

Abstract
Ambra1 is considered an autophagy and trafficking protein with roles in neurogenesis and cancer cell invasion. Here, we report that Ambra1 also localizes to the nucleus of cancer cells, where it has a novel nuclear scaffolding function that controls gene expression. Using biochemical fractionation and proteomics, we found that Ambra1 binds to multiple classes of proteins in the nucleus, including nuclear pore proteins, adaptor proteins such as FAK and Akap8, chromatin-modifying proteins, and transcriptional regulators like Brg1 and Atf2. We identified biologically important genes, such as Angpt1, Tgfb2, Tgfb3, Itga8, and Itgb7, whose transcription is regulated by Ambra1-scaffolded complexes, likely by altering histone modifications and Atf2 activity. Therefore, in addition to its recognized roles in autophagy and trafficking, Ambra1 scaffolds protein complexes at chromatin, regulating transcriptional signaling in the nucleus. This novel function for Ambra1, and the specific genes impacted, may help to explain the wider role of Ambra1 in cancer cell biology.
Keywords: Ambra1, trafficking, autophagy, nucleus, transcription, Atf2, chromatin, Akap8, Cdk9, DNA transcription
Ambra1 (activating molecule in Beclin1-regulated autophagy) is already known to be an important protein in physiology, e.g. in the development of the central nervous system, vertebrate embryogenesis, adult neurogenesis, and cancer cell invasion (1–5). However, the full range of functions of this crucial cellular regulator is not understood. As an important autophagy regulator, Ambra1 binds Beclin1 and is involved in the initiation of autophagy that is needed for neurogenesis (2). In the absence of autophagy, the Ambra1–Beclin1–Vps34 complex is bound to the dynein motor complex; when autophagy is induced, the kinase ULK1 phosphorylates Ambra1, resulting in its release from the dynein complex (6, 7). Additionally, the function of Ambra1 is negatively regulated by mTOR, which suppresses its binding to the E3-ligase TRAF1 and the ubiquitylation of ULK1, thereby controlling the stability and function of ULK1 (8). During apoptosis, caspases and calpains mediate cleavage as well as degradation of Ambra1 (9). Furthermore, Ambra1 expression is regulated by RNF2-dependent ubiquitylation, resulting in degradation (10). Ambra1 is also involved in the regulation of mitophagy (11).
Ambra1 has been both positively and negatively implicated in cancer. Thus far, it has been proposed as a tumor suppressor, supporting the binding of c-Myc to the phosphatase PP2A, resulting in c-Myc degradation as well as reduced proliferation and tumorigenesis (12). Ambra1 has been positively implicated in cholangiocarcinoma, where overexpression is correlated with invasion and poor survival (5). In addition, through its ability to bind PP2A, Ambra1 stabilizes the transcription factor FOXO3, triggering FOXP3-mediated transcription, and T-cell differentiation and homeostasis (13).
We reported previously that in squamous cell carcinoma (SCC) cells derived from the mutated oncogenic H-Ras-driven DMBA/TPA model of carcinogenesis, Ambra1 is a focal adhesion kinase (FAK)- and Src-binding partner, regulating cancer cell polarization and chemotactic invasion (4, 14). In FAK-depleted SCC cells, Ambra1 is involved in the targeting of active Src to intracellular autophagic puncta, whereas an Ambra1-binding-impaired FAK mutant retains more active FAK and Src at focal adhesions, resulting in increased cell adhesion and invasion. We concluded that Ambra1 lies at the heart of an intracellular trafficking network in SCC cells, regulating the localization of active FAK and Src required for cancer processes (4).
Here, we investigate a nuclear function of Ambra1 and show that it binds to FAK in the nucleus, as well as to other nuclear adaptor proteins, nuclear pore components, histone-modifying enzymes, and regulators of transcription, in some cases regulating their recruitment to chromatin. Specifically, Ambra1 forms complexes with Akap8, Brg1, and Atf2 and is responsible for the recruitment of Akap8, Bgr1, the Mediator complex component Cdk9, and p-Atf2 T71 to chromatin. Both Ambra1 and its binding protein Akap8 regulate the binding of transcriptional proteins to chromatin, especially p-Atf2 T71, and proteins that modulate histone modifications. The binding of Atf2/p-Atf2 T71 to chromatin is most likely regulated by the Ambra1-interacting protein Cdk9. Therefore, Ambra1 acts as a scaffold protein in the nucleus, recruiting transcriptional regulators to chromatin. This creates an Ambra1-dependent nuclear microdomain that regulates gene expression.
Results
Ambra1 localizes to the nucleus
Here, we show that Ambra1 locates not only at focal adhesions and in intracellular autophagic puncta in SCC cells but also in the nucleus (Fig. 1A). Staining with only secondary antibodies (anti-rabbit 488 and anti-mouse 594) ruled out unspecific false-positive nuclear staining (Fig. S1A). To confirm Ambra1 was nuclear biochemically and to investigate whether this localization depended on FAK, fractions of SCC FAK-deficient (−/−) cells and the same cells reexpressing WT FAK to levels similar to those of parental SCC cells (FAK-WT) were isolated and subjected to Western blotting (Fig. 1B) (15, 16). Biochemical nuclear isolations were checked by blotting with anti-GAPDH, anti-Lamin A/C, and anti-H4. In the nuclear fractions of these SCC cells, we could detect Ambra1 as well as FAK, with the latter being in line with our previous reports (17, 18). The nuclear localization of Ambra1 was not dependent on FAK, as nuclear Ambra1 was present at indistinguishable levels in nuclear fractions from both FAK −/− and FAK-WT SCC cells and the same cells reexpressing WT FAK to levels similar to those of parental SCC cells (FAK-WT) (15, 16). In more highly purified cellular fractions of SCC FAK-WT and −/− cell lysates, extracting cytosolic (C), perinuclear (PN), and nuclear (N) fractions (see the supporting text and Fig. S1B), Ambra1 was present at comparable levels in the cytosolic and nuclear fractions and at even higher levels in the perinuclear fraction. Fraction purities were confirmed by blotting respective fractions with anti-GM130, anti-PDI, anti-Lamin A/C, and anti-GAPDH (Fig. S1B). Ambra1 could also be detected in the nucleus of a human SCC cell line (Fig. S1C). Furthermore, in contrast to FAK, Ambra1 was also detected in nuclear extracts from primary mouse keratinocytes (Fig. S1D) (18).
Figure 1.
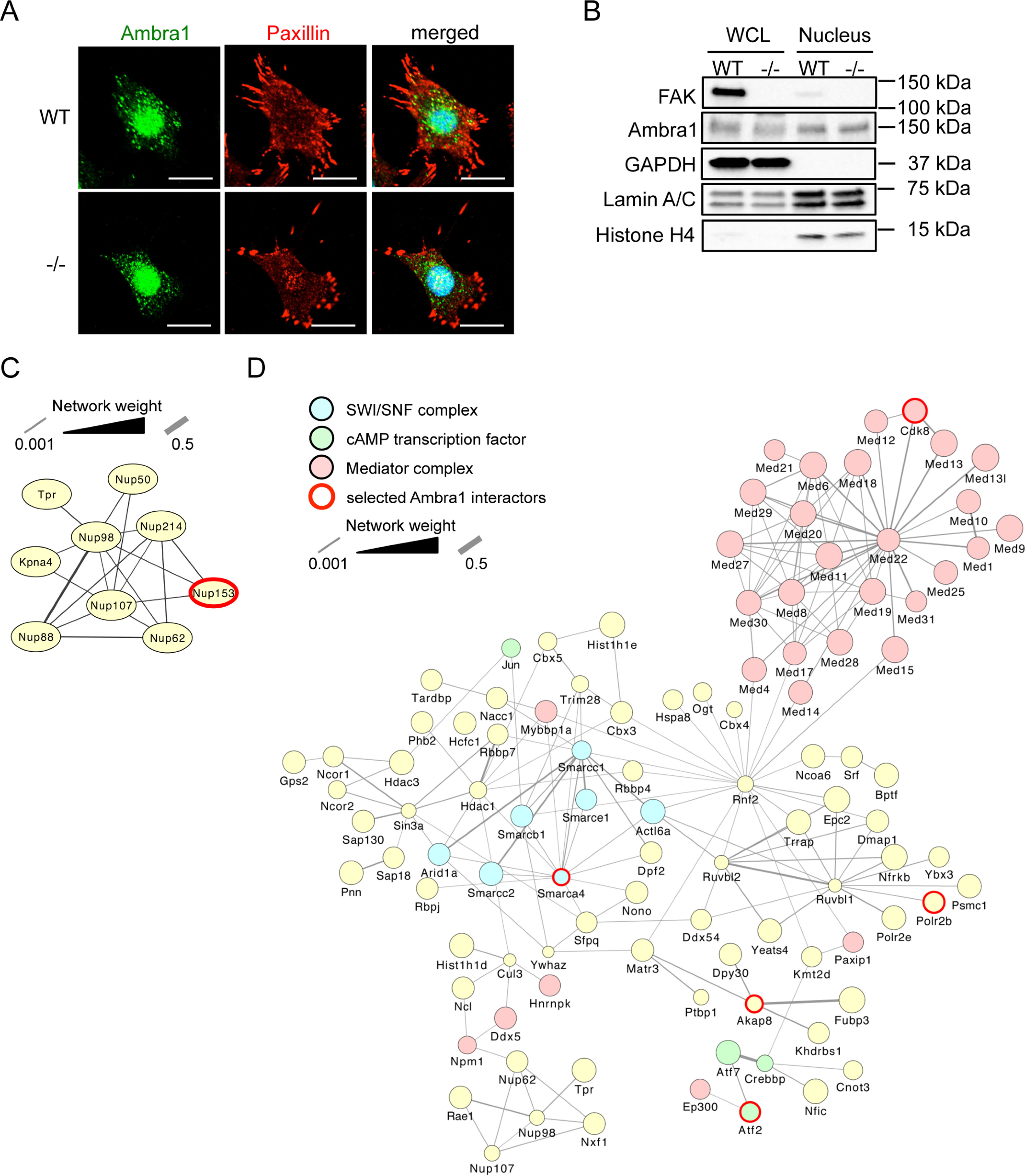
Nuclear Ambra1 binds chromatin modifiers and transcriptional regulators. A, representative immunofluorescence images of SCC FAK-WT and −/− cells, which were grown on glass coverslips for 24 h, fixed, and stained with anti-Ambra1, anti-Paxillin, and DAPI. Scale bars, 20 μm. B, whole-cell and nuclear lysates of SCC FAK-WT and −/− cells were subjected to Western blot analysis with anti-Ambra1 and anti-FAK. Anti-GAPDH, anti-Lamin A/C, and anti-H4 served as controls for the purity of the nuclear extracts as well as loading controls. C and D, interaction network analysis of nuclear Ambra1-binding proteins in SCC FAK-WT and −/− cells associated with the nuclear pore complex (C) and being involved in transcription (D), as determined by gene ontology enrichment analysis of biological processes. Hits were filtered for statistically significant (p < 0.05) 2-fold enrichment over the IgG control and used to build a protein interaction network based on direct physical interactions (gray lines). Components of various complexes involved in transcription are highlighted: SWI/SNF complex (blue nodes), cAMP-regulated transcription factor (green nodes), and Mediator complex (light red nodes). Ambra1-interacting proteins selected for further investigation are highlighted with a red border.
