

Abstract
Background
Recent impressive advances in cancer immunotherapy have been largely derived from cellular immunity. The role of humoral immunity in carcinogenesis has been less understood. Based on our previous observations we hypothesize that an immunoglobulin subtype IgG4 plays an essential role in cancer immune evasion.
Methods
The distribution, abundance, actions, properties and possible mechanisms of IgG4 were investigated with human cancer samples and animal tumor models with an extensive array of techniques both in vitro and in vivo.
Results
In a cohort of patients with esophageal cancer we found that IgG4-containing B lymphocytes and IgG4 concentration were significantly increased in cancer tissue and IgG4 concentrations increased in serum of patients with cancer. Both were positively related to increased cancer malignancy and poor prognoses, that is, more IgG4 appeared to associate with more aggressive cancer growth. We further found that IgG4, regardless of its antigen specificity, inhibited the classic immune reactions of antibody-dependent cell-mediated cytotoxicity, antibody-dependent cellular phagocytosis and complement-dependent cytotoxicity against cancer cells in vitro, and these effects were obtained through its Fc fragment reacting to the Fc fragments of cancer-specific IgG1 that has been bound to cancer antigens. We also found that IgG4 competed with IgG1 in reacting to Fc receptors of immune effector cells. Therefore, locally increased IgG4 in cancer microenvironment should inhibit antibody-mediated anticancer responses and help cancer to evade local immune attack and indirectly promote cancer growth. This hypothesis was verified in three different immune potent mouse models. We found that local application of IgG4 significantly accelerated growth of inoculated breast and colorectal cancers and carcinogen-induced skin papilloma. We also tested the antibody drug for cancer immunotherapy nivolumab, which was IgG4 in nature with a stabilizing S228P mutation, and found that it significantly promoted cancer growth in mice. This may provide an explanation to the newly appeared hyperprogressive disease sometimes associated with cancer immunotherapy.
Conclusion
There appears to be a previously unrecognized immune evasion mechanism with IgG4 playing an essential role in cancer microenvironment with implications in cancer diagnosis and immunotherapy.
Keywords: immunotherapy, immune evasion, antibody specificity, tumor microenvironment
Introduction
While new immune therapy for cancer focuses mostly on manipulating cellular immunity, humoral immunity also holds great promise for cancer therapy.1 2 IgG4 is a unique antibody that has the lowest concentration among IgG subtypes in healthy individuals, and its function has not been well understood.3–5 IgG4 was regarded as a ‘blocking antibody’ because of its reduced ability to trigger effector immune reactions.6 7 Therefore whatever molecules IgG4 reacts to, the subsequent immune reaction was subdued.8 IgG4 has a unique structure of Fab arm exchange (FAE) in which the two heavy and light chains of two different antibodies with different specificities are exchanged, resulting in an asymmetric bispecific antibody with reduced ability to bind to antigen and to form immune complexes.9 Another unique feature of IgG4 is that it can react to other IgGs via its Fc fragment, and the significance of this property has not been well understood. Evidence suggests that FAE and Fc-Fc reactivity may involve the same molecular structure on IgG4 molecule.10 Davies et al11 12 resolved the crystal structure of IgG4 Fc fragment revealing a unique molecular conformation supporting its Fc binding property. Recent interests in IgG4-related diseases unveiled a wide range of pathologies with a common phenomenon of often increased IgG4 concentration in the serum and IgG4-postive plasma cells in the affected organs accompanied by local inflammation and fibrosis, but its pathogenic mechanism is still poorly understood.13–15
The possible functions of IgG4 in cancer, and also in the immune system, have not been well elucidated. Increases of IgG4-positive plasma cells were reported in gastrocarcinoma,16 extrahepatic cholangiocarcinoma17 and melanoma.18 The above studies were performed on limited number of cases without convincing explanation of mechanism or significance. Wu et al19 reported that serum IgG4:IgG ratio could predict recurrence of hepatocellular carcinoma after surgery. The most extensive study on IgG4 and cancer was performed by Karagiannis et al,20 who investigated the possible effect of cancer-specific IgG4 in inhibiting cancer immunity in melanomas and suggested competition between cancer-specific IgG4 and IgG1 in binding to cancer antigens as the cause for the inhibition. A recent report raised the concept of cancer-educated B cells and toxic IgG produced by these cells in facilitating lymph node metastasis for breast cancer in a mouse model.21 We performed a multidimensional investigation of IgG4 in a wide array of patients with cancer and tissues with both in vitro and in vivo experiments. Extensive new evidence led us to hypothesize that there is a potent humoral immune editing mechanism in cancer microenvironment, with IgG4 playing an essential role. We propose that Fc-Fc reaction could be the basic mechanism of this immune inhibition. We validated this in three immune potent animal models. This property was also found applicable to cancer immunotherapy drug nivolumab, which was IgG4 in nature. Our study points to a so far little appreciated mechanism of immune evasion in cancer.
Materials and methods
Key resources
Detailed information about key resources including antibodies, biological samples, chemicals, assay kits, cell lines and software are shown in online supplementary table 1.
jitc-2020-000661supp001.pdf (12.9MB, pdf)
Experimental model and subject details
Patients and healthy volunteers
We collected tissue and blood samples from over 100 patients with cancer in Shantou University Affiliated Tumor Hospital and the East Guangdong Provincial Pathological Consultation Center. Details of the human samples are shown in online supplementary table 2.
Immunohistochemistry
Details of the protocols and the antibodies are shown in online supplementary data.
SDS technique
The expressions of IgG1, IgG2, IgG3 and IgG4 in esophageal cancer were detected at the histological level. The stain-decolorize-stain (SDS) technology22 was performed on the same slide sequentially with four different antibodies to demonstrate the four antigens. The distribution pattern, abundance and relationship of the four antigens were processed with an image software (Photoshop) to achieve an integrated figure. The proximity of different cell types on the tissue sections was measured with an image analyzing software Image-Pro Plus V.6.0. Details of the protocol are presented in the online supplementary data.
Immunofluorescence double staining
Two antibodies from different species were incubated on the same tissue section. Primary antibodies were detected with goat antimouse 555/antirabbit 488 IgG or goat antimouse 488/antirabbit 555 IgG (Alexa Fluor, Life Sciences), and immunoreactivity was visualized with a fluorescence microscope. Antigens were detected and demonstrated with two fluorescence signals in red and green, and the blue signal of 4',6-diamidino-2-phenylindole (DAPI) as for cell nuclei. Images were acquired with the EVOS FL fluorescence microscope (Life Technologies, USA).
IgG4 immunohistochemistry
To verify that IgG4 could react to IgG1 that had been immobilized to tissue sections, we used human pancreas and brain and antibodies to insulin, glucagon and neurofilament as models. Detailed protocol of this experiment is shown in the online supplementary data.
Immunoglobulin preparations
Fab and Fc fragments of IgG, IgG1 and IgG4 were prepared with papain digestion. In the presence of cysteine, IgGs were cleaved at a position above the hinge region by papain at 37°C for 5 hours. After purification with Protein A affinity chromatography, pure Fab and Fc fragments were isolated. With elution buffer, IgG Fc fragment was washed down from Protein A column. After centrifugation and concentration measurement, IgG preparations were collected and stored at 4°C before use.
Western blot
IgG subclasses were resolved on 10% sodium dodecyl sulfate-polyacrylamide gels under reducing conditions and then transferred onto nitrocellulose membranes (GE Healthcare, Life Sciences). The membranes were blocked for 1 hour with 5% bovine serum albumin (BSA) in Tris-buffered saline, with Tween-20, pH 7.6, and then incubated with primary antibody (Biotin Labeling Kit, AnaSpec) overnight at 4°C. It was then incubated with secondary antibody for 1 hour at room temperature. Finally reaction density was measured with Odyssey western at 680 nm and 800 nm detection channel.
Serum IgG4 and IgG assessment
Sera samples collected from 82 patients with esophageal cancer and 70 healthy volunteers were sent to Golden Field Medical Test Company (Guangzhou, China), and Roche immune turbidimetry method was used to measure serum IgG4 and total IgG concentrations. All serum samples were stored at −80°C freezer immediately before analysis. All quantitative data were treated statistically.
Measurement of IgG4 concentrations in tumor and tumor-adjacent normal tissues with ELISA
Concentrations of IgG4 in 46 pairs of esophageal cancer and adjacent normal tissues (5 cm from the edge of the cancer mass) were measured with ELISA. Detailed protocol of this experiment is shown in the online supplementary data.
