

Abstract
Background and Purpose
The macrocyclic tetrapeptide natural product CJ‐15,208 (cyclo[Phe‐d‐Pro‐Phe‐Trp]) is a multifunctional μ‐opioid receptor and κ‐opioid receptor agonist and κ‐opioid receptor antagonist that produces antinociception and prevents stress‐induced reinstatement of extinguished cocaine‐conditioned place preference (CPP). We hypothesized that an analogue of CJ‐15,208, cyclo[Pro‐Sar‐Phe‐d‐Phe], would demonstrate multifunctional μ‐opioid receptor and κ‐opioid receptor ligand activity, producing potent antinociception with fewer liabilities than selective μ‐opioid receptor agonists, while preventing both drug‐ and stress‐induced reinstatement of morphine‐induced CPP.
Experimental Approach
The opioid receptor agonist and antagonist activity of cyclo[Pro‐Sar‐Phe‐d‐Phe] was characterized after i.c.v. and i.p. administration to C57BL/6J or transgenic opioid receptor “knockout” mice using the 55°C warm‐water tail‐withdrawal assay. Liabilities of locomotor coordination, respiration and spontaneous ambulation, and direct rewarding or aversive properties were assessed. Finally, the ability of cyclo[Pro‐Sar‐Phe‐d‐Phe] to block morphine‐ and stress‐induced reinstatement of extinguished CPP was determined.
Key Results
cyclo[Pro‐Sar‐Phe‐d‐Phe] demonstrated dose‐dependent, short‐lasting antinociception, with an ED50 (and 95% confidence interval) of 0.15 (0.05–0.21) nmol i.c.v. and 1.91 (0.40–3.54) mg·kg−1 i.p., mediated by μ‐ and κ‐opioid receptors. The macrocyclic tetrapeptide also demonstrated potent dose‐dependent κ‐opioid receptor antagonist‐like activity at 2.5, but not at 4.5, h after administration. cyclo[Pro‐Sar‐Phe‐d‐Phe] displayed reduced liabiities compared with morphine, attributed to its additional activity at κ‐receptors. Pretreatment with cyclo[Pro‐Sar‐Phe‐d‐Phe] prevented stress‐ and drug‐induced reinstatement of extinguished morphine‐place preference responses in a time‐dependent manner.
Conclusions and Implications
These data suggest that cyclo[Pro‐Sar‐Phe‐d‐Phe] is a promising lead compound for both the treatment of pain with reduced sideeffects and preventing both drug‐ and stress‐induced relapse in morphine‐abstinent subjects.
Abbreviations
- CI
confidence interval
- CJ‐15,208
cyclo[Phe‐d‐Pro‐Phe‐Trp]
- CLAMS
Comprehensive Lab Animal Monitoring System
- CPA
conditioned place aversion
- CPP
conditioned place preference
- DOPrKO
δ‐opioid receptor gene‐disrupted mice
- [d‐Trp]CJ‐15,208
cyclo[Phe‐d‐Pro‐Phe‐D‐Trp]
- FSS
forced swim stress
- KOPrKO
κ‐opioid receptor gene‐disrupted mice
- MOPr KO
μ‐opioid receptor gene‐disrupted mice
- nor‐BNI
nor‐binaltorphimine
- U50,488
(±)‐trans‐3,4‐dichloro‐N‐methyl‐N‐[2‐(1‐pyrrolidinyl)cyclohexyl]benzeneacetamide
- SNC‐80
(+)‐4‐[(αR)‐α‐((2S,5R)‐4‐allyl‐2,5‐dimethyl‐1‐piperazinyl)‐3‐methoxybenzyl]‐N,N‐diethylbenzamide
- Sar
sarcosine, N‐methylglycine
- β‐FNA
β‐funaltrexamine
What is already known
Agonists targeting κ‐opioid receptors to treat pain and addiction have been hindered by undesirable side‐effects.
Agonists at μ‐opioid receptors are valuable analgesics, but produce respiratory depression and have abuse liability.
What this study adds
A multifunctional ligand acting at κ‐ and μ‐opioid receptors produces antinociception without the liabilities of μ‐opioid agonists.
This macrocyclic tetrapeptide prevents both drug‐ and stress‐induced reinstatement of morphine‐seeking behaviour.
What is the clinical significance
Multifunctional κ‐ and μ‐opioid receptor ligands retain desired therapeutic effects, with fewer κ‐ or μ‐opioid receptor liabilities.
1. INTRODUCTION
An estimated 100 million Americans suffer from chronic pain (Dahlhamer et al., 2018), for which opioids are often prescribed. While clinically used opioids produce profound analgesia, they also generate tolerance, respiratory depression, abuse and overdose, contributing to 47,000 deaths in 2017 (Center for Behavioral Health Statistics and Quality, 2018). An estimated 3 million US citizens suffer from an opioid use disorder (Huecker, Azadfard, & Leaming, 2019). Thus, there is a clear and urgent need for improved treatments for both pain and opioid use disorder.
The endogenous opioid system, consisting of μ‐, κ‐, and δ‐opioid receptors and their respective endogenous peptides, modulates both nociception and reward systems (Darcq & Kieffer, 2018), making it a valuable target for the treatment of pain and substance abuse. Unfortunately, receptor‐selective opioid ligands produce adverse outcomes, limiting their therapeutic utility. Selective agonists of μ‐opioid receptors remain in clinical use for pain management but produce undesired and potentially fatal side effects, complicating their continuous use (Buntin‐Mushock, Phillip, Moriyama, & Palmer, 2005; Williams, Christie, & Manzoni, 2001). Attempts to circumvent these limitations of µ‐opioid receptor agonists (e.g., biased ligands) have met with disappointment to date (Hill et al., 2018; US Food and Drug Administration [FDA], 2018). Agonists of κ‐opioid receptors also produce analgesia (Corder, Castro, Bruchas, & Scherrer, 2018) and have been shown to counter stimulant‐induced dopaminergic signalling in the reward pathways (Graziane, Polter, Briand, Pierce, & Kauer, 2013; Shippenberg, LeFevour, & Heidbreder, 1996). However, clinical use of κ‐opioid receptor‐selective agonists has been hindered by their CNS‐mediated dysphoric effects (Pfeiffer, Brantl, Herz, & Emrich, 1986). Moreover, dysregulation of the κ‐opioid receptor system in the nucleus accumbens and reward circuits resulting from prolonged psychostimulant exposure or stress is theorized to paradoxically increase drug‐seeking behaviour (Bruchas, Land, & Chavkin, 2010). Antagonists of κ‐opioid receptors have shown promise as treatments to prevent stress‐induced relapse to drug‐seeking behaviour (Aldrich & McLaughlin, 2009; Beardsley, Howard, Shelton, & Carroll, 2005). Thus, new ligands targeting the opioid system may provide much needed improved medications for substance abuse and pain.
Of interest, opioid receptor ligands possessing activity at more than one receptor (designated “multifunctional” opioids; see Schiller, 2010) may produce potent therapeutic activity with fewer undesired effects. The co‐administration of μ‐ and κ‐opioid receptor agonists demonstrates additive or synergistic increases in antinociception (Ko & Husbands, 2009; Negus, Schrode, & Stevenson, 2008; Sutters, Miaskowski, Taiwo, & Levine, 1990). The μ‐ and κ‐opioid receptors are both expressed in the peripheral sensory neurons, spinal cord, and supraspinal regions such as the rostral ventromedial medulla and periaqueductal gray that regulate nociception (Corder et al., 2018), supporting the hypothesis that the activation of both these opioid receptors could produce additive or synergistic antinociception. Interestingly, the contrasting side effects of μ‐ and κ‐opioid receptor agonists may further counteract one another's undesired effects, improving the therapeutic index. Agonists of κ‐opioid receptors have been shown to reverse μ‐opioid receptor‐mediated respiratory depression (Dosaka‐Akita, Tortella, Holaday, & Long, 1993; Haji & Takeda, 2001). Consistent with this, nalbuphine, a κ‐ and μ‐opioid receptor agonist, demonstrates similar analgesic efficacy to morphine, but with negligible respiratory effects (Miller, 1980; Schmidt et al., 1985). Likewise, the addition of μ‐opioid receptor agonist activity might improve the therapeutic utility of κ‐opioid agonists by mitigating dysphoric effects (Anand & Montgomery, 2018).
Together, multifunctional activity at both μ‐ and κ‐opioid receptors may enhance analgesic potency while offsetting major opioid‐related liabilities such as respiratory depression and abuse potential. This desirable multifunctional opioid activity profile was found with a macrocyclic tetrapeptide natural product, CJ‐15,208 (cyclo[Phe‐d‐Pro‐Phe‐Trp]). CJ‐15,208 exhibits antinociception mediated by μ‐ and κ‐opioid receptor agonism, followed by κ‐opioid receptor antagonism of finite duration (lasting hours, rather than days) that prevented stress‐induced reinstatement of extinguished cocaine‐conditioned place preference (CPP) (Ross, Reilley, Murray, Aldrich, & McLaughlin, 2012). To explore structure–activity relationships of CJ‐15,208 and enhance opioid activity, the novel peptide analogue cyclo[Pro‐Sar‐Phe‐d‐Phe] (Figure 1) was designed and synthesized (Ferracane et al., 2020). We hypothesized that cyclo[Pro‐Sar‐Phe‐d‐Phe] would demonstrate similar multifunctional opioid receptor activity, producing antinociception and preventing reinstatement of extinguished morphine‐CPP. Here, we characterize the opioid activity profile in vivo of this novel macrocyclic tetrapeptide, assess potential opioid liabilities such as respiratory depression, and report its ability to prevent drug‐induced as well as stress‐induced reinstatement of extinguished morphine‐CPP in mice.
FIGURE 1.