Nuclear Ambra1 binds proteins involved in transcription
Next, we investigated the nature of protein binding partners of Ambra1 in the nucleus. Highly purified nuclear extracts of SCC FAK-WT and −/− cells were obtained by sucrose gradient centrifugation and used for Ambra1 immunoprecipitations (anti-rabbit IgG served as a negative control), and specific nuclear binding proteins were determined by quantitative label-free MS (see Materials and methods) (Table S1). In total, 456 Ambra1-interacting proteins were identified, among which were several proteins that form part of the nuclear pore complex (interaction network shown in Fig. 1C; Table S2). Indeed, we had previously observed that the nuclear pore protein Tpr was an Ambra1-interacting protein using whole-cell lysates (4), together implying that Ambra1 was associated with nuclear pore proteins either during entry into the nucleus or as part of its nuclear functions. Gene ontology enrichment analysis of biological processes attributed to proteins that bind Ambra1 in nuclear fractions revealed transcription, mRNA processing, histone modification, as well as chromatin modification and remodeling as the most highly overrepresented categories (Fig. S2A). As a result of these analyses, we filtered all identified Ambra1-binding proteins for Ambra1 interactors likely to be involved in the regulation of transcription and used these hits to build a protein interaction network based on known direct physical interactions (Fig. 1D and Table S2). Among these were several components of the Mediator complex, a multiprotein complex that functions as a transcriptional coactivator for RNA polymerase II (highlighted in light red in Fig. 1D) (19, 20). Also present were several components of the SWI/SNF (SWItch/sucrose nonfermentable) nucleosome remodeling complex that allows transcription factor binding by opening up the chromatin structure, e.g. the catalytic subunit SMARCA4 (Brg1), which allows ATP-dependent chromatin remodeling (represented in blue in Fig. 1D) (21–23). Nuclear Ambra1 was also found to interact with several members of the cAMP-dependent AP-1 complex, including c-Jun, Fosl2, Atf2 (a member of the CREB [cAMP response element binding] family of leucine zipper proteins), and Atf7, which also binds to nuclear FAK (represented in green in Fig. 1D) (18, 24). As cAMP-regulated transcription factors were among nuclear Ambra1-binding proteins, we also noted that nuclear Ambra1 binds to Akap8 (A kinase anchor protein 8, also known as Akap95), a scaffold that targets PKA to cAMP-responsive elements in gene promoters that is linked to chromatin status and retention of p90 S6K in the nucleus (25–28). Interestingly, both Atf2 and Akap8 were also identified by proteomics as Ambra1-binding proteins using whole-cell lysates in our previous experiments (4).
We next selected a number of proteins identified by mass spectrometric analyses, i.e. Nup153, Akap8, Brg1, Atf2, and the RNA polymerase II Rpb1 (all highlighted with red borders in Fig. 1, C and D) and confirmed their binding to Ambra1 in the nucleus by coimmunoprecipitation in nuclear fractions (Fig. 2, A–E; in contrast, we show an example of a nuclear protein, PARP, that does not bind Ambra1 in coimmunoprecipitation experiments in Fig. S2B). One question we had, given our previously reported cofunctioning of Ambra1 and FAK in the cytoplasm, was whether or not FAK regulates the nuclear translocation of its binding partners, such as Ambra1, which also locates to the nucleus. We did not find any significant difference in the nuclear levels of Ambra1 or its interaction with the nuclear binding partners examined between SCC cells expressing FAK-WT compared with FAK-deficient (−/−) cells; therefore, we conclude that FAK does not regulate the trafficking of Ambra1 to the nucleus or Ambra1 interactions there. Therefore, in future experiments, we have generally only presented data from SCC cells expressing FAK-WT.
Figure 2.

Nuclear Ambra1 binding to MS-identified interaction partners. Ambra1 was immunoprecipitated from whole-cell and nuclear lysates of SCC FAK-WT and −/− cells using anti-Ambra1, followed by Western blot analysis with anti-Ambra1 and anti-Nup153 (A), anti-Akap8 (B), anti-Brg1 (C), anti-Atf2 (D), and anti-Rpb1 (E). Anti-Lamin A/C and anti-GAPDH were used as a control for the purity of the nuclear lysates as well as a loading control.
Loss of Ambra1 causes reduced association of interacting partners with chromatin
Because Ambra1 has predominantly been defined as a scaffold protein involved in intracellular trafficking/autophagy thus far, we hypothesized that Ambra1 also serves as a scaffold protein in the nucleus. Using efficient siRNA pool-mediated depletion that we have used previously (4), we found that reducing Ambra1 levels only modestly altered levels of its binding proteins in the nucleus (Brg1, Cdk9, and p-Atf2 T71) (Fig. 3, A and B). We next isolated chromatin from SCC FAK-WT cells after Ambra1 depletion and probed for FAK, Brg1, Akap8, Atf2, and its phosphorylated (and activated) form, p-Atf2 T71 (Fig. 3, C and D). Reduced expression of Ambra1 suppressed the binding of FAK, Brg1, Akap8, and p-Atf2 (but not visibly total Atf2) to chromatin to a greater or lesser extent. This suggests that, in addition to Ambra1-mediated chromatin recruitment, there are likely other routes by which binding partners are recruited to chromatin because their recruitment is reduced, but not ablated, upon Ambra1 loss. In these analyses, we also included the Cdk8 and Cdk9 components of the Mediator complex that modulates RNA polymerase II-mediated transcription (29–31). These were included because they were 1) identified as binding to Ambra1 in MS experiments (highlighted with a red border [for Cdk8] in Fig. 1D; Cdk9 was inferred from a single peptide identification, Fig. S2C and Table S2) and 2) predicted to be enzymes with the potential to phosphorylate Atf2 at residue T71 on chromatin, which we observed in the presence of Ambra1 (Fig. 3; predictions via the Biocuckoo phosphorylation prediction site for Cdk8 [score, 40.329; cutoff, 29.727] and Cdk9 [score, 7.393; cutoff, 2.931]). In keeping with other Ambra1-binding proteins studied here, we found that Cdk9, but not Cdk8, binding to chromatin was reduced upon depletion of Ambra1 (Fig. 3, C and D). In this regard, Cdk9 has been reported to bind another SWI/SNF complex component, SMARCB1, which has also been identified as a nuclear Ambra1 binding protein (32), implying there are other components of Ambra1 complexes that link to chromatin remodeling. Taken together, our data imply that Ambra1 forms complexes in the nucleus with other protein scaffolds, such as the PKA scaffold Akap8, chromatin modifiers, and transcription factors, including Atf2.
Figure 3.
