

Abstract
Disorganized vessels in the tumor vasculature lead to impaired perfusion, resulting in reduced accessibility to immune cells and chemotherapeutic drugs. In the breast tumor–stroma interplay, paracrine factors such as interleukin-6 (IL-6) often facilitate disordered angiogenesis. We show here that epigenetic mechanisms regulate the crosstalk between IL-6 and vascular endothelial growth factor receptor 2 (VEGFR2) signaling pathways in myoepithelial (CD10+) and endothelial (CD31+, CD105+, CD146+, and CD133−) cells isolated from malignant and nonmalignant tissues of clinically characterized human breast tumors. Tumor endothelial (Endo-T) cells in 3D cultures exhibited higher VEGFR2 expression levels, accelerated migration, invasion, and disorganized sprout formation in response to elevated IL-6 levels secreted by tumor myoepithelial (Epi-T) cells. Constitutively, compared with normal endothelial (Endo-N) cells, Endo-T cells differentially expressed DNA methyltransferase isoforms and had increased levels of IL-6 signaling intermediates such as IL-6R and signal transducer and activator of transcription 3 (STAT3). Upon IL-6 treatment, Endo-N and Endo-T cells displayed altered expression of the DNA methyltransferase 1 (DNMT1) isoform. Mechanistic studies revealed that IL-6 induced proteasomal degradation of DNMT1, but not of DNMT3A and DNMT3B and subsequently led to promoter hypomethylation and expression/activation of VEGFR2. IL-6–induced VEGFR2 up-regulation was inhibited by overexpression of DNMT1. Transfection of a dominant-negative STAT3 mutant, but not of STAT1, abrogated VEGFR2 expression. Our results indicate that in the breast tumor microenvironment, IL-6 secreted from myoepithelial cells influences DNMT1 stability, induces the expression of VEGFR2 in endothelial cells via a promoter methylation–dependent mechanism, and leads to disordered angiogenesis.
Keywords: tumor microenvironment, breast cancer, IL-6, myoepithelial cells, DNA methyltransferase 1 (DNMT1), vascular endothelial growth factor receptor 2, angiogenesis, epigenetics, gene regulation interleukin 6 (IL-6), DNA methyltransferase, signal transduction, VEGFR2
Pathologically activated tissue microenvironment has been shown to shape breast tumor growth, progression, nature, evolution and response to therapy. Persistent proliferation, immune cell recruitment, and angiogenesis are the key determinants of tumor growth (1). In tumor ecosystem, secreted factors such as IL-6 significantly contribute to inflammation associated with tumor progression by eliciting angiogenic program in endothelial cells (2). Myoepithelial cells which surround the luminal epithelial cells are recognized as “natural tumor suppressor cells” and “gate keepers” (3, 4). Alterations in gene expression patterns in tumor-associated myoepithelial cells result in secretion of paracrine molecules including IL-6 and promote tumor growth (5).
In models of breast, renal, and cervical cancers, IL-6 activates JAK2/STAT3-dependent HIF-1α and leads to elevated VEGF expression (6–8). Cooperative effects of IL-6 with RANTES or VEGF results in aggressiveness of breast cancer cells, both in vitro and in vivo models (9). IL-6 activates STAT3 in tumor-associated endothelial cells, macrophages, and myeloid-derived suppressor cells to enhance their ability to express VEGF in a feed-forward loop which subsequently regulates tumor angiogenesis (10). Clinical studies have shown that neutralizing IL-6 in ovarian cancer subjects results in decreased VEGF expression, and further, xenograft experiments have shown reduced neovascularization upon blocking IL-6 effects (11). IL-6 induced endothelial proliferation and migration and is associated with Jagged1/angiopoietin-dependent defective coverage of pericytes, thus contributing to vessel perforation (12). Blocking of IL-6 results in restoration of pericyte coverage in vessels (12).
Epigenetic mechanisms including DNA methylation and histone acetylation are reported as key regulators of endothelial gene expression and functions including tumor induced neovascularization (13, 14). Earlier studies have shown that critical genes such as VEGF and its receptors, eNOS, MMPs, TMP, TSP, and RECK in maintaining angiogenic homeostasis are influenced by promoter DNA methylation. Inhibitors of DNA methyltransferases (DNMTs) exhibit significant angiostatic effects in various tumor models (14, 15). Our earlier studies have revealed IL-6 as potential modulator of DNMT isoforms and significantly altered epigenome of endothelial cells (16). Microarray analysis indicated IL-6–induced significant hypomethylation of VEGFR2 promoter in HUVECs. These findings led us to investigate the paracrine effects of IL-6 derived from CD10+ myoepithelial cells on endothelial cells (CD31+, CD105+; CD133−) in human breast tissue microenvironment. From clinically characterized breast tumors, we isolated four cell types: (a) myoepithelial cells from malignant tissues (Epi-T), (b) myoepithelial cells from nonmalignant tissues (Epi-N), (c) endothelial cells from malignant tissues (Endo-T), and (d) endothelial cells from nonmalignant tissues (Endo-N). In these cell types, we examined the impact of IL-6 on epigenetic control of VEGFR2 expression and its role in modulating normal and tumor associated angiogenesis.
Results
Tumor myoepithelial cells secrete increased IL-6 and VEGFA and show paracrine effects on endothelial cells to induce VEGFR2 expression and sprout formation
In HUVECs, IL-6 elevated STAT3Tyr-705 phosphorylation to nearly 6-fold by 6 h, which was sustained until 24 h and subsequently increased to 8-fold at 36 h. The total VEGFR2 levels were increased to 2-fold after 6 h and further increased to 8-fold by 36 h of IL-6 treatment (Fig. 1A). We then isolated myoepithelial and endothelial cells from the malignant and nonmalignant part of human breast tissues to examine paracrine effects of IL-6 between these cell types (Fig. S1A). Endothelial and myoepithelial cells were extensively characterized from four individuals diagnosed with infiltrative ductal carcinoma graded and staged as ER+/PR+/Her2/neu+ based on immunohistochemistry analysis. Endo-N and Endo-T cells were characterized based on abundance of CD31 (>95%) and CD105 (>95%) expression Fig. S1B. Further characterization of Endo-T cells revealed increased expression of endothelial cell specifc markers such as total and phospho-eNOS, VE-cadherin, and CD146 compared with Endo-N cells. Endo-T, Endo-N, and HUVEC cells were negative for cytokeratins, E-cadherin, and smooth muscle α actin precluding the contamination of nonEC cell types (Fig. S2). These cells when cultured for seven consecutive passages showed persistent expression of CD31 and CD105 (Fig. S3). However, we observed phenotype change in seventh passage cells. Endo-T cells did not express CD133 (Fig. S1B) and karyotype (Fig. S4) matched with Endo-N cells, suggesting these cells were not tumor derived and of host origin. Further, Epi-N and Epi-T cells were characterized by presence of CD10 (>95%) and keratin by flow cytomtery and immunoblotting, respectively (Fig. S1, C and D).
Figure 1.

