

Abstract
The gut microbiome profoundly affects human health and disease, and their infecting viruses are likely as important, but often missed because of reference database limitations. Here, we (1) built a human Gut Virome Database (GVD) from 2,697 viral particle or microbial metagenomes from 1,986 individuals representing 16 countries, (2) assess its effectiveness, and (3) report a meta-analysis that reveals age-dependent patterns across healthy Westerners. The GVD contains 33,242 unique viral populations (approximately species-level taxa) and improves average viral detection rates over viral RefSeq and IMG/VR nearly 182-fold and 2.6-fold, respectively. GVD meta-analyses show highly personalized viromes, reveal that inter-study variability from technical artifacts is larger than any “disease” effect at the population level, and document how viral diversity changes from human infancy into senescence. Together, this compact foundational resource, these standardization guidelines, and these meta-analysis findings provide a systematic toolkit to help maximize our understanding of viral roles in health and disease.
Keywords: gut microbiome, virome, database, bacteriophage, human health, dysbiosis, lifespan, virus
Graphical Abstract
Highlights
-
•
Assembly of 2,697 gut metagenomes from 32 studies exposed 33,242 viral populations
-
•
Inter-study analyses reveal strong study biases at the viral population-level
-
•
Viral population detection was higher in bulk versus VLP-enriched metagenomes
-
•
Gut viral diversity is age-dependent across healthy, Western people
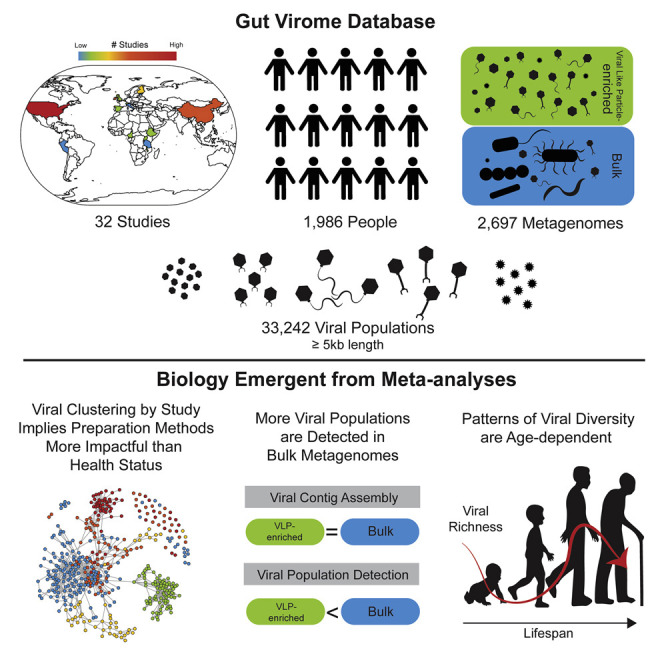
At least 32 studies to date have looked at the human gut virome but with limited consistency. Gregory and Zablocki et al. curate and aggregate these data to provide a systematic virome database; use it to assess study biases, global ecological patterns; and show how viromes evolve throughout the human lifespan.
Introduction
The human gut microbiome is now thought to play an integral role in health and disease (Clemente et al., 2012; Gilbert et al., 2018; Lynch and Pedersen, 2016; Schmidt et al., 2018). Persistent alterations in the structure, diversity, and function of gut microbial communities—dysbiosis—are increasingly recognized as key contributors in the establishment and maintenance of a growing number of disease states (Frank et al., 2007; Human Microbiome Project Consortium, 2012; Qin et al., 2012), including obesity (Turnbaugh et al., 2006) and cancer (Yoshimoto et al., 2013). Gut dysbiosis can develop from complex interplays between host, cognate microbiota, and external environmental factors (Mirzaei and Maurice, 2017; Shreiner et al., 2015). Within the gut microbial consortium, the bacteriome has been the most extensively studied, where significant shifts in population dynamics have been observed between healthy and diseased individuals (Zhang et al., 2015). However, emerging views (Mirzaei and Maurice, 2017; Ogilvie and Jones, 2015; Tetz et al., 2017) suggest that the gut virome plays an important role in homeostatic regulation and disease progression through multiple interaction paths with the co-occurring bacteriome and even directly with human immune system components (Keen and Dantas, 2018).
The first step in studying viruses in complex communities is being able to detect them. Problematically, identifying viral sequences in large, mixed-community datasets is notoriously challenging. Because viruses lack a universal viral marker (Rohwer and Edwards, 2002), as opposed to bacterial 16S rRNA for example, human gut microbiome studies have most commonly used sequence homology searches with BLAST or Kraken (Wood and Salzberg, 2014) against NCBI viral Reference Sequence Database (RefSeq) (https://www.ncbi.nlm.nih.gov/genome/viruses/), ACLAME (a mobile element genome database [Leplae et al., 2009]) or custom hidden Markov model (HMM) databases (e.g., Prokaryotic Virus Orthologous Groups [pVOGs] [Grazziotin et al., 2017]). Although there is now a suite of virus identification tools available, including DeepVirFinder (Ren et al., 2018), MARVEL (Amgarten et al., 2018), VIBRANT (Kieft et al., 2019), and VirSorter (Roux et al., 2015), only the latter has been used in the human gut microbiome literature to date and all are dependent upon reference genome databases to some degree. Further, once viruses are detected there is no standard applied on how viral contigs translate into “species”-level sequences that are to be used as a “working” virus pool for downstream analysis. The lack of viral analysis standards could partly explain the estimated, highly variable (14%–87%) (Mirzaei and Maurice, 2017) rates of virus detection between studies. In addition, factors such as differences in sample processing (Shkoporov et al., 2018), broad under-representation of viral genome space in reference databases (Wang, 2020), lack of culturable host gut microbes (Wang, 2020), and inter-individual variation add further variability (Shkoporov et al., 2019). Further, although viral reference datasets are being generated at unprecedented rates (Roux et al., 2019), these new data are rarely incorporated for cross-comparisons, which would inflate virus novelty in new datasets and/or leaves many virus sequences undetected. In response to these challenges and to enable virome-centric research in health and disease, we sought to establish a comprehensive, easy-access database dedicated to human gut viruses. This effort would enable future gut microbiome research by augmenting virus detection and helping establish processing standards for human gut viruses.
Here, we (1) collected and curated 2,697 human gut metagenomes previously studied for viruses and published as of October 2019 to build the human Gut Virome Database (GVD), (2) evaluated its utility against the best available databases (National Center for Biotechnology Information [NCBI] viral RefSeq and Integrated Microbial Genome/Virus [IMG/VR] [Paez-Espino et al., 2018]), and (3) used it in meta-analyses to assess methodological effects and establish large-scale patterns of gut virome diversity during the course of the human lifespan. The GVD’s 2,697 human gut metagenomic datasets derive from 32 studies and encompass 1,986 individuals from 16 countries that originated either from virus-like particles (VLPs) or whole microbial communities (bulk), as well as several datasets that included RNA sequencing data derived from VLPs. All these datasets were previously studied for viruses, but by using highly variable methods. For the GVD, we in silico re-processed these data to identify viral populations and rigorously remove contamination. This GVD resource is now available on iVirus (Bolduc et al., 2017a) and will be regularly updated.
Results and Discussion
The GVD Contains 33,242 Unique Viral Populations, Dominated by Phages
To build the GVD, 2,697 metagenomic samples from 1,986 individuals were processed from datasets publicly available as of December 2019 (n = 32) (see Table S1), along with one unpublished dataset where access was granted prior to publication. These studies represent 5.35 Tbp of sequence data, derived from a spectrum of gut virome study areas including the following: (1) healthy gut viromes of infants (Lim et al., 2015; Reyes et al., 2010) and adults (Ly et al., 2016; Manrique et al., 2016; Minot et al., 2011, 2012, 2013; Rampelli et al., 2017), as well as individuals experiencing (2) fecal microbiota transplant (FMT) for autism and Clostridium difficile infection (Broecker et al., 2016, 2017; Chehoud et al., 2016; Draper et al., 2018; Kang et al., 2017; Zuo et al., 2018), (3) inflammatory bowel disease (IBD) (Fernandes et al., 2019; Norman et al., 2015; Pérez-Brocal et al., 2013; Zuo et al., 2019), (4) HIV infection (Monaco et al., 2016), (5) type I and II diabetes (Aiemjoy et al., 2019; Kramná et al., 2015; Ma et al., 2018; Zhao et al., 2017), (6) malnutrition (Reyes et al., 2015), and (7) chronic fatigue syndrome (Giloteaux et al., 2016) and hypertension (Han et al., 2018) (see Table S1). These datasets were globally distributed (Figure 1 ). However, most of the studies originated from the United States (38% of GVD studies), and the highest number of sampled individuals and base pairs (bps) sequenced came from Chinese cohorts (44% of individuals and 75% bp sequenced in the GVD). All reads from both bulk and VLP metagenomes (48% and 52% of the GVD, respectively) were processed consistently, assembled into contigs, and viral-like sequences were identified by using three independent methods (Figure S1; see Method Details). Each low-scoring prediction was validated by cross-comparisons between methods and subsequently evaluated for false positives by detecting whether candidate virus sequences contained benchmarking universal single-copy orthologs (BUSCO)-related host single-copy genes (Simão et al., 2015) and for the presence of viral family proteins (VPFs) (Paez-Espino et al., 2018)). Confidence scores associated with each virus detection method and potential false positives are reported in Table S2. To avoid duplicate viral genomes and/or partial virus genomes across the datasets, contigs were de-replicated by clustering sequences according to percentage of average nucleotide identity (ANI) and sequence length. Multiple reports (Brum et al., 2015a; Duhaime and Sullivan, 2012; Duhaime et al., 2017; Gregory et al., 2019a, 2016; Roux et al., 2019) have revealed that >95% ANI was a suitable threshold for defining a set of closely related discrete “viral populations”; follow-on studies suggest that this cut-off establishes populations that are largely concordant with a biologically relevant viral species definition (Bobay and Ochman, 2018; Gregory et al., 2019a, 2016). Using this clustering strategy, we identified highly variable numbers of unique viral populations per study (range: 0–14,018 viral populations; mean = 1,581) (Figure S2A). The GVD comprises 57,605 viral contigs and 33,242 viral populations (≥5 kb or ≥1.5 kb and circular contigs; N50 = 15,395 bp; L50 = 105,286 bp) and mostly bacteriophages (97.7% of GVD). For context, NCBI’s viral RefSeq (v98, released January 2020) database holds 12,183 viruses of eukaryotes, bacteria, and archaea from all environments, combined. Specifically for bacteriophages, the GVD contains 12-fold more than the entire set of cultured phage isolates in viral RefSeq to date. Thus, the GVD greatly augments the repertoire of known phages in the human gut. Importantly, due to a lack of negative controls across 31 out of the 32 studies in the GVD, there is a chance that some of the viral populations included in the GVD might result from contamination. This paucity of negative controls is currently a limitation to gut virome studies.
Figure 1.

Overview of Studies Comprising the Gut Virome Database (GVD)
Global heatmap of the world showing the number and distribution of studies per country. Each white box represents a different continent and contains information about the number of individuals sampled represented by the filled human pictograms and percentage of the total GVD sequencing effort for VLP-enriched (red pie charts) and bulk metagenomes (yellow pie charts) of each country studied within that continent.
See also Table S1.
Taxonomically, 97.7% of GVD viral populations are bacterial viruses (i.e., phages), 2.1% are eukaryotic viruses, and 0.1% are archaeal viruses (Figure 2 A). The 712 eukaryotic viruses were taxonomically diverse (from 23 families), dominated by single-stranded DNA (ssDNA) families Anelloviridae (71%), Genomoviridae (8%), and Circoviridae (8%), all of which have been previously reported in the datasets underlying the GVD (Monaco et al., 2016), with the exception of Genomoviridae. Three single-stranded, positive-sense RNA virus families were detected (Table S3), represented by 34 viral populations (0.1% of the GVD). The human Picornaviridae was the most represented (parechoviruses, coxsackievirus, cosaviruses, enterovirus, and hepatovirus), along with 8 plant or fungal viruses of the Alphaflexviridae and Virgaviridae and one putative member of Cruliviridae. Detection of plant viruses has been reported before (Zhang et al., 2006) and is likely the result of transient passage through dietary habits. Human picornaviruses associated with gastrointestinal tract disorders were to be expected, and most derived from a Cameroonian patient cohort selected for gastroenteritis symptoms, in which the study design included RNA sequencing (Yinda et al., 2019). The low number of recovered RNA viruses (0.1% of the GVD) (see Tables S2 and S3) in the GVD might stem in part from having a few studies (6 out of 32) that included viral RNA sequencing. More importantly, the likely biggest factor contributing to low RNA virus detection is that de novo RNA virus identification method development is an ongoing effort (Shi et al., 2016; Starr et al., 2019), such that RNA virus diversity in gut viromes (and generally in viral metagenomes) is likely vastly undersampled and that our detection is limited to homology to well-characterized pathogens (Zhang et al., 2019). Among the phages, 88% did not have International Committee on Taxonomy of Viruses (ICTV) classification, and the remaining fraction comprised of double-stranded DNA (dsDNA) tailed phage families (Siphoviridae, Myoviridae, Podoviridae, and Ackermannviridae), Microviridae, and Inoviridae (see Table S2). Twenty-four unknown archaeal viral populations were detected, but none with close genome and/or gene homology to any of the classified archaeal viruses. Notably, our naive viral taxonomic assignments using “a majority-rules approach” (see Method Details) led to taxonomic assignments that recent literature has shown are erroneous and due to methodological artefacts, such as Phycodnaviridae and Mimiviridae (Sutton et al., 2019), so we manually removed such taxa. Thus, given that most of the viral populations are represented by fragments of their genomes, taxonomic assignments using the “a majority-rules approach” will improve and be refined as more complete genome representatives are sequenced and assembled. Nonetheless, the high number of unclassified phages likely results from the underrepresentation of human gut phages in reference databases and further highlights how much viral diversity remains to be characterized in the human gut.
Figure 2.

The Gut Virome Database (GVD)
(A) Pie charts showing the number of bacteriophages, eukaryotic viruses, and archaeal viruses in the GVD (center) and their familial taxonomic composition by the bacteriophages (left) and the eukaryotic viruses (right).
(B) Gene-sharing taxonomic network of the GVD, including viral RefSeq viruses v88. RefSeq viruses are highlighted in red. Every node represents a virus genome, whereas connecting edges identify significant gene-sharing between genomes, which form the basis for their clustering in genus-level taxonomy.
