

Abstract
Fast-developing single-cell multi-modal omics (scMulti-omics) technologies enable the measurement of multiple modalities, such as DNA methylation, chromatin accessibility, RNA expression, protein abundance, gene perturbation, and spatial information, from the same cell. scMulti-omics can comprehensively explore and identify cell characteristics, while also presenting challenges in developing computational methods and tools for integrative analyses. We review the integrative methods and summarize the existing tools for studying a variety of scMulti-omics data. The various functionalities and practical challenges in using the available tools in the public domain are explored through several case studies. Finally, we identify remaining challenges and future trends in scMulti-omics modeling and analyses.
Keywords: single-cell sequencing technology, single-cell multi-modality, integrative methods, analysis tools
Single-cell sequencing technologies and multi-omics data
An individual cell maintains genetic heterogeneity that manifests unique cell functions and states [1]. With rapid developments in cell isolation and high-throughput sequencing technologies, single-cell omics (see Glossary) profiling can significantly benefit the study of characteristics and heterogeneity of individual cells, which has historically been confounded by bulk measurements[2]. Opportunities have arisen at the single-cell level to generate molecular information about whole-genome gene copies [3, 4], gene expression [5, 6], epigenome [7, 8], CRISPR perturbation [9, 10], spatial information [11, 12], and protein abundance [13, 14], among other phenomena. However, different molecules in a cell work synergistically to determine the cell state, and such individual profiling provides only a partial landscape of the entire cell heterogeneity [15, 16].
Single-cell multi-modal omics (scMulti-omics) sequencing technologies have recently emerged and showed advantages of simultaneously measuring multiple modalities from the same individual cell, which enables a more comprehensive exploration of cell behavior and identity [2, 16–19]. These technologies have tremendous potential in precision treatments, drug resistance, and relapse potential for specific tumors since they are particularly useful for studying the cells undergoing rapid differentiation (e.g. cancer and Alzheimer diseases) or evolving highly-diverse sub-populations (e.g. immune cells)[20, 21]. Specifically, scMulti-omics can greatly impact clinical application by identifying novel disease mechanisms. The underlying modalities can help predict drug sensitivities in tumor cells, prior to any in vivo/vitro drug doses, in order to exclude low-sensitivity drugs and decrease the diagnostic cost. By 2025, the global scMulti-omics market is anticipated to be $5.32 billion, mainly driven by the increasing need for non-invasive diagnosis and personalized medicine [22].
Intuitively, one omics profile can recover the missing values lost in another. Dropout issues in single-cell RNA-sequencing (scRNA-Seq) are prevalent. This problem stems from the inherent issue with sampling, in which samples do not fully represent their target population, especially for low expressed genes. Sampling inefficiencies coupled with the inherent uncertainty that follows sampling procedures lead to loss of data. The gene expression values missing from scRNA-Seq may be recoverable by integrating more omics levels; hence, the integrative analysis of scMulti-omics can theoretically lead to cell state prediction, cell trajectory elucidation, and other functional analyses with high accuracy and low bias [23]. However, considering the intrinsic challenges in sequencing multiple levels of information from the same cell, the sequencing technology accuracy, and the relevant cost, it is not currently feasible to comprehensively profile all aspects of cellular sequence types. Current approaches allow up to four types of omics data to be measured simultaneously, leading to 13 available combinations in the public domain (9 with double-modality sequencing, 3 with triple-modality sequencing, and 1 with quad-modality sequencing). For example, one technique combines measurements of RNA expression, chromatin accessibility, and DNA methylation; another combines RNA expression with spatial information (more details can be found in Figure 1).
Figure 1.
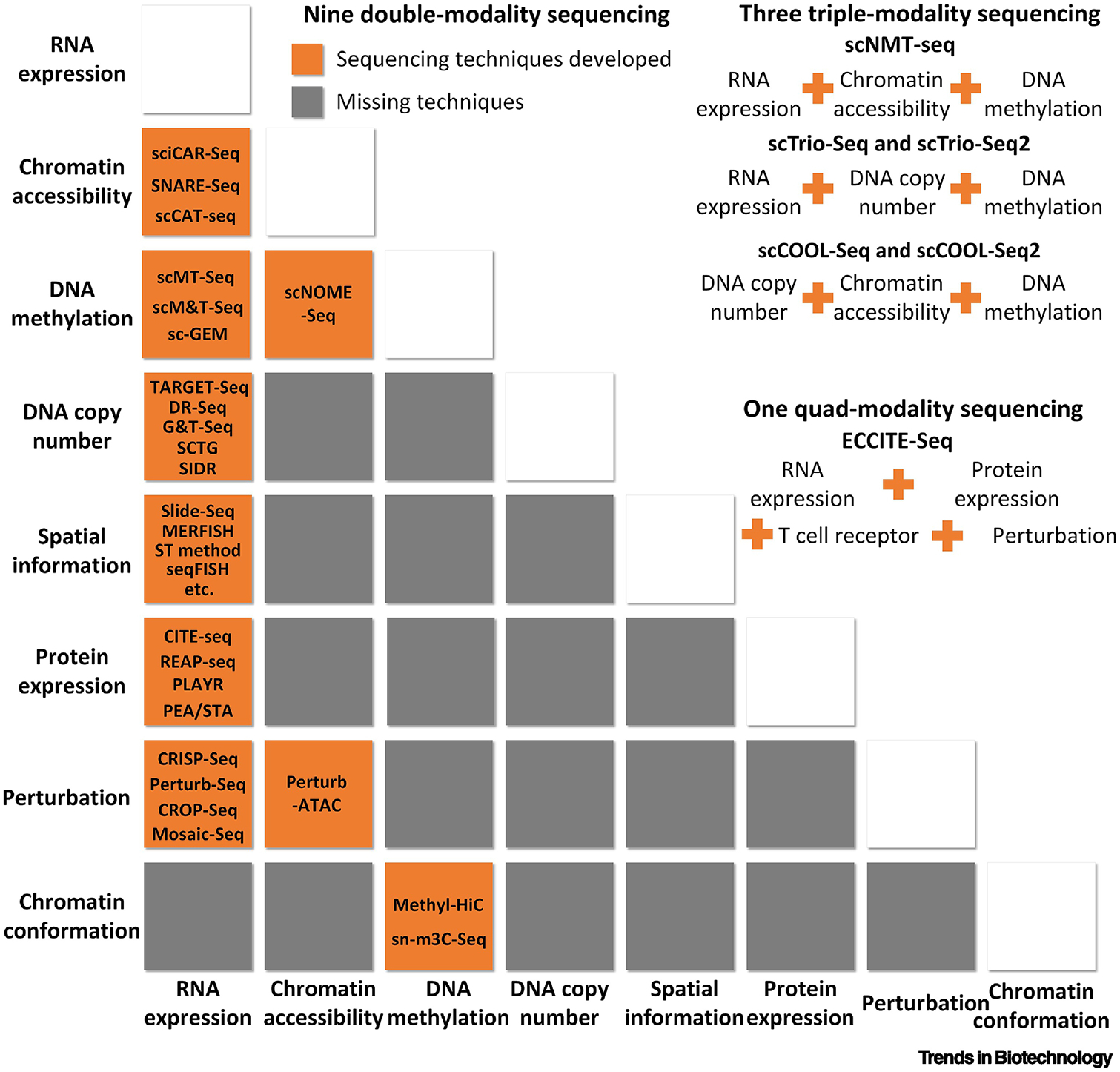
Existing scMulti-omics combinations and representative sequencing techniques. By December 2019, multiple sequencing techniques have been developed to acquire nine combinations of two modalities from a single cell, and three for triple-modality and 1 for quad-modality. Names of sequencing methods are listed accordingly, and most of them are not commercialized.
It is not surprising that scRNA-Seq serves as a general mediator that is included in most scMulti-omics studies. Not only does RNA’s intermediate position in the central dogma allow for proximity to multiple molecular processes, but also, the application of scRNA-Seq is commercialized and routinely available [24, 25]. At least 43 papers in the past five years published scMulti-omics datasets that were profiled within the same cell (i.e., matched data) (Table 1). By jointly analyzing these matched data, integrative studies may reveal relationships and interactions between various types of molecular information that are otherwise missing in traditional single-omics studies. However, most existing scMulti-omics tools analyze data obtained across single-cell experiments (i.e., unmatched data), which provide opportunities to utilize large amounts of existing single-cell omics data to discover novel insights by properly aligning cross-experimental datasets for integrative analysis. We review integrative methods and their affiliated computational tools designed for matched and unmatched scMulti-omics data. The derived knowledge and insights can provide guidance to choose proper tools regarding supported data types, expected analytical outcomes, functionalities, and performances.