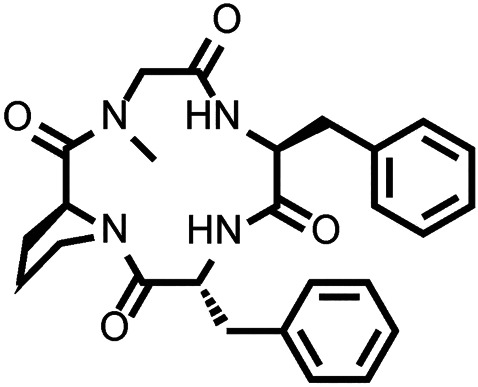
Structure of cyclo[Pro‐Sar‐Phe‐d‐Phe]
2. METHODS
2.1. Experimental design
The objective of this study was to test cyclo[Pro‐Sar‐Phe‐d‐Phe] for antinociceptive activity along with characteristic liabilities of clinically used opioids and for the ability to prevent both drug‐ and stress‐induced reinstatement of morphine‐seeking behaviour. We characterized the opioid receptor agonist and antagonist activity of cyclo[Pro‐Sar‐Phe‐d‐Phe] after intracerebroventricular (i.c.v.) and intraperitoneal (i.p.) administration to C57BL/6J or transgenic opioid receptor “knockout” (KO) mice using the 55°C warm‐water tail‐withdrawal assay. Liabilities of evoked locomotor coordination, respiration, spontaneous ambulation, and rewarding or aversive properties were also assessed. Finally, the ability of cyclo[Pro‐Sar‐Phe‐d‐Phe] to block morphine‐ and stress‐induced reinstatement of extinguished morphine‐CPP was determined. Sample sizes were chosen on their basis to detect statistically significant results, with statistical analysis detailed throughout the study.
2.2. Animals
All animal care and experimental procedures complied with the laws of the United States and regulations of the Department of Agriculture, and were approved by the University of Florida (Gainesville, FL, USA) Institutional Animal Care and Use Committee, in accordance with the 2011 National Institute of Health Guide for the Care and Use of Laboratory Animals. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010; McGrath & Lilley, 2015) and with the recommendations made by the British Journal of Pharmacology.
A total of 739 adult male C57BL/6J mice weighing 22–26 g were obtained from Jackson Labs (Bar Harbor, ME, USA; RRID:IMSR_JAX:000664). C57BL/6J mice are established and validated subjects in the tail‐withdrawal, rotarod, respiratory, and place conditioning assays. Forty‐nine male, μ‐opioid receptor gene‐disrupted “knockout” (MOPrKO), 44 κ‐opioid receptor gene‐disrupted “knockout” (KOPrKO), and eight δ‐opioid receptor gene‐disrupted “knockout” (DOPrKO) mice were obtained from homozygous breeding pairs from colonies at the University of Florida, started from progenitors obtained from Jackson Labs (MOPrKO and KOPrKO) or as a generous gift from the laboratory of Dr. Gregory Scherrer of Stanford University (DOPrKO mice). All mice were used at an age of 7–11 weeks at the initiation of testing and were group housed in ventilated cages (maximum of five animals per cage) in a temperature‐controlled specific pathogen‐free room kept on a 12‐h light–dark cycle. Upon the completion of testing, all mice were euthanized by inhalation of carbon dioxide, followed by cervical dislocation as a secondary measure, as recommended by the American Veterinary Medical Association.
Initial sample sizes were approximated by Power analysis, with animals assigned to groups randomly. Drug treatment experiments were conducted in a blinded fashion. Eight animals were excluded from presentation and data analysis due to their failure to meet the extinction criteria for final reinstatement testing (as discussed below). Fifteen animals across the study were considered statistical outliers, by predefined criteria, and were excluded from presentation and data analysis (see Section 2.11).
2.3. Preparation of drug solutions
cyclo[Pro‐Sar‐Phe‐d‐Phe] administered through the i.c.v. route was initially dissolved in DMSO to give a 40‐mM stock solution and then diluted daily prior to use in sufficient warm (40°C) sterile saline to give a final mixture of 50% DMSO/50% saline (0.9%). cyclo[Pro‐Sar‐Phe‐d‐Phe] administered through the i.p. route was initially dissolved daily prior to use in DMSO and Solutol HS 15 (Solutol) and diluted with warm sterile saline to provide a final mixture of 5% DMSO/10% Solutol/85% saline (0.9%). SNC‐80 was administered in 100% DMSO; all other compounds were administered in 0.9% sterile saline. For i.p. administration, 0.5, 1, 3, 5, and 10 mg·kg−1 doses were delivered in solutions containing 0.05, 0.1, 0.3, 0.5, or 1 mg of test compound per 1 mL of vehicle, respectively.
2.4. Intracerebroventricular drug administration
cyclo[Pro‐Sar‐Phe‐d‐Phe] and other drugs were administered through the i.c.v. route in a volume of 5 μL directly into the lateral ventricle according to the modified method of Haley and McCormick (1957) and as previously described (McLaughlin et al., 1999). Mice were lightly anaesthetized with isoflurane (0.4%), and a small (<0.3 cm) incision was made in the scalp. Using a 10‐μL Hamilton microliter syringe, the injection was made 2 mm lateral and 2 mm caudal to bregma at a depth of 3 mm. After injection, the incision site was sealed with tissue adhesive. Mice typically recover consciousness within 30 s after the injection. cyclo[Pro‐Sar‐Phe‐d‐Phe] and other drugs were also administered i.p. in a volume of 250 μL·25−1 g body weight.
2.5. Antinociceptive testing
The 55°C warm‐water tail‐withdrawal assay was performed as previously described (McLaughlin et al., 1999). Warm (55°C) water in a 1.5‐L heated water bath was used as the thermal nociceptive stimulus, with the latency of the mouse to withdraw its tail from the water taken as the endpoint. After determining baseline tail‐withdrawal latencies (1.79 ± 0.03 s across all tested subjects), mice were administered vehicle alone or a single graded dose of established opioids or cyclo[Pro‐Sar‐Phe‐d‐Phe] through the i.c.v. or i.p. route. To determine agonist activity, the tail‐withdrawal latency was determined repeatedly every 10 min following administration of the compounds for 1 h or until latencies returned to baseline values. Testing of opioid receptor mediation of antinociception utilized MOPrKO, KOPrKO, DOPrKO, or KOPrKO mice pretreated 24 h with the μ‐opioid receptor antagonist β‐funaltrexamine (β‐FNA, 10 mg·kg−1, i.p.), followed by subsequent treatment with a single dose of cyclo[Pro‐Sar‐Phe‐d‐Phe], and testing as described. To determine antagonist activity, a single graded dose of cyclo[Pro‐Sar‐Phe‐d‐Phe] was administered 2.5 h prior to the μ‐opioid receptor agonist morphine or the κ‐opioid receptor agonist U50,488 (each at 10 mg·kg−1, i.p.) or the δ‐opioid receptor agonist SNC‐80 (100 nmol, i.c.v.), and antinociception measured 40 min after their administration. A cut‐off time of 15 s was used in this study; if the mouse failed to display a tail‐withdrawal response during that time, the tail was removed from the water, and the animal was assigned a maximal antinociceptive score of 100%. At each time point, antinociception was calculated according to the following formula:
2.6. Rotarod assay to assess coordinated locomotor activity
Possible impairment of coordinated locomotor activity by cyclo[Pro‐Sar‐Phe‐d‐Phe] was assessed via rotarod performance as described previously (Armishaw et al., 2013; Eans et al., 2013). Locomotor activity was recorded using an automated, computer‐controlled rotarod apparatus (San Diego Instruments, San Diego, CA, USA). Mice were first habituated to the rotarod over seven trials, with the last trial serving as the baseline response. Mice habituated to the rotarod were then administered (i.p.) saline, vehicle (5% DMSO/10% Solutol in saline), U50,488 (10 mg·kg−1), or cyclo[Pro‐Sar‐Phe‐d‐Phe] (10 mg·kg−1) 15 min prior to assessment in accelerated speed trials (180s max latency at 0–20 rpm) performed every 10 min over a 60‐min period. Mice were tested a total of 14 trials (seven habituation trials prior to treatment + seven drug trials). Decreased latencies to fall in the rotarod test indicate impaired motor coordination/sedation.
2.7. Respiration and ambulation
Respiration rates (in breaths per minute) and animal locomotive activity (as ambulations) were assessed using the Comprehensive Laboratory Animal Monitoring System (CLAMS)/Oxymax (Columbus Instruments, Columbus, OH) as described previously (Armishaw et al., 2013; Hoot et al., 2013). Mice were habituated to their individual sealed housing chambers for 60 min before testing. Mice were administered cyclo[Pro‐Sar‐Phe‐d‐Phe] (10 mg·kg−1, i.p.), morphine (10 mg·kg−1, i.p.), or vehicle, as indicated, and 5 min later confined to the CLAMS testing chambers. In order to assess the possible contributions of κ‐ and μ‐opioid receptor activity on respiratory and locomotor activity, KOPrKO and MOPrKO mice were also administered cyclo[Pro‐Sar‐Phe‐d‐Phe] or vehicle and tested in the CLAMS chambers. Pressure monitoring within the sealed chambers measured frequency of respiration. Infrared beams located in the floor measured locomotion as number of beam breaks. Respiration and locomotive data were averaged over 20‐min periods for 120 min post‐injection of the test compound. In order to account for natural changes in respiration and ambulation over time due to initial exploratory behaviour and sleeping at later time points, treatment groups were run with their respective vehicle control group. Data are presented as % vehicle response ± SEM for ambulations or breaths per minute.
2.8. Place conditioning to determine CPP and conditioned place aversion
CPP is a well‐established model in mice, where drugs such as morphine with abuse liabilities produce CPP (Bardo, Rowlett, & Harris, 1995) and dysphoric agents such as κ‐opioid receptor agonists produce aversion (Shippenberg & Herz, 1986; Spetea et al., 2017). An automated, balanced three‐compartment place conditioning apparatus (San Diego Instruments) and a 2‐ or 4‐day counterbalanced morphine‐place conditioning design were used, similar to methods previously described (Eans et al., 2013). The amount of time subjects spent in each of the three compartments was measured over a 30‐min testing period. Prior to place conditioning, an initial preference test was performed in which the animals could freely explore all open compartments; the animals did not demonstrate significant differences in their time spent exploring the outer left (638 ± 13 s) versus right (625 ± 12 s) compartments (P > 0.05, Student's t‐test). To perform place conditioning, mice were administered 0.9% saline and consistently confined in a randomly assigned outer compartment: half of each group in the right chamber and half in the left chamber. Four hours later, mice were administered test compound and confined to the opposite compartment for 40 min. To determine if cyclo[Pro‐Sar‐Phe‐d‐Phe] (0.1, 1, or 10 nmol, i.c.v.) produced CPP or conditioned place aversion (CPA), mice were place conditioned in this way for 2 days, with a final preference test performed on the fourth day, as this has been shown to produce dependable morphine‐CPP and U50,488‐induced CPA (Spetea et al., 2017). Additional mice were place conditioned 4 days with morphine (10 mg·kg−1, i.p.) for use in reinstatement testing as described previously for cocaine conditioning (Ross et al., 2012). Data are plotted as the difference in time spent in the eventual conditioning drug‐paired compartment versus the vehicle‐paired compartment. Accordingly, a negative value reflects conditioned place aversion, while a positive value reflects a conditioned preference for the drug‐paired side.