Cell culture for Fc receptor binding assays
U937 cell line was bought from Procell Life Science & Technology China (#CL-0239), and cells were cultured in RPMI 1640 (Gibco, #C22400500BT) supplemented with 10% fetal bovine serum (Gibco, #10 099–141) and 100 U/mL penicillin-streptomycin (Gibco, # 15 140–122) at 1×106/mL in 25 mL cell culture bottle.
Protein preparations
Details of the protocols for protein preparation, separation and papain digestion are shown in online supplementary data.
Fluorescence activating cell sorter (FACS) for Fc receptor assays
Details of the relevant protocols are shown in online supplementary data.
ADCC, ADCP and CDC tests
The classic antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP) and complement-dependent cytotoxicity (CDC) tests were performed based on previously reported protocols.23–31 Non-specific IgG4, instead of cancer-specific IgG4, was used to inhibit these reactions. IgG1 was used as control. Details of the protocols are shown in online supplementary data.
Animal models
Breast cancer and colon cancer models
BALB/c mice were obtained from Vital River Technical (Beijing, China). Mice aged between 6 and 8 weeks and weighed 20±2 g were used in all experiments. All mice were inoculated with 4T1 mouse breast cancer cells or CT26 mouse colon cancer cells subcutaneously under the left forearm, 1×105 4T1 cells per mouse to build cancer models. The mice were divided into different groups and were treated with IgG1 or IgG4 derived from intravenous IgG (IVIG), IVIG without IgG4, nivolumab and Fc of nivolumab, respectively. Details of the protocols are shown in online supplementary data.
Carcinogen-induced skin tumor model
We employed a well-established carcinogen-induced skin tumor model to study the effect of IgG4 and IVIG without IgG4 in comparison with controls (phosphate buffered saline (PBS)). Detailed protocol is shown in online supplementary data.
Quantification and statistical analysis
Data were analyzed in GraphPad Prism. All reported p values were derived from two-sided comparisons, with values of less than 0.05 considered to be statistically significant.
Results
IgG4 was significantly increased in the serum of patients with cancer and this increase was related to cancer stage and patient prognosis
We first measured the concentrations of IgG1, IgG2, IgG3 and IgG4 in sera of patients with esophageal cancer (n=82). IgG4 was significantly increased in patients with cancer in comparison with normal healthy subjects (n=70). The concentration of IgG4 was increased from about 3% to 11%. The ratio of IgG4:IgGtotal was also significantly increased. The statistical significance of both reached p<0.0001. The increase of IgG4 was also positively correlated to the stages of cancer, with late stage cancers having more obvious increases. Higher IgG4 serum concentrations were associated with worse prognosis (figure 1A–D).
Figure 1.

Significant increase of IgG4 and IgG4:IgGtotal in serum and IgG4-positive B lymphocytes in esophageal cancer. (A) IgG4 in serum of esophageal squamous cell cancer (ESCC) (n=82) was significantly higher when compared with healthy controls (n=70) (p<0.0001). (B) IgG4:IgGtotal ratio in ESCC (n=82) was significantly higher than that in matching healthy adults (n=70) (p<0.0001). (C) IgG4 in stage Ⅳ (n=18) was significantly higher than those in stages Ⅰ and Ⅱ (n=16) (p<0.01). (D) IgG4:IgGtotal in serum of stage IV ESCC (n=18) was significantly higher than those in stages Ⅰ and Ⅱ (n=16) (p<0.05). (E) Scatter diagram of IgG4-positive cell numbers in cancer (cancer), adjacent normal tissue (adjacent) and normal tissues. IgG4-positive lymphocytes in and around the esophageal cancer mass (n=110) are significantly more abundant than those in the adjacent normal tissue (n=60) and normal lymphoid tissues (n=63) (***p<0.001 for both). Increases of IgG4-positive lymphocytes were most abundant in areas of cancer cell proliferation. (F) The increase of IgG4-positive cell numbers was related to the prognoses of the patients. More IgG4-positive cells were associated with worse outcome (p<0.05). (G) IgG4 concentration in cancer tissue (n=46) was significantly higher than that in adjacent normal tissue (n=46) (p<0.01). (H) Immunohistochemistry of IgG4 in esophageal cancer tissues. From left to right are IgG4 in cancer tissues, cancer-adjacent tissue and normal lymphoid tissue (tonsil). It clearly demonstrates that IgG4-positive lymphocytes (red) were markedly increased (left) in comparison with normal lymphoid tissue (right) and with tumor-adjacent normal tissue (middle). Scale bar: 100 µm. (I) Demonstration of four subpopulations of IgG-containing plasma cells with multiple immunostaining (SDS method) in cancer. Each subclass has its own distribution pattern and one plasma cell only produces one subclass of IgG. IgG1: yellow; IgG2: green; IgG3: purple; IgG4: red. (J) On the same tissue section, a triple immunostaining was performed with the SDS method to demonstrate the distribution and relationship among CD3-positive T cells (yellow), IgG1-positive B cells (greens) and IgG4-positive B cells (red). Each cell type has its own distribution, and no overlap between different cell types is observed. SDS, stain-decolorize-stain.
IgG4-containing lymphocytes and IgG4 concentration were significantly increased in cancer microenvironment and this increase was associated with cancer cell infiltration
IgG4-positive lymphocytes were significantly increased in cancer microenvironment in comparison with tissue more distant to the cancer mass. IgG4-positive lymphocytes were barely detectable in tumor-adjacent normal tissue (figure 1H). On the same tissue sections employing the SDS technique22 to simultaneously visualize the four subtypes of IgG with four distinct colors, we found that one plasma cell only contained one IgG subtype, that is, IgG1, IgG2, IgG3 and IgG4 were all contained in their own plasma cell populations separately (figure 1I). The marked increase of IgG4-containing plasma cells in comparison with other subtypes was clearly visualized. On cancer tissue sections, IgG4-positive plasma cells appeared to accumulate more in tissues with extensive cancer cell infiltration than in other areas (figure 1E, H). In addition, IgG4-positive cells are often in close proximity to cells containing IgG1 and IgG2 but not to IgG3. This phenomenon was not seen among other IgG subtypes apart from IgG4. Quantitative data of the proximity of different cell types are presented in online supplementary figure S2. With the multiple immunostaining method, we also demonstrated that IgG1-containing and IgG4-containing lymphocytes were distinct from CD3-positive T lymphocytes in the same region of the cancer tissue (figure 1J). In addition, we measured the concentrations of IgG4 in cancer tissue and cancer-adjacent normal tissue (n=46 pairs). The average concentration of IgG4 in cancer tissue was about four times higher than that in the adjacent normal tissue, and the difference between these two groups was statistically significant (p<0.01) (figure 1G).
IgG1 extracted from patients with cancer reacted to cancer cells of the same patients but IgG4 extracted from the same patients did not
We extracted IgG1 and IgG4 from the serum of 10 patients with esophageal cancer, 10 with breast cancer and 2 with colon cancer with respective specific antibody columns. We then labeled the antibodies with biotin and tested the reactivity of these extracted antibodies to cancer tissue sections of the same patients. In all cases IgG1 reacted to cancer cells from the same patients but IgG4 did not (figure 2A). It appeared that the increased IgG4 in cancer microenvironment and in the patient’s serum was not reactive to cancer antigens, while IgG1 extracted from the same patients reacted to the cancer antigens.
Figure 2.