Table 1:
Publicly available matched scMulti-omics data.
| Single-cell Profiling | Seq tech | Accession | Ref |
|---|---|---|---|
|
G&T-seq | EGAS00001001204 | [82, 83] |
| E-ERAD-381 | |||
| SIDR | PRJEB20144 | [84] | |
| TARGET-Seq | GSE105454 | [85] | |
| SCTG | SRP040646 | [86] | |
| DR-seq | GSE62952 | [87] | |
|
sc-GEM | SRR3748387 | [51] |
| scM&T-seq | GSE74535 | [50] | |
| scMT-seq | GSE76483 | [88] | |
|
sci-CAR | GSE117089 | [29] |
| scCAT-seq | Along with the paper | [89] | |
| SNARE-seq | GSE126074 | [90] | |
| CITE-seq + scATAC-Seq | GSE139369 | [91] | |
|
scNOME-seq | GSE83882 | [92] |
|
CITE-seq | GSE100866 | [13] |
| GSE128639 | [24] | ||
| REAP-seq | GSE100501 | [14] | |
| PLAYR | Along with the paper | [93] | |
| PEA/STA | Along with the paper | [94] | |
|
Methyl-HiC | GSE119171 | [56] |
| sn-m3C-Seq | GSE124391 | [95] | |
|
Perturb-seq | GSE90546 | [37] |
| GSE90063 | [52] | ||
| CRISP-seq | GSE90486 | [96] | |
| CROP-seq | GSE92872 | [97] | |
| GSE108699 | [98] | ||
| Mosaic-Seq | GSE81884 | [99] | |
|
osmFISH | http://linnarssonlab.org/osmFISH | [11] |
| STARmap | https://www.starmapresources.com/data/ | [12] | |
| seqFISH | Along with the paper | [100] | |
| MERFISH | Along with the paper | [101] | |
| GSE113576 | [102] | ||
| Slide-Seq | https://singlecell.broadinstitute.org/single_cell/study/SCP354/slide-seq-study | [58] | |
| Spatial transcriptomics (10X spatial) | https://github.com/SpatialTranscriptomicsResearch/st_pipeline | [103] | |
| https://www.10xgenomics.com/solutions/spatial-gene-expression/ | [104–106] | ||
| MASC-Seq | Along with the paper | [107] | |
| ST-RNA-Seq | SRP100428 | [108] | |
|
Perturb-ATAC | GSE116297 | [10] |
|
|||
| scNMT-seq | GSE109262 | [57] | |
|
scTrio-seq | GSE65364 | [109] |
| scTrio-seq2 | GSE97693 | [110] | |
|
scCOOL-seq | GSE78140 | [111] |
| GSE100272 | [112] | ||
| iscCOOL-seq | GSE114822 | [113] | |
|
ECCITE-Seq | GSE126310 | [114] |
Integrative methods for scMulti-omics data
Typically, single-omics data is represented by a matrix, with rows representing genes, columns representing cells, and each element of the matrix representing the specific omics information about a feature in the corresponding cell (e.g., gene expression). Joint analyses of scMulti-omics data simultaneously consider both the cross-modality variations and cross-cell correlations. The challenges in developing such integrative methods for scMulti-omics data include unifying different modalities, batch effects between experiments (especially for unmatched data), low sequencing depth, and high-modality interactions. Based on the underlying method design, we summarize the existing integrative methods into the following four broad categories: feature projection, Bayesian modeling, regression modeling, and decomposition (Figure 2).
Figure 2.
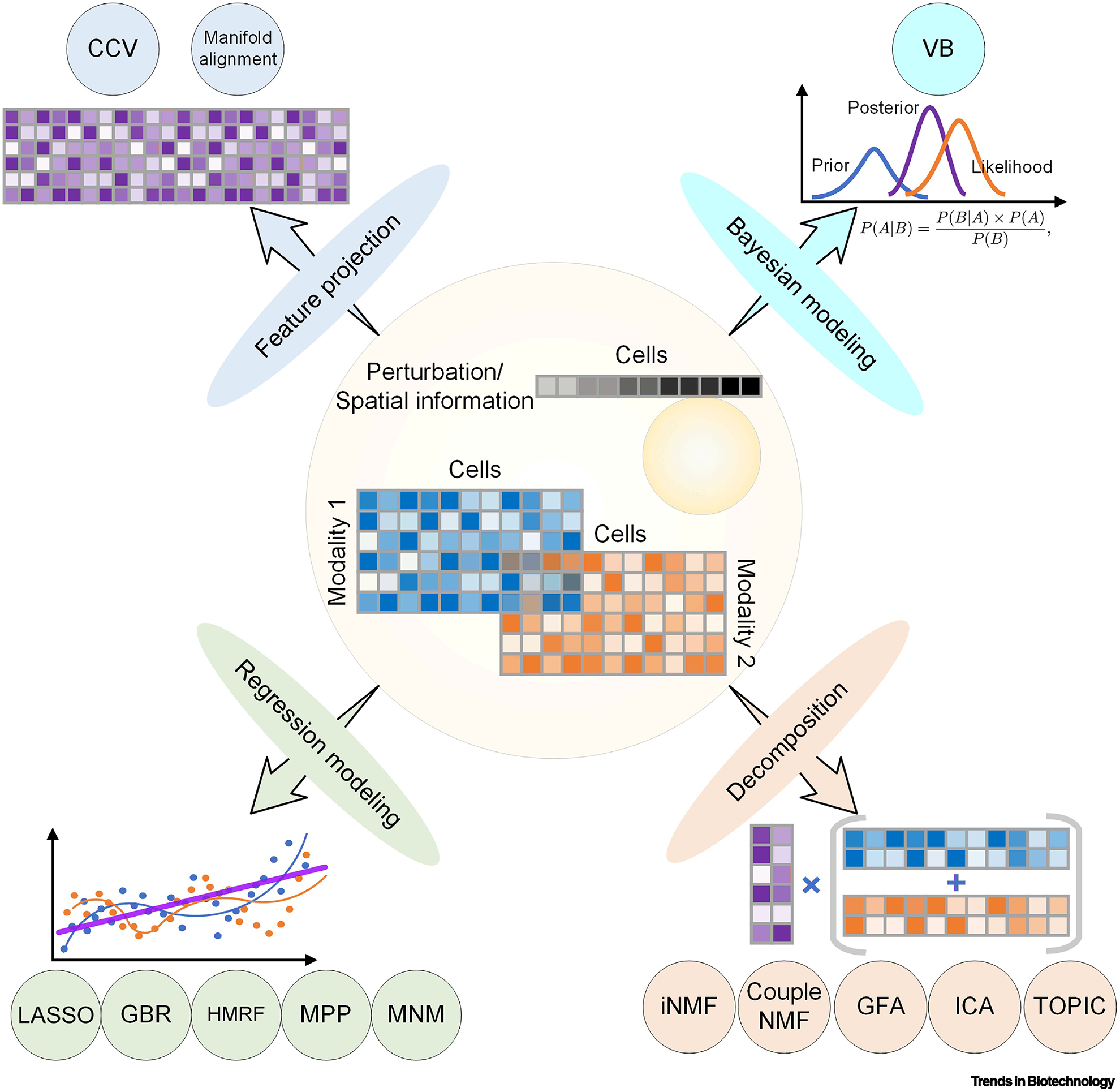
Integrative methods for scMulti-omics. We summarized all 13 integrative algorithms applied on scMulti-omics analysis into four categories, and each of which is followed by a graph for understanding.
First, data from different profiling methods can be jointly analyzed through feature projection. Canonical correlation vectorization (CCV), modified from canonical correlation analysis, investigates the relationship between variables by capturing anchors that are maximally correlated across the datasets [24]. For instance, given matched scRNA-Seq and single-cell ATAC-Sequencing (scATAC-Seq) data, CCV identifies the gene features shared between the two matrices by projecting cells. A single matrix can then be generated by projecting expression values and chromatin accessibility of each gene onto a common basis space and normalizing values by penalization and regularization. Manifold alignment is another method that has been applied to unmatched scRNA-Seq and epigenomics data to unravel the pseudo-time correlation between, e.g., gene expression and DNA methylation [26, 27]. However, how to legitimately account for batch effects is one of the major difficulties with the projection approach. CCV and Manifold alignment are both feature projection-based dimension reduction techniques. These have the theoretical ability to reduce high-dimensional data down to its critical components in non-linear spaces, ideally emphasizing the features that differentiate cell types. When these methods are applied to multi-omics data, they can denoise each individual dataset and highlight cell-type-specific features, resulting links between multi-omics datasets.