2.9. Extinction
Preference tests were performed twice weekly, during which the animals were allowed to access all open compartments to test for a loss of preference for the drug‐paired chamber. This occurred over an 8‐week period until extinction was established (see Figure 8a). Extinction is defined as a statistically significant decrease in the time spent in the drug‐paired compartment during the extinction trial as compared to the postconditioning response after the initial 4 days of morphine‐place conditioning.
FIGURE 8.
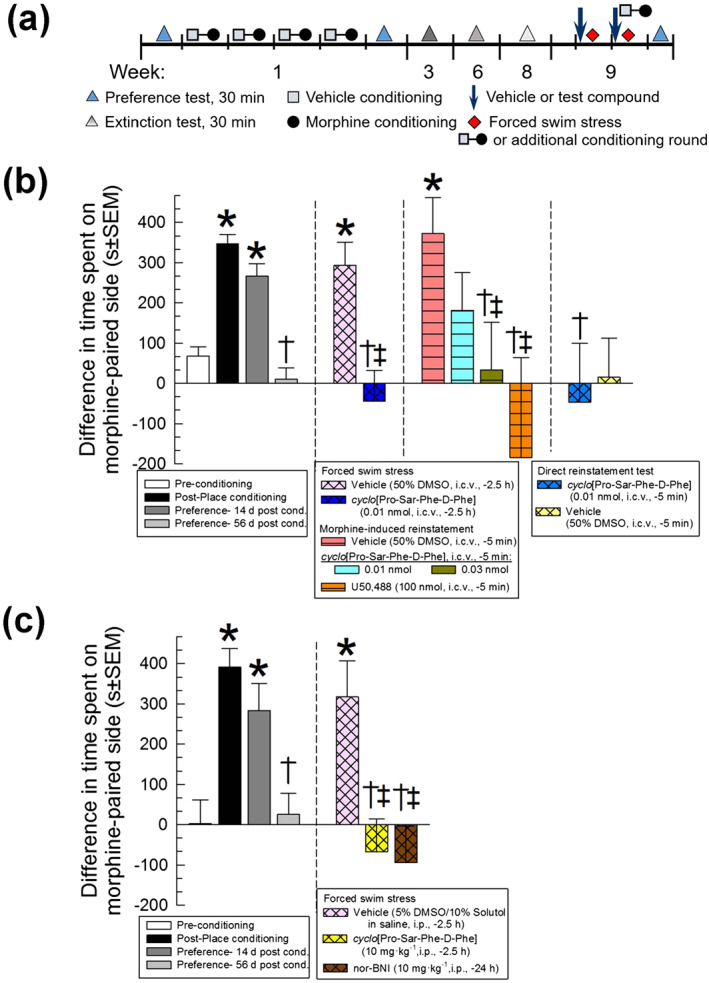
Time‐dependent prevention of stress‐ and drug‐induced reinstatement of extinguished morphine‐CPP by cyclo[Pro‐Sar‐Phe‐d‐Phe]. (a) Schematic representation of reinstatement and testing protocol. During week 1, preference tests were recorded before and after a 4‐day morphine conditioning paradigm to establish the development of morphine‐CPP. Over the next 7 weeks, extinction tests were performed until extinction of morphine‐CPP was established in week 8. For reinstatement testing in week 9, vehicle (50% DMSO, i.c.v. or 5% DMSO/10% Solutol, i.p.), cyclo[Pro‐Sar‐Phe‐d‐Phe] (0.01 or 0.03 nmol, i.c.v., or 10 mg·kg−1, i.p.), or nor‐BNI (10 mg·kg−1, i.p.) were administered on days 63 and 64 before the exposure to FSS; for morphine‐place conditioning additional mice were treated on days 63 and 64 with vehicle, cyclo[Pro‐Sar‐Phe‐d‐Phe], or U50,488 (100 nmol, i.c.v.), followed by morphine‐place conditioning 5 min after the injection on day 64. An additional preference test was then performed on day 65 to assess for the reinstatement of CPP. (b) Following 4 days of morphine administration (10 mg·kg−1, i.p. daily), mice exhibited significant preference for the morphine‐paired environment, with extinction occurring at the end of week 8. Mice were then exposed to FSS or another round of morphine‐place conditioning, reinstating preference. Pretreatment with the cyclo[Pro‐Sar‐Phe‐d‐Phe] (0.01 nmol, i.c.v.) prevented stress‐induced reinstatement of place preference, and pretreatment with the cyclo[Pro‐Sar‐Phe‐d‐Phe] (0.03 nmol, i.c.v.) prevented morphine‐induced reinstatement of place preference. Further, a round of conditioning with cyclo[Pro‐Sar‐Phe‐d‐Phe] (0.01 nmol, i.c.v.) did not in itself cause reinstatement of place preference; peptide‐treated mice were similar to mice conditioned with vehicle alone. (c) Peripheral pretreatment with cyclo[Pro‐Sar‐Phe‐d‐Phe] or nor‐BNI (each at 10 mg·kg−1, i.p.) prevented stress‐induced reinstatement of place preference. Data shown are the means ± SEM of n = 12–46 mice (b) or n = 14–21 mice (c); morphine‐place conditioning data in the left panel represent combined responses of all 202 mice (b) or 55 mice tested (c). * P < .05, significantly different from preconditioning place preference response. † P < .05, significantly different from post‐conditioning place preference response. ‡ P < .05, significantly different from respective vehicle control group‐induced reinstatement of place preference response; one‐way ANOVA followed by Tukey's multiple comparison post hoc test. Data from 257 mice are shown in this figure
2.10. Reinstatement
The week following extinction, reinstatement of morphine‐CPP was examined after either exposure to forced swim stress (FSS) or an additional cycle of morphine‐place conditioning, as described previously for cocaine‐place conditioning (Aldrich et al., 2013; Ross et al., 2012). Mice were pretreated daily (−2.5 h·day−1) with vehicle (50% DMSO, i.c.v., or 5% DMSO/10% Solutol, i.p.) or graded doses of cyclo[Pro‐Sar‐Phe‐d‐Phe] (i.c.v. or i.p.) prior to a 2‐day FSS protocol used to produce stress‐induced reinstatement of extinguished CPP. As a control, additional mice were pretreated with the established κ‐receptor antagonist nor‐binaltorphimine (nor‐BNI, once −24 h, i.p.) prior to the first day of swim stress. To evaluate prevention of morphine‐induced reinstatement, mice were administered vehicle, or the standard κ‐opioid agonist, U50,488, or graded doses of cyclo[Pro‐Sar‐Phe‐d‐Phe] (i.c.v. or i.p.) 5 min prior to morphine (10 mg·kg−1, i.p.) for an additional day of place conditioning. A subset of mice were conditioned with either vehicle or cyclo[Pro‐Sar‐Phe‐d‐Phe] (i.c.v.) and saline rather than morphine to test for possible direct effects of vehicle or cyclo[Pro‐Sar‐Phe‐d‐Phe] in reinstatement procedures. On the day following the completion of stress exposure or place conditioning, mice were tested for place preference to determine possible reinstatement (Figure 8).
2.11. Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are presented as means ± SEM with significance set at P < 0.05. Statistical analysis was undertaken only for data sets where each group size was at least n = 5. All tail‐withdrawal latency data are reported as percent antinociception to control for each animal's baseline latency response. All dose–response lines were analysed by regression, and ED50 (dose producing 50% antinociception) values and 95% confidence limits determined using each individual data point with Prism 8.0 software (GraphPad Software, La Jolla, CA; RRID:SCR_002798) and compared via linear or non‐linear regression modelling. Significant differences in behavioural data were analysed by ANOVA (one‐way or two‐way with repeated measures [RM], as appropriate), with significant results further analysed with Dunnett's, Tukey's, or Sidak's multiple comparison post hoc tests, as appropriate. For direct comparisons between data points, a Student's t‐test was used. In order to account for heterogeneity of variance across the groups in CLAMS experiments, a Greenhouse–Geisser correction was utilized in the statistical analysis. Animals responding above or below two SDs from the mean within their experimental group were excluded from statistical analysis; 15 such mice were excluded.
2.12. Materials
All chemicals other than cyclo[Pro‐Sar‐Phe‐d‐Phe] (Figure 1) were obtained from Sigma‐Aldrich (St. Louis, MO, USA). The design and synthesis of cyclo[Pro‐Sar‐Phe‐d‐Phe] is described separately (Ferracane et al., 2020).
2.13. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. Characterization of cyclo[Pro‐Sar‐Phe‐d‐Phe]‐mediated antinociception in the 55°C warm‐water tail‐withdrawal assay
There was a significant dose‐dependent antinociceptive effect of cyclo[Pro‐Sar‐Phe‐d‐Phe] over time (Figure2a). The macrocyclic tetrapeptide demonstrated significant antinociception after i.c.v. administration lasting up to 90 min after administration of a 10nmol dose. Likewise, i.p. administration yielded significant time‐ and dose‐dependent antinociception, although this was of shorter duration, lasting up to 40 min following treatment with a 10 mg·kg−1 dose (Figure 2b). Comparison of the ED50 values calculated from cyclo[Pro‐Sar‐Phe‐d‐Phe] at collective peak antinociceptive effect for each dose demonstrated potency equivalent to, or greater than, morphine when administered through the i.p. (10 and 30 min postadministration, respectively) or i.c.v. (20 min postadministration) routes (Table 1; see also Figure S1).
FIGURE 2.