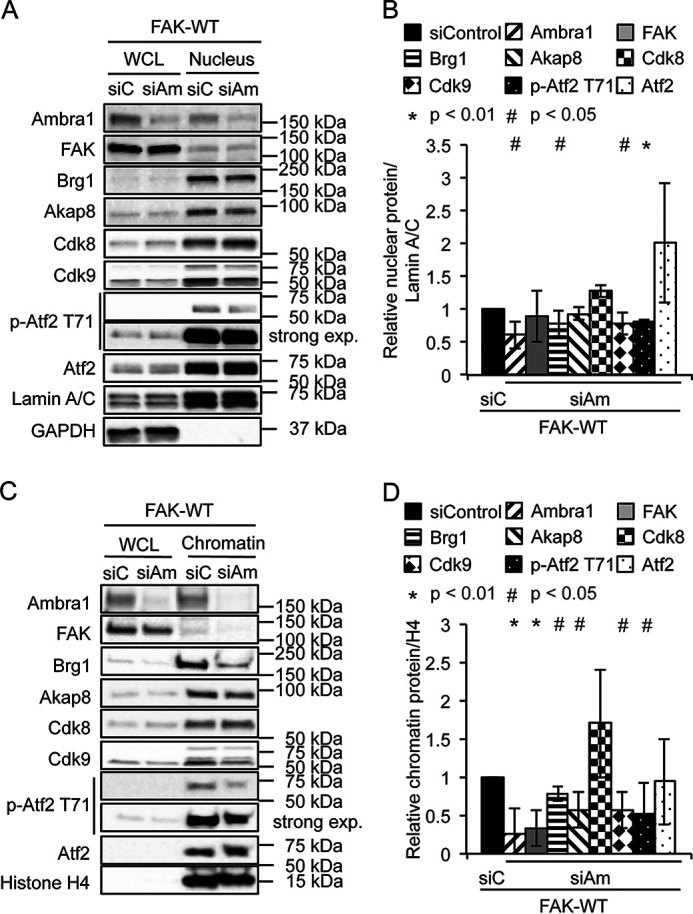
Loss of Ambra1 leads to reduced association of interacting proteins with chromatin. A, SCC FAK-WT cells were transfected with siControl and siAmbra1 (siGENOME pool). After 48 h, whole-cell and nuclear lysates were analyzed by Western blotting using anti-Ambra1, anti-FAK, anti-Brg1, anti-Akap8, anti-Cdk8, anti-Cdk9, anti-p-Atf2 T71, and anti-Atf2. Anti-Lamin A/C and anti-GAPDH were used as controls for the purity of the nuclear lysates as well as loading controls. B, the graph shows relative nuclear protein levels normalized to Lamin A/C. Error bars represent S.D. *, p < 0.01; #, p < 0.05. C, SCC FAK-WT cells were transfected with siControl and siAmbra1. After 48 h, whole-cell lysates and chromatin extracts were analyzed by Western blotting using anti-Ambra1, anti-FAK, anti-Brg1, anti-Akap8, anti-Cdk8, anti-Cdk9, anti-p-Atf2 T71, and anti-Atf2. Anti-histone H4 served as a marker for chromatin as well as a loading control. D, the graph shows relative chromatin protein levels normalized to histone H4. Error bars represent S.D. *, p < 0.01; #, p < 0.05.
Akap8 also regulates the level of p-Atf2 at chromatin
Ambra1 binds the PKA scaffold Akap8 and is required for its optimal binding to chromatin. Upon depletion of Akap8 by pooled siRNA, we found that while neither Ambra1 nor FAK binding to chromatin was affected, showing it was downstream of FAK/Ambra1, the level of p-Atf2 T71 associated with chromatin was reduced (Fig. 4, A and B). This implies a model (Fig. 4E) whereby Ambra1 is upstream of recruitment of Akap8 to chromatin, and Akap8, in turn, is required for optimal chromatin association of active p-Atf2 that is also Ambra1 dependent. Total Atf2 recruitment was not reduced by depletion of Akap8, suggesting that a specific function of Akap8 is to recruit the enzyme that phosphorylates Atf2 at chromatin. We noted that the activity of Atf2 is proposed to be regulated by phosphorylation at several residues, including T71, by kinases such as ERK, JNK, p38, and PLK3, promoting Atf2 heterodimerization and increased transcription and histone acetyl transferase activity (24, 33–39). However, using inhibitors of the kinases proposed above to phosphorylate Atf2 on T71, we did not find evidence for an obvious key role for any of these in regulating Atf2 phosphorylation at chromatin in SCC cells used here (not shown). Therefore, we examined the Mediator complex kinases Cdk8 and Cdk9, which, as mentioned previously according to the Biocuckoo phosphorylation prediction site, potentially phosphorylate Atf2 at T71. Therefore, we depleted Cdk8 and Cdk9 in SCC FAK-WT cells and prepared nuclear and chromatin fractions to probe for p-Atf2 T71. We found that depletion of Cdk9 resulted in reduced chromatin-associated p-Atf2 T71 (Fig. 4, C and D) that is also Ambra1 and Akap8 dependent; however, in this case, total Atf2 recruited to chromatin was also reduced. These findings imply that the Mediator complex component Cdk9, which binds to Ambra1 in the nucleus, controls recruitment of Atf2 to chromatin downstream of Ambra1 and also Atf2 phosphorylation/activation.
Figure 4.

Depletion of Akap8 and Cdk9 reduce p-Atf2 T71 binding to chromatin. A, SCC FAK-WT cells were transfected with siControl and siAkap8 (siGENOME pool). After 48 h, whole-cell lysates and chromatin extracts were analyzed by Western blotting using anti-Ambra1, anti-FAK, anti-p-Atf2 T71, anti-Atf2, and anti-Akap8. Anti-histone H4 served as a marker for chromatin as well as a loading control. B, the graph shows relative chromatin protein levels normalized to histone H4. Error bars represent S.D. *, p < 0.01; #, p < 0.05. C, SCC FAK-WT cells were transfected with siControl, siCdk8, and siCdk9 (siGENOME pool). After 48 h, whole-cell lysates and nuclear and chromatin extracts were analyzed by Western blotting using anti-pAtf2 T71, anti-Atf2, anti-Cdk8, and anti-Cdk9. Anti-Lamin A/C, anti-GAPDH, and anti-histone H4 served as controls for the purity of nuclear and chromatin extracts as well as loading controls. D, the graph shows relative nuclear or chromatin protein levels normalized to Lamin A/C or histone H4, respectively. Error bars represent S.D. *, p < 0.01; #, p < 0.05. E, hypothesis model. In the nucleus of SCC cells, Ambra1 and Akap8 form a complex and contribute to the recruitment of active Atf2 (p-Atf2 T71) to chromatin, most likely downstream via the Mediator complex component Cdk9. This strongly suggests that Ambra1 is involved in chromatin remodeling and transcription. Further, together with Akap8 and Atf2, it might coregulate the expression of a subset of genes.
Thus, Ambra1 and Akap8, which form a complex in the nucleus of SCC cells, both contribute to the recruitment of active Atf2 (p-Atf2 T71) to chromatin, likely, at least in part, via the Mediator complex component Cdk9 downstream (see the model in Fig. 4E). Therefore, an obvious question that follows is whether Ambra1, Akap8, and Atf2 coregulate the expression of a subset of genes.
Ambra1, Akap8, CDK9, and Atf2 coregulate a subset of genes
The data presented to this point showed that Ambra1 localizes to the nucleus, associates with chromatin, and interacts with nuclear proteins that regulate transcription. Both Ambra1 and its binding partner, Akap8, recruit transcription factors, such as the active form of Atf2, p-Atf2 T71, which is proposed to result in histone modifications and altered chromatin accessibility, leading to transcriptional changes (40). To address whether there were genes whose transcription was coregulated by Ambra1, Akap8, and Atf2, SCC FAK-WT cells were transfected with siControl, siAmbra1, siAkap8, or siAtf2 siRNA. A subset of genes whose expression was changed by all three protein depletions was identified using the nCounter PanCancer Pathways Panel. In total, we identified 94 genes that were significantly (p < 0.05) at least 2-fold up- or downregulated compared with control siRNA (Fig. 5, A and B, and Fig. S3, A–F). Ambra1, Atf2, or Akap8 depletion significantly altered the expression of 18 genes from this panel (Fig. 5, A and B). To validate the gene expression changes, we performed qRT-PCR for the coregulated genes Angpt1, Tgfb2, Tgfb3, Itga8, and Itgb7 (Fig. 5, D and E). For Angpt1, Tgfb2, Tgfb3, and Itga8, we confirmed the downregulation upon siRNA transfection (Fig. 5E), whereas upregulation of Itgb7 was also confirmed (Fig. 5E). In addition, pathway analysis of Ambra1-, Akap8-, and Atf2-regulated genes revealed the top enriched signaling pathway gene sets, “PI3K-Akt signaling pathway,” “pathways in cancer,” “focal adhesion,” and “MAPK signaling pathway,” were in common (Fig. 5C; for a full list, see Fig. S4), strongly suggesting an overlap in the functions of the genes regulated by Ambra1, Akap8, and Atf2 complexes.
Figure 5.
