

Abstract
DNA polymerase (pol) β catalyzes two reactions at DNA gaps generated during base excision repair, gap-filling DNA synthesis and lyase-dependent 5´-end deoxyribose phosphate removal. The lyase domain of pol β has been proposed to function in DNA gap recognition and to facilitate DNA scanning during substrate search. However, the mechanisms and molecular interactions used by pol β for substrate search and recognition are not clear. To provide insight into this process, a comparison was made of the DNA binding affinities of WT pol β, pol λ, and pol μ, and several variants of pol β, for 1-nt-gap-containing and undamaged DNA. Surprisingly, this analysis revealed that mutation of three lysine residues in the lyase active site of pol β, 35, 68, and 72, to alanine (pol β KΔ3A) increased the binding affinity for nonspecific DNA ∼11-fold compared with that of the WT. WT pol μ, lacking homologous lysines, displayed nonspecific DNA binding behavior similar to that of pol β KΔ3A, in line with previous data demonstrating both enzymes were deficient in processive searching. In fluorescent microscopy experiments using mouse fibroblasts deficient in PARP-1, the ability of pol β KΔ3A to localize to sites of laser-induced DNA damage was strongly decreased compared with that of WT pol β. These data suggest that the three lysines in the lyase active site destabilize pol β when bound to DNA nonspecifically, promoting DNA scanning and providing binding specificity for gapped DNA.
Keywords: DNA binding proteins, facilitated diffusion, processive search, DNA polymerase, DNA repair, nonspecific DNA binding, DNA binding protein, DNA damage, base excision repair (BER), DNA–protein interaction
Small base lesions, nicks, gaps, and abasic sites are among the most abundant forms of DNA damage, and these lesions are repaired predominantly by the base excision repair (BER) pathway (1). The canonical BER pathway is often initiated by a DNA glycosylase that catalyzes the removal of a damaged base, generating an abasic site in dsDNA. The repair pathway is completed through abasic site cleavage by APE1, dRP removal and gap-filling DNA synthesis by polymerase (pol) β, and ligation by ligase I or III (2). This multistep process has been hypothesized to occur in a coordinated manner, where the previous enzyme remains bound to its product DNA and is displaced by the next enzyme in the pathway (3–5). This presumably minimizes the time the reactive DNA repair intermediate is exposed and decreases the likelihood of exacerbating the damage. Because DNA damage occurs throughout the 6 billion base-pair human genome, DNA repair enzymes must search DNA with incredible efficiency to find sites of damage. The mechanisms that allow BER enzymes to efficiently search and find damage among this vast excess of undamaged DNA and, within a timely manner, remain an area of active investigation.
One mechanism employed by many BER enzymes to locate DNA damage is known as processive searching (5). In processive searching, also known as facilitated diffusion, DNA repair enzymes bind DNA nonspecifically and then scan in search of damage (6, 7). This mechanism effectively increases the quantity of DNA searched after one macromolecular DNA binding event. Many BER enzymes are capable of such a mechanism, and we previously examined the ability of gap-filing DNA polymerases to perform DNA scanning using a processive search assay (8). Among the three gap-filling human pol X- family members tested, pol β has the highest fraction processive value, followed by pol λ and then pol μ (8). The fraction processive value is the probability of the enzyme to locate and catalyze product formation at two substrate sites on one DNA molecule in a single binding event (9). Processive searching among the pol X family members correlates with the positive charge of the lyase domains, and this may reflect different strategies of how the enzymes are localized to DNA damage (8).
The pol X-family members β, λ, and μ are gap-filling polymerases that lack exonuclease domains (10). These pols are defined by having at least two domains: the 31-kDa (31K) and 8-kDa (8K) domains. The 31-kDa pol domain catalyzes DNA polymerization and is composed of the canonical pol subdomains (11). The 8K domains in pols β and λ have lyase activity on 5´-dRP, the product of APE1 incision, generating a 5´-phosphate end suitable as a DNA ligase substrate (12, 13). The 8K domain also is important for DNA gap recognition, in keeping with specific interactions observed in crystal structures. These interactions anchor the lyase domain to a 1-nt gap and occur through lysine side chains interacting with the 5´-phosphate group and base-stacking interactions. For pol β, the positively charged 8K domain is important for processive searching and is likely the first domain to specifically recognize the 1-nt-gap substrate (14, 15).
During a processive search, an enzyme whose substrate is embedded in DNA must first make nonspecific interactions with undamaged DNA. To provide mechanistic insight into substrate search and recognition, we determined binding affinities for specific and nonspecific DNA and uncovered a correlation between processive searching ability and binding specificity among WT pol β and pol β mutants and also for pol μ and pol λ. In vivo recruitment studies in living cells demonstrated the importance of the dRP lyase lysines for the direct localization of pol β to DNA damage.