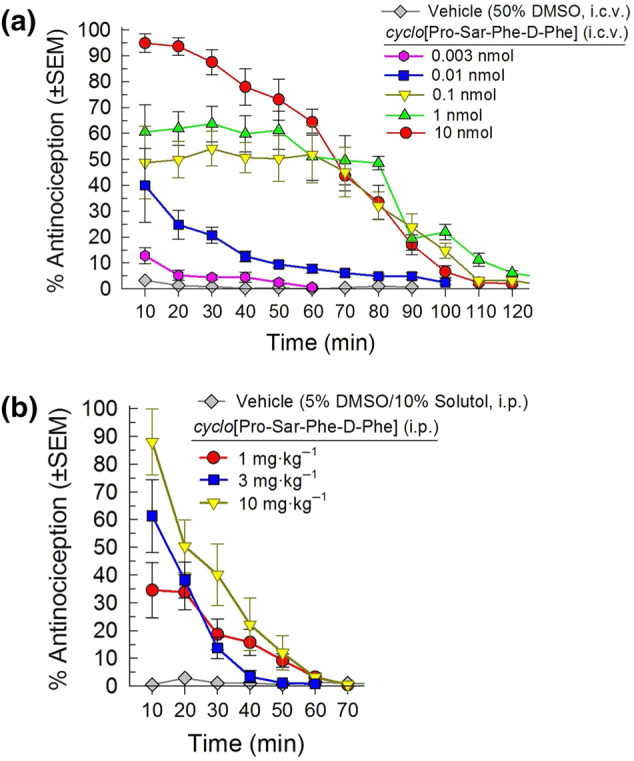
Antinociceptive activity of cyclo[Pro‐Sar‐Phe‐d‐Phe] following administration i.c.v. (a) or i.p. (b) in the 55°C warm‐water tail‐withdrawal assay in C57BL/6J mice. cyclo[Pro‐Sar‐Phe‐d‐Phe] demonstrated significant time‐ and dose‐dependent antinociception with repeated measurement over time. Data shown are the % antinociception (as means ± SEM) from six to 24 mice for each set presented; overall, results from 129 mice are presented
TABLE 1.
ED50 (with 95% confidence interval (CI)) values for antinociception produced by cyclo[Pro‐Sar‐Phe‐d‐Phe] or morphine following i.c.v. or i.p. administration
| Route of administration | ED50 (with 95% CI values) | |
|---|---|---|
| cyclo[Pro‐Sar‐Phe‐d‐Phe] | Morphine | |
| i.c.v. | 0.15 (0.08–0.29) nmol | 2.35 (1.13–5.03) nmol |
| i.p. | 1.91 (0.40–3.54) mg·kg−1 | 3.91 (2.92–5.17) mg·kg−1 |
The opioid receptor selectivity of cyclo[Pro‐Sar‐Phe‐d‐Phe]‐induced antinociception at peak effect after i.c.v. administration (20 min) was examined in opioid receptor gene‐disrupted (KO) mice (Figure 3). Antinociception produced by a 30nmol dose of cyclo[Pro‐Sar‐Phe‐d‐Phe] was partially but significantly reduced in both MOPrKO and KOPrKO mice, but not in DOPrKO mice. Complete inhibition of cyclo[Pro‐Sar‐Phe‐d‐Phe]‐induced antinociception was achieved only by pretreatment of KOPrKO mice with the μ‐opioid receptor‐selective antagonist β‐FNA (rightmost bar, Figure 3), restoring the response to baseline values (1.66 ± 0.08 s vs. the baseline response of 1.43 ± 0.10 s).
FIGURE 3.
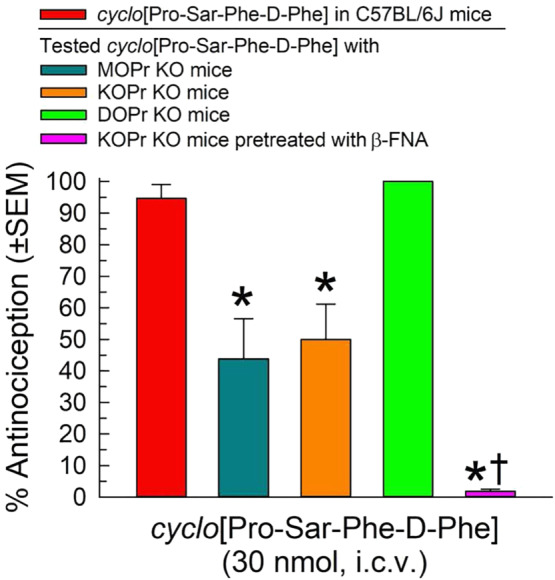
Opioid receptor involvement in cyclo[Pro‐Sar‐Phe‐d‐Phe]‐induced antinociception in the 55°C warm‐water tail‐withdrawal assay 20 min after i.c.v. administration of a 30nmol dose using MOPrKO, KOPrKO, or DOPrKO mice. A subset of KOPrKO mice received β‐FNA (10 mg·kg−1, i.p.) 24 h prior to testing of the cyclic tetrapeptide. Data shown are the % antinociception (as means ± SEM) from eight mice for each group, for a total of 40 mice. *P < .05, significantly different from C57BL/6J mice; one‐way ANOVA with Tukey's multiple comparison post hoc test). †P > .05, not significantly different from baseline tail‐withdrawal latency; Student's t‐test
3.2. Antagonist activity against opioid receptor‐selective agonists
The in vivo effects of cyclo[Pro‐Sar‐Phe‐d‐Phe] were next evaluated at a time point after the dissipation of agonist activity. Intracerebroventricular pretreatment with cyclo[Pro‐Sar‐Phe‐d‐Phe] (0.01 nmol, i.c.v.) 150 min prior to agonist administration demonstrated significant antagonism of only the κ‐opioid receptor selective agonist U50,488 (Figure 4a). A 2.5 h pretreatment with a much higher dose (30 nmol, i.c.v.) of cyclo[Pro‐Sar‐Phe‐d‐Phe] also did not significantly antagonize morphine‐ or SNC‐80‐mediated antinociception (100 ± 0% and 87.1 ± 7.7%, respectively), confirming κ‐opioid receptor‐selective antagonist activity.
FIGURE 4.
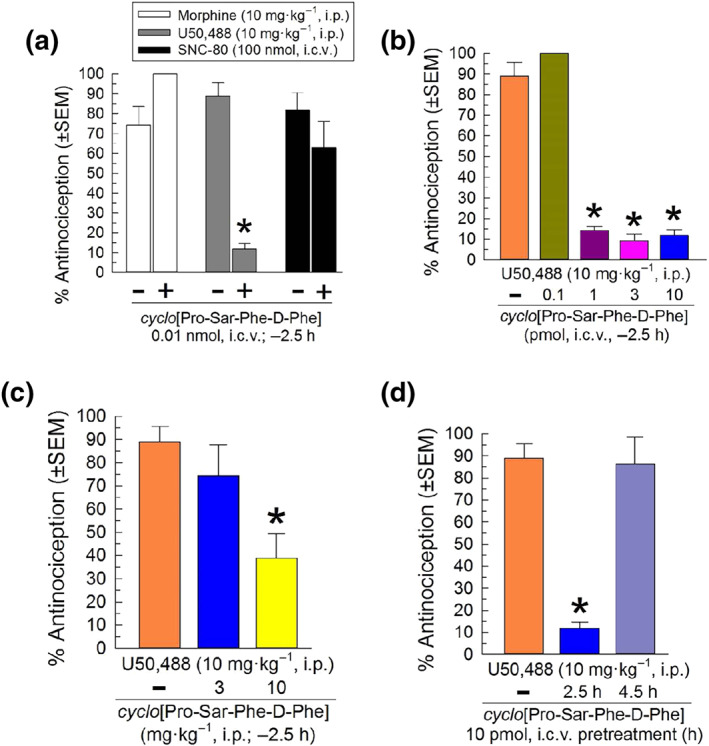
Characterization of opioid receptor antagonist activity of cyclo[Pro‐Sar‐Phe‐d‐Phe] in the mouse 55°C warm‐water tail‐withdrawal assay. (a) Opioid receptor selectivity. A 2.5 h pretreatment with cyclo[Pro‐Sar‐Phe‐d‐Phe (0.01 nmol, i.c.v.) prevented U50,488‐induced antinociception (10 mg·kg−1, i.p.), but not that of morphine (10 mg·kg−1, i.p.) or SNC‐80 (100 nmol, i.c.v.). Mice pretreated (2.5 h) with cyclo[Pro‐Sar‐Phe‐d‐Phe] (b) i.c.v. or (c) i.p. demonstrate dose‐dependent antagonism of U50,488‐induced antinociception. (d) Time‐dependent κ‐receptor antagonism by cyclo[Pro‐Sar‐Phe‐d‐Phe]. The antinociceptive effect of U50,488 (10 mg·kg−1, i.p.) was determined in mice pretreated for 2.5 or 4.5 h with cyclo[Pro‐Sar‐Phe‐d‐Phe] (0.01 nmol, i.c.v). Data shown are the % antinociception (as means ± SEM) from eight to 18 mice. P < .05, significantly different from response of agonist alone; one‐way ANOVA with Dunnett's or Tukey's multiple comparison post hoc tests. Overall, results from 143 mice are presented. The same data from 16 mice for the U50,488 (10 mg·kg−1, i.p.) control are displayed in Figure 4a–d
A 2.5 h pretreatment with cyclo[Pro‐Sar‐Phe‐d‐Phe] significantly antagonized κ‐opioid receptors in a dose‐dependent manner after i.c.v. (Figure 4b) or i.p. (Figure 4c) administration, with significant antagonism at doses of either 1 pmol i.c.v., or 10 mg·kg−1 i.p. The duration of κ‐opioid receptor antagonism produced by a single dose (10 pmol i.c.v.) of cyclo[Pro‐Sar‐Phe‐d‐Phe] lasted at least 2.5 h, but less than 4.5 h (Figure 4d).
3.3. Evaluation of potential side‐effects of cyclo[Pro‐Sar‐Phe‐d‐Phe]
The effects of cyclo[Pro‐Sar‐Phe‐d‐Phe] or the κ‐opioid receptor‐selective agonist U50,488 (10 mg·kg−1, i.p., each) on locomotor coordination were assessed in the mouse rotarod assay. Control mice received vehicle (either 5% DMSO/10% Solutol/85% saline or saline alone) via i.p. injection. While U50,488 significantly impaired coordinated locomotor activity over time as compared with saline, cyclo[Pro‐Sar‐Phe‐d‐Phe] did not significantly impair locomotor coordination, as compared to vehicle (Figure 5).
FIGURE 5.