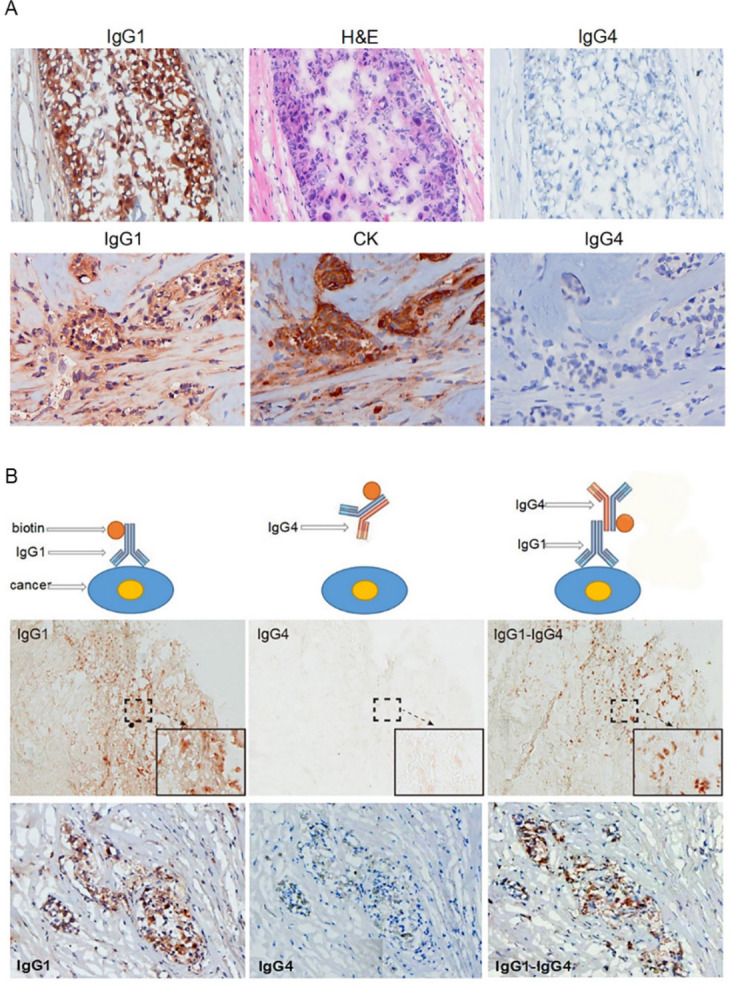
IgG4 extracted from a patient with cancer reacted to cancer-bound IgG1 and blocked antibody-mediated cancer immunity. (A) Upper panel: These photos serve as an example of the reactivity of IgG1 and IgG4 extracted from patients with cancer. IgG1 from the serum of a patient with breast cancer was labeled with biotin and stained a frozen cancer tissue section of the same patient. Cancer cells were positively stained by IgG1 (left). The cancer cells were confirmed by their characteristic histopathology of H&E staining (middle). IgG4 from serum of the same patient labeled with biotin and applied on the same cancer on a consecutive section was completely negative (right). Lower panel: Another breast cancer positively stained by IgG1 from the patient’s serum (left). The cancer cells were identified by positive immunostaining of cytokeratin (CK) on a consecutive section (middle). IgG4 from the same patient was not reactive to the same cancer on a consecutive section (right). (B) The upper panel illustrates the principle of the experimental reactions, and the middle and lower panels show staining results from two patients with breast cancer. Left: IgG1 from a patient with cancer positively reacted to frozen cancer tissue of the same patient (brown cells). Middle: IgG4 from the same patient with cancer applied to consecutive sections, but did not react to the same cancer. Right: However, when unlabeled IgG1 was applied to the same cancer tissue section followed by biotin-labeled IgG4, the cancer cells were positively stained (brown cells).
IgG4 reacted to cancer-specific IgG1 that was bound to cancer cells
Although IgG4 extracted from patients with cancer did not react to cancer antigen, it did react to cancer-specific IgG1 that was bound to cancer antigen on tissue sections. As shown in figure 2B, when cancer-specific IgG1 that was not labeled with biotin was applied to cancer tissue sections followed by biotin-labeled IgG4, the cancer cells became positive, while when the same biotin-labeled IgG4 was applied to the same cancer tissue section without prior application of IgG1 it did not react. This reaction of IgG4 to cancer-specific IgG1 was not via the antigen-specific variable region of IgG4, as such IgG4 was neither specifically against IgG1 nor was it specifically against cancer antigen with its antigen recognizing Fab variable region, as shown in figure 2A. The only explanation was that IgG4 reacted to IgG1 through its Fc region. This was validated by subsequent experiments with western blot analysis, as shown in figure 3A–E.
Figure 3.

IgG4 reacted to IgG1 in western blot and tissue sections in an Fc-Fc fashion. (A) In western blot, non-cancer-specific IgG4 from a patient with breast cancer reacted to IgG1 and IgG4 from the same patient with cancer (right panel, arrows). However, when IgG1 and IgG4 were run on the gel and biotin-labeled IgG1 was used as the primary antibody, no band was seen (left panel). These are the same antibodies used in figure 2A, B, providing support to explain the reaction between IgG4 and IgG1 seen on cancer tissue. (B) Western blot demonstrated that IgG4 reacted with IgG1, IgG2, IgG3 and IgG4. (C) IgG4 reacted with IgG Fc fragment but not with Fab arm. (D) IgG4 reacted with IgG1 Fc fragment but not with Fab arm. (E) Biotin-labeled IgG4 Fc fragment reacted to IgG1 and IgG4 Fc fragments but not with IgG1 or IgG4 Fab. Biotin-labeled IgG4 Fab did not react to IgG1 or IgG4 Fc or Fab. These results demonstrate that it is the Fc region of IgG4 that reacted to Fc of IgG1.
In western blot IgG4 was found to react to other IgGs (IgG1, IgG2, IgG3 and IgG4) via an Fc-Fc mechanism, and this reaction was across species, but IgG4 did not react to other Ig subtypes (IgM, IgE, IgA or IgD)
To test if and how IgG4 could react to IgG1, we performed western blot with IgG4 from different sources (extracted from patients with cancer, from normal adults and commercially purchased). IgG4 was found to react to IgG1, IgG2, IgG3 and IgG4 at the molecular weight (MW) of about 50 KD, and this reaction was not seen when IgG1, IgG2 or IgG3 was used as the antibody and IgG4 as the target molecule running on the gel. The above phenomenon was observed in western blot of both reducing and non-reducing conditions (figure 3A; online supplementary figure S3). However, human IgG4 did not react to human IgM, IgA, IgD or IgE (online supplementary figure S4). Nevertheless we found that this reaction was across species, that is, human IgG4 reacted to IgGs of human, mouse, rabbit and goat (online supplementary figure S5). We further digested human IgG4 and IgG1 into Fab and Fc fragments with papain. It was found that it was the Fc fragment of IgG4 reacting to Fc of IgG1 (figure 3B–E). This reaction was easy to occur as only 5 min incubation resulted in a clear band. Therefore, the positive reaction obtained by sequential applications with cancer-specific IgG1 followed by non-cancer-specific IgG4 on cancer tissue (figure 2B) had to take place between the Fc of IgG4 and the Fc of IgG1, as shown in figure 3.
The Fc-Fc reaction between IgG4 and IgG1 bound to tissue sections was further tested and validated with a number of antibodies and tissue types apart from cancer
Following the same logic, we tested the reactivity between the Fc fragments of IgG4 and IgG1, already demonstrated in western blot, on tissue sections. We used IgG1 primary antibodies to insulin and glucagon in normal human pancreas and antibody to neurofilament in human brain for this test. The same principle was established with these normal tissues. Detailed results and figures are shown in online supplementary figure S6.
IgG4 competed with IgG1 to bind to Fc receptors of PBMC and macrophages
We performed immunoglobulin and Fc receptor binding assays with peripheral blood monocytes (PBMC) and with a human monocyte cell line U937 (Procell Life Science & Technology China, #CL-0239). IgG1 and IgG4 were extracted from the serum of patients with cancer and PBMCs were isolated from the same human subjects. The extracted and purchased IgG1 and IgG4 were labeled with biotin or FITC. In the IgG1 and IgG4 binding assay, we found that IgG1 and IgG4 competed with one another in binding to PBMC, and this reaction could be completely blocked by Fc receptor blocker. This competition was concentration-dependent, that is, as the concentration of IgG4 increased, more IgG1 bound to PBMC was replaced. The reverse was also true, that is, IgG1 could also replace IgG4 in this competition assay. This phenomenon was demonstrated on cytospin slides of PBMC preparation (online supplementary figure S7).
In addition, flow cytometry was performed to examine the binding properties of IgG1 and IgG4 to Fc receptors of monocytes. The ability of IgG1 and IgG4 to bind to all three subtypes of Fc gamma receptor (FcγR)—FcγRⅠ (CD64), FcγRⅡ (CD32) and FcγRⅢ (CD16)—was examined with corresponding antibodies. We found that IgG4 could compete with IgG1 in binding to the Fc receptor of monocytes (U937). We further found that the binding force of IgG1 was about twice as strong as that of IgG4. For individual receptor subtypes, IgG1 could bind to all three receptor subtypes, that is, FcγRⅠ (CD64) > FcγRⅡ (CD32) > FcγRⅢ (CD16). In contrast, IgG4 could only bind to FcγRⅠ (CD64). Although their binding sites were different, IgG4 could completely block IgG1. We also found that it was necessary for a relatively high concentration of IgG4 to be present in the solution in order to compete with IgG1 in binding to Fc receptors (online supplementary figure S8).
IgG4 inhibited the classic immune reactions of ADCC, ADCP and CDC mediated via cancer-specific IgG1 and effector immune cells/complements
We first verified that non-cancer-specific IgG4 indeed reacted to IgG1 (cetuximab) used in ADCC, ADCP and CDC (figure 4A). We then found that IgG4 inhibited ADCC elicited cytotoxicity with cancer-specific IgG1 antibody and PBMC (figure 4B). The IgG4 used in our test was not directed against cancer antigen or to lymphocytes. Non-cancer-specific IgG1 could also inhibit ADCC but to a much lesser extent (also reached statistical significance). We obtained evidence to show that inhibition of ADCC appeared to take place at the site of the cancer-specific antibody, that is, IgG4 reacted to the IgG1 antibody bound to cancer cells (figure 2B) and the site of immune effector Fc receptors. The latter effect could be abolished with the addition of Fc receptor blocker (Human TruStain FcX, BioLegend, China) to the PBMC.