(C) Concentric pie chart showing the number of annotated bacterial host phyla (inner) and family (outer) of the GVD viruses. Host taxonomy follows the GTDB database taxonomic classifications, and putative host information per each viral population is listed in Table S2. See also Figures S1 and S2 and Tables S2, S3, and S6.
To fill this phage and archaeal virus taxonomic classification gap, we used an extensively validated (Adriaenssens et al., 2020; Bolduc et al., 2017b; Jang Bin et al., 2019), genome-based, gene-sharing network strategy that de novo predicts genus-level groupings (“viral clusters” [VCs]) from viral population data. A network (Figure 2B) computed from 15,330 GVD phage genomes (only those >10 kb in length; 46% of GVD) and 2,191 reference phage genomes (from NCBI Viral RefSeq version 88) revealed 2,048 VCs. Of these, 1,666 VCs were exclusively composed of GVD genomes (7,055 viral genomes or ∼46% of GVD genomes), whereas 125 VCs contained genomes from both RefSeq and the GVD (600 viral genomes or ∼4% of GVD genomes) and 257 VCs were exclusively composed of RefSeq taxa. Thus, the GVD augments the current number of ICTV-recognized phage genera approximately 3.5-fold. Although not explored here, given that our goals focused on taxonomic classification, the shared protein content within and between VCs calculated in our network analyses could be used to guide qPCR assays for next-generation sequencing validation (Monaco and Kwon, 2017) and/or tracking of viruses at either the viral population or genera level under changing conditions (Kramná et al., 2015).
Next, we sought to link the GVD phage and archaeal viral populations to their hosts by using in silico strategies (see Method Details). In total, we were able to identify the hosts down to the microbial taxonomic family (Genome Taxonomy Database [GTDB] taxonomy) (Parks et al., 2018) of ∼42% (n = 13,954) of the viral populations (see Table S2). The most common identifiable hosts (Figure 2C) across GVD viral populations belonged to the bacterial phyla Firmicutes (GTDB Firmicutes, Firmicutes_A, and Firmicutes_C combined; 49.3%) and Bacteroidetes (GTDB Bacteroidota; 33.7%), consistent with our knowledge that Firmicutes and Bacteroides are the most prominent bacterial phyla in the human gastrointestinal tract (Eckburg et al., 2005). Notably, Firmicutes typically outnumber Bacteroidetes in unhealthy individuals with metabolic and digestive disorders (Broecker et al., 2016; Chehoud et al., 2016; Ley et al., 2005; Nicholson et al., 2012; Norman et al., 2015; Ott et al., 2004; Zhao et al., 2017) and GVD metagenomes are biased toward unhealthy individuals (>60% of the metagenomes comprising >83% of the bps sequenced), which might account for the increased Firmicutes viral populations in GVD.
The GVD Significantly Improves Virus Detection over Current Viral Genome Databases
To assess the value of the GVD, we quantitatively evaluated virus identification sensitivity between multiple databases by comparing the number of viral populations identifiable by read recruitment against GVD, NCBI’s viral RefSeq v96, DOE’s IMG/VR v4 (Paez-Espino et al., 2018) and the individual virome databases from each study (Figure 3 ; see Method Details). To control for assembly improvements since the original metagenome and/or virome datasets were published, for the latter, we individually assembled the original viromes into viral populations for read recruitment. NCBI viral RefSeq was the most commonly used viral genome database across the studies surveyed here, being used in 23 of 29 studies where the specific database used was documented (information on the genome database used was unavailable for three studies; see Table S1), and hosted 9,294 virus genomes already de-replicated (as of v96, November 2019, used here). In comparison, the IMG/VR database was not documented as being used by any of the 32 studies gathered, despite the latest release (v4, July 2018, used in this study) containing nearly two orders of magnitude more virus genomes and genome fragments (760,453 virus contigs, though not de-replicated). For comparison purposes to the GVD (see Method Details), we de-replicated the IMG/VR contigs the same way as we did the GVD to obtain viral-population-level genomes. This yielded 359,826 viral populations for the IMG/VR database.
Figure 3.

GVD As a Reference Database Increases Viral Population Detection
Boxplots showing median and quartiles of the number of viral populations detected per study using the individual virome, Viral Refseq v96, JGI IMG/VR v4, or GVD databases. All pairwise comparisons were performed by using Mann-Whitney U tests. Non-significant p values are denoted as “ns.”
In 31 out of the 32 total studies tested (Figure 3), the GVD enabled the detection of significantly more viruses than viral RefSeq v96 (Mann-Whitney U tests; p < 0.05; 182 [average] ± 390 [standard deviation]-fold increase) and individual viromes (Mann-Whitney U tests; p < 0.05; 6-fold ± 40-fold increase). Notably, the proportion of the metagenome mapping to the GVD was highly variable between studies (Figure S2B) and, as expected, a higher proportion of VLP-enriched metagenomes mapped to the GVD than did bulk metagenomes (Figure S2C). There was a single study (Reyes et al., 2010) in which no viruses were detected (see Method Details) in all databases queried in this analysis. In comparison to IMG/VR, we detected more viruses with the GVD in all studies, 15 (47% from total) of which were in a significant manner (Mann-Whitney U tests; p < 0.05; 2.6-fold ± 2.1-fold increase) (Figure 3). Five of the remaining fourteen studies had too low of a sample size and/or number of detected viruses to statistically compare the GVD and IMG/VR. Additionally, we tested the ability of the GVD to increase the number of viral populations detected in a study not included in the GVD (Clooney et al., 2019) (Figure S3A). We saw similar results, and GVD significantly outperformed viral RefSeq v96 and the individual virome while having a non-significant higher median number of viral populations detected than IMG/VR.
When we considered the number of reads that recruited across the different databases across all studies, significantly more reads (Mann-Whitney U tests; p < 0.05) were recruited to the GVD than to any other database across 19 out of the 32 studies (Figure S3B). After GVD, IMG/VR was the next best performing database for viral detection in the human gut, given that our tests showed an average of 64-fold ± 120-fold increase over viral RefSeq (Mann-Whitney U tests; p < 0.05). IMG/VR was expected to surpass viral RefSeq because it aggregates both cultivated reference virus genomes from RefSeq, >12,000 prophages, and >700,000 uncultivated virus genomes and/or fragments from many environments, including multiple human body sites (Paez-Espino et al., 2018). Overall, the significant increase in virus detection by the GVD over other databases highlights the low representation of gut viruses in RefSeq and thus demonstrates the value of the GVD for sequence-based virus identification in human gut microbiome datasets. Thus, given that the GVD significantly improves viral detection over current viral genome databases, we used the GVD as the database for all remaining analyses in this study.
The Human Gut Virome Is Highly Person Specific
In light of the current hypothesis of a “core” gut virome (Manrique et al., 2016), we were first curious whether any GVD viral population was found across a high percentage or all metagenomes in the GVD. On average, 542 ± 726 (average ± SD; range: 0–6,420) viral populations were detected per metagenome, but not a single viral population was found across all metagenomes. In fact, the most ubiquitous viral population in the GVD was found in only 39% of the metagenomes, 128 viral populations occurred in more than 20% of the metagenomes, and most (69% or 22,913) of the viral populations were only sporadically detected at all (<0.5% of the metagenomes) (Figures S4A and S4B; Table S4). Further, we specifically looked at the prevalence of crAssphages, a well-recognized, multi-genera family of phages known to be widespread in gut viromes (Guerin et al., 2018) (Figure S4B). In total, we identified 70 crAssphage populations (see Method Details), 30 of which had genomes >10 kb that clustered into 12 VCs (i.e., genus-level grouping) by genome-based, gene-sharing networks (Jang Bin et al., 2019). Although together these 70 crAssphage populations are ubiquitous across the GVD samples, there was not one crAssphage viral population found universally, and the most widespread crAssphage population occurred in only 12% of samples. These findings support the mounting evidence for highly personal gut viromes, as recently highlighted in twins (Moreno-Gallego et al., 2019) and in ten healthy adults during a year-long monitoring period (Shkoporov et al., 2019). Although the latter study pointed to the potential of a core virome at higher taxonomic levels, we failed to recover any universally shared viral VCs (approximately genus level taxonomy) (Figures S4C and S4D), given that the most ubiquitous VC was only present in 40% of the metagenomes. That same study suggesting the potential of a higher-taxon core only looked at ten healthy, Western adults, and the metagenomes in the GVD represented a diversity of people from different geographical regions and ages. Thus, further studies are necessary to resolve whether a core virome does exists at higher taxonomic levels.
Varied Processing Methodology Prevents Population-Level β-Diversity Inter-Study Comparisons
Next, given a systematically processed GVD and its demonstrably improved virus detection capability, we sought to determine whether global clustering patterns would emerge via a GVD β-diversity (between-sample changes in population composition) meta-analysis. To this end, we performed population co-occurrence analyses at two levels of resolution (per study and across metagenomes within studies) and then evaluated what level of metadata best captured the resultant variation (methodology, disease state, etc.). To assess population-overlap between studies, we counted the number of GVD viral populations that recruited reads within and between different GVD studies (i.e., the same viral population was detected in >1 metagenome in both studies compared). We expected studies exploring similar disease states would share the greatest number of viral populations. However, hierarchical clustering of the studies on the basis of the number of shared viral populations revealed that this was rare and mostly not the case, and studies exploring the viromes of diseased individuals (e.g., IBD, Clostridium difficile infection, and diabetes) did not cluster together (Figure 4 , top left—heatmap). Instead, we saw that the different studies hierarchically clustered into four groups (I–IV), and that this clustering was weakly driven around metagenome type; many bulk metagenomes clustered together into group III.
Figure 4.

Individual Viromes Study Databases and Cross-Study Comparisons
Shown at the top left is a hierarchically clustered heatmap showing the number of viral populations shared within and between studies clustered into four groups (I–IV). Viral population co-occurrence network per individual within each study per group. Shown on the bottom right is a hierarchically clustered heatmap showing the number of viral genera shared within and between studies clustered into three groups. Viral genus cluster co-occurrence networks per metagenome within each study per group. Colored dots and pictograms next to study names in heatmaps represent metagenome type and a common disease studies across all 32 studies in GVD, respectively.
See also Figure S4.
Because the GVD studies did cluster into four distinct groups (Figure 4, top left—heatmap), we next tested whether any metadata best captured the resultant variation across the metagenomes within each study within each group. Using an inverse covariance analysis (i.e. combined partial correlations across shared viral population between metagenomes) to sub-cluster the different metagenomes across the studies in each group, we found that the metagenomes within a study sub-clustered together irrespective of geographical origin, health status, and/or diet (Figure 4, top left—networks). Notably, the group III metagenomes derived mostly from bulk metagenomes were more closely sub-clustered, but they still sub-clustered strongly by study. This per-study sub-clustering implies that even within these grouped studies, metagenomes from different studies are not comparable because the inter-study variation is driven by methodological impacts. These results reveal that although methodology does not affect the number of viral contigs recovered, it does affect the recovered types of viruses (see upcoming findings comparing methodological effects). Interestingly, when we looked at genus-level (i.e., VC-level) co-occurrence, we saw that there are still strong groupings (A–C) at the study level, but within each group, metagenomes across these studies share many VCs (Figure 4, bottom right). Thus, β-diversity meta-analysis across all studies exploring the effect of “disease” across GVD studies is not possible at the population level, but within similarly processed studies, it might be possible at the genus level.
To Enrich or Not to Enrich? Viruses Recovered from Bulk Versus Virus-Particle-Enriched Metagenomes
From a pragmatic point of view, we next wondered whether GVD datasets could inform experimental design. Specifically, to study viruses, is sequencing effort better put into metagenomes of bulk or purified VLPs? The GVD’s 2,697 gut metagenomes are roughly evenly divided across these two metagenome types with bulk and VLP metagenomes contributing 2.7 Tbp (∼51.4% of GVD) and 2.6 Tbp (∼48.6% of GVD) of data, respectively. Although most samples only have one or the other data, one study (10 samples) (Shkoporov et al., 2019) provided both bulk and VLP metagenomes for 10 samples.
We first assessed whether there was a difference in de novo viral recovery between VLP and bulk metagenomes (Figure 5 A). We measured viral recovery by using the number of viral contigs (>5 kb or >1.5 kb and circular in length; i.e., not de-replicated viral populations) assembled per bp sequenced per study given that the viral contigs assembled from samples within the same study are often pooled. These analyses revealed no significant difference (Mann-Whitney U test; p = 0.25) in the number of viral contigs assembled per bp sequenced between VLP and bulk metagenomes, which contrasts viral recovery results from permafrost soils, where VLP metagenomes outperform bulk metagenomes by 2-fold (Trubl et al., 2018). However, viral recovery from the GVD’s VLP metagenomes was heterogeneous, so we evaluated how VLP methodology affected viral recovery. First, although multiple displacement amplification (MDA) is known to provide non-quantitative metagenomic datasets with both systematic and stochastic biases (Solonenko et al., 2013; Yilmaz et al., 2010), we found no significant difference (Mann-Whitney U test; p = 0.75) in viral recovery between non-MDA and MDA-treated metagenomes (Figure 5B). Nonetheless, it was notable that MDA-treated VLP studies were significantly enriched in eukaryotic, ssDNA viruses (Mann-Whitney U tests; p < 0.05), a known bias of MDA (Figure S5A). Second, we tested the effect of VLP enrichment strategies, which ranged from removing human and bacterial cells to enrich for VLPs (centrifugation, filtration, CsCl gradients, and nucleases) to concentrating the VLPs (centricon concentration and PEG precipitation). Again, we found no significant difference in the number of viral contigs recovered (Kruskal-Wallis test; p = 0.47) across the different VLP enrichment strategies (Figure 5C). Further, we found that contig sizes were not significantly different either between VLP and bulk metagenomes (Mann-Whitney U test; p = 1) (Figure S5B) or across VLP-enrichment strategies (Kruskal-Wallis test; p = 0.18) (Figure S5C). Although surprising given that prior work with seawater showed VLP-enrichment methods, especially at the concentration step (tangential flow filtration versus FeCl3) can have large effects on the number of viral types recovered (Hurwitz et al., 2013), we note that the concentration steps tested here are much more similar, with both being physical steps, in contrast to the physical and chemical steps tested on marine samples. Overall, we found sequencing depth (i.e. number of bps sequenced) was the only major driver that increased viral recovery because it was strongly correlated to the number of assembled contigs in fecal samples (linear regression; R2 = 0.89 (all), R2 = 0.95 (bulk), R2 = 0.45 (VLP) Figures 5D, S5D, and S5E). The non-fecal study of colon biopsies was an outlier (Zuo et al., 2019).