Results
To examine specific gap binding and nonspecific DNA binding properties of pol β and several of its mutants, and to make comparisons with pol µ and pol λ, model “dumbbell” DNA substrates were used as DNA binding ligands (Fig. 1A). These substrates were used to exclude any end-binding effects and are based on previous studies performed with pol β and PARP-1 (16, 17). Electrophoretic mobility shift assays (EMSAs) were used to characterize the DNA binding properties of the purified proteins. EMSAs are not considered ideal for measuring equilibrium binding in solution, as the technique depends on separating bound from unbound DNA during gel electrophoresis, which may disrupt the binding equilibrium. However, the EMSA allows for straightforward interpretation of multiple proteins binding to DNA, and differing mobilities may infer different conformations of the DNA-protein binary complexes.
Figure 1.

A, representation of the DNA substrates used in this study. Substrates contain a fluorescein moiety (FL) covalently attached to the designated thymine base. The 1-nt-gap dumbbell DNA substrate (used to evaluate specific binding) contains a 5´-phosphate (not shown). The undamaged DNA substrate (used to evaluate nonspecific binding) was produced as described in Materials and methods. B, cartoon schematic of the DNA pol β domain organization (top) and the crystal structure of pol β bound to 1-nt-gap DNA (PDB entry 3isb). DNA pol β comprises two domains: the 8-kDa (8K) N-terminal dRP lyase domain, which catalyzes 5´-dRP removal, and the 31-kDa (31K) polymerase domain, which catalyzes gap-filling DNA polymerization during BER. The pol β mutations in the 8K lyase domain studied here, K35, K68, and K72, are illustrated; these lysine side chains are proximal to the 5´-phosphate in the 1-nt-gap.
WT and mutant enzyme preparations of pol β were first characterized for binding to a 1-nt-gap-containing DNA (i.e. specific binding) and undamaged DNA (i.e. nonspecific binding). The WT and mutants included the full-length (FL) enzyme, the KΔ3A mutant (with lyase active site lysines 35, 68, and 72 mutated to alanine), the 8-kDa (8K) domain from the WT enzyme and KΔ3A mutant, and the 31K domain (Fig. 1B). The KΔ3A mutation disrupts the interaction of the lyase domain with the 5´-phosphate group of a 1-nt-gap substrate (18).
Differing EMSA mobilities of pol β variants bound to specific and nonspecific DNA
The native gel mobilities of these DNA-bound pol β mutants differed significantly compared with the binding against 1-nt-gap and undamaged DNA (Fig. 2A). As expected, the binary complex of the WT enzyme and 1-nt-gap DNA migrated slower than the 8K domain binary complex bound to 1-nt-gap DNA. However, the KΔ3A mutant complex migrated to a position similar to that of the KΔ3A-8K construct. This behavior was observed with nonspecific DNA as well. Nonspecific DNA binding by both FL WT and 8K WT proteins showed mainly a smear, suggesting that stable distinct complexes were not formed under these conditions. However, testing the KΔ3A mutation in both the FL enzyme and the 8K domain revealed a discrete complex, and addition of more enzyme to the binding incubation resulted in multiple distinct binding species resolvable in the gel, indicating up to six bound species (Fig. 2 and Fig. S2 and S4).
Figure 2.

A, EMSA representing the different protein-DNA complex mobilities of WT pol β, the WT 8K domain, and two corresponding mutant enzymes, as designated, with 3 lysine to alanine substitutions (KΔ3A). The left panel represents mobilities of complexes bound to 1-nt-gap dumbbell DNA, and the right panel represents mobilities of complexes bound to undamaged dumbbell DNA. The KΔ3A mutants used here included substitutions in full-length (FL) WT and the 8K, as illustrated in red. The concentrations of proteins used were based on Kd measurements, such that about one-half the DNA would be bound: 20 nm FL WT, 1.5 μm 31K, 20 nm 8K WT, 100 nm FL KΔ3A, and 1.5 μm 8K KΔ3A for 1-nt-gap-containing DNA and 500 nm FL WT, 1.5 μm 31K, 1.5 μm 8K WT, 100 nm FL KΔ3A, 1.25 μm, 8K KΔ3A, and 5 μm 8K KΔ3A for undamaged DNA. In panel A, FL+ and 8K+ indicate binding reactions where additional protein was added. This was performed to illustrate that multiple distinct complexes can be detected with these mutants (up to 6 discrete bands [*] were detected by EMSA). Gel quantification is reported in Table S3. B, apparent binding affinities as determined by EMSA for indicated protein and DNA. No binding was observed for 31K with either DNA construct under the concentration range examined (Kd > 3 μm). See the supporting information for gel images and binding isotherms. Data from 3 and 2 independent experiments are shown for FL WT and FL KΔ3A binding, respectively.
Apparent binding affinities of pol β variants for specific and nonspecific DNA
Examination of the apparent binding affinities of these variants for 1-nt-gap and nonspecific DNA revealed two surprising results (Fig. 2B and Fig. S1–4 and Table S1). First, the mutation to KΔ3A within the full-length enzyme had a modest effect on the apparent binding affinity for the 1-nt-gap DNA compared with the WT. Second, the DNA binding affinity of the KΔ3A mutant to nonspecific DNA was similar to that of the 1-nt-gap DNA binding affinity. This indicates that the KΔ3A mutant was not able to distinguish between specific and nonspecific DNA. Additionally, these data suggest that the three lysine residues (35, 68, and 72) in WT pol β are involved in destabilizing nonspecific DNA binding, as their replacement with alanine leads to an increase in DNA binding affinity for nonspecific DNA.