Figure 4.

Non-cancer-specific IgG4 inhibited classic ADCC, ADCP and CDC reactions against cancer but had no direct effect on cancer cell growth. (A) On western blot, the chimeric antibody cetuximab (IgG1 against EGFR) was run on the gel, and IgG4 and IgG1 at concentrations of 3, 5 and 10 µg/mL were used as the primary antibodies. IgG4 reacted to cetuximab at a concentration-dependent manner, but IgG1 did not react. (B) Left: In a classic ADCC experiment, cetuximab (IgG1) was incubated with an EGFR-expressing lung cancer cell line (A549) and then with PBMC from normal healthy adult. Cancer cell activity was significantly reduced (n=12). Non-cancer-specific IgG1 and HSA were used as controls showing that they had no direct effect on the cancer cells (n=12). Middle: When non-cancer-specific IgG4 was added to the mixture, the effect of cetuximab was significantly reversed demonstrating an inhibitory effect of IgG4 in ADCC (n=12). Non-cancer-specific IgG1 had a much smaller, but also significant, effect in inhibiting ADCC action (n=12). Right: When Fc receptor blocker was incubated with PBMC, the effect of cytotoxicity was blocked. (C–E) ADCP was performed with a lung cancer cell line A549 (expressing EGFR) as the targets, human peripheral monocyte-derived macrophages as the effector cells and the antibody cetuximab (IgG1) against EGFR as the mediating antibody. The tumor cells were stained with CFDA-SE fluorescence probes (green). Macrophages derived from PBMC were stained with DiI fluorescent probes (orange). Blue fluorescence is the nuclei stained with DAPI. (D) Higher magnification of (C). The orange-colored macrophages were in close contact with green tumor cells. Tumor debris ingested by macrophages appeared yellow in the cytoplasm of macrophages. (E) Bar chart showing the effect of ADCP and its inhibition by IgG4. (F) Left: In 10 µg/mL rituximab-mediated ADCP model, Giemsa staining results of phagocytosis of Raji cells by macrophages after the addition of 100 µg/mL IgG1 and IgG4, respectively. Right: IgG4 significantly inhibited rituximab-mediated ADCP in phagocytosis by macrophage, but IgG1 could not inhibit the ADCP effect. Scale bar=30 µm (*p<0.05, **p<0.01, ***p<0.001). (G) In a classic CDC experiment, cetuximab anti-EGFR antibody was incubated with an EGFR-expressing lung cancer cell line (A549) and then with complement (Co) from serum of a normal healthy adult. The cancer cell activity was significantly reduced. (H) When non-cancer-specific IgG4 was added to the mixture in the above CDC experiment, the effect of cetuximab was significantly reversed. (I) IgG4 and IgG1 were incubated with KYSE150 for 24 hours and no effect on cell growth was found. ADCC, antibody-dependent cell-mediated cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; CDC, complement-dependent cytotoxicity; CFSE-DA, carboxyfluorescein diacetate succinimidyl ester; DAPI, 4',6-diamidino-2-phenylindole; EGFR, epidermal growth factor receptor; HSA, human serum albumin; PBMC, peripheral blood mononuclear cell.
We also performed an ADCP experiment employing human monocyte-derived macrophages and esophageal cancer cells. Cetuximab (IgG1) was used as the mediating antibody. This was performed employing a coculture and cell-counting method and FITC-labeled antibody flow cytometry. Two models were employed and both methods showed that non-cancer-specific IgG4 was able to reduce the effect of ADCP mediated by cancer-specific IgG1 (figure 4C–F).
In a classic CDC assay, we used cancer cell line A549 (ATCC, USA, #C4215) as the target cancer cells, cetuximab as the cancer-specific IgG1 mediating antibody and human plasma as the source of complements, and demonstrated the destructive effect on cancer cells. We then used non-cancer-specific IgG4 or IgG1 to inhibit the effect. We found that the CDC effect was partially inhibited by non-cancer-specific IgG4 but not by IgG1 (figure 4G, H).
For comparison, we added IgG4 or IgG1 at various concentrations in cancer cell culture for different periods of time and found no direct effect of these proteins on cancer cell growth (figure 4I).
IgG4 (including nivolumab) significantly accelerated breast cancer cell and colon cancer cell growth in two immune potent mouse models in vivo
The above results point to a mechanism that IgG4 plays an important role in local immune evasion by blocking immune responses mediated by cancer-specific IgG antibodies. To further examine this mechanism mediated by such antibodies, we performed in vivo studies to verify this hypothesis with immune competent mouse models. In one model, we injected non-cancer-specific IgG4 into a location where breast cancer cells were inoculated subcutaneously. In this group of mice, cancer cell growth was significantly increased, resulting in a much larger cancer mass by 21 days in comparison with other groups (injections of PBS or IgG without IgG4) (figure 5A, B). As there is no direct effect of IgG4 on cancer cell growth (figure 4I), these results unequivocally confirmed that IgG4 can inhibit local immune reaction and thereby promote cancer growth in vivo through immune evasion.
Figure 5.

IgG4 accelerated cancer cell growth in three immune potent mouse models. (A, B) Non-cancer-specific IgG4 accelerated breast cancer cell growth. Local inoculation of mouse breast cancer cells (4T1, 1×105 cells per mouse) into immune competent mice (BALB/c, body weight 20 g, n=5) resulted in sizeable tumor masses in 21 days. Local injections of non-cancer-specific IgG4 resulted in tumor masses that doubled the size of those similarly injected with ‘IgG without IgG4’ or with PBS. As there is no direct effect of IgG4 on cancer cell growth (figure 4I), these results clearly demonstrate that IgG4 can effectively promote tumor growth by inhibiting local immunity. (C, D) IgG4, anti-PD-1 (nivolumab) and anti-PD-1-Fc induced significant tumor (CT26 mouse colon cancer) progression in mice. Five groups of mice were tested by injecting IgG4, nivolumab (IgG4 subtype), nivolumab-Fc, IgG1 and PBS. By 19 days, IgG4, nivolumab and nivolumab-Fc induced significant progression of tumor size in comparison with the other groups. (E–G) IgG4 significantly accelerated skin papilloma formation in a carcinogen-induced skin tumor model in immune potent mice. IgG4 significantly accelerated tumor development and growth in comparison with control (PBS), which in turn had more tumor formation than the group treated with IVIG without IgG4. By 12 weeks, the total tumor volume in IgG4-treated group was more than 12 times larger than the group that was treated with IVIG without IgG4, and was six times larger than the group treated with PBS. (E) The removed back skin of mice from the three groups (at 15 weeks). The tumors are shown in dark-brown color. (F) Tumors at higher magnification. (G) Changes of tumor numbers and volume over time of the three groups. (H) Total tumor volumes of the three groups at 12 weeks after treatment. *p<0.05, **p<0.01, ***p<0.001. IVIG, intravenous IgG; PBS, phosphate buffered saline; PD-1, programmed cell death-1.
In a separate but similar experiment of a colon cancer mouse model, we injected antibody to programmed cell death-1 (PD-1) (nivolumab), which is a widely used drug in cancer immune therapy and is also an IgG4 isotype with S228P mutation, which replaces a serine residue in the hinge region with a proline residue to prevent FAE and stabilize the protein. We also injected the Fc fragment of anti-PD-1 and compared the results with IgG4, IgG1, and PBS groups. We found that IgG4, anti-PD-1 and in particular the Fc of anti-PD-1 groups produced much larger cancer masses in comparison with the groups with PBS and IgG1. The differences were statistically significant (figure 5C, D). The group injected with non-cancer-specific IgG1 also resulted in enlarged cancer mass to a significant extent. Before the animal experiment, we tested the reactivity of nivolumab to react to IgG1 in a western blot assay. We found that the Fc of nivolumab reacted to IgG1 (online supplementary figure S3), and the S228P mutation did not seem to prevent Fc-Fc reaction taking place between nivolumab and other IgGs. In addition, we examined T lymphocytes in the different groups of mice and found that local injection of IgG4 led to significant reduction of T lymphocytes in tumor in this group but not in the control groups (online supplementary figure S9).