Figure 5.

VLP-Enriched (VLP) and Bulk Metagenomes Comparisons for Studying Viruses in the Human Gut
(A–C) Boxplots showing median and quartiles of the number of assembled contigs per base pair sequenced per study (A) of VLP and bulk metagenomes, (B) of VLP metagenomes with and without MDA, and (C) of the different VLP-enrichment methodologies across the studies. Outlier dots were removed from plot (C) to better show the range of values. The n value above each box plot represents the number of studies using each VLP-enrichment method.
(D) Scatter plot with a linear regression line showing the number of assembled viral contigs per bp sequenced per study with VLP and bulk metagenome studies identified by different colors. In the inset is a Venn diagram showing the number of GVD viral populations that originated from VLP or bulk or both types of metagenomes.
(E) Boxplots showing median and quartiles of the number of viral populations detected per bp sequenced per individual of VLP and bulk metagenomes.
(F) Boxplots showing median and quartiles of the number of assembled contigs per bp sequenced (top left) and the median contig length (top right) for VLP and bulk metagenomes processed for the same samples in the Shkoporov et al, (2019) (bottom). Connected dot plot showing the number of viral populations detected per bp sequencedby using VLP and bulk metagenomes for each individual in the Shkoporov et al, (2019) study. All pairwise comparisons were performed by using Mann-Whitney U tests. Non-significant p values are denoted as “ns.”
See also Figure S4.
Importantly, although the number of viral contigs recovered does not vary across the treatments evaluated here, there are clear differences between the viruses that are captured by VLP and bulk metagenomes. In fact, only 10% of the GVD viral populations (Figure 5D, Venn diagram inset) were recovered in both VLP and bulk metagenomes, indicating that the different methods enrich for different virus populations. Analyses of the 10 samples processed by using both VLP and bulk methods (Shkoporov et al., 2019) revealed a similar overlap (8.5%) of the viral populations being recovered from both metagenome types. Mechanistically, this presumably results from bulk metagenomes primarily capturing actively infecting viruses or integrated prophages, whereas VLP metagenomes target free viral particles that would have long residence times in seawater, but are perhaps much more transient in the gut (Neil and Cadwell, 2018; Shkoporov et al., 2019). Thus, despite no significant difference in the number of viruses recovered, the two methods are clearly capturing different subsets of the gut viral community such that combined VLP and bulk metagenomes can increase the number of viral populations recovered. Further, increasing sequencing efforts will increase the number of viral contigs assembled in fecal samples regardless of enrichment method.
Next, we assessed viral detection differences between VLP and bulk metagenomes. Although not all viruses readily assemble because of low abundances or hypervariable genomic regions (Pop, 2009), once reference genomes are available, viral populations outside those de novo assemblies can be detected via read mapping. We used the GVD as a reference database and recruited reads from all GVD metagenomes. Because read mapping is mostly done per metagenome, we evaluated viral detection by using the number of viral populations detected per bp sequenced per metagenome (Figure 5E). We found that detection performance by using bulk metagenomes was significantly higher (Mann-Whitney U test; p = 2.22e-16) than in VLP metagenomes. These results suggest that bulk metagenomes provide a clear advantage for viral detection if searched with a well-furnished database like GVD. To our knowledge, a quantitative estimate of viral detection rates between VLP and bulk metagenomes has not been reported previously in any ecosystem.
To further validate these results, we applied the same analysis by using only samples in which both bulk and VLP metagenomes were generated from the same 10 samples, and outside the viral particle purification step they were identically processed (Shkoporov et al., 2019). As in our aforementioned results, we found no significant difference between VLP and bulk number of viral contigs assembled per bp sequenced (Mann-Whitney U test; p = 0.48) (Figure 5F, top left) and higher virus detection was observed in bulk datasets than in VLP datasets (Figure 5F, bottom). When we looked at the median assembled contig lengths, VLP contigs were significantly longer than bulk contigs (Mann-Whitney U test; p = 0.0011) (Figure 5F, top right). This higher median contig length contrasts our findings above at the study level. Nonetheless, we hypothesize that VLP enrichment, in the absence of more contigs recovered, should assemble longer contigs when comparing identical samples.
Altogether these findings suggest that, for human gut viruses, sequencing-effort-normalized viral recovery efficiency is similar across the suite of commonly used preparation methods, so bulk metagenomes might be the best choice for future work because of their ease of preparation compared with that of VLP metagenomes and because of their higher viral detection rates. Nonetheless, combining both VLP and bulk metagenomes can improve de novo viral recovery. We hypothesize that the increase in detection performance in bulk metagenomes might be driven by the fact that the gut virome is enriched in temperate phages (reviewed in Mirzaei and Maurice, 2017), such that when integrated into their hosts genomes as prophages, these viruses would likely be removed from the VLP metagenomes in the VLP enrichment process. Analysis of the number of detectable temperate phages in the GVD, soil, and marine viral datasets has revealed that gut viruses have ∼2.6- and 4.3-fold more detectable temperate phages than soil (Emerson et al., 2018; Trubl et al., 2018) and marine (Gregory et al., 2019a), respectively (Figure 6 ), indicating that we most likely are losing more viruses from VLP enrichment than in other systems.
Figure 6.

More Gut Viruses Are Temperate Phages than in the Soil and Oceans
Pie charts showing the percentages of temperate phages found in the human gut (GVD dataset), soils (IsoGenie dataset), and oceans (Global Oceans Viromes 2 dataset).
Human Gut Virome Richness Is Also Impacted by Methodology, but Is Still Comparable among Some Studies
Because of the differences in viral detection across VLP and bulk metagenomes and the difficulty in exploring cross-study β-diversity, we next wanted to determine whether it was even possible to compare α-diversity (local diversity) across studies. Notably, α- and β-diversity were theoretically proposed as components of γ-diversity (regional diversity), meaning that they should scale together (Whittaker, 1960). Nonetheless, using these theoretical definitions in practice, it is impossible because full species inventories at local and regional scales are difficult to survey (Chao et al., 2006; Colwell and Coddington, 1994; Plotkin and Muller-Landau, 2002). Thus, most β-diversity metrics try to be independent of α-diversity to account for compositional sampling (Barwell et al., 2015; Jost, 2010), resulting in uncoupled α-diversity and β-diversity metrics often driven by completely different ecological drivers. This phenomenon has been seen in marine viruses (Gregory et al., 2019b), soil microbes (Prober et al., 2015), soil fungi (Chen et al., 2018), and at global scales looking at conservation across different ecosystems (Hillebrand et al., 2018). Given this uncoupling between α- and β-diversity, we evaluated whether α-diversity could be comparable between studies after removing confounders.
Given that 96% of the studies in the GVD used MDA, we used viral richness as our α-diversity metric because it is more insensitive to compositional changes and thus less affected by the population-abundance-skewing effects of MDA. Further, because of unequal sequencing depth, we chose to use the number of viral populations per bp sequenced per individual as a proxy for viral richness (with viral richness being averaged across time points for individuals with more than one metagenome). Importantly, MDA might also result in low abundance populations not even being amplified, which could lead to decreased viral richness. An initial exploration of this viral richness across studies revealed discordance among studies, with many studies having median viral richness across individuals above and below the 75% and 25% quantiles, respectively, of viral richness across all individuals (Figure S6A), and viral richness strongly correlated by study (Kruskal-Wallis test; p < 2.2e-16).
Across the GVD, the vast majority of studies were Illumina sequenced (84%), VLP enriched (84%), and MDA treated (96%) (Table S1). Thus, we hypothesized that studies that did not have the aforementioned characteristics were most likely outliers. We sequentially and additively tested the effect of sequencing platform, enrichment type (bulk or VLP), and MDA and found that 454 sequenced metagenomes were significantly different (Figure S6B) (Mann-Whitney U test; p ≤ 9.5e-06) and, of the remaining non-454 studies, bulk metagenomes were also significantly different (Figure S6C) (Mann-Whitney U test; p < 2.22e-16). Thus, 454 and bulk metagenomic studies were removed. Although non-454, VLP-enriched, non-MDA, and MDA-treated metagenomes were not significantly different (Figure S6D; Mann-Whitney U test; p = 0.12), non-MDA studies, which only account for 4% of the studies, were also removed to maintain consistency among studies and to ensure that the potential biases introduced by MDA are universal across the metagenomes assessed. Analyses of viral richness across studies, nonetheless, still revealed discordance between studies with viral richness still strongly correlating by study (Kruskal-Wallis test; p < 2.2e-16).
Geographic origin and health status can also have a huge effect on the gut virome (Broecker et al., 2016; Ma et al., 2018; Monaco et al., 2016; Norman et al., 2015). Thus, we tested the effect of geographic origin (Western or non-Western) and health status (healthy or diseased) on viral richness (geographic origin and disease state for each metagenome can be found in Table S5). We found that non-Western individuals have significantly higher viral richness than Western individuals (Figure S6E) (Mann-Whitney U test; p = 8.6e-07). This supports previous findings of higher viral richness in non-Western individuals (Rampelli et al., 2017) and parallels findings of bacterial richness in Western versus non-Western individuals (Obregon-Tito et al., 2015; Schnorr et al., 2014; Yatsunenko et al., 2012). Next, among the Western individuals, we found higher viral richness among healthy individuals than among individuals with disease (Figure S6F) (Mann-Whitney U test; p = 8.5e-13). This supports previous findings that show healthy individuals have higher viral richness than do individuals with Clostridium difficile infection (Zuo et al., 2018) and IDB in one study (Pérez-Brocal et al., 2013), but contrasts findings that show viral richness is higher in patients with diabetes (Ma et al., 2018) and IBD in other studies (Fernandes et al., 2019; Norman et al., 2015). Thus, we filtered out non-Western, diseased individuals. The remaining individuals from 11 studies represented non-454-sequenced, VLP-enriched, MDA-treated metagenomes from healthy, Western individuals. Across these studies, the median number of viral richness all fell within the 75% and 25% quantiles of viral richness across all remaining individuals (Figure S6G), resulting in a non-significant association between viral richness and study (Kruskal-Wallis test; p < 0.09745) and indicating that the viral richness values across these individuals in these studies were comparable.
Gut Virome Diversity Is Age-Dependent in Healthy, Western Cohorts
Beyond using GVD meta-analyses to re-assess existing human gut virome study conclusions, we next wanted to leverage the subset of data for which viral richness was identified to be comparable (see above), and used it to assess a near-completely open question: how does age affect gut viral richness? This filtered dataset (151 healthy, Western individuals, non-454, VLP-enriched, and MDA-treated) represented individuals whose ages spanned the different life stages (0–65+ years old) within the GVD. In total, there were 27 infants (0–3 years old [Lim et al., 2015; McCann et al., 2018]), 11 children (3–18 years old [Fernandes et al., 2019]), 93 adults (18–65 years old [Chehoud et al., 2016; Draper et al., 2018; Ly et al., 2016; Minot et al., 2012, 2013; Moreno-Gallego et al., 2019; Shkoporov et al., 2019]), and 20 elderly (>65 years old [Stockdale et al., 2018]) defined as healthy in their respective datasets. Mounting evidence suggests that the human gut bacteriome displays distinct, age-dependent patterns of diversity (i.e., species richness, assessed with 16S rRNA sequencing), in response to an array of factors including immune status fluctuations during life (Claesson et al., 2012; Odamaki et al., 2016; Scepanovic et al., 2019). However, there are no comparable estimates for the human gut virome across the lifespan, except in babies (Liang et al., 2020; Lim et al., 2015).
Using these healthy, Western individuals, we looked at viral richness across the human life stages (Figure 7 A). Across the human lifespan in the GVD, highest overall viral richness was observed in infants and adults, and there were significant increases between children and adults (Mann-Whitney U test; p = 0.034) and significant decreases between adults and elderly individuals (Mann-Whitney U test; p = 0.014) (Figure 7B, left). The latter trend was also true for a Chinese cohort (Mann-Whitney U test; p = 0.00048) (Figure 7B, right) (Ma et al., 2018).
Figure 7.

Viral Diversity across Lifespan in Healthy, Western Individuals
(A) Composite plot showing (from top to bottom) the number of bacterial operational taxonomic unit (OTU) trends across the life stages derived from a literature review; a map highlighting the origin of the healthy, Western individuals; the number of healthy, Western individuals per life stage; Loess smoothing plots of the number of viral populations; the number of viral populations by type; the number of viral populations by viral family; and the number of crAssphage populations per bp sequenced across the life stages in healthy, Western individuals. Box plots showing median and quartiles and Mann-Whitney U test results between the different life stages can be found in Figure S7.
(B) Box plots showing median and quartiles of the number of viral populations per bp sequenced across the life stages across healthy, Western individuals (left) and across adults and elderly individuals from non-Western Chinese individuals (right). All pairwise comparisons were performed by using Mann-Whitney U tests.
(C) Presence absence plot showing the distribution of the 70 crAssphage populations in the GVD across the healthy, Western individuals.
See also Figures S6 and S7 and Table S5.
These overall trends, however, did not apply evenly across virus types. For example, eukaryotic (mostly human Annelloviruses) virus richness (Figure 7A, red curve) is high at infancy, presumably driven by an underdeveloped immune system, and then decreases into childhood and remains constant and low through the rest of life (Figure S7A). In contrast, bacteriophages mirrored the overall viral richness trend, with the bacteriophage family Siphoviridae mirroring the overall viral richness trend the best (Figures 7A and S7A). This follows our basic understanding of the gut virome, which suggests that most viruses are temperate bacteriophages, of which many are Siphoviridae viruses (Mirzaei and Maurice, 2017). Curiously, Microviridae richness per bp sequenced peaked modestly in infancy, dropped in childhood and then slowly increased across the rest of the lifespan (Figures 7A and S7A).