Second, variational Bayes (VB) is a stochastic variational inference method based on Bayesian modeling. The underlying method was designed using the hypothesis that the gene copy number (single-cell genome sequencing) is positively correlated with measures corresponding to gene expression (scRNA-Seq) [28]. VB fits the RNA expression matrix to a set of GxC sub-matrices (generated from gene phylogenic analysis, with genes as rows, cells as columns, and gene copy numbers as elements) and integrates the two modalities by finding the lowest variance between the fitted approximate distribution and the true distribution in the original GxC matrix. While the underlying hypothesis for VB is naively intuitive, there are many complex interactions that dictate the level of gene expression. Assuming the hypothesis of a strong positive correlation between gene copy number and gene expression leads to the potential of inconsistent or unreliable results.
The third type of integrative analysis uses various regression models to accurately characterize the high heterogeneity among individual cells and multi-layer data types, simultaneously. Duan and colleagues described a method using a linear regression model to integrate covariates between spatial information and RNA-expression combined with an expectation-maximization approach to fit the two models. The least absolute shrinkage and selection operator (LASSO) regression method has been applied to correlate gene expression with chromatin accessible sites [29]. Gradient boosting regression (GBR) iteratively fits “weak” models to estimate response variables and align matched gene expression and chromatin accessibility patterns to predict differentially accessible genomics sites for cell type prediction [30]. Hidden Markov random field (HMRF) is a graph-based model widely used for pattern recognition in image data analysis. HMRF first builds connections between cells and spatial coordinates and clusters cell groups, integrating gene expression patterns and cell positions [31]. Marked point process (MPP) is a nonparametric statistical framework to identify the dependency of spatial distribution and gene expression levels [32]. A more complex multivariate normal model (MNM) for spatial transcription analysis defines spatial dependence by combining gene expression profiles with cell locations considering both spatial covariance and nonspatial variance [33]. These regression-based integrative methods vary widely, as does their applicability to integrating multi-omics datasets. The underlying assumptions for regression models are often an issue when applying them to new areas. Specifically, linear regression has strict assumptions that would make it difficult to apply such a parametric approach to genetic data that often does not fit the parametric requirements. However, nonparametric approaches such as MPP do not suffer from this limitation. These types of robust methods have great potential for the integration of multi-omics datasets. In particular, parallels between longitudinal data analysis and spatial omics methods make these types of regression approaches a strong fit.
Lastly, decomposition is a straightforward method for unsupervised data integration. The basic idea is to decompose the original matrices into two low-dimensional submatrices: a coefficient submatrix records signature information (e.g. genes) and a residue submatrix records cell information. One widely used decomposition method in scMulti-omics studies is matrix factorization. The input matrices (e.g. scRNA-Seq and scATAC-Seq data) are decomposed into (i) an amplitude matrix (or coefficient matrix) composing genes as rows and factors as columns to reflect gene co-regulation patterns; and (ii) a pattern matrix (or residue matrix) composing factors as rows and cells as columns for further cell clusters. Four modified methods have been proposed: integrative nonnegative matrix factorization (iNMF) [34], coupled nonnegative matrix factorizations (coupleNMF) [35], group factor analysis (GFA) [36], and independent component analysis (ICA) [37]. TOPIC modeling is another decomposition method to discover the shared latent information among input matrices. It has been applied to derive the functional topics of each cell with specific perturbation by integrative analysis of matched scRNA-Seq and single-cell CRISPR screening data [38]. Similar to the feature project-based dimension reduction approaches, the decomposition methods allow for denoising of datasets through dimension reduction in an attempt to emphasize the key features of each cell. When implemented correctly, with the necessary assumptions met, decomposition has the potential to reliably integrate different omics datasets.
Apart from these integrative methods, various advanced methods—such as network simulated annealing and deep learning (e.g. autoencoder)—have emerged in the last three years for bulk multi-omics studies [39]. However, these approaches have yet to be applied at the single-cell level. Theoretically, all these models can be applied to scMulti-omics analysis with proper optimization, and we believe that the knowledge gained from these methods could be transferred from the bulk level to the single-cell level in the near future.
Analytical tools for scMulti-omics data
To decipher the secrets hidden in scMulti-omics data, a powerful integrative method is necessary, yet not entirely sufficient. An efficient tool should also be able to properly process the massive and chaotic data before analysis for a more accurate and reliable result. A proper combination of preprocessing steps is necessary to decrease bias and generate meaningful analytical results, including but not limited to data setup, removing low-quality genes and cells, normalizing values across modalities and cells, imputing missing values, and imaging digitalization to generate 2D coordination. In this section, we comprehensively review ten computational integrative tools for scMulti-omics analysis that are equipped with multiple functionalities and various analytical interpretations as mature pipelines (Figure 3, Key Figure).
Figure 3, Key Figure.
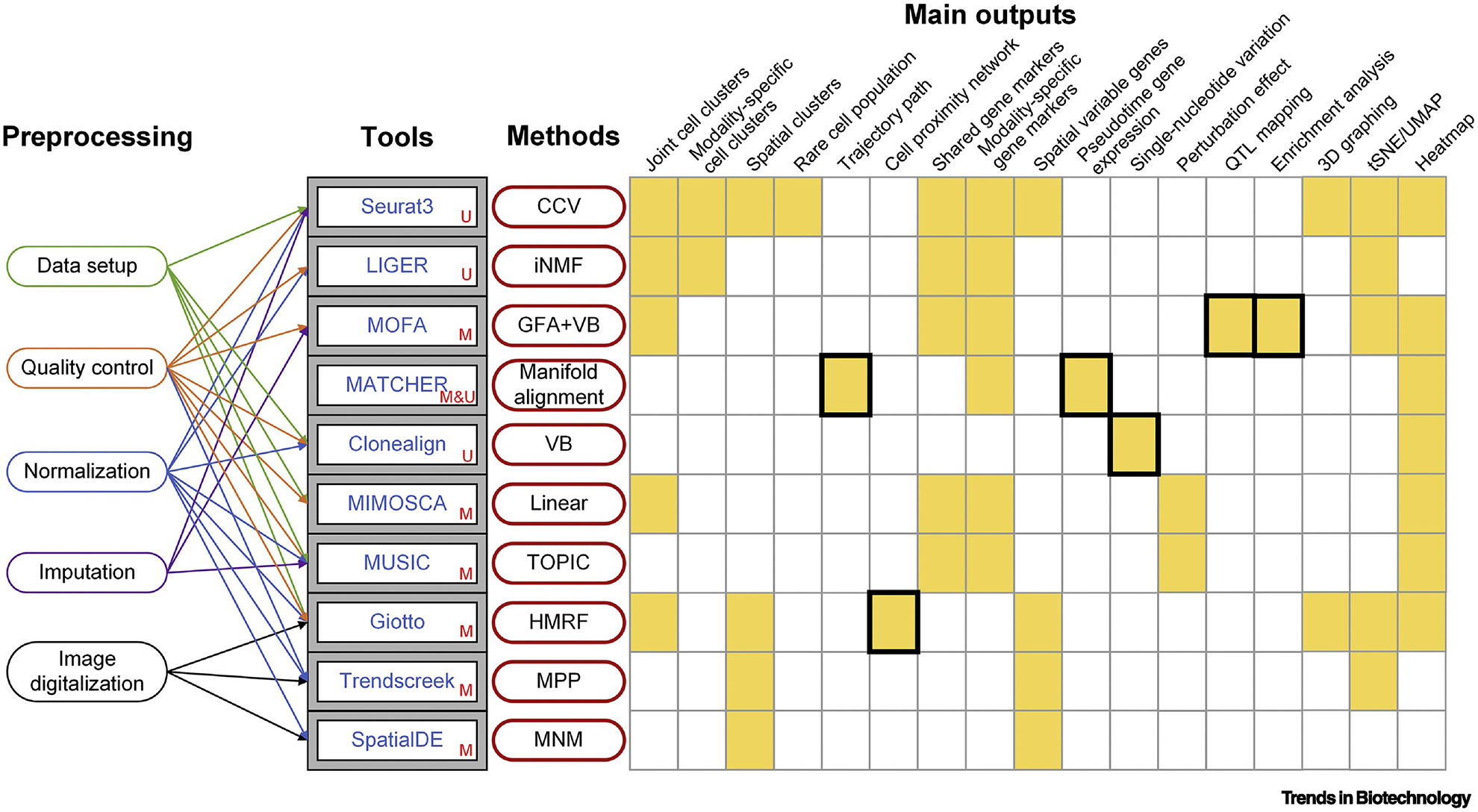
Ten tools integrated with proper preprocessing steps, core integrative methods, and adequate results interpretations for integrative analysis of scMulti-omics. U (unmatched), M (matched), and M&U (both matched and unmatched) represent the data type that the original tool’s paper claimed to support. The main outputs are summarized based on original papers and tool tutorials from our investigations. Black frames indicate unique outputs.
Seurat3—which is based upon a CCV method—is one of the best-developed tools for scRNA-Seq and scMulti-omics data analyses with supreme functionalities and well-written documentation [24, 40]. Released in 2019, the authors demonstrated the use of Seurat3 by (i) co-embedding scRNA-Seq and unmatched scATAC-Seq data to reveal cell-type-specific regulatory loci, (ii) applying matched RNA expression and cell-surface protein expressions to leverage deeper connections between protein abundance and gene expression, and (iii) applying spatial transcriptomic data to predict spatial gene expression patterns and classify subpopulations [24]. Seurat3 inherited all functionalities from its previous version in comprehensive single-cell RNA-Seq analysis [41, 42], cross-experimental single-cell data analysis [43], and integrative bulk and single-cell RNA-Seq data analysis [44], resulting in a wide application range. Its key feature of analyzing multiple modalities at the single-cell level has also been widely applied by other independent labs. More applications of Seurat3 can be found in Table 2.
Table 2.
Tools for scMulti-omics analysis and application examples.
| Toolsa | Implemented inb | Datac | Number of Cells | Main biological insights | Ref |
|---|---|---|---|---|---|
| MATCHER | Py | RE+DM+CA | 5,151 | Establish a continuum that ranges from pluripotency to a differentiation primed state; while the shared master time was highly correlated in the matched dataset which was useful for determining the overall reprogramming progress of each cell | [26] |
| MIMOSCA | Py | RE+GP | 200,000 | Predict the regulation of anti-parasitic response genes Gbp2,2b,3,4,5 and 7 by inducing CRISPR/Cas9 knockout, targeting 24 transcriptome factors; suggest Stat2’s impact on Gpb genes may be mediated through Irf8. | [52] |
| MOFA | R/Py | RE+DM | 87 | Reveal the cooperation between the transcriptome and methylation sites during the transition from naive to primed pluripotent states. Factor-specific markers were identified, such as Rex1/Zpf42, Tbx3, and Fbxo15. | [36] |
| RE+PE | 200,000 | Identified 44 genes whose activation induces a zygotic genome activation-like transcriptional response, including 40 novel maternal proteins. | [45] | ||
| Clonealign | R | RE+DC | 1,152 | Build single-cell phylogeny with four distinct clades and eight sub-clades. The intra-clonal clustering identified cell cycle corresponding clusters. | [28] |
| Trendsceek | R | RE+SI | ~10,000 | Identify 35 significant genes with expression primarily in nongranular cells including Ptn, Nr2f2, and Fabp7 in mouse olfactory bulb tissue. | [32] |
| SpatialDE | Py | RE+SI | ~10,000 | Identify 67 spatial variable genes with spatial dependencies of the gene expression variance and showed clear spatial substructure, consistent with matched tissues in mouse olfactory bulb. | [33] |
| Seurat3 | R | RE+CA | 14,249+100,000 | Reveal cell-type-specific regulatory loci whose accessibility profiles were consistent with expected patterns. | [24] |
| 35,882 | Identify 91,601 putative peak-to-gene linkages and inferred the potential oncogene RUNX1. | [91] | |||
| 7,846 | Reveal cell-state transcriptional regulators and lineage relationships in mammary gland cells. | [115] | |||
| RE+PE | 33,454 | Matched RNA expression and 25 cell-surface protein expressions, leveraging connections between protein abundance and gene expression. | [24] | ||
| RE+SI | 14,249 | Predict spatial gene expression patterns and spatial subpopulations. | [24] | ||
| LIGER | R | RE+DM | 55,803+3378 | Resulted in 37 neuron clusters and identified methylation regions that were anticorrelated with Arx expression, including a validated Arx enhancer. | [49] |
| MUSIC | R | RE+GP | 32,777 | Identify a novel knockout effect on cell migration impacted by the perturbation of Cebpb on immune cell activation, and gene-gene perturbation associations between Cebpb and other gene perturbations. | [38] |
| Giotto | R | RE+SI | 913 | Identify six global and distinctive clusters including excitatory neurons (Icam), GABAergic neurons (Slc32a1), and four smaller groups; Visualize both single-cell resolution heterogeneity in both expression and spatial space representations. | [53] |
Tools that are equipped with multiple functions; developed methods with only scripts are not considered in this table; tools are sorted by publishing year from old to new.
The platform of each tool, either as an R or Python (Py) package.
Data type combination examples using the corresponding tool. RE=RNA expression, DM=DNA methylation, CA=chromatin accessibility, PE=protein expression, SI=Spatial information, DC=DNA copy, GP=Gene perturbation, HM=HiChiP.
MOFA was developed by Argelaguet and lab members as a statistically rigorous tool that decomposes modality and captures sources of variability between different datasets [36]. MOFA was originally applied on a study with parallel profiled DNA methylation and gene expression revealing the cooperation between the transcriptome and methylation sites. Each factor captured signatures that reflect a specific cell state and can be used to impute missing information in modalities [36]. Catalinas and colleagues applied MOFA on a perturbation-transcription dataset and showed the advantage in capturing relevant gene signatures, of both coding and non-coding transcription compared to the conventional gene differential analysis [45]. MOFA has also been applied to reveal connections between the transcriptome and the epigenetic state in enhancers during germ layer formation and capture the global cell-to-cell variability via factors inferred from triple-modalities [46, 47]. Its new version, MOFA+, claims to have better performance and more functionalities for discovering the association among gene expression, epigenetic variations, and cell fates commitment [48].
Welch’s lab recently developed a tool called LIGER (linked inference of genomics experimental relationships) that uses the iNFM method to best approximate the original data and identify dataset-specific and shared factors across different datasets. LIGER was originally applied to an unmatched scRNA-Seq and DNA methylation dataset showing the ability in identifying methylation regions that were anticorrelated specific expressions. As with Seurat3, LIGER can also be applied to analyze, e.g. cross-experimental single-cell RNA-Seq data analysis [49].
MATCHER aligns the underlying manifolds of diverse single-cell modalities (epigenome and transcriptome) to create an equivalent pseudo-time representation and has been applied to both matched and unmatched single-cell data [26]. MATCHER was initially applied on two publicly available datasets: (i) unmatched gene expression, DNA methylation, and chromatin accessibility data from mouse embryonic stem cells, and (ii) matched gene expression and DNA methylation data from human-induced pluripotent stem cells [50, 51] (Table 2). As a result, MATCHER identifies sequential changes and reveals the connections of trajectory changes among multiple modalities which provides a great potential for reprogramming and differentiation studies.
Clonealign is a tool uniquely designed for the integrative study of scRNA-Seq and gene copy numbers from single-cell DNA sequencing, based on the assumption that the increasing gene copy number will result in an increased expression of the corresponding gene [28]. It was applied to matched scDNA-Seq and 10X scRNA-Seq datasets to identify clone-specific gene expression patterns and the correlation of single-nucleotide variation and expression. However, Clonealign is not suitable for cancers that have quiescent genomes and are devoid of copy number changes, such as sarcomas and karyotypically normal acute myeloid leukemia. As an extension, Clonealign holds the potential to apply to other scMulti-omics studies, such as methylation-transcription and chromatin accessibility-transcription [28].
Pooled CRISPR knockout screening is used to evaluate gene biological functions by comparing gene perturbations with phenotypes (e.g. cell growth). Combining CRISPR screening with scRNA-Seq can elucidate the effects of perturbation on the RNA expression level. MIMOSCA is used for perturbation-expression analysis which deciphers the effect of individual perturbations and the marginal contributions of genetic interactions using a maximum likelihood approach and linear model [52]. MUSIC is also a tool newly developed for analyzing CRISPR perturbation and RNA expression [38]. It utilizes the TOPIC method to model the perturbation and cell functions from the annotated expression matrix and shows better performance than MIMOSCA [38].
Spatial transcriptomics links differentially expressed genes to the actual cell position in the interactions intra- or inter-subtypes and is critical for understanding cell identity and function in the context of tissue. By including spatial information, researchers can broadly define cell types and subtle alterations without the requirement of direct gene measurement. Giotto [53], Trendscreek [32], and SpatialDE [33] are three well-rounded tools for analyzing spatial transcriptomics data. These pipelines first process the photo image and scRNA-Seq data separately to generate a spatial coordinate figure and cell clusters, respectively. The predicted clusters are then correlated with spatial coordinates to map the spatial-specific cell cluster patterns. A cell spatial network can be built considering both the actual spatial distance between each cell pair and the cell clusters predicted from gene expressions. The spatial variable gene plot intuitively shows the expression patterns at different locations which are intuitively useful to construct the whole tissue regulatory landscape by combining with epigenetic features. Future studies could utilize other omics data to infer relationships between cell positions and causal genetic patterns, and a specific case study for spatial transcriptomics is showcased in our case study.