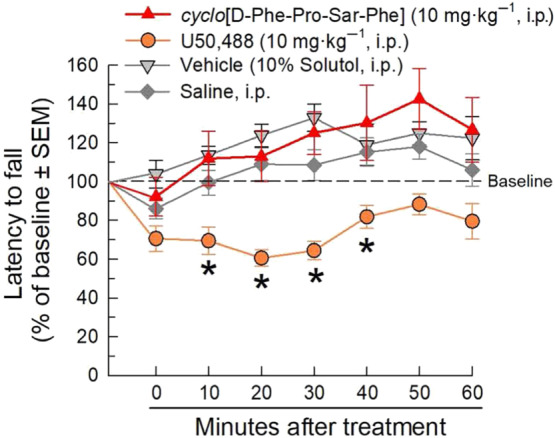
Effect of cyclo[Pro‐Sar‐Phe‐d‐Phe] on locomotor coordination after i.p. administration to C57BL/6J mice in the rotarod assay. Mice received i.p. cyclo[Pro‐Sar‐Phe‐d‐Phe] (10 mg·kg−1), U50,488 (10 mg·kg−1), vehicle (5% DMSO/10% Solutol in 0.9% saline), or 0.9% saline and were tested on the rotarod apparatus with repeated measurements over time. Latencies to fall are given as the mean % change from baseline (100%) performance ± SEM. Data from eight to 16 mice are presented. A total of 48 animals were used in this experiment. P < .05, significantly different from response of saline alone; two‐way RM ANOVA with Tukey's multiple comparison post hoc test
The effect of cyclo[Pro‐Sar‐Phe‐d‐Phe] or the μ‐opioid receptor‐preferring agonist morphine on spontaneous locomotor activity and respiration rate were assessed over 2 h after administration of a 10 mg·kg−1 (i.p.) dose using the Comprehensive Laboratory Animal Monitoring System (CLAMS). Treatment produced significant, time‐dependent effects on ambulation (Figure 6a). Morphine produced sustained, significant increases in ambulation at all time points. The cyclic tetrapeptide induced significantly less effects on ambulation in wild‐type, KOPrKO, and MOPrKO mice compared to morphine treated wild‐type mice. In wild‐type mice, cyclo[Pro‐Sar‐Phe‐d‐Phe] had no significant effect on ambulation in comparison to vehicle‐treated wild‐type mice. The peptide induced a significant increase in ambulation in KOPrKO mice compared to wild‐type cyclo[Pro‐Sar‐Phe‐d‐Phe]‐treated mice and the KOPrKO vehicle‐treated control group (20–40 min). Interestingly, cyclo[Pro‐Sar‐Phe‐d‐Phe]‐treated MOPrKO animals demonstrated several points of significant hypolocomotion, compared to their respective vehicle control group (0–40 and 80–120 min).
FIGURE 6.
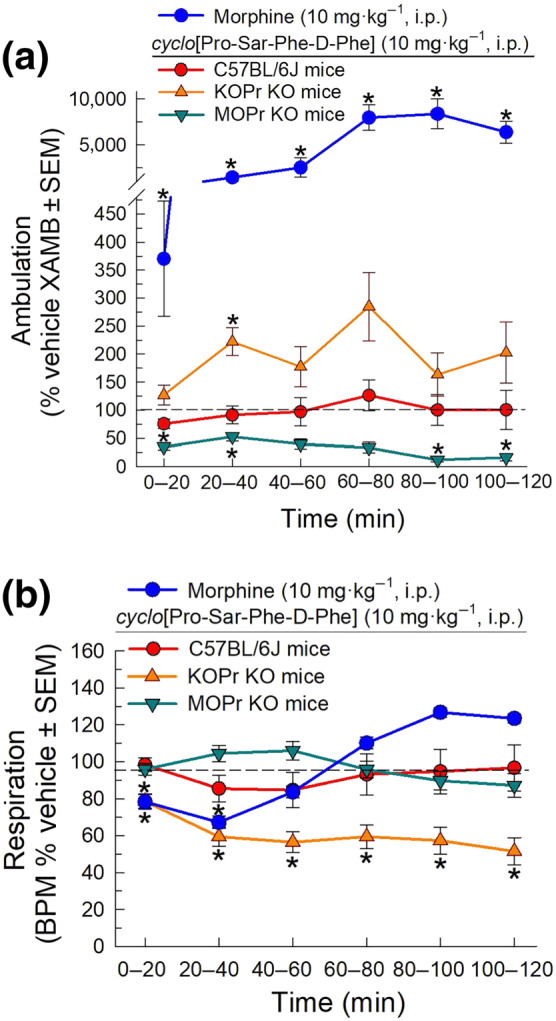
Effects of cyclo[Pro‐Sar‐Phe‐d‐Phe] on (a) ambulation and (b) respiration in C57BL/6J wild‐type, MOPrKO, or KOPrKO mice tested in the CLAMS/Oxymax system. Ambulation and respiration were monitored after i.p. administration of cyclo[Pro‐Sar‐Phe‐d‐Phe] (10 mg·kg−1) or morphine (10 mg·kg−1). Data are presented as % vehicle response ± SEM, ambulation, XAMB (a) or breaths per minute, BPM (b). P < .05, significantly different from vehicle control response; two‐way RM ANOVA with Tukey's multiple comparison post hoc test. Data from 154 animals are shown in this figure
Treatment also had a generally significant effect on respiration over time (Figure 6b). Morphine significantly reduced respiration for the first 40 min compared to vehicle. cyclo[Pro‐Sar‐Phe‐d‐Phe] was without significant effect at any time point tested in both wild‐type and MOPrKO mice compared to their respective vehicle groups. However, the cyclic tetrapeptideinduced significant respiratory depression sustained over the 2‐h test period in KOPrKO mice, compared to KOPrKO vehicle‐treated mice. Notably, the respiratory depressant effect of cyclo[Pro‐Sar‐Phe‐d‐Phe] in KOPrKO mice was significantly greater and longer lasting than that in morphine‐treated wild‐type mice at later time points (40–120 min).
To assess for potential rewarding or aversive effects without possible confounds related to compound distribution to the brain after peripheral administration, cyclo[Pro‐Sar‐Phe‐d‐Phe] was evaluated in a place conditioning paradigm after central administration. Six groups of mice demonstrated no significant differences in baseline chamber preference prior to place conditioning (Figure 7). Following a 2‐day place conditioning paradigm, mice conditioned with morphine demonstrated a significant place preference response whereas U50,488 produced significant conditioned place aversion (Figure 7). In contrast, mice conditioned with cyclo[Pro‐Sar‐Phe‐d‐Phe] (0.1, 1, or 10 nmol, i.c.v.) demonstrated neither a significant preference for, nor aversion to, the drug paired chamber.
FIGURE 7.
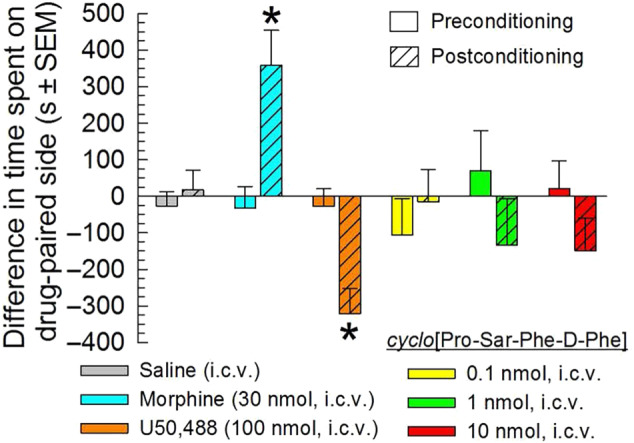
Evaluation of potential rewarding or aversive properties of cyclo[Pro‐Sar‐Phe‐d‐Phe]. cyclo[Pro‐Sar‐Phe‐d‐Phe] alone did not produce conditioned place preference or aversion. After determination of initial preconditioning preferences, C57BL/6J mice were place conditioned daily for 2 days with 0.9% saline (i.c.v.), morphine (30 nmol, i.c.v.), U50,488 (100 nmol, i.c.v.), or cyclo[Pro‐Sar‐Phe‐d‐Phe] (0.1–10 nmol, i.c.v.) with a counterbalanced design as described in Section 2. Data are presented as mean difference in time spent in the drug‐paired chamber ± SEM (n = 16–28). P < .05, significantly different from matching preconditioning preference; two‐way ANOVA with Sidak's multiple comparison post hoc test. Data from 109 animals are shown in this figure
3.4. Administration of cyclo[Pro‐Sar‐Phe‐d‐Phe] prevents reinstatement of extinguished morphine‐CPP
Following 4 days of morphine‐place conditioning (Figure 8a), mice demonstrated a significant preference for the morphine‐paired chamber (Figure 8b). Extinction of this preference was observed after repeated preference testing over 8 weeks following conditioning (Figure 8b, light grey bar). Mice were then pretreated once daily for 2 days with vehicle (50% DMSO in 0.9% saline, i.c.v.), cyclo[Pro‐Sar‐Phe‐d‐Phe] (0.01 or 0.03 nmol, i.c.v.), or a κ‐opioid receptor agonist (U50,488, 100 nmol, i.c.v.) as a positive control, and exposed to FSS or an additional cycle of morphine‐place conditioning (see protocol in Figure 8a). An additional subset of mice underwent direct reinstatement testing to assess if cyclo[Pro‐Sar‐Phe‐d‐Phe] alone had any effect on reinstatement. Mice pretreated with vehicle displayed significant reinstatement of morphine‐CPP after exposure to forced swimming or morphine‐place conditioning (Figure 8b). When pretreated with a dose and duration producing κ‐opioid receptor antagonist‐like activity (0.01 nmol, i.c.v., −2.5 h), cyclo[Pro‐Sar‐Phe‐d‐Phe] prevented stress‐induced reinstatement of morphine‐CPP. In contrast, when pretreated for 5 min, a period when the peptide exhibits agonist‐like activity in the antinociception assay, cyclo[Pro‐Sar‐Phe‐d‐Phe] prevented morphine‐induced reinstatement of morphine‐CPP at a dose of 0.03 nmol, much as did the κ‐opioid agonist U50,488 (100 nmol, i.c.v.; Figure 8b). The cyclo[Pro‐Sar‐Phe‐d‐Phe]‐induced prevention of morphine‐induced reinstatement of morphine‐CPP was dose dependent, as pretreatment with a lower dose (0.01 nmol, i.c.v.) did not have significant effects. Notably, treatment with cyclo[Pro‐Sar‐Phe‐d‐Phe] alone at this dose did not directly reinstate extinguished CPP (second rightmost bar, Figure 8b). This effect was similar to a vehicle‐treated reinstatement control (rightmost bar, Figure 8b), discounting any confounding effect of the cyclic tetrapeptide in this testing and supporting the conclusion that when administered alone, the effects of cyclo[Pro‐Sar‐Phe‐d‐Phe] were not different from those of vehicle.