Given the importance of crAssphages in the human gut virome literature, we next assessed how its populations per bp sequenced varied with age. This revealed a relatively constant upward trend from infancy to elderhood, and the largest shift occurred between childhood and adulthood (Figures 7A, white curve, and S7). We were then curious whether this increase was because of the acquisition of additional crAssphage species through life or because of the initial crAssphage populations expanding their proportional niche in the gut virome (Figure 7C). These analyses revealed that crAssphage were not detectable in infants (except in one individual) or children, which contrasts findings from recent studies (Guerin et al., 2018; Liang et al., 2020). This implies that the large increase in crAssphage populations per bp sequenced from children to adults was because of the acquisition of crAssphage. For the increase observed between adults and the elderly, we saw no significant difference in the number of crAssphage populations and no changes in the crAssphage populations detected between adults and elderly individuals (Mann-Whitney U tests; p > 0.05). Notably, because we were using the number of viral populations per bp sequenced as a proxy for viral richness, changes in this value can represent an increase in the number of viral populations or an increase in the proportion of the total virome the viral populations make up. Thus, we hypothesize that the increase in crAssphage from adult to elderly is most likely because of crAssphage populations taking up a larger proportion of the total gut virome. All age-dependent viral richness patterns were upheld even after a stringent removal of 19,551 potential contaminants, defined here as any population that was rare in any study and only found in one study (Figure S7B; see Method Details).
Lastly, we wanted to see how these age-dependent viral richness trends compared with overall bacterial richness trends in the gut. There are two major paradigms for the life stages of gut bacterial richness. The first paradigm is that the commensal gut bacterial richness increases into adulthood and then decreases into old age (reviewed in Nagpal et al., 2018). The second paradigm is that bacterial richness slowly increases throughout the lifespan from infancy into old age (reviewed in Santoro et al., 2018). Some studies attribute this paradigm split at old age to whether an elderly person is living in a nursing home or in the broader community, with those living in nursing homes seeing a decrease in bacterial richness (e.g. Claesson et al., 2012) (Figure 7A). Prior analyses of the viral richness nonetheless revealed no difference between elderly individuals living in nursing homes or the community (Stockdale et al., 2018); thus, all of the GVD elderly individuals, who coincidentally were from that study, were included regardless of where they lived. Here, we found the viral richness fluctuations broadly related to bacterial richness trends in bacterial richness paradigm one, but with a strong deviation at infancy, where most likely the weak, underdeveloped immune system and lack of epithelium-protecting commensal bacterial allows for viral infection of human cells (Figure 7A). The presence of many eukaryotic viruses was also previously found in babies (Liang et al., 2020; Lim et al., 2015). Analyses of human cohorts after the first paradigm suggest that increased bacterial inter-species competition over the lifespan induces the establishment of more successful strains of the same species, thus reducing richness into old age (Aleman and Valenzano, 2019). It remains unclear whether this inter-species competition affects viral richness, but given the parallels between bacterial and viral richness, we hypothesize it most likely plays a role for both. This inter-species competition might also help explain the crAssphage trend, in which Bacteroides (crAssphage’s host) gain a stronger foothold into older years, thus increasing crAssphage abundance. Overall, these results suggest that, like gut bacterial richness, gut viral richness is also age dependent.
Conclusions
The lack of a curated database for the detection of viral sequences in the human gut has been identified as the most critical shortcoming of applying metagenomic approaches to studying the human gut virome (Shkoporov and Hill, 2019). Although sample preparation standards are emerging for human gut viromics (Shkoporov et al., 2018), the field currently lacks an equivalent for in silico virus analytics. The GVD and its associated contig processing methods are geared towards filling this standardization gap and performs well beyond “classical” databases used across the field.
However, the GVD dataset currently suffers from several limitations. First, the geographic and ethnic representation across the dataset is not very broad. Meta-analyses will benefit from more broadly representative datasets as they become available. Second, there are many more human gut and other human-associated bulk metagenomic datasets and, if mined for viruses, these could be a rich source for virus reference genomes as found for soils (Emerson et al., 2018) and the large-scale Earth Virome study (Paez-Espino et al., 2016). In addition, given the current challenges in RNA virus discovery in metagenomic datasets (Greninger, 2018), the extent of RNA viruses in the human gut is likely underestimated. Lastly, GVD viral contigs, even though a conservatively determined dataset, might contain other non-viral mobile elements that possess phage-like characteristics, such as gene transfer agents and defective prophages.
The GVD, combined with the means to classify uncultivated virus genomes (Jang Bin et al., 2019), are prime starting requirements for enabling ecosystem-wide examinations (Roux et al., 2016) of the dynamics and effects of the virome within the human gut. For example, here, we used the GVD database to uncover the age-dependent patterns of virome diversity in healthy, Western individuals. However, the GVD could also have much broader implications including helping better classify individuals’ native gut microbiomes and viromes to determine how it affects a person’s predisposition to diseases like COVID-19 (Gou et al., 2020). Outside of the human ecosystem, the GVD could have potential use to increase viral detection in a broader context, such as animal gut microbiomes or aquatic samples being analyzed for fecal contamination monitoring. Other environmental advances also invite such studies to include assessing the role of micro- and macro-diversity on virus persistence (Gregory et al., 2019a), and metabolic reprogramming via virus-encoded auxiliary metabolic genes (Emerson et al., 2018; Roux et al., 2016) and without that could drastically alter the ecosystem outputs of any infected cell (Howard-Varona et al., 2020). These combined eco-systems biology efforts are critical to enable studies of the human gut virome to advance from “stamp collecting” diversity studies towards the kinds of comprehensive efforts needed to incorporate viruses into mechanistic, predictive models. Such efforts, with future viral mapping outside the gut to parallel efforts for the “non-gut” human microbiome (Pasolli et al., 2019), should help transform personalized medicine and lead to a better understanding of human ecosystems.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| Aiemjoy et al., 2019 sequencing reads | Aiemjoy et al., 2019 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Broecker et al., 2016 sequencing reads | Broecker et al., 2016 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Chehoud et al., 2016 sequencing reads | Chehoud et al., 2016 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Clooney et al., 2019 sequencing reads | Clooney et al., 2019 | NCBI Sequence Read Archive (SRA) - PRJNA552463 |
| Draper et al., 2018 sequencing reads | Draper et al., 2018 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Fernandes et al., 2019 sequencing reads | Fernandes et al., 2019 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Giloteaux et al., 2016 sequencing reads | Giloteaux et al., 2016 | MG-RAST - see Table S1 for details |
| Han et al., 2018 sequencing reads | Han et al., 2018 (originally from Li et al., 2017) | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Kang et al., 2017 sequencing reads | Kang et al., 2017 | iVirus - see Table S1 for details |
| Kramná et al., 2015 sequencing reads | Kramná et al., 2015 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Lim et al., 2015 sequencing reads | Lim et al., 2015 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Ly et al., 2016 sequencing reads | Ly et al., 2016 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Ma et al., 2019 sequencing reads | Ma et al., 2018 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Manrique et al., 2016 sequencing reads | Manrique et al., 2016 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| McCann et al., 2018 sequencing reads | McCann et al., 2018 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Minot et al., 2011 sequencing reads | Minot et al., 2011 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Minot et al., 2012 sequencing reads | Minot et al., 2012 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Minot et al., 2013 sequencing reads | Minot et al., 2013 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Monaco et al., 2016 sequencing reads | Monaco et al., 2016 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Moreno-Gallego et al., 2019 sequencing reads | Moreno-Gallego et al., 2019 | European Nucleotide Archive (ENA) - see Table S1 for details |
| Neto et al. (unpublished) sequencing reads | Unpublished data | iVirus -- we were given some of the reads before publication |
| Norman et al., 2015 sequencing reads | Norman et al., 2015 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Pérez-Brocal et al., 2013 sequencing reads | Pérez-Brocal et al., 2013 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Rampelli et al., 2017 sequencing reads | Rampelli et al., 2017 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Reyes et al., 2010 sequencing reads | Reyes et al., 2010 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Reyes et al., 2015 sequencing reads | Reyes et al., 2015 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Shkoporov et al., 2018 sequencing reads | Shkoporov et al., 2018 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Shkoporov et al., 2019 sequencing reads | Shkoporov et al., 2019 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Stockdale et al., 2018 sequencing reads | Stockdale et al., 2018 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Yinda et al., 2019 sequencing reads | Yinda et al., 2019 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Zhao et al., 2017 sequencing reads | Zhao et al., 2017 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Zuo et al., 2018 sequencing reads | Zuo et al., 2018 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Zuo et al., 2019 sequencing reads | Zuo et al., 2019 | NCBI Sequence Read Archive (SRA) - see Table S1 for details |
| Software and Algorithms | ||
| nucmer (MUMmer3.23) | Kurtz et al., 2004 | https://sourceforge.net/projects/mummer/ |
| bbmap 37.57 | https://jgi.doe.gov/data-and-tools/bbtools/ | https://jgi.doe.gov/data-and-tools/bbtools/ |
| metaSPAdes 3.11 | Nurk et al., 2017 | https://github.com/ablab/spades/releases |
| prodigal 2.6.1 | Hyatt et al., 2010 | https://github.com/hyattpd/Prodigal |
| diamond | Buchfink et al., 2014 | https://github.com/bbuchfink/diamond |
| VirSorter v2 | Roux et al., 2015 | https://github.com/simroux/VirSorter |
| VirFinder | Ren et al., 2017 | https://github.com/jessieren/VirFinder |
| CAT | Cambuy et al., 2016 | https://github.com/dutilh/CAT |
| BUSCO | Simão et al., 2015 | https://busco.ezlab.org/ |
| Viral protein families (VPFs) | Paez-Espino et al., 2017 | http://portal.nersc.gov/dna/microbial/prokpubs/EarthVirome_DP/final_list.hmms |
| hmmmr | Finn et al., 2015 | http://www.hmmer.org/ |
| blast 2.4.0+ | ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/ | ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/ |
| IMG/VR v4 | Paez-Espino et al., 2017 | https://img.jgi.doe.gov/cgi-bin/vr/main.cgi |
| Viral Refseq v96 | https://ftp.ncbi.nlm.nih.gov/refseq/release/viral/ | https://ftp.ncbi.nlm.nih.gov/refseq/release/viral/ |
| vConTACT2 | Bin Jang et al., 2019 | https://bitbucket.org/MAVERICLab/vcontact2 |
| minced | Bland et al., 2007 | https://github.com/ctSkennerton/minced |
| tRNA-scan | Lowe and Eddy, 1997 | http://lowelab.ucsc.edu/tRNAscan-SE/ |
| MArVD | Vik et al., 2017 | https://bitbucket.org/MAVERICLab/marvd |
| WIsH | Galiez et al., 2017 | https://github.com/soedinglab/WIsH |
| MCL | Enright et al., 2002 | https://micans.org/mcl/ |
| bowtie2 | Langmead and Salzberg, 2012 | https://github.com/BenLangmead/bowtie2 |
| coverM | https://github.com/wwood/CoverM | https://github.com/wwood/CoverM |
| bedtools | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| GTDB-Tk v1.1 | Parks et al., 2020 | https://github.com/Ecogenomics/GTDBTk |
| vegan (R package) | Dixon, 2003 | https://cran.r-project.org/web/packages/vegan/index.html |
| maps (R package) | https://cran.r-project.org/web/packages/maps/index.html | https://cran.r-project.org/web/packages/maps/index.html |
| pheatmap (R package) | https://cran.r-project.org/web/packages/pheatmap/index.html | https://cran.r-project.org/web/packages/pheatmap/index.html |
| SpiecEasi (R package) | https://www.rdocumentation.org/packages/SpiecEasi/versions/0.1.4 | https://www.rdocumentation.org/packages/SpiecEasi/versions/0.1.4 |
| igragh (R package) | https://cran.r-project.org/web/packages/igraph/ | https://cran.r-project.org/web/packages/igraph/ |
| ggplot2 (R package) | https://cran.r-project.org/web/packages/ggplot2/index.html | https://cran.r-project.org/web/packages/ggplot2/index.html |
| ggpubr (R package) | https://cran.r-project.org/web/packages/ggpubr/index.html | https://cran.r-project.org/web/packages/ggpubr/index.html |
| gtools (R package) | https://cran.r-project.org/web/packages/gtools/index.html | https://cran.r-project.org/web/packages/gtools/index.html |
| biomod2 (R package) | https://cran.r-project.org/web/packages/biomod2/index.html | https://cran.r-project.org/web/packages/biomod2/index.html |
| BiodiveristyR (R package) | https://cran.r-project.org/web/packages/BiodiversityR/index.html | https://cran.r-project.org/web/packages/BiodiversityR/index.html |
| Analyses scripts and input data (per Figure) | This paper | https://bitbucket.org/MAVERICLab/GVD |
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the corresponding contact, Matthew Sullivan (sullivan.948@osu.edu).
Materials Availability
Gut virome database (GVD) studies were selected by doing a thorough and manually curated search of the Web of Science Core Collection of Thomson Reuters for studies looking at gut viruses published until October 2019. All studies that used next-generation sequencing and looked for viruses within the gut microbiome were selected to be part of GVD (see full list of studies in Table S1). Additionally, we were given access to the reads of one study that is unpublished (unpublished data) and are available upon request. Lastly, we used the reads from another gut virome study not included in GVD ((Clooney et al., 2019); SRA: PRJNA552463).
Data and Code Availability
Scripts used in this manuscript are available on the Sullivan laboratory bitbucket under ‘gvd’ (https://bitbucket.org/MAVERICLab/gvd/). All raw reads are available through SRA, iVirus, or MG-RAST using the identifiers listed in Table S1. GVD viral populations and all IV databases per study can be downloaded directly from iVirus through the following DOI link: https://doi.org/10.25739/12sq-k039.
Method Details
Viral Contig Assembly and Identification
Previously published GVD reads and their associated metadata were downloaded from their respective hosting databases (e.g. SRA, iVirus, or MG-RAST). The reads for one study were given to us prior to publication (unpublished data). Each individual downloaded library was counted as a metagenome and processed independently, with the exception of four studies that were previously processed in the lab and were pooled per individual for the assembly process (Chehoud et al 2016, Lim et al., 2015, Minot et al., 2013, Zhao et al., 2017) based on the knowledge that the gut virome is fairly consistent throughout time (Minot et al., 2013). Reads were cleaned by filtering for quality, trimming, and removing adaptors and ϕx174 reads using bbduk (ktrim=r k=23 mink=11 hdist=1) and removing the reads that mapped to the human genome using bbmap (minid=0.95 maxindel=3 bwr=0.16 bw=12 quickmatch fast minhits=2) from the bbmap suite (Bushnell, 2015). All references to the number of base pairs sequenced is based on the cleaned, quality-controlled reads. A global map showing the number of studies originating from each country was created using the R packages ‘rworldmap.’ In total, there were 2,697 metagenomes from 1,986 individuals across 32 studies.