IgG4 significantly accelerated skin tumor formation in a carcinogen-induced papilloma mouse model
We also tested IgG4 in an established carcinogen-induced skin cancer mouse model.32 A two-stage skin carcinogenesis procedure was performed first with 7,12-dimethylbenz[a]anthracene (DMBA) and then with 12-O-tetradecanoylphorbol-13-acetate (TPA) on a mouse’s posterior dorsal skin as described previously.33 The animals were divided into three groups (nine in each group) according to subcutaneous treatment with IgG4, IVIG without IgG4 or PBS. The animals were followed for 12 weeks. On day 29, mice treated with IgG4 began to grow tumors, followed by the PBS (control) group on day 50 and IVIG without IgG4 group on day 64. On day 64, the IgG4 treated group developed seven papillomas in five mice, while the PBS group had four papillomas in four mice, and the IVIG without IgG4 group had two papillomas in two mice. On day 84 when the observation was terminated, the IgG4 treated group had 19 papillomas in all nine mice, the control group had 12 papillomas in seven mice and the IVIG without IgG4 group had 6 papillomas in six mice. The volume of each tumor was also larger in the IgG4-treated group than the PBS group, which in turn is larger than the IVIG without IgG4 group. At termination on day 84, the total tumor volume of the IgG4 treated group was 12 times larger than the group treated with IVIG without IgG4 and was about six times larger than the control (PBS) group (figure 5E–H, online supplementary figure S10).
All the above effects were inducible by IgG4 from different sources regardless of its antigen specificity
We tested IgG4 extracted from the serum of patients with cancer and healthy individuals, isolated from IVIG, and genetically engineered IgG4 purchased from different commercial sources, in all cases without knowing their antigen specificity. They all gave the same inhibitory effect. This included a monoclonal antibody to PD-1 of which the S228 region of the Fc fragment was mutated to prevent FAE and stabilize the antibody. It produced the same effect too.
Discussion
A number of original observations were made in this study that unveiled previously unknown properties of cancer immunity. Collectively these findings point to an immune evasion mechanism with IgG4 playing a crucial role in coordinating an inhibitory effect of humoral and cellular immune responses against cancer.
The finding of increased IgG4 concentration in the serum of patients with cancer was in agreement with those reported by Karagiannis et al18 and Miyatani et al,16 who found increased IgG4 in melanoma and gastric cancers. In agreement with Wu et al,19 we further found that the extent of increase was proportionally related to cancer malignancy and patient prognoses. Late stage cancers were accompanied with more obvious IgG4 increase. The marked increases of IgG4 positive lymphocytes and IgG4 concentration in cancer microenvironment were in agreement with a previous report by Harada et al, who found such an increase in extrahepatic cholangiocarcinoma.17 Our observation was derived from a much larger cohort of patients in comparison with previous reports.16–19 34 We also found that IgG4-positive lymphocytes were distinct from CD3-positive T lymphocytes, and this increase was mainly seen in areas of cancer cell proliferation.
An important observation in our study was that the IgG4 in cancer microenvironment and in the patient’s serum was not reactive to cancer antigens but it reacted to IgG1 that was specifically against cancer. In addition the IgG4 to IgG1 reaction was via Fc-Fc reaction rather than Fab fragments. We also demonstrated that non-cancer-specific IgG4 could inhibit ADCC, ADCP and CDC reactions mediated by cancer-specific IgG1 against cancer.
It should be noted that the increase of IgG4 in cancer microenvironment was significant, by at least tenfold (IgG4-containing plasma cells) and by about fourfold (IgG4 concentration) in comparison with adjacent normal tissue. This remarkable increase would likely allow the binding of IgG4 to all or most cancer-bound IgGs and block the subsequent immune effector reaction as shown in our in vitro experiments. In addition, from the Fc receptor competition experiments, it showed that considerable amount of IgG4 is required to compete with IgG1 in reacting to Fc receptors of PBMC and macrophages. Therefore the inhibitory effect observed in our experiments should be operative in cancer microenvironment.
In this study, we found that IgG4 reacted to IgG1 not only on western blot but also on cancer tissue sections. We demonstrated that non-cancer-specific IgG4 reacted to cancer-specific IgG1 that was bound to cancer cells. This would allow IgG4 to block the subsequent immune effector response that would otherwise detect and destroy cancer cells. Our study was the first to demonstrate in cancer tissue that non-cancer-specific IgG4 was able to bind to cancer-bound IgG1, thereby blocking cancer-specific antibody-elicited cancer targeting immunity.
The ability of IgG4 to react to IgG1 and other subtypes through Fc-Fc reaction was noticed over 20 years ago by Zack et al.35 This only occurred when IgG1 was immobilized to solid phase, but the biological implication of this ‘unusual’ phenomenon has not been explained adequately.5 We found in this study that this Fc-Fc reaction was across species and this possibly indicates that such a property was preserved during evolution and therefore likely to have significant biological functions. Rispens et al10 reported that Fc-Fc interaction was CH3-mediated. They further speculated that this interaction might be an intermediate step of FAE that was stabilized at this stage because the target IgG was coupled to a solid phase.10 FAE only takes place between two IgG4s in liquid phase because of weakened covalent (hinge disulfide bonds) and non-covalent (CH3-CH3) interactions of IgG4 molecules.36 37 This would not occur between IgG4 and IgG1 in liquid phase as IgG1 has more stable intra-heavy chain bonds and would not be available for Fc-Fc binding with IgG4. When IgG1 is bound to solid phase, however, such as immobilized to cancer antigens, its Fc heavy chains may be partially dissociated and undergo conformational changes allowing IgG4 to bind to.10 38 In cancer, this denaturation of immobilized IgG1 is likely to be facilitated by increased antioxidant glutathione (GSH) in cancer tissue10 39 or cleaved by proteases rich in cancer.40 41 Therefore, IgG4 would react to cancer-antigen bound IgG1 only but not to IgG1 in circulation. Thus this immune inhibition by IgG4 only occurs in cancer microenvironment where anticancer IgG1 has bound to cancer tissue but not in other parts of the body including in circulation.
The ability of IgG4 to inhibit ADCC, ADCP and CDC was reported previously.42–45 However, the IgG4 they used was specifically engineered against a cancer antigen. They concluded that the inhibition was due to competition between cancer-specific IgG1 and cancer-specific IgG4 on the same cancer antigens.20 46–49 One study reported that non-cancer-specific IgG4 also had inhibitory effect on ADCC without giving any explanation of the mechanism and its possible significance.20 Here we found that non-specific IgG4 can inhibit ADCC, ADCP and CDC against cancer. We also obtained evidence to show that this inhibition took place in two sites, that is, with its ability to react to Fc fragment of cancer-bound IgG1 and Fc receptor on effector immune cells. Both will lead to disconjunction between immune effector cells and cancer cells resulting in immune escape. As for complement, it was reported previously that IgG4 did not react to C1q.50 Therefore no competition of binding is needed for IgG4 to inhibit CDC.
In this study, we found that IgG4 extracted from a patient with cancer did not react to the same cancer tissue but IgG1 did. We speculate that this could be due to the fact that most IgG4s were bispecific, that is, its two Fabs reacted to different antigens due to FAE, and therefore its reactivity to cancer antigens was unstable, which could not be demonstrated with a biotin-labeled IgG4 applied on cancer tissue sections in our experiment. Previously it was shown that IgG4 could evolve from other IgG subtypes, and it could be also directly produced by plasma cells46 without being specific to cancer antigens.
There have been extensive studies on Fc receptors on immune effector cells. Such receptors are divided into three major types and at least six subtypes with FcγRIIa as the inhibitory type and the rest as the activating types.45 51 52 All four types of IgG were found to react to these receptors with their Fc fragments at different strengths. In this study, we blocked the three receptor subtypes with respective antibodies and identified the specific receptors for IgG1 and IgG4. We found that IgG4 could completely block IgG1 from binding to Fc receptors, although each of the two antibodies has its own preferred Fc receptor subtypes. This could be due to interfering or shielding of the receptor binding sites by stereo structures of IgG4. We also found that a relatively high concentration of IgG4 in the environment is necessary for it to effectively block IgG1 binding. Such a concentration would be available in cancer as we found that IgG4 was markedly elevated in cancer microenvironment. It was interesting to note that not only non-cancer-specific IgG4 could reverse the effect of ADCC but non-cancer-specific IgG1 also reversed the ADCC effect but to a lesser extent. The difference between the two was statistically significant and both were also statistically different from that of the control with human albumin. The likely explanation for this observation was that IgG4 was able to block the ADCC reaction at two levels, that is, one at the cancer-bound IgG1 level and another at the Fc receptor of the immune effector cell level, while non-cancer-specific IgG1 only interfered the reaction at one level, that is, the Fc receptor level. As the affinity of IgG1 for Fc receptors is higher than that of IgG4,45 more IgG4 is needed to block IgG1 binding to Fc receptors, as demonstrated in our experiments.