Epi-T cells secrete increased IL-6 and induce VEGFR2 expression and sprout formation in endothelial cells. A, HUVECs were treated with IL-6 (25 ng/ml) for indicated time points and cells were lysed and processed for immunoblotting. Representative images of phospho- and total STAT3, VEGFR2, and β-actin are shown. Data are represented as -fold change with respect to untreated (U) control. B, IL-6 levels were estimated in the conditioned medium of Epi-N and Epi-T cells from study subjects and breast cancer cell lines. Levels of IL-6 are represented as pg/105cells. C, VEGFA levels were estimated in the conditioned medium of Epi-N and Epi-T cells along with breast cancer cell lines by immunoblotting assays. VEGFA levels were plotted as -fold change versus Epi-N levels. Representative blots are shown in Fig. S5. D, HUVECs were treated with conditioned medium of either Epi-T or Epi-N cells with or without IL-6R neutralizing antibody (2 µg/ml) for 36 h. Control cells were treated with nonimmune IgG. Upon indicated treatment, cells were lysed and processed for immunoblotting. Representative blots for phospho- and total STAT3, VEGFR2, and β-actin are shown. Data are represented as -fold change of phospho-/total VEGFR2 and STAT3 with respect to untreated control. Data corresponding to subject 1 is shown and data of other two subjects are provided as supporting images (Fig. S6). E, collagen 3D cultures were prepared from HUVECs and treated with conditioned medium of either Epi-T or Epi-N cells with or without IL-6R neutralizing antibody (2 µg/ml) and sorafenib (5 μm) for 24 h. F and G, Sprout outgrowth was monitored in 3D cultures after 24 h indicated treatment and data are represented as sprout number and sprout length. Scale bar = 50 μm. Statistical significance is represented by asterisk. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
IL-6 quantification from culture supernatants showed a 4-fold increase in IL-6 levels in Epi-T cells (656 ± 20 pg/105 cells) compared with Epi-N cells (138 ± 9 pg/105 cells) (Fig. 1B). Breast tumor cell lines such as MCF-10A, MCF-7, MDA-MB-231, and MDA-MB-468 also secreted IL-6, albeit at lower levels than primary cells. Immunoblotting analysis indicated significanlty elevated levels of VEGFA-121 (6- to 8-fold) in Epi-T cell–conditioned medium compared with that of Epi-N cells (Fig. 1C and Fig. S5). We further examined influence of Epi-N and Epi-T cell–conditioned medium on HUVECs for the effect on STAT3/VEGFR2 expression. Treatment with Epi-T cell–conditioned medium showed nearly 4-fold higher STAT3 phosphorylation. Further, IL-6R neutralizing antibody significantly reduced both phospho-STAT3Tyr-705 (nearly 75%) and total VEGFR2 and phospho-VEGFR2Tyr-1175 (nearly 50%) levels (Fig. 1D and Fig. S6) suggesting that IL-6 in epithelial cell conditioned medium is the key to induce STAT3 phsophorylation and VEGFR2 expression. We next prepared 3D spheriods from HUVECs and cultured them in presence of Epi-T cell conditioned media and compared with Epi-N cell medium. The results showed an increase in sprout length and numbers (Fig. 1E) when cultured in presence of Epi-T cell medium than Epi-N cell medium (p < 0.001). IL-6R antibody (p < 0.001) and VEGFR2 inhibitor (tyrosine kinase) sorafenib (p < 0.001) significantly reduced sprout numbers and length (Fig. 1, F and G), suggesting that IL-6 and VEGFA secreted by Epi-T cells induced VEGFR2-dependent angiogenesis.
IL-6–dependent VEGFR2 expression is a consequence of DNMT1 degradation
We further investigated the underlying mechanisms for IL-6–dependent VEGFR2 expression in endothelial cells. As has been observed in our earlier studies (16), treatment of HUVECs with IL-6 increased DNMT1 levels at early time points to nearly 4- to 5-fold, which was sustained up to 12 h and subsequently decreased to less than the control levels by 24 h (Fig. 2A). HUVECs when cultured with conditioned medium of Epi-N and Epi-T cells in presence of IL-6R neutralizing antibody, we observed a significant increase in DNMT1 expression. The effects of IL-6R antibody was more robust in endothelial cells treated with conditioned medium from Epi-T cells (p < 0.01) than Epi-N cells (Fig. 2B and Fig. S6). Therefore, Epi-T cell secretome containing IL-6 may be responsible for the changes in DNMT1 levels in endothelial cells in tumor microenvironment.
Figure 2.

IL-6 induces proteasomal degradation of DNMT1 and elevates VEGFR2 expression. A, HUVECs were treated with IL-6 (25 ng/ml) for indicated time points and cells were lysed and processed for immunoblotting. Representative images of DNMT1 and β-actin are shown. Data are represented as -fold change of untreated control. B, HUVECs were treated with conditioned medium from cultures of either with Epi-N or Epi-T cells in presence or absence of IL-6 R neutralizing antibody for 36 h and lysed. The blot was probed for DNMT1 and actin. Representative blots are shown and data are represented as -fold change. C, HUVECS were treated with IL-6 for 3 h followed by actinomycin D (10 µg/ml) for indicated time points. RNA was isolated and transcripts of DNMT1 and actin were measured by real time PCR. D, HUVECs were treated with IL-6, cycloheximide alone, or pretreated with IL-6 for 3 h and cycloheximide (10 μm) for indicated time points. Lysates were processed for immunoblotting and stained for DNMT1 and actin. E, HUVECs were treated with IL-6 in either absence or presence of MG132 (5 μm) for 36 h and lysates were processed for immunoblotting and stained for phospho- and total STAT3 and VEGFR2 and DNMT1. Statistical significance is represented as asterisk. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Treatment of HUVECs with actinomycin D, a transcription inhibitor, showed nearly 50% degradation of DNMT1 transcripts by 2 h. However, pretreatment of HUVECs with IL-6 for 3 h prior to blocking of transcription did not alter DNMT1 levels (Fig. 2C). Inhibition of protein translation by cycloheximide alone or in cells pretreated with IL-6 showed significant reduction in the levels of DNMT1 by 60 min (Fig. 2D). Taken together, these results indicate that regulation of DNMT1 expression by IL-6 is not at the level of transcription but probably a post-transcriptional or post-translational event.
To test whether increased expression of VEGFR2 in response to IL-6 is as a consequence of DNMT1 degradation, we cultured HUVECs in presence of MG132 (carbobenzoxyleu-leu-leucinal), a potent inhibitor of 16S subunit of proteasomal complex. MG132 restored IL-6–induced degradation of DNMT1 to nearly 50% and, at the same time, enhanced both total and phospho-VEGFR2 expression to normalized basal levels. Thus, IL-6 facilitated proteasomal degradation of DNMT1, which subsequently led to elevation of VEGFR2 expression (Fig. 2E).
Expression of DNMT isoforms are not uniform in normal and tumor endothelial cells and responded differently to IL-6
Next, we examined the constitutive levels of IL-6 signaling intermediates, DNMT isoforms, and total/phospho-VEGFR2 levels in Endo-N and Endo-T cells (Fig. 3). Endo-T cells showed consitutively elevated expression of IL-6 signaling intermediates such as IL-6R, total and phospho-STAT3Tyr-705. Interestingly, phosphorylation of STAT1Tyr-701 were significantly reduced in Endo-T cells compared with Endo-N cells. However, levels of total and phospho-STAT1Ser-727 did not alter between Endo-N and Endo-T cells. Levels of DNMT protein isoforms such as DNMT1 and DNMT3A were significantly elevated in Endo-T cells as opposed to DNMT3B which was down-regulated in Endo-T cells compared with its normal counterpart. Therefore, aberrant endothelial cell function in tumor microenvironment might be associated with dynamic reprogramming in methylome. Basal levels of phospho- and total VEGFR2 were also significantly increased in Endo-T cells.
Figure 3.

Constitutive levels of IL-6 signaling intermediates, DNMT isoforms, and VEGFR2 in Endo-N and Endo-T cells. Cell lysates from Endo-N (N) and Endo-T (T) from four subjects were grown under normal culture conditions, processed for immunoblotting, and stained for IL-6 signaling intermediates and DNMT isoforms. Fold change values in Endo-T cells with respect to normal counterpart is shown.
Next, we examined the differential response of Endo-N and Endo-T cells to IL-6 signaling pathway proteins (Fig. S7, A and B). IL-6 induced 2-fold increase in IL-6R levels in Endo-N cells but not in Endo-T cells. However, Endo-T cells showed constitutively higher levels of IL-6R. Kinetic analysis revealed both endothelial cell types responded to IL-6 treatment which led to induction of STAT3Tyr-705 phosphorylation at similar magnitude (2- to 3-fold increase). IL-6 also facilitated an increase in total STAT3 expression. Similar to HUVECs, Endo-N cells showed significant down-regulation of DNMT1 in response to chronic IL-6 treatment although the reduction in levels of DNMT1 varied among the three individual isolates. Endo-T cells expressed constitutively higher levels of DNMT1 and treatment with IL-6 induced its degradation. However, IL-6 effects on DNMT1 down-regulation was more prominent in Endo-N cells than in Endo-T cells. Both the cell types showed 2- to 3-fold increase in VEGFR2 expression upon IL-6 treatment.