Reads were then assembled using metaSPAdes 3.11.1 (Nurk et al., 2017), due to its performance in sensitivity analyses (Roux et al., 2017; Sutton et al., 2019) and assembly of large-scale virome datasets (Guerin et al., 2018; Paez-Espino et al., 2016). Following assembly, contigs ≥1.5kb were piped through VirSorter (Roux et al., 2015) and VirFinder (Ren et al., 2017) and those that mapped to the human, cat or dog genomes were removed, as well as known spike-in contaminants (lactococcal phage Q33 and coliphage Qβ) from the Shkoporov et al. 2018 and Shkoporov et al. 2019 studies. Contigs ≥5kb or ≥1.5kb and circular that were sorted as VirSorter categories 1-6 and/or VirFinder score ≥0.7 and p <0.05 were pulled for further investigation. Of these contigs, those sorted as VirSorter categories 1 and 2, VirFinder score ≥0.9 and p <0.05 or were identified as viral by both VirSorter (categories 1-6) and VirFinder (score ≥0.7 and p <0.05) were classified as viral. The remaining contigs were run through CAT (Cambuy et al., 2016) and those with <40% (based on an average gene size of 1000) of the contig classified as bacterial, archaeal, or eukaryotic were considered viral contigs. Contigs ≥5kb or ≥1.5kb and circular that were classified as eukaryotic viral contigs by CAT were also considered viral contigs. In total, 57,605 putative viral contigs were identified.
Assessing Potential False Positives and Dereplication
All putative viral contigs were then assessed to determine whether they could be a bacterial false positive by the level of bacterial and viral gene enrichment in each contig. Assessing whether a putative viral contig could be bacterial is extremely hard given that viruses often pick up their host genes, even ribosomal proteins (Mizuno et al., 2019). Further, bacterial genomes also pick up phage genomes and genes (e.g., intact and degraded prophages). Thus, a BLAST search of a viral contig against a database of all bacterial proteins would undeniably result in hits. To limit this problem, we chose to assess the level of bacterial gene enrichment using the number of hits to bacterial universal single-copy orthologs (i.e., BUSCO; (Waterhouse et al., 2017)) because these genes are highly conserved and the more of these genes present, the more likely it is bacterial. We used hmmsearch (Finn et al., 2011) to search the 148 BUSCO gene HMMs and then used the BUSCO provided HMM score cut-offs to filter our results for “hits.” A hit was defined by an e < 0.05 and a score ≥ scores cut-offs identified by BUSCO. Because some of these genes could still be present in viruses (Mizuno et al., 2019), we wanted a way to establish a level of BUSCO genes that was “acceptable” for a viral genome. In order to establish this acceptable baseline, we assessed the number of BUSCO genes present within prokaryotic viral genomes in Viral RefSeq v96, which are genomes that are derived from viral isolates. Because most of the putative viral genomes within GVD are not full genomes, we wanted to know the rate of BUSCO hits per total number of genes in each Viral RefSeq genome (BUSCO ratio). This established a range of BUSCO ratios values of 0-0.067 that were derived from known virus genomes, and so were considered ‘acceptable’. We then assessed the BUSCO ratios values for all GVD putative viral contigs and compared it to the Viral RefSeq BUSCO ratio values (see Table S2). To assess the level of viral gene enrichment, an hmmsearch of all GVD viral contigs against the curated viral protein family modules (VPFs) (Paez-Espino et al., 2017) was performed with hits being defined as any matches with an e-value < 0.05. The number of VPF hits are available in Table S2. To remove potential false positives and decontaminate the set of GVD viral contigs, only the GVD contigs that had a BUSCO ratio < 0.067 or had a BUSCO ratio > 0.067 and at least 3 VFP hits were kept in the remaining database.
The remaining GVD viral contigs that were from known ssDNA or RNA viral families using CAT were grouped into populations if they shared ≥95% nucleotide identity across 3100% of the genome. Because there are no benchmarked metagenomic population boundaries for ssDNA and RNA viral families, we chose to not use stringent dereplication. All other contigs were considered double-stranded DNA and were grouped into populations if they shared ≥95% nucleotide identity across ≥70% of the genome (sensu (Brum et al., 2015b)) using nucmer (Kurtz et al., 2004). All the viral contigs that were assembled were dereplicated per study to create the individual virome (IV) databases and across all of GVD (see Figure S2A and Table S6). For GVD, this resulted in 33,242 total viral populations found in GVD (see Table S2 for VirSorter, VirFinder, and CAT results), of which 15,330 were ≥10 kb in length.
Viral Taxonomy
For each viral population, ORFs were called using Prodigal (Hyatt et al., 2010) and the resulting protein sequences were used as input for vConTACT2 (Bin Jang et al., 2019) and for BLASTp. Double-stranded DNA viral populations represented by contigs >10kb were clustered with Viral RefSeq release 88 viral genomes using vConTACT2. Those that clustered with a virus from RefSeq based on amino acid homology based on DIAMOND (Buchfink et al., 2014) alignments were able to be assigned to a known viral taxonomic genera. The gene-sharing network was processed using igraph's python package. After the initial import, networks were cleaned to remove duplicate edges, and all VCs with fewer than 5 members were discarded. Afterwards, the network layout was calculated using the Fruchterman-Reingold algorithm, with RefSeq phage references genomes (red nodes in the network) having a fixed position based on their positions originally published in the initial vConTACT2 paper (Bin Jang et al., 2019). For viral dsDNA populations that could not be assigned taxonomy or were <10kb, family level taxonomy was assigned using a majority-rules approach, where if >50% of a genome’s proteins were assigned to the same viral family using a BLASTp bitscore ≥50 with a Viral RefSeq virus, it was considered part of that viral family (see Table S2 for family-level taxonomy). For eukaryotic, ssDNA and RNA viruses, CAT was used to assign the viral family (see Table S2 for family-level taxonomy).
Identifying Temperate Phages across Datasets
VIBRANT (Kieft et al., 2019) was run using its default settings on the GVD, Global Oceans Viromes 2 (Gregory et al., 2019a) and IsoGenie (Emerson et al., 2018; Trubl et al., 2018) viral populations. The viruses identified as lysogenic were pulled as the detected temperate phages across the different datasets.
Virus-Host Predictions
Microbial hosts for the GVD viral populations were predicted using a variety of bioinformatic methods that include viral exact matches (or close similarity) to (i) host CRISPR-spacers, (ii) integrated prophages in host genomes, (iii) host tRNA genes, and (iv) host k-mer signatures calculated by WisH (Galiez et al., 2017). Two host databases were used to establish these virus-host linkages: (i) 239,583 assembled prokaryotic genomes from Refseq (downloaded March 2020) which were employed for the first three bioinformatic approaches above, and (ii) 4644 species-level prokaryotic genomes from the Unified Human Gastrointestinal Genome (UHGG) catalogue (Almeida et al., 2020) which were employed for all of the four bioinformatic approaches. All genomes across the two databases were taxonomically annotated using the Genome Taxonomy Database (GTDB) taxonomy system (Parks et al., 2020) either by the curators of the two databases (UHGG and GTDB) or by us (using GTDB-Tk v1.1 in the “classify_wf” mode). CRISPR spacers were predicted from the host genomes with MinCED (Bland et al., 2007) using the “-minNR 2” parameter (https://github.com/ctSkennerton/minced) and a BLASTn was used to assess matches between the CRISPR spacers and viral populations in GVD. The number of exact spacer matches to the viral genome were recorded for each viral population-host pair along with the cases where there is a single base difference at the spacer end when aligned against the viral genome. We then assigned scores for all the virus-host pairs so that multiple spacer matches would score higher (perfect score) than a single spacer exact match (high score) than a single spacer with a base difference at its end (intermediate score). For prophage blasts, a BLASTn (-task megablast) of the viral population against the two databases was performed. A microbial genome with ≥2500bp regions of their genome matching at 90% ID with a viral population genome were kept for further consideration (see Roux et al., 2016). These matches were then further filtered by both viral contig coverage (requiring at least 30% viral coverage) and host contig coverage (requiring at least 30% of the host contig to be outside the prophage region alignment to avoid mis-binned viral fragments in host metagenome assembled genomes). Finally, the remaining matches were scored based on viral contig coverage so that 90% coverage would score higher (perfect score) than 75% (high score) than 50% (intermediate score) than 30% (low score). Viral and host tRNA genes were predicted using tRNA-scan (Lowe and Eddy, 1997) (using the general and bacterial/archeal models, respectively) and then a BLASTn was performed between the viral and bacterial tRNA genes. Viral tRNA genes were also searched with BLASTn against the tRNA sequences from the Earth virome dataset (Paez-Espino et al., 2016) and all the promiscuous tRNAs were removed from further analyses. The tRNA matches between the viruses and the hosts in our dataset were then scored so that an exact match would score higher (high score) than a host tRNA with a single base difference (intermediate score) than a host tRNA with two bases difference (low score). Lastly, WIsH was used to predict hosts after masking tRNA sequences on the viral genomes to improve performance (Galiez et al., 2017). Viral Refseq was used as a decoy database after conservatively excluding viruses that are known to infect the genus of a host under prediction at any given instance. For each viral population, the predicted host with the lowest p was kept for further investigation. We then assigned these linkages scores so that the lower the p, the higher the score, with a p of zero given a (high score) and a p of 1e-05 given an (intermediate score). In order to conservatively show family-level host assignments here, we chose to only include predictions with perfect and high scores. Note that perfect scores were only given to CRISPR and prophage matches to allow them priority host assignment over WIsH and tRNA results. Viruses with putative archaeal hosts were also predicted using MarVD (Vik et al., 2017). Viruses with predicted eukaryotic hosts were assigned based on their assigned taxonomic viral family.
Detecting Viral Populations and Calculating Their Raw Abundances per Each Assembled Metagenome or Assembled Pooled Read Set
To calculate the raw abundances of the different viral populations in each sample, reads from each GVD metagenome or pooled read sets for the four previously processed studies (Chehoud et al 2016, Lim et al., 2015, Minot et al., 2013, Zhao et al., 2017) were non-deterministically mapped to the GVD viral populations using bowtie2 (Langmead and Salzberg, 2012). CoverM (https://github.com/wwood/CoverM) was used to remove reads that mapped at <95% nucleotide identity to the contigs, bedtools genomecov (Quinlan and Hall, 2010) was used to determine how many positions across each genome were covered by reads, and custom Perl scripts were used to further filter out contigs without enough coverage across the length of the contig. All contigs <5kb in length with ≥70% of the contig covered were considered detected in the sample. Contigs ≥5 kb in length with ≥5 kb in length covered were also considered detected in the sample (Gregory et al., 2019a). CoverM was used to calculate the average read depth (‘tpmean’- i.e. mean minus the top and bottom 5% depths) across each detected contig. The average read depth was considered the raw abundance of each viral population in each study.
Comparisons to IMG/VR, Viral RefSeq v96, and Individual Virome Databases
The latest IMG/VR release (v4, July 2018) was downloaded, and included all viral contigs, not dereplicated into populations or vOTUs. All of the viral contigs in GVD, Viral Refseq v96, and individual virome databases are dereplicated at the population level. In order to make IMG/VR comparable to GVD, Viral Refseq and individual virome databases, we needed to dereplicate the IMG/VR database. IMG/VR v4 is composed of 760,453 contigs. Because the database is so large, we first used BLASTn to compare homology between all IMG/VR contigs using a word size = 100. The BLASTn results were then used to cluster the genomes using MCL (Enright et al., 2002) and the clustering similarity graphs encoded in BLAST methodology (https://micans.org/mcl/). The clustered genomes based on MCL clustering were then dereplicated if they shared ≥95% nucleotide identity across ≥70% of the genome (sensu (Brum et al., 2015b)) using nucmer. In total, all of the IMG/VR viral contigs were dereplicated into 359,826 viral populations. GVD metagenomes were then mapped to this IMG/VR human gut viral population database, Viral RefSeq v96, and their respective IV databases for each individual study in GVD. The raw abundances of the different IMG/VR, Viral RefSeq, and IV viral populations in each sample were calculated the same way as described in the previous section. The total number of viral populations detected per sample per study was calculated using the ‘vegan’ package (Dixon, 2003) in R. These values were then plotted and comparative statistics were generated using the ‘ggboxplot’ function from the ‘ggpubr’ package in R. Importantly, ggboxplot plots the median and quartiles and calculates Mann-Whitney U tests between groupings. Fold-change differences were calculated using the ‘gtools’ package in R.
The number of unique reads mapped from each GVD sample to GVD, IMG/VR v4,Viral Refseq v96, and IV databases was calculated by counting the number of reads mapped following removal of reads mapped at <95% nucleotide identity. The total number of reads mapped per sample using the different databases were then plotted and comparative statistics were generated using the ‘ggboxplot’ function from the ‘ggpubr’ package in R.
To test if GVD was also useful for gut virome studies not included in GVD, reads were also downloaded from a recent gut virome study not included in GVD (Clooney et al., 2019; SRA PRJNA552463), processed, and viral contigs identified and assessed for false-positives using the same method described above. In total, we identified 1,299 viral populations. The number of viral populations detected using GVD, IMG/VR, Viral RefSeq, and the Clooney et al. 2019 individual virome database using the same methods described above.
Detecting Viral Populations and Calculating Their Raw Abundances by Metagenome
To calculate the raw abundances of the different viral populations in each sample, reads from each GVD metagenome included the unpooled read sets (Chehoud et al 2016, Lim et al., 2015, Minot et al., 2013, Zhao et al., 2017) were non-deterministically mapped to the GVD viral populations using bowtie2 and processed as detailed above in Detecting viral populations and calculating their raw abundances per each assembled metagenome or assembled pooled read set. The raw abundances for each GVD viral population in each metagenome are available in Table S4.
Assessing VLP-Enriched and Bulk Metagenomes
Metagenomes were divided into VLP-enriched (VLP) and bulk metagenomes (information per metagenome can be found in Table S5). To assess whether there was a difference between viral recovery in VLP versus bulk metagenomes, the number of assembled contigs per study was divided by the total number of clean base pairs sequenced in the study. For the Shkoporov et al., 2019 study, the viral contigs assembled from the VLP and bulk were kept separate and divided by the respective number of base pairs sequenced. The VLP and bulk studies were then plotted in boxplots and comparative statistics were performed using the ‘ggboxplot’ function from the ‘ggpubr’ package in R. Of the VLP studies, the number of viral contigs assembled per base pair sequenced per study with and without MDA-treatment and the different VLP-enrichment strategies were also plotted in boxplots and comparative statistics were performed using the ‘ggboxplot’ function from the ‘ggpubr’ package in R. The median contig length per study between VLP and bulk and across the different VLP-enrichment strategies were also plotted in boxplots and comparative statistics were performed using the ‘ggboxplot’ function from the ‘ggpubr’ package in R. To assess the impact of sequencing depth on viral contig assembly, a scatterplot of the number of assembled contigs per study were plotted against the total number of clean sequenced base pairs per study and linear regression run using the package ‘ggplot2’ in R. This was repeated in solely the VLP metagenome studies and solely the bulk metagenome studies.