Despite these ten well-developed tools (Table 2), several other packages were available for specific scMulti-omics data analysis, yet with only scripts and limited functionalities. MOGSA claims to perform gene-set analysis from single-cell transcriptomics and proteomics data to yield insights into the complex molecular machinery of biological systems [54]. Duren and others developed De-Convolution and Coupled-Clustering using the coupleNMF integrative method to intake scHi-C and bulk HiChIP data in order to deconvolve the 3D gene contacts into cluster-specific profiles [55]; An R script integrates LASSO to jointly analyze matched RNA expression and chromatin accessibility data from scCAR-Seq [29]; Li and coworkers analyzed matched DNA methylation and chromatin architecture in single cells using an assay, called Methyl-HiC, to reveal coordinated DNA methylation status between distal genomics segments [56]; Clark and teammates developed the scNMT-Seq technique to simultaneously measure matched chromatin accessibility, DNA methylation, and RNA expression, and developed a Bernoulli likelihood-based regression assay to show how parallel profiling of the transcriptome and epigenome could reveal dynamic changes in how chromatin accessibility and DNA methylation interact during differentiation [57]; Adamson and colleagues developed an ICA dependent method named LRICA for Perturb-seq that analyzes gene perturbation and RNA expression in single-cell data [37]. Overall, these tools were only developed as prototypes without executable implementations and user-friendly interfaces. The robustness and usability of these tools need to be further improved for a wider application to other analytical studies.
Case studies and practical challenges
We applied four existing tools on four publicly available datasets downloaded online to evaluate their compatibilities and functions. We first reproduced the analysis of a spatial transcription dataset from Giotto to showcase the unique outcomes of such an integrative study [53, 58] (Figure 4A). Three signature features were reported: spatially correlated clusters, spatial cell networks, and spatial variable genes. Overall, Giotto is a user-friendly and powerful tool for spatial transcriptome analysis with concise and reproducible tutorials. The unique spatial network can be constructed by associating gene expression levels with cell neighborhoods and intuitively presented on the spatial map. Giotto is one of the few tools that provide web-based interactive visualization and exploration for scMulti-omics analysis.
Figure 4.
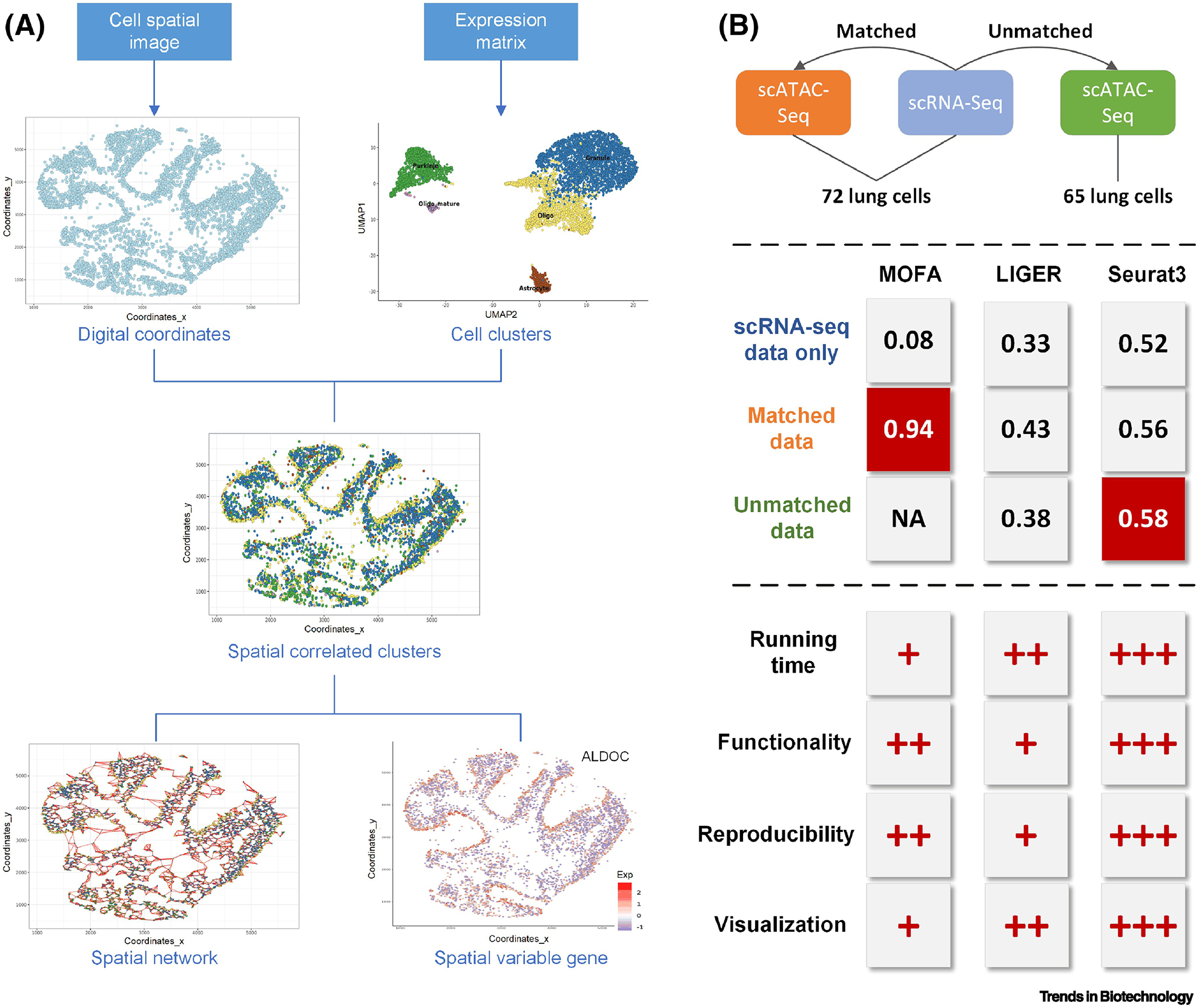
Two case studies for scMulti-omics analysis. (A) Main outcomes of Giotto. (B) Assessment of cell type prediction using MOFA, LIGER, and Seurat3, in terms of ARI (adjusted rand index) scores on three datasets. The three tools were further systematically evaluated and ranked, where +++ represents the most efficient tool in terms of CPU running time, provides the best practical support in analytical functionalities and visualizations, and has the most robustness in reproducibility of results; and + represents less efficiency, practical support, and robustness.
We further applied three well-cited and widely-used tools (MOFA, LIGER, and Seurat3) on three datasets (scRNA-Seq only, matched scRNA-Seq and scATAC-Seq, unmatched scRNA-Seq and scATAC-Seq) with default parameters (Figure 4B). The performance evaluation and comparison in terms of the cell type prediction led to the following three insights. First, the joint analysis of scRNA-Seq and scATAC-Seq showed better performance in predicting cell clusters than using scRNA-Seq alone, in terms of adjusted rand index, which evaluates the similarity between predicted cell labels and benchmarked cell types (higher score means better clustering performance). Second, the clustering result using matched data was better than that using unmatched data. Third, MOFA was the best-performing tool to analyze matched data and Seurat3 was the best-performing tool for unmatched data. We further evaluated the three tools from four perspectives: CPU running time, enrichment of analytical functionalities, reproducibility of software tutorial, and diversity of result interpretations and visualizations. We conclude that Seurat3 is the most robust and easy-to-use tool among the three (ranked as +++ in Figure 4B). However, the above three tools, as well as most existing integrative tools for scMulti-omics analysis, require different input file formats. For example, Seurat3 requires a gtf file recording gene structure information to create an activity matrix to transfer ATAC peak regions to corresponding genes, while LIGER needs a BED file. MOFA failed to clearly state data preprocessing steps. Such multiple input files and vague tutorials greatly increase the difficulty of the application and reproducibility of tools. More challenges of scMulti-omics analyses can be found in the following section.
Remaining challenges
While recently developed methods for integrating scMulti-omics data provide new opportunities to jointly analyze different types of single-cell data, there remain several challenges and issues to address for the future of scMulti-omics studies and integrative methods. One challenge is related to the computational issues in dealing with large data. Analyzing massive amounts of data from technologies such as RNA-seq, whole-genome sequencing, and ChIP-seq is an established problem due to their sheer size. Furthermore, single-cell methods have the computational burden of including information from hundreds or thousands of cells. These issues, combined with the multi-omics paradigm of integrating two or more technologies, will exacerbate the problem of dealing with big data. As a result, an important computational challenge to address is to determine how to more efficiently generate, manage, store, and analyze large datasets from a practical and economics standpoint. Since this point is not unique to omics analyses, let alone scMulti-omics methods, there is strong evidence that computational resources will increase. However, technologies allowing for more samples and more levels of information will also be developed, requiring further computational resources. Nevertheless, our capabilities to handle the currently available data will only increase.