Apparent binding affinities of pols β, µ, and λ for specific and nonspecific DNA
The ability of the X-family DNA pols β, λ, and µ to perform a processive search varies among the three enzymes (Table S2) (8). To determine whether processive searching correlates with binding affinities for specific or nonspecific DNA and to provide further insight into substrate recognition, we measured the apparent binding affinities of pols β, λ, and µ for the 1-nt-gap dumbbell and undamaged DNA (Fig. 3 and Fig. S6). The apparent binding affinities for 1-nt-gap DNA were comparable among the three enzymes: 30, 90, and 40 nm for pols β, λ, and µ, respectively. However, the pattern of nonspecific DNA binding by pol µ was different from that observed with pols β and λ. Pol µ bound nonspecific DNA with affinity similar to that of 1-nt-gap DNA, suggesting this polymerase lacks specificity for gapped DNA (Table S2). In contrast, pols β and λ exhibited much weaker binding to nonspecific DNA (apparent Kd of ∼500–700 μm) than to the specific DNA.
Figure 3.
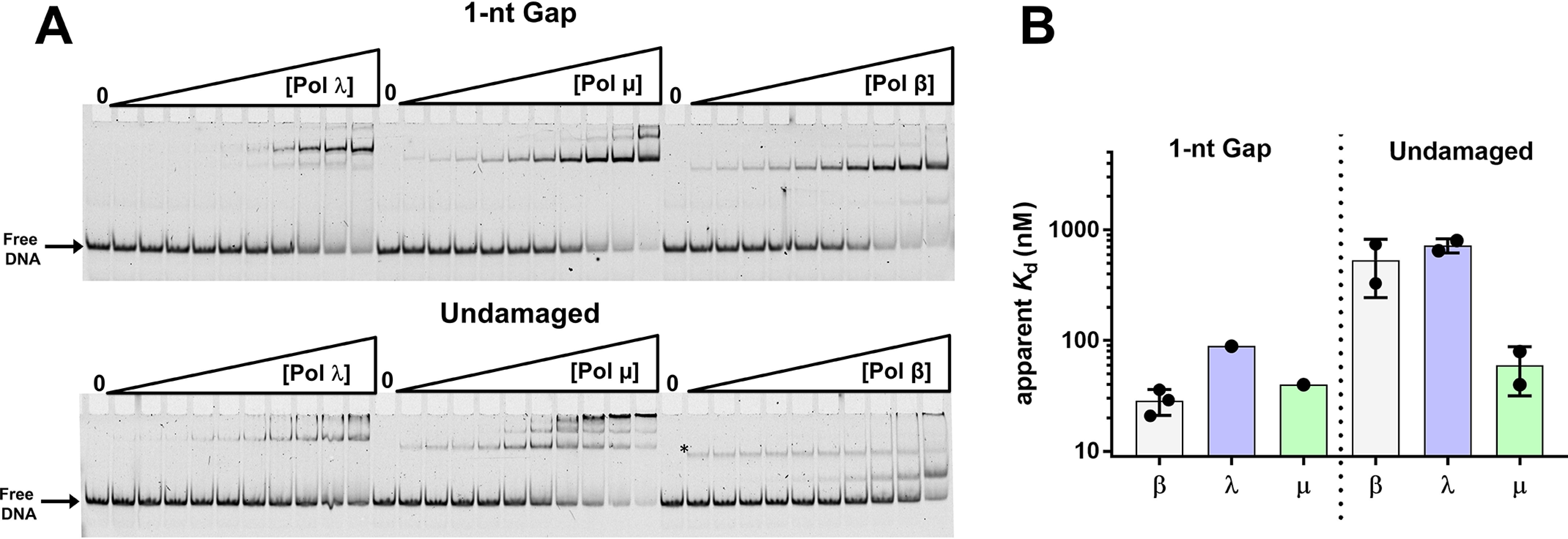
A, EMSA illustrating binding of pols γ, µ, and β to 1-nt-gap (top) and undamaged (bottom) DNA. Experiments were performed as described in Materials and methods. The asterisk in the reactions performed with pol β and undamaged DNA indicates a band that was not included in the quantification of the gel. Its intensity remains about the same with increasing enzyme concentration and was not observed in other assays under similar conditions (Fig. 2B). B, summary of apparent binding constants of pols to the 1-nt-gap DNA and undamaged DNA. Data in panel A were plotted and fit to a hyperbolic binding equation (Fig. S5 and Table S2).
Recruitment of WT pol β-GFP and pol β KΔ3A-GFP to laser-induced DNA damage in vivo
To determine whether the KΔ3A mutation has an effect on substrate recognition in vivo, the ability of WT pol β-GFP and lyase domain mutant pol β KΔ3A-GFP to localize to laser-induced DNA damage in living cells was examined. Previous studies had demonstrated recruitment of GFP-tagged BER proteins to sites of laser-induced damage (19, 20), for example, recruitment of pol β-GFP in MEF cells. Using this system, WT pol β-GFP and mutant pol β KΔ3A-GFP were separately transfected into a pol β null mouse embryo fibroblast (MEF) cell line, and then the ability of these two tagged enzymes to localize to microirradiation-induced DNA damage was examined by confocal fluorescence microscopy (Fig. 4A).