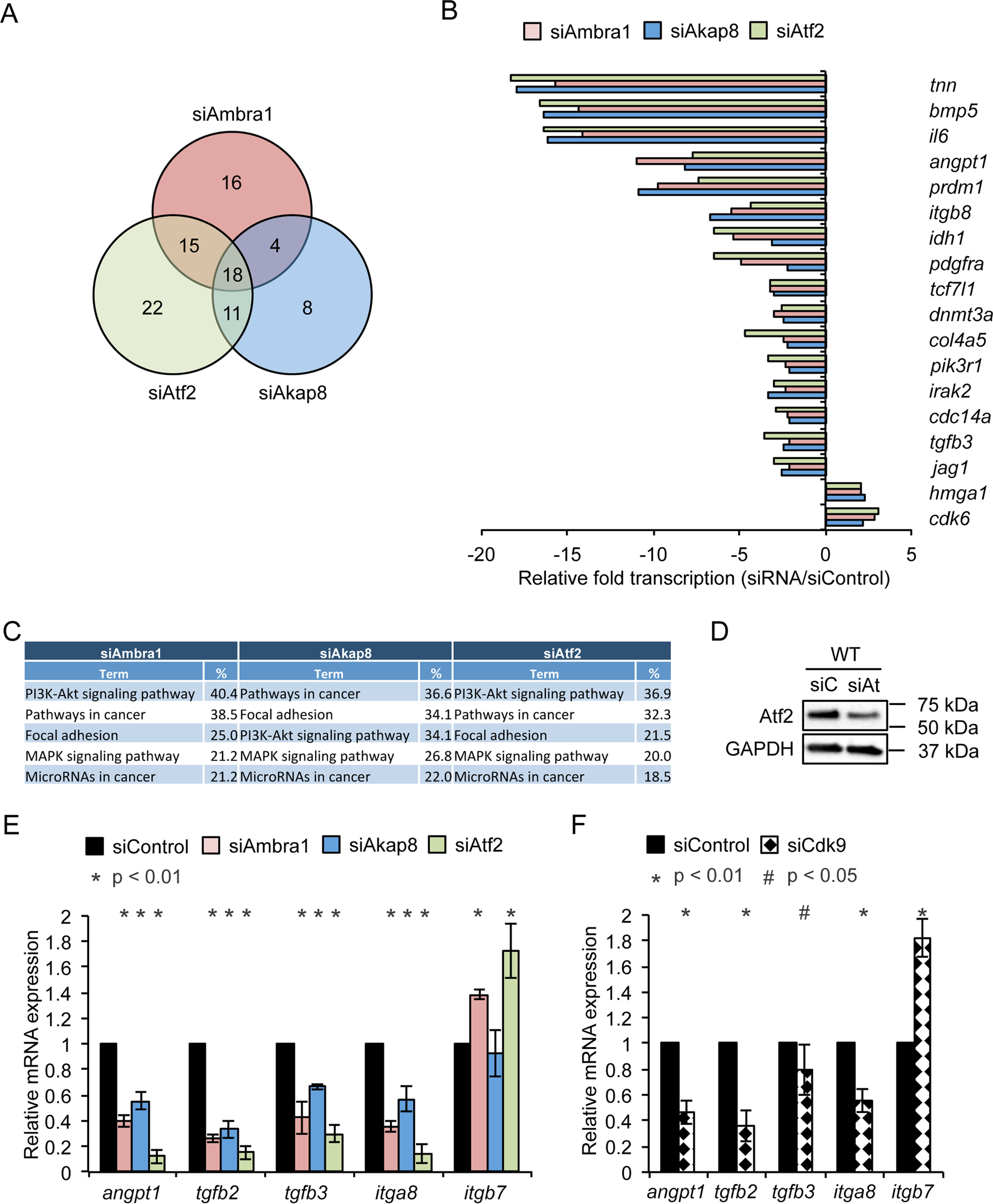
Ambra1, Akap8, Cdk9, and Atf2 coregulate a subset of genes. SCC FAK-WT cells were transfected with siControl, siAmbra1, siAkap8, and siAtf2 (siGENOME pool). RNA was isolated 48 h posttransfection and subjected to gene expression analysis using the mouse nCounter PanCancer Pathways panel. A, Venn diagram of significantly altered genes compared with siControl (p < 0.05, 2-fold difference compared with siControl). B, relative gene expression of 18 genes coregulated by Ambra1, Akap8, and Atf2 knockdown compared with siControl. C, table of the top five enriched signaling pathway gene sets according to KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis of Ambra1-, Akap8-, and Atf2-regulated genes. D, Western blotting showing Atf2 knockdown by siRNA in SCC FAK-WT cells. Anti-GAPDH served as a loading control. E and F, validation of Ambra1-, Akap8-, and Atf2-mediated Angpt1, Tgfb2, Tgfb3, Itga8, and Itgb7 expression changes by qRT-PCR using RNA isolated 48 h posttransfection of SCC FAK-WT cells with siControl, siAmbra1, siAkap8, and siAtf2 (E) as well as siControl and siCdk9 (F). Error bars represent S.D. *, p < 0.01; #, p < 0.05.
Because we found the nuclear Ambra1-binding Mediator complex protein Cdk9 to be part of the regulation of p-Atf2 at chromatin in SCC cells (Fig. 4, C and D), we next addressed whether Cdk9 was also implicated in the transcriptional regulation of the above coregulated genes. Depletion of Cdk9 by siRNA resulted in gene expression changes broadly similar to that observed upon depletion of Ambra1, Akap8, or Atf2, e.g. expression of Angpt1, Tgfb2, Tgfb3, and Itga8 was reduced (Fig. 5F), whereas Itgb7 expression was increased, in qualitative agreement with the effects of depleting Ambra1, Akap8, or Atf2 (Fig. 5F), suggesting that Ambra1, Akap8, Atf2, and Cdk9 bind to, and regulate, these same gene promoters.
Taken together, these results imply that Ambra1, Akap8, Atf2, and, most likely, Cdk9 are in the same chromatin complexes, likely at the promoters of coregulated genes described above, and potentially many more are not represented in the mouse nCounter PanCancer Pathways Panel used here. The presence of chromatin modifiers in the nuclear Ambra1 interactome, validated in the case of Brg1 (Fig. 1 and 2C), suggested the intriguing possibility that the novel transcriptional regulatory pathway we describe is, at least in part, regulated by chromatin accessibility. In keeping with this, the functioning of Atf2, which is phosphorylated at T71 downstream of Ambra1 and Akap8, is known to promote histone acetyl transferase activity and transcription (35, 38).
Ambra1 and Akap8 regulate histone modifications
Functional interaction network analysis of nuclear Ambra1 binding partners identified by MS revealed several components of histone modification complexes, including histone acetylation complexes NSL (nonspecific lethal complex), NuA4 (nucleosome acetyltransferase of H4), and PCAF (p300/CBP-associated factor), as well as the histone methylation complex MLL1/MLL (mixed-lineage leukemia 1) (Fig. 6A). Further, gene ontology enrichment analysis of biological processes attributed to nuclear Ambra1 interactors identified covalent chromatin and histone modifications as well as chromatin remodeling as overrepresented categories (Fig. S2). In addition, Akap8 has been reported to bind to Dpy30, a core subunit of H3K4 histone methyltransferases (41). Therefore, we next addressed whether Ambra1 and Akap8 influence histone modifications by transfecting SCC FAK-WT and −/− cells with siControl, siAmbra1 (Fig. 6, B and C), or siAkap8 (Fig. 6, D and E) siRNA and examined the histone modifications H3K4me2, H3K4me3, and H3K27Ac by Western blotting. Trimethylation of lysine-4 and acetylation of lysine-27 on histone 3 are generally regarded as positive indicators of transcriptional activation, promoting gene expression (42–45). Depletion of Ambra1 or Akap8 reduced both di- and trimethylation of H3K4 as well as acetylation of H3K27 (irrespective of whether FAK was present or not). This indicates that both Ambra1 and Akap8 can influence histone modifications, likely as a result of interacting histone-modifying enzymes, which could result in chromatin remodeling and altered accessibility of transcription factors.
Figure 6.
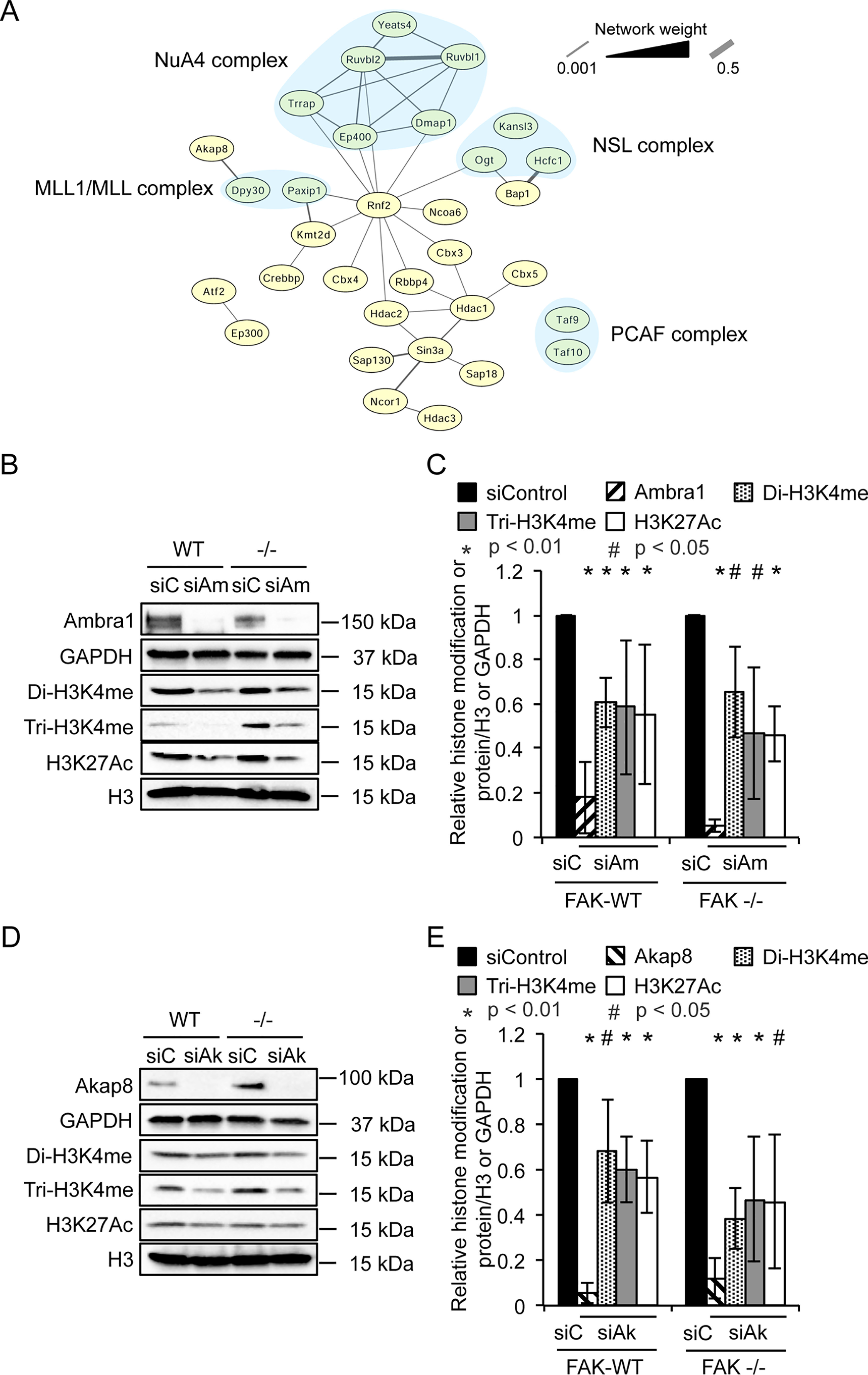
Ambra1 or Akap8 depletion decreases histone modifications. A, functional interaction network analysis of nuclear Ambra1 binding partners identified by MS were filtered for statistically significant (p < 0.05) 2-fold enrichment over IgG control. Components of histone modification complexes were used to build a protein interaction network based on direct physical interaction (gray lines). These complexes include histone acetylation complexes (NSL, nonspecific lethal; NuA4, nucleosome acetyltransferase of H4; PCAF, p300/CBP-associated factor) and a histone methylation complex (MLL1/MLL, mixed-lineage leukemia 1) (all highlighted by light blue background). B–E, SCC FAK-WT and −/− cells were transfected with siControl and siAmbra1 (B and C) or siControl and siAkap8 (D and E) (siGENOME pool). After 48 h, whole-cell lysates were subjected to Western blot analysis using anti-Ambra1, anti-Akap8, anti-di-H3K4me, anti-tri-H3K4me, and anti-H3K27Ac. Anti-GAPDH and anti-histone H3 served as loading controls. C and E, the graphs show relative histone modification levels normalized to histone H3 or protein levels normalized to GAPDH upon Ambra1 (C) or Akap8 (E) knockdown. Error bars represent S.D. *, p < 0.01; #, p < 0.05.