It has been shown that class switching of lymphocyte toward production of IgG4 can be promoted by a T helper 2-biased response with expressions of cytokines interleukin (IL)-4, IL-13,3 53 IL-10, vascular endothelial growth factor (VEGF),20 54 55 IL-12 and IL-21.56 57 IgG4 was increased in parasite infections and chronic antigen stimulation in autoimmune diseases.54 58 59 IgG4 would then suppress the chronic inflammation to confine tissue damage.47 It appears that cancer cells are chronic antigens that continuously stimulate the local immune cells leading to increase of IgG4-producing plasma cells in cancer. This in turn causes IgG4 to increase first locally and then in circulation.
The in vivo tumor model with immune potent mice performed in this study clearly demonstrated that IgG4 accelerated cancer growth. In the breast cancer model, this led to almost doubled tumor size in 21 days in comparison with controls without IgG4. In the carcinogen-induced skin tumor model, application of IgG4 for 12 weeks led to a total tumor volume 12 times larger than the group treated with IVIG without IgG4 and about six times larger than the group treated with PBS alone. As there is no direct effect of IgG4 on cancer cell growth (figure 4I), the only explanation was that IgG4 inhibited local immune response against cancer resulting in immune escape. The decreased lymphocytes in the IgG4 group of our animal model supports this notion. A similar experiment was performed20 on NSG (NOD/SCID-gamma) mice engrafted with human immune effector cells. They found that cancer-specific IgG4 (anti-CSPG4) antibody could block the cancer killing effect on subcutaneous melanoma mediated by cancer-specific IgG1. The difference of our finding was that the IgG4 did not need to be cancer-specific. Local increase of IgG4 molecule would lead to local immune inhibition that indirectly promotes cancer growth, and this effect was likely rendered by the Fc fragment of IgG4 rather than antigen-specific Fab fragment as claimed by others.47
A previous study reported that IVIG could prevent the development and inhibit the growth of cancers in mice.60 In this study, treatment with IVIG deprived of IgG4 significantly inhibited the development of inoculated cancer in comparison with the control and the IgG4-treated groups. It appears that the immune inhibitory effect of IgG4, regardless of its antigen specificity, demonstrated in vitro is also effective in vivo. It should be noted that in these animal models, we used human IVIG and IgG4 instead of mouse IgG because the volume of mouse IgG was too small to be extracted from and applied to the small animals. We demonstrated in this study that human IgG4 could effectively react to mouse IgG via the Fc-Fc reaction (online supplementary figure S5).
From our observations, a new humoral immune editing mechanism emerges with non-cancer-specific IgG4 playing a central role. The following mechanism is likely at work. Growing cancer cells act as a chronic antigen stimulant that induces marked increase of IgG4-producing B lymphocytes resulting in elevated concentration of IgG4 in cancer microenvironment and in patients’ serum. IgG4 then reacts to cancer-specific IgG (mostly IgG1) that has bound to cancer antigens, with its inherited ability of Fc to react to the Fc of immobilized IgG. This reaction is likely facilitated by increased concentration of GSH and increased protease found in most cancer tissues. At the same time significantly increased IgG4 in cancer microenvironment also reacts with Fc receptors of effector leukocytes, thereby blocking effector immune response against cancer. This new mechanism was validated by the results of the mouse models where locally administered IgG4 significantly increased cancer growth in comparison with the control groups in which no IgG4 was applied. This proposed IgG4-mediated immune evasion mechanism is illustrated diagrammatically in figure 6.
Figure 6.
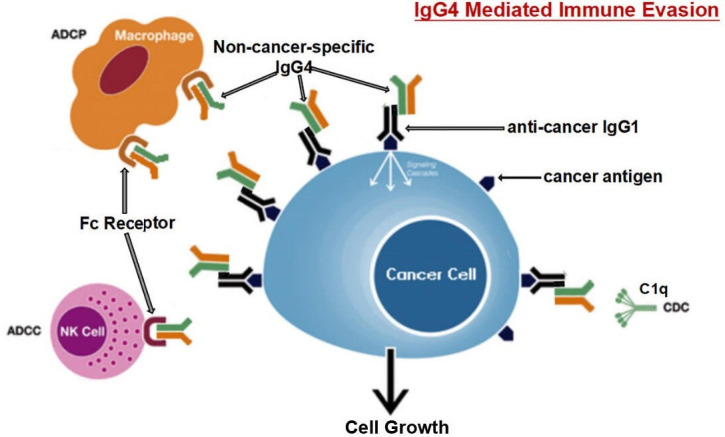
Diagrammatic illustration of proposed mechanism of cancer-initiated B lymphocyte-derived IgG4-mediated immune evasion. Chronic stimulation by cancer antigens induces class switch of B lymphocytes to produce IgG4. Such increased IgG4 can react to cancer-bound IgG with its Fc-Fc binding property and also to Fc receptors of immune effector cells. With its unique structural and biological property, increased IgG4 in cancer microenvironment mediates an effective immune escape for cancer. ADCC, antibody-dependent cell-mediated cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; CDC, complement-dependent cytotoxicity; NK, natural killer cells.
We also tested a Food and Drug Administration-approved antibody drug to PD-1 (nivolumab), which has been widely used in cancer immune therapy, in the animal model as it is also of the IgG4 subtype. We found that anti-PD-1 and its Fc fragment led to accelerated cancer growth in comparison with the controls. Recent awareness of hyperprogressive disease (HPD) associated with anti-PD-1 and anti-PD-L1 monoclonal antibody treatment for cancer has caught widespread attention,61–65 but no consensual explanation for this phenomenon has arrived. HPD appeared to be a common complication for immunotherapy with nivolumab in many cancer types, including head and neck squamous cell carcinoma,61 non-small cell lung cancer,62 gastric cancer66 and so on. Our findings suggest that these IgG4 antibody drugs might have undesired side effects of inhibiting local immune responses and indirectly promote cancer growth. When the specific target molecule is present in cancer, these IgG4 antibody drugs might be effective. However, when the targets are absent or scanty, the IgG4’s immune inhibitory effect might prevail and accelerate cancer growth. This possible detrimental effect of IgG4 might contribute to HPD in patients treated with PD-1 targeting drugs with IgG4 structure.6 These drugs should possess the properties of IgG4 described in this and other reports.4 8 10 23 38 To stabilize the hinge region and prevent FAE, the IgG4 used in clinic immunotherapy usually carries a mutation at position 228 of the heavy chain at the hinge region.6 However, in our experiments this mutation did not seem to prevent Fc-Fc reaction (online supplementary figure S3) and the immune blockage effect of nivolumab (IgG4) Fc leading to the largest cancer size in our animal experiment (figure 5C, D). We hope that this result will draw attention of the oncology community to this phenomenon in cancer immune therapy. The clinical and biological implications of this observation warrant further investigation.
What was intriguing in our findings is that the increased IgG4 in patients with cancer was not necessarily cancer-specific, that is, it did not seem to react to cancer antigen of the same patient, although we could not rule out that there were cancer-specific IgG4 antibodies in the cancer microenvironment. Because of the often bispecific nature of IgG4’s Fabs, their binding to antigens, even if it exists, might not be able to withstand the procedure of immunohistochemistry to be visualized. Nevertheless, it reacted to cancer-bound IgG1 and competed with Fc receptors on immune effector cells. Both reactions were accomplished with its Fc fragment. In addition, when we used IgG4 from different sources, they all gave the same exact results both in vitro and in vivo. Therefore, IgG4 molecule itself without its specific antigen-recognizing variable region is sufficient to exert the inhibitory effect on local immunity.
These actions of IgG4 found in this study represent a previously unrecognized dimension of cancer immunology. In addition to going through specific antigens and receptors, this response uses the Fc fragment of IgG4 to react with the Fc of other antibodies and to Fc receptors of immune effector cells. This mechanism may provide new insights into the immunopathology of cancer, IgG4-related diseases and immune tolerance in general. Manipulation of B cells and the concentration of IgG4 in cancer may bring benefits to cancer immunotherapy.
Acknowledgments
We thank the Clinical Laboratories of Affiliated Cancer Hospital and Second affiliated Hospital of Shantou University Medical College for providing serum samples. We thank the Departments of Affiliated Cancer Hospital and Yuedong Pathology Center of Shantou University Medical College for providing tumor tissue samples. We also appreciate the help of the Departments of Pathology and Forensic Medicine, Shantou University Medical College for providing human tissue samples.