To assess whether there was a difference between viral population detection in VLP versus bulk metagenomes, the total number of viral populations detected per base pair sequenced was calculated for each metagenome and plotted in boxplots and statistically compared by VLP or bulk metagenome status using the ‘ggboxplot’ function from the ‘ggpubr’ package in R. For the Shkoporov et al., 2019 study, one time point (T8) of the ten individuals in the study was processed and sequenced using both VLP-enrichment and bulk methods. There were two VLP metagenomes and one bulk metagenome per individual for that time point. The number of viral contigs assembled per base pair sequenced and the median contig lengths per individual were also plotted in boxplots and comparative statistics were performed using the ‘ggboxplot’ function from the ‘ggpubr’ package in R. The total number of viral populations detected per base pair sequenced was calculated and, for the VLP samples, the values were averaged. The averaged VLP value and the bulk metagenome were then plotted using ‘ggplot2.’
Clustering Studies Based on Shared Viral Populations
To test how studies clustered together, the viral population presence-absence data from individuals (or pooled read sets) within a study were merged. In Study 1, individual A had viral populations 1, 2, 4, 5 and individual B had viral populations 3, then Study 1 had viral populations 1, 2, 3, 4, and 5. The different studies were then assessed for the number of shared viral populations that were present in both studies. These values were then displayed and hierarchically clustered using the R ‘pheatmap’ package. The resulting hierarchical clusters were used as guides to divide the studies into four groups (I-IV). The number of shared viral populations in metagenomes within each study in each group were clustered using the R ‘SPIEC-EASI’ package (method='mb',lambda.min.ratio=1e-2,nlambda=20,icov.select.params=list(rep.num=50; Kurtz et al., 2015) to infer associations between samples based on the shared number of viral populations. Each network for each group was plotted using the R ‘igraph’ package.
Identifying crAssphage Populations
CrAssphage viral populations in GVD were identified by using BLASTn against the crAssphage genomes identified in (Guerin et al., 2018). Those with >80% ID across ≥50% the length of the GVD viral genome were classified as crAssphage. In total, there were 70 unique crAssphage populations.
Core Viral Population Analyses
To explore if there were any core viral populations, the abundance table was turned into a binary presence-absence matrix using the ‘biomod2’ package in R. The number of GVD samples that each viral population was detected within was then calculated using R and divided by the total number (2,697) of metagenomes to get the percentage of metagenomic samples. Each viral population’s percentage was plotted in hive plot using ‘geom_curve’ in ggplot2 (Ginestet, 2011). CrAssphage populations were replotted on top of the all viral populations to differentiate them. The number of viral populations that were present across different percentages were calculated using R and their distributions plotted using ‘geom_histogram’ in ggplot2.
Assessing the Impact of Age on Viral Diversity in the Gut Virome
Because no single study in GVD has samples that spanned all of the human life stages (infancy, childhood, adulthood, and senescence), we needed to combine samples from multiple different studies. Due to unequal sequencing and MDA, which skews population abundances, across GVD, we chose to use the number of viral populations per clean base pair sequenced as a proxy for viral richness. If multiple metagenomes were collected for a single individual the number of viral populations per base pair sequenced per metagenome was averaged. We the ran a Kruskal-Wallis test in R between viral populations per base pair sequenced and study which shows revealed that study origin was driving significant differences in viral richness values. Next, to visualize potential study outliers that could be driving this correlation, the number of viral populations per base pair sequenced per study was plotted using the ‘ggboxplot’ function from the ‘ggpubr’ package in R. The 75% and 25% quantiles number of viral populations per base pair sequenced across all metagenomes were calculated using base R and plotted over the boxplots. Studies with medians that fell outside of 25-75% quantile range were considered outliers. To assess what was driving the outlier status of these studies, we tested all the parameters that we had information across all the studies including sequencing platform, enrichment type (bulk or VLP), and MDA-treatment. First, we sequentially and additively tested the impact of sequencing platform, enrichment type (bulk or VLP), and MDA using comparative statistics from the ‘ggpubr’ package in R and plotted the results using ‘ggboxplot’ function in the same package in R. The 454, bulk, and non-MDA studies were removed as outliers.
Next, we assessed what could be driving potential confounders among individuals in these remaining studies by testing the impact of geographic origin (Western or non-Western) and health status (healthy or diseased) on the number of viral populations per base pair sequenced. Again, comparative statistics were calculated using the ‘ggpubr’ package in R and plotted using the ‘ggboxplot’ function in the same package in R. Because the non-Western, diseased individuals were significantly different, they were removed as potential confounders for when looking at the impact of age. The remaining individuals represented 151 healthy Western individuals across 11 different studies. The number of viral populations per base pair sequenced for these remaining individuals were plotted by study using the ‘ggboxplot’ function from the ‘ggpubr’ package in R. The 75% and 25% quantiles number of viral populations per base pair sequenced across all metagenomes were calculated using base R and plotted over the boxplots. All the studies now had medians that fell within the 25-75% quantile range and were kept for further analyses. A final Kruskal-Wallis test in R revealed that study origin was no longer significantly driving differences in viral richness values.
The remaining individuals were partitioned into life stages based on age: infancy (0-3 years old), childhood (3-18 years old), adulthood (18-65 years old), and senescence (65+ years old). In total, there were 28 infants, 12 children, 95 adults, and 20 elderly individuals. We next removed outlier individuals per life stage by removing the individuals that had number of viral populations per base pair sequenced that was greater than 1.5 times the interquartile range. After removal of these life stage outliers, there were 27 infants, 11 children, 93 adults, and 20 elderly individuals. We also were curious about how different viral types (bacteriophage and eukaryotic viruses) and different viral families including crAssphage varied across the life stages. Using the taxonomy as a guide, we pulled out the total number of each of the aforementioned categories per individual and divided by the total base pair sequenced. We plotted the data two ways. The first way was using boxplots to statistically assess differences between the life stages using the ‘ggboxplot’ function from the ‘ggpubr’ package in R. The second way was using Loess smoothing. To perform the Loess smoothing, each life stage was counted as a unit of 1, so infancy was 1, childhood 2, adulthood 3, and senescence 4. The number of viral populations per base pair sequenced per life stage was then plotted using Loess smoothing (span = 1) in the ‘ggplot2’ package in R. The Loess curves were then put on the same axis from their maximum to the minimum value in order to better visualize each curve and compare trends in Figure 7A. The binary presence-absence data for the crAssphage populations across all 151 healthy Western individuals were plotted using pheatmap in R. Lastly, because we also had a single study that had non-Western, Chinese adults and elderly individuals (Ma et al., 2018), we also statistically evaluated if they were different and plotted the number of viral populations per base pair sequenced using the ‘ggboxplot’ function from the ‘ggpubr’ package in R.
Removing Potential Contaminants and Validating the Impact of Age on Viral Diversity in the Gut Virome
Given that only 1 of the 32 studies within GVD sequenced and publicly provided the data for blank, negative controls, removing potential contaminants was difficult. Contaminants by definition should be in low abundance in a study and most likely are found only in on study. Thus, to identify potential contaminant viral populations, we took a very liberal approach (i.e. we identified and removed all populations that had the potential to be contamination). We first normalized the raw abundances per study using the number of base pairs sequenced. Thus, samples were scaled to the sample with the most base pairs sequenced. We then ran rank abundance curves on all the detected GVD viral populations per study using the BiodiveristyR package in R. Populations in the rare-tail of the rank abundance curves (proportion < 0.1) were putatively considered contaminants. These initial putative contaminants per study were checked to see if they were detected in any other study. If they were detected, they were removed from the contaminant list. All other rare-tail viral populations were considered contamination. In total, there were 19,551 putative contaminant viral populations using this liberal approach. These viral populations were removed from further analyses and the same analyses described in the methods section “Assessing the Impact of Age on Viral Diversity in the Gut Virome” was repeated with the putative contaminants removed.
Quantification and Statistical Analyses
Please refer to figure legends and Method Details for full details on statistical analysis.
Acknowledgements
Computational support was provided by an award from the Ohio Supercomputer Center (OSC) to M.B.S. Study design and manuscript comments from Shini Sunagawa, Miguelangel Cuenca Vera, Bas E. Dutilh, Ksenia Arkhipova, Pedro Meirelles, Simon Roux, and Christine Sun are gratefully acknowledged. For help digging into the literature of bacterial diversity across the human life stages, we thank Jiyeon Si. We also thank Jelle Matthijnssens’ lab for granting us early access to the reads for the Neto et al. study. Funding was provided by the Gordon and Betty Moore Foundation (3790), an NIH grant (R01HG010318) (PI Sanggu Kim), and the Ohio State University Center of Microbiome Science to M.B.S.; and an NIH T32 training grant fellowship (AI112542) awarded to A.C.G.
Author Contributions
A.C.G. collected all datasets and performed the meta-analyses for the study. A.C.G. and A.H. curated metadata for the study. A.A.Z. conducted the in silico host analyses. A.C.G., O.Z., A.A.Z., B.B., and M.B.S created the study design, analyzed the data, and wrote the manuscript. All authors approved the final manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: August 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.chom.2020.08.003.
Supplemental Information
References
- Adriaenssens E.M., Sullivan M.B., Knezevic P., Van Zyl L.J. 2020. Taxonomy of prokaryotic viruses: 2018 – 2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. [DOI] [PubMed] [Google Scholar]
- Aiemjoy K., Altan E., Aragie S., Fry D.M., Phan T.G., Deng X., Chanyalew M., Tadesse Z., Callahan E.K., Delwart E., Keenan J.D. Viral species richness and composition in young children with loose or watery stool in Ethiopia. BMC Infect. Dis. 2019;19:53. doi: 10.1186/s12879-019-3674-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleman F.D.D., Valenzano D.R. Microbiome evolution during host aging. PLoS Pathog. 2019;15:e1007727. doi: 10.1371/journal.ppat.1007727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A., Nayfach S., Boland M., Strozzi F., Beracochea M., Shi Z.J., Pollard K.S., Sakharova E., Parks D.H., Hugenholtz P. A unified sequence catalogue of over 200,000 genomes obtained from the human gut microbiome. Nat. Biotechnol. 2020 doi: 10.1038/s41587-020-0603-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amgarten D., Braga L.P.P., da Silva A.M., Setubal J.C. MARVEL, a tool for prediction of bacteriophage sequences in metagenomic bins. Front. Genet. 2018;9:304. doi: 10.3389/fgene.2018.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barwell L.J., Isaac N.J.B., Kunin W.E. Measuring β-diversity with species abundance data. J. Anim. Ecol. 2015;84:1112–1122. doi: 10.1111/1365-2656.12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland C., Ramsey T.L., Sabree F., Lowe M., Brown K., Kyrpides N.C., Hugenholtz P. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007;8:209. doi: 10.1186/1471-2105-8-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobay L.-M., Ochman H. Biological species in the viral world. Proc Natl Acad Sci U S A. 2018 doi: 10.1073/pnas.1717593115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc B., Bin Jang H., Doulcier G., You Z.-Q., Roux S., Sullivan M.B. vConTACT: an iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ. 2017;5:e3243. doi: 10.7717/peerj.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc B., Youens-Clark K., Roux S., Hurwitz B.L., Sullivan M.B. iVirus: facilitating new insights in viral ecology with software and community data sets imbedded in a cyberinfrastructure. ISME J. 2017;11:7–14. doi: 10.1038/ismej.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broecker F., Klumpp J., Moelling K. Long-term microbiota and virome in a Zürich patient after fecal transplantation against Clostridium difficile infection. Ann. N Y Acad. Sci. 2016;1372:29–41. doi: 10.1111/nyas.13100. [DOI] [PubMed] [Google Scholar]
- Broecker F., Russo G., Klumpp J., Moelling K. Stable core virome despite variable microbiome after fecal transfer. Gut Microbes. 2017;8:214–220. doi: 10.1080/19490976.2016.1265196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum J.R., Ignacio-espinoza J.C., Roux S., Doulcier G., Acinas S.G., Alberti A., Chaffron S. Ocean Viral Communities. Science. 2015;348:1261498. doi: 10.1126/science.1261498. [DOI] [PubMed] [Google Scholar]
- Brum J.R., Ignacio-Espinoza J.C., Roux S., Doulcier G., Acinas S.G., Alberti A., Chaffron S., Cruaud C., de Vargas C., Gasol J.M., Tara Oceans Coordinators Ocean plankton. Patterns and ecological drivers of ocean viral communities. Science. 2015;348:1261498. doi: 10.1126/science.1261498. [DOI] [PubMed] [Google Scholar]
- Buchfink B., Xie C., Huson D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2014;1:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- Bushnell B. Lawrence Berkeley National Lab; 2015. BBMap, 9th Annual Genomics of Energy & Environment Meeting.https://www.osti.gov/biblio/1241166-bbmap-fast-accurate-splice-aware-aligner [Google Scholar]