Another challenge is related to the analytical capabilities of integrative tools. We have described methods with a variety of functionalities when integrating multi-omics data from both within the same cells and across experiments. However, to our knowledge, existing computational methods cannot perform several important functionalities. For example, from the standpoint of matched data, functionalities including identifying cis-regulatory motifs, finding cell-type-specific regulons, and inferring gene regulatory networks are not yet covered by existing integrative methods [59, 60]. While these functionalities may be addressed by methods that integrate data across experiments or even methods that analyze single-omics data [59, 61], there is a lack of integrative methods for matched data. With this type of integrative analysis still in its infancy, it is likely that more integrative methods will be developed to answer a wider range of complex biological questions. Furthermore, while there are options for integrating specific types of single-cell data, drawing inferences from the results is still troubling. Co-inference, the process of simultaneously making inferences from multiple single-cell data types, is quite a difficult challenge. Methods such as co-biclustering, co-imputation, and co-deconvolution [62] are not as straightforward as their singular counterparts. For scMulti-omics to truly provide comprehensive insights, these co-inference methods need to be explored and validated.
The need for a robust benchmarking pipeline is an even bigger concern for scMulti-omics methods than computational challenges. As the number of applicable methods grows, so does the potential benefit of an established benchmarking pipeline. Such a pipeline could retrospectively benchmark established methods or evaluate recently developed methods, allowing for robust scenario-specific validation of each approach. Furthermore, benchmarking pipelines can provide valuable insight into areas in which current methods underperform, highlighting areas of interest for future research and method improvement. Currently, there are benchmarking frameworks and evaluations of various computational methods for single-cell single-omics [63–65], however, no current method is available to scMulti-omics.
Concluding remarks and future perspectives
Integrative methods for scMulti-omics data provide the opportunity for researchers to jointly analyze different types of molecular information at the single-cell level, producing a more comprehensive view of cellular function. Rather than the traditional single-omics approach of studying biological processes from the genomics or transcriptomics perspective, multi-omics methods allow researchers to explore how two or multiple other realms interact and jointly produce biological observations. We pose several outstanding questions to be answered in the field of scMulti-omics analysis (see Outstanding Questions). Several future trends in scMulti-omics data analyses are listed below.
OUTSTANDING QUESTIONS.
How do the challenges in designing computational methods, from both infrastructure and algorithmic points of view, limit our understanding of single-cell multi-omics processes?
How reliably do methods that integrate data across experiments replicate the connections between data types?
What techniques would allow researchers to accurately conclude the reliable performance of integrating methods?
Would efforts toward a generation of a robust single-cell multi-omics tool that can integrate any type of omics data be more beneficial to the single-cell multi-omics field than explicit work towards detailed improvements and innovations in focused on defined data types?
To what extent can AI/ML be used to impute or infer the spatial information from single-cell multi-omics data?
How can AI/ML methods be used to effectively integrate numerous multi-omics data types across numerous experiments?
What challenges does the “black box” problem in AI/ML pose in determining the effectiveness of linking cross-experiment data?
Will single-cell multi-omics methods become so widespread that biologists begin to routinely collect data with these types of methods in mind?
How will single-cell multi-omics drive insights into heterogeneous regulation across the same or different cell states and tissue types?
How much insight from single-cell multi-omics can be gained in the scope of genomic variation—specifically intronic variation—and its impact on gene expression and regulation?
How will single-cell multi-omics’ inevitable implementation in the realm of precision medicine aid in the elucidation of specific mechanisms behind cancer cell development and aid in targeted therapies?
The application of new methods will inevitably lead to substantial improvements in scMulti-omics. One trend in single-omics single-cell analyses is the use of bulk RNA-seq data to impute scRNA-seq data [66–68]. The sparsity of scRNA-seq data stemming from low coverage and inefficiencies in sequencing methods means that the imputation of gene expression is a consistent hurdle in scRNA-seq analyses. Nevertheless, these issues will persist in scMulti-omics and must be dealt with accordingly.
Another trend throughout much of the computational sciences is the development of machine learning (ML) and artificial intelligence (AI) methods [69–72]. Within the scope of multi-omics, these approaches most presumably benefit the areas of imputation and matching of data across experiments [73–77]. Using ML/AI methods, researchers can potentially construct tools that will capture intricacies between data across experiments that cannot be manually identified and programmed into methods. This would allow for a more reliable linkage of multi-omics data, which will prove especially beneficial for benchmarking. One example is the autoencoder, which compresses input data and filters it through a bottleneck, followed by the decoding of the same data in an attempt to preserve the key defining features of the data.
Lastly, the nature of all technologies is to advance. From Sanger sequencing in the 1970s to next-generation sequencing of the late 1990s and early 2000s, methods for profiling omics data have continually evolved [78]. More recently, so-called third-generation sequencing provides an even greater opportunity to understand complex systems biology, although there is some difficulty in separating the generations of sequencing due to the rapid advances [79]. The most advanced methods are now considered fourth-generation sequencing, and they allow for in situ profiling, preserving the available spatial context of the data [80]. These advancing technologies will inevitably be integrated within multi-omics methods, requiring further development of technologies to integrate the new levels of information gained.
HIGHLIGHTS.
Applying integrative methods to single-cell multi-omics data opens a new window into the understanding of heterogeneous mechanism landscapes and cell-cell interactions.
Integration of cross-experiment data poses a special challenge.
A comprehensive understanding of the underlying methods is necessary to determine which pipeline is appropriate for a given single-cell multi-omic dataset.
We designed and implemented two case studies to demonstrate the application of available single-cell multi-omic tools, where new insights and practical challenges are generated.
Among the numerous remaining challenges in single-cell multi-omics, establishing a robust benchmarking pipeline is paramount.
Trends observed in traditional multi-omics, including machine learning, artificial intelligence, and evolving technologies, are paralleled in single-cell multi-omics methods.
Acknowledgments
This work was supported by an R01 award #1R01GM131399-01 from the National Institute of General Medical Sciences of the National Institutes of Health. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation #ACI-1548562. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Science Foundation and the National Institutes of Health. We would like to highly appreciate Mr. Cankun Wang and Mr. Zichun Zhang for their efforts in generating the case study.
GLOSSARY
- Artificial intelligence (AI)
a kind of machine algorithm processing data analysis with mimic cognitive functions.
- Autoencoder
is a type of multi-layer artificial neural network for unsupervised and efficient data coding and specifically useful for dimension reduction.
- Canonical correlation analysis
is a statistical method for investigating relationships between two data sets aiming to identify shared sources of variation in a pair of data sets (e.g. two scRNA-Seq data from different sources).
- Canonical correlation vectorization (CCV)
is a dimensional reduction method based on augmented implicitly restarted Lanczos bidiagonalization algorithm capturing features that are maximally correlated across multiple datasets. It takes datasets X1 … Xn and finds the projection vectors W = w1 … wn that optimize with .
- Coupled nonnegative matrix factorization (coupleNMF)
finds a subset of genes in unmatched data in which one modality is highly predictable from another. It considers a regression model and estimates parameters by fitting a penalized least-square problem based on the two modalities.
- CRISPR perturbation
is an artificial technique that induces specific genetic perturbations to the cells. It allows the controllable repression of gene expression by knocking out the corresponding genes or single nucleotides.
- Dimensional reduction
is the process of reducing the variables by identifying the principle features that explain data variations.
- Gradient boosting regression (GBR)
uses the idea of iteratively fitting “weak” models in order to build a “stronger” model that can more accurately estimate a response variable.
- Group factor analysis (GFA)
is an unsupervised dimension reduction approach that decomposes input matrices into a product of matrices. A set of m data matrices can be decomposed as Ym = ZWmT + εm where Ym are input matrices and Z denotes latent factors. Wm is amplitude matrix reflecting feature patterns, and εm denotes cell clusters.
- Hidden Markov random field (HMRF)
is a graph-based model for transfer pixel intensities over a 2D image using estimated variables from other modalities to reduce the spatial constraints and build connections between neighbor cells.
- Independent component analysis (ICA)
is a dimensional reduction method that decomposes observed data before ICA is applied to a low-rank matrix to capture underlying processes. The ICA model can be defined as modeling Y = AS where Y is the input, A is the mixing matrix, and S contains the independent components.
- Integrative non-negative matrix factorization (iNMF)
can be defined as Ei ≈ Hi (W + Vi) where Ei is the ith dataset, Hi contains the dataset-specific components, Vi approximates dataset-specific effects, W approximates the shared effects.
