

Abstract
The cytosolic class IIb histone deacetylase HDAC10 is an emerging target for drug design. As an inducer of autophagy, its selective inhibition suppresses the autophagic response that otherwise attenuates the efficacy of cytotoxic cancer chemotherapy drugs. HDAC10 is a zinc-dependent polyamine deacetylase exhibiting maximal catalytic activity against N8-acetylspermidine. As revealed in the structure of Danio rerio (zebrafish) HDAC10, two conserved structural motifs direct this narrow substrate specificity: a 310 helix containing the P(E,A)CE motif that sterically constricts the active site, and an electrostatic “gatekeeper”, E274, that confers selectivity for cationic polyamine substrates. To accelerate drug design efforts targeting human HDAC10, we now report the preparation of “humanized” zebrafish HDAC10 in which two amino acid substitutions, A24E and D94A, yield an active site contour more similar to that of human HDAC10. X-ray crystal structures of this HDAC10 variant complexed with Tubastatin A and indole analogues bearing pendant tertiary amines reveal that inhibitors capable of hydrogen bonding with gatekeeper E274 exhibit high affinity and selectivity for HDAC10 over HDAC6 (the other class IIb isozyme). Moreover, these structures reveal that the P(E,A)CE motif helix can shift by up to 2 Å to accommodate the binding of bulky inhibitors. Thus, slender polyamine-like inhibitor structures are not exclusively required for selective, high affinity binding to HDAC10. Indeed, the flexibility of the P(E,A)CE motif helix could conceivably enable the binding of certain protein substrates.
Graphical Abstract

Among the 11 metal-dependent histone deacetylases (HDACs),1–3 the class IIb isozyme HDAC10 is perhaps the most intriguing in terms of its structure and catalytic function.4 The recently determined X-ray crystal structure of HDAC10 from Danio rerio (zebrafish)5 reveals that the catalytic domain adopts the characteristic arginase-deacetylase fold,6–9 but it is paired with a smaller, catalytically-inactive domain of identical topology, referred to as the pseudo-deacetylase domain (Figure 1a). Additionally, HDAC10 is a polyamine deacetylase: both zebrafish and human HDAC10 enzymes exhibit poor catalytic activity against acetyl-L-lysine peptide substrates in vitro, instead exhibiting optimal catalytic activity against N8-acetylspermidine, acetylputrescine, and acetylcadaverine, but not N1-acetylspermidine.5 Given the enrichment of HDAC10 in the cytosol of eukaryotic cells,10 HDAC10 is likely responsible for the N8-acetylspermidine hydrolase activity first observed in mammalian cells more than 40 years ago.11–14
Figure 1.
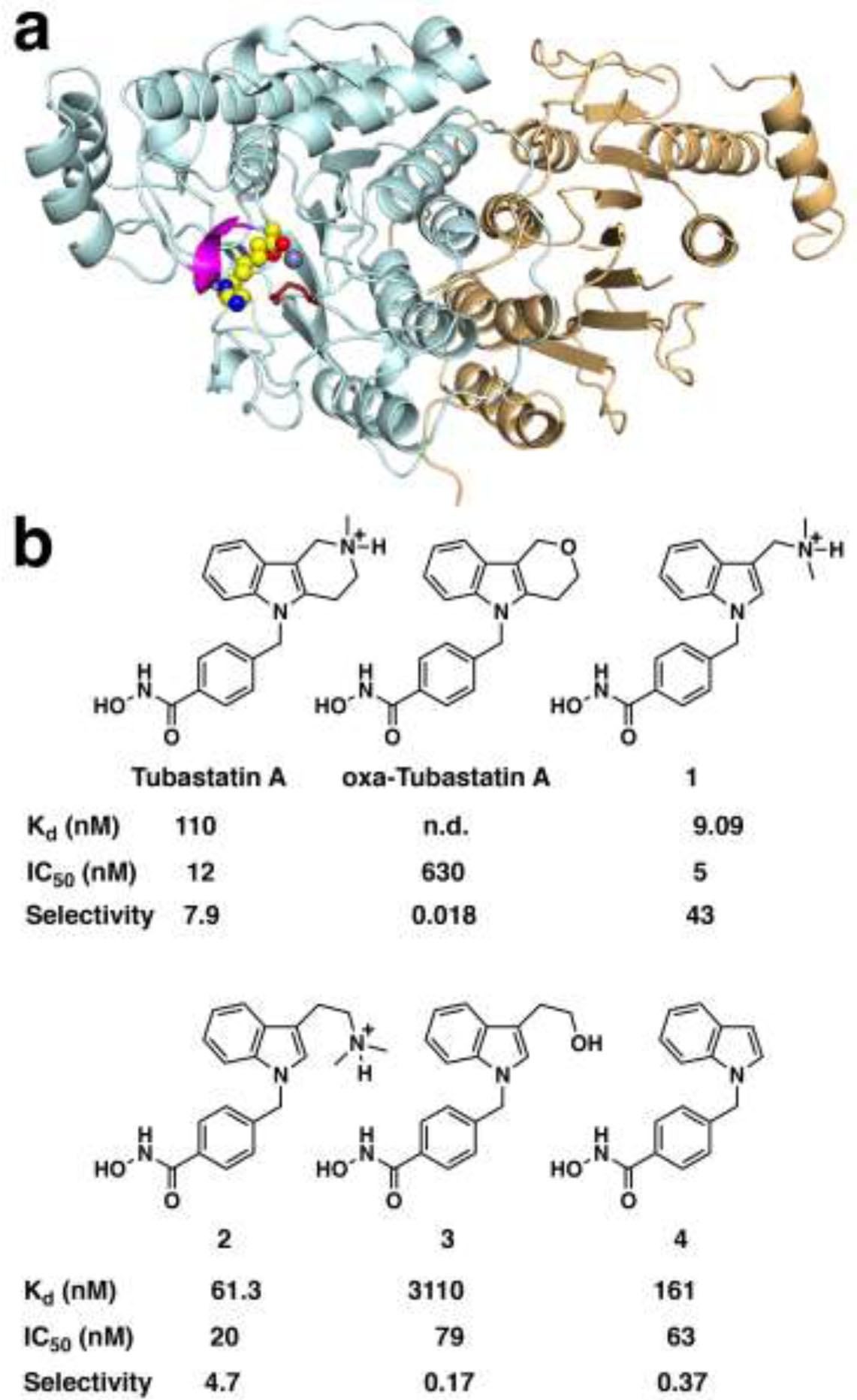
(a) Ribbonplot of HDAC10 showing the catalytically active deacetylase domain (cyan) with a bound analogue of substrate N8-acetylspermidine (CPK model, C = yellow, N = blue, O = red) coordinating to the active site Zn2+ ion (gray sphere). The “PEACE” motif (P23(E,A)CE26, magenta) is a 310 helix unique to HDAC10 that sterically constricts the approach to the active site so as to favor the binding of long, slender polyamine substrates. Opposite the PEACE motif is E274 (red stick figure), which provides electrostatic complementarity to cationic polyamine substrates. The catalytically inactive pseudo-deacetylase domain is shown in tan. (b) HDAC10 inhibitors Tubastatin A and recently reported analogues. Dissociation constants (Kd) were measured here by isothermal titration calorimetry using “humanized” A24E-D94A zebrafish HDAC10 (n.d., not determined). Human HDAC10 IC50 values and HDAC10/HDAC6 selectivities were previously reported.16 The tertiary amino groups of Tubastatin A, inhibitor 1, and inhibitor 2 are drawn in their protonated forms as they would exist at physiological pH.