Figure 4.
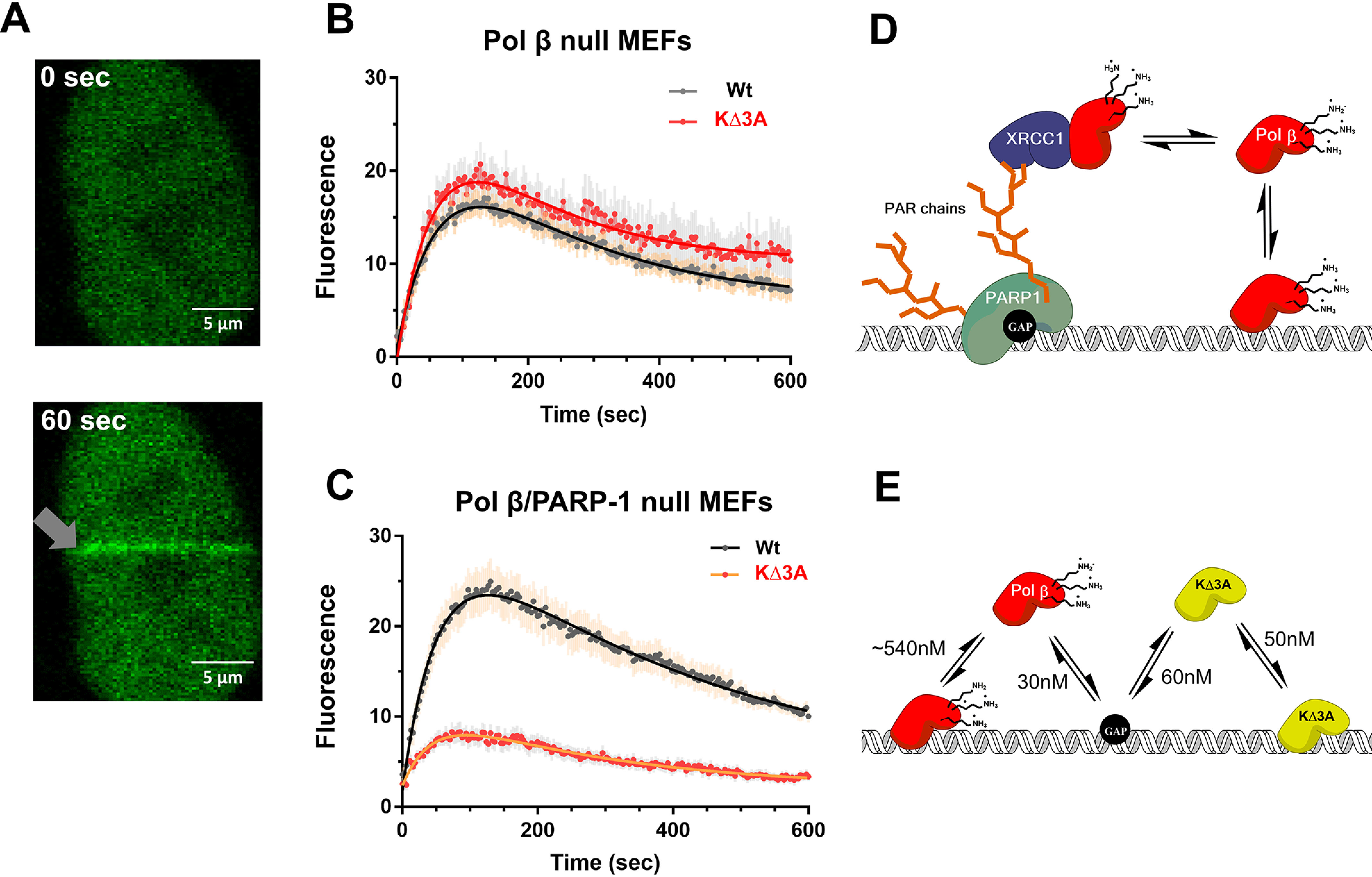
In vivo recruitment of pol β to DNA damage. A, representative confocal fluorescent images of nuclear localized GFP-WT pol β expressed in MEFs at time zero and 60 s after laser-induced DNA damage (gray arrow). B, recruitment of WT (n = 25) and the KΔ3A mutant (n = 17) to laser-induced DNA damage in the cell background of pol β deficiency by virtue of pol β gene deletion. Error bars represent the standard error of the mean. The resulting observed rate constants from a double exponential fit are kobs1 = 0.015 s−1 and kobs2 = 0.005 s−1 as well as kobs1 = 0.017 s−1 and kobs2 = 0.006 s−1 for WT and KΔ3A, respectively. C, recruitment of WT (16 cells) and KΔ3A mutant (21 cells) to laser-induced DNA damage in the cell background of deficiency of both pol β and PARP-1. Error bars represent the standard error of the mean. The resulting observed rate constants from a double exponential fit are kobs1 = 0.02 s−1 and kobs2 = 0.002 s−1 as well as kobs1 = 0.02 s−1 and kobs2 = 0.004 s−1 for WT and KΔ3A, respectively. D, working model explaining in vivo results on pol β localization to DNA damage as shown in panel B. Pol β may localize to DNA damage through an indirect pathway that relies on PARP-1 recognition of the gap, PARylation, and protein–protein interactions with such scaffolding factors as XRCC1; in this model, PARylation signals recruitment and binding of XRCC1, which in turn is a binding partner of pol β, facilitating its recruitment. E, proposed model for results shown in panel C. In the direct model, pol β interacts directly with DNA. The lyase domain lysines destabilize nonspecific DNA binding, allowing the polymerase to scan DNA for damage and provide binding specificity for gapped DNA. The absence of these lysines (KΔ3A) leads to tight nonspecific DNA binding. The black circle on the DNA represents a 1-nt gap. WT pol β is shown in red and KΔ3A in yellow.