- LASSO regression
is a type of linear regression that shrinks regression coefficients by subjecting the coefficients to a constraint, allowing for feature selection and regularization.
- Machine learning (ML)
is an application of AI that automatically train parameters from known samples to predicting patterns from unknown samples, without explicit programming.
- Manifold alignment
infers pseudotime patterns by simultaneously measuring shared latent variables from multiple modalities. Each modality fits in a linear or non-linear model embedded to a squared exponential kernel, and the latent time variable can be found by maximizing the posterior distribution of a multivariate Gaussian.
- Multivariate normal modeling (MNM)
decomposes a data vector into spatial and nonspatial components, and can be defined as: y = f((x1, x2, …)) + ψ, where y is the spatial covariance, f is the spatial variance component that parametrizes covariance based on pairwise distances of samples of interest, (x1, x2, …) are the spatial coordinates, and ψ is an independent observation term that models the nonspatial variance component.
- Network simulated annealing
is a probability technique for approximating the global optimum to solve the combinatory problems in network construction.
- Omics is
a collection of comprehensive data reflecting the attribution of a particular molecular type, e.g. genomics, transcriptomics, proteomics, etc.
- Pseudotime
represents a theoretical timeline that estimated from the progression of input modality patterns.
- scHi-C
uses specific restriction enzymes to digest genome per individual cell allowing the examination of genome 3D organization [81]. It is very useful for predicting the topologically associated domains.
- Single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-Seq)
identifies the open regions on chromatins to indicate whether the corresponding gene is accessible to be bound by the transcriptome factors.
- Variational Bayes (VB)
derives a lower bound estimator (a stochastic objective function) for a variety of directed graphical models with continuous latent variables.
- Topic modeling
is a probabilistic generative model that used to aim to detect “topics” across a collection of documents. When applying in the bioinformatics area, it identifies a shared topic distribution across each input modality.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Lab webpage: https://u.osu.edu/bmbl/
GitHub webpage: https://github.com/OSU-BMBL
Twitter: Qin Ma (@QinMaBMBL), Anjun Ma (@anjunma90)
REFERENCES
- 1.Arendt D et al. (2016) The origin and evolution of cell types. Nat Rev Genet 17 (12), 744–757. [DOI] [PubMed] [Google Scholar]
- 2.S Teichmann ME (2020) Method of the Year 2019: Single-cell multimodal omics. Nature Methods 17 (1), 1–1. [DOI] [PubMed] [Google Scholar]
- 3.Gawad C et al. (2016) Single-cell genome sequencing: current state of the science. Nat Rev Genet 17 (3), 175–88. [DOI] [PubMed] [Google Scholar]
- 4.Linnarsson S and Teichmann SA (2016) Single-cell genomics: coming of age. Genome Biol 17 (1), 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang B et al. (2018) Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med 50 (8), 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haque A et al. (2017) A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med 9 (1), 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelsey G et al. (2017) Single-cell epigenomics: Recording the past and predicting the future. Science 358 (6359), 69–75. [DOI] [PubMed] [Google Scholar]
- 8.Lo PK and Zhou Q (2018) Emerging techniques in single-cell epigenomics and their applications to cancer research. J Clin Genom 1 (1), 10.4172/JCG.1000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fang Z et al. (2019) Single-Cell Heterogeneity Analysis and CRISPR Screen Identify Key beta-Cell-Specific Disease Genes. Cell Rep 26 (11), 3132–3144 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubin AJ et al. (2019) Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks. Cell 176 (1–2), 361–376 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Codeluppi S et al. (2018) Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods 15 (11), 932–935. [DOI] [PubMed] [Google Scholar]
- 12.Wang X et al. (2018) Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 361 (6400), eaat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stoeckius M et al. (2017) Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 14 (9), 865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peterson VM et al. (2017) Multiplexed quantification of proteins and transcripts in single cells. Nat Biotechnol 35 (10), 936–939. [DOI] [PubMed] [Google Scholar]
- 15.Leonavicius K et al. (2019) Multi-omics at single-cell resolution: comparison of experimental and data fusion approaches. Curr Opin Biotechnol 55, 159–166. [DOI] [PubMed] [Google Scholar]
- 16.Macaulay IC et al. (2017) Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends Genet 33 (2), 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Y et al. (2018) Single Cell Multi-Omics Technology: Methodology and Application. Front Cell Dev Biol 6, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Packer J and Trapnell C (2018) Single-Cell Multi-omics: An Engine for New Quantitative Models of Gene Regulation. Trends Genet 34 (9), 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stuart T and Satija R (2019) Integrative single-cell analysis. Nat Rev Genet 20 (5), 257–272. [DOI] [PubMed] [Google Scholar]
- 20.Tang X et al. (2019) The single-cell sequencing: new developments and medical applications. Cell & Bioscience 9 (1), 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keener A.B.J.N.m. (2019) Single-cell sequencing edges into clinical trials. 25 (9), 1322. [DOI] [PubMed] [Google Scholar]
- 22.BISResearch, Global Single Cell Multi-Omics Market: Focus on Global Single Cell Multi-Omics Market by Product, Type, Workflow, End-User 15 Countries Mapping, and Competitive Landscape - Analysis and Forecast: 2019–2025, Genomics Proteomics and Enabling Technology Market Trends, BIS Research, 2019, pp. 1–331. [Google Scholar]
- 23.Colomé-Tatché M and Theis FJ (2018) Statistical single cell multi-omics integration. Current Opinion in Systems Biology 7, 54–59. [Google Scholar]
- 24.Stuart T et al. (2019) Comprehensive Integration of Single-Cell Data. Cell 177 (7), 1888–1902 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.La Manno G (2019) From single-cell RNA-seq to transcriptional regulation. Nature Biotechnology 37 (12), 1421–1422. [DOI] [PubMed] [Google Scholar]
- 26.Welch JD et al. (2017) MATCHER: manifold alignment reveals correspondence between single cell transcriptome and epigenome dynamics. Genome Biol 18 (1), 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J et al. (2019) Jointly embedding multiple single-cell omics measurements. 644310. [DOI] [PMC free article] [PubMed]
- 28.Campbell KR et al. (2019) clonealign: statistical integration of independent single-cell RNA and DNA sequencing data from human cancers. Genome Biol 20 (1), 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao J et al. (2018) Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 361 (6409), 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lake BB et al. (2018) Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol 36 (1), 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu Q et al. (2018) Identification of spatially associated subpopulations by combining scRNAseq and sequential fluorescence in situ hybridization data. Nat Biotechnol 36, 1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edsgard D et al. (2018) Identification of spatial expression trends in single-cell gene expression data. Nat Methods 15 (5), 339–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svensson V et al. (2018) SpatialDE: identification of spatially variable genes. Nat Methods 15 (5), 343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Setty M et al. (2016) Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat Biotechnol 34 (6), 637–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duren Z et al. (2018) Integrative analysis of single-cell genomics data by coupled nonnegative matrix factorizations. Proc Natl Acad Sci U S A 115 (30), 7723–7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Argelaguet R et al. (2018) Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol 14 (6), e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adamson B et al. (2016) A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167 (7), 1867–1882 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duan B et al. (2019) Model-based understanding of single-cell CRISPR screening. Nat Commun 10 (1), 2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang S et al. (2017) More Is Better: Recent Progress in Multi-Omics Data Integration Methods. Front Genet 8, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finotello F et al. (2019) Next-generation computational tools for interrogating cancer immunity. Nat Rev Genet 20 (12), 724–746. [DOI] [PubMed] [Google Scholar]
- 41.Penaloza JS et al. (2019) Single-cell RNA-seq analysis of Mesp1-induced skeletal myogenic development. Biochem Biophys Res Commun 520 (2), 284–290. [DOI] [PubMed] [Google Scholar]
- 42.Mancuso R et al. (2019) Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat Neurosci 22 (12), 2111–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Z et al. (2019) More than one antibody of individual B cells revealed by single-cell immune profiling. Cell Discov 5 (1), 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morag S and Salmon-Divon M (2019) Characterizing Human Cell Types and Tissue Origin Using the Benford Law. Cells 8 (9), 1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alda-Catalinas C et al. (2019) A single-cell transcriptomics CRISPR-activation screen identifies new epigenetic regulators of zygotic genome activation. 741371. [DOI] [PMC free article] [PubMed]
- 46.Argelaguet R et al. (2019) Single cell multi-omics profiling reveals a hierarchical epigenetic landscape during mammalian germ layer specification. 519207.
- 47.Argelaguet R et al. (2019) Multi-omics profiling of mouse gastrulation at single-cell resolution. Nature 576 (7787), 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Argelaguet R et al. (2019) MOFA+: a probabilistic framework for comprehensive integration of structured single-cell data. 837104.