IL-6–induced DNMT1 down-regulation and VEGFR2 expression are STAT3 dependent
In response to IL-6, we observed nearly 50% reduction in DNMT1 protein levels and an increase in nearly 2- to 3-fold of VEGFR2 levels by 36 h in HUVECs. These effects were abrogated upon treatment with IL-6R neutralizing antibody, where DNMT1 levels were restored and VEGFR2 levels remained similar to that of control (Fig. S8). Further, transient overexpression of WT STAT3 in HUVECs enhanced the effects of IL-6 on VEGFR2 expression and activation (Fig. 4A). We observed 50% reduction in DNMT1 levels in IL-6–treated cells in control group transfected with empty vector (p < 0.001) and, interestingly, cells overexpressing STAT3 showed further 10% reduction in DNMT1 levels in response to IL-6 (p < 0.001). IL-6–induced VEGFR2 levels in empty vector and STAT3 overexpressing cells remained unaltered. However, IL-6 increased the phosphorylation of VEGFR2Tyr-1175 to nearly 3-fold in cells overexpressing STAT3, suggesting IL-6 dependent STAT3 phosphorylation is the key determinant for increased activation of VEGFR2 (Fig. 4A). Role of STAT3 in regulating VEGFR2 expression was further confirmed by transfection of plasmid with dominant negative mutatnt of STAT3(Y705F). Overexpression of dominant negative STAT3 revealed significant abrogation of total and phospho-VEGFR2Tyr-1175 levels in presence or absence of IL-6 (Fig. 4B). As IL-6 has been demonstrated to activate STAT1 phosphorylation, we examined effects of dominant negative STAT1(Y701F) on VEGFR2 expression. We observed overexpression of STAT1(Y701F) did not influence expression of VEGFR2 in both presence and absence of IL-6, suggesting STAT3 but not STAT1 as a key regulator of VEGFR2 (Fig. 4C). Interestingly, overexpression of WT, dominant negative STAT3, and dominant negative STAT1 did not show any influence on DNMT1 levels. These results suggest that IL-6–induced reduction in DNMT1 levels were mediated by proteasomal degradation.
Figure 4.

IL-6 induces promoter DNA hypomethylation of VEGFR2 gene and increases its expression. A–D, HUVECs were transiently transfected with STAT3 pcDNA 3.1+ and pcDNA 3.1+ vector (A); dominant negative STAT3 (B); dominant negative STAT1 (C); and DNMT1, DNMT3A, DNMT3B, and pcDNA3.1+ plasmids (D) and further treated with IL-6 for 36 h (A, B, and C). Cells were lysed and processed for immunoblotting and stained for phospho- and total STAT3, VEGFR2, DNMT1, DNMT3A, and DNMT3B. Data are represented as -fold change of respective untreated pcDNA3 transfected cells. Statistical significance is denoted by asterisk. *, p < 0.05; **, p < 0.01; ***, p < 0.001. E, HUVECs were treated with 5-aza cytidine for 5 days and cell lysates were processed for immunoblotting. Membrane was probed for VEGFR2 and actin. F, DNA extracted from HUVECs treated with IL-6 (25 ng/ml) for 36 h, Endo-N and Endo-T cells isolated from three individuals were subjected to bisulfite DNA sequencing. Graphical representation for percentage of methylation of VEGFR2 promoter region is shown. Inset, representative electropherogram of chromosome 4q2 55083113–55083501 coordinates is shown.
DNMT1 down-regulation facilitated DNA hypomethylation of VEGFR2 promoter and led to elevated expression
As we observed differential expression of DNMT isoforms in tumor and normal endothelial cells, we tested influence of DNMT isoforms on VEGFR2 gene regulation and protein expression in HUVECs. DNMT1 overexpression completely abrogated the VEGFR2 expression independent of IL-6. However, VEGFR2 levels remained unaltered in DNMT3A and DNMT3B transfected cells under normal culture condition and in response to IL-6 treatment (Fig. 4D). This suggested DNMT1 but not DNMT3A or DNMT3B are essential for regulating VEGFR2 expression. Further, inhibition of DNMT isoforms by 5′-aza-deoxycytidine resulted in 3-fold increase in VEGFR2 expression (Fig. 4E). Therefore, blocking of DNMTs has similar effects to that of IL-6 on VEGFR2 expression.
Further, we determined whether IL-6 modulates DNA methylation levels of VEGFR2 promoter in HUVECs. The bisulfite DNA sequencing of the promoter region spanning –408 to +6 corresponding to 55083071–55083531 coordinates on chromosome 4 showed significant hypomethylation of VEGFR2 promoter region in response to IL-6 treatment for 36 h (Fig. 4F). Compared with basal levels of CpG methylation, a significant demethylation was observed at −54 position (80 versus 10–15%) and a complete demethylation at −75 (40 versus 0%) position relative to the transcription start site in the promoter region. Subsequently, we examined status of DNA promoter methylation of VEGFR2 gene in Endo-N and Endo-T cells. Endo-T cells from all three subjects showed significant constitutive hypomethylation when compared with controls in VEGFR2 promoter at −54 (45% versus 0; 42 versus 2%; and 30% versus 0) and −75 (15% versus 0; 12% versus 0; 10% versus 0) positions (Fig. 4F).
Tumor endothelial cells show constitutively higher rate of proliferation and did not further respond to IL-6
We observed Endo-T cells displayed increased expression of CD105/endoglin (Fig. S1B) and proliferated more robustly than Endo-N cells (Fig. S9). Cell cycle analysis showed that 60.23 ± 9.6% of Endo-T cells were in G0/G1 phase and 29.04 ± 6.63% in G2/M phase or proliferative phase constitutively, whereas 84.13 ± 3% of Endo-N cells were in G0/G1 phase and 4.28 ± 2.6% were in G2/M phase (Fig. S10, A–C). The percentages of cell cycle phase distribution for each subject are given in Table S1. Further, we tested the response of IL-6 in serum-depleted Endo-N and Endo-T cells. The 24 h serum starvation arrested 3–4% more cells in G0/G1 phase and 2–3% fewer cells in G2/M phase in both cell types (Fig. S10, A–C). Addition of IL-6 did not significantly effect the redistribution of cells into various phases of cell cycle both in Endo-N and Endo-T cells (Fig. S10, A–C). This indicated that the Endo-T cells possessed increased inherent ability to proliferate and IL-6 signaling does not induce abnormal proliferation.
IL-6 accelerates invasive and migratory property of tumor endothelial cells
In conventional scratch assays, Endo-T cells displayed inherent pro-migratory properties by closing the wound in 12 h and in presence of IL-6, the migration was complete in 6 h. The Endo-N cells required 18 h to close the wound and in response to IL-6, for these cells, complete migration was observed in 12 h (Fig. 5, A–C, and Fig. S11). Similar results were observed when HUVECs were treated with supernatants of myoepithelial cells. The HUVEC treated with Epi-N cell culture supernatant for 12 h showed 80% of gap closure which was significantly inhibited upon inclusion of IL-6R neutralizing antibody. When treated with Epi-T cell supernatant, complete wound closure was observed within 12 h and these effects were inhibited in presence of IL-6R neutralizing antibody (Fig. S12). This indicated IL-6 secreted from the Epi-T cell–facilitated endothelial cell migration.
Figure 5.