Footnotes
Contributors: HW: performed most of the experiments. QX: performed the animal experiments. CZ: performed some of the animal models. ZZ: performed the experiments of IgG4 immunohistochemistry. SZ: performed the Fc receptor assays. JZ, BZ: performed most of the experiments. TY, XZ: performed the ADCC experiments. JL: performed some experiment and tissue collection. MY: performed some experiments. RL: performed some of the Fc receptor assays. CM, YQ, YZ, CC, SC, DL, YC: performed tissue collection and processing. WZ, YG: result and literature analysis. DT, MS: collected and analyzed some pathological tissue samples. XC: helped in organizing and editing the manuscript. JG: designed and directed the experiments, analyzed the results and wrote the manuscript.
Funding: This study was supported by the National Natural Science Foundation of China (no. 81872334) and the Li Ka Shing Foundation.
Competing interests: None declared.
Patient consent for publication: Not required.
Ethics approval: All experiments were conducted based on the protocols approved by the Animal Care and Use Committee of Shantou University Medical College according to the guidelines for Laboratory Animal Sciences.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Data sharing not applicable as no datasets generated and/or analyzed for this study. All data relevant to the study are included in the article or uploaded as supplementary information.
References
- 1.Miller JFAP, Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell 2015;27:439–49. 10.1016/j.ccell.2015.03.007 [DOI] [PubMed] [Google Scholar]
- 2.Coulie PG, Van den Eynde BJ, van der Bruggen P, et al. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 2014;14:135–46. 10.1038/nrc3670 [DOI] [PubMed] [Google Scholar]
- 3.Agresti A, Vercelli D. Analysis of gamma4 germline transcription in human B cells. Int Arch Allergy Immunol 1999;118:279–81. 10.1159/000024099 [DOI] [PubMed] [Google Scholar]
- 4.Aalberse RC, Schuurman J. IgG4 breaking the rules. Immunology 2002;105:9–19. 10.1046/j.0019-2805.2001.01341.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aalberse RC, Stapel SO, Schuurman J, et al. Immunoglobulin G4: an odd antibody. Clin Exp Allergy 2009;39:469–77. 10.1111/j.1365-2222.2009.03207.x [DOI] [PubMed] [Google Scholar]
- 6.Yang X, Wang F, Zhang Y, et al. Comprehensive analysis of the therapeutic IgG4 antibody pembrolizumab: hinge modification blocks half molecule exchange in vitro and in vivo. J Pharm Sci 2015;104:4002–14. 10.1002/jps.24620 [DOI] [PubMed] [Google Scholar]
- 7.Schuurman J, Van Ree R, Perdok GJ, et al. Normal human immunoglobulin G4 is bispecific: it has two different antigen-combining sites. Immunology 1999;97:693–8. 10.1046/j.1365-2567.1999.00845.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Neut Kolfschoten M, Schuurman J, Losen M, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 2007;317:1554–7. 10.1126/science.1144603 [DOI] [PubMed] [Google Scholar]
- 9.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol 2014;5:520. 10.3389/fimmu.2014.00520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rispens T, Meesters J, den Bleker TH, et al. Fc-Fc interactions of human IgG4 require dissociation of heavy chains and are formed predominantly by the intra-chain hinge isomer. Mol Immunol 2013;53:35–42. 10.1016/j.molimm.2012.06.012 [DOI] [PubMed] [Google Scholar]
- 11.Davies AM, Rispens T, Ooijevaar-de Heer P, et al. Room temperature structure of human IgG4-Fc from crystals analysed in situ. Mol Immunol 2017;81:85–91. 10.1016/j.molimm.2016.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies AM, Jefferis R, Sutton BJ. Crystal structure of deglycosylated human IgG4-Fc. Mol Immunol 2014;62:46–53. 10.1016/j.molimm.2014.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawakami H, Zen Y, Kuwatani M, et al. IgG4-related sclerosing cholangitis and autoimmune pancreatitis: histological assessment of biopsies from Vater's ampulla and the bile duct. J Gastroenterol Hepatol 2010;25:1648–55. 10.1111/j.1440-1746.2010.06346.x [DOI] [PubMed] [Google Scholar]
- 14.Zen Y. The pathology of IgG4-related disease in the bile duct and pancreas. Semin Liver Dis 2016;36:242–56. 10.1055/s-0036-1584319 [DOI] [PubMed] [Google Scholar]
- 15.Guma M, Firestein GS. IgG4-related diseases. Best Pract Res Clin Rheumatol 2012;26:425–38. 10.1016/j.berh.2012.07.001 [DOI] [PubMed] [Google Scholar]
- 16.Miyatani K, Saito H, Murakami Y, et al. A high number of IgG4-positive cells in gastric cancer tissue is associated with tumor progression and poor prognosis. Virchows Arch 2016;468:549–57. 10.1007/s00428-016-1914-0 [DOI] [PubMed] [Google Scholar]
- 17.Harada K, Shimoda S, Kimura Y, et al. Significance of immunoglobulin G4 (IgG4)-positive cells in extrahepatic cholangiocarcinoma: molecular mechanism of IgG4 reaction in cancer tissue. Hepatology 2012;56:157–64. 10.1002/hep.25627 [DOI] [PubMed] [Google Scholar]
- 18.Karagiannis P, Villanova F, Josephs DH, et al. Elevated IgG4 in patient circulation is associated with the risk of disease progression in melanoma. Oncoimmunology 2015;4:e1032492. 10.1080/2162402X.2015.1032492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu J, Ma X-L, Tian L, et al. Serum IgG4:IgG Ratio Predicts Recurrence of Patients with Hepatocellular Carcinoma after Curative Resection. J Cancer 2017;8:1338–46. 10.7150/jca.18030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karagiannis P, Gilbert AE, Josephs DH, et al. IgG4 subclass antibodies impair antitumor immunity in melanoma. J Clin Invest 2013;123:1457–74. 10.1172/JCI65579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu Y, Liu Y, Fu L, et al. Tumor-educated B cells selectively promote breast cancer lymph node metastasis by HSPA4-targeting IgG. Nat Med 2019;25:312–22. 10.1038/s41591-018-0309-y [DOI] [PubMed] [Google Scholar]
- 22.Li J, Zhou Y, Gu J. Stain-Decolorize-Stain (SDS): a new technique for multiple staining. Histochem Cell Biol 2014;141:251–62. 10.1007/s00418-013-1177-7 [DOI] [PubMed] [Google Scholar]
- 23.Shinkawa T, Nakamura K, Yamane N, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem 2003;278:3466–73. 10.1074/jbc.M210665200 [DOI] [PubMed] [Google Scholar]
- 24.Watanabe M, Wallace PK, Keler T, et al. Antibody dependent cellular phagocytosis (ADCP) and antibody dependent cellular cytotoxicity (ADCC) of breast cancer cells mediated by bispecific antibody, MDX-210. Breast Cancer Res Treat 1999;53:199–207. 10.1023/A:1006145507567 [DOI] [PubMed] [Google Scholar]
- 25.Keler T, Graziano RF, Mandal A, et al. Bispecific antibody-dependent cellular cytotoxicity of HER2/neu-overexpressing tumor cells by Fc gamma receptor type I-expressing effector cells. Cancer Res 1997;57:4008–14. [PubMed] [Google Scholar]
- 26.Overdijk MB, Verploegen S, Bögels M, et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs 2015;7:311–20. 10.1080/19420862.2015.1007813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golay J, Da Roit F, Bologna L, et al. Glycoengineered CD20 antibody obinutuzumab activates neutrophils and mediates phagocytosis through CD16B more efficiently than rituximab. Blood 2013;122:3482–91. 10.1182/blood-2013-05-504043 [DOI] [PubMed] [Google Scholar]
- 28.Richards JO, Karki S, Lazar GA, et al. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther 2008;7:2517–27. 10.1158/1535-7163.MCT-08-0201 [DOI] [PubMed] [Google Scholar]
- 29.Klausz K, Berger S, Lammerts van Bueren JJ, et al. Complement-mediated tumor-specific cell lysis by antibody combinations targeting epidermal growth factor receptor (EGFR) and its variant III (EGFRvIII). Cancer Sci 2011;102:1761–8. 10.1111/j.1349-7006.2011.02019.x [DOI] [PubMed] [Google Scholar]
