SQOR (5 μM, solid red line) upon treatment with DHLA (200 μM, solid blue line). The data are representative of three independent experiments.
To further test this model, the reaction of DTT with SQOR was characterized by stopped-flow spectroscopy. Upon mixing SQOR rapidly with DTT, FADH2 formation (kohs = 0.36 ± 0.04 s−1) was observed without accumulation of a CT complex intermediate (Figure 6A). This contrasted with the reaction of other nucleophiles with SQOR, and suggested that resolution of the mixed disulfide, via an intramolecular reaction, is more rapid than its formation (Figure 6B). Next, we tested whether the intact cysteine trisulfide in SQOR is required for FAD reduction by DTT. Pretreatment of SQOR with cyanide prevented FAD reduction by DTT (Figure 6C). In contrast to DTT, the monothiol, β-mercaptoethanol, was unable to drive FAD reduction, and formed a stable CT complex instead (Figure 6C). Like DTT, the dithiol dihydrolipoic acid (DHLA), a physiological reductant, also led to FAD reduction, but only when the cysteine trisulfide was intact (Figure 6D).
Structure of SQOR-CoQ1 soaked with cyanide–
To obtain structural insights into the interaction of cyanide with SQOR, crystals of human SQOR-CoQ1 were soaked with cyanide. The 2.25 Å resolution structure was obtained by molecular replacement using coordinates for the SQOR-CoQ1 structure (PDB ID: 60IB) (Table 1). The overall structure (Figure 7A) is similar to that reported previously for SQOR-CoQ1.24 Surprisingly, strong and continuous electron density was observed between Cys-201 and Cys-379 (Figure 7B, C). The electron density in the presence of cyanide was more extended than for the trisulfide in the native SQOR-CoQ1 + sulfide structure (Figure 7D). We interpret the additional electron density as evidence for the insertion of a cyanide molecule in the trisulfide bridge, forming an N-(20lCys-disulfanyl)-methanimido thioate intermediate, 201Cys-S-S-N=CH-S-379Cys. The relevance of this species to the spectral intermediates observed in the presence of cyanide is discussed later.
Table 1.
Crystallographic data collection and refinement statistics*
| SQOR-CoQ1 with cyanide | |
|---|---|
| Space group | P212121 |
| Unit cell parameters (Å) | a=78.39 |
| b=111.75 | |
| c=134.05 | |
| α=β=γ=90° | |
| Wavelength (Å) | 1.12723 |
| Data collection statistics | |
| Resolution range (Å) | 50.00-2.25 (2.29-2.25) |
| Number of unique reflections | 57050 (2696) |
| Completeness (%) | 99.7 (96.0) |
| Rmerge | 0.175 (0.974) |
| Rpim | 0.059 (0.359) |
| Redundancy | 8.6 (6.6) |
| Mean I/σ | 14.0 (2.3) |
| Refinement statistics | |
| Resolution range (Å) | 39.20-2.24 |
| Rwork/Rfree (%) | 17.24/21.60 |
| RMSD bonds (Å) | 0.008 |
| RMSD angles (deg) | 0.922 |
| Average B factor (Å2) | 36.62 |
| Number of water molecules | 125 |
| Ramachandran | |
| favored (%) | 96.51 |
| allowed (%) | 3.49 |
| not allowed (%) | 0 |
| PDB: | 6WH6 |
Values in parentheses are for highest-resolution shell.
Figure 7.
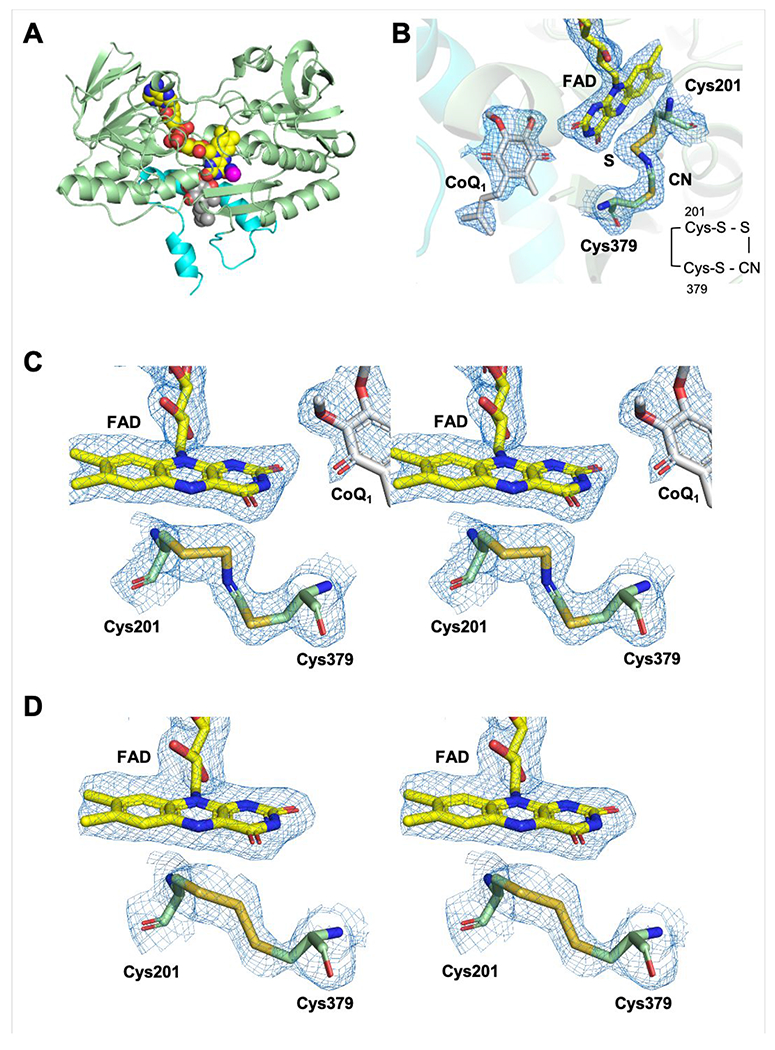