Analysis of the HDAC10 structure reveals two principal features unique to this isozyme that account for polyamine substrate specificity:5 molecular gatekeeper E274 (zebrafish HDAC10 numbering), which establishes electrostatic complementarity for cationic polyamine substrates, and a 310 helix with consensus sequence P23(E,A)CE26 (the “PEACE” motif) that sterically constricts the active site so as to favor the binding of long, slender polyamines (Figure 1a). The E274L substitution diminishes N8-acetylspermidine deacetylase activity 20-fold and increases acetyl-L-lysine deacetylase activity 100-fold; deletion of the PEACE motif diminishes N8-acetylspermidine deacetylase activity 15-fold and increases acetyl-L-lysine deacetylase activity 16-fold.5
Given that high HDAC10 expression levels correlate with poor survival in children diagnosed with late-stage neuroblastoma, HDAC10 is emerging as a possible target for cancer chemotherapy.15 As an inducer of autophagy, HDAC10 protects cancer cells from the cytotoxic chemotherapy drug doxorubicin; however, knock-down or specific inhibition of HDAC10 renders cancer cells more susceptible to doxorubicin.15,16 Inhibition of HDAC10 also impedes drug efflux mechanisms and induces double-stranded breaks in neuroblastoma cells but not non-malignant cells.17 Since the polyamine spermidine is also implicated as an inducer of autophagy,18,19 HDAC10 may represent a link between polyamine metabolism and autophagic cellular survival mechanisms.
Despite the emerging importance of HDAC10 as a therapeutic target, the exploration of isozyme-selective inhibition has received relatively little attention until recently. Surprisingly, Tubastatin A, designed to selectively inhibit HDAC6, the other class IIb isozyme,20 is more selective for the inhibition of HDAC10 by nearly 8-fold.16 Tubastatin A is sterically bulky in comparison with the slender polyamine substrates that are more complementary in size and shape to the narrow active site cleft of HDAC10, so the potency of Tubastatin A is surprising and suggests that conformational changes of the PEACE motif helix may be required to enable binding. Using this discovery as a starting point, Miller and colleagues recently reported a new class of bicyclic Tubastatin A analogues, some of which exhibit nanomolar inhibitory potencies and up to 43-fold selectivity for HDAC10.16 These investigators also determined that HDAC10/HDAC6 inhibitor selectivity profiles measured in vitro correlate with in vivo measurements made in a neuroblastoma cell line.
Here, we demonstrate the functional importance of the tertiary amino group of Tubastatin A by comparing its binding with an oxa analogue, and we present the X-ray crystal structure of a “humanized” form of zebrafish HDAC10 complexed with Tubastatin A and bicyclic Tubastatin A analogues 1–4 (Figure 1b). With their catalytic domains related by 63% amino acid sequence identity and 79% similarity, the amino acid compositions of the zebrafish and human HDAC10 active sites are essentially identical except for A24 and D94 in zebrafish HDAC10, which appear as glutamate and alanine, respectively, in human HDAC10. The active site of A24E-D94A zebrafish HDAC10 thus mimics the active site of human HDAC10, the actual drug target. Interestingly, crystals of A24E-D94A zebrafish HDAC10 (henceforth simply “HDAC10”) generally diffract X-rays to higher resolution than crystals of the wild-type enzyme. These structures reveal the electrostatic contribution of gatekeeper residue E274 as well as the steric contribution of the humanized PEACE motif helix in the binding of selective inhibitors.
RESULTS AND DISCUSSION
Thermal Shift Measurements.
We first aimed to confirm the reported16 52-fold difference in HDAC10 binding affinity between Tubastatin A and its oxa analogue with an independent biophysical technique. In oxa-Tubastatin A, the tertiary amino group is replaced by an oxygen atom to form a tetrahydropyranoindole ring. Differential scanning fluorimetry21 was used to measure protein stability of recombinant human HDAC10, expressed as a fusion protein with an N-terminal TwinStrep tag (TwinStrep-HDAC10). The TwinStrep-HDAC10 protein (3 μM) had a melting temperature (Tm) of 65.77 °C when measured alone and a Tm of 65.90 °C in the presence of 0.6% DMSO (Figure 2). When incubated with Tubastatin A (6 μM), a Tm of 72.94 ± 0.08 °C was measured, a thermal shift of 7.04 °C. This is consistent with strong binding of Tubastatin A to HDAC10, which can be expected at the concentrations tested. Incubation of TwinStrep-HDAC10 (3 μM) with oxa-Tubastatin A (6 μM) resulted in a Tm of 67.27 ± 0.01 °C, a thermal shift of 1.37 °C. The much smaller thermal shift observed with oxa-Tubastatin A in comparison with that measured for Tubastatin A indicates that substitution of the tertiary amino group of Tubastatin A with an oxygen atom substantially compromises HDAC10 binding affinity. Due to its poor affinity, we did not pursue the crystal structure determination of the oxa analogue in complex with HDAC10, but instead focused on the structure determination of the HDAC10–Tubastatin A complex.
Figure 2.
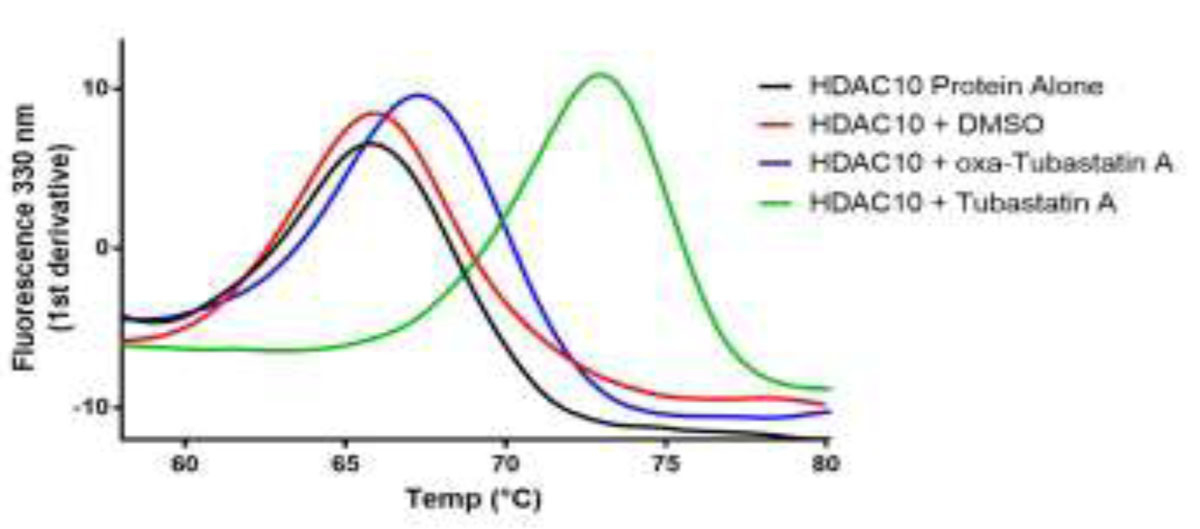
Measurements using recombinant TwinStrep-HDAC10 fusion protein alone (black), with DMSO (red), Tubastatin A (green), and oxa-Tubastatin A (blue). Protein was tested at 3 μM and inhibitors at 6 μM, giving a protein:inhibitor ratio of 1:2. For the two control experiments (black and red), n = 1. For the inhibitor experiments (blue and green), n = 2; the data shown represent the mean of the two experiments.
Isothermal Titration Calorimetry (ITC).
Because previously reported inhibitory potencies were measured using human HDAC10 and the crystal structures reported herein utilized humanized A24E-D94A zebrafish HDAC10, we sought to confirm inhibitor binding affinity trends to the humanized zebrafish enzyme using ITC. Dissociation constants (Kd) are recorded in Figure 1 and individual ITC enthalpograms are found in Figure S1. Inhibitory potency trends observed with human HDAC10 generally correlate with inhibitor affinity trends measured with the humanized zebrafish enzyme used for X-ray crystallographic structure determinations. Compound 1 is the most potent inhibitor with IC50 = 5 nM and is the tightest binding inhibitor with Kd = 9.09 nM.
Structure of the HDAC10–Tubastatin A complex.
The crystal structure of the HDAC10–Tubastatin A complex was determined at 2.00 Å resolution. The overall HDAC10 protein structure is quite similar to that of wild-type HDAC10 complexed with a trifluoromethylketone inhibitor determined at 2.85 Å resolution (PDB 6UIL; root-mean-squared (rms) deviation = 0.32 Å for 524 Cα atoms).22 The hydroxamate moiety of Tubastatin A chelates the catalytic Zn2+ ion with C=O---Zn2+ and N–O–---Zn2+ separations of 2.1 Å each (Figure 3a). The hydroxamate carbonyl accepts a hydrogen bond from Y307, the hydroxamate NH group donates a hydrogen bond to H137, and the hydroxamate N–O– group accepts a hydrogen bond from H136. This constellation of hydrogen bond interactions suggests that H136 is the positively charged imidazolium cation, and the side chain of H137 is the neutral imidazole.
Figure 3.
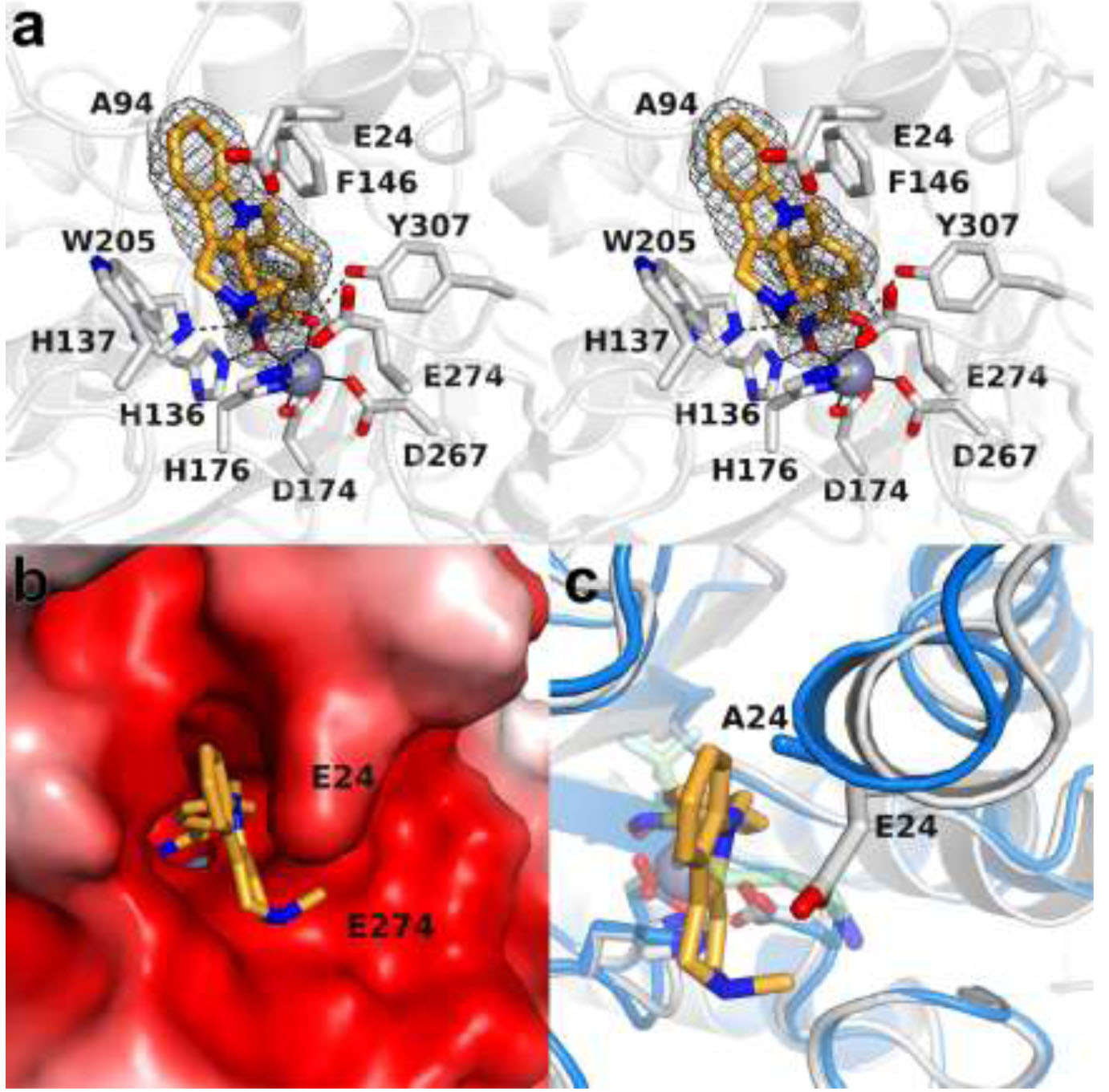
(a) Stereoview of a Polder omit map of Tubastatin A bound in the active site of HDAC10 (contoured at 5σ). Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. (b) Active site surface of HDAC10 color-coded by electrostatic potential (−5kT (red) to +5kT (blue)). The side chain of E24 in the humanized PEACE motif plays an important role in accommodating the tricyclic tetrahydro-γ-carboline capping group. (c) Superposition of the HDAC10–Tubastatin A complex (gray) and the HDAC10–AAT complex (blue) reveals the significant shift (up to 1.9 Å) of the PEACE motif helix containing E24/A24 that accommodates the bulky capping group of Tubastatin A.
The phenyl group of the phenylhydroxamate moiety nestles in an aromatic crevice defined by F146 and W205, where it makes favorable offset π−π stacking interactions. This aromatic crevice is conserved in HDAC6, where it is similarly occupied by the aromatic rings of phenylhydroxamate inhibitors.23–26 This interaction presumably contributes to inhibitor potency but not inhibitor selectivity, given the general conservation of this crevice among HDAC isozymes.
The tricyclic tetrahydro-γ-carboline capping group packs against the indole side chain of W205, also making favorable offset π−π stacking interactions. The tetrahydro-γ-carboline ring also packs against E24, the “humanized” residue in the PEACE motif that sterically constricts the active site. While neither E24 nor E274 forms hydrogen bonds with the tertiary amino group of the Tubastatin A capping group, these residues establish a complementary electrostatic environment that favors the binding of the protonated form of this amino group. The electrostatic interaction between the tertiary amine of Tubastatin A and E274 appears to pull E274 close enough to the interior of the active site allowing for a direct hydrogen bond to zinc ligand H176. Taken together, W205, E24, and E274 seem to be perfectly situated to clamp the tetrahydro-γ-carboline capping group in place in a cavity that is complementary in shape, size, and electrostatics to the tricyclic ring system (Figure 3b).
Conformational changes are required in the HDAC10 active site to accommodate the binding of the bulky tetrahydro-γ-carboline capping group of Tubastatin A. Specifically, the PEACE motif shifts by nearly 2 Å to lock the methylpiperidine ring in place (Figure 3c). Ordinarily, the PEACE motif serves as a steric block to favor the binding of long, slender polyamine substrates, but the structure of this enzyme-inhibitor complex reveals that this helix can shift to accommodate a bulky inhibitor as long as steric complementarity can be achieved.
It is interesting that the N-methyl group of the tetrahydro-γ-carboline moiety adopts an axial conformation, similar to that observed in the HDAC6–Tubastatin A complex (Figure 4).27 While an equatorial conformation for the N-methyl moiety is thought to be more energetically favorable, destabilizing 1,3-diaxial interactions of the axial conformation are minimized due to the unsaturation of the tetrahydro-γ-carboline ring system. Furthermore, the axial conformation of the N-methyl group in N-methylpiperidinium hydrochlorides is destabilized by only 1.4–2.2 kcal/mol relative to the equatorial conformation.28 Thus, an axial N-methyl conformation in the tetrahydro-γ-carboline moiety is thermodynamically accessible, especially if an equatorial N-methyl conformation would make unfavorable steric interactions in the active site.
Figure 4.

(a) Stereoview of Tubastatin A complexed with catalytic domain 2 of HDAC6 (PDB 6THV). Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. (b) Stereoview of Tubastatin A complexed with HDAC10 oriented in identical fashion to HDAC6 in plate (a). Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively.
Other features of Tubastatin A binding differ between HDAC6 and HDAC10 (Figure 4). Although Tubastatin A was originally determined to be an HDAC6-selective inhibitor,20 more recent studies indicate that Tubastatin A is nearly 8-fold more selective for inhibition of HDAC10 compared to HDAC6.16,29 Hydroxamate-Zn2+ coordination is monodentate in HDAC6 but bidentate in HDAC10, and the conformation of the tetrahydro-γ-carboline capping group is substantially different in HDAC10 due to packing interactions with E24 in the PEACE motif and the polypeptide segment containing A94 (recall that these residues were engineered to make the active site of zebrafish HDAC6 more like that of human HDAC6). The active site of HDAC6 is not as sterically constricted, so here the tetrahydro-γ-carboline capping group of the inhibitor makes van der Waals contacts with just F643 and L712 in the L1 pocket and is otherwise exposed to solvent. These structural features – energetically more favorable bidentate hydroxamate-Zn2+ coordination and more extensive packing interactions of the tetrahydro-γ-carboline capping group – likely account for improved inhibitory potency of Tubastatin A against HDAC10 compared with HDAC6.
Structure of the HDAC10–1 complex.
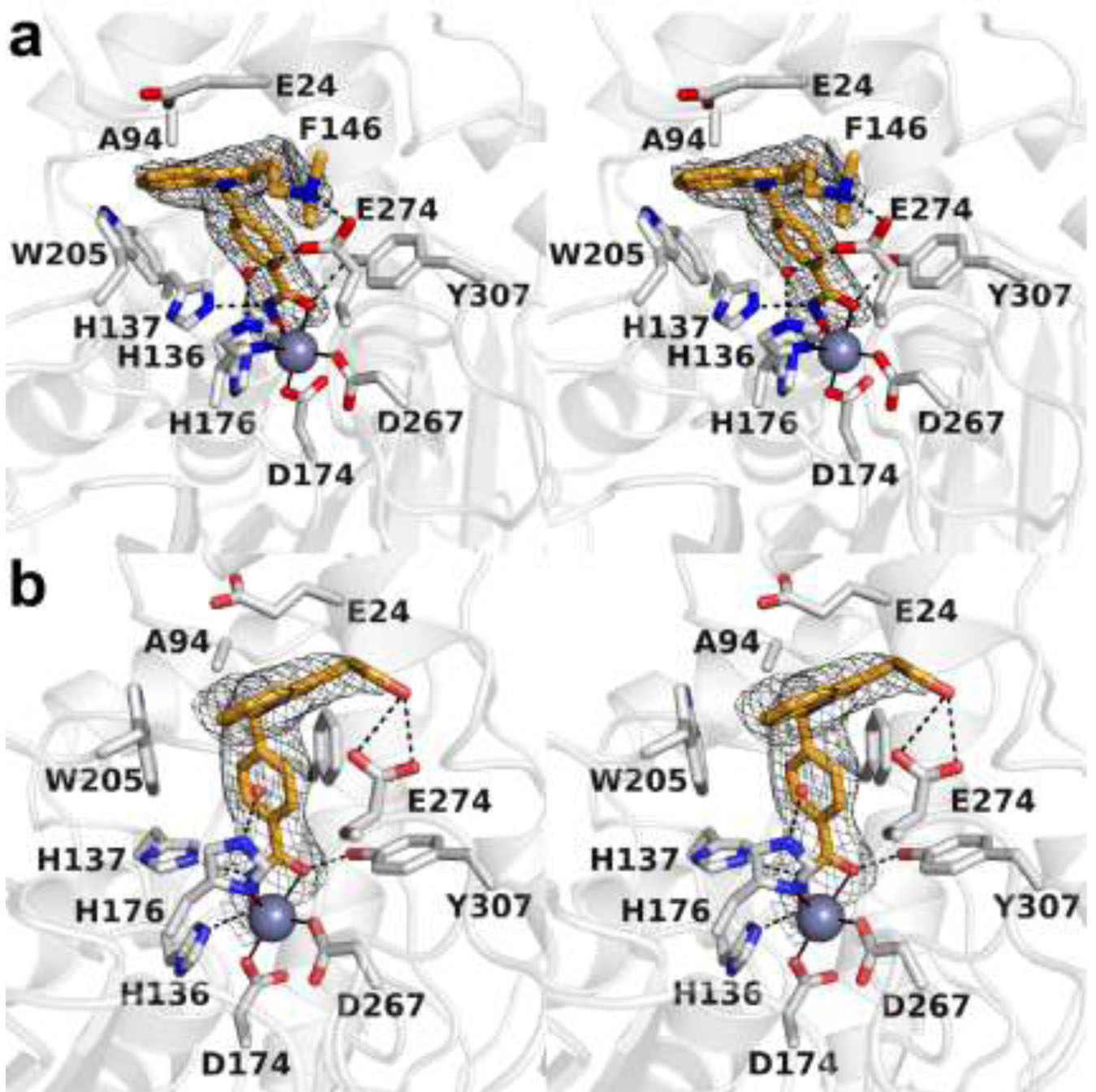
The dimethylaminomethyl group of inhibitor 1 is isosteric with the amino group of Tubastatin A, but it is not conformationally constrained within a 6-membered ring. This simplification of the Tubastatin A structure results in a 2.4-fold enhancement of inhibitory potency against HDAC10 and a 5.4-fold enhancement of selectivity against HDAC10 compared with HDAC6.16 The 2.40 Å-resolution crystal structure of the HDAC10–1 complex reveals generally similar enzyme-inhibitor interactions: the inhibitor hydroxamate group chelates the catalytic Zn2+ ion with a C=O---Zn2+ separation of 2.5 Å and a N–O–---Zn2+ separation of 2.0 Å; the hydroxamate carbonyl accepts a hydrogen bond from Y307, the hydroxamate NH group donates a hydrogen bond to H137, and the hydroxamate N–O– group accepts a hydrogen bond from H136. Additionally, the phenylhydroxamate moiety engages in favorable offset π–π interactions with F146 and W205 (Figure 5a). Notably, a water molecule forms hydrogen bonds with E274 and zinc ligand H176.
Figure 5.

(a) Stereoview of polder omit maps of inhibitor 1 bound in the active site of HDAC10 (contoured at 5.5σ). Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. (b) Stereoview of the superposition of the HDAC10–1 complex (colored as in plate a) and the HDAC10–Tubastatin A complex (protein = blue, inhibitor = mauve). Note the conformational changes of active site gatekeeper E274 and PEACE motif residue E24 that accompany the binding of each inhibitor.
In contrast with the binding of Tubastatin A, the capping group of 1 is oriented away from the PEACE motif and is roughly perpendicular to W205 (Figure 5b). The dimethylaminomethyl moiety is presumably protonated and donates a salt linked hydrogen bond to the carboxylate side chain of the HDAC10 gatekeeper, E274. This hydrogen bond has excellent geometry, with the tertiary amino nitrogen close to the plane of the carboxylate and a N---O separation of 2.8 Å. The corresponding hydrogen bond cannot be formed in the HDAC10–Tubastatin A complex due to the conformational constraints of the N-methyl tetrahydro-γ-carboline ring system. Additionally, the PEACE motif helix does not shift to the extent observed in the HDAC10–Tubastatin A complex to accommodate the binding conformation of the inhibitor. Since E274 is unique to HDAC10 among all HDAC isozymes, an enzyme-inhibitor hydrogen bond involving this residue makes a critical contribution to the enhanced inhibitory potency and selectivity of 1 for binding to HDAC10 compared with Tubastatin A.
Structure of the HDAC10–2 complex.
The pendant dimethylaminoethyl substituent of the indole capping group of inhibitor 2 is just one methylene group longer than the pendant dimethylaminomethyl substituent of the indole capping group of inhibitor 1. However, inhibitor 2 exhibits decreased potency and selectivity for HDAC10 compared with Tubastatin A.
The 2.20 Å-resolution crystal structure of the HDAC10–2 complex reveals a single conformer (Figure 6a) in which the indole moiety is oriented away from the PEACE motif in similar fashion to that observed in the HDAC10–1 complex. This orientation positions the protonated dimethylaminoethyl moiety to donate a salt linked hydrogen bond to E274. However, due to the longer pendant chain containing the tertiary amino group, this hydrogen bond is less well-oriented – although the N---O separation is 2.7 Å, the tertiary amino nitrogen lies well out of the plane of the carboxylate. This feature may contribute to the compromised affinity and selectivity of inhibitor 2 relative to inhibitor 1.
Figure 6.
(a) Stereoview of a Polder omit map of inhibitor 2 bound in the active site of HDAC10 (contoured at 5σ). (b) Stereoview of a Polder omit map of inhibitor 3 bound in the active site of HDAC10 (contoured at 4σ). In each plate, metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively.
Other aspects of inhibitor binding are generally similar to those observed in the HDAC10–1 complex, including hydroxamate-Zn2+ coordination geometry, hydroxamate-protein hydrogen bonds, and the binding of the phenylhydroxamate moiety in the aromatic crevice defined by F146 and W205. Additionally, a water molecule forms a hydrogen bond between E274 and zinc ligand H176, as observed in the HDAC10–1 complex. Finally, the inhibitor is accommodated in the active site without a significant shift of the PEACE motif helix.
Structure of the HDAC10–3 complex.
The pendant hydroxyethyl substituent of the indole capping group of inhibitor 3 is isosteric with the pendant dimethylaminoethyl group of inhibitor 2. However, the inhibitory properties of inhibitor 3 are substantially compromised in comparison with its tertiary amino analogue. Compromised affinity and selectivity for inhibitor 3 may arise from the weaker dipole-charge hydrogen bond interaction between its hydroxyl group and E274 compared with the stronger charge-charge hydrogen bond interactions between E274 and the dimethylammonium moieties of inhibitors 1 and 2.
The 2.65 Å-resolution crystal structure of the HDAC10–3 complex reveals a similar orientation of the indole capping group to that observed for inhibitors 1 and 2, allowing for the pendant hydroxyethyl moiety to make a bifurcated hydrogen bond with E274. However, weak electron density for the hydroxyl group suggests some degree of conformational disorder (Figure 6b).
The hydroxamate-zinc coordination geometry of inhibitor 3 is bidentate, as observed for inhibitors 1 and 2. The hydroxamate C=O and N–O– moieties chelate the catalytic Zn2+ ion with interatomic separations of 2.4 Å and 2.1 Å, respectively. The aromatic ring of the phenylhydroxamate moiety of inhibitor 3 binds in the aromatic crevice defined by F146 and W205, as observed for other phenylhydroxamate derivatives. In contrast with inhibitors 1 and 2, the water molecule hydrogen bonded to H176 does not hydrogen bond with E274 due to the more favorably oriented hydrogen bond between E274 and the hydroxyl group of the inhibitor. It appears that the E274–H2O–H176 hydrogen bond network helps to stabilize active site side chain conformations; disruption of this network correlates with compromised affinity and selectivity. Finally, the inhibitor is accommodated in the active site without a significant shift of the PEACE motif helix.
Structure of the HDAC10–4 complex.
Inhibitor 4 contains only an indole capping group. Its compromised inhibitory properties highlight the importance of the tertiary amino groups of Tubastatin A, inhibitor 1, and inhibitor 2 for hydrogen bond and/or electrostatic interactions with E274. The modest increase in potency and selectivity of inhibitor 4 over inhibitor 3 may arise from the mode by which the PEACE motif clamps the indole capping group of 4, highlighting the importance of this function in inhibitor design. The 2.65 Å-resolution crystal structure of the HDAC10–4 complex (Figure 7a) reveals that the indole capping group is flipped by approximately 180° compared to the indole moiety of Tubastatin A in its complex with HDAC10. This reorientation appears to result from the absence of a pendant tertiary amino group that would otherwise interact with E274 (Figure 7b). Furthermore, and in contrast with the structure of the HDAC10–Tubastatin A complex, a water molecule makes bridging hydrogen bonds between E274 and zinc ligand H176.
Figure 7.
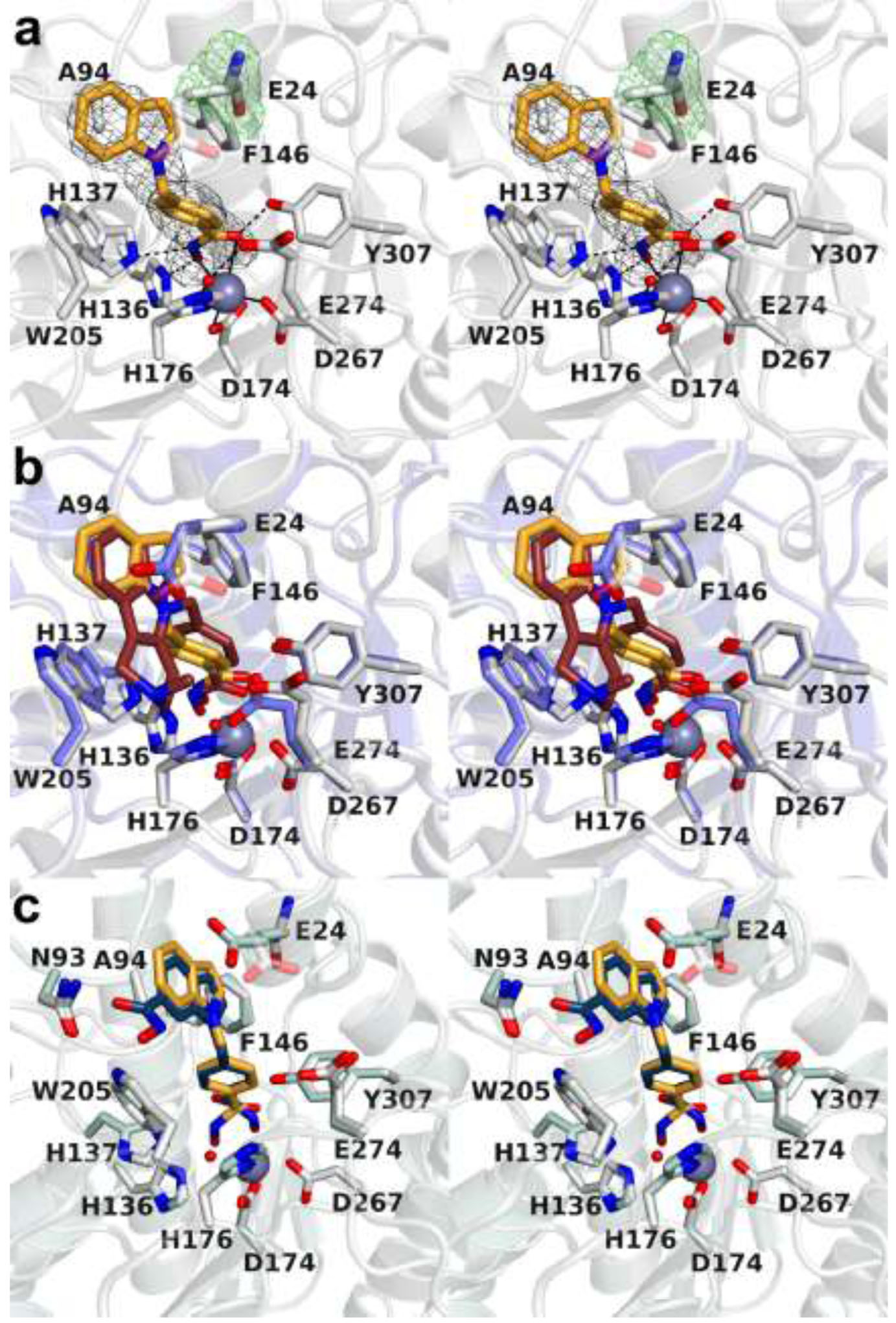
(a) Stereoview of Polder omit maps of inhibitor 4 and E24 in the active site of HDAC10 (contoured at 5.5σ). Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. Electron density for the E24 side chain is weak or absent, indicative of conformational disorder. (b) Stereoview of the superposition of the HDAC10–4 complex (colored as in plate a; a modeled conformation of the disordered E24 side chain is shown for reference as a semi-transparent image) and the HDAC10–Tubastatin A complex (protein = blue, inhibitor = mauve). Note the conformational change of active site gatekeeper E274 that accompanies the binding of each inhibitor. (c) Stereoview of the superposition of the HDAC10–4 complex (colored as in plates a and b) and the recently reported structure of the HDAC10 complex with a bis-hydroxamate derivative.29
Similar to the HDAC10–Tubastatin A complex, a substantial shift in the PEACE motif accommodates the binding of inhibitor 4. Interestingly, electron density for the side chain of E24 in the PEACE motif is weak or absent, suggesting disorder for this residue (Figure 7a); accordingly, the E24 side chain is not modeled. It is conceivable that this is a consequence of the lack of a hydrogen bonding partner with the bound inhibitor. Hydroxamate-Zn2+ coordination geometry is comparable to that observed in the HDAC10–1 complex. The hydroxamate C=O---Zn2+ separation is 2.8 Å, which is too long to be considered inner sphere coordination; however, given the 2.65 Å resolution of the HDAC10–4 complex, weak coordination of the hydroxamate C=O moiety is feasible within the coordinate error of a structure determined at this resolution. The hydroxamate N–O– moiety also coordinates to Zn2+ with a N–O–---Zn2+ separation of 2.4 Å. The hydroxamate C=O group accepts a strong hydrogen bond from Y307, the hydroxamate N–O– group accepts a hydrogen bond from H136, and the hydroxamate NH group donates a hydrogen bond to H137.
Structural determinants of inhibitor affinity and selectivity.
The initial X-ray crystal structure determination of HDAC10 revealed that the active site is sterically constricted so as to favor the binding of long, slender substrates and inhibitors.5 Structural features constricting the HDAC10 active site are distinct from those constricting the active sites of bacterial polyamine deacetylases.4,30 Accordingly, the structural determinants of HDAC10–inhibitor affinity have been explored for various analogues of the slender polyamine substrate N8-acetylspermidine.21 In these analogues, the N8-acetylspermidine-like core structure is held constant while the zinc-binding group is varied. Among the different metal-binding groups studied in these complexes, the hydroxamate moiety coordinates to the catalytic Zn2+ ion with bidentate geometry, forming a 5-membered ring chelate.
In the current study, the hydroxamate zinc-binding group is the common feature of Tubastatin A and inhibitors 1–4, and the bulky capping group is varied to probe interactions with the PEACE motif helix that sterically constricts the active site. Given the nanomolar inhibitory potency of these inhibitors, it is clear that effective HDAC10 inhibition does not always require a slender inhibitor structure. Crystal structures of complexes with Tubastatin A and inhibitors 1–4 conclusively demonstrate that a bulky inhibitor capping group can be accommodated in the HDAC10 active site if it is complementary in shape to the PEACE motif helix, which can shift by up to 2 Å to accommodate inhibitor binding. Of note, the PEACE motif helix is observed to shift substantially only in the complexes with Tubastatin A and inhibitor 4.
Inhibitor 4 is similar in structure to a recently reported dihydroxamate derivative that contains an additional hydroxamate substituent at the C6 position of the indole capping group.29 In contrast to inhibitor 4, the dihydroxamate inhibitor binds with two alternate hydroxamate-Zn2+ coordination modes;23,24 the major conformer is the monodentate coordination mode (Figure 7c). Even so, the phenyl group of the phenylhydroxamate moiety and the indole capping group of each inhibitor bind with similar conformations regardless of whether the indole hydroxamate substituent is present or absent. Interestingly, though, the indole hydroxamate forms a hydrogen bond with N93. Formation of this hydrogen bond could conceivably influence the equilibrium between bidentate and monodentate hydroxamate-Zn2+ coordination. The dihydroxamate inhibitor is more selective for HDAC10 as compared to inhibitor 4, so the hydrogen bond between the indole hydroxamate and N93 may contribute to increased selectivity.
Interactions with the active site gatekeeper, E274, were proposed to confer HDAC10 selectivity in this series of inhibitors,16 and the array of structures reported herein verify this proposal. HDAC10 complexes with Tubastatin A and inhibitors 1–3 reveal electrostatic and/or hydrogen bond interactions between the tertiary amino moieties in their respective capping groups and E274. Inhibitor 4 lacks a pendant amino moiety in its capping group and hence cannot interact with E274, leading to compromised inhibitory potency.
Importantly, E24 in the humanized zebrafish HDAC10 PEACE motif and A94 play an important role in defining the active site contour and directing the binding conformation of inhibitor capping groups, for example, as observed in the HDAC10–Tubastatin A complex (Figures 3 and 4). Both of these residues correspond to the “humanizing” mutations introduced so that the active site of zebrafish HDAC10 would better mimic that of human HDAC10. All other active site residues interacting with Tubastatin A are conserved between zebrafish HDAC10 and human HDAC10. Thus, the humanized zebrafish enzyme presented herein will serve as a useful and readily studied surrogate of the human enzyme for X-ray crystallographic studies.
Of the five compounds structurally characterized in the current study, inhibitor 1 exhibits the highest affinity and selectivity for HDAC10. The pendant tertiary amino moiety in 1 forms a direct hydrogen bond with gatekeeper residue E274, thereby contributing to high affinity and selectivity. The capping group of inhibitor 2 also allows for a hydrogen bond between the pendant tertiary amino moiety and gatekeeper E274. However, inhibitor 2 exhibits compromised affinity and selectivity. Structural features contributing to activity differences between inhibitors 1 and 2 are the geometry of the hydrogen bond between E274 and the tertiary amino group of the inhibitor, and the apparent strength of the E274–H2O–H176 hydrogen bond network.
The imidazole side chain of H176 donates a hydrogen bond to the bridging water molecule in the E274–H2O–H176 network. Due to zinc coordination, the proton on H176 must have a lower pKa and thus be a stronger hydrogen bond donor, which in turn strengthens the hydrogen bond network. Shorter and stronger hydrogen bonds are observed in the HDAC10–1 complex, with an exceptionally short separation of 2.3 Å for the E274–H2O hydrogen bond and 2.6 Å for the H2O–H176 hydrogen bond. In contrast, longer hydrogen bonds are observed for this network in the structure of the HDAC10–2 complex (2.6 Å and 3.0 Å, respectively). The lengths of these hydrogen bonds seem to correlate with the location of the pendant dimethylammonium moiety, since the shorter methylene linker of inhibitor 1 allows the E274 carboxylate to make a closer hydrogen bond contact with the bridging water molecule in comparison with the longer ethylene linker of inhibitor 2.
The role of E274 as a structural determinant of inhibitory potency and selectivity is further illustrated in the HDAC10–3 complex. Of the inhibitors studied here, inhibitor 3 has the lowest potency and HDAC10/HDAC6 selectivity even though its pendant hydroxyl group donates a bifurcated hydrogen bond to E274. However, this is a weaker dipole-charge interaction compared to the stronger charge-charge interactions between E274 and the dimethylammonium moieties of inhibitors 1 and 2. Additionally, the water molecule in the E274–H2O–H176 network does not form a hydrogen bond with E274. Thus, two strong hydrogen bonds with E274 – one with the pendant inhibitor dimethylammonium group and one with the water molecule hydrogen bonded to zinc ligand H176 – comprise the hallmarks of highly potent and selective inhibitors.
Concluding remarks.
A key lesson emanating from the current study is that inhibitors with bulky capping groups can be accommodated in the narrow HDAC10 active site as long as they have a proper size and shape to fit in the narrow crevice, and as long as the PEACE motif helix can move sufficiently if needed to accommodate binding, e.g., as observed for Tubastatin A. It is conceivable that the flexibility of the PEACE motif helix could similarly accommodate the binding of large protein substrates for lysine deacetylation, which could account for the observed effects of HDAC10 on acetylation levels of Hsp70 family proteins in neuroblastoma cells.15 Future studies exploring this possibility will be reported in due course.
METHODS
All chemicals, buffers, and general reagents, including Tubastatin A, were purchased from Sigma-Aldrich and used without further purification. Oxa-Tubastatin A and inhibitors 1–4 were synthesized and assayed as previously described.16
PCR mutagenesis (Supplementary Table S1) was used to introduce the A24E mutation in the D94A zebrafish HDAC10 plasmid previously reported by Hai and colleagues5 to generate a plasmid encoding “humanized” zebrafish HDAC10 (A24E-D94A zebrafish HDAC10, henceforth simply “HDAC10”). Following mutagenesis, the plasmid was transformed into DH5α Escherichia coli, confirmed by sequencing, and transformed into BL21(DE3) E. coli (NE Biolabs). Protein was expressed and purified as described in the Supporting Information.
Crystallization of HDAC10-inhibitor complexes was achieved by the sitting-drop vapor diffusion method. Crystals diffracted to 2.00–2.65 Å resolution using synchrotron radiation, which represents an improvement over the diffraction properties of crystals generated from the wild-type enzyme. Initial electron density maps of each enzyme-inhibitor complex were phased by molecular replacement using the atomic coordinates of the Y307F HDAC10-trifluoromethylketone complex (PDB 5TD7)5 as a search probe. Crystallographic refinement of each enzyme-inhibitor complex converged smoothly to Rwork values ranging 0.1818–0.2028 and Rfree values ranging 0.2263–0.2597. Detailed methods are outlined in the Supporting Information. Disordered polypeptide segments in each final model are listed in Supplementary Table S2. Data collection and refinement statistics are recorded in Supplementary Table S3.
Supplementary Material
ACKNOWLEDGMENTS
We thank K. Remans and K. Perez of the Protein Expression and Purification Core Facility of the European Molecular Biology Laboratory (EMBL, Heidelberg) for TwinStrep-HDAC10 protein production and nano-DSF support. Additionally, this work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Pilatus 6M detector on beamline 24-ID-C is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Finally, this work is also based on research conducted at beamline 17-ID-1 (AMX) of the National Synchrotron Light Source II, a DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract DE-SC0012704. The Center for BioMolecular Structure (CBMS) is primarily supported by the National Institutes of Health, NIGMS, through a Center Core P30 Grant (P30GM133893) and by the DOE Office of Biological and Environmental Research (KP1605010).
Funding
We thank the National Institutes of Health for grant GM49758 in support of this research.
Footnotes
The authors declare no competing financial interests.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: https://pubs.acs.org/doi/10.1021/acschembio.0c00362.
Detailed methods and tables; ITC enthalpograms (PDF)
Accession Codes
The atomic coordinates and crystallographic structure factors of HDAC10 complexes with the inhibitors shown in Figure 1b have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes as follows: Tubastatin A, 6WBQ; 1, 6WDV; 2, 6WDW; 3, 6WDX; 4, 6WDY.
REFERENCES
- 1.Gregoretti IV, Lee YM, and Goodson HV (2004) Molecular evolution of the histone deacetylase family: functional implication of phylogenetic analysis. J. Mol. Biol 338, 17–31. [DOI] [PubMed] [Google Scholar]
- 2.Haberland M, Montgomery RL, and Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet 10, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porter NJ, and Christianson DW (2019) Structure, mechanism, and inhibition of the zinc-dependent histone deacetylases. Curr. Opin. Struct. Biol 59, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shinsky SA, and Christianson DW (2018) Polyamine deacetylase structure and catalysis: prokaryotic acetylpolyamine amidohydrolase and eukaryotic HDAC10. Biochemistry 57, 3105–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hai Y, Shinsky SA, Porter NJ, and Christianson DW (2017) Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat. Commun 8, 15368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanyo ZF, Scolnick LR, Ash DE, and Christianson DW (1996) Structure of a unique binuclear manganese cluster in arginase. Nature. 383, 554–557. [DOI] [PubMed] [Google Scholar]
- 7.Finnin MS, Donigina JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, and Pavletich NP (1996) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 401, 188–193. [DOI] [PubMed] [Google Scholar]
- 8.Christianson DW (2005) Arginase: structure, mechanism, and physiological role in male and female sexual arousal. Acc. Chem. Res 38, 191–201. [DOI] [PubMed] [Google Scholar]
- 9.Lombardi PM, Cole KE, Dowling DP, and Christianson DW (2011) Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol 21, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong JJ, Liu J, Bertos NR, and Yang XJ (2002) Identification of HDAC10, a novel class II human histone deacetylase containing a leucine-rich domain. Nucleic Acids Res 30, 1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Libby PR (1978) Properties of an acetylspermidine deacetylase from rat liver. Arch. Biochem. Biophys 188, 360–363. [DOI] [PubMed] [Google Scholar]
- 12.Blankenship J (1978) Deacetylation of N8-acetylspermidine by subcellular fractions of rat tissues. Arch. Biochem. Biophys 189, 20–27. [DOI] [PubMed] [Google Scholar]
- 13.Libby PR (1980) Rat liver nuclear N-acetyltransferases: separation of two enzymes with both histone and spermidine acetyltransferase activity. Arch. Biochem. Biophys 203, 384–389. [DOI] [PubMed] [Google Scholar]
- 14.Marchant P, Dredar S, Manneh V, Alshabanah O, Matthews H, Fries D, and Blankenship J (1989) A selective inhibitor of N8-acetylspermidine deacetylation in mice and HeLa cells without effects on histone deacetylation. Arch. Biochem. Biophys 273, 128–136. [DOI] [PubMed] [Google Scholar]
- 15.Oehme I, Linke J-P, Böck BC, Milde T, Lodrini M, Hartenstein B, Wiegand I, Eckert C, Roth W, Kool M, Kaden S, Gröne H-J, Schulte JH, Lindner S, Hamacher-Brady A, Brady NR, Deubzer HE, and Witt O (2013) Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Nat. Acad. Sci. USA 110, E2592–E2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Géraldy M, Morgen M, Sehr P, Steimbach RR, Moi D, Ridinger J, Oehme I, Witt O, Malz M, Nogueira MS, Koch O, Gunkel N, and Miller AK (2019) Selective inhibition of histone deacetylase 10: hydrogen bonding to the gatekeeper residue is implicated. J. Med. Chem 62, 4426–4443. [DOI] [PubMed] [Google Scholar]
- 17.Ridinger J, Koeneke E, Kolbinger FR, Koerholz K, Mahboobi S, Hellweg L, Gunkel N, Miller AK, Peterziel H, Schmezer P, Hamacher-Brady A, Witt O, and Oehme I (2018) Dual role of HDAC10 in lysosomal exocytosis and DNA repair promotes neuroblastoma chemoresistance. Sci. Rep 8, 10039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eisenberg T, et al. (2009) Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol 11, 1305–1314. [DOI] [PubMed] [Google Scholar]
- 19.Minois N (2014) Molecular basis of the ‘anti-aging’ effect of spermidine and other natural polyamines – a mini-review. Gerontology 60, 319–326. [DOI] [PubMed] [Google Scholar]
- 20.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, and Kozikowski AP (2010) Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, Tubastatin A. J. Am. Chem. Soc 132, 10842–10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Senisterra G, Chau I, Vedadi M (2012) Thermal denaturation assays in chemical biology. Assay Drug Dev. Techn 10, 128–136. [DOI] [PubMed] [Google Scholar]
- 22.Herbst-Gervasoni CJ, Christianson DW (2019) Binding of N8-acetylspermidine analogues to histone deacetylase 10 reveals molecular strategies for blocking polyamine deacetylation. Biochemistry 58, 4957–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hai Y, and Christianson DW (2016) Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol 12, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porter NJ, Mahendran A, Breslow R, and Christianson DW (2017) Unusual zinc binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. USA 114, 13459–13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Porter NJ, Wagner FF, and Christianson DW (2018) Entropy as a driver of selectivity for inhibitor binding to histone deacetylase 6. Biochemistry 57, 3916–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackwitz MKW, Hamacher A, Osko JD, Held J, Schöler A, Christianson DW, Kassack MU, and Hansen FK (2018) Multicomponent synthesis and binding mode of imidazo[1,2- α]pyridine-capped selective HDAC6 inhibitors. Org. Lett 20, 3255–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen S, Svoboda M, Zhang G, Cavasin MA, Motlova L, McKinsey TA, Eubanks JH, Barinka C, Kozikowski AP (2020) Structural and in vivo characterization of Tubastatin A, a widely used histone deacetylase 6 inhibitor. ACS Med. Chem. Lett, 10.1021/acsmedchemlett.9b00560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eliel EL, Kandasamy D, Yen C. y., Hargrave KD (1980) Conformational analysis. 39. 13C NMR spectra of saturated heterocycles. 9. Piperidine and N-methylpiperidine. J. Am. Chem. Soc 102, 3698–3707. [Google Scholar]
- 29.Morgen M, Steimbach RR, Géraldy M, Hellweg L, Sehr P, Ridinger J, Witt O, Oehme I, Herbst-Gervasoni CJ, Osko JD, Porter NJ, Christianson DW, Gunkel N, Miller AK (2020) Design and synthesis of dihydroxamic acids as HDAC6/8/10 inhibitors. ChemMedChem 15, 1163–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Osko JD, Roose BW, Shinsky SA, Christianson DW (2019) Structure and function of the acetylpolyamine amidohydrolase from the deep earth halophile Marinobacter subterrani. Biochemistry 58, 3755–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.