- 30.Wirt T, Rosskopf S, Rösner T, et al. An Fc Double-Engineered CD20 antibody with enhanced ability to trigger complement-dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity. Transfus Med Hemother 2017;44:292–300. 10.1159/000479978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goswami MT, Reka AK, Kurapati H, et al. Regulation of complement-dependent cytotoxicity by TGF-β-induced epithelial-mesenchymal transition. Oncogene 2016;35:1888–98. 10.1038/onc.2015.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abel EL, Angel JM, Kiguchi K, et al. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc 2009;4:1350–62. 10.1038/nprot.2009.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monga J, Chauhan CS, Sharma M. Human epithelial carcinoma cytotoxicity and inhibition of DMBA/TPA induced squamous cell carcinoma in Balb/c mice by Acacia catechu heartwood. J Pharm Pharmacol 2011;63:1470–82. 10.1111/j.2042-7158.2011.01354.x [DOI] [PubMed] [Google Scholar]
- 34.Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med 2001;344:732–8. 10.1056/NEJM200103083441005 [DOI] [PubMed] [Google Scholar]
- 35.Zack DJ, Stempniak M, Wong AL, et al. Localization of an Fc-binding reactivity to the constant region of human IgG4. Implications for the pathogenesis of rheumatoid arthritis. J Immunol 1995;155:5057–63. [PubMed] [Google Scholar]
- 36.Labrijn AF, Buijsse AO, van den Bremer ETJ, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol 2009;27:767–71. 10.1038/nbt.1553 [DOI] [PubMed] [Google Scholar]
- 37.Angal S, King DJ, Bodmer MW, et al. A single amino acid substitution abolishes the heterogeneity of chimeric mouse/human (IgG4) antibody. Mol Immunol 1993;30:105–8. 10.1016/0161-5890(93)90432-B [DOI] [PubMed] [Google Scholar]
- 38.Rispens T, Ooievaar-De Heer P, Vermeulen E, et al. Human IgG4 binds to IgG4 and conformationally altered IgG1 via Fc-Fc interactions. J Immunol 2009;182:4275–81. 10.4049/jimmunol.0804338 [DOI] [PubMed] [Google Scholar]
- 39.Bansal A, Simon MC. Glutathione metabolism in cancer progression and treatment resistance. J Cell Biol 2018;217:2291–8. 10.1083/jcb.201804161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brezski RJ, Jordan RE. Cleavage of IgGs by proteases associated with invasive diseases: an evasion tactic against host immunity? MAbs 2010;2:212–20. 10.4161/mabs.2.3.11780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brezski RJ, Vafa O, Petrone D, et al. Tumor-associated and microbial proteases compromise host IgG effector functions by a single cleavage proximal to the hinge. Proc Natl Acad Sci U S A 2009;106:17864–9. 10.1073/pnas.0904174106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Overdijk MB, Verploegen S, Ortiz Buijsse A, et al. Crosstalk between human IgG isotypes and murine effector cells. J Immunol 2012;189:3430–8. 10.4049/jimmunol.1200356 [DOI] [PubMed] [Google Scholar]
- 43.Warncke M, Calzascia T, Coulot M, et al. Different adaptations of IgG effector function in human and nonhuman primates and implications for therapeutic antibody treatment. J Immunol 2012;188:4405–11. 10.4049/jimmunol.1200090 [DOI] [PubMed] [Google Scholar]
- 44.Niwa R, Natsume A, Uehara A, et al. IgG subclass-independent improvement of antibody-dependent cellular cytotoxicity by fucose removal from Asn297-linked oligosaccharides. J Immunol Methods 2005;306:151–60. 10.1016/j.jim.2005.08.009 [DOI] [PubMed] [Google Scholar]
- 45.Bruhns P, Iannascoli B, England P, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009;113:3716–25. 10.1182/blood-2008-09-179754 [DOI] [PubMed] [Google Scholar]
- 46.Collins AM, Jackson KJL. A temporal model of human IgE and IgG antibody function. Front Immunol 2013;4:235. 10.3389/fimmu.2013.00235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crescioli S, Correa I, Karagiannis P, et al. IgG4 characteristics and functions in cancer immunity. Curr Allergy Asthma Rep 2016;16:7. 10.1007/s11882-015-0580-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jackson KJL, Wang Y, Collins AM. Human immunoglobulin classes and subclasses show variability in VDJ gene mutation levels. Immunol Cell Biol 2014;92:729–33. 10.1038/icb.2014.44 [DOI] [PubMed] [Google Scholar]
- 49.Rogosch T, Kerzel S, Dey F, et al. IgG4 and IgE transcripts in childhood allergic asthma reflect divergent antigen-driven selection. J Immunol 2014;193:5801–8. 10.4049/jimmunol.1401409 [DOI] [PubMed] [Google Scholar]
- 50.Davies AM, Rispens T, Ooijevaar-de Heer P, et al. Structural determinants of unique properties of human IgG4-Fc. J Mol Biol 2014;426:630–44. 10.1016/j.jmb.2013.10.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohen-Solal JFG, Cassard L, Fridman W-H, et al. Fc gamma receptors. Immunol Lett 2004;92:199–205. 10.1016/j.imlet.2004.01.012 [DOI] [PubMed] [Google Scholar]
- 52.Clynes RA, Towers TL, Presta LG, et al. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med 2000;6:443–6. 10.1038/74704 [DOI] [PubMed] [Google Scholar]
- 53.Punnonen J, Aversa G, Cocks BG, et al. Interleukin 13 induces interleukin 4-independent IgG4 and IgE synthesis and CD23 expression by human B cells. Proc Natl Acad Sci U S A 1993;90:3730–4. 10.1073/pnas.90.8.3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Francis JN, James LK, Paraskevopoulos G, et al. Grass pollen immunotherapy: IL-10 induction and suppression of late responses precedes IgG4 inhibitory antibody activity. J Allergy Clin Immunol 2008;121:1120–5. 10.1016/j.jaci.2008.01.072 [DOI] [PubMed] [Google Scholar]
- 55.Akdis CA, Akdis M. Mechanisms of immune tolerance to allergens: role of IL-10 and Tregs. J Clin Invest 2014;124:4678–80. 10.1172/JCI78891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Boer BA, Kruize YC, Rotmans PJ, et al. Interleukin-12 suppresses immunoglobulin E production but enhances immunoglobulin G4 production by human peripheral blood mononuclear cells. Infect Immun 1997;65:1122–5. 10.1128/IAI.65.3.1122-1125.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maehara T, Moriyama M, Nakashima H, et al. Interleukin-21 contributes to germinal centre formation and immunoglobulin G4 production in IgG4-related Dacryoadenitis and sialoadenitis, so-called Mikulicz's disease. Ann Rheum Dis 2012;71:2011–20. 10.1136/annrheumdis-2012-201477 [DOI] [PubMed] [Google Scholar]
- 58.Shamji MH, Ljørring C, Francis JN, et al. Functional rather than immunoreactive levels of IgG4 correlate closely with clinical response to grass pollen immunotherapy. Allergy 2012;67:217–26. 10.1111/j.1398-9995.2011.02745.x [DOI] [PubMed] [Google Scholar]
- 59.Platts-Mills TAE, Woodfolk JA, Erwin EA, et al. Mechanisms of tolerance to inhalant allergens: the relevance of a modified Th2 response to allergens from domestic animals. Springer Semin Immunopathol 2004;25:271–9. 10.1007/s00281-003-0149-8 [DOI] [PubMed] [Google Scholar]
- 60.Xu Q, Zhang Z, Chen Z, et al. Nonspecific immunoglobulin G is effective in preventing and treating cancer in mice. Cancer Manag Res 2019;11:2073–85. 10.2147/CMAR.S188172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saâda-Bouzid E, Defaucheux C, Karabajakian A, et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol 2017;28:1605–11. 10.1093/annonc/mdx178 [DOI] [PubMed] [Google Scholar]
- 62.Ferrara R, Mezquita L, Texier M, et al. Hyperprogressive disease in patients with advanced non-small cell lung cancer treated with PD-1/PD-L1 inhibitors or with single-agent chemotherapy. JAMA Oncol 2018;4:1543–52. 10.1001/jamaoncol.2018.3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Champiat S, Dercle L, Ammari S, et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res 2017;23:1920–8. 10.1158/1078-0432.CCR-16-1741 [DOI] [PubMed] [Google Scholar]
- 64.Brower V. Hyperprogressive disease with anti-PD-1 and anti-PD-L1. Lancet Oncol 2016;17:e527. 10.1016/S1470-2045(16)30590-3 [DOI] [PubMed] [Google Scholar]
- 65.Kato S, Goodman A, Walavalkar V, et al. Hyperprogressors after immunotherapy: analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res 2017;23:4242–50. 10.1158/1078-0432.CCR-16-3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sasaki A, Nakamura Y, Mishima S, et al. Predictive factors for hyperprogressive disease during nivolumab as anti-PD1 treatment in patients with advanced gastric cancer. Gastric Cancer 2019;22:793–802. 10.1007/s10120-018-00922-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2020-000661supp001.pdf (12.9MB, pdf)