An additional set of mice place conditioned 4 days with morphine subsequently demonstrated extinction (Figure 8c), and were then treated in week 9 with peripheral (i.p.) doses of vehicle, cyclo[Pro‐Sar‐Phe‐d‐Phe], or nor‐BNI (10 mg·kg−1) prior to 2 days of forced swimming. Mice pretreated with vehicle demonstrated significant reinstatement of morphine‐CPP after exposure to forced swimming (Figure 8c). Pretreatment with nor‐BNI (i.p., once 24 h prior to the start of the first day of forced swimming) or cyclo[Pro‐Sar‐Phe‐d‐Phe] (−2.5 h·day−1 prior to forced swimming) prevented stress‐induced reinstatement (Figure 8c, rightmost bars).
4. DISCUSSION AND CONCLUSIONS
Opioids remain the gold standard for treating pain, but use of established opioids such as morphine gives rise to serious liabilities and the risk of abuse and opioid addiction (Volkow & McLellan, 2016). The present data show that cyclo[Pro‐Sar‐Phe‐d‐Phe] produces initial opioid receptor agonist‐like effects with antagonism of κ‐opioid receptors apparent at later time points, demonstrating its potent antinociceptive properties without apparent adverse opioid side effects and preventing both morphine‐ and stress‐induced reinstatement of extinguished morphine‐conditioned place preference. A novel lead agent that can simultaneously manage pain while promoting abstinence from drug‐seeking behaviour may have therapeutic value in treating patients suffering from pain and/or an opioid use disorder.
The present results provide evidence that cyclo[Pro‐Sar‐Phe‐d‐Phe] is functioning through both μ‐ and κ‐opioid receptors, but how this activity is produced remains uncertain. Initial studies found that cyclo[Pro‐Sar‐Phe‐d‐Phe] showed poor affinity for opioid receptors in traditional radioligand binding assays (Ferracane et al., 2020). Molecular modelling in that study suggested that cyclo[Pro‐Sar‐Phe‐d‐Phe] adopts a variety of conformations depending on the polarity of its environment, with different conformations potentially exhibiting distinct mechanisms of action that could possibly account for the observed range of activity. It is feasible that cyclo[Pro‐Sar‐Phe‐d‐Phe] may owe its pharmacological activity to other less conventional mechanisms that are not captured in standard in vitro assays, such as active metabolites. Moreover, opioid receptor agonists activate a wide range of receptor mechanisms and downstream signalling pathways, some of which may contribute to the activity of this macrocyclic tetrapeptide. While beyond the scope of the current study, these possibilities are the subject of ongoing study.
cyclo[Pro‐Sar‐Phe‐d‐Phe] demonstrates a multifunctional μ‐opioid receptor agonist and κ‐receptor ligand profile in vivo, similar to that of its parent macrocyclic tetrapeptide CJ‐15,208. cyclo[Pro‐Sar‐Phe‐d‐Phe] displayed dose‐ and time‐dependent initial agonist‐like activity seen from 10 to 110 min after administration, with κ‐opioid receptor‐antagonist‐like effects detected subsequently 160, but not 270, min after administration. Notably, cyclo[Pro‐Sar‐Phe‐d‐Phe] produced more potent antinociception than morphine following i.c.v. administration, possibly as the result of synergistic agonist activity at μ‐ and κ‐opioid receptors, as antinociception was eliminated in KOPrKO mice pretreated with the μ‐opioid receptor antagonist β‐FNA. Multifunctional opioids appear to produce potent antinociception as a result of synergistic analgesic activity, as demonstrated by co‐administration of the μ‐opioid receptor agonist DAMGO and the κ‐opioid receptor agonist U50,488 that generated dose‐dependent, synergistic increases in antinociception (Sutters et al., 1990).
Multifunctional opioids possessing both agonist and antagonist activity have previously been reported that demonstrate reduced side effect profiles while producing potent analgesia. For instance, multifunctional μ‐opioid receptor agonists/ δ‐opioid receptor antagonists have demonstrated potent antinociception with less tolerance and dependence (Schiller, 2010). Likewise, the κ‐opioid receptor partial agonist/ μ‐opioid receptor antagonist nalbuphine also has demonstrated potent antinociception with reduced abuse liability (Miller, 1980; Schmidt et al., 1985). In addition, multifunctional opioids have shown therapeutic benefit against drug self‐administration. For instance, nalbuphine decreased cocaine intake in animals trained to self‐administer cocaine (Negus & Mello, 2002). This result is consistent with the present demonstration that a multifunctional opioid may have several therapeutic applications, specifically analgesia and counteracting substance abuse.
Notably, cyclo[Pro‐Sar‐Phe‐d‐Phe] lacks some of the characteristic side effects of established μ‐opioid receptor agonists, such as respiratory depression, hyperlocomotion, and conditioned place preference in rodents. Of particular interest, a handful of reports suggest that κ‐opioid receptor agonists may increase respiration (Haji & Takeda, 2001; Johnson, Kinney, & Wiegel, 2008) even as they suppress locomotion, counteracting the respiratory depressant effects of μ‐receptor agonists (Dosaka‐Akita et al., 1993). The present results with cyclo[Pro‐Sar‐Phe‐d‐Phe] are consistent with these findings. The absence of respiratory depression appears to be due to the κ‐receptor agonist‐like activity of cyclo[Pro‐Sar‐Phe‐d‐Phe], as significant, sustained respiratory depression was displayed by KOPrKO mice administered cyclo[Pro‐Sar‐Phe‐d‐Phe]. Notably, cyclo[Pro‐Sar‐Phe‐d‐Phe] did not produce CPP, in contrast to the established μ‐opioid receptor agonist morphine which produced robust CPP under the same conditions, consistent with morphine's reinforcing properties (Bardo et al., 1995). It should be acknowledged, however, that cyclo[Pro‐Sar‐Phe‐d‐Phe] may display reinforcing or aversive properties under different conditions; further testing (i.e., with prolonged place conditioning, or in other assays such as self‐administration) are needed to confirm these initial findings. Together, the combination of multifunctional μ‐ and κ‐opioid receptor agonist‐like activity in cyclo[Pro‐Sar‐Phe‐d‐Phe] seems to counter typical side effects produced by μ‐opioid receptor selective agonists, thereby improving the side effect profile of this peptide. Further work exploring the ratio of μ‐opioid receptor to κ‐opioid receptor agonism required to mitigate common liabilities of μ‐opioid receptor‐selective agonists is needed to better explore this relationship.
While our results suggest that the κ‐opioid receptor agonist activity produced by cyclo[Pro‐Sar‐Phe‐d‐Phe] can offset the μ‐opioid receptor agonist side effects, the same may also be true for μ‐opioid receptor agonist activity on κ‐opioid receptor‐associated side effects. κ‐opioid agonists are known to have contrasting effects to those of μ‐opioid agonists, with κ‐opioid receptor‐selective agonists (such asthe U50,488 used here as a control) suppressing coordinated locomotion (Dykstra, Gmerek, Winger, & Woods, 1987) and causing CPA (Shippenberg & Herz, 1986; Spetea et al., 2017). The μ‐opioid receptor agonist activity of cyclo[Pro‐Sar‐Phe‐d‐Phe] appears to offset the κ‐opioid receptor agonist‐mediated hypolocomotor effects, with MOPrKO mice displaying hypolocomotion when given cyclo[Pro‐Sar‐Phe‐d‐Phe]. The peptide also lacks effects on coordinated locomotion seen with the κ‐opioid receptor‐selective agonist U50,488 in the rotarod assay. Although possible, it does not seem likely that the reversal of μ‐opioid receptor agonist effects are due to the activity of cyclo[Pro‐Sar‐Phe‐d‐Phe] at a different, non‐opioid target, since the antinociceptive effects of cyclo[Pro‐Sar‐Phe‐d‐Phe] were fully abolished under conditions eliminating both μ‐ and κ‐opioid receptor activity.
cyclo[Pro‐Sar‐Phe‐d‐Phe] prevented both morphine‐ and stress‐induced reinstatement of morphine‐CPP in a time‐dependent manner. A short (5 min) pretreatment with cyclo[Pro‐Sar‐Phe‐d‐Phe] dose‐dependently ameliorated reinstatement of extinguished drug‐seeking behaviour induced by an additional round of morphine‐place conditioning. The ability to block acute drug‐induced reinstatement is characteristic of κ‐opioid receptor agonists (Freeman, Naylor, Prisinzano, & Woolverton, 2014; Shippenberg et al., 1996), and the present results are consistent with the interpretation that cyclo[Pro‐Sar‐Phe‐d‐Phe] produces κ‐opioid receptor agonist‐like effects as a component of its multifunctional activity. Additionally, cyclo[Pro‐Sar‐Phe‐d‐Phe] blocked stress‐induced reinstatement of morphine‐CPP after a pretreatment time (2.5 h) that takes advantage of its potent κ‐opioid receptor antagonist‐like properties. These latter results are consistent with those for the κ‐opioid receptor antagonists CJ‐15,208, [d‐Trp]‐CJ‐15,208, arodyn and zyklophin, each of which prevented stress‐induced reinstatement of extinguished cocaine‐CPP (Aldrich et al., 2013; Aldrich, Patkar, & McLaughlin, 2009; Carey, Borozny, Aldrich, & McLaughlin, 2007; Eans et al., 2013), as well as other κ‐opioid receptor antagonists that prevent stress‐induced drug‐seeking behaviour (Beardsley et al., 2005; for review, see Aldrich & McLaughlin, 2009). Moreover, application of a “functional κ‐opioid receptor antagonist” utilizing co‐administration of buprenorphine and naltrexone was shown to block the relapse of rats to cocaine and morphine in a CPP paradigm (Cordery et al., 2014), and heroin‐dependent patients treated for 12 weeks showed significant improvements in drug abstinence relative to patients treated only with naltrexone (Gerra, Fantoma, & Zaimovic, 2006). Furthermore, the effective prevention of stress‐induced relapse during the relatively short duration of cyclo[Pro‐Sar‐Phe‐d‐Phe] κ‐opioid receptor antagonism suggests long‐lasting antagonism of κ‐opioid receptors, such as that produced by nor‐BNI, is not necessary to prevent relapse as long as the κ‐opioid receptor antagonism coincides with the time of exposure to the stressor, protecting against the activation of κ‐opioid receptors by released dynorphin. The present results add to the growing body of evidence for the potential of κ‐opioid receptor antagonists as maintenance medications to prevent relapse in abstinent substance use disorder patients by preventing the stress‐induced activation of the endogenous κ‐opioid receptor system that is implicated in relapse risk and craving (Sinha et al., 2009; Wee & Koob, 2010).