Time courses for both recruitment and dissociation of these tagged enzymes are shown in Fig. 4. No significant differences were observed between the WT enzyme and the KΔ3A mutant in pol β null MEFs (Fig. 4B). However, a major mode of recruitment to laser-induced DNA damage may be through a PARP-1 dependent pathway (19, 21). In this case, PARylation and protein–protein interactions may be the dominant mode of localization rather than a direct DNA and pol β interaction. To investigate this possibility, we examined the ability of WT pol β-GFP and pol β KΔ3A-GFP to localize to laser-induced DNA damage in the presence of the PARP inhibitor veliparb (Fig. S10). Cells treated with PARP inhibitor did not show recruitment of GFP-pol β to laser-induced DNA damage (Fig. S10). These data suggest that PARylation catalyzed by PARPs at sites of laser-induced DNA damage is an important event for GFP-pol β recruitment to damage.
To study the localization of pol β in the absence of PARP-1, pol β-PARP-1 double null cells were used (Fig. 4C). In this cell line, the recruitment of the GFP-tagged pol β KΔ3A mutant was strongly reduced compared with that of GFP-tagged WT pol β. These data suggest that, in the absence of PARP-1, robust localization of pol β to DNA damage depends on lysines in the lyase domain.
Discussion
Nonspecific DNA binding must occur for an enzyme to scan DNA in search of its specific substrate target. Understanding the interactions that are made between the enzyme and DNA in the nonspecifically bound mode and substrate-bound mode should provide insight into the mechanisms of searching and substrate recognition. Use of pol β and its variants and the other two pol X family members revealed that the 8K domain of pol X family controls nonspecific DNA binding affinity. These studies also suggest that the 8K lyase domain not only stabilizes binding to 1-nt-gap DNA but also actively destabilizes nonspecific DNA binding. Importantly, we hypothesize that this destabilization is critical for processive searching, as it promotes microdissociation of the nonspecifically bound pol β from DNA. We extended our mechanistic studies to live cells and demonstrated the importance of processive searching for substrate recognition in vivo by making use of a DNA damage recruitment assay and a processive searching-deficient pol β mutant (pol β KΔ3A).
A surprising finding was that the KΔ3A mutation of pol β increases the binding affinity for nonspecific DNA ∼11-fold compared with the WT. The binding affinities between gap and nonspecific DNA are comparable with this mutant. Although the KΔ3A mutation has no effect on DNA binding affinity, compared with the WT, the KΔ3A mutation in the 8K domain alone does (Fig. 2B). This suggests that for DNA binding with the full-length KΔ3A mutant, the 31K region of the enzyme stabilizes the nonspecifically bound state (Fig. S7). Although WT and KΔ3A have similar binding affinities for 1-nt-gapped DNA, the mobilities of the bound complexes are strikingly different (Fig. 2A). This result is consistent with the existence of two different binding modes for WT and 3ΔKA on 1-nt-gapped DNA. Indeed, FRET studies of pol β binary complexes in the presence and absence of a 5´-phosphate have different FRET efficiencies, suggesting differential binding modes (22).
Pol µ has DNA binding behavior similar to that of the pol β KΔ3A mutant (Fig. 2 and 3). Interestingly, pol µ has a relatively neutral 8K domain and lacks the lysine interactions with the 5´-phosphate in the gap compared with pol β (Fig. S8). The lack of specificity in DNA binding with pol β KΔ3A and pol µ correlates with their lack of ability to perform a processive search (Table S2). This suggests that, contrary to our previous hypothesis, the positively charged lyase domain is important for electrostatic interactions during the search process (15); the 8K lyase domain actually destabilizes nonspecific binding. This leads to a new model where the lysines in the 8K domain promote processive searching by encouraging microdissociation of the nonspecifically bound state, allowing the polymerase to search neighboring regions and providing binding specificity for gapped DNA. Additionally, the affinity for undamaged DNA is similar to that for damaged DNA, and there seems to be no favorable interaction to promote gap binding over nonspecific binding, implying that Pol β KΔ3A and pol µ have flat DNA binding energetic landscapes. These results uncover the complex nature of interpreting DNA binding mutations and highlight the interplay between nonspecific and specific binding among processive searching enzymes.