Discussion
Here, we describe a completely novel transcriptional signaling pathway controlled by the scaffold protein Ambra1 in the nucleus. Like Ambra1, other proteins involved in autophagy have been reported in the nucleus, e.g. LC3B binds to Lamin B1, mediating the degradation of the nuclear lamina and Beclin 1, promoting autophagy-independent DNA damage repair (46, 47). No typical nuclear localization sequence is evident for Ambra1; hence, the mechanism of nuclear translocation is unknown; however, as nuclear Ambra1 interacts with components of nuclear pore complexes and importins (Fig. 1C), it is likely that nuclear import of Ambra1 occurs via binding to these in some way. Nuclear Ambra1 interacts with chromatin modifiers and transcriptional regulators in the nucleus, including those also identified as proteins that bind to FAK and IL33, e.g. SMARCC1, Ruvbl1, and Ruvbl2 (17), suggesting there is a link between Ambra1 and FAK functions in the nucleus as well as in the cytoplasm (4). In this regard, we did find that the proteins associate in the nucleus and that depletion of Ambra1 leads to reduced FAK recruitment to chromatin.
The PKA scaffold Akap8, which binds to Ambra1 in the nucleus, has itself previously been strongly linked to histone modifications and chromatin changes. Indeed, by interacting with the MLL1/MLL complex via Dpy30, Akap8 regulates histone H3K4 methyltransferase complexes and binds to the nuclear matrix, nucleoporin component Tpr, as well as chromatin; in turn, this contributes to chromosome condensation and transcription, effects that are important for the mitotic checkpoint (41, 48–51). Akap8 also binds the histone deacetylase HDAC3 and influences mitosis (52). Therefore, existing studies had already suggested a scaffolding function for Akap8 in assembly of chromatin modification complexes. Akap8 dissociates from chromatin and the nuclear matrix as a result of nuclear tyrosine phosphorylation, and it may have a role in regulation of chromatin structural changes (53). Finally, a recent study confirmed the scaffold function of Akap8 in organizing nuclear microdomains, thereby controlling local cAMP for nuclear PKA regulation (54), although it is not clear whether this is a chromatin-associated function of Akap8.
Nuclear envelope and nuclear pore components, like Nup153, associate with chromatin and regulate genome organization and gene expression via nuclear pore complexes, acting as scaffold platforms to allow the assembly and recruitment of transcription factors to the nuclear periphery (55, 56). Therefore, Ambra1 might serve as a molecular scaffold that links chromatin to the nuclear pore complex, allowing active transcription factor binding (such as p-Atf2 T71) and resulting gene expression. The most likely explanation for our data is that Atf2 is phosphorylated by a kinase associated with chromatin. Therefore, we examined nuclear kinases interacting with Ambra1, e.g. Cdk9. However, we could not detect decreased Atf2 phosphorylation upon Cdk9 knockdown. We note that Cdk9 is also present in additional complexes besides the Mediator complex; therefore, it is possible that the effect of Cdk9 knockdown on the recruitment of p-Atf2 is independent of the Mediator complex. When we looked at potential Atf2-binding sequences in the genes that were altered after depletion of Ambra1, Akap8, and Atf2, we found that (according to RRID:SCR_008027) Tgfb2 and Tgfb3 both have CRE_TATA boxes that might serve as Atf2 binding sites. Moreover, it is likely that some of the cancer-associated functions of Ambra1 in SCC cells (such as cancer cell invasion) are associated with the nuclear transcription signaling effects we report here, e.g. on TGFβ isoforms as well as its trafficking effects that we reported previously (4).
Taken together, our data lead us to propose the following model (depicted in Fig. 7). Ambra1 was already known to localize to autophagosomes in the cytoplasm and to focal adhesions, where it regulates the removal of untethered tyrosine kinases via an autophagy mechanism (2, 4). We now show that Ambra1 also interacts with nuclear pore proteins and locates to the nucleus, where it is part of a complex network of interlinked chromatin modifiers and transcriptional regulators, including a set of interacting proteins whose recruitment to chromatin is influenced by Ambra1. This includes the PKA-scaffold Akap8, the Mediator complex component Cdk9, and the transcription factor Atf2 in its active form. Moreover, Ambra1, Akap8, Cdk9, and Atf2 coregulate the expression of a subset of genes. Both Ambra1 and Akap8 influence cellular histone modifications that could contribute to their transcriptional effects. Therefore, we have uncovered a completely novel function for the autophagy and trafficking protein Ambra1, which acts as a nuclear scaffold to recruit other scaffold proteins, chromatin modifiers, and transcriptional regulators to elicit gene expression changes via Atf2. A similar scaffolding mechanism creating nuclear transcription signaling hubs has been described for FAK, controlling Ccl5, Il33, Tgfb2, and Igfbp3 transcription, as well as for mAKAPβ, which creates nuclear signalosomes and binds the transcriptional regulators NFAT, MEF2, and HIF1α (18, 57, 58).
Figure 7.

Model depicting Ambra1-dependent transcriptional regulation. In mouse SCC cells, Ambra1 is already known to localize to autophagosomes and focal adhesions, where it binds FAK and Src and regulates the removal of untethered kinases via autophagy. Ambra1 can also interact with nuclear pore components and is translocated into the nucleus most likely via nuclear pores and importins. Nuclear Ambra1 is part of a network consisting of chromatin modifiers and transcriptional regulators, some of which are recruited to chromatin in an Ambra1-mediated manner, including the PKA scaffold Akap8, Cdk9, and active Atf2 (p-Atf2 T71). Further, Ambra1, Akap8, Cdk9, and Atf2 coregulate the expression of a subset of genes, like Angpt1, Tgfb2, Tgfb3, Itga8, and Itgb7. Both Ambra1 and Akap8 influence cellular histone modifications, which could contribute to their transcriptional effects. Overall, the autophagy protein Ambra1 also acts as a nuclear platform to recruit key scaffolds, chromatin modifiers, and transcriptional regulators to elicit gene expression changes via Atf2.
Materials and methods
Antibodies
Antibodies used were anti-Paxillin and anti-GM130 antibodies (BD Transduction Laboratories, New Jersey, USA), anti-Akap8 and anti-Nup153 (Abcam, Cambridge, UK), anti-FAK, anti-PDI, anti-p-Atf2 T71, anti-Atf2, anti-Brg1, anti-Rpb1, anti-Cdk8, anti-Cdk9, anti-histone H4, anti-H3K4me2, anti-H3K4me3, anti-H3K27Ac, anti-histone H3, anti-Lamin A/C, and anti-GAPDH (Cell Signaling Technologies, Danvers, MA, USA), as well as anti-Ambra1 antibody (Millipore, Billerica, MA, USA). Anti-rabbit or -mouse peroxidase-conjugated secondary antibodies were purchased from Cell Signaling Technologies.
Cell culture
FAK-deficient SCC cell lines were generated as described previously (16). SCC cells were maintained in Glasgow MEM containing 10% FCS, 2 mm l-glutamine, nonessential amino acids, sodium pyruvate, and MEM vitamins at 37 °C, 5% CO2. SCC FAK-WT cells were maintained in 1 mg/ml hygromycin B.
siRNA
FAK-WT or FAK −/− SCC cells were transiently transfected using HiPerFect (Qiagen, Manchester, UK) according to manufacturer's protocol, with a final concentration of 80 or 100 nm siRNA, respectively (Table S3). Cells were analyzed at 48 h posttransfection.