While these initial results hint at the therapeutic potential for cyclo[Pro‐Sar‐Phe‐d‐Phe] and similar multifunctional compounds, additional translational studies of this macrocyclic tetrapeptide still remain to be carried out. Further work is needed to investigate efficacy against multiple types of nociception (e.g., in models of neuropathic pain, which were not evaluated here) and in a battery of additional models of addiction (such as rodent self‐administration). The ameliorating effects on reinstatement demonstrated here were time‐dependent, but were not evaluated in the context of important factors such as opioid withdrawal and the prolonged dysphoria over time that is associated with reinstatement to morphine‐seeking behaviour (Garcia‐Carmona, Baroja‐Mazo, Milanes, & Laorden, 2015). Further studies evaluating the effects of cyclo[Pro‐Sar‐Phe‐d‐Phe] treatment in drug‐responding animals still actively seeking drugs are also important, although beyond the scope of this initial evaluation. Still, the unmet medical need is clear. While medications exist to manage opioid misuse, the relapse rate after opioid detoxification and in individuals undergoing substance use disorder treatment is reportedly as high as 60%–80% (Chalana, Kundal, Gupta, & Malhari, 2016; McLellan, Lewis, O'Brien, & Kleber, 2000; Viswanathan et al., 2012), emphasizing the need for additional therapeutic options throughout the recovery process. The favourable antinociceptive potency of cyclo[Pro‐Sar‐Phe‐d‐Phe] in the warm‐water tail‐withdrawal assay and its ability to prevent relapse to morphine‐seeking behaviour after both stress‐ and drug‐induced reinstatement paradigms suggests that this macrocyclic tetrapeptide could have therapeutic potential for the treatment of both pain and opioid abuse, specifically for preventing relapse.
In conclusion, our results provide evidence that multifunctional opioids, particularly those with mixed, multifunctional μ‐opioid receptor agonist and κ‐opioid receptor ligand activity, offer promise as effective analgesics and potential treatments for substance abuse. cyclo[Pro‐Sar‐Phe‐d‐Phe]'s apparent lack of the negative side effects found with opioid receptor‐selective agonists makes it a valuable probe for better understanding the therapeutic benefit of multifunctional opioids combining μ‐ and κ‐opioid receptor activity.
AUTHOR CONTRIBUTIONS
J.V.A. and J.P.M. provided project planning. A.C.B.‐T., J.V.A., and J.P.M. designed the experiments. G.G.S., J.S.C., and M.J.F. synthesized cyclo[Pro‐Sar‐Phe‐d‐Phe]. A.C.B.‐T., L.L.W., S.O.E., H.M.S., and C.A.S. performed the experiments. A.C.B.‐T. and J.P.M. performed data analysis. A.C.B.‐T., M.J.F., J.V.A., and J.P.M. wrote the manuscript. All authors provided active and valuable feedback on the manuscript.
CONFLICT OF INTEREST
M.J.F. and J.V.A. are inventors on a patent application filed by the University of Florida on this compound. The remaining authors state they have no competing interests.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design and Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Data S1 Supporting information
ACKNOWLEDGEMENTS
This research was supported by grants from the National Institute on Drug Abuse R01 DA023924 and the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under Award W81XWH‐15‐1‐0452 (to J.V.A.) and W81XWH‐15‐1‐0464 (to J.P.M.). Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the Department of Defense.
Brice‐Tutt AC, Wilson LL, Eans SO, et al. Multifunctional opioid receptor agonism and antagonism by a novel macrocyclic tetrapeptide prevents reinstatement of morphine‐seeking behaviour. Br J Pharmacol. 2020;177:4209–4222. 10.1111/bph.15165
Present address: Michael J. Ferracane, Department of Chemistry, University of Redlands, Redlands, CA 92373, USA.
Note that amino acids are the l‐isomer unless otherwise specified; abbreviations for standard amino acids follow IUPAC‐IUB Joint Commission of Biochemical Nomenclature (Eur J Biochem, 1984, 138, 9–37).
DATA AVAILABILITY STATEMENT
All data needed to evaluate the conclusions in the paper are present in the paper and supporting information. Data related to this manuscript may be requested from the authors.
REFERENCES
- Aldrich, J. V. , & McLaughlin, J. P. (2009). Peptide kappa opioid receptor ligands: Potential for drug development. The AAPS Journal, 11(2), 312–322. 10.1208/s12248-009-9105-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich, J. V. , Patkar, K. A. , & McLaughlin, J. P. (2009). Zyklophin, a systemically active selective kappa opioid receptor peptide antagonist with short duration of action. Proceedings of the National Academy of Sciences, USA, 106(43), 18396–18401. 10.1073/pnas.0910180106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich, J. V. , Senadheera, S. N. , Ross, N. C. , Ganno, M. L. , Eans, S. O. , & McLaughlin, J. P. (2013). The macrocyclic peptide natural product CJ‐15,208 is orally active and prevents reinstatement of extinguished cocaine‐seeking behavior. Journal of Natural Products, 76(3), 433–438. 10.1021/np300697k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie, A. , Peters, J. A. , … Pawson, A. J. (2019). The Concise Guide to PHARMACOLOGY 2019/20: G protein‐coupled receptors. British Journal of Pharmacology, 176, S104–S106. [Google Scholar]
- Anand, J. P. , & Montgomery, D. (2018). Multifunctional opioid ligands In Jutkiewicz E. (Ed.), Delta opioid receptor pharmacology and therapeutic applications. Handbook of experimental pharmacology (Vol. 247) (pp. 21–51). [DOI] [PubMed] [Google Scholar]
- Armishaw, C. J. , Banerjee, J. , Ganno, M. L. , Reilley, K. J. , Eans, S. O. , Mizrachi, E. , … McLaughlin, J. P. (2013). Discovery of novel antinociceptive α‐conotoxin analogues from the direct in vivo screening of a synthetic mixture‐based combinatorial library. ACS Combinatorial Science, 15(3), 153–161. 10.1021/co300152x [DOI] [PubMed] [Google Scholar]
- Bardo, M. T. , Rowlett, J. K. , & Harris, M. J. (1995). Conditioned place preference using opiate and stimulant drugs: A meta‐analysis. Neuroscience & Biobehavioral Reviews, 19(1), 39–51. 10.1016/0149-7634(94)00021-R [DOI] [PubMed] [Google Scholar]
- Beardsley, P. M. , Howard, J. L. , Shelton, K. L. , & Carroll, F. I. (2005). Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine‐seeking induced by footshock stressors vs cocaine primes and its antidepressant‐like effects in rats. Psychopharmacology, 183(1), 118–126. 10.1007/s00213-005-0167-4 [DOI] [PubMed] [Google Scholar]
- Bruchas, M. R. , Land, B. B. , & Chavkin, C. (2010). The dynorphin/kappa opioid system as a modulator of stress‐induced and pro‐addictive behaviors. Brain Research, 1314, 44–55. 10.1016/j.brainres.2009.08.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buntin‐Mushock, C. , Phillip, L. , Moriyama, K. , & Palmer, P. P. (2005). Age‐dependent opioid escalation in chronic pain patients. Anesthesia & Analgesia, 100(6), 1740–1745. 10.1213/01.ANE.0000152191.29311.9B [DOI] [PubMed] [Google Scholar]
- Carey, A. N. , Borozny, K. , Aldrich, J. V. , & McLaughlin, J. P. (2007). Reinstatement of cocaine place‐conditioning prevented by the peptide kappa‐opioid receptor antagonist arodyn. European Journal of Pharmacology, 569(1–2), 84–89. 10.1016/j.ejphar.2007.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Behavioral Health Statistics and Quality . (2018). 2017 National Survey on Drug Use and Health: Detailed tables. Rockville, MD: Substance Abuse and Mental Health Services Administration.