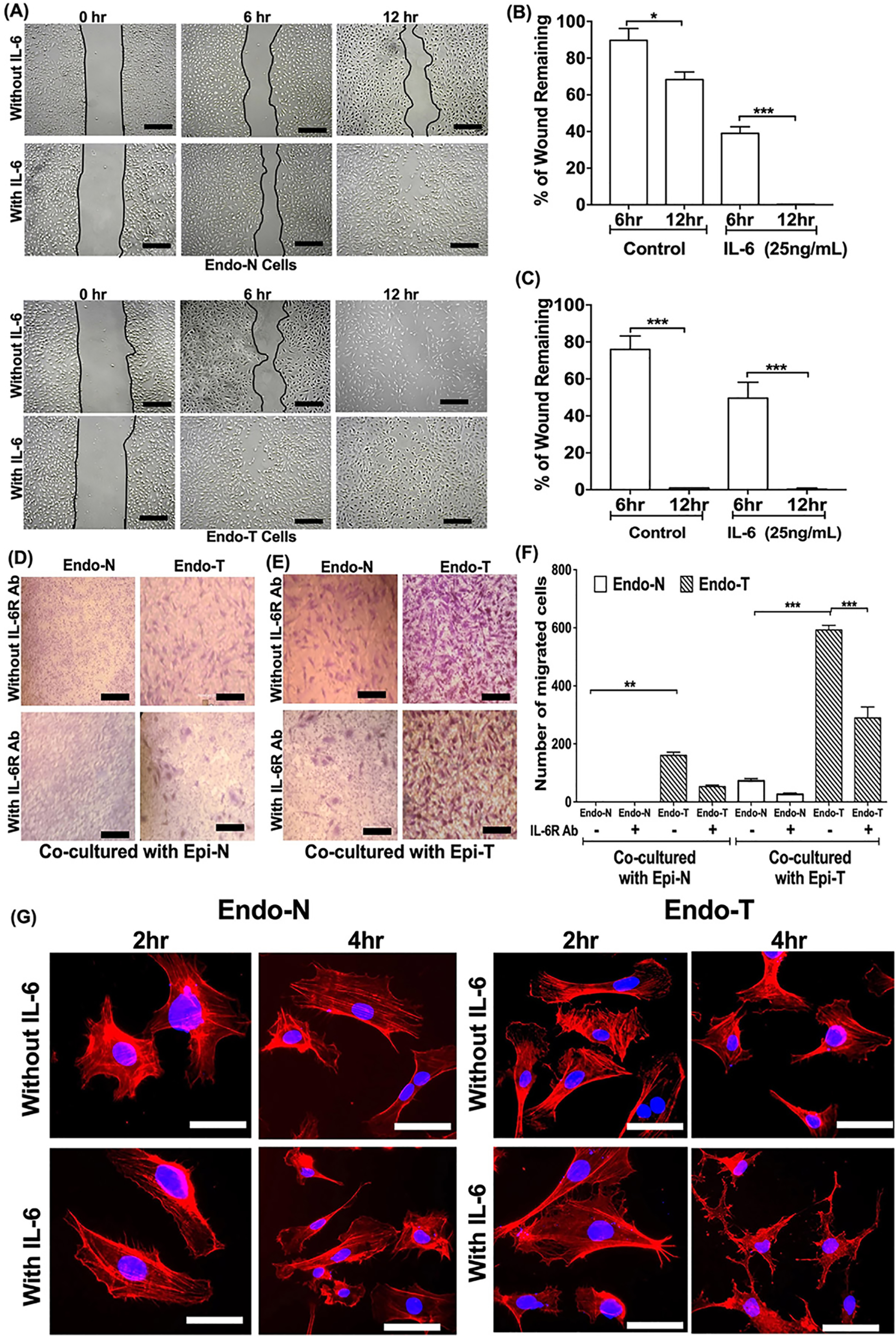
IL-6 accelerates migration and invasive ability of tumor endothelial cells. A, Endo-N and Endo-T cells were grown under normal culture conditions and monolayers were scratch-wounded and subsequently treated with or without IL-6 for indicated time points. Migration of cells into the wound area was monitored. Bold line indicates wound closure. Scale =100 μm. B and C, data are represented as percentage wound area for Endo-N and Endo-T separately. D and E, the Endo-N and Endo-T were co-cultured with Epi-N and Epi-T with or without IL-6R neutralizing antibody. F, the migrated cells were stained with crystal violet and counted and number of migrated cells were plotted. Statistical significance is denoted as asterisk. *, p < 0.1; **, p < 0.01; ***, p < 0.001. G, the Endo-N and Endo-T cells were treated with IL-6 for 2 and 4 h. Cells were fixed and stained with actin phalloidin. Scale = 25 1 μm.
To examine the invasive properties, endothelial cell types were co-cultured with epithelial cells with or without IL-6R neutralizing antibody. In transwell invasion assays, the cells were co-cultured in four different combinations as indicated in Fig. 5, D and E. The Endo-N cells when co-cultured with Epi-N derived from the same patient did not invade the membrane and IL-6R neutralizing antibody did not show any effect. The Endo-N cells when co-cultured with Epi-T cells showed a 75% increase in invasion and the percentage of migration was decreased to 26% in presence of IL-6R neutralizing antibody (Fig. 5, D and F). The Endo-T cells showed increased invasion (140%) when cultured with Epi-N cells and in presence of IL-6R neutralizing antibody, 53% inhibition of invasion was observed. Interestingly, in the Endo-T cells cultured with Epi-T, the invasive ability was significantly higher (600%) and IL-6R neutralizing antibody decreased it to 290% (Fig. 5, E and F). This indicated that the IL-6 secreted by Epi-T cells induced invasiveness of endothelial cells in tumor environment.
Further, we examined cytoskeletal changes in IL-6–treated endothelial cell types. Actin phalloidin staining showed that Endo-T cells possessed constitutively higher number of cells with lamellipodia than Endo-N cells which correlated with the invasion assay. In response to IL-6, Endo-N cells formed lamellipodia, which is a characteristic migratory phenotype. On the other hand, IL-6 induced stellate phenotype in the Endo-T cells in 6 h, facilitating an aggressive migratory behavior (Fig. 5G).
Tumor endothelial cells failed to make intact sprouts in response to IL-6
We then tested the angiogenic behavior of endothelial cell types in 3D culture/sprout assays and Matrigel tube-forming assays. Endo-T cells showed inherently formed higher number of sprouts (2-fold) and with increased length (1.5-fold) than Endo-N cells in the absence of any stimulant. Upon IL-6 treatment, Endo-N cells formed 3-fold increase in number of sprouts compared with untreated spheroids and was completely inhibited in presence of IL-6R neutralizing antibody. In response to IL-6, the tumor endothelial cells failed to form intact sprouts which was associated with the deflection of cells from core indicating their invasive behavior (Fig. 6, A and B). The addition of IL-6R neutralizing antibody reversed the effect of IL-6 on tumor endothelial cells and showed formation of rudimentary sprouts. The spheriods made from Endo-T cells when treated with bFGF resulted in significantly enhanced and intact sprouts, unlike IL-6, which showed pro-invasive phenotype (Fig. S13).
Figure 6.

IL-6 induces aberrant angiogenesis in tumor-derived endothelial cells. A and C, Endo-N and Endo-T cells were cultured in either (A) collagen spheroids or (C) Matrigel in presence or absence of IL-6 and IL-6R neutralizing antibody (2 µg/ml). Sprout outgrowth from spheroids was evaluated after 24 h in culture. Representative images of whole spheroids are shown. Scale = 50 μm. B, histograms show morphometric analysis of spheroids for total sprout number and total sprout length. Statistical significance is denoted as asterisk. *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, representative images of tubular formation at 6 h after the indicated treatment is shown. The tubular formation of endothelial cells was monitored for every 2 h. D, data are represented as number of intact tubes in HUVEC, Endo-N, and Endo-T.
In Matrigel tube-forming assays, we observed Endo-T cells started rearranging themselves without any stimulation to form tube like structures by 2 h, indicating inherent reprogramming of Endo-T cells leading to angiogenic switch. Both Endo-N cells and HUVECs showed 2.5-fold increased number of tube formation in response to IL-6 treatment which was abrogated by the addition of IL-6R neutralizing antibody. In response to IL-6, Endo-T cells lost their ability to form intact hexagonal tubes and IL-6R neutralizing antibody resensitized tube formation in these cells (Fig. 6, C and D, and Fig. S14). This clearly suggested that IL-6 might be one of the potential molecules to induce disorganized vessels in the tumor microenvironment.