- 49.Welch JD et al. (2019) Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 177 (7), 1873–1887 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Angermueller C et al. (2016) Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods 13 (3), 229–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheow LF et al. (2016) Single-cell multimodal profiling reveals cellular epigenetic heterogeneity. Nat Methods 13 (10), 833–6. [DOI] [PubMed] [Google Scholar]
- 52.Dixit A et al. (2016) Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167 (7), 1853–1866 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dries R et al. (2019) Giotto, a pipeline for integrative analysis and visualization of single-cell spatial transcriptomic data. 701680.
- 54.Meng C et al. (2019) MOGSA: Integrative Single Sample Gene-set Analysis of Multiple Omics Data. Mol Cell Proteomics 18 (8 suppl 1), S153–S168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zeng W et al. (2019) DC3 is a method for deconvolution and coupled clustering from bulk and single-cell genomics data. Nat Commun 10 (1), 4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li G et al. (2019) Joint profiling of DNA methylation and chromatin architecture in single cells. Nat Methods 16 (10), 991–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clark SJ et al. (2018) scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun 9 (1), 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rodriques SG et al. (2019) Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. 363 (6434), 1463–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aibar S et al. (2017) SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14 (11), 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hawe JS et al. (2019) Inferring Interaction Networks From Multi-Omics Data. Front Genet 10 (535), 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Angermueller C et al. (2017) DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol 18 (1), 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chang W et al. (2019) A semi-supervised approach for cell phenotypic and functional estimation in tissue microenvironment. 426593.
- 63.Tian L et al. (2019) Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat Methods 16 (6), 479–487. [DOI] [PubMed] [Google Scholar]
- 64.Soneson C and Robinson MD (2018) Bias, robustness and scalability in single-cell differential expression analysis. Nat Methods 15 (4), 255–261. [DOI] [PubMed] [Google Scholar]
- 65.Saelens W et al. (2019) A comparison of single-cell trajectory inference methods. Nat Biotechnol 37 (5), 547–554. [DOI] [PubMed] [Google Scholar]
- 66.Peng T et al. (2019) SCRABBLE: single-cell RNA-seq imputation constrained by bulk RNA-seq data. Genome Biol 20 (1), 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li WV and Li JJ (2018) An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat Commun 9 (1), 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mongia A et al. (2019) McImpute: Matrix Completion Based Imputation for Single Cell RNA-seq Data. Front Genet 10 (9), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McDermaid A et al. (2018) A New Machine Learning-Based Framework for Mapping Uncertainty Analysis in RNA-Seq Read Alignment and Gene Expression Estimation. Front Genet 9 (313), 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ching T et al. (2018) Opportunities and obstacles for deep learning in biology and medicine. J R Soc Interface 15 (141). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tomasev N et al. (2019) A clinically applicable approach to continuous prediction of future acute kidney injury. Nature 572 (7767), 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu L and Zheng WJ (2018) Informatics, Data Science, and Artificial Intelligence. JAMA 320 (11), 1103–1104. [DOI] [PubMed] [Google Scholar]
- 73.Grapov D et al. (2018) Rise of Deep Learning for Genomic, Proteomic, and Metabolomic Data Integration in Precision Medicine. OMICS 22 (10), 630–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang L et al. (2018) Deep Learning-Based Multi-Omics Data Integration Reveals Two Prognostic Subtypes in High-Risk Neuroblastoma. Front Genet 9, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chaudhary K et al. (2018) Deep Learning-Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin Cancer Res 24 (6), 1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perakakis N et al. (2018) Omics, big data and machine learning as tools to propel understanding of biological mechanisms and to discover novel diagnostics and therapeutics. Metabolism 87, A1–A9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu T et al. (2018) A comprehensive review of computational prediction of genome-wide features. Briefings in bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sanger F and Coulson AR (1975) A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol 94 (3), 441–8. [DOI] [PubMed] [Google Scholar]
- 79.Schadt EE et al. (2010) A window into third-generation sequencing. Human Molecular Genetics 19 (R2), R227–R240. [DOI] [PubMed] [Google Scholar]
- 80.Ke R et al. (2016) Fourth Generation of Next-Generation Sequencing Technologies: Promise and Consequences. Hum Mutat 37 (12), 1363–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu J et al. (2018) Unsupervised embedding of single-cell Hi-C data. Bioinformatics 34 (13), i96–i104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Macaulay IC et al. (2015) G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods 12 (6), 519–22. [DOI] [PubMed] [Google Scholar]
- 83.Macaulay IC et al. (2016) Separation and parallel sequencing of the genomes and transcriptomes of single cells using G&T-seq. Nature Protocols 11, 2081. [DOI] [PubMed] [Google Scholar]
- 84.Han KY et al. (2018) SIDR: simultaneous isolation and parallel sequencing of genomic DNA and total RNA from single cells. Genome Res 28 (1), 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rodriguez-Meira A et al. (2019) Unravelling Intratumoral Heterogeneity through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol Cell 73 (6), 1292–1305 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li W et al. (2015) Single-cell transcriptogenomics reveals transcriptional exclusion of ENU-mutated alleles. Mutat Res 772, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dey SS et al. (2015) Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol 33 (3), 285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hu Y et al. (2016) Simultaneous profiling of transcriptome and DNA methylome from a single cell. Genome Biol 17 (1), 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu L et al. (2019) Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat Commun 10 (1), 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen S et al. (2019) High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nature Biotechnology 37 (12), 1452–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Granja JM et al. (2019) Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat Biotechnol 37 (12), 1458–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pott S (2017) Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. Elife 6, e23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Frei AP et al. (2016) Highly multiplexed simultaneous detection of RNAs and proteins in single cells. Nat Methods 13 (3), 269–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Genshaft AS et al. (2016) Multiplexed, targeted profiling of single-cell proteomes and transcriptomes in a single reaction. Genome Biol 17 (1), 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee D-S et al. (2018) Single-cell multi-omic profiling of chromatin conformation and DNA methylome. 503235.
- 96.Jaitin DA et al. (2016) Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 167 (7), 1883–1896 e15. [DOI] [PubMed] [Google Scholar]
- 97.Datlinger P et al. (2017) Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods 14 (3), 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hill AJ et al. (2018) On the design of CRISPR-based single-cell molecular screens. Nat Methods 15 (4), 271–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xie S et al. (2017) Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol Cell 66 (2), 285–299 e5. [DOI] [PubMed] [Google Scholar]
- 100.Shah S et al. (2018) Dynamics and Spatial Genomics of the Nascent Transcriptome by Intron seqFISH. Cell 174 (2), 363–376 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xia C et al. (2019) Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc Natl Acad Sci U S A 116 (39), 19490–19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moffitt JR et al. (2018) Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 362 (6416), eaau5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Asp M et al. (2017) Spatial detection of fetal marker genes expressed at low level in adult human heart tissue. Sci Rep 7 (1), 12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Moncada R et al. (2019) Integrating single-cell RNA-Seq with spatial transcriptomics in pancreatic ductal adenocarcinoma using multimodal intersection analysis. 254375.
- 105.Salmen F et al. (2018) Barcoded solid-phase RNA capture for Spatial Transcriptomics profiling in mammalian tissue sections. Nat Protoc 13 (11), 2501–2534. [DOI] [PubMed] [Google Scholar]
- 106.Ståhl PL et al. (2016) Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. 353 (6294), 78–82. [DOI] [PubMed] [Google Scholar]
- 107.Vickovic S et al. (2016) Massive and parallel expression profiling using microarrayed single-cell sequencing. Nat Commun 7 (1), 13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Giacomello S et al. (2017) Spatially resolved transcriptome profiling in model plant species. Nat Plants 3 (6), 17061. [DOI] [PubMed] [Google Scholar]
- 109.Hou Y et al. (2016) Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res 26 (3), 304–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bian S et al. (2018) Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 362 (6418), 1060–1063. [DOI] [PubMed] [Google Scholar]
- 111.Guo F et al. (2017) Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res 27 (8), 967–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li L et al. (2018) Single-cell multi-omics sequencing of human early embryos. Nat Cell Biol 20 (7), 847–858. [DOI] [PubMed] [Google Scholar]
- 113.Gu C et al. (2019) Integrative single-cell analysis of transcriptome, DNA methylome and chromatin accessibility in mouse oocytes. Cell Res 29 (2), 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mimitou EP et al. (2019) Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat Methods 16 (5), 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chung CY et al. (2019) Single-Cell Chromatin Analysis of Mammary Gland Development Reveals Cell-State Transcriptional Regulators and Lineage Relationships. Cell Rep 29 (2), 495–510 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]