Whole-cell lysates
Cells were washed twice in ice-cold PBS and then lysed in radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Triton X-100, 0.1% SDS, and 0.5% sodium deoxycholate), supplemented with PhosSTOP and cOmplete Ultra phosphatase and protease inhibitor cocktails (Roche, Welwyn Garden City, UK), and cleared by centrifugation.
Nuclear fractionation
Cells were washed twice in ice-cold PBS and then lysed in DET buffer (150 mm NaCl, 25 mm Hepes, pH 7.5, 1 mm β-mercaptoethanol, 0.2 mm CaCl2, 0.5 mm MgCl2, 0.5% NP-40) supplemented with PhosSTOP and cOmplete Ultra phosphatase and protease inhibitor cocktails. Lysates were incubated on ice for 10 min and centrifuged. The resulting pellets were washed twice in DET buffer, resuspended in radioimmune precipitation assay buffer, and cleared by centrifugation.
For MS, nuclear lysates were prepared using a Nuclei PURE Prep isolation kit (Sigma, Gillingham, UK).
Chromatin isolation
The protocol was adapted from McAndrew et al. (59). All buffers were supplemented with PhosSTOP and cOmplete Ultra phosphatase and protease inhibitor cocktails. Briefly, cells were washed twice in ice-cold PBS, lysed in extraction buffer (10 mm Hepes, pH 7.9, 10 mm KCl, 1.5 mm MgCl2, 0.34 m sucrose, 10% glycerol, 0.2% NP-40), and centrifuged for 5 min at 6500 × g. Nuclear pellets were washed in extraction buffer without NP-40 and centrifuged for 5 min at 6500 × g. Pellets were resuspended in low-salt buffer (10 mm Hepes, pH 7.9, 3 mm EDTA, 0.2 mm EGTA) and incubated for 30 min at 4 °C with rotation before centrifugation for 5 min at 6500 × g. Pellets were resuspended in high-salt buffer (50 mm Tris-HCl, pH 8.0, 2.5 m NaCl, 0.05% NP-40) and incubated for 30 min at 4 °C with rotation. Supernatants containing chromatin fractions were cleared by centrifugation. Proteins were precipitated by adding TCA to a final volume of 10% and incubating on ice for 15 min. Precipitated proteins were pelleted by centrifugation, washed twice with ice-cold acetone, and then resuspended in 2× sample buffer prior to analysis by Western blotting.
Immunoprecipitation
Protein concentration was calculated using a BCA protein assay kit (Thermo Fisher Scientific, Loughborough, UK). For analysis by Western blotting, 1 mg lysates was incubated with 2 μg of unconjugated antibodies at 4 °C overnight with agitation. Unconjugated antibody samples were incubated with 10 μl of Protein A agarose for 1 h at 4 °C. Beads were washed three times in lysis buffer and once in 0.6 M LiCl, resuspended in 20 μl 2× sample buffer (130 mm Tris, pH 6.8, 20% glycerol, 5% SDS, 8% β-mercaptoethanol, bromphenol blue), and heated for 5 min at 95 °C. Samples were then subjected to SDS-PAGE analysis as described elsewhere (4).
For MS, Ambra1 was immunoprecipitated from 2 mg nuclear lysates of SCC FAK-WT and −/− cells (samples in triplicates) using a KingFisher Duo liquid handling station (Thermo Fisher Scientific). Nuclear lysates were incubated with 2 μg of antibody and 5 μl Protein G Mag-Sepharose (GE Healthcare) for 2 h at 4 °C. Beads were washed twice in lysis buffer and three times in TBS and then processed by proteolytic digestion.
Proteolytic digestion
For MS, immunoprecipitated proteins were digested using 5 μg/ml trypsin in 2 M urea, 50 mm Tris-HCl, pH 7.5, as reported previously (60). Digestion was performed for 30 min at 27 °C. After the initial digestion, the beads were removed using a KingFisher Duo, and the supernatants were left to continue to digest at 37 °C for 8 h. After digestion, samples were incubated with 50 mg/ml iodoacetamide for 30 min in the dark. Samples were treated with 1 μl TFA to stop the digestion and then desalted using C18 StageTips. Briefly, the tips were prepared by placing a small disc of 3M Empore solid-phase extraction material (Sigma-Aldrich) in an ordinary 200-μl-capacity pipette tip, preparing a single tip for each sample. Tips were activated with 80% acetonitrile, 0.1% TFA buffer and washed with 0.1% TFA. Samples (100 μl) were added to each column and washed twice with 0.1% TFA. Liquid was passed through the pipette tip manually with the aid of a syringe or with a light centrifugation step. Peptides were then eluted using 80% acetonitrile, 0.1% TFA. Samples were evaporated in a SpeedVac vacuum concentrator (Thermo Fisher Scientific), resuspended in 12 μl 0.1% TFA buffer, and analyzed by MS.
Mass spectrometric data acquisition
Tryptic peptides were analyzed on a Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer connected to an UltiMate Ultra3000 chromatography system (both from Thermo Fisher Scientific) incorporating an autosampler. Peptides (5 μl) for each sample were loaded on a homemade column (250-mm length, 75-μm inside diameter) packed with 1.8 μm UChrom C18 (nanoLCMS Solutions) and separated by an increasing acetonitrile gradient, using a 40-min reverse-phase gradient (from 3%–32% acetonitrile) at a flow rate of 250 nl/min. The mass spectrometer was operated in positive ion mode with a capillary temperature of 220 °C, with a potential of 2000 V applied to the column. Data were acquired with the mass spectrometer operating in automatic data-dependent switching mode, selecting the 12 most intense ions prior to tandem mass spectrometric analysis. All spectra were acquired with 1 microscan and without lockmass.
Mass spectrometric data analysis
Label-free quantitative analysis of mass spectrometric data was performed using MaxQuant (version 1.5.7.4). All the experimental conditions (Ambra1 and IgG control immunoprecipitations from SCC FAK-WT and −/− cells) were analyzed in biological triplicate. Each raw data file was considered separate in the experimental design; the replicates of each experimental condition were grouped for the subsequent statistical analysis. Raw data files were searched against the mouse UniProtKB database (version 2017_05; 86,453 entries) and a common contaminants database using the Andromeda search engine (packaged with MaxQuant, version 1.5.7.4). A mass accuracy of 4.5 ppm was applied, and a false discovery rate of 1%, applying a target-decoy search strategy using MaxQuant, was set at both the peptide and protein levels. Enzyme specificity was set as C terminal to arginine and lysine, except when followed by proline, and a maximum of two missed cleavages were allowed in the database search. Cysteine carbamidomethylation was specified as a fixed modification; methionine oxidation and acetylation of protein N termini were specified as variable modifications. Minimum peptide length was seven amino acids, and at least one peptide ratio was required for label-free quantification. Peptide identifications in one or more sample runs not identified in other samples were matched and transferred between runs (0.7-min time window). Proteins matching to the reversed or common contaminants databases were omitted, and ribosomal proteins were omitted as putative contaminants. Missing values were replaced by a constant (1), and significant protein interactors were determined based on average ratio (fold change over IgG control) and Student's t test. Interaction network analysis was performed using Cytoscape.
Immunofluorescence microscopy and image analysis
Cells were fixed, stained, and imaged as described in Schoenherr et al. (4).
qRT-PCR
RNA from cells was isolated using the RNeasy mini kit (Qiagen, Manchester, UK). 500 ng total RNA was reverse transcribed using the SuperScript first-strand cDNA synthesis kit (Life Technology, Paisley, UK). For the PCR amplification in a StepOne Plus real-time PCR system (Life Technology, Paisley, UK), 25 ng cDNA was used in a total reaction mix of 20 μl containing 10 μl Sensi Fast SYBR Green Hi-Rox (Bioline, London, UK) as well as 400 nm forward and reverse primers (Table S4). Gapdh was used to control for differences in cDNA input. Relative expression was calculated according to the ΔΔCT quantification method. Each sample within an experiment was analyzed in triplicate, and the experiment was carried out three times.
nCounter gene expression analysis
SCC FAK-WT cells were transfected with siControl, siAmbra1, siAkap8, and siAtf2 siRNA. RNA from cells was isolated 48 h posttransfection using the RNeasy mini kit (Qiagen, Manchester, UK) and diluted to 20 ng/μl. Samples (in triplicates) were subjected to gene expression analysis using the mouse nCounter PanCancer Pathways panel (Nanostring, Amersham Biosciences, UK). Analysis was performed using nSolver analysis software (Nanostring). The cutoff point of statistically significant relative changes (siRNA/siControl, p < 0.05) was set to 2-fold.
Statistical tests
For all experiments shown, n = 3–5. Error bars for the graphs show S.D. Student's t test was carried out to calculate the statistical significance.
Data availability
The MS-based proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD018745.
Supplementary Material
Acknowledgments
We thank Charlotte Proby and Gareth Inman for human SCC cells.
This article contains supporting information.
Author contributions—C. S., and M. C. F. conceptualization; C. S., J. C. W., A. F. M., and A. V. K. data curation; C. S. formal analysis; C. S. validation; C. S., B. G., A. L., and M. C. F. investigation; C. S. and A. B. visualization; C. S. methodology; C. S. and M. C. F. writing-original draft; M. C. F. resources; M. C. F. supervision; M. C. F. funding acquisition; M. C. F. project administration.
Funding and additional information—This work was funded by a Cancer Research UK Programme Grant (C157/A24837) to M. C. Frame and V. G. Brunton.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- SSC
- squamous cell carcinoma
- FAK
- focal adhesion kinase.
References
- 1. Benato F., Skobo T., Gioacchini G., Moro I., Ciccosanti F., Piacentini M., Fimia G. M., Carnevali O., and Dalla Valle L. (2013) Ambra1 knockdown in zebrafish leads to incomplete development due to severe defects in organogenesis. Autophagy 9, 476–495 10.4161/auto.23278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fimia G. M., Stoykova A., Romagnoli A., Giunta L., Di Bartolomeo S., Nardacci R., Corazzari M., Fuoco C., Ucar A., Schwartz P., Gruss P., Piacentini M., Chowdhury K., and Cecconi F. (2007) Ambra1 regulates autophagy and development of the nervous system. Nature 447, 1121–1125 10.1038/nature05925 [DOI] [PubMed] [Google Scholar]
- 3. Yazdankhah M., Farioli-Vecchioli S., Tonchev A. B., Stoykova A., and Cecconi F. (2014) The autophagy regulators Ambra1 and Beclin 1 are required for adult neurogenesis in the brain subventricular zone. Cell Death Dis. 5, e1403 10.1038/cddis.2014.358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schoenherr C., Byron A., Sandilands E., Paliashvili K., Baillie G. S., Garcia-Munoz A., Valacca C., Cecconi F., Serrels B., and Frame M. C. (2017) Ambra1 spatially regulates Src activity and Src/FAK-mediated cancer cell invasion via trafficking networks. Elife 6, e23172 10.7554/eLife.23172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nitta T., Sato Y., Ren X. S., Harada K., Sasaki M., Hirano S., and Nakanuma Y. (2014) Autophagy may promote carcinoma cell invasion and correlate with poor prognosis in cholangiocarcinoma. Int. J. Clin. Exp. Pathol. 7, 4913–4921 [PMC free article] [PubMed] [Google Scholar]
- 6. Di Bartolomeo S., Corazzari M., Nazio F., Oliverio S., Lisi G., Antonioli M., Pagliarini V., Matteoni S., Fuoco C., Giunta L., D'Amelio M., Nardacci R., Romagnoli A., Piacentini M., Cecconi F., et al. (2010) The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 191, 155–168 10.1083/jcb.201002100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Strappazzon F., Vietri-Rudan M., Campello S., Nazio F., Florenzano F., Fimia G. M., Piacentini M., Levine B., and Cecconi F. (2011) Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 30, 1195–1208 10.1038/emboj.2011.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nazio F., Strappazzon F., Antonioli M., Bielli P., Cianfanelli V., Bordi M., Gretzmeier C., Dengjel J., Piacentini M., Fimia G. M., and Cecconi F. (2013) mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 15, 406–416 10.1038/ncb2708 [DOI] [PubMed] [Google Scholar]
- 9. Pagliarini V., Wirawan E., Romagnoli A., Ciccosanti F., Lisi G., Lippens S., Cecconi F., Fimia G. M., Vandenabeele P., Corazzari M., and Piacentini M. (2012) Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differ. 19, 1495–1504 10.1038/cdd.2012.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xia P., Wang S., Huang G., Du Y., Zhu P., Li M., and Fan Z. (2014) RNF2 is recruited by WASH to ubiquitinate AMBRA1 leading to downregulation of autophagy. Cell Res. 24, 943–958 10.1038/cr.2014.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Strappazzon F., Nazio F., Corrado M., Cianfanelli V., Romagnoli A., Fimia G. M., Campello S., Nardacci R., Piacentini M., Campanella M., and Cecconi F. (2015) AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 22, 419–432 10.1038/cdd.2014.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cianfanelli V., Fuoco C., Lorente M., Salazar M., Quondamatteo F., Gherardini P. F., De Zio D., Nazio F., Antonioli M., D'Orazio M., Skobo T., Bordi M., Rohde M., Dalla Valle L., Helmer-Citterich M., et al. (2015) AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 17, 20–30 10.1038/ncb3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Becher J., Simula L., Volpe E., Procaccini C., La Rocca C., D'Acunzo P., Cianfanelli V., Strappazzon F., Caruana I., Nazio F., Weber G., Gigantino V., Botti G., Ciccosanti F., Borsellino G., et al. (2018) AMBRA1 controls regulatory T-cell differentiation and homeostasis upstream of the FOXO3-FOXP3 axis. Dev. Cell 47, 592–607 10.1016/j.devcel.2018.11.010 [DOI] [PubMed] [Google Scholar]
- 14. Quintanilla M., Brown K., Ramsden M., and Balmain A. (1986) Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 322, 78–80 10.1038/322078a0 [DOI] [PubMed] [Google Scholar]
- 15. Sandilands E., Serrels B., Wilkinson S., and Frame M. C. (2012) Src-dependent autophagic degradation of Ret in FAK-signalling-defective cancer cells. EMBO Rep. 13, 733–740 10.1038/embor.2012.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Serrels B., Sandilands E., Serrels A., Baillie G., Houslay M. D., Brunton V. G., Canel M., Machesky L. M., Anderson K. I., and Frame M. C. (2010) A complex between FAK, RACK1, and PDE4D5 controls spreading initiation and cancer cell polarity. Curr. Biol. 20, 1086–1092 10.1016/j.cub.2010.04.042 [DOI] [PubMed] [Google Scholar]
- 17. Serrels B., McGivern N., Canel M., Byron A., Johnson S. C., McSorley H. J., Quinn N., Taggart D., Von Kreigsheim A., Anderton S. M., Serrels A., and Frame M. C. (2017) IL-33 and ST2 mediate FAK-dependent antitumor immune evasion through transcriptional networks. Sci. Signal. 10, eaan8355 10.1126/scisignal.aan8355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Serrels A., Lund T., Serrels B., Byron A., McPherson R. C., von Kriegsheim A., Gomez-Cuadrado L., Canel M., Muir M., Ring J. E., Maniati E., Sims A. H., Pachter J. A., Brunton V. G., Gilbert N., et al. (2015) Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 163, 160–173 10.1016/j.cell.2015.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kelleher R. J. 3rd, Flanagan P. M., and Kornberg R. D. (1990) A novel mediator between activator proteins and the RNA polymerase II transcription apparatus. Cell 61, 1209–1215 10.1016/0092-8674(90)90685-8 [DOI] [PubMed] [Google Scholar]
- 20. Flanagan P. M., Kelleher R. J. 3rd, Sayre M. H., Tschochner H., and Kornberg R. D. (1991) A mediator required for activation of RNA polymerase II transcription in vitro. Nature 350, 436–438 10.1038/350436a0 [DOI] [PubMed] [Google Scholar]
- 21. Kwon H., Imbalzano A. N., Khavari P. A., Kingston R. E., and Green M. R. (1994) Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature 370, 477–481 10.1038/370477a0 [DOI] [PubMed] [Google Scholar]
- 22. Phelan M. L., Sif S., Narlikar G. J., and Kingston R. E. (1999) Reconstitution of a core chromatin remodeling complex from SWI/SNF subunits. Mol. Cell 3, 247–253 10.1016/S1097-2765(00)80315-9 [DOI] [PubMed] [Google Scholar]
- 23. Malik S., and Roeder R. G. (2010) The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat. Rev. Genet. 11, 761–772 10.1038/nrg2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abdel-Hafiz H. A., Heasley L. E., Kyriakis J. M., Avruch J., Kroll D. J., Johnson G. L., and Hoeffler J. P. (1992) Activating transcription factor-2 DNA-binding activity is stimulated by phosphorylation catalyzed by p42 and p54 microtubule-associated protein kinases. Mol. Endocrinol. 6, 2079–2089 10.1210/mend.6.12.1337144 [DOI] [PubMed] [Google Scholar]
- 25. Lu J., Wang W., Mi Y., Zhang C., Ying H., Wang L., Wang Y., Myatt L., and Sun K. (2017) AKAP95-mediated nuclear anchoring of PKA mediates cortisol-induced PTGS2 expression in human amnion fibroblasts. Sci. Signal. 10, eaac6160 10.1126/scisignal.aac6160 [DOI] [PubMed] [Google Scholar]
- 26. Eide T., Coghlan V., Orstavik S., Holsve C., Solberg R., Skalhegg B. S., Lamb N. J., Langeberg L., Fernandez A., Scott J. D., Jahnsen T., and Tasken K. (1998) Molecular cloning, chromosomal localization, and cell cycle-dependent subcellular distribution of the A-kinase anchoring protein, AKAP95. Exp. Cell Res. 238, 305–316 10.1006/excr.1997.3855 [DOI] [PubMed] [Google Scholar]
- 27. Collas P., Le Guellec K., and Tasken K. (1999) The A-kinase-anchoring protein AKAP95 is a multivalent protein with a key role in chromatin condensation at mitosis. J. Cell Biol. 147, 1167–1180 10.1083/jcb.147.6.1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gao X., Chaturvedi D., and Patel T. B. (2012) Localization and retention of p90 ribosomal S6 kinase 1 in the nucleus: implications for its function. Mol. Biol. Cell. 23, 503–515 10.1091/mbc.E11-07-0658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Knuesel M. T., Meyer K. D., Donner A. J., Espinosa J. M., and Taatjes D. J. (2009) The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of mediator. Mol. Cell. Biol. 29, 650–661 10.1128/MCB.00993-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Knuesel M. T., Meyer K. D., Bernecky C., and Taatjes D. J. (2009) The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 23, 439–451 10.1101/gad.1767009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. De Falco G., and Giordano A. (2002) CDK9: from basal transcription to cancer and AIDS. Cancer Biol. Ther. 1, 342–347 10.4161/cbt.1.4.6113 [DOI] [PubMed] [Google Scholar]
- 32. Shen J. P., Zhao D., Sasik R., Luebeck J., Birmingham A., Bojorquez-Gomez A., Licon K., Klepper K., Pekin D., Beckett A. N., Sanchez K. S., Thomas A., Kuo C. C., Du D., Roguev A., et al. (2017) Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 14, 573–576 10.1038/nmeth.4225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gupta S., Campbell D., Derijard B., and Davis R. J. (1995) Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267, 389–393 10.1126/science.7824938 [DOI] [PubMed] [Google Scholar]
- 34. van Dam H., Wilhelm D., Herr I., Steffen A., Herrlich P., and Angel P. (1995) ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J. 14, 1798–1811 10.1002/j.1460-2075.1995.tb07168.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Livingstone C., Patel G., and Jones N. (1995) ATF-2 contains a phosphorylation-dependent transcriptional activation domain. EMBO J. 14, 1785–1797 10.1002/j.1460-2075.1995.tb07167.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ouwens D. M., de Ruiter N. D., van der Zon G. C., Carter A. P., Schouten J., van der Burgt C., Kooistra K., Bos J. L., Maassen J. A., and van Dam H. (2002) Growth factors can activate ATF2 via a two-step mechanism: phosphorylation of Thr71 through the Ras-MEK-ERK pathway and of Thr69 through RalGDS-Src-p38. EMBO J. 21, 3782–3793 10.1093/emboj/cdf361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song B., Xie B., Wang C., and Li M. (2011) Caspase-3 is a target gene of c-Jun:ATF2 heterodimers during apoptosis induced by activity deprivation in cerebellar granule neurons. Neurosci. Lett. 505, 76–81 10.1016/j.neulet.2011.09.060 [DOI] [PubMed] [Google Scholar]
- 38. Kawasaki H., Schiltz L., Chiu R., Itakura K., Taira K., Nakatani Y., and Yokoyama K. K. (2000) ATF-2 has intrinsic histone acetyltransferase activity which is modulated by phosphorylation. Nature 405, 195–200 10.1038/35012097 [DOI] [PubMed] [Google Scholar]
- 39. Valdez B. C., Zander A. R., Song G., Murray D., Nieto Y., Li Y., Champlin R. E., and Andersson B. S. (2014) Synergistic cytotoxicity of gemcitabine, clofarabine and edelfosine in lymphoma cell lines. Blood Cancer J. 4, e171 10.1038/bcj.2013.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Roth S. Y., Denu J. M., and Allis C. D. (2001) Histone acetyltransferases. Annu. Rev. Biochem. 70, 81–120 10.1146/annurev.biochem.70.1.81 [DOI] [PubMed] [Google Scholar]
- 41. Bieluszewska A., Weglewska M., Bieluszewski T., Lesniewicz K., and Poreba E. (2018) PKA-binding domain of AKAP8 is essential for direct interaction with DPY30 protein. FEBS J. 285, 947–964 10.1111/febs.14378 [DOI] [PubMed] [Google Scholar]
- 42. Santos-Rosa H., Schneider R., Bannister A. J., Sherriff J., Bernstein B. E., Emre N. C., Schreiber S. L., Mellor J., and Kouzarides T. (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419, 407–411 10.1038/nature01080 [DOI] [PubMed] [Google Scholar]
- 43. Liang G., Lin J. C., Wei V., Yoo C., Cheng J. C., Nguyen C. T., Weisenberger D. J., Egger G., Takai D., Gonzales F. A., and Jones P. A. (2004) Distinct localization of histone H3 acetylation and H3-K4 methylation to the transcription start sites in the human genome. Proc. Natl. Acad. Sci. U S A 101, 7357–7362 10.1073/pnas.0401866101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tie F., Banerjee R., Stratton C. A., Prasad-Sinha J., Stepanik V., Zlobin A., Diaz M. O., Scacheri P. C., and Harte P. J. (2009) CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 136, 3131–3141 10.1242/dev.037127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Creyghton M. P., Cheng A. W., Welstead G. G., Kooistra T., Carey B. W., Steine E. J., Hanna J., Lodato M. A., Frampton G. M., Sharp P. A., Boyer L. A., Young R. A., and Jaenisch R. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U S A 107, 21931–21936 10.1073/pnas.1016071107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dou Z., Xu C., Donahue G., Shimi T., Pan J. A., Zhu J., Ivanov A., Capell B. C., Drake A. M., Shah P. P., Catanzaro J. M., Ricketts M. D., Lamark T., Adam S. A., Marmorstein R., et al. (2015) Autophagy mediates degradation of nuclear lamina. Nature 527, 105–109 10.1038/nature15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu F., Fang Y., Yan L., Xu L., Zhang S., Cao Y., Xu L., Zhang X., Xie J., Jiang G., Ge C., An N., Zhou D., Yuan N., and Wang J. (2017) Nuclear localization of Beclin 1 promotes radiation-induced DNA damage repair independent of autophagy. Sci. Rep. 7, 45385 10.1038/srep45385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lopez-Soop G., Ronningen T., Rogala A., Richartz N., Blomhoff H. K., Thiede B., Collas P., and Kuntziger T. (2017) AKAP95 interacts with nucleoporin TPR in mitosis and is important for the spindle assembly checkpoint. Cell Cycle 16, 947–956 10.1080/15384101.2017.1310350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Akileswaran L., Taraska J. W., Sayer J. A., Gettemy J. M., and Coghlan V. M. (2001) A-kinase-anchoring protein AKAP95 is targeted to the nuclear matrix and associates with p68 RNA helicase. J. Biol. Chem. 276, 17448–17454 10.1074/jbc.M101171200 [DOI] [PubMed] [Google Scholar]
- 50. Eide T., Carlson C., Tasken K. A., Hirano T., Tasken K., and Collas P. (2002) Distinct but overlapping domains of AKAP95 are implicated in chromosome condensation and condensin targeting. EMBO Rep. 3, 426–432 10.1093/embo-reports/kvf089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jiang H., Lu X., Shimada M., Dou Y., Tang Z., and Roeder R. G. (2013) Regulation of transcription by the MLL2 complex and MLL complex-associated AKAP95. Nat. Struct. Mol. Biol. 20, 1156–1163 10.1038/nsmb.2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li Y., Kao G. D., Garcia B. A., Shabanowitz J., Hunt D. F., Qin J., Phelan C., and Lazar M. A. (2006) A novel histone deacetylase pathway regulates mitosis by modulating Aurora B kinase activity. Genes Dev. 20, 2566–2579 10.1101/gad.1455006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kubota S., Morii M., Yuki R., Yamaguchi N., Yamaguchi H., Aoyama K., Kuga T., Tomonaga T., and Yamaguchi N. (2015) Role for tyrosine phosphorylation of A-kinase anchoring protein 8 (AKAP8) in its dissociation from chromatin and the nuclear matrix. J. Biol. Chem. 290, 10891–10904 10.1074/jbc.M115.643882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Clister T., Greenwald E. C., Baillie G. S., and Zhang J. (2019) AKAP95 organizes a nuclear microdomain to control local cAMP for regulating nuclear PKA. Cell Chem. Biol. 26, 885–891 10.1016/j.chembiol.2019.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nanni S., Re A., Ripoli C., Gowran A., Nigro P., D'Amario D., Amodeo A., Crea F., Grassi C., Pontecorvi A., Farsetti A., and Colussi C. (2016) The nuclear pore protein Nup153 associates with chromatin and regulates cardiac gene expression in dystrophic mdx hearts. Cardiovasc. Res. 112, 555–567 10.1093/cvr/cvw204 [DOI] [PubMed] [Google Scholar]
- 56. Van de Vosse D. W., Wan Y., Wozniak R. W., and Aitchison J. D. (2011) Role of the nuclear envelope in genome organization and gene expression. Wiley Interdiscip. Rev. Syst. Biol. Med. 3, 147–166 10.1002/wsbm.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Canel M., Byron A., Sims A. H., Cartier J., Patel H., Frame M. C., Brunton V. G., Serrels B., and Serrels A. (2017) Nuclear FAK and Runx1 cooperate to regulate IGFBP3, cell-cycle progression, and tumor growth. Cancer Res. 77, 5301–5312 10.1158/0008-5472.CAN-17-0418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dodge-Kafka K., Gildart M., Tokarski K., and Kapiloff M. S. (2019) mAKAPbeta signalosomes–a nodal regulator of gene transcription associated with pathological cardiac remodeling. Cell Signal. 63, 109357 10.1016/j.cellsig.2019.109357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McAndrew M. J., Gjidoda A., Tagore M., Miksanek T., and Floer M. (2016) Chromatin remodeler recruitment during macrophage differentiation facilitates transcription factor binding to enhancers in mature cells. J. Biol. Chem. 291, 18058–18071 10.1074/jbc.M116.734186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Turriziani B., Garcia-Munoz A., Pilkington R., Raso C., Kolch W., and von Kriegsheim A. (2014) On-beads digestion in conjunction with data-dependent mass spectrometry: a shortcut to quantitative and dynamic interaction proteomics. Biology 3, 320–332 10.3390/biology3020320 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS-based proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD018745.