- Cambuy D.D., Coutinho F.H., Dutilh B.E. Contig annotation tool CAT robustly classifies assembled metagenomic contigs and long sequences. bioRxiv. 2016 doi: 10.1101/072868. [DOI] [Google Scholar]
- Chao A., Chazdon R.L., Colwell R.K., Shen T.J. Abundance-based similarity indices and their estimation when there are unseen species in samples. Biometrics. 2006;62:361–371. doi: 10.1111/j.1541-0420.2005.00489.x. [DOI] [PubMed] [Google Scholar]
- Chehoud C., Dryga A., Hwang Y., Nagy-Szakal D., Hollister E.B., Luna R.A., Versalovic J., Kellermayer R., Bushman F.D. Transfer of Viral Communities between Human Individuals during Fecal Microbiota Transplantation. MBio. 2016;7:e00322. doi: 10.1128/mBio.00322-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W., Xu R., Wu Y., Chen J., Zhang Y., Hu T., Yuan X., Zhou L., Tan T., Fan J. Plant diversity is coupled with beta not alpha diversity of soil fungal communities following N enrichment in a semi-arid grassland. Soil Biol. Biochem. 2018 doi: 10.1016/j.soilbio.2017.10.039. [DOI] [Google Scholar]
- Claesson M.J., Jeffery I.B., Conde S., Power S.E., O’Connor E.M., Cusack S., Harris H.M.B., Coakley M., Lakshminarayanan B., O’Sullivan O. Gut microbiota composition correlates with diet and health in the elderly. Nature. 2012;488:178–184. doi: 10.1038/nature11319. [DOI] [PubMed] [Google Scholar]
- Clemente J.C., Ursell L.K., Parfrey L.W., Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clooney A.G., Sutton T.D.S., Shkoporov A.N., Holohan R.K., Daly K.M., O’Regan O., Ryan F.J., Draper L.A., Plevy S.E., Ross R.P., Hill C. Whole-Virome Analysis Sheds Light on Viral Dark Matter in Inflammatory Bowel Disease. Cell Host Microbe. 2019;26:764–778.e5. doi: 10.1016/j.chom.2019.10.009. [DOI] [PubMed] [Google Scholar]
- Colwell R.K., Coddington J.A. Estimating terrestrial biodiversity through extrapolation. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1994 doi: 10.1098/rstb.1994.0091. [DOI] [PubMed] [Google Scholar]
- Dixon P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003 doi: 10.1111/j.1654-1103.2003.tb02228.x. [DOI] [Google Scholar]
- Draper L.A., Ryan F.J., Smith M.K., Jalanka J., Mattila E., Arkkila P.A., Ross R.P., Satokari R., Hill C. Long-term colonisation with donor bacteriophages following successful faecal microbial transplantation. Microbiome. 2018;6:220. doi: 10.1186/s40168-018-0598-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duhaime M.B., Sullivan M.B. Ocean viruses: rigorously evaluating the metagenomic sample-to-sequence pipeline. Virology. 2012;434:181–186. doi: 10.1016/j.virol.2012.09.036. [DOI] [PubMed] [Google Scholar]
- Duhaime M.B., Solonenko N., Roux S., Verberkmoes N.C., Wichels A., Sullivan M.B. Comparative omics and trait analyses of marine Pseudoalteromonas phages advance the phage OTU concept. Front. Microbiol. 2017;8:1241. doi: 10.3389/fmicb.2017.01241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg P.B., Bik E.M., Bernstein C.N., Purdom E., Dethlefsen L., Sargent M., Gill S.R., Nelson K.E., Relman D.A. Diversity of the human intestinal microbial flora. Science. 2005;308:80. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson J.B., Roux S., Brum J.R., Bolduc B., Woodcroft B.J., Jang H.B., Singleton C.M., Solden L.M., Naas A.E., Boyd J.A. Host-linked soil viral ecology along a permafrost thaw gradient. Nat. Microbiol. 2018;3:870–880. doi: 10.1038/s41564-018-0190-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright A.J., Van Dongen S., Ouzounis C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes M.A., Verstraete S.G., Phan T.G., Deng X., Stekol E., LaMere B., Lynch S.V., Heyman M.B., Delwart E. Enteric Virome and Bacterial Microbiota in Children With Ulcerative Colitis and Crohn Disease. J. Pediatr. Gastroenterol. Nutr. 2019;68:30–36. doi: 10.1097/MPG.0000000000002140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D., Clements J., Arndt W., Miller B.L., Wheeler T.J., Schreiber F., Bateman A., Eddy S.R. HMMER web server: 2015 update. Nucleic Acids Research. 2015;43:W30–W38. doi: 10.1093/nar/gkv397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D., Clements J., Eddy S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29-37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank D.N., St Amand A.L., Feldman R.A., Boedeker E.C., Harpaz N., Pace N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiez C., Siebert M., Enault F., Vincent J., Söding J. WIsH: who is the host? Predicting prokaryotic hosts from metagenomic phage contigs. Bioinformatics. 2017;33:3113–3114. doi: 10.1093/bioinformatics/btx383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J.A., Blaser M.J., Caporaso J.G., Jansson J.K., Lynch S.V., Knight R. Current understanding of the human microbiome. Nat. Med. 2018;24:392–400. doi: 10.1038/nm.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giloteaux L., Hanson M.R., Keller B.A. A pair of identical twins discordant for myalgic encephalomyelitis/chronic fatigue syndrome differ in physiological parameters and gut microbiome composition. Am. J. Case Rep. 2016;17:720–729. doi: 10.12659/AJCR.900314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestet C. ggplot2: Elegant Graphics for Data Analysis. Royal Statistics Society. 2011 doi: 10.1111/j.1467-985X.2010.00676_9.x. [DOI] [Google Scholar]
- Gou W., Fu Y., Yue L., Chen G., Cai X., Shuai M., Xu F., Yi X., Chen H., Zhu Y.J. Gut microbiota may underlie the predisposition of healthy individuals to COVID-19. MedRxiv. 2020 doi: 10.1101/2020.04.22.20076091. [DOI] [Google Scholar]
- Grazziotin A.L., Koonin E.V., Kristensen D.M. Prokaryotic Virus Orthologous Groups (pVOGs): a resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017;45(D1):D491–D498. doi: 10.1093/nar/gkw975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory A., Zayed A., Conceição-Neto N., Temperton B., Bolduc B., Alberti A., Ardyna M., Arkhipova K., Carmicheal M., Cruaud C. Marine DNA Viral Macro-and Micro-Diversity From Pole to Pole. SSRN Electron. J. 2019:1–15. doi: 10.1016/j.cell.2019.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory A.C., Solonenko S.A., Ignacio-Espinoza J.C., LaButti K., Copeland A., Sudek S., Maitland A., Chittick L., Dos Santos F., Weitz J.S. Genomic differentiation among wild cyanophages despite widespread horizontal gene transfer. BMC Genomics. 2016;17:930. doi: 10.1186/s12864-016-3286-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory A.C., Zayed A.A., Conceição-Neto N., Temperton B., Bolduc B., Alberti A., Ardyna M., Arkhipova K., Carmichael M., Cruaud C., Tara Oceans Coordinators Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell. 2019;177:1109–1123.e14. doi: 10.1016/j.cell.2019.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greninger A.L. A decade of RNA virus metagenomics is (not) enough. Virus Res. 2018;244:218–229. doi: 10.1016/j.virusres.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerin E., Shkoporov A., Stockdale S.R., Clooney A.G., Ryan F.J., Sutton T.D.S., Draper L.A., Gonzalez-Tortuero E., Ross R.P., Hill C. Biology and Taxonomy of crAss-like Bacteriophages, the Most Abundant Virus in the Human Gut. Cell Host Microbe. 2018;24:653–664.e6. doi: 10.1016/j.chom.2018.10.002. [DOI] [PubMed] [Google Scholar]
- Han M., Yang P., Zhong C., Ning K. The Human Gut Virome in Hypertension. Front. Microbiol. 2018;9:3150. doi: 10.3389/fmicb.2018.03150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillebrand H., Blasius B., Borer E.T., Chase J.M., Downing J.A., Eriksson B.K., Filstrup C.T., Harpole W.S., Hodapp D., Larsen S. Biodiversity change is uncoupled from species richness trends: Consequences for conservation and monitoring. J. Appl. Ecol. 2018 doi: 10.1111/1365-2664.12959. [DOI] [Google Scholar]
- Howard-Varona C., Lindback M.M., Bastien G.E., Solonenko N., Zayed A.A., Jang H., Andreopoulos B., Brewer H.M., Glavina Del Rio T., Adkins J.N. Phage-specific metabolic reprogramming of virocells. ISME J. 2020;14:881–895. doi: 10.1038/s41396-019-0580-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz B.L., Deng L., Poulos B.T., Sullivan M.B. Evaluation of methods to concentrate and purify ocean virus communities through comparative, replicated metagenomics. Environ. Microbiol. 2013;15:1428–1440. doi: 10.1111/j.1462-2920.2012.02836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D., Chen G.-L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Bin H., Bolduc B., Zablocki O., Kuhn J.H., Roux S., Adriaenssens E.M., Brister J.R., Kropinski A.M., Krupovic M., Lavigne R. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019;37:632–639. doi: 10.1038/s41587-019-0100-8. [DOI] [PubMed] [Google Scholar]
- Jost L. Independence of alpha and beta diversities. Ecology. 2010;91:1969–1974. doi: 10.1890/09-0368.1. [DOI] [PubMed] [Google Scholar]
- Kang D.W., Adams J.B., Gregory A.C., Borody T., Chittick L., Fasano A., Khoruts A., Geis E., Maldonado J., McDonough-Means S. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Microbiome. 2017;5:10. doi: 10.1186/s40168-016-0225-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen E.C., Dantas G. Close Encounters of Three Kinds: Bacteriophages, Commensal Bacteria, and Host Immunity. Trends Microbiol. 2018;26:943–954. doi: 10.1016/j.tim.2018.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieft K., Zhou Z., Anantharaman K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of virome function from genomic sequences. bioRxiv. 2019 doi: 10.1101/855387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramná L., Kolářová K., Oikarinen S., Pursiheimo J.P., Ilonen J., Simell O., Knip M., Veijola R., Hyöty H., Cinek O. Gut virome sequencing in children with early islet autoimmunity. Diabetes Care. 2015;38:930–933. doi: 10.2337/dc14-2490. [DOI] [PubMed] [Google Scholar]
- Kurtz S., Phillippy A., Delcher A.L., Smoot M., Shumway M., Antonescu C., Salzberg S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004 doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz Z.D., Müller C.L., Miraldi E.R., Littman D.R., Blaser M.J., Bonneau R.A. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLoS Comput. Biol. 2015 doi: 10.1371/journal.pcbi.1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leplae R., Lima-Mendez G., Toussaint A. ACLAME: A CLAssification of mobile genetic elements, update 2010. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkp938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley R.E., Bäckhed F., Turnbaugh P., Lozupone C.A., Knight R.D., Gordon J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. U S A. 2005 doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Zhao F., Wang Y., Chen J., Tao J., Tian G., Wu S., Liu W., Cui Q., Geng B. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. 2017 doi: 10.1186/s40168-016-0222-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G., Zhao C., Zhang H., Mattei L., Sherrill-Mix S., Bittinger K., Kessler L.R., Wu G.D., Baldassano R.N., DeRusso P. The stepwise assembly of the neonatal virome is modulated by breastfeeding. Nature. 2020;581:470–474. doi: 10.1038/s41586-020-2192-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E.S., Zhou Y., Zhao G., Bauer I.K., Droit L., Ndao I.M., Warner B.B., Tarr P.I., Wang D., Holtz L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015;21:1228–1234. doi: 10.1038/nm.3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe T.M., Eddy S.R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly M., Jones M.B., Abeles S.R., Santiago-Rodriguez T.M., Gao J., Chan I.C., Ghose C., Pride D.T. Transmission of viruses via our microbiomes. Microbiome. 2016;4:64. doi: 10.1186/s40168-016-0212-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch S.V., Pedersen O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016;375:2369–2379. doi: 10.1056/NEJMra1600266. [DOI] [PubMed] [Google Scholar]
- Ma Y., You X., Mai G., Tokuyasu T., Liu C. A human gut phage catalog correlates the gut phageome with type 2 diabetes. Microbiome. 2018;6:24. doi: 10.1186/s40168-018-0410-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manrique P., Bolduc B., Walk S.T., van der Oost J., de Vos W.M., Young M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA. 2016;113:10400–10405. doi: 10.1073/pnas.1601060113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann A., Ryan F.J., Stockdale S.R., Dalmasso M., Blake T., Ryan C.A., Stanton C., Mills S., Ross P.R., Hill C. Viromes of one year old infants reveal the impact of birth mode on microbiome diversity. PeerJ. 2018;6:e4694. doi: 10.7717/peerj.4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot S., Sinha R., Chen J., Li H., Keilbaugh S.A., Wu G.D., Lewis J.D., Bushman F.D. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res. 2011;21:1616–1625. doi: 10.1101/gr.122705.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot S., Grunberg S., Wu G.D., Lewis J.D., Bushman F.D. Hypervariable loci in the human gut virome. Proc. Natl. Acad. Sci. U S A. 2012;109:3962–3966. doi: 10.1073/pnas.1119061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minot S., Bryson A., Chehoud C., Wu G.D., Lewis J.D., Bushman F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. U S A. 2013;110:12450–12455. doi: 10.1073/pnas.1300833110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzaei M.K., Maurice C.F. Ménage à trois in the human gut: interactions between host, bacteria and phages. Nat. Rev. Microbiol. 2017;15:397–408. doi: 10.1038/nrmicro.2017.30. [DOI] [PubMed] [Google Scholar]