- Chalana, H. , Kundal, T. , Gupta, V. , & Malhari, A. S. (2016). Predictors of relapse after inpatient opioid detoxification during 1‐year follow‐up. Journal of Addiction, 1–7, 7620860 10.1155/2016/7620860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder G., Castro D. C., Bruchas M. R., Scherrer G. (2018). Endogenous and Exogenous Opioids in Pain. Annual Review of Neuroscience, 41, (1), 453–473. 10.1146/annurev-neuro-080317-061522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordery, S. F. , Taverner, A. , Ridzwan, I. E. , Guy, R. H. , Delgado‐Charro, M. B. , Husbands, S. M. , & Bailey, C. P. (2014). A non‐rewarding, non‐aversive buprenorphine/naltrexone combination attenuates drug‐primed reinstatement to cocaine and morphine in rats in a conditioned place preference paradigm. Addiction Biology, 19, 575–586. [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Bond, R. A. , Spina, D. , Ahluwalia, A. , Alexander, S. P. , Giembycz, M. A. , … Lawrence, A. J. (2015). Experimental design and analysis and their reporting: New guidance for publication in BJP . British Journal of Pharmacology, 172(14), 3461–3471. 10.1111/bph.12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlhamer, J. , Lucas, J. , Zelaya, C. , Nahin, R. , Mackey, S. , DeBar, L. , … Helmick, C. (2018). Prevalence of chronic pain and high‐impact chronic pain among adults—United States, 2016. Morbidity and Mortality Weekly Report, 67(36), 1001–1006. 10.15585/mmwr.mm6736a2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darcq, E. , & Kieffer, B. L. (2018). Opioid receptors: Drivers to addiction? Nature Reviews Neuroscience, 19(8), 499–514. 10.1038/s41583-018-0028-x [DOI] [PubMed] [Google Scholar]
- Dosaka‐Akita, K. , Tortella, F. C. , Holaday, J. W. , & Long, J. B. (1993). The kappa opioid agonist U‐50,488 H antagonizes respiratory effects of mu opioid receptor agonists in conscious rats. Journal of Pharmacology and Experimental Therapeutics, 264(2), 631–637. 10.21236/ada263043 [DOI] [PubMed] [Google Scholar]
- Dykstra, L. A. , Gmerek, D. E. , Winger, G. A. I. L. , & Woods, J. H. (1987). Kappa opioids in rhesus monkeys. I. Diuresis, sedation, analgesia and discriminative stimulus effects. Journal of Pharmacology and Experimental Therapeutics, 242(2), 413–420. [PubMed] [Google Scholar]
- Eans, S. O. , Ganno, M. L. , Reilley, K. J. , Patkar, K. A. , Senadheera, S. N. , Aldrich, J. V. , & McLaughlin, J. P. (2013). The macrocyclic tetrapeptide [D‐Trp]CJ‐15,208 produces short‐acting κ opioid receptor antagonism in the CNS after oral administration. British Journal of Pharmacology, 169(2), 426–436. 10.1111/bph.12132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferracane, M. J. , Brice‐Tutt, A. C. , Coleman, J. , Simpson, G. G. , Wilson, L. , Eans, S. O. , … Aldrich, J. V. (2020). Design, synthesis, and characterization of the macrocyclic tetrapeptide cyclo[Pro‐Sar‐Phe‐D‐Phe]: A mixed opioid receptor agonist‐antagonist following oral administration. ACS Chemical Neuroscience, 11(9), 1324–1336. 10.1021/acschemneuro.0c00086 [DOI] [PubMed] [Google Scholar]
- Freeman, K. B. , Naylor, J. E. , Prisinzano, T. E. , & Woolverton, W. L. (2014). Assessment of the kappa opioid agonist, salvinorin A, as a punisher of drug self‐administration in monkeys. Psychopharmacology, 231(14), 2751–2758. 10.1007/s00213-014-3436-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Carmona, J. A. , Baroja‐Mazo, A. , Milanes, M. V. , & Laorden, M. L. (2015). Sex differences between CRF1 receptor deficient mice following naloxone‐precipitated morphine withdrawal in a conditioned place aversion paradigm: Implication of HPA axis. PLoS ONE, 10(4), 1–16, e0121125 10.1371/journal.pone.0121125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerra, G. , Fantoma, A. , & Zaimovic, A. (2006). Naltrexone and buprenorphine combination in the treatment of opioid dependence. Journal of Psychopharmacology, 20, 806–814. 10.1177/0269881106060835 [DOI] [PubMed] [Google Scholar]
- Graziane, N. M. , Polter, A. M. , Briand, L. A. , Pierce, R. C. , & Kauer, J. A. (2013). Kappa opioid receptors regulate stress‐induced cocaine seeking and synaptic plasticity. Neuron, 77(5), 942–954. 10.1016/j.neuron.2012.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haji, A. , & Takeda, R. (2001). Effects of a κ‐receptor agonist U‐50488 on bulbar respiratory neurons and its antagonistic action against the μ receptor‐induced respiratory depression in decerebrate cats. The Japanese Journal of Pharmacology, 87(4), 333–337. 10.1254/jjp.87.333 [DOI] [PubMed] [Google Scholar]
- Haley, T. J. , & McCormick, W. G. (1957). Pharmacological effects produced by intracerebral injection of drugs in the conscious mouse. British Journal of Pharmacology and Chemotherapy, 12(1), 12–15. 10.1111/j.1476-5381.1957.tb01354.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, R. , Disney, A. , Conibear, A. , Sutcliffe, K. , Dewey, W. , Husbands, S. , … Henderson, G. (2018). The novel μ‐opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. British Journal of Pharmacology, 175(13), 2653–2661. 10.1111/bph.14224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoot, M. R. , Sypek, E. I. , Reilley, K. J. , Carey, A. N. , Bidlack, J. M. , & McLaughlin, J. P. (2013). Inhibition of Gβγ‐subunit signaling potentiates morphine‐induced antinociception but not respiratory depression, constipation, locomotion, and reward. Behavioural Pharmacology, 24(2), 144–152. 10.1097/FBP.0b013e32835f3d2f [DOI] [PubMed] [Google Scholar]
- Huecker, M. R. , Azadfard, M. , & Leaming, J. M. (2019). Opioid addiction. StatPearls. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK448203/ [PubMed]
- Johnson, S. M. , Kinney, M. E. , & Wiegel, L. M. (2008). Inhibitory and excitatory effects of μ‐, δ‐, and κ‐opioid receptor activation on breathing in awake turtles, Trachemys scripta . American Journal of Physiology‐Regulatory, Integrative and Comparative Physiology, 295(5), R1599–R1612. [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , & Altman, D. G. (2010). Animal research: Reporting in vivo experiments: The ARRIVE guidelines. British Journal of Pharmacology, 160(7), 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko, M. C. , & Husbands, S. M. (2009). Effects of atypical κ‐opioid receptor agonists on intrathecal morphine‐induced itch and analgesia in primates. Journal of Pharmacology and Experimental Therapeutics, 328(1), 193–200. 10.1124/jpet.108.143925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath, J. C. , & Lilley, E. (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP . British Journal of Pharmacology, 172(13), 3189–3193. 10.1111/bph.12955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin, J. P. , Hill, K. P. , Jiang, Q. , Sebastian, A. , Archer, S. , & Bidlack, J. M. (1999). Nitrocinnamoyl and chlorocinnamoyl derivatives of dihydrocodeinone: in vivo and in vitro characterization of μ‐selective agonist and antagonist activity. Journal of Pharmacology and Experimental Therapeutics, 289(1), 304–311. [PubMed] [Google Scholar]
- McLellan, A. T. , Lewis, D. C. , O'Brien, C. P. , & Kleber, H. D. (2000). Drug dependence, a chronic medical illness: Implications for treatment, insurance, and outcomes evaluation. JAMA, 284(13), 1689–1695. 10.1001/jama.284.13.1689 [DOI] [PubMed] [Google Scholar]
- Miller, R. R. (1980). Evaluation of nalbuphine hydrochloride. American Journal of Hospital Pharmacy, 37(7), 942–949. [PubMed] [Google Scholar]
- Negus, S. S. , & Mello, N. K. (2002). Effects of μ‐opioid agonists on cocaine‐and food‐maintained responding and cocaine discrimination in rhesus monkeys: Role of μ‐agonist efficacy. Journal of Pharmacology and Experimental Therapeutics, 300(3), 1111–1121. 10.1124/jpet.300.3.1111 [DOI] [PubMed] [Google Scholar]
- Negus, S. S. , Schrode, K. , & Stevenson, G. W. (2008). Mu/kappa opioid interactions in rhesus monkeys: Implications for analgesia and abuse liability. Experimental and Clinical Psychopharmacology, 16(5), 386–399. 10.1037/a0013088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer, A. , Brantl, V. , Herz, A. , & Emrich, H. M. (1986). Psychotomimesis mediated by kappa opiate receptors. Science, 233(4765), 774–776. 10.1126/science.3016896 [DOI] [PubMed] [Google Scholar]
- Ross, N. C. , Reilley, K. J. , Murray, T. F. , Aldrich, J. V. , & McLaughlin, J. P. (2012). Novel opioid cyclic tetrapeptides: Trp isomers of CJ‐15,208 exhibit distinct opioid receptor agonism and short‐acting κ opioid receptor antagonism. British Journal of Pharmacology, 165(4b), 1097–1108. 10.1111/j.1476-5381.2011.01544.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller, P. W. (2010). Bi‐or multifunctional opioid peptide drugs. Life Sciences, 86(15–16), 598–603. 10.1016/j.lfs.2009.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, W. K. , Tam, S. W. , Shotzberger, G. S. , Smith, D. H. Jr. , Clark, R. , & Vernier, V. G. (1985). Nalbuphine. Drug and Alcohol Dependence, 14(3‐4), 339–362. 10.1016/0376-8716(85)90066-3 [DOI] [PubMed] [Google Scholar]
- Shippenberg, T. S. , & Herz, A. (1986). Differential effects of mu and kappa opioid systems on motivational processes. NIDA Research Monograph, 75, 563–566. [PubMed] [Google Scholar]
- Shippenberg, T. S. , LeFevour, A. , & Heidbreder, C. H. (1996). kappa‐Opioid receptor agonists prevent sensitization to the conditioned rewarding effects of cocaine. Journal of Pharmacology and Experimental Therapeutics, 276(2), 545–554. [PubMed] [Google Scholar]
- Sinha, R. , Fox, H. C. , Hong, K. A. , Bergquist, K. , Bhagwagar, Z. , & Siedlarz, K. M. (2009). Enhanced negative emotion and alcohol craving, and altered physiological responses following stress and cue exposure in alcohol dependent individuals. Neuropsychopharmacology, 34(5), 1198–1208. 10.1038/npp.2008.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spetea, M. , Eans, S. O. , Ganno, M. L. , Lantero, A. , Mairegger, M. , Toll, L. , … McLaughlin, J. P. (2017). Selective κ receptor partial agonist HS666 produces potent antinociception without inducing aversion after icv administration in mice. British Journal of Pharmacology, 174(15), 2444–2456. 10.1111/bph.13854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutters, K. A. , Miaskowski, C. , Taiwo, Y. O. , & Levine, J. D. (1990). Analgesic synergy and improved motor function produced by combinations of μ‐δ‐and μ‐κ‐opioids. Brain Research, 530(2), 290–294. 10.1016/0006-8993(90)91297-T [DOI] [PubMed] [Google Scholar]
- US Food and Drug Administration/Center for Drug Evaluation and Research . (2018). Anesthetic and Analgesic Drug Products Advisory Committee (AADPAC): FDA briefing document. Silver Spring, MD. https://www.fda.gov/advisory-committees/advisory-committee-calendar/updated-public-participation-information-october-11-2018-meeting-anesthetic-and-analgesic-drug#event-materials
- Viswanathan, M. , Golin, C. E. , Jones, C. D. , Ashok, M. , Blalock, S. J. , Wines, R. C. , … Lohr, K. N. (2012). Interventions to improve adherence to self‐administered medications for chronic diseases in the United States: A systematic review. Annals of Internal Medicine, 157(11), 785–795. 10.7326/0003-4819-157-11-201212040-00538 [DOI] [PubMed] [Google Scholar]
- Volkow, N. D. , & McLellan, A. T. (2016). Opioid abuse in chronic pain—Misconceptions and mitigation strategies. New England Journal of Medicine, 374(13), 1253–1263. 10.1056/NEJMra1507771 [DOI] [PubMed] [Google Scholar]
- Wee, S. , & Koob, G. F. (2010). The role of the dynorphin‐κ opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology, 210, 121–135. 10.1007/s00213-010-1825-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, J. T. , Christie, M. J. , & Manzoni, O. (2001). Cellular and synaptic adaptations mediating opioid dependence. Physiological Reviews, 81(1), 299–343. 10.1152/physrev.2001.81.1.299 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supporting information
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and supporting information. Data related to this manuscript may be requested from the authors.