IL-6–induced abnormal angiogenesis is normalized by VEGFR2 blocking antibody
Because IL-6 induces VEGFR2 up-regulation and aberrant angiogenesis (Fig. 6, A and B), we tested the influence of VEGFR2 antibody in spheriod models. IL-6 effects on angiogenesis in HUVECs were inhibited by addition of VEGFR2 blocking antibody in dose-dependent manner (Fig. 7, A and D). Endo-N cells showed 3-fold increase in sprout numbers and length in response to IL-6 (Fig. 7, B and D), and VEGFR2 antibody at 1.5 μg/ml was sufficient to block the IL-6 effects. In contrast, the Endo-T cells formed higher number of sprouts constitutively (3-fold increase versus normal) with increased length (4-fold increase). In response to IL-6, the tumor-derived endothelial cells failed to form the additional sprouts and deflected away from the spheroid core (Fig. 7, C and D). The VEGFR2 antibody concentrations at 1.0 μg/ml and 1.5 μg/ml were able to restore the sprout formation in Endo-T cells in presence of IL-6, and further increase in the concentration of VEGFR2 antibody (2.0 μg/ml) deteriorated the spheroid architecture. Taken together, this indicated excess VEGFR2 facilitates disorganized angiogenesis as a functional consequence of IL-6 effects.
Figure 7.

IL-6 induced aberrant sprout formation in tumor endothelial cells is abrogated by VEGFR2 antibody. A–C, HUVEC (A), Endo-N (B), and Endo-T (C) were cultured in spheroids and embedded in collagen gels in presence or absence of IL-6 along with indicated concentrations of either nonimmune IgG or anti-VEGFR2 antibody. Sprout outgrowth from spheroids was evaluated after 24 h in culture. Representative images of intact spheroids are shown. Scale = 50 μm. D, histograms show morphometric analysis of spheroids for total sprout number. Statistical significance for the effects of VEGFR2 antibody on IL-6 induced sprout formation in Endo-N cells is shown as # p < 0.05 and restoration of sprouts in Endo-T cells is represented as an asterisk. *, p < 0.05.
Discussion
Our study deciphered key molecules and mechanisms that cause disordered angiogenesis in breast tumors. Paracrine signaling and interrelationship between IL-6 and VEGFR2 in myoepithelial and endothelial cells in human breast tumors were responsible for epigenetic control of VEGFR2 gene and its contribution toward disordered angiogenesis. We observed that tumor and normal endothelial cells show (a) significant differences in their biological behaviors and in response to IL-6, (b) differential expression of IL-6 intermediates and DNMT isoforms, (c) distinct patterns of promoter DNA methylation of VEGFR2 gene and increased expression as a consequence of DNMT1 proteasomal degradation. Subsequently, IL-6 impelled migratory and invasive properties of tumor-associated endothelial cells and facilitated disorganized sprouting which was restored upon blocking of VEGFR2 (Fig. S16).
Various cellular and molecular mechanisms have been attributed for inducing neovascularization in tumors and these include (a) classic angiogenic switch, (b) vasculogenic mimicry, and (c) recruitment of progenitor endothelial cells into the tumors (17). Vasculogenic mimicry–associated breast tumor cells were characterized by CD31 and CD133 expression (18). Akino et al. (19), using human renal carcinoma models, demonstrated genetic instability of tumor endothelial cells which were characterized by aneuploidy compared with diploid nature of normal cells. In xenograft mouse models, endothelial cells which were CD133+ displayed abnormal chromosome numbers than that of cells which were CD133−, suggesting that the stem cell–derived endothelial cells were associated with aneuploidy. However, Endo-T cells used in our study model did not express CD133 and showed unaltered karyotype, suggesting that these cells were not as a consequence of either vasculogenic mimicry or because of recruitment of progenitor endothelial cells and represented pools of pre-existing host capillaries. Further, increased expression of CD105/endoglin in Endo-T cells was in agreement with earlier studies (20).
Promoter DNA methylome changes have been significantly correlated with dynamic gene expression in various cancers (21). Endothelial cells isolated from malignant and nonmalignant parts of prostate tumors showed distinct DNA methylation patterns in genes such as AREG, JMY, EPB41, GMNN, and FAM53C, which are involved in angiogenesis (22). Endo-T cells showed varied expression of DNMT isoforms, and in all subjects these cells showed significant promoter DNA hypomethylation that was associated with elevated VEGFR2 expression. Bioinformatics analysis of VEGFR2 promoter revealed putative regulatory elements for binding transcription factors such as STAT1, STAT3, EGR1, EGR2, AP2β, SP1, SP2, RUNX1, FOXA1, FOXC2, MYC GATA2, E2F, YY1, USF, EGR1, EGR2, and EGR3 (Fig. S15). Earlier studies have reported binding and interaction of GATA2, E2F, and SP1 necessary for transcriptional activation of VEGFR2 during angiogenesis (23–26). Further, expression of dominant negative STAT3 mutant reduced expression of VEGFR2, suggesting STAT3 as one of the critical transcriptional factors that regulate VEGFR2. VEGFR2 is one of the key regulators of physiological and pathological angiogenesis and VEGFR2 overexpression was demonstrated in solid tumors of lung, colon, liver, stomach, and breast tissue (27). Previous studies have addressed various mechanisms to support epigenetic regulation of VEGFR2. Xu et al. (28) have demonstrated hypomethylation of VEGFR2 promoter was because of increased adenosine levels and knockdown of adenosine kinase led to accumulation of intracellular adenosine. This effect was attributed to increased endothelial proliferation, migration, and angiogenesis. In mouse model of ischemia, it was reported that VEGFR2/Akt/eNOS signaling was down-regulated because of physical interaction of methyl CpG binding domain protein 2 (MBD2) at the promoter sequences of VEGFR2 gene (29). Authors demonstrated MBD2 knockdown in HUVECs resulted in increased VEGFR2 expression and led to enhanced proliferation, migration, and angiogenesis (29). We hypothesize that IL-6–induced DNMT1 degradation and IL-6/STAT3-dependent VEGFR2 expression are two independent processes as a consequence of elevated IL-6 levels in tumor microenvironment. However, IL-6–induced reduction in DNMT1 levels might lead to promoter DNA hypomethylation of VEGFR2, thus facilitating STAT3 occupancy to activate VEGFR2 expression. However, further promoter characterization using reporter assays and CHIP assays are warranted to understand interplay between DNMT1 and STAT3 during IL-6–mediated regulation of VEGFR2.
IL-6 is the key determinant of tumor microenvironment and contributes to tumorigenesis. For example, upper gastrointestinal tract tumors showed IL-6–mediated crosstalk between tumor cells and cancer-associated fibroblasts which led to fibroblast activation and promoted tumor cell growth (30). Authors also showed the suppression of tumor growth in organotypic models by blocking IL-6R with tocilizumab and pharmacological inhibition of STAT3/MEK1/2 (30). In preclinical mucoepidermoid carcinoma, inhibition of IL-6R by tocilizumab resulted in decreased VEGF production which led to reduced tumor microvessel density and, further, sensitized the cells for cisplatin and paclitaxel cytotoxicity (31). Using breast tumor models, Bharti et al. (32) showed inhibition of IL-6 signaling increased monoamino oxidase-A (MAO-A) expression which resulted in decreased angiogenesis. Further, treatment with 5-azacytidine induced expression of MAO-A and modulated IL-6–dependent angiogenesis which was associated with VEGF expression. We show that IL-6 accelerated migratory behavior and led to disorganized sprouts in Endo-T cells which were characterized by invasive phenotype of cells deflecting from core of spheroids. Both IL-6R and VEGFR2 neutralizing antibodies restored these processes.
Over the years, studies have demonstrated tumor-associated vessel patterns as complex, nonhierarchical, proangiogenic, and disorganized because of reprogramming of tumor endothelial cells (33). The process of vessel normalization in solid tumors has been demonstrated to improve oxygen levels, drug delivery, and immune cell infiltration facilitating efficient utilization of anti-angiogenic therapies (34). Inefficiency of anti-VEGF therapy in cancer has been correlated to elevated IL-6 levels. Obese breast cancer subjects harboring elevated systemic IL-6 levels failed to respond for bevacizumab. Mouse experiments showed administration of metformin reduced IL-6 levels and subsequently restored tumor sensitivity to anti-VEGF therapy (35). This prompted us to examine whether blocking IL-6–induced VEGFR2 improves sprout formation in Endo-T cells. Upon titrating anti-VEGFR2 antibodies, we observed restoration of intact sprout formation even in the presence of IL-6 in Endo-T cells.
Conclusion
For the first time, our study shows interplay between tumor myoepithelial and endothelial cells mediated by IL-6 resulting in DNMT1 instability and increased VEGFR2 expression and disorganized sprout formation in breast tumor microenvironment. Hence, our study suggests therapies targeting both IL-6 and VEGFR2 signaling axis in combination might ameliorate anti-angiogenic therapies resulting in better clinical outcome.
Experimental Procedures
Cell lines, chemicals, reagents, and plasmids
Breast cancer cell lines (MCF-7, MDA-MB-231, MDA-MB-468, and MCF-10A) were procured from American Type Culture Collection (ATCC) and cultured following ATCC instructions. Human umbilical vein endothelial cells (HUVECs) were cultured in endothelial cell growth medium (ECGM) (PromoCell, Germany) with growth supplements as described earlier (16). Recombinant IL-6, SYBR Green, and Lipofectamine transfection kit were purchased from Invitrogen. Human IL-6 ELISA kit was purchased from BioLegend (USA). Actin-phalloidin dye was purchased from Cytoskeleton, Inc. (USA). DAPI and ECL substrate kit were from Thermo Fisher Scientific. Collagen R solution was procured from Serva Electrophoresis (Germany), basic fibroblast growth factor (bFGF) from R&D Systems (USA) and Matrigel was purchased from Corning (USA). A list of antibodies and plasmids along with vendor details and RRID is provided as Table S2.
Human tissues
The study was approved by Institutional Ethics Committee, Kasturba Medical College, Manipal Academy of Higher Education, Manipal, in accordance with the declaration of Helsinki and Indian Council of Medical Research (ICMR), Government of India, ethical guidelines. The breast tissues were collected after surgical removal from the patients with prior informed and written consent.
Isolation, separation, and maintenance of primary cells
The breast tissue at the site of tumor (malignant part) and minimum 5 cm away from the tumor (nonmalignant part) were processed separately. The tissues were minced into 1-mm3 pieces and digested using collagenase at 37°C for 1 h and passed through 100-μm cell strainer to obtain single cell suspension. The cells were grown on gelatin precoated plates with ECGM until it reached 80% confluency. The endothelial cells were separated from the stromal cells using EasySepTM magnet, EasySepTM Human PE positive selection kit (StemCell Technologies) and PE mouse anti-human CD31. Endothelial cells were characterized using PE mouse anti-human CD31 using flow cytometry and VEGFR2 by immunoblotting. The cells were used for two to six passages for all the experiments and were continuously monitored for the stable expression of CD31 and CD105 by flow cytometry. The myoepithelial cells were isolated from the same individuals and characterized based on CD10 and cytokeratins expression using flow cytometry and Western blotting, respectively. The isolated cells were grown and maintained in Mammary Epithelial Cell Expansion Medium (MECEM, Himedia, India). The cells between two and four passages were used for the experiments. Endothelial cells isolated from all four individuals were considered for examining constitutive levels of IL-6 signaling molecules and cells from three individuals were used for functional assays.
Immunoblotting
Endothelial cells seeded onto gelatin precoated plates were subjected to different conditions as indicated in figure legends and lysed using RIPA buffer (50 mm Tris, pH 7.4, 150 mm sodium chloride, 5 mm EGTA, 0.1% SDS, 1% sodium deoxycholate, 1% Nonidet P-40, 10 mm sodium pyrophosphate, 0.5 mm sodium fluoride, 1 mm PMSF, 100 μm sodium vanadate, and 1× protease inhibitor mixture). Proteins resolved on SDS-PAGE gels were transferred onto nitrocellulose membrane (GE Healthcare). Prior to hybridization with primary and secondary antibodies, nonspecific binding was blocked with 5% BSA (The Jackson Laboratory). The substrate Enhanced Chemiluminescence (ECL) was used to detect immunoreactive proteins. The images were captured in Image-Quant LAS4000 (GE Healthcare) and intensity of expression were quantified using ImageJ (version 1.49) densitometry software.
Transfection
HUVECs (1 × 105/ml) were transfected with 1.0 μg/ml of either STAT3-pcDNA3/Myc-DNMT1, pcDNA3/Myc-DNMT3A, pcDNA3/Myc-DNMT3B, pCDNA3-Empty, STAT3DN.Ubc.GFP, pLV-Y701F-STAT1 or pLVX-puro using Lipofectamine LTX 3000 and Plus Reagent for 8 h and processed for further experiments. The plasmids were procured from Addgene.
Bisulfite sequencing
DNA extracted from cells were subjected to bisulfite conversion using EZ DNA methylation kit (Zymo Research). VEGFR2 bisulfite primers (Forward, GATTTAGTGTAGGGTGGGAG; Reverse, ATAACTCCAAACTACTACAAATTCTC) were used to amplify the region 55083071–55083531 coordinates on chromosome 4. DNA sequencing was performed using big dye terminator kit in 3130 Genetic Analyzer (Applied Biosystems, USA) according to manufacturer's instructions. The percentage of methylation at each CpG site was calculated by comparing the peak height of cytosine with sum of peak heights of the cytosine and thymine.
Karyotyping
Endo-T and Endo-N cells were grown under normal culture conditions and were treated with 0.01% colchicine for 30 min at 37°C and 5% CO2. Harvested cell pellet was resuspended in KCl (0.075M) and incubated for 25 min at room temperature. A fixative (3:1 ratio of methanol:acetic acid) was added and incubated for 2 min followed by centrifugation for 5 min at 1000 rpm and pellet was resuspended in fixative and incubated overnight at 4°C. The cell suspension was dropped on glass slides at 50°C. The cells were then allowed to dry and stained with 4% Giemsa. More than 30 metaphases were analyzed using Ikaros software (MetaSystems Hard & Software, Germany).
Cell cycle analysis
The cells were harvested by trypsinization, washed twice with 1× PBS, and fixed using 70% chilled ethanol. Fixative was removed by centrifuging and cells were washed with PBS. The pellet was treated with RNase (10 mg/ml) and after 1–2 h, cells were suspended in 500 μl of 1× PBS and 10 μl of propidium iodide (1 mg/ml) was added and incubated in ice for 30 min. The amount of DNA stained was analyzed using flow cytometry.
Quantitative real time PCR
Total RNA was isolated and purified from HUVECs using TRI reagent (MRC, Cincinnati, OH, USA). Complementary cDNA was synthesized from 500 ng of purified RNA using high-capacity cDNA reverse transcription kit (Applied Biosystems, USA) according to supplier's protocol. Real time PCR was carried out with the use of SYBR Green PCR Master Mix using following primers: 5′-GACGACATGGAGAAAATCTG-3′ and 5′-ATGATCTGGGTCATCTTCTC-3′ for β-actin and 5′-CAGAGAACGAGTTGCTAGACC-3′ and 5′-GGGTGTTGGTTCTTTGGTTTG-3′ for DNMT1 in 7500 Fast Real-Time PCR System (Applied Biosystems). The relative quantification of target gene expression was calculated after normalizing with actin using comparative cycle threshold method.
Migration assay
Endothelial cells were seeded at the density of 80,000 cells/ml per well in a 12-well plate and grown up to 80% confluence. A scratch was made using 20 μl tip to simulate migration. The cells were monitored and images were captured for every 2 h.
Invasion assay
In collagen precoated Transwell inserts (pore size: 8 μm, diameter: 24 mm, Corning), endothelial cells (1 × 104 cells/well) were seeded on top and myoepithelial cells (1 × 105 cells/well) were seeded in the bottom along with various combinations of treatments as indicated in figures. Invasion of cells under different conditions was measured by staining underneath surface of Transwell with crystal violet (0.2% in methanol).
3D collagen culture
Endothelial cells were suspended in the ECGM containing 20% methyl cellulose and cell suspension was then distributed as drops containing 500–1000 cells on a Petri dish and incubated in inverted position overnight. The spheroids were collected next day, embedded on collagen matrix in 48-well plate and analyzed for sprout formation. The number and length of the sprouts formed after 24 h were analyzed using the AnalySIS software.
Tube formation assay
Assays were carried out in 96-well plates coated with 60 μl of Matrigel for 1 h followed by plating of endothelial cells at the density of 2 × 104 cells/well. Cells were stimulated and incubated for 6 h. Rearrangement of cells was monitored every 2 h until 6 h. Number of tubes formed were manually counted and kinetics of tube formation was analyzed.
ELISA
Cells were seeded at the density of 1 × 105 cells/ml in a 6-well plate in triplicate, cultured for 24 h; supernatants were collected and IL-6 levels were measured using ELISA (BioLegend) as per manufacturer's instructions. The absorbance of each well after the reaction was read at 450 nm using Varioskan Flash (Thermo Scientific).
Immunofluorescence
Endo-N and Endo-T cells were seeded on coverslips, treated with IL-6 (25 ng/ml) for indicated time points, fixed using 4% paraformaldehyde, and stained with actin-phalloidin (100 nm) and DAPI (1 μg/ml). The images were captured using Olympus 1×-51 inverted fluorescence microscope with EMC2 Rolera camera (Q imaging, Canada) and analyzed using Image Pro Plus software 7.0.
Statistical analysis
All the experiments were repeated thrice at independent occasions. The data were represented as mean ± S.D. Data were analyzed using either two-tailed Student's t test or one-way analysis of variance (ANOVA) followed by post hoc Bonferroni's multiple comparison tests using GraphPad Prism 7. A p value less than 0.05 was considered as statistically significant.
Data availability
All data are provided in manuscript.
Supplementary Material
Acknowledgments
We thank Technology Information Forecasting and Assessment Council-center of Relevance and Excellence in Pharmacogenomics (TIFAC-CORE) and Manipal Academy of Higher Education Manipal for infrastructure support. We also thank Prof. Meenhard Herlyn, The Wistar Institute Cancer Center, Philadelphia, PA, USA, and Dr. Kishore Parsa, Dr. Reddy's Institute for Life Sciences, Hyderabad for gifting anti-CD146 and anti–phospho-STAT1 antibodies, respectively; Prof. Moka Rajasekhar for assistance in cytogenetic analysis; Jyoti, Sowmya and Ganesh for technical support. We also thank patients for the consent to provide the specimens.
This article contains supporting information.
Author contributions—M. H. and M. B. J. data curation; M. H., K. P. G., and L. R. formal analysis; M. H., K. P. G., and L. R. methodology; M. H. writing-original draft; K. S. and M. B. J. supervision; K. S. and M. B. J. writing-review and editing; M. B. J. conceptualization; M. B. J. funding acquisition; M. B. J. project administration.
Funding and additional information—This work was supported by Dr. TMA Pai Foundation scholarship by Manipal Academy of Higher Education, Manipal and Senior Research Fellowship by Council of Scientific and Industrial Research, Government of India (to M. H.). This work is also funded by Pilot Project Grant 6242-P81/RGCB/PMD/DBT/MNBJ/2015 for Young Investigators in Cancer Biology, Dept. of Biotechnology (DBT), Government of India (to M. B. J.).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this manuscript.
- VEGF
- vascular endothelial growth factor
- Endo-T
- tumor endothelial
- Epi-T
- tumor myoepithelial
- Endo-N
- normal endothelial
- Epi-N
- normal epithelial
- DNMT
- DNA methyltransferase
- HUVEC
- human umbilical vein endothelial cell
- MAO-A
- monoamino oxidase-A
- ECGM
- endothelial cell growth medium.
References
- 1. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: The next generation. Cell 144, 646–674 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 2. Chen X., Xu C., Hong S., Xia X., Cao Y., McDermott J., Mu Y., and Han J. D. J. (2019) Immune cell types and secreted factors contributing to inflammation-to-cancer transition and immune therapy response. Cell Rep. 26, 1965–1977.e4 10.1016/j.celrep.2019.01.080 [DOI] [PubMed] [Google Scholar]
- 3. Sarper M., Allen M. D., Gomm J., Haywood L., Decock J., Thirkettle S., Ustaoglu A., Sarker S. J., Marshall J., Edwards D. R., and Jones J. L. (2017) Loss of MMP-8 in ductal carcinoma in situ (DCIS)-associated myoepithelial cells contributes to tumour promotion through altered adhesive and proteolytic function. Breast Cancer Res. 19, 33 10.1186/s13058-017-0822-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Katarina Sirka O., Shamir E. R., and Ewald A. J. (2018) Myoepithelial cells are a dynamic barrier to epithelial dissemination. J. Cell Biol. 217, 3368–3381 10.1083/jcb.201802144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martinez E. F., Napimoga M. H., Montalli V. A. M., de Araújo N. S., and de Araújo A. V. (2013) In vitro cytokine expression in in situ-like areas of malignant neoplasia. Arch. Oral Biol. 58, 552–557 10.1016/j.archoralbio.2012.06.001 [DOI] [PubMed] [Google Scholar]
- 6. Wei L.-H., Kuo M.-L., Chen C.-A., Chou C.-H., Lai K.-B., Lee C.-N., and Hsieh C.-Y. (2003) Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene 22, 1517–1527 10.1038/sj.onc.1206226 [DOI] [PubMed] [Google Scholar]
- 7. Jung J. E., Lee H. G., Cho I. H., Chung D. H., Yoon S.-H., Yang Y. M., Lee J. W., Choi S., Park J.-W., Ye S.-K., and Chung M.-H. (2005) STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 19, 1296–1298 10.1096/fj.04-3099fje [DOI] [PubMed] [Google Scholar]
- 8. Xu Q., Briggs J., Park S., Niu G., Kortylewski M., Zhang S., Gritsko T., Turkson J., Kay H., Semenza G. L., Cheng J. Q., Jove R., and Yu H. (2005) Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 24, 5552–5560 10.1038/sj.onc.1208719 [DOI] [PubMed] [Google Scholar]
- 9. Gallo M., Frezzetti D., Roma C., Chicchinelli N., Barbieri A., Arra C., Scognamiglio G., Botti G., De Luca A., and Normanno N. (2018) RANTES and IL-6 cooperate in inducing a more aggressive phenotype in breast cancer cells. Oncotarget 9, 17543–17553 10.18632/oncotarget.24784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kujawski M., Kortylewski M., Lee H., Herrmann A., Kay H., and Yu H. (2008) Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Invest. 118, 3367–3377 10.1172/JCI35213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coward J., Kulbe H., Chakravarty P., Leader D., Vassileva V., Leinster D. A., Thompson R., Schioppa T., Nemeth J., Vermeulen J., Singh N., Avril N., Cummings J., Rexhepaj E., Jirström K., et al. (2011) Interleukin-6 as a therapeutic target in human ovarian cancer. Clin. Cancer Res. 17, 6083–6096 10.1158/1078-0432.CCR-11-0945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gopinathan G., Milagre C., Pearce O. M., Reynolds L. E., Hodivala-Dilke K., Leinster D. A., Zhong H., Hollingsworth R. E., Thompson R., Whiteford J. R., and Balkwill F. (2015) Interleukin-6 stimulates defective angiogenesis. Cancer Res. 75, 3098–3107 10.1158/0008-5472.CAN-15-1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yan M. S., Turgeon P. J., Man H. S. J., Dubinsky M. K., Ho J. J. D., El-Rass S., Wang Y. D., Wen X. Y., and Marsden P. A. (2018) Histone acetyltransferase 7 (KAT7)-dependent intragenic histone acetylation regulates endothelial cell gene regulation. J. Biol. Chem. 293, 4381–4402 10.1074/jbc.RA117.001383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pirola L., Ciesielski O., and B A. (2018) The methylation status of the epigenome: Its emerging role in the regulation of tumor angiogenesis and tumor growth, and potential for drug targeting. Cancers (Basel) 10, 268 10.3390/cancers10080268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hellebrekers D. M. E. I., Jair K.-W., Viré E., Eguchi S., Hoebers N. T. H., Fraga M. F., Esteller M., Fuks F., Baylin S. B., van Engeland M., and Griffioen A. W. (2006) Angiostatic activity of DNA methyltransferase inhibitors. Mol. Cancer Ther. 5, 467–475 10.1158/1535-7163.MCT-05-0417 [DOI] [PubMed] [Google Scholar]
- 16. Balakrishnan A., Guruprasad K. P., Satyamoorthy K., and Joshi M. (2018) Interleukin-6 determines protein stabilization of DNA methyltransferases and alters DNA promoter methylation of genes associated with insulin signaling and angiogenesis. Lab. Invest. 98, 1143–1158 10.1038/s41374-018-0079-7 [DOI] [PubMed] [Google Scholar]
- 17. Saaristo A., Karpanen T., and Alitalo K. (2000) Mechanisms of angiogenesis and their use in the inhibition of tumor growth and metastasis. Oncogene 19, 6122–6129 10.1038/sj.onc.1203969 [DOI] [PubMed] [Google Scholar]
- 18. Liu T. J., Sun B. C., Zhao X. L., Zhao X. M., Sun T., Gu Q., Yao Z., Dong X. Y., Zhao N., and Liu N. (2013) CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple-negative breast cancer. Oncogene 32, 544–553 10.1038/onc.2012.85 [DOI] [PubMed] [Google Scholar]
- 19. Akino T., Hida K., Hida Y., Tsuchiya K., Freedman D., Muraki C., Ohga N., Matsuda K., Akiyama K., Harabayashi T., Shinohara N., Nonomura K., Klagsbrun M., and Shindoh M. (2009) Cytogenetic abnormalities of tumor-associated endothelial cells in human malignant tumors. Am. J. Pathol. 175, 2657–2667 10.2353/ajpath.2009.090202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paauwe M., Heijkants R. C., Oudt C. H., van Pelt G. W., Cui C., Theuer C. P., Hardwick J. C., Sier C. F., and Hawinkels L. J. A. C. (2016) Endoglin targeting inhibits tumor angiogenesis and metastatic spread in breast cancer. Oncogene 35, 4069–4079 10.1038/onc.2015.509 [DOI] [PubMed] [Google Scholar]
- 21. Liu Y., Liu Y., Huang R., Song W., Wang J., Xiao Z., Dong S., Yang Y., and Yang X. (2019) Dependency of the cancer-specific transcriptional regulation circuitry on the promoter DNA methylome. Cell Rep. 26, 3461–3474 10.1016/j.celrep.2019.02.084 [DOI] [PubMed] [Google Scholar]
- 22. Luo W., Hu Q., Wang D., Deeb K. K., Ma Y., Morrison C. D., Liu S., Johnson C. S., and Trump D. L. (2013) Isolation and genome-wide expression and methylation characterization of CD31+ cells from normal and malignant human prostate tissue. Oncotarget 4, 1472–1483 10.18632/oncotarget.1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Y., Hoeppner L. H., Angom R. S., Wang E., Dutta S., Doeppler H. R., Wang F., Shen T., Scarisbrick I. A., Guha S., Storz P., Bhattacharya R., and Mukhopadhyay D. (2019) Protein kinase D up-regulates transcription of VEGF receptor-2 in endothelial cells by suppressing nuclear localization of the transcription factor AP2β. J. Biol. Chem. 294, 15759–15767 10.1074/jbc.RA119.010152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mammoto A., Connor K. M., Mammoto T., Yung C. W., Huh D., Aderman C. M., Mostoslavsky G., Smith L. E. H., and Ingber D. E. (2009) A mechanosensitive transcriptional mechanism that controls angiogenesis. Nature 457, 1103–1108 10.1038/nature07765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bakker W. J., Weijts B. G. M. W., Westendorp B., and de Bruin A. (2013) HIF proteins connect the RB-E2F factors to angiogenesis. Transcription 4, 62–66 10.4161/trns.23680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carmeliet P., and J R. (2000) Angiogenesis in cancer and other diseases. Nature 407, 249–257 10.1038/35025220 [DOI] [PubMed] [Google Scholar]
- 27. Kim J., Hwang J., Jeong H., Song H. J., Shin J., Hur G., Park Y. W., Lee S. H., and Kim J. (2012) Promoter methylation status of VEGF receptor genes—a possible epigenetic biomarker to anticipate the efficacy of intracellular-acting VEGF-targeted drugs in cancer cells. Epigenetics 7, 191–200 10.4161/epi.7.2.18973 [DOI] [PubMed] [Google Scholar]
- 28. Xu Y., Wang Y., Yan S., Zhou Y., Yang Q., Pan Y., Zeng X., An X., Liu Z., Wang L., Xu J., Cao Y., Fulton D. J., Weintraub N. L., Bagi Z., et al. (2017) Intracellular adenosine regulates epigenetic programming in endothelial cells to promote angiogenesis. EMBO Mol. Med. 9, 1263–1278 10.15252/emmm.201607066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rao X., Zhong J., Zhang S., Zhang Y., Yu Q., Yang P., Wang M.-H., Fulton D. J., Shi H., Dong Z., Wang D., and Wang C.-Y. (2011) Loss of methyl-CpG–binding domain protein 2 enhances endothelial angiogenesis and protects mice against hind-limb ischemic injury. Circulation 123, 2964–2974 10.1161/CIRCULATIONAHA.110.966408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karakasheva T. A., Lin E. W., Tang Q., Qiao E., Waldron T. J., Soni M., Klein-Szanto A. J., Sahu V., Basu D., Ohashi S., Baba K., Giaccone Z. T., Walker S. R., Frank D. A., Wileyto E. P., et al. (2018) IL-6 mediates cross-talk between tumor cells and activated fibroblasts in the tumor microenvironment. Cancer Res. 78, 4957–4970 10.1158/0008-5472.CAN-17-2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mochizuki D., Adams A., Warner K. A., Zhang Z., Pearson A. T., Misawa K., Mclean S. A., Wolf G. T., and Nör J. E. (2015) Anti-tumor effect of inhibition of IL-6 signaling in mucoepidermoid carcinoma. Oncotarget 6, 22822–22835 10.18632/oncotarget.4477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bharti R., Dey G., Das A. K., and Mandal M. (2018) Differential expression of IL-6/IL-6R and MAO-A regulates invasion/angiogenesis in breast cancer. Br. J. Cancer 118, 1442–1452 10.1038/s41416-018-0078-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maishi N., and Hida K. (2017) Tumor endothelial cells accelerate tumor metastasis. Cancer Sci. 108, 1921–1926 10.1111/cas.13336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jain R. K. (2014) Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 26, 605–622 10.1016/j.ccell.2014.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Incio J., Ligibel J. A., McManus D. T., Suboj P., Jung K., Kawaguchi K., Pinter M., Babykutty S., Chin S. M., Vardam T. D., Huang Y., Rahbari N. N., Roberge S., Wang D., Gomes-Santos I. L., et al. (2018) Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci. Transl. Med. 10, eaag0945 10.1126/scitranslmed.aag0945 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are provided in manuscript.