To determine how the measured apparent binding affinities might correlate with processive searching, the fraction processive was plotted versus the binding affinity for 1-nt-gap DNA (Fig. S12), undamaged DNA (Fig. S12), and the discrimination ratio (nonspecific divided by the specific affinity) (Fig. S12). Processive searching ability (fraction processive) correlated directly with the discrimination ratio, indicating that a balance between specific and nonspecific DNA binding affinity is a key component of processive searching.
The observation that the KΔ3A enzyme is deficient in processive searching and a lesser amount localizes to DNA damage in pol β/PARP-1 null cells suggests that processive searching is important for efficient substrate recognition in vivo. Based on previous studies, we have hypothesized two main modes for the recruitment of pol β to laser-induced DNA damage (Fig. 4, D and E). The first, referred to as indirect, involves PARylation catalyzed by PARP-1. In this pathway, PARP-1 binds DNA strand breaks and catalyzes PARylation of itself and neighboring proteins. These PAR chains act as recruitment scaffolds for other proteins that can specifically bind PAR. A binding partner of pol β, XRCC1, has a PAR binding motif (23, 24). Therefore, recruitment of XRCC1 to PAR enables the recruitment of pol β (Fig. 4D). Because PARP-1 is responsible for the majority of PARylation at strand breaks (25), a PARP-1 null cell line was used to examine the other mode of pol β recruitment to DNA damage, referred to as direct. In this direct mode, we hypothesized that the recruitment of pol β to DNA damage would depend on direct interactions of pol β with DNA. Accordingly, any mutations that disrupt the ability of pol β to search and scan DNA should disrupt recruitment to DNA damage sites. Indeed, the ability of the KΔ3A mutant to accumulate at sites of DNA damage was reduced compared with the WT in PARP-1 null cells. These data highlight the importance of the relationship between nonspecific and specific DNA binding for in vivo substrate search and recognition.
Materials and methods
Proteins
Human pols β, μ, and λ were prepared as described previously (8). The extinction coefficients used to calculate the concentration of the proteins are 23,380, 47,440, and 57,870 M−1 cm−1 for pol β, pol μ, and pol λ, respectively. The 8K and 31K domains contain amino acids 1–87 and 88–335, respectively, and were prepared as previously described (26). Extinction coefficients of 4470 and 19,000 M−1cm−1 were used to quantify the 8-kDa and 31-kDa domains, respectively. The purity of the proteins was assessed by SDS-PAGE and visually determined to be greater than 90%.
Substrates
Oligonucleotides were purchased from TriLink Biotechnologies and purified by PAGE and HPLC. DNA sequences are shown in Fig. 2. The undamaged dumbbell DNA was produced by ligation of a nick containing a dumbbell oligonucleotide and then gel purified. To ensure proper folding of DNA dumbbells, oligonucleotides were heated at 95 °C for 1 min and then set at room temperature for 15 min before use. Extinction coefficients provided by the supplier were used for determining the concentration.
EMSA
Binding reactions were prepared at room temperature in reaction buffer: 50 mm Tris (pH 7.4 at 37 °C), 100 mm KCl, 5 mm MgCl2, 1 mm DTT, 0.1 mg/ml BSA, and 10% glycerol. Indicated proteins were serially diluted 2-fold in reaction buffer before addition of fluorescently labeled oligonucleotide to a final concentration of 10 nm. Reaction mixtures were incubated for ∼5 min before loading onto a 10% native gel (acrylamide:bis-acrylamide, 37.5:1) at 4 °C. The gel was equilibrated and prerun at 4 °C for at least 30 min before samples were loaded. The gel dimensions were 33 cm (width) by 10 cm (height) and 0.75 mm (thick). The gel was run at 200 V for ∼45 min in 0.5× TBE and scanned on a typhoon gel imaging system. Images were quantified using the rolling ball method in ImageQuant and fit to a hyperbolic binding equation in Prism. The binding equation included a variable for cooperativity, a Hill coefficient (n). The fitted binding parameters are reported in Tables S1 and S2. Reported error is from least-squares fitting unless otherwise indicated.
In vivo recruitment
Cell lines
Pol β null MEFs have been described previously (27). Spontaneously immortalized PARP1−/−/pol β −/− double knockout MEFs (28) were a kind gift from Dr. Robert W. Sobol (University of Pittsburgh). Knockout of pol β and PARP-1 was confirmed by Western blot analysis (Fig. S9). Cells were maintained in high-glucose DMEM (HyClone) supplemented with glutamine and 10% FBS (HyClone). All cells were routinely tested and found to be free of mycoplasma contamination.