Structure and active site of SQOR-CoQ1 + cyanide. A, The overall structure of SQOR-CoQ1 + cyanide is shown with FAD, CoQ1, and cyanide in yellow, grey, and magenta spheres, respectively. The C-terminal membraneanchoring helices are highlighted in cyan. B, Electron density maps (2Fo-Fc) of the active site shown in mesh contoured at 1.0 σ. Cys-201, Cys-379, FAD, CoQ1, sulfur derived from the trisulfide, and cyanide are shown in stick display. C, Stereo image of the active site of SQOR-CoQ1 treated with cyanide. The electron densities (2Fo-Fc) are contoured at 1.0 σ. D, Stereo image of the active site in SQOR-CoQ1 treated with sulfide (PDB ID: 6O16) showing the resting trisulfide. Chain A is shown in tins figure.
MD simulations and QM/MM reactivity predictors for a disulfide versus trisulfide cofactor–
Direct comparison of the active site architecture in representative SQOR structures was extracted from 600 ns trajectories (RMSDs shown in Figure S2). The simulations provided insights into the structural and dynamical differences between the trisulfide versus a modeled disulfide state, and a chemical rationale for the use of the trisulfide cofactor by SQOR. In the disulfide structure (Figure 8A, right) the sulfur atoms of Cys-201 and Cys-379 are buried (Figure S3, right), and not in contact with solvent molecules. The Sγ atoms in the two cysteines exhibit differences in their distance to the C4a in FAD, with Cys-379 being closer (Figure 8B, right) at distances of 3.4 Ǻ versus 3.9 Ǻ for Cys-201 in the representative structure. These data argue against a catalytic disulfide configuration in SQOR, which is reinforced by considerations of the intrinsic electrophilicity of the sulfur atoms calculated at the QM/MM level.
Figure 8.
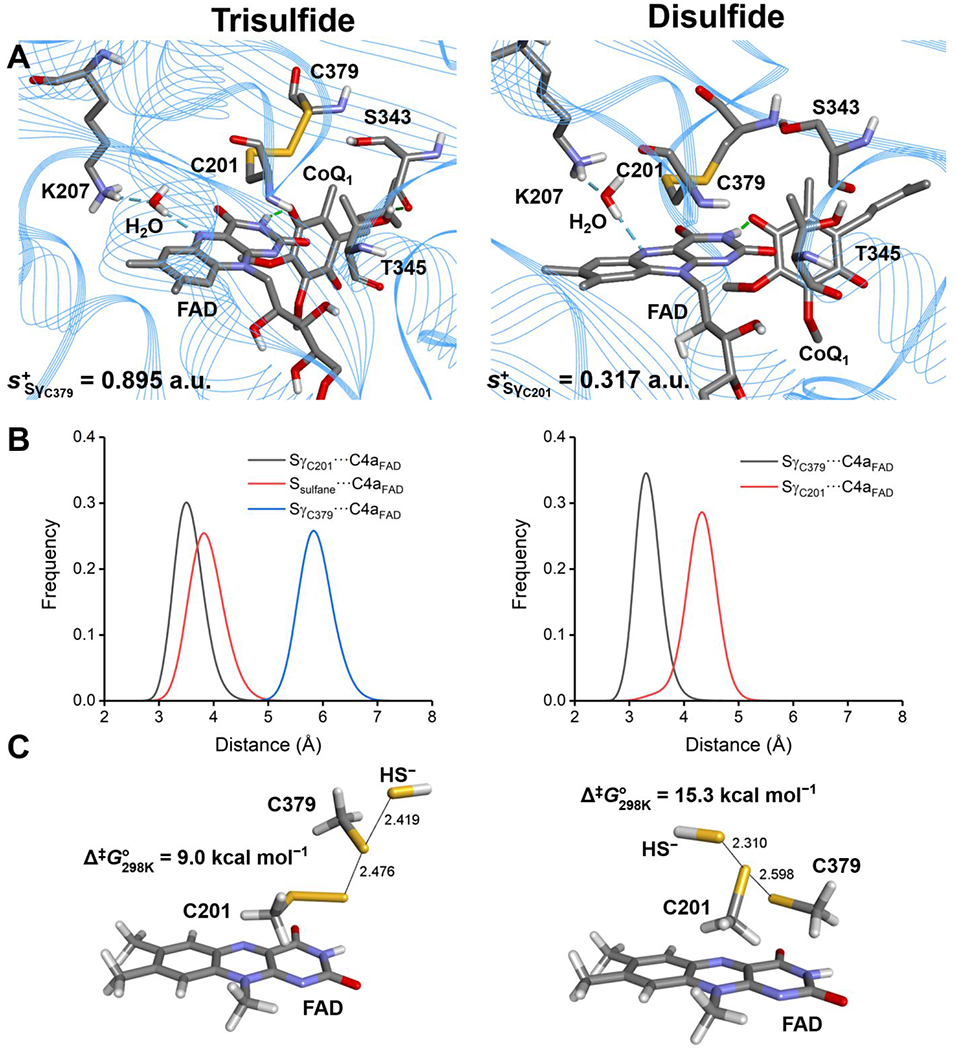
MD simulations and computational modeling of SQOR. A, Active site architecture in representative structures corresponding to the most populated cluster from 600 ns MD simulations of SQOR in the trisulfide (left panel) or disulfide state (right panel). Condensed local softness for the most electrophilic Sγ atom between Cys-201 and Cys-379 is reported for each system in atomic units (a.u.). B, Sulfur-to-C4a FAD distances for SγC201–C4aFAD/Ssulfane–C4aFAD/SγC379–C4aFAD (trisulfide, left panel) and SγC201–C4aFAD/SγC379–C4aFAD (disulfide, right panel) monitored along the corresponding MD trajectories. C, Structure of the transition states (TS) located for the sulfide anion attack on the trisulfide (left panel) or disulfide (right panel) using a reduced model of the active site of SQOR at the IEFPCM-DFT level of theory in a dielectric of ε = 10.125. Data correspond to interatomic distances in Å and Gibbs free-energy associated barriers at 298 K in kcal mol−1.
In the trisulfide model, the Sγ atom of Cys-201 is estimated to be intrinsically less electrophilic than Cys-379 (s+ = 0.317 versus 0.895 atomic units). These results support the proposed attack of the sulfide anion on the Sγ atom of Cys-379 as the first step in the catalytic mechanism (Figure 1). In the trisulfide structure (Figure 8A, left) the sulfur atom of Cys-379 is located in a small cavity and is slightly solvent exposed (Figure S3, left, Figure S4). The sulfane sulfur in the trisulfide points inward, sits at the apex of a 109° S-S-S angle, and is almost equidistant from the C4a atom in FAD as the Sγ of Cys-201. The 4.9 Å (sulfane sulfur) and 4.2 Å (Sγ of Cys-201) distances to C4a in FAD in the representative structure from the most populated cluster in solution (Figure 8A, left), are comparable to the 4.3 and 3.3 Å distances observed in the trisulfide-containing crystal structure of SQOR-CoQ1 + sulfide.24 Inspection of the distribution of values for SC201-C4aFAD and for the sulfane sulfur-C4aFAD distances along the simulation (Figure 8B, left) reveal broader histograms compared to the disulfide ones and a 0.5 Å difference between the maxima.
The atomic charges calculated for the sulfur atoms in Cys-201, Cys-379 and the sulfane sulfur in the trisulfide, reveal a slightly electropositive reaction zone, particularly over Cys-379 (+0.112 atomic units Table S1), favoring attack of the negatively charged sulfide anion. On the other hand, the local softness for gaining electron density reveals that the Sγ of Cys-379 and the sulfane sulfur are similar and significantly more reactive than the Sγ of Cys-201.
Density Functional Theory (DFT) in a Polarized Continuum Model (PCM) characterization of the sulfide addition step–
To gain further insights into the specific reactivities of the electrophilic sulfurs in a disulfide (Cys-201) versus a trisulfide (Cys-379) cofactor, we modeled the detailed mechanism using a DFT/PCM level of theory as described under Experimental Procedures. The SN2 mechanism involved the attack of a sulfide anion on the trisulfide or a hypothetical disulfide, with a persulfide or a thiolate anion, respectively serving as the leaving group (Figure 8C). The transition states are quasi-linear, late, and quite synchronic, both in terms of heavy atom reorganization (HAR) and CT complex, with HAR/CT complex being more advanced in the disulfide compared to the trisulfide, as evidenced by the Wiberg Bond indices (WBI) and natural population analysis (NPA) charges (Tables S2–S5). The computed free energy barriers for the reaction of sulfide anion with the trisulfide versus disulfide cofactor in SQOR are 9.0 and 15.3 kcal mol−1 at 25 °C and 1 atm, respectively. These results provide strong supporting evidence for the significantly greater reactivity of the trisulfide over the disulfide, accounting for much of the 107-fold difference in the second order rate constant for the reaction of sulfide anion with SQOR versus with a disulfide in solution.28 The structure of the resulting CT product complex (Figure S5) confirms completion of the SN2 reaction, and that the persulfide (or thiolate) can proceed to the next step in the reaction mechanism, concentrating excess negative charge at the 201Cys-SS− or (379Cys-S−).
DISCUSSION
Members of the flavoprotein disulfide reductase superfamily have a signature two redox cofactor active site constellation. While the flavin is common to all members, the second cofactor can be a cysteine disulfide, which is the most common theme in the superfamily, a cysteine sulfenic acid, or a mixed disulfide (e.g. Cys-S-S-CoA).13 Recently, a fourth variation on the redox active cysteine cofactor theme, i.e., a trisulfide, was discovered in human SQOR.23–24
Examples of cysteine bridged by sulfane sulfur in proteins are relatively rare. In bacteria, the transcriptional repressors CstR and SqrR form cysteine di-, tri-, or tetrasulfides upon exposure to reactive sulfur species, with tetrasulfide representing the major configuration.54–56 Formation of the polysulfide species inhibits repressor activity by physically hindering binding to promoter regions. Stable trisulfides have been observed primarily as artifacts in recombinant human growth hormone preparations.57–60 Trisulfide intermediates have been postulated as intermediates in dissimilatory sulfite reduction61 and in SQOR-catalyzed polysulfide formation in Acidianus ambivalens19 and Aquifex aeolicus.18 The role of the cysteine trisulfide in human SQOR is controversial.23–24 Initially, it was proposed to result from a dead-end reaction with sulfide under anaerobic conditions in the absence of a sulfur acceptor.14 In this model, the active site cysteine disulfide in SQOR would be regenerated via a chemically unusual mechanism that necessitates the elimination of Sγ of Cys-379 as an oxidized product, and replaces it with the sulfur atom derived from the trisulfide bridge.23 In addition to the unusual chemistry, the mechanism would require a significant conformational change to shorten the ~3.5 Å distance between the Sγ of Cys-379 and Cys-201 to allow cysteine disulfide formation. Recent biochemical data from our laboratory have however, indicated that the trisulfide in SQOR likely represents the active form of the enzyme.24 In this study, we disassembled and then reassembled the active site trisulfide by cyanolysis followed by sulfuration, and demonstrated that these processes led to the restoration of SQOR activity.
The presence of the cysteine trisulfide in SQOR was previously confirmed biochemically via cold cyanolysis,24 which extracts the bridging sulfur as thiocyanate.62 During the cyanolysis reaction, we had observed a rapid color change from yellow to blue, followed by a slow reversion to yellow, indicating the transient formation of a cyanide-induced CT complex followed by its decay. In the current study, this mechanism was supported by spectral and kinetic analyses, which demonstrate that cyanide, acting as a nucleophile, adds into the cysteine trisulfide (Figure 2). We propose that cyanide attacks at the solvent-accessible Cys-379, forming a 379Cys-S-C≡N organic thiocyanate and a 201Cys-SS’ persulfide-to-FAD CT complex (Figure 9, 2). The intense CT complex is similar to those seen with alternative nucleophiles such as sulfite or methanethiol adding to human SQOR25, 53 and also resembles the CT complex induced by coenzyme A persulfide in short-chain acyl-CoA dehydrogenase.24 The off-rate constant (koff = 3.7 ± 0.6 s−1 at 4 °C) indicates that cyanide-induced CT complex formation is reversible, and that cyanide dissociation regenerates the trisulfide. Notably, cyanide can also act as a sulfur acceptor in the SQOR reaction forming thiocyanate,14 and can thus contribute to the sulfide-mediated FAD reduction when both sulfide and cyanide are present. The ability of cyanide treated SQOR to support catalysis indicated that the oxidation status of the active site cysteines was preserved in cyanolyzed enzyme.
Figure 9.
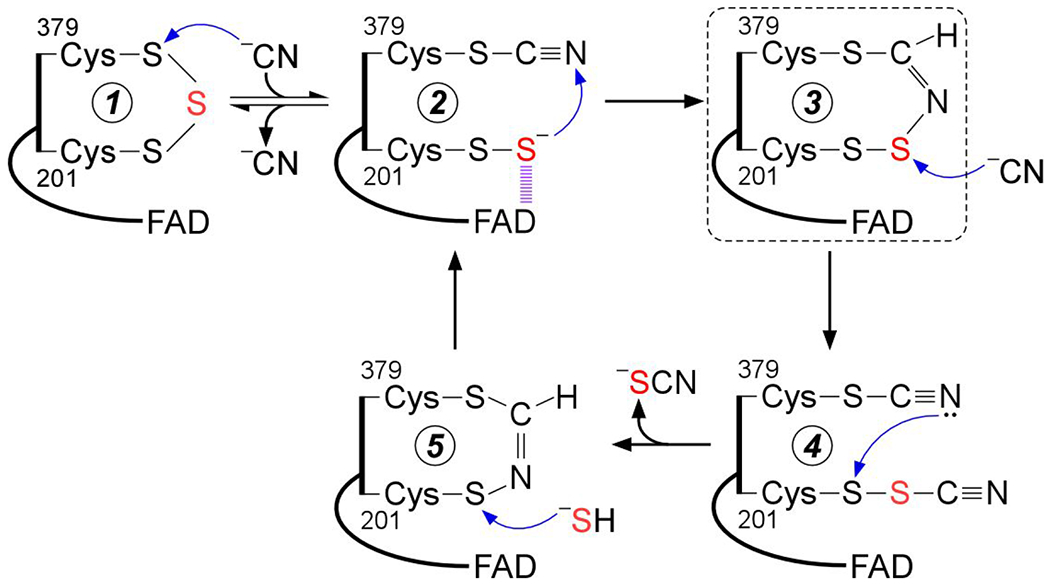
Proposed mechanism for cyanolysis and cysteine trisulfide rebuilding in SQOR. Cyanide adds into the resting cysteine trisulfide (1) to generate a 379Cys-S-CN organic thiocyanate while the bridging sulfur is retained in the 201Cys-SS− persulfide that participates in a CT complex with FAD (2). Conversion to the 379Cys N-(201Cys-disulfanyl)-methanimido thioate intennediate (3) leads to loss of the CT complex. Addition by a second cyanide at the sulfane sulfur of Cys-201 leads to intennediate (4), which can cyclize and eliminate thiocyanate (5), completing the cyanolysis reaction. Addition of sulfide to the Sγ of Cys-201 in the 379Cys N-(201Cys-sulfanyl)-methanimido thioate intennediate (5) regenerates the CT complex (2). Elimination of cyanide regenerates the resting trisulfide fonn of the enzyme. The bridging sulfur of the cysteine trisulfide is labeled in red. The dashed box highlights the intennediate observed in the crystal structure.
The crystal structure of SQOR provided a clue as to how the redox state of the cysteines is maintained upon cyanide treatment by revealing a bridging 379Cys N-(201Cys-disulfanyl)-methanimido thioate intermediate (Figure 7). We propose that this intermediate is formed by attack of the 201Cys-SS− persulfide on the nitrogen atom in 379Cys-S-C≡N thiocyanate (Figure 9, 3). While attack on the electrophilic carbon is also a feasible, the presence of the 379Cys A-(201Cys-disulfanyl)-methanimido thioate intermediate is not consistent with its formation and suggests that stereoelectronic constraints in the SQOR active site preclude this alternative pathway. While the bridging A-(disulfanyl)-methanimido thioate intermediate is stabilized in crystallo, it is susceptible to attack by a second equivalent of cyanide (Figure 9, 4), leading to thiocyanate elimination, which was detected by the cold cyanolysis reaction. The resulting 201Cys-S-N=CH-S-379Cys intermediate (Figure 9, 5) preserves the redox state of the active site cysteines. It does not however, support generation of a sulfite-induced CT complex (Figure 3C), or FAD reduction in the presence of dithiols (Figure 6C, D), and it destabilizes SQOR (Figure 5). The absorption spectrum of FAD in the presence of this intermediate is subtly different from that in the native enzyme with the 450 nm peak blue shifted to 447 nm, and the 380 nm peak being better resolved.
We propose that the trisulfide is rebuilt by the nucleophilic attack of sulfide on the Sγ of Cys-201, leading to a CT complex and then, to the resting enzyme (Figure 9, 5→2→1). We attribute the lag phase that was seen by stopped flow spectroscopy when sulfide was mixed with cyanide treated versus untreated SQOR, to the time needed to rebuild the active enzyme trisulfide (Figure 4A, B). Once rebuilt, the enzyme cycles through multiple catalytic turnovers and a difference in specific activities was not seen in cyanide treated versus untreated SQOR under steady-state assay conditions (Figure S1).
The difference in the active site configurations of untreated versus cyanide treated SQOR was further demonstrated by their differential reactivity to the dithiols DTT and DHLA. Both dithiols can substitute for sulfide in the oxidative half reaction, transferring electrons to FAD to form FADH2 (Figure 6). Notably, the bridging sulfur in the SQOR cysteine trisulfide is retained in the presence of DTT and DHLA, which contrasts with other examples of protein trisulfides57 and tetrasulfides63 that are readily reduced to cysteine dithiols. Neither DTT nor DHLA reduced FAD in cyanide pre-treated SQOR, supporting the proposed mechanism (Figure 6B).
Computational QM/MM and QM modeling provide strong evidence for the catalytic relevance of the trisulfide versus the disulfide form of the cofactor in SQOR (Figure 8). Based on accessibility, electrostatics and local softness considerations, the combination of MD simulations and QM/MM modeling predicted that Cys-379 in the trisulfide is the electrophilic target in the first step of SQOR mechanism. Based on DFT/PCM modeling of the first step in the reaction mechanism, i.e. the attack by a sulfide anion, it was estimated that the trisulfide configuration affords an ~105-fold rate enhancement over a disulfide cofactor in the active site of SQOR.
In summary, we have demonstrated that the cysteine trisulfide in human SQOR can be reversibly dismantled and reassembled. The trisulfide not only contributes to a significant rate enhancement over a disulfide for the nucleophilic addition of sulfide, but also stabilizes the enzyme. Studies are underway in our laboratory to investigate whether assembly of the trisulfide is enzyme catalyzed.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (GM130183 to R.B.). We thank Drs. David P. Ballou and Markus Ruetz for insightful discussions on the cysteine trisulfide cyanolysis and rebuilding mechanisms. MD simulations were carried out using Uruguayan supercomputer resources from ClusterUY (https://cluster.uy). Continuous support from PEDECIBA-Uruguay is gratefully acknowledged by JB and ELC who are active members of the Uruguayan National Research System (SNI-ANII).
Footnotes
Steady-state kinetic traces and supporting MD simulation data
The authors declare no competing financial interest.
REFERENCES
- 1.Kimura H, Hydrogen sulfide: from brain to gut. Antioxid. Redox Signal 2010, 12 (9), 1111–23. [DOI] [PubMed] [Google Scholar]
- 2.Kabil O; Banerjee R, Redox biochemistry of hydrogen sulfide. J. Biol. Chem 2010, 285 (29), 21903–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filipovic MR; Zivanovic J; Alvarez B; Banerjee R, Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev 2018, 118 (3), 1253–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh S; Banerjee R, PLP-dependent H(2)S biogenesis. Biochim. Biophys. Acta 2011, 1814 (11), 1518–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiku T; Padovani D; Zhu W; Singh S; Vitvitsky V; Banerjee R, H2S biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem 2009, 284 (17), 11601–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yadav PK; Yamada K; Chiku T; Koutmos M; Banerjee R, Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem 2013, 288 (27), 20002–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yadav PK; Vitvitsky V; Carballal S; Seravalli J; Banerjee R, Thioredoxin regulates human mercaptopyruvate sulfurtransferase at physiologically-relevant concentrations. J. Biol. Chem 2020, 295 (19), 6299–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furne J; Saeed A; Levitt MD, Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am. J. Physiol. Regul. Integr. Comp. Physiol 2008, 295 (5), R1479–85. [DOI] [PubMed] [Google Scholar]
- 9.Levitt MD; Abdel-Rehim MS; Furne J, Free and acid-labile hydrogen sulfide concentrations in mouse tissues: anomalously high free hydrogen sulfide in aortic tissue. Antioxid. Redox Signal 2011, 15 (2), 373–8. [DOI] [PubMed] [Google Scholar]
- 10.Vitvitsky V; Kabil O; Banerjee R, High turnover rates for hydrogen sulfide allow for rapid regulation of its tissue concentrations. Antioxid. Redox Signal 2012, 17 (1), 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouillaud F; Blachier F, Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling? Antioxid. Redox Signal 2011, 15 (2), 379–91. [DOI] [PubMed] [Google Scholar]
- 12.Hildebrandt TM; Grieshaber MK, Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008, 275 (13), 3352–61. [DOI] [PubMed] [Google Scholar]
- 13.Argyrou A; Blanchard JS, Flavoprotein disulfide reductases: advances in chemistry and function. Prog. Nucleic Acid Res. Mol. Biol 2004, 78, 89–142. [DOI] [PubMed] [Google Scholar]
- 14.Jackson MR; Melideo SL; Jorns MS, Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 2012, 51 (34), 6804–15. [DOI] [PubMed] [Google Scholar]
- 15.Libiad M; Yadav PK; Vitvitsky V; Martinov M; Banerjee R, Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem 2014, 289 (45), 30901–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landry AP; Ballou DP; Banerjee R, H2S oxidation by nanodisc-embedded human sulfide quinone oxidoreductase. J. Biol. Chem 2017, 292 (28), 11641–11649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friederich MW; Elias AF; Kuster A; Laugwitz L; Larson AA; Landry AP; Ellwood-Digel L; Mirsky DM; Dimmock D; Haven J, Pathogenic variants in SQOR encoding sulfide: quinone oxidoreductase are a potentially treatable cause of Leigh disease. J. Inherit. Metab. Dis 2020. doi: 10.1002/jimd.12232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcia M; Ermler U; Peng G; Michel H, The structure of Aquifex aeolicus sulfide:quinone oxidoreductase, a basis to understand sulfide detoxification and respiration. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (24), 9625–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brito JA; Sousa FL; Stelter M; Bandeiras TM; Vonrhein C; Teixeira M; Pereira MM; Archer M, Structural and functional insights into sulfide:quinone oxidoreductase. Biochemistry 2009, 48 (24), 5613–22. [DOI] [PubMed] [Google Scholar]
- 20.Cherney MM; Zhang Y; Solomonson M; Weiner JH; James MN, Crystal structure of sulfide:quinone oxidoreductase from Acidithiobacillus ferrooxidans: insights into sulfidotrophic respiration and detoxification. J. Mol. Biol 2010, 398 (2), 292–305. [DOI] [PubMed] [Google Scholar]
- 21.Chen ZW; Koh M; Van Driessche G; Van Beeumen JJ; Bartsch RG; Meyer TE; Cusanovich MA; Mathews FS, The structure of flavocytochrome c sulfide dehydrogenase from a purple phototrophic bacterium. Science 1994, 266 (5184), 430–2. [DOI] [PubMed] [Google Scholar]
- 22.Shen J; Peng H; Zhang Y; Trinidad JC; Giedroc DP, Staphylococcus aureus sqr Encodes a Type II Sulfide:Quinone Oxidoreductase and Impacts Reactive Sulfur Speciation in Cells. Biochemistry 2016, 55 (47), 6524–6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson MR; Loll PJ; Jorns MS, X-Ray Structure of Human Sulfide:Quinone Oxidoreductase: Insights into the Mechanism of Mitochondrial Hydrogen Sulfide Oxidation. Structure 2019, 27 (5), 794–805 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Landry AP; Moon S; Kim H; Yadav PK; Guha A; Cho US; Banerjee R, A Catalytic Trisulfide in Human Sulfide Quinone Oxidoreductase Catalyzes Coenzyme A Persulfide Synthesis and Inhibits Butyrate Oxidation. Cell Chem. Biol 2019, 26 (11), 1515–1525 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mishanina TV; Yadav PK; Ballou DP; Banerjee R, Transient Kinetic Analysis of Hydrogen Sulfide Oxidation Catalyzed by Human Sulfide Quinone Oxidoreductase. J. Biol. Chem 2015, 290 (41), 25072–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goubern M; Andriamihaja M; Nubel T; Blachier F; Bouillaud F, Sulfide, the first inorganic substrate for human cells. FASEB J. 2007, 21 (8), 1699–706. [DOI] [PubMed] [Google Scholar]
- 27.Benchoam D; Cuevasanta E; Moller MN; Alvarez B, Persulfides, at the crossroads between hydrogen sulfide and thiols. Essays Biochem. 2020, 64 (1), 155–168. [DOI] [PubMed] [Google Scholar]
- 28.Cuevasanta E; Lange M; Bonanata J; Coitino EL; Ferrer-Sueta G; Filipovic MR; Alvarez B, Reaction of Hydrogen Sulfide with Disulfide and Sulfenic Acid to Form the Strongly Nucleophilic Persulfide. J. Biol. Chem 2015, 290 (45), 26866–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Senisterra G; Chau I; Vedadi M, Thermal denaturation assays in chemical biology. Assay Drug Dev. Technol 2012, 10 (2), 128–36. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z; Minor W, Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–26. [DOI] [PubMed] [Google Scholar]
- 31.Emsley P; Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 2004, 60 (Pt 12 Pt 1), 2126–32. [DOI] [PubMed] [Google Scholar]
- 32.Murshudov GN; Vagin AA; Dodson EJ, Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr 1997, 53 (Pt 3), 240–55. [DOI] [PubMed] [Google Scholar]
- 33.Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 2010, 66 (Pt 1), 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bietz S; Urbaczek S; Schulz B; Rarey M, Protoss: a holistic approach to predict tautomers and protonation states in protein-ligand complexes. J. Cheminform 2014, 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Case DA; Cerutti DS; Cheatham III TE; Darden TA; Duke RE; Giese TJ; Gohlke H; A.W. G; Greene D; Homeyer N; Izadi S; Kovalenko A; Lee TS; LeGrand S; Li P; Lin C; Liu J; Luchko T; Luo R; Mermelstein D; Merz KM; Monard G; Nguyen H; Omelyan I; Onufriev A; Pan F; Qi R; Roe DR; Roitberg A; Sagui C; Simmerling CL; Botello-Smith WM; Swails J; Walker RC; Wang J; Wolf RM; Wu X; Xiao L; York DM; Kollman PA., AMBER 2017. University of California, San Francisco: 2017. [Google Scholar]
- 36.Bayly CI; Cieplak P; Cornell W; Kollman PA, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem 1993, 97 (40), 10269–10280. [Google Scholar]
- 37.Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG, A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103 (19), 8577–8593. [Google Scholar]
- 38.Loncharich RJ; Brooks BR; Pastor RW, Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N’-methylamide. Biopolymers 1992, 32 (5), 523–35. [DOI] [PubMed] [Google Scholar]
- 39.Åqvist J; Wennerström P; Nervall M; Bjelic S; Brandsdal BO, Molecular dynamics simulations of water and biomolecules with a Monte Carlo constant pressure algorithm. Chem. Phys. Lett 2004, 384 (4), 288–294. [Google Scholar]
- 40.Ryckaert J-P; Ciccotti G; Berendsen HJC, Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys 1977, 23 (3), 327–341. [Google Scholar]
- 41.Geerlings P; De Proft F; Langenaeker W, Conceptual density functional theory. Chem. Rev 2003, 103 (5), 1793–873. [DOI] [PubMed] [Google Scholar]
- 42.Francl MM; Pietro WJ; Hehre WJ; Binkley JS; Gordon MS; DeFrees DJ; Pople JA, Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys 1982, 77 (7), 3654–3665. [Google Scholar]
- 43.Grimme S; Antony J; Ehrlich S; Krieg H, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys 2010, 132 (15), 154104. [DOI] [PubMed] [Google Scholar]
- 44.Zhao Y; Truhlar DG, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc 2008, 120 (1-3), 215–241. [Google Scholar]
- 45.Jones RO; Gunnarsson O, The density functional formalism, its applications and prospects. Rev. Mod. Phys 1989, 61 (3), 689. [Google Scholar]
- 46.Foster a. J. P.; Weinhold F, Natural hybrid orbitals. J. Am. Chem. Soc 1980, 102 (24), 7211–7218. [Google Scholar]
- 47.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd J; Brothers EN; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam NJ; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ, Gaussian 09 Revision D.01. Gaussian, Inc., Wallingford, CT. 2013. [Google Scholar]
- 48.Bonanata J; Coitino EL, Understanding the mechanism of H2S oxidation by flavindependent sulfide oxidases: a DFT/IEF-PCM study. J. Mol. Model 2019, 25 (10), 308. [DOI] [PubMed] [Google Scholar]
- 49.Mennucci B, Polarizable continuum model. Wiley Interdiscip. Rev. Comput. Mol. Sci 2012, 2 (3), 386–404. [Google Scholar]
- 50.Bondi A. v., van der Waals volumes and radii. J. Phys. Chem 1964, 68 (3), 441–451. [Google Scholar]
- 51.Fukui K, The path of chemical reactions-the IRC approach. Acc. Chem. Res 1981, 14 (12), 363–368. [Google Scholar]
- 52.Hratchian HP; Schlegel HB, Using Hessian updating to increase the efficiency of a Hessian based predictor-corrector reaction path following method. J. Chem. Theory Comput 2005, 1 (1), 61–69. [DOI] [PubMed] [Google Scholar]
- 53.Landry AP; Ballou DP; Banerjee R, Modulation of Catalytic Promiscuity during Hydrogen Sulfide Oxidation. ACS Chem. Biol 2018, 13 (6), 1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Luebke JL; Shen J; Bruce KE; Kehl-Fie TE; Peng H; Skaar EP; Giedroc DP, The CsoR-like sulfurtransferase repressor (CstR) is a persulfide sensor in Staphylococcus aureus. Mol. Microbiol 2014, 94 (6), 1343–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shimizu T; Masuda S, Characterization of redox-active cysteine residues of persulfide-responsive transcriptional repressor SqrR. Commun. Integr. Biol 2017, 10 (4), e1329786. [Google Scholar]
- 56.Shimizu T; Shen J; Fang M; Zhang Y; Hori K; Trinidad JC; Bauer CE; Giedroc DP; Masuda S, Sulfide-responsive transcriptional repressor SqrR functions as a master regulator of sulfide-dependent photosynthesis. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (9), 2355–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jespersen AM; Christensen T; Klausen NK; Nielsen F; Sorensen HH, Characterisation of a trisulphide derivative of biosynthetic human growth hormone produced in Escherichia coli. Eur. J. Biochem 1994, 219 (1–2), 365–73. [DOI] [PubMed] [Google Scholar]
- 58.Thomsen MK; Hansen BS; Nilsson P; Nowak J; Johansen PB; Thomsen PD; Christiansen J, Pharmacological characterization of a biosynthetic trisulfide-containing hydrophobic derivative of human growth hormone: comparison with standard 22 K growth hormone. Pharmacol. Toxicol 1994, 74 (6), 351–8. [DOI] [PubMed] [Google Scholar]
- 59.Andersson C; Edlund PO; Gellerfors P; Hansson Y; Holmberg E; Hult C; Johansson S; Kordel J; Lundin R; Mendel-Hartvig IB; Noren B; Wehler T; Widmalm G; Ohman J, Isolation and characterization of a trisulfide variant of recombinant human growth hormone formed during expression in Escherichia coli. Int. J. Pept. Protein Res 1996, 47 (4), 311–21. [DOI] [PubMed] [Google Scholar]
- 60.Canova-Davis E; Baldonado IP; Chloupek RC; Ling VT; Gehant R; Olson K; Gillece-Castro BL, Confirmation by mass spectrometry of a trisulfide variant in methionyl human growth hormone biosynthesized in Escherichia coli. Anal Chem 1996, 68 (22), 4044–51. [DOI] [PubMed] [Google Scholar]
- 61.Santos AA; Venceslau SS; Grein F; Leavitt WD; Dahl C; Johnston DT; Pereira IA, A protein trisulfide couples dissimilatory sulfate reduction to energy conservation. Science 2015, 350 (6267), 1541–5. [DOI] [PubMed] [Google Scholar]
- 62.Wood JL, Sulfane sulfur. Methods Enzymol. 1987, 143, 25–9. [DOI] [PubMed] [Google Scholar]
- 63.Kofler S; Ackaert C; Samonig M; Asam C; Briza P; Horejs-Hoeck J; Cabrele C; Ferreira F; Duschl A; Huber C, Stabilization of the dimeric birch pollen allergen Bet v 1 impacts its immunological properties. J. Biol. Chem 2014, 289 (1), 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.