

Abstract
Arteriovenous malformation (AVM) is a locally destructive congenital vascular anomaly caused by somatic mutations in MAP2K1. The mutation is isolated to endothelial cells (ECs). The purpose of this study was to determine the effects of mutant MAP2K1 on EC signaling and vascular network formation. Pathway effects were studied using both mutant MAP2K1 (K57N) human AVM tissue and human umbilical vein endothelial cells (HUVECs) engineered to overexpress the MAP2K1 (K57N) mutation. Western blot was used to determine cell signaling along the RAS/MAPK pathway. Geltrex tube formation assays were performed to assess EC vascular network formation. Cells were treated with a MAP2K1 inhibitor (Trametinib) to determine its effect on signaling and vascular tube formation. Human mutant MAP2K1-AVM ECs had similar baseline MEK1 and ERK1/2 expression with controls; however, mutant MAP2K1-AVM ECs produced significantly more phosphorylated ERK1/2 than wild-type ECs. Mutant MAP2K1 HUVECs demonstrated significantly more phosphorylated ERK1/2 than control HUVECs. Trametinib reduced the phosphorylation of ERK1/2 in mutant cells and prevented the ability of ECs to form vascular networks. AVM MAP2K1 mutations activate RAS/MAPK signaling in ECs. ERK activation and vascular network formation are reduced with Trametinib. Pharmacotherapy using MAP2K1 inhibitors may prevent the formation or progression of AVMs.
Keywords: arteriovenous malformation, malformation, mechanism, MAP2K1, RAS, MAPK, signaling, vascular
INTRODUCTION
Arteriovenous malformation (AVM) is a congenital vascular anomaly consisting of abnormal connections between arteries and veins through either a fistula or nidus instead of a normal capillary bed. Extracranial lesions enlarge and cause deformity, pain, ulceration, bleeding, and occasionally heart failure. We previously showed that most sporadic extracranial AVMs contain a mutation in MAP2K1 that is isolated to the endothelial cell (EC) [1]. MAP2K1 mutations in non-ECs may cause neoplasms (melanoma, lung, hematopoietic) and can increase MEK1 activity [2-11]. The effects of MAP2K1 mutations on EC function and AVM formation, however, are unknown. The purpose of this study was to determine if the most common AVM MAP2K1 mutation, MAP2K1-K57N, influences EC signaling and whether or not a MEK inhibitor affects mutant cells. Understanding how MAP2K1 mutations alter EC biology will provide insight into mechanisms by which AVMs form and enlarge.
MATERIALS AND METHODS
Specimen collection
The Committee on Clinical Investigation approved this study and informed consent was obtained. AVM tissue was collected during a clinically-indicated procedure and processed to separate endothelial cells (ECs) from non-ECs as we have previously described [1]. AVM specimen was washed in PBS to remove blood cell contaminants, digested with collagenase A (2.5 mg/mL) (Roche) for 1 hour at 37°C, then filtered through a 100 μm strainer to produce a single cell suspension. Cells were placed on fibronectin-coated (1 μg/cm2) tissue culture plates (Olympus Plastics) in endothelial growth medium-2 (EGM-2, PromoCell) supplemented with 10% fetal bovine serum (FBS, Gibco, Life Technologies). After 5-7 days of expansion, cells were fractionated into 2 populations (endothelial and non-endothelial) using anti-human CD31 (endothelial cell marker) magnetic beads (DynaBeads™, Life Technologies). DNA was extracted from each cell population using the DNeasy Blood & Tissue kit (Qiagen) and the mutant allele frequency (MAF) was determined using ddPCR as previously described [1]. CD31+ ECs and CD31- non-ECs were grown in endothelial cell growth medium (PromoCell) and mesenchymal stem cell growth medium (Lonza), respectively.
Plasmids
A pReceiver-M14 plasmid containing the human MAP2K1 ORF was obtained from Genecopoeia (Cat# EX-A0826-M14). The K57N mutation was introduced into the MAP2K1 ORF using the Agilent QuikChange II XL site directed mutagenesis kit (Cat# 200521) (FP: 5’-ctttcttacccagaatcagaaggtgggagaac RP: 5’-gttctcccaccttctgattctgggtaagaaag-3’). pLVX-puro-IreszsGreen1 lentiviral overexpression plasmids for wild-type and K57N MAP2K1 were generated using Infusion HD (Takara) in combination with a high fidelity DNA polymerase (Clone Amp, Takara). 3 PCR fragments were generated: a pLVX-Puro backbone (FP: 5’-gtcgacggtaccgcgggcccgggatc-3’; RP: 5’-tgcagaattcgaagcttgagctcg-3’; template pLVX-Puro (Takara), a MAP2K1 ORF (FP: 5’-gcttcgaattctgcatccaaaatgcccaagaagaagccgacgcccatc-3’; RP: 5’gagaggggttagacgccagcagcatgggttg; template: pReceiver-M14-MAP2K1 (wild-type or K57N)) and an IRES-zsGreen1 reporter ORF (FP: 5’-cgtctaacccctctccctcccccccccctaac-3’; RP: cgcggtaccgtcgactcagggcaaggcggagccggag-3’; template: pLVX-IRES-zsGreen1 (Takara)). The pLVX-puro-zsGreen1 empty vector control was generated with Infusion HD merging 2 PCR fragments: a pLVX-Puro backbone (FP 5’- gaattctgcagtcgacggtaccgcg-3’ RP: 5’-gaattcgaagcttgagctcgagatc-3’; template pLVX-Puro) and an IRES-zsGreen1 reporter ORF (FP: 5’- ctcaagcttcgaattccccctctccctcccccccccctaac −3’; RP: 5’-gtcgactgcagaattctcagggcaaggcggagccggag; template: pLVX-IRES-zsGreen1 (Takara). Lentiviral stocks were generated by the Massachusetts General Hospital Viral Vector Core.
Cell Lines
Endothelial colony forming cells (ECFCs) were isolated from human white adipose tissue as previously described [12]. ECFCs were cultured in fibronectin-coated flasks or plates maintained in EGM2 complete medium (EGM2 + endothelial growth supplement (PromoCell) + 20% FBS). Human umbilical vein endothelial cells (HUVECs) were obtained from ThermoFisher Scientific (Cat # C01510C) and were cultured in fibronectin-coated flasks or plates and maintained in EGM2 complete medium. The HUVEC-pLVX, HUVEC-pLVX-WT and HUVEC-pLVX-K57N cell lines were generated using lentiviral infection. 300,000 HUVECs were plated in one well of a 6-well plate. The next day the medium was replaced with 3 ml of EGM2 complete medium containing 8 μg/ml Hexadimethine Bromide (Sigma). 2.4 μl of lentiviral preparation [2.0 ×109 IU (Infectious Units)/mL] was added to the wells. 20 hrs later the lentivirus containing medium was replaced and cells were cultured for another 72 hours. Selection of infected cells was then performed using EGM2 complete medium containing 2 μg/mL puromycin (Invitrogen). On reaching confluency, HUVECs were passaged to a 25 cm2 flask, kept under puromycin selection, and allowed to reach confluency. Cells then were collected using 0.25% Trypsine-EDTA and sorted for zsGreen1 expression (Green fluorescent). Cells expressing zsGreen1 were plated in a 25 cm2 flask, kept in culture under puromycin selection, and expanded to a 75 cm2 flask. A fraction of the cells then was used to analyze the overexpression of wild type and K57N mutant MAP2K1, while the remainder of the cells were stored in liquid nitrogen. These cells were designated passage 1 and were not used later than passage 5 for our experiments.
Trametinib Treatment
Patient-derived AVM MAP2K1 ECs (MAF of 39%) and MAP2K1 engineered HUVECs were seeded on fibronectin coated dishes at 10,000 cells/cm2 in complete growth medium. After 24 hours, cells were incubated for 18 hours with DMSO (vehicle, 1:1000 in complete growth medium) or Trametinib (SelleckChem) at concentrations of 0.1μM, 1μM, or 10μM. Cells were lysed in the culture dish on ice using mammalian lysis buffer (Promega) containing protease and phosphatase inhibitor (Roche) for 10 minutes. Experiments were repeated 3 times.
Western Blot Analysis
Protein concentration was determined using a Pierce BCA Protein Assay Kit (Thermo Scientific). 15 μg of protein was separated on 4-20% gradient SDS-PAGE gels (Biorad; cat#: 456-1093). Because the protein components of the MAP2K1 signaling pathway (MEK1, ERK, P-ERK) and the loading controls (GAPDH and ACTIN) all have a similar molecular weight, a master loading mix was made for each sample. 20 μl (containing 15 μg protein) of this mastermix was loaded on 4 separate gels. The gels were transferred to a PVDF membrane (Invitrolon 0.45 μm pore; Life Technologies) and each membrane was detected using an antibody against one specific protein. Membranes were blocked in TBST containing 5% non-fat dry milk for 30 minutes at room temperature. Primary antibodies were diluted in blocking buffer and incubated with the membranes for 1 hour at room temperature. Membranes were washed 3 times for 5 minutes each with TBST buffer. Alkaline phosphate-coupled secondary antibodies were diluted in blocking buffer and incubated with the membranes for 30 minutes at room temperature followed by 3 washes with TBST. Membranes then were rinsed two times with water and incubated for 5 minutes with Tropix CDP star substrate (Applied Biosystems). Immuno-reactive bands were visualized using Hyblot CL autoradiography film (Denville). In order to determine the MAP2K1 mutation effects on cell signaling, the downstream target of MAP2K1 (ERK) was targeted. Activation of ERK was determined by its phosphorylation (PERK). Primary antibodies used were: anti-p44/42-ERK1/2 (Cell Signaling: #9102; 1/1000); anti-Phospho-p44/42 ERK1/2 (Cell Signaling: #9101; 1/1000); anti-MEK1 (MAP2K1) (Cell Signaling: 9124; 1/1000); anti-GAPDH (Cell signaling: #5174; 1/1000), and anti-beta-ACTIN (Sigma Aldrich: #A1978; 1/15,000). Secondary antibodies included: Goat anti-Rabbit IgG (H+L)-AP conjugated (Invitrogen: #31340; 1/10,000) and Goat anti-Mouse IgG (H+L)-AP conjugated (Invitrogen: #31320; 1/10,000). Western blots were repeated a minimum of 3 times.
Endothelial Cell Tube Formation Assay
Tube formation assays were performed using GelTrex (ThermoFisher Scientific, cat#: A1413202). GelTrex was thawed on ice and wells of a 24 well plate were coated with 300 μl GelTrex matrix and heated to 37°C for 30 minutes to allow the GelTrex to solidify. HUVECs then were plated in EGM2 complete medium on the surface of the GelTrex matrix at a density of 9000 cells/well (500 μl of an 18,000 cells/ml stock). Cells were placed in a 37°C CO2 incubator and the formation of a cell tube network was analyzed after 16 hours using an inverted microscope. GelTrex assays were done in triplicate.
Statistical analysis
Computation was performed used VassarStats [13]. Mean and standard deviation for pERK densitometry data was calculated. Groups were compared using the Mann-Whitney U-test. Statistical significance was defined as a p-value < 0.05.
RESULTS
Human AVM ECs were obtained from a patient who underwent resection of an AVM of the hand which had a confirmed MAP2K1-K57N mutation in ECs (MAF = 39%, passage 5). We found that MAP2K1-K57N mutant ECs do not have a proliferative advantage because continuous cultivation revealed a loss of mutant ECs, and by passage 10 mutant ECs were lost from the cultures. Thus, we only used cells up to passage 5 for our experiments.
Because it is not possible to obtain ECs from a non-affected part of a patient undergoing resection of an AVM, we used human white adipose tissue (HWAT) extracted ECFCs as a control. These cells display a stable endothelial phenotype and have robust in vivo blood vessel-forming capacity [12]. Western blot analysis showed that MAP2K1-K57N-AVM ECs had similar baseline MEK1 and ERK1/2 expression as compared to control HWAT-ECFCs. However, MAP2K1-K57N AVM ECs had a significant increase in the levels of phosphorylated and thus active ERK1/2 protein compared to ECFCs (mean increase 622% ±244%, p<0.05) (Figure 1).
Figure 1:

The MAP2K1 K57N mutation increases ERK1/2 phosphorylation in endothelial cells (ECs). (A) Western blot illustrates MEK, ERK, and P-ERK protein levels in control cells (HWAT ECFCs) and AVM ECs containing a MAP2K1 K57N mutation. Note the higher levels of phosphorylated ERK1/2 in the mutant ECs. (B) (Left) Schematic of lentiviral plasmids used to generate engineered HUVECs that overexpress either wild-type or MAP2K1 K57N mutant protein. (Right) Western blot depicting MEK1, ERK, and P-ERK expression in lentiviral infected HUVECs. Higher expression levels of P-ERK protein are detected in HUVECs infected with K57N MAP2K1 overexpressing lentivirus compared to HUVECs infected with empty (pLVX) lentivirus or wild-type MAP2K1 overexpressing lentivirus (WT).
To independently verify that the higher P-ERK levels in mutant AVM-ECs were the result of the MAP2K1-K57N mutation, we used lentiviral infection to overexpress either wild-type MAP2K1 or K57N mutant MAP2K1 in HUVECs. HUVECs infected with the empty lentiviral vector were used as control cells. Western analysis showed a moderate increase in MAP2K1 protein levels in cells infected with either wild-type or mutant MAP2K1 overexpressing vectors. While P-ERK levels in cells overexpressing wild type MAP2K1 was only slightly upregulated, a strong increase in P-ERK proteins levels was found in HUVECs overexpressing MAP2K1-K57N compared to both empty vector (mean increase 426%±164%, p<0.05) and overexpressing wild-type (mean increase 345%±135%, p<0.05) confirming the results obtained in patient derived AVM-ECs.
The FDA approved MEK1(MAP2K1) inhibitor Trametinib has been used to treat neoplasms that contain activating MAP2K1 mutations [14] and has been used off-label to treat AVMs [15]. To investigate whether Trametinib was able to counteract the increased phosphorylated ERK1/2 protein level in mutant cells, we exposed AVM derived ECs to increasing concentrations of Trametinib (Figure 2). A Trametinib concentration of 0.1 μM was able to reduce the P-ERK level in AVM ECs to the level of the control ECFCs. Finally, we tested whether Trametinib could influence vessel formation. Exposure of both AVM-derived ECs and MAP2K1-K57N overexpressing HUVECs to Trametinib reduced the ability of mutant ECs to form vascular networks (Figure 3).
Figure 2:
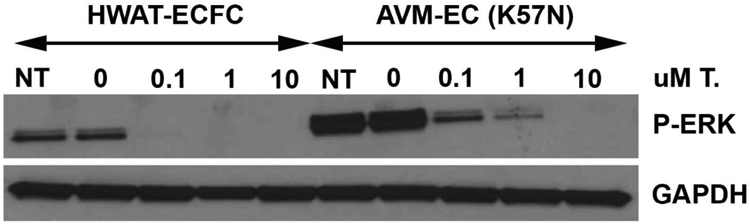
Western blot shows that a MAP2K1 inhibitor (Trametinib) reduces P-ERK activation in both ECFCs and human mutant MAP2K1 AVM ECs (K57N) in a dose-dependent manner. Note that elevated doses are necessary to inhibit the upregulated ERK signaling in mutant cells.
Figure 3:

Trametinib inhibits MAP2K1 mutant endothelial cell vascular network formation. (A) GelTrex assay using mutant human AVM ECs containing the MAP2K1-K57N mutation. (Above) Vascular network formation without treatment. (Below) Failed network development when treated with Trametinib (10 μM). (B) GelTrex assay using lentiviral infected HUVECs with the MAP2K1-K57N mutation. (Above) Vascular network formation in the absence of treatment. (Below) Inhibition of network formation in the presence of Trametinib (10 μM).
DISCUSSION
Our data show that the most common AVM mutation, MAP2K1-K57N, over-activates the RAS/MAPK signaling pathway in ECs. This finding is consistent with previous reports that the MAP2K1 mutation is activating in other cell types that cause cancer [8,9], and when overexpressed in HEK293T embryonic kidney cells [11]. MAPK signaling is enhanced by receptor tyrosine kinases, integrins, and G-protein coupled receptors; MAP2K1 phosphorylates ERK1 and ERK2 [16].
In mammals, this cascade plays a crucial role in development, including fate determination, differentiation, proliferation, survival, migration, growth and apoptosis [17-19]. Interestingly, we have consistently found that prolonged cultivation of AVM derived ECs lowers the MAF in the cultures. This suggests that mutant ECs do not have a proliferative advantage despite their increased RAS/MAP2K1 signaling. We hypothesize that stimulation of RAS/MAPK signaling by mutant ECs might lead to abnormal coordination of artery-capillary-vein formation. The fundamental pathological finding in AVMs is the connection of arteries to veins through a nidus or fistula instead of a normal capillary bed [20]. MAP2K1 mutant ECs affecting normal vascular development might be the initiating stimulus that causes the pathological connection of arteries to veins. Absence of capillaries in AVMs reduces oxygen delivery to tissues leading to ischemia, ulceration, bleeding, and pain. Reactive neovascularization then contributes to enlargement of the AVM.
Our finding that Trametinib blocks mutant MAP2K1 upregulated signaling suggests that it might prove effective for AVMs, similar to its role in treating MAP2K1 dependent neoplasms (i.e., melanoma, lung adenocarcinoma) [21,22]. Trametinib also stopped vascular network formation. MAP2K1 inhibition of upregulated EC signaling might prevent the formation and progression of AVMs; regression of lesions also might occur. This hypothesis is supported by a recent case report showing reduction in the size of an AVM treated with Trametinib [15].
Highlights.
MAP2K1 mutation (K57N) increased activation of MAP2K1 in endothelial cells as measured by phosphorylation of its downstream target ERK
Inhibition of MAP2K1 with the FDA-approved drug Trametinib restored normal MAP2K1 activity
Mutant vascular networks did not form in the presence of a MAP2K1 inhibitor
ACKNOWLEDGEMENTS
Research reported in this publication was supported by the Eunice Kennedy Shriver National Institute Of Child Health & Human Development of the National Institutes of Health under Award Number R01HD093735 (AKG) and the Translational Research Program Boston Children’s Hospital (AKG). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- [1].Couto JA, Huang AY, Konczyk DJ, Goss JA, Fishman SJ, Mulliken JB, Warman ML, Greene AK, Somatic MAP2K1 Mutations Are Associated with Extracranial Arteriovenous Malformation, Am. J. Hum. Genet 100 (2017) 546–554. 10.1016/j.ajhg.2017.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Arcila ME, Drilon A, Sylvester BE, Lovly CM, Borsu L, Reva B, Kris MG, Solit DB, Ladanyi M, MAP2K1 (MEK1) mutations define a distinct subset of lung adenocarcinoma associated with smoking, Clin. Cancer Res 21 (2015) 1935–1943. 10.1158/1078-0432.CCR-14-2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bansal A, Ramirez RD, Minna JD, Mutation analysis of the coding sequences of MEK1 and MEK-2 genes in human lung cancer cell lines, Oncogene. 14 (1997) 1231–1234. 10.1038/sj.onc.1200947. [DOI] [PubMed] [Google Scholar]
- [4].Chakraborty R, Hampton OA, Shen X, Simko SJ, Shih A, Abhyankar H, Lim KPH, Covington KR, Trevino L, Dewal N, Muzny DM, Doddapaneni H, Hu J, Wang L, Lupo PJ, Hicks MJ, Bonilla DL, Dwyer KC, Berres ML, Poulikakos PI, Merad M, McClain KL, Wheeler DA, Allen CE, Parsons DW, Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis, Blood. 124 (2014) 3007–3015. 10.1182/blood-2014-05-577825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Choi YL, Soda M, Ueno T, Hamada T, Haruta H, Yamato A, Fukumura K, Ando M, Kawazu M, Yamashita Y, Mano H, Oncogenic MAP2K1 mutations in human epithelial tumors, Carcinogenesis. 33 (2012) 956–961. 10.1093/carcin/bgs099. [DOI] [PubMed] [Google Scholar]
- [6].Estep AL, Palmer C, McCormick F, Rauen KA, Mutation analysis of BRAF, MEK1 and MEK2 in 15 Ovarian Cancer cell lines: Implications for therapy, PLoS One. 2 (2007) e1279 10.1371/journal.pone.0001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, Harshman K, Guipponi M, Bukach O, Zoete V, Michielin O, Muehlethaler K, Speiser D, Beckmann JS, Xenarios I, Halazonetis TD, Jongeneel CV, Stevenson BJ, Antonarakis SE, Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma, Nat. Genet 44 (2012) 133–139. 10.1038/ng.1026. [DOI] [PubMed] [Google Scholar]
- [8].Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Vande Woude GF, Ahn NG, Transformation of mammalian cells by constitutively active MAP kinase kinase, Science (80-. ). 265 (1994) 966–970. 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- [9].Marks JL, Gong Y, Chitale D, Golas B, McLellan MD, Kasai Y, Ding L, Mardis ER, Wilson RK, Solit D, Levine R, Michel K, Thomas RK, Rusch VW, Ladanyi M, Pao W, Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma, Cancer Res. 68 (2008) 5524–5528. 10.1158/0008-5472.CAN-08-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sherry ST, Ward M, Sirotkin K, Use of molecular variation in the NCBI dbSNP database, Hum. Mutat 15 (2000) 68–75. . [DOI] [PubMed] [Google Scholar]
- [11].Al-Olabi L, Polubothu S, Dowsett K, Andrews KA, Stadnik P, Joseph AP, Knox R, Pittman A, Clark G, Baird W, Bulstrode N, Glover M, Gordon K, Hargrave D, Huson SM, Jacques TS, James G, Kondolf H, Kangesu L, Keppler-Noreuil KM, Khan A, Lindhurst MJ, Lipson M, Mansour S, O’Hara J, Mahon C, Mosica A, Moss C, Murthy A, Ong J, Parker VE, Rivière JB, Sapp JC, Sebire NJ, Shah R, Sivakumar B, Thomas A, Virasami A, Waelchli R, Zeng Z, Biesecker LG, Barnacle A, Topf M, Semple RK, Patton EE, Kinsler VA, Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy, J. Clin. Invest 128 (2018) 1496–1508. 10.1172/JCI98589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lin RZ, Moreno-Luna R, Muñoz-Hernandez R, Li D, Jaminet SCS, Greene AK, Melero-Martin JM, Human white adipose tissue vasculature contains endothelial colony-forming cells with robust in vivo vasculogenic potential, Angiogenesis. 16 (2013) 735–744. 10.1007/s10456-013-9350-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lowry R, VassarStats: Website for Statistical Computation, (2019). http://vassarstats.net/.
- [14].Caunt CJ, Sale MJ, Smith PD, Cook SJ, MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road, Nat. Rev. Cancer 15 (2015) 577–592. 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- [15].Lekwuttikarn R, Lim YH, Admani S, Choate KA, Teng JMC, Genotype-Guided Medical Treatment of an Arteriovenous Malformation in a Child, JAMA Dermatology. 155 (2019) 256–257. 10.1001/jamadermatol.2018.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Greene AK, Goss JA, Vascular Anomalies: From a Clinicohistologic to a Genetic Framework, Plast. Reconstr. Surg 141 (2018) 709e–717e. 10.1097/PRS.0000000000004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fischer AM, Katayama CD, Pagès G, Pouysségur J, Hedrick SM, The role of Erk1 and Erk2 in multiple stages of T cell development, Immunity. 23 (2005) 431–443. 10.1016/j.immuni.2005.08.013. [DOI] [PubMed] [Google Scholar]
- [18].Johnson GL, Vaillancourt RR, Sequential protein kinase reactions controlling cell growth and differentiation, Curr. Opin. Cell Biol 6 (1994) 230–238. 10.1016/0955-0674(94)90141-4. [DOI] [PubMed] [Google Scholar]
- [19].Ussar S, Voss T, MEK1 and MEK2, different regulators of the G1/S transition, J. Biol. Chem 279 (2004) 43861–43869. 10.1074/jbc.M406240200. [DOI] [PubMed] [Google Scholar]
- [20].Gupta A, Kozakewich H, Histopathology of vascular anomalies, Clin. Plast. Surg 38 (2011) 31–44. 10.1016/j.cps.2010.08.007. [DOI] [PubMed] [Google Scholar]
- [21].Hirsch FR, Suda K, Wiens J, Bunn PA, New and emerging targeted treatments in advanced non-small-cell lung cancer, Lancet. 388 (2016) 1012–1024. 10.1016/S0140-6736(16)31473-8. [DOI] [PubMed] [Google Scholar]
- [22].Zhu Z, Liu W, Gotlieb V, The rapidly evolving therapies for advanced melanoma-Towards immunotherapy, molecular targeted therapy, and beyond, Crit. Rev. Oncol. Hematol 99 (2016) 91–99. 10.1016/j.critrevonc.2015.12.002. [DOI] [PubMed] [Google Scholar]