Transfection and laser-induced damage analysis
Human C-terminal TurboGFP pol β (WT) was from Origene (RG210765), and KΔ3A pol β-GFP was prepared by sequential rounds of site-directed mutagenesis (29). All constructs were confirmed by DNA sequencing (Genewiz). Recruitment of WT and KΔ3A were assessed in pol β null MEF cells following transient transfection. Cells were transfected 24 h after plating using LipofectamineTM 2000 in medium supplemented with 10 μm 5-bromo-2'-deoxyuridine (BrdUrd) (Sigma-Aldrich). 24 h after transfection, medium was changed to fresh room temperature growth medium without BrdUrd. Only cells with similar low to moderate fluorescence intensities were analyzed (Fig. S11). DNA SSBs were introduced by microirradiation with a fiber-coupled 355-nm coherent laser at maximum power (60 milliwatts, 100% output), with a one-pixel-width strip (0.45 μm) region of interest (ROI) per nucleus and an irradiation time of 200 μs (100 scanning iterations), using the 40× C-Apochromat NA 1.2 Korr FCS M27 water immersion objective coupled to a Zeiss LSM780 confocal microscope (Carl Zeiss MicroImaging).
For image acquisition in live-cell experiments with GFP-tagged reporters, a 488-nm laser was used at 1% intensity to minimalize photobleaching. Fluorescence emission was detected in the range of 491–580 nm using a pinhole of 40 μm. Samples were imaged using the same 40× water immersion objective coupled to a Zeiss LSM780 confocal microscope. Three prebleach images were taken to establish a baseline prior to damage induction. Images were acquired at room temperature every 3 s after the bleaching event for 10 min. Relative recruitment of pol β-GFP was determined by measuring the signal intensity of GFP at the induced damage site using a manually drawn fixed 2.25-μm-width ROI and data were analyzed using GraphPad. Time-lapse recruitment curves were corrected by subtracting the postbleach signal intensity of the entire nucleus excluding the ROI.
Data availability
All data are contained within the manuscript.
Supplementary Material
Acknowledgments
We thank Agnes Janoshazi of the NIEHS Fluorescence Imaging Center for expert assistance in microirradiation studies. We thank Lars Pedersen and Andrea Moon of the x-ray crystallography core laboratory at NIEHS and Kasia Bebenek for the generous gift of purified pols µ and λ.
This article contains supporting information.
Author contributions—M. J. H., and S. H. W. conceptualization; M. J. H., J. K. H., and M.-L. Z. data curation; M. J. H. formal analysis; M. J. H., J. K. H., and M.-L. Z. methodology; M. J. H. writing-original draft; M. J. H., J. K. H., and S. H. W. writing-review and editing; J. K. H. and S. H. W. supervision; J. K. H. and M.-L. Z. investigation; S. H. W. resources; S. H. W. funding acquisition; S. H. W. project administration.
Funding and additional information—This work was funded by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences project numbers Z01 ES050158 and Z01 ES050159 to S.H.W. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- pol
- polymerase
- BER
- base excision repair
- EMSA
- electrophoretic mobility shift assays
- FL
- full-length
- MEF
- mouse embryo fibroblast
- ROI
- region of interest.
References
- 1. Krokan H. E., and Bjoras M. (2013) Base excision repair. Cold Spring Harb. Perspect. Biol. 5, a012583 10.1101/cshperspect.a012583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Beard W. A., Horton J. K., Prasad R., and Wilson S. H. (2019) Eukaryotic base excision repair: new approaches shine light on mechanism. Annu. Rev. Biochem. 88, 137–162 10.1146/annurev-biochem-013118-111315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wilson S. H., and Kunkel T. A. (2000) Passing the baton in base excision repair. Nat. Struct. Biol. 7, 176–178 10.1038/73260 [DOI] [PubMed] [Google Scholar]
- 4. Prasad R., Shock D. D., Beard W. A., and Wilson S. H. (2010) Substrate channeling in mammalian base excision repair pathways: passing the baton. J. Biol. Chem. 285, 40479–40488 10.1074/jbc.M110.155267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Howard M. J., and Wilson S. H. (2018) DNA scanning by base excision repair enzymes and implications for pathway coordination. DNA Repair 71, 101–107 10.1016/j.dnarep.2018.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berg O. G., Winter R. B., and von Hippel P. H. (1981) Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry 20, 6929–6948 10.1021/bi00527a028 [DOI] [PubMed] [Google Scholar]
- 7. Halford S. E., and Marko J. F. (2004) How do site-specific DNA-binding proteins find their targets?. Nucleic Acids Res. 32, 3040–3052 10.1093/nar/gkh624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Howard M. J., and Wilson S. H. (2017) Processive searching ability varies among members of the gap-filling DNA polymerase X family. J. Biol. Chem. 292, 17473–17481 10.1074/jbc.M117.801860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hedglin M., and O'Brien P. J. (2008) Human alkyladenine DNA glycosylase employs a processive search for DNA damage. Biochemistry 47, 11434–11445 10.1021/bi801046y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beard W. A., and Wilson S. H. (2019) DNA polymerase beta and other gap-filling enzymes in mammalian base excision repair. Enzymes 45, 1–26 10.1016/bs.enz.2019.08.002 [DOI] [PubMed] [Google Scholar]