- Mizuno C.M., Guyomar C., Roux S., Lavigne R., Rodriguez-Valera F., Sullivan M.B., Gillet R., Forterre P., Krupovic M. Numerous cultivated and uncultivated viruses encode ribosomal proteins. Nat. Commun. 2019;10:752. doi: 10.1038/s41467-019-08672-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco C.L., Kwon D.S. Next-generation Sequencing of the DNA Virome from Fecal Samples. Bio Protoc. 2017;7:e2159. doi: 10.21769/BioProtoc.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco C.L., Gootenberg D.B., Zhao G., Handley S.A., Ghebremichael M.S., Lim E.S., Lankowski A., Baldridge M.T., Wilen C.B., Flagg M. Altered Virome and Bacterial Microbiome in Human Immunodeficiency Virus-Associated Acquired Immunodeficiency Syndrome. Cell Host Microbe. 2016;19:311–322. doi: 10.1016/j.chom.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Gallego J.L., Chou S.P., Di Rienzi S.C., Goodrich J.K., Spector T.D., Bell J.T., Youngblut N.D., Hewson I., Reyes A., Ley R.E. Virome Diversity Correlates with Intestinal Microbiome Diversity in Adult Monozygotic Twins. Cell Host Microbe. 2019;25:261–272.e5. doi: 10.1016/j.chom.2019.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagpal R., Mainali R., Ahmadi S., Wang S., Singh R., Kavanagh K., Kitzman D.W., Kushugulova A., Marotta F., Yadav H. Nutr. Heal. Aging; 2018. Gut microbiome and aging: Physiological and mechanistic insights. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neil J.A., Cadwell K. The Intestinal Virome and Immunity. J. Immunol. 2018;201:1615–1624. doi: 10.4049/jimmunol.1800631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson J.K., Holmes E., Kinross J., Burcelin R., Gibson G., Jia W., Pettersson S. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- Norman J.M., Handley S.A., Baldridge M.T., Droit L., Liu C.Y., Keller B.C., Kambal A., Monaco C.L., Zhao G., Fleshner P. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160:447–460. doi: 10.1016/j.cell.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S., Meleshko D., Korobeynikov A., Pevzner P.A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obregon-Tito A.J., Tito R.Y., Metcalf J., Sankaranarayanan K., Clemente J.C., Ursell L.K., Zech Xu Z., Van Treuren W., Knight R., Gaffney P.M. Subsistence strategies in traditional societies distinguish gut microbiomes. Nat. Commun. 2015;6:6505. doi: 10.1038/ncomms7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odamaki T., Kato K., Sugahara H., Hashikura N., Takahashi S., Xiao J.Z., Abe F., Osawa R. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. 2016;16:90. doi: 10.1186/s12866-016-0708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvie L.A., Jones B.V. The human gut virome: a multifaceted majority. Front. Microbiol. 2015;6:918. doi: 10.3389/fmicb.2015.00918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott S.J., Musfeldt M., Wenderoth D.F., Hampe J., Brant O., Fölsch U.R., Timmis K.N., Schreiber S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–693. doi: 10.1136/gut.2003.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Espino D., Eloe-Fadrosh E.A., Pavlopoulos G.A., Thomas A.D., Huntemann M., Mikhailova N., Rubin E., Ivanova N.N., Kyrpides N.C. Uncovering Earth’s virome. Nature. 2016;536:425–430. doi: 10.1038/nature19094. [DOI] [PubMed] [Google Scholar]
- Paez-Espino D., Pavlopoulos G.A., Ivanova N.N., Kyrpides N.C. Nontargeted virus sequence discovery pipeline and virus clustering for metagenomic data. Nat. Protoc. 2017;12:1673–1682. doi: 10.1038/nprot.2017.063. [DOI] [PubMed] [Google Scholar]
- Paez-Espino D., Roux S., Chen I.-M.A., Palaniappan K., Ratner A., Chu K., Huntemann M., Reddy T.B.K., Pons J.C., Llabrés M. IMG/VR v.2.0: an integrated data management and analysis system for cultivated and environmental viral genomes. Nucleic Acids Res. 2018;47:gky1127. doi: 10.1093/nar/gky1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks D.H., Chuvochina M., Waite D.W., Rinke C., Skarshewski A., Chaumeil P.A., Hugenholtz P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018;36:996–1004. doi: 10.1038/nbt.4229. [DOI] [PubMed] [Google Scholar]
- Parks D.H., Chuvochina M., Chaumeil P.-A., Rinke C., Mussig A.J., Hugenholtz P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 2020 doi: 10.1038/s41587-020-0501-8. [DOI] [PubMed] [Google Scholar]
- Pasolli E., Asnicar F., Manara S., Zolfo M., Karcher N., Armanini F., Beghini F., Manghi P., Tett A., Ghensi P. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell. 2019;176:649–662.e20. doi: 10.1016/j.cell.2019.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Brocal V., García-López R., Vázquez-Castellanos J.F., Nos P., Beltrán B., Latorre A., Moya A. Study of the viral and microbial communities associated with Crohn’s disease: a metagenomic approach. Clin. Transl. Gastroenterol. 2013;4:e36. doi: 10.1038/ctg.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin J.B., Muller-Landau H.C. Sampling the species composition of a landscape. Ecology. 2002 [Google Scholar]
- Pop M. Genome assembly reborn: recent computational challenges. Brief. Bioinform. 2009;10:354–366. doi: 10.1093/bib/bbp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prober S.M., Leff J.W., Bates S.T., Borer E.T., Firn J., Harpole W.S., Lind E.M., Seabloom E.W., Adler P.B., Bakker J.D. Plant diversity predicts beta but not alpha diversity of soil microbes across grasslands worldwide. Ecol. Lett. 2015;18:85–95. doi: 10.1111/ele.12381. [DOI] [PubMed] [Google Scholar]
- Qin J., Li Y., Cai Z., Li S., Zhu J., Zhang F., Liang S., Zhang W., Guan Y., Shen D. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampelli S., Turroni S., Schnorr S.L., Soverini M., Quercia S., Barone M., Castagnetti A., Biagi E., Gallinella G., Brigidi P., Candela M. Characterization of the human DNA gut virome across populations with different subsistence strategies and geographical origin. Environ. Microbiol. 2017;19:4728–4735. doi: 10.1111/1462-2920.13938. [DOI] [PubMed] [Google Scholar]
- Ren J., Ahlgren N.A., Lu Y.Y., Fuhrman J.A., Sun F. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome. 2017;5:69. doi: 10.1186/s40168-017-0283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J., Song K., Deng C., Ahlgren N.A., Fuhrman J.A., Li Y., Xie X., Sun F. Identifying viruses from metagenomic data by deep learning. Quant Biol. 2018;8:64–77. doi: 10.1007/s40484-019-0187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A., Haynes M., Hanson N., Angly F.E., Heath A.C., Rohwer F., Gordon J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature. 2010;466:334–338. doi: 10.1038/nature09199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A., Blanton L.V., Cao S., Zhao G., Manary M., Trehan I., Smith M.I., Wang D., Virgin H.W., Rohwer F., Gordon J.I. Gut DNA viromes of Malawian twins discordant for severe acute malnutrition. Proc. Natl. Acad. Sci. U S A. 2015;112:11941–11946. doi: 10.1073/pnas.1514285112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohwer F., Edwards R. The Phage Proteomic Tree: a genome-based taxonomy for phage. J. Bacteriol. 2002;184:4529–4535. doi: 10.1128/JB.184.16.4529-4535.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S., Enault F., Hurwitz B.L., Sullivan M.B. VirSorter: mining viral signal from microbial genomic data. PeerJ. 2015;3:e985. doi: 10.7717/peerj.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S., Brum J.R., Dutilh B.E., Sunagawa S., Duhaime M.B., Loy A., Poulos B.T., Solonenko N., Lara E., Poulain J., Tara Oceans Coordinators Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature. 2016;537:689–693. doi: 10.1038/nature19366. [DOI] [PubMed] [Google Scholar]
- Roux S., Emerson J.B., Eloe-Fadrosh E.A., Sullivan M.B. Benchmarking viromics: an in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ. 2017;5:e3817. doi: 10.7717/peerj.3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux S., Adriaenssens E.M., Dutilh B.E., Koonin E.V., Kropinski A.M., Krupovic M., Kuhn J.H., Lavigne R., Brister J.R., Varsani A. Minimum Information about an Uncultivated Virus Genome (MIUViG): a community consensus on standards and best practices for describing genome sequences from uncultivated viruses. Nat. Biotechnol. 2019;37:29–37. doi: 10.1038/nbt.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro A., Ostan R., Candela M., Biagi E., Brigidi P., Capri M., Franceschi C. Gut microbiota changes in the extreme decades of human life: a focus on centenarians. Cell. Mol. Life Sci. 2018;75:129–148. doi: 10.1007/s00018-017-2674-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scepanovic P., Hodel F., Mondot S., Partula V., Byrd A., Hammer C., Alanio C., Bergstedt J., Patin E., Touvier M., Milieu Intérieur Consortium A comprehensive assessment of demographic, environmental, and host genetic associations with gut microbiome diversity in healthy individuals. Microbiome. 2019;7:130. doi: 10.1186/s40168-019-0747-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt T.S.B., Raes J., Bork P. The Human Gut Microbiome: From Association to Modulation. Cell. 2018;172:1198–1215. doi: 10.1016/j.cell.2018.02.044. [DOI] [PubMed] [Google Scholar]
- Schnorr S.L., Candela M., Rampelli S., Centanni M., Consolandi C., Basaglia G., Turroni S., Biagi E., Peano C., Severgnini M. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 2014;5:3654. doi: 10.1038/ncomms4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M., Lin X.D., Tian J.H., Chen L.J., Chen X., Li C.X., Qin X.C., Li J., Cao J.P., Eden J.S. Redefining the invertebrate RNA virosphere. Nature. 2016;540:539–543. doi: 10.1038/nature20167. [DOI] [PubMed] [Google Scholar]
- Shkoporov A.N., Hill C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe. 2019;25:195–209. doi: 10.1016/j.chom.2019.01.017. [DOI] [PubMed] [Google Scholar]
- Shkoporov A.N., Ryan F.J., Draper L.A., Forde A., Stockdale S.R., Daly K.M., McDonnell S.A., Nolan J.A., Sutton T.D.S., Dalmasso M. Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome. 2018;6:68. doi: 10.1186/s40168-018-0446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shkoporov A.N., Clooney A.G., Sutton T.D.S., Ryan F.J., Daly K.M., Nolan J.A., McDonnell S.A., Khokhlova E.V., Draper L.A., Forde A. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe. 2019;26:527–541.e5. doi: 10.1016/j.chom.2019.09.009. [DOI] [PubMed] [Google Scholar]
- Shreiner A.B., Kao J.Y., Young V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015;31:69–75. doi: 10.1097/MOG.0000000000000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Solonenko S.A., Ignacio-Espinoza J.C., Alberti A., Cruaud C., Hallam S., Konstantinidis K., Tyson G., Wincker P., Sullivan M.B. Sequencing platform and library preparation choices impact viral metagenomes. BMC Genomics. 2013;14:320. doi: 10.1186/1471-2164-14-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr E.P., Nuccio E.E., Pett-Ridge J., Banfield J.F., Firestone M.K. Metatranscriptomic reconstruction reveals RNA viruses with the potential to shape carbon cycling in soil. Proc. Natl. Acad. Sci. U S A. 2019 doi: 10.1073/pnas.1908291116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockdale S.R., Ryan F.J., Mccann A., Dalmasso M., Ross P.R., Hill C. 2018. Viral Dark Matter in the Gut Virome of Elderly Humans. [Google Scholar]
- Sutton T.D.S., Clooney A.G., Ryan F.J., Ross R.P., Hill C. Choice of assembly software has a critical impact on virome characterisation. Microbiome. 2019;7:12. doi: 10.1186/s40168-019-0626-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetz G.V., Ruggles K.V., Zhou H., Heguy A., Tsirigos A., Tetz V. Bacteriophages as potential new mammalian pathogens. Sci. Rep. 2017;7:7043. doi: 10.1038/s41598-017-07278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trubl G., Jang H.B., Roux S., Emerson J.B., Solonenko N., Vik D.R., Solden L., Ellenbogen J., Runyon A.T., Bolduc B. Soil Viruses Are Underexplored Players in Ecosystem Carbon Processing. mSystems. 2018;3:1–21. doi: 10.1128/mSystems.00076-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh P.J., Ley R.E., Mahowald M.A., Magrini V., Mardis E.R., Gordon J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Vik D.R., Roux S., Brum J.R., Bolduc B., Emerson J.B., Padilla C.C., Stewart F.J., Sullivan M.B. Putative archaeal viruses from the mesopelagic ocean. PeerJ. 2017;5:e3428. doi: 10.7717/peerj.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D. 5 challenges in understanding the role of the virome in health and disease. PLoS Pathog. 2020;16:e1008318. doi: 10.1371/journal.ppat.1008318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse R.M., Seppey M., Sim F.A., Ioannidis P., Simão F.A., Manni M., Ioannidis P., Klioutchnikov G., Kriventseva E.V., Zdobnov E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2017 doi: 10.1093/molbev/msx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker R.H. Vegetation of the Siskiyou Mountains, Oregon and California. Ecol. Monogr. 1960 doi: 10.2307/1943563. [DOI] [PubMed] [Google Scholar]
- Wood D.E., Salzberg S.L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15:R46. doi: 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T., Rey F.E., Manary M.J., Trehan I., Dominguez-Bello M.G., Contreras M., Magris M., Hidalgo G., Baldassano R.N., Anokhin A.P. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz S., Allgaier M., Hugenholtz P. Multiple displacement amplification compromises quantitative analysis of metagenomes. Nat. Methods. 2010;7:943–944. doi: 10.1038/nmeth1210-943. [DOI] [PubMed] [Google Scholar]
- Yinda C.K., Vanhulle E., Conceição-Neto N., Beller L., Deboutte W., Shi C., Ghogomu S.M., Maes P., Van Ranst M., Matthijnssens J. Gut Virome Analysis of Cameroonians Reveals High Diversity of Enteric Viruses, Including Potential Interspecies Transmitted Viruses. MSphere. 2019;4:4. doi: 10.1128/mSphere.00585-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S., Loo T.M., Atarashi K., Kanda H., Sato S., Oyadomari S., Iwakura Y., Oshima K., Morita H., Hattori M. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- Zhang T., Breitbart M., Lee W.H., Run J.Q., Wei C.L., Soh S.W.L., Hibberd M.L., Liu E.T., Rohwer F., Ruan Y. RNA viral community in human feces: Prevalence of plant pathogenic viruses. PLoS Biol. 2006;4:0108–0118. doi: 10.1371/journal.pbio.0040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-J., Li S., Gan R.-Y., Zhou T., Xu D.-P., Li H.-B. Impacts of gut bacteria on human health and diseases. Int. J. Mol. Sci. 2015;16:7493–7519. doi: 10.3390/ijms16047493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-Z., Chen Y.-M., Wang W., Qin X.-C., Holmes E.C. Expanding the RNA Virosphere by Unbiased Metagenomics. Annu. Rev. Virol. 2019;6:119–139. doi: 10.1146/annurev-virology-092818-015851. [DOI] [PubMed] [Google Scholar]
- Zhao G., Vatanen T., Droit L., Park A., Kostic A.D., Poon T.W., Vlamakis H., Siljander H., Härkönen T., Hämäläinen A.-M. Intestinal virome changes precede autoimmunity in type I diabetes-susceptible children. Proc. Natl. Acad. Sci. USA. 2017;114:E6166–E6175. doi: 10.1073/pnas.1706359114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo T., Wong S.H., Lam K., Lui R., Cheung K., Tang W., Ching J.Y.L., Chan P.K.S., Chan M.C.W., Wu J.C.Y. Bacteriophage transfer during faecal microbiota transplantation in Clostridium difficile infection is associated with treatment outcome. Gut. 2018;67:634–643. doi: 10.1136/gutjnl-2017-313952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo T., Lu X.J., Zhang Y., Cheung C.P., Lam S., Zhang F., Tang W., Ching J.Y.L., Zhao R., Chan P.K.S. Gut mucosal virome alterations in ulcerative colitis. Gut. 2019;68:1169–1179. doi: 10.1136/gutjnl-2018-318131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Scripts used in this manuscript are available on the Sullivan laboratory bitbucket under ‘gvd’ (https://bitbucket.org/MAVERICLab/gvd/). All raw reads are available through SRA, iVirus, or MG-RAST using the identifiers listed in Table S1. GVD viral populations and all IV databases per study can be downloaded directly from iVirus through the following DOI link: https://doi.org/10.25739/12sq-k039.