- 11. Beard W. A., and Wilson S. H. (2014) Structure and mechanism of DNA polymerase beta. Biochemistry 53, 2768–2780 10.1021/bi500139h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matsumoto Y., and Kim K. (1995) Excision of deoxyribose phosphate residues by DNA polymerase beta during DNA repair. Science 269, 699–702 10.1126/science.7624801 [DOI] [PubMed] [Google Scholar]
- 13. Garcia-Diaz M., Bebenek K., Kunkel T. A., and Blanco L. (2001) Identification of an intrinsic 5'-deoxyribose-5-phosphate lyase activity in human DNA polymerase lambda: a possible role in base excision repair. J. Biol. Chem. 276, 34659–34663 10.1074/jbc.M106336200 [DOI] [PubMed] [Google Scholar]
- 14. Jezewska M. J., Galletto R., and Bujalowski W. (2002) Dynamics of gapped DNA recognition by human polymerase beta. J. Biol. Chem. 277, 20316–20327 10.1074/jbc.M200918200 [DOI] [PubMed] [Google Scholar]
- 15. Howard M. J., Rodriguez Y., and Wilson S. H. (2017) DNA polymerase beta uses its lyase domain in a processive search for DNA damage. Nucleic Acids Res. 45, 3822–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kirby T. W., DeRose E. F., Cavanaugh N. A., Beard W. A., Shock D. D., Mueller G. A., Wilson S. H., and London R. E. (2012) Metal-induced DNA translocation leads to DNA polymerase conformational activation. Nucleic Acids Res. 40, 2974–2983 10.1093/nar/gkr1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eustermann S., Videler H., Yang J. C., Cole P. T., Gruszka D., Veprintsev D., and Neuhaus D. (2011) The DNA-binding domain of human PARP-1 interacts with DNA single-strand breaks as a monomer through its second zinc finger. J. Mol. Biol. 407, 149–170 10.1016/j.jmb.2011.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Prasad R., Beard W. A., Chyan J. Y., Maciejewski M. W., Mullen G. P., and Wilson S. H. (1998) Functional analysis of the amino-terminal 8-kDa domain of DNA polymerase beta as revealed by site-directed mutagenesis. DNA binding and 5'-deoxyribose phosphate lyase activities. J. Biol. Chem. 273, 11121–11126 10.1074/jbc.273.18.11121 [DOI] [PubMed] [Google Scholar]
- 19. Horton J. K., Gassman N. R., Dunigan B. D., Stefanick D. F., and Wilson S. H. (2015) DNA polymerase beta-dependent cell survival independent of XRCC1 expression. DNA Repair 26, 23–29 10.1016/j.dnarep.2014.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Horton J. K., Seddon H. J., Zhao M. L., Gassman N. R., Janoshazi A. K., Stefanick D. F., and Wilson S. H. (2017) Role of the oxidized form of XRCC1 in protection against extreme oxidative stress. Free Radic. Biol. Med. 107, 292–300 10.1016/j.freeradbiomed.2017.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horton J. K., Stefanick D. F., Gassman N. R., Williams J. G., Gabel S. A., Cuneo M. J., Prasad R., Kedar P. S., Derose E. F., Hou E. W., London R. E., and Wilson S. H. (2013) Preventing oxidation of cellular XRCC1 affects PARP-mediated DNA damage responses. DNA Repair 12, 774–785 10.1016/j.dnarep.2013.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang J., Alnajjar K. S., Mahmoud M. M., Eckenroth B., Doublie S., and Sweasy J. B. (2018) The nature of the DNA substrate influences pre-catalytic conformational changes of DNA polymerase beta. J. Biol. Chem. 293, 15084–15094 10.1074/jbc.RA118.004564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li M., Lu L. Y., Yang C. Y., Wang S., and Yu X. (2013) The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes Dev. 27, 1752–1768 10.1101/gad.226357.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Breslin C., Hornyak P., Ridley A., Rulten S. L., Hanzlikova H., Oliver A. W., and Caldecott K. W. (2015) The XRCC1 phosphate-binding pocket binds poly (ADP-ribose) and is required for XRCC1 function. Nucleic Acids Res. 43, 6934–6944 10.1093/nar/gkv623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hanzlikova H., Gittens W., Krejcikova K., Zeng Z., and Caldecott K. W. (2017) Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 45, 2546–2557 10.1093/nar/gkw1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beard W. A., and Wilson S. H. (1995) Purification and domain-mapping of mammalian DNA polymerase beta. Methods Enzymol. 262, 98–107 10.1016/0076-6879(95)62013-3 [DOI] [PubMed] [Google Scholar]
- 27. Sobol R. W., Prasad R., Evenski A., Baker A., Yang X. P., Horton J. K., and Wilson S. H. (2000) The lyase activity of the DNA repair protein beta-polymerase protects from DNA-damage-induced cytotoxicity. Nature 405, 807–810 10.1038/35015598 [DOI] [PubMed] [Google Scholar]
- 28. Dantzer F., de La Rubia G., Menissier-De Murcia J., Hostomsky Z., de Murcia G., and Schreiber V. (2000) Base excision repair is impaired in mammalian cells lacking poly(ADP-ribose) polymerase-1. Biochemistry 39, 7559–7569 10.1021/bi0003442 [DOI] [PubMed] [Google Scholar]
- 29. Hutchison C. A. 3rd, Phillips S., Edgell M. H., Gillam S., Jahnke P., and Smith M. (1978) Mutagenesis at a specific position in a DNA sequence. J. Biol. Chem. 253, 6551–6560 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript.