

Abstract
In December 2019, a COVID-19 epidemic was discovered in Wuhan, China, and since has disseminated around the world impacting human health for millions. Herein, in-silico drug discovery approaches have been utilized to identify potential natural products (NPs) as Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV-2) main protease (Mpro) inhibitors. The MolPort database that contains over 100,000 NPs was screened and filtered using molecular docking techniques. Based on calculated docking scores, the top 5,000 NPs/natural-like products (NLPs) were selected and subjected to molecular dynamics (MD) simulations followed by molecular mechanics–generalized Born surface area (MM-GBSA) binding energy calculations. Combined 50 ns MD simulations and MM-GBSA calculations revealed nine potent NLPs with binding affinities (ΔGbinding) > −48.0 kcal/mol. Interestingly, among the identified NLPs, four bis([1,3]dioxolo)pyran-5-carboxamide derivatives showed ΔGbinding > −56.0 kcal/mol, forming essential short hydrogen bonds with HIS163 and GLY143 amino acids via dioxolane oxygen atoms. Structural and energetic analyses over 50 ns MD simulation demonstrated NLP-Mpro complex stability. Drug-likeness predictions revealed the prospects of the identified NLPs as potential drug candidates. The findings are expected to provide a novel contribution to the field of COVID-19 drug discovery.
Communicated by Ramaswamy H. Sarma
Keywords: COVID-19, main protease, natural-like product, molecular docking, molecular dynamics, drug-likeness

Introduction
The recent pandemic due to the worldwide spread of COVID-19 caused by a new Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV-2) is a great concern for global public health (Lu et al., 2020; Zhu et al., 2020). The WHO declared a global emergency in January 2020, and by March COVID-19 had expanded into a pandemic due to its rapid propagation (Cucinotta & Vanelli, 2020). Since then the U.S. Food and Drug Administration (FDA) has issued an emergency use authorization of remdesivir for treatment of suspected or laboratory-confirmed COVID-19 cases. In addition, clinical trials are being conducted for several potential drugs to treat COVID-19 (Thorlund et al., 2020). Identification of small molecules as potential SARS-CoV-2 inhibitors continues to be a major research focus within the scientific and pharmaceutical communities (Al-Khafaji et al., 2020; Liu et al., 2020; Sanders et al., 2020). The mechanism of SARS-CoV-2 inhibition depends on a main protease (Mpro) that represents a potential target for terminating viral replication (Boopathi et al., 2020). SARS-CoV-2 Mpro with a peptidomimetic inhibitor (N3) has been recently crystallized providing structural information for in-silico studies to screen for novel SARS-CoV-2 inhibitors (Jin et al., 2020).
Natural products (NPs) have been the source of most of the active ingredients of medicines, as over 50% of the approved drugs are based on NPs (Harvey, 2008). Therefore, NPs have hitherto received great attention by researchers that seek to discover potential drugs for the treatment of various diseases such as malaria (Clark, 1996; Wells, 2011), HIV (Kurapati et al., 2015), cardiovascular disease (Mashour et al., 1998), and neoplastic disease (Cragg et al., 1997). Recently, NPs screenings have been reported to identify anti-COVID-19 inhibitors based on NPs/NLPs (Gentile et al., 2020; Gonzalez-Paz et al., 2020; Ul Qamar et al., 2020). The long lead time and high cost of isolating and identifying NPs have prompted investigators to design natural-like products (NLPs) libraries based on small molecules taking into account the stability improvement and bioavailability (Quinn et al., 2008; Thomas & Johannes, 2011). One of the most common enriched databases is MolPort database containing 113,756 NPs/NLPs (https://www.molport.com). The current study set out to screen the MolPort database against potential small molecules that would bind in the SARS-CoV-2 Mpro active site and act as an inhibitor. Based on estimated docking scores, the top 5,000 NPs/NLPs were selected and subjected to molecular dynamics simulations combined with binding energy calculations using molecular mechanics–generalized Born surface area (MM-GBSA) approach. The stability, binding affinity and interactions of the identified NPs/NLPs with the SARS-CoV-2 Mpro active site were investigated with 50 ns molecular dynamics simulations. Drug likeness parameters were predicted for the proposed NPs/NLPs. The obtained results provide implications of new drug development in the prevention or treatment of COVID-19.
Computational methodology
Database preparation
The MolPort database that contains 113,756 natural and natural-like products was downloaded and prepared for molecular docking calculations (https://www.molport.com). The 3 D chemical structures of the compounds were generated using Omega2 software (Hawkins et al., 2010; OMEGA, 2013). The geometrical structures of the compounds were then optimized by MMFF94S force field using SZYBKI (SZYBKI, 2016). Gasteiger method was used to assign partial atomic charges (Gasteiger & Marsili, 1980). Prepared files may be accessed at the publicly-available site CompChem-DataBase (www.compchem.net/ccdb).
Mpro preparation
The crystal structure of SARS-CoV-2 main protease (Mpro) complexed with peptidomimetic inhibitor (N3) (PDB code: 6LU7 (Jin et al., 2020) was considered as a template for all molecular docking and molecular dynamics calculations. Water molecules and ions were removed. The protonation state of Mpro was examined using H++ server, and all missing hydrogen atoms were added (Gordon et al., 2005). Finally, the pdbqt file for Mpro was prepared according to AutoDock protocol (Forli et al., 2016).
Molecular docking
In this study, two molecular docking calculations were conducted—namely, standard and expensive. In the two calculations, all docking parameters were kept to default values, except the number of genetic algorithm (GA) runs and the maximum number of energy evaluations (eval). The latter two variables were set to 25 and 100, and 2,500,000 and 10,000,000 for standard and expensive molecular docking calculations, respectively. The docking grid was purposed to embrace the active site of the SARS-CoV-2 Mpro receptor with a grid size of 60 Å × 60 Å × 60 Å and a spacing value of 0.375 Å. The grid center was located at −13.069, 9.74, 68.49 (XYZ coordinates). All molecular docking calculations were carried out using Autodock4.2 software (Morris et al., 2009).
Molecular dynamics simulations
Molecular dynamics (MD) simulations were executed on top potent NPs/NLPs in complex with SARS-CoV-2 Mpro using AMBER16 software (Case et al., 2016). In MD simulations, NPs/NLPs and Mpro were described using General AMBER force field (GAFF) and AMBER force field 14SB, respectively (Maier et al., 2015; Wang et al., 2004). Two types of MD simulations were performed—namely, implicit and explicit MDs. In implicit MD, the atomic partial charges of the NPs/NLPs were assigned using am1-bcc method with the help of Antechamber tool implemented inside AMBER16 (Jakalian et al., 2002). A non-bonded cutoff of 999 Å and no periodic boundary conditions were applied. Moreover, the solvation effect was captured with the igb = 1 implicit solvent model (Roux & Simonson, 1999). The docked NP/NLP-Mpro complexes were then minimized for 500 steps and heated from 0 K to 300 K over 10 ps. Finally, 100 ps and 1,000 ps production stages were performed and snapshots were collected every 1 ps, giving 100 and 1,000 snapshots, respectively. All implicit MD simulations were performed with the CPU version of pmemd (pmemd.MPI) in AMBER16. In explicit MD, the atomic partial charges of the studied NPs/NLPs were assigned using the restrained electrostatic potential (RESP) approach at the HF/6-31G* level with the help of Gaussian09 software (Bayly et al., 1993; Frisch et al., 2009). The docked NP/NLP-Mpro complexes were solvated in a cubic water box with 15 Å distances between the edges of the box and any atom of NP/NLP or NP/NLP-Mpro complexes. The solvated NP/NLP-Mpro complexes were then minimized for 5000 steps and subsequently gently annealed from 0 K to 300 K over 50 ps. The systems were equilibrated for 1 ns and production stages were conducted over simulation times of 10 and 50 ns. The snapshots were collected every 10 ps, giving 1,000 and 5,000 snapshots, respectively. Periodic boundary conditions and the NPT ensemble were adopted in all explicit MD simulations, including both the equilibration and production stages. Long-range electrostatic interactions under periodic conditions with a direct space cut-off of 12 Å were treated with Particle Mesh Ewald (PME) method (Darden et al., 1993). Langevin dynamics with the collision frequency gamma_ln set to 1.0 was used to keep the temperature constant at 298 K. Berendsen barostat with a relaxation time of 2 ps was employed to control the pressure of system (Berendsen et al., 1984). A time step of 2 fs and the SHAKE option to constrain all bonds involving hydrogen atoms were used. All explicit MD simulations were performed with the GPU version of pmemd (pmemd.cuda) in AMBER16.
MM-GBSA binding energy
Snapshots were collected every 10 ps over the production stage of 50 ns MD simulation, giving 5,000 snapshots. Based on the collected snapshots, molecular mechanics–generalized Born surface area (MM-GBSA) approach was used to evaluate the binding energy of the studied NPs/NLPs with SARS-CoV-2 Mpro (Massova & Kollman, 2000). The MM-GBSA binding free energies were estimated as follows:
where the energy term (G) is estimated as:
with and as the van der Waals, electrostatic, General Born solvation, and surface area energies, respectively. For the NPs/NLPs, entropy contributions were neglected.
Drug likeness
The online Molinspiration cheminformatics software (http://www.molinspiration.com) was utilized to evaluate the physicochemical parameters of the identified potent NPs and NLPs. Lipinski’s parameters including topological polar surface area (TPSA), relative molecular weight (MW), number of hydrogen bond donors (nOHNH), number of hydrogen bond acceptors (nON) and the partition coefficient log P (miLog P) were assessed.
Molecular target prediction and network analysis
Based on the structural similarity of known ligands-target integrations, target prediction of the most promising NPs/NLPs was performed using the online website-based tools of SwissTargetPredicition (http://www.swisstargetprediction.ch). Additionally, the available database for Severe Acute Respiratory Syndrome diseases was collected from the online database DisGeNET (https://www.disgenet.org). Venn diagram was plotted using InteractiVenn online tool (Heberle et al., 2015). Protein-protein interaction (PPI) network was generated using a functional database of STRING for top predicted targets (Li et al., 2019). The PPI network was diagramed and analyzed using Cytoscape 3.8.0 (Shannon et al., 2003).
Results and discussion
Unavailability of therapies as yet indicates an urgent need for drug exploration against SARS-CoV-2. The main protease (Mpro) is a likely targeted for enzyme inhibition due to the polypeptides' essential role in viral replication. In this study, the expansive MolPort database for natural products (NPs) and natural-like products (NLPs) was screened and filtered for the identification of potential SARS-CoV-2 Mpro inhibitors.
Molecular docking
Two levels of molecular docking calculations were carried out to minimize computational-costs and time. Initially, all NPs/NLPs in the MolPort database were screened against Mpro with standard docking parameters of GA = 25 and eval = 2,500,000. The NPs/NLPs were then sorted based on their docking scores. In terms of the calculated docking scores, almost three-fourths of the screened NPs and NLPs (≈78%) showed binding energies less than −8.7 kcal/mol with SARS-CoV-2 Mpro. Therefore, only the top 25,000 NPs/NLPs were selected for further docking calculations. Using expensive docking parameters of GA = 100 and eval = 10,000,000, the selected 25,000 NPs/NLPs were re-docked against Mpro. Docking scores and binding features, as well as 2 D chemical structures, of nine potent NLPs with SARS-CoV-2 Mpro are listed in Table 1. The 2 D and 3 D representations of interactions of the top nine potent NLPs with important amino acid residues of SARS-CoV-2 Mpro are depicted in Supporting Information Figure S1.
Table 1.
Calculated standard and expansive docking scores (in kcal/mol) and binding features for nine potent natural-like products (NLPs) against SARS-CoV-2 main protease (Mpro).
| No. | MolPort code | 2D-chemical structure | Docking score
(kcal/mol) |
Binding features (Hydrogen bond length in Å) | |||
|---|---|---|---|---|---|---|---|
| Standard | Expensive | ||||||
| 1 | MolPort-000-708-794 | 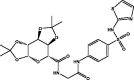 |
−10.4 | −11.0 | GLU166 (1.80 Å), GLY143 (1.72 Å), HIS163 (2.35 Å), HIS 164 (1.83 Å) | ||
| 2 | MolPort-044-179-844 | 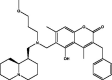 |
−10.0 | −10.9 | GLN189 (1.80 Å), THR190 (2.39 Å), GLN192 (2.45 Å), HIS163 (2.07 Å) | ||
| 3 | MolPort-000-702-646 | 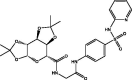 |
−10.4 | −10.8 | HIS163 (2.26), HIS164 (2.05 Å), THR190 (1.66 Å), GLN192 (2.84 Å), GLN189 (2.94 Å), GLY143 (1.95 Å) | ||
| 4 | MolPort-002-513-915 | 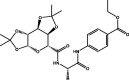 |
−9.6 | −10.5 | HIS164 (2.15 Å), GLN192 (2.38 Å), GLN189 (2,55 Å), GLY143 (2.09 Å), HIS163 (2.57 Å) | ||
| 5 | MolPort-005-948-349 | 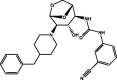 |
−10.2 | −10.4 | GLN189 (2.10, 1.89 Å), ASN142 (2.15 Å), GLU166 (2.85 Å) | ||
| 6 | MolPort-039-056-062 | 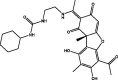 |
−10.0 | −10.4 | GLN192 (2.24 Å), THR190 (1.97 Å), GLU166 (2.04 Å), GLN189 (2.18, 2.18 Å), ASN142 (2.54 Å) | ||
| 7 | MolPort-004-849-765 | 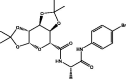 |
−9.8 | −10.4 | HIS164 (2.07 Å), GLN189 (2.51 Å), GLY143 (1.97 Å), HIS163 (2.44 Å) | ||
| 8 | MolPort-046-158-375 | 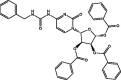 |
−9.7 | −10.3 | PHE140 (1.68, 2.34 Å), GLU166 (2.38 Å), CYS145 (2.51 Å), HIS163 (2.85, 1.78 Å), | ||
| 9 | MolPort-001-751-850 | 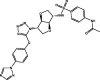 |
−9.6 | −10.1 | GLN192 (2.14 Å), HIS163 (2.10 Å), ASN142 (1.94 Å), GLU166 (2.09 Å), CYS145 (2.35 Å), HIS41 (2.84 Å) | ||
The most promising NLPs (shown in Table 1 and Supporting Information Figure S1) share the same binding mode inside the active site of SARS-CoV-2 Mpro, forming essential hydrogen bonds with key amino acid residues including HIS164, HIS163, and GLU166. Further interactions including van der Waals and hydrophobic interactions were identified, giving a docking score higher than ≥ −9.6 kcal/mol (Supporting Information Figure S1). For instance, the potency of MolPort-000-708-794 with a binding energy of −11.0 kcal/mol may be attributed to its ability to form four hydrogen bonds with GLU166, GLY143, HIS163, and HIS164 with bond lengths of 1.80, 1.72, 2.35 and 1.83 Å, respectively (Figure 1). Two out of the latter four hydrogen bonds were formed via dioxolane oxygen atoms, revealing a significant contribution of dioxolane rings in NLPs-Mpro binding mode and affinity. Based on calculated expensive docking scores, the top 5,000 potent NPs/NLPs were closely investigated using molecular dynamics (MD) calculations.
Figure 1.

2 D and 3 D representation of predicted binding mode of MolPort-000-708-794 as potent natural-like products (NLPs) inside the active site of SARS-CoV-2 main protease (Mpro).
Molecular dynamics simulations
Conformational flexibilities of drug-receptor complexes, solvent effects, and dynamics must be considered to achieve reliable drug-receptor binding affinities (De Vivo et al., 2016; Kerrigan, 2013). Therefore, molecular dynamics (MD) simulations combined with binding energy calculations over reasonable simulation time were performed for the top 5,000 potent NPs/NLPs in complex with SARS-CoV-2 Mpro. Considering computational-costs and time, MD simulations for the 5,000 docked NPs/NLPs-Mpro complexes were conducted in implicit solvent for 100 ps and the corresponding binding energies were estimated using MM-GBSA approach (see Computational Methodology section for details). The calculated MM-GBSA binding energies of the top 1,000 compounds are listed in Supporting Information Table S1.
Interestingly, of the 5,000 investigated NPs/NLPs, only 86 NPs/NLPs showed significant MM-GBSA binding energies with values in range −45.0 to −56.3 kcal/mol. To achieve a greater degree of accuracy, molecular dynamics of 86 NPs/NLPs in complex with Mpro were further investigated over a longer implicit MD simulation with a time of 1 ns. Estimated MD//MM-GBSA binding energies are presented in Supporting Information Table S2. The corresponding MM-GBSA binding energies for nine potent NLPs are presented in Figure 2.
Figure 2.

Calculated MM-GBSA binding energies for the top nine potent natural-like products (NLPs) as SARS-CoV-2 main protease (Mpro) inhibitors over 100 ps and 1 ns implicit MD and 10 ns and 50 ns explicit MD simulations.
On the basis of the calculated MM-GBSA binding energies over 1 ns (Supporting Information Table S2), 46 NPs/NLPs exhibited instability in molecular dynamics and their corresponding MM-GBSA binding energies with Mpro were decreased to be less than −45.0 kcal/mol, while the other 40 NPs/NLPs showed significant binding affinities with MM-GBSA energies ≥ −45.0 kcal/mol. Therefore, we focused on these potent 40 NPs/NLPs over a reasonable MD simulation time in explicit solvent. Consequently, 10 ns MD simulations for the potent 40 NPs/NLPs-Mpro complexes were conducted in explicit solvent and followed by MM-GBSA binding energies calculation. The estimated 10 ns MD//MM-GBSA binding energies are noted in Supporting Information Table S3, and the corresponding binding energies for nine potent NLPs are presented in Figure 2.
Interestingly, only nine of the investigated NPs/NLPs showed promising binding affinities > −50.0 kcal/mol towards SARS-CoV-2 Mpro over 10 ns MD simulation time (Supporting Information Table S3). To increase the reliability of the observed results, each one of these nine NLPs in complex with Mpro was further simulated for 50 ns MD, and the corresponding binding energies were calculated (Figure 2).
As can be seen from Figure 2, there was no significant difference between the estimated MM-GBSA binding energies over 10 ns MD and those over 50 ns MD, reflecting the tight binding of the identified NLPs with SARS-CoV-2 Mpro. Surprisingly, four of the nine NLPs—namely, MolPort-004-849-765, MolPort-000-708-794, MolPort-002-513-915 and MolPort-000-702-646—are bis([1,3]dioxolo)pyran-5-carboxamide derivatives and showed outperformance affinity towards SARS-CoV-2 Mpro with binding energies > −56.0 kcal/mol. Therefore, considerable interest was given in the following sections to investigate the identified dioxolo-derivatives as potential SARS-CoV-2 Mpro inhibitors.
Post-dynamics analyses
The purpose of the post-dynamics analyses was to evaluate the interaction nature and stability of the identified dioxolo-derivatives inside the SARS-CoV-2 Mpro active site. Structural and energetic analyses for the four promising dioxolo-derivatives were conducted over 50 ns explicit MD simulations.
Binding energy decomposition
Decomposition of average MM-GBSA binding energy over 50 ns MD simulation was performed to reveal the nature of dominant interactions in NLPs-Mpro complexes (Table 2). Energy decomposition results showed that Evdw was the dominant force in NP/NLP-Mpro binding affinities with a contribution value of ≈−63.0 kcal/mol for the four dioxolo-derivatives. Eele was favorable with values of −35.1, −47.1, −34.5 and −43.6 kcal/mol for MolPort-004-849-765, MolPort-000-708-794, MolPort-002-513-915 and MolPort-000-702-646, respectively.
Table 2.
Decomposition of MM-GBSA binding energies for four dioxolo-derivatives in complex with SARS-CoV-2 main protease (Mpro) through 50 ns MD simulations.
| MolPort code | Calculated
MM-GBSA binding energy (kcal/mol) |
||||||
|---|---|---|---|---|---|---|---|
| ΔEVDW | ΔEele | ΔEGB | ΔESUR | ΔGgas | ΔGSolv | ΔGbinding | |
| MolPort-004-849-765 | −62.4 | −35.1 | 45.7 | −6.6 | −97.5 | 39.1 | −58.4 |
| MolPort-000-708-794 | −63.7 | −47.1 | 60.4 | −6.8 | −110.8 | 53.6 | −57.3 |
| MolPort-002-513-915 | −63.0 | −34.5 | 47.6 | −6.7 | −97.5 | 40.9 | −56.6 |
| MolPort-000-702-646 | −63.1 | −43.6 | 57.4 | −6.9 | −105.7 | 50.5 | −56.2 |
Binding energy per frame
The stability of dioxolo-derivatives inside the Mpro active site was investigated using the correlation between the binding-energy and time. Therefore, MM-GBSA binding energy was estimated per-frame for each of MolPort-004-849-765, MolPort-000-708-794, MolPort-002-513-915, and MolPort-000-702-646 with Mpro and presented in Figure 3. According to data in Figure 3, there was overall stability for the four dioxolo-derivatives till the end of the simulations with average values of −58.4, −57.3, −56.6, and −56.2 (kcal/mol), respectively. These findings indicated the promising stability of NLP-Mpro complexes over the simulated MD time of 50 ns.
Figure 3.

Variations in the MM-GBSA binding energies for MolPort-004-849-765 (in cyan), MolPort-000-708-794 (in red), MolPort-002-513-915 (in blue) and MolPort-000-702-646, (in black) with SARS-CoV-2 main protease (Mpro) during the 50 ns MD simulation.
Hydrogen bond length and center-of-mass distance
Inspecting the hydrogen bond length and center-of-mass (CoM) distance between the dioxolo-derivatives and the key HIS164 amino acid residue over the 50 ns MD simulation would reflect an indication of NLPs-Mpro stability. Therefore, the desired hydrogen bond lengths and CoM distances were measured over the 50 ns MD simulations and depicted in Figure 4.
Figure 4.

Hydrogen bond lengths and center-of-mass (CoM) distances between MolPort-004-849-765 (in cyan), MolPort-000-708-794 (in red), MolPort-002-513-915 (in blue) and MolPort-000-702-646 (in black) with the HIS164 amino acid residue inside the active site of SARS-CoV-2 main protease (Mpro) over the 50 ns MD simulation.
The most obvious finding to emerge from data plotted in Figure 4 was that MolPort-004-849-765, MolPort-000-708-794, MolPort-002-513-915 and MolPort-000-702-646 showed high stability inside the active site with average hydrogen bond lengths of 1.93, 1.98, 1.92, and 1.99 Å, respectively. In terms of the measured CoM distances, the average CoM distance between NLP and HIS164 was nearly constant around 8 Å during 50 ns MD simulations for the four investigated dioxolo-derivatives.
Overall, these post-dynamics results provided evidence for the stability of the identified dioxolo-derivatives in complex with SARS-CoV-2 Mpro, forming hydrogen bond interactions with the key amino acids.
Root-mean-square deviation
Root-mean-square deviation (RMSD) was used to investigate the structural changes in the NLP-Mpro complexes. RMSD for the backbone atoms of the MolPort-004-849-765, MolPort-000-708-794, MolPort-002-513-915 and MolPort-000-702-646 in complex with Mpro relative to the starting structures throughout the 50 ns MD simulations were evaluated and presented in Figure 5.
Figure 5.

Root-mean-square-deviation (RMSD) of the backbone from the initial structure for MolPort-004-849-765 (in cyan), MolPort-000-708-794 (in red), MolPort-002-513-915 (in blue) and MolPort-000-702-646 (in black) with SARS-CoV-2 main protease (Mpro) through 50 ns MD simulation.
From the data in Figure 5, it is apparent that the backbone of NLP-Mpro complexes exhibited stability over 50 ns MD simulation, giving RMSD with less than 0.35 nm. These results emphasize that four investigated dioxolo-derivatives are tightly bonded in the active site and do not impact the overall topology of Mpro. Finally, these energetic and structural analyses demonstrated the high stability of the four investigated dioxolo-derivatives-Mpro complexes through 50 ns MD simulations.
Drug likeness
Lipinski’s rule of five is commonly used in drug discovery and development to evaluate the oral bioavailability of active drug in humans. In this study, physicochemical parameters of the promising NLPs as potential SARS-CoV-2 Mpro inhibitors were predicted using Molinspiration cheminformatics, (http://www.molinspiration.com) online software calculation toolkit. The predicted parameters included Lipinski’s parameters, topological polar surface area (TPSA), and percentage of absorption (%ABS). The predicted parameters are listed in Table 3.
Table 3.
Predicted physiochemical parameters of the four dioxolo-derivatives as potent SARS-CoV-2 main protease (Mpro) inhibitors and their different structural descriptors.
| MolPort code | miLog Pa | TPSAb | nONc | nOHNHd | nviolation | Nrotbe | MolVolf | MWtg | %ABSh |
|---|---|---|---|---|---|---|---|---|---|
| MolPort-004-849-765 | 2.38 | 104.37 | 9 | 2 | 0 | 4 | 393.88 | 499.36 | 72.9% |
| MolPort-000-708-794 | 1.43 | 163.43 | 13 | 3 | 2 | 7 | 461.20 | 568.63 | 52.6% |
| MolPort-002-513-915 | 2.12 | 130.67 | 11 | 2 | 1 | 7 | 437.32 | 492.52 | 63.9% |
| MolPort-000-702-646 | 1.48 | 163.43 | 13 | 3 | 2 | 7 | 470.49 | 562.60 | 52.6% |
aLogarithm of partition coefficient between n-octanol and water (miLogP).
bTopological polar surface area (TPSA).
cNumber of hydrogen bond acceptors (nON).
dNumber of hydrogen bond donors (nOHNH).
eNumber of rotatable bonds (nrotb).
fMolecular volume (Mol Vol).
gMolecular weight (MWt).
h%ABS = 109 − [0.345× TPSA] (Zhao et al., 2002).
From data given in Table 3, the milog P values of the four dioxolo-derivatives were found to be below five (calc. in range 1.43 to 2.38), suggesting that these NLPs have good permeability across the cell membrane. Molecular weight was found to be more or less than 500 (calc. in range 492.52 to 568.63), predicting the compounds to be easily transported, diffused and absorbed. Besides, the number of hydrogen bond donors (nOHNH) was less than 5 in accordance with Lipinski’s rules, and the number of hydrogen bond acceptors (nON) was in range 9 to 13. It is worth mentioning that this slight increase in molecular weight and hydrogen bond acceptors will not have a significant impact on compound transportation and diffusion, where it has been shown that several FDA-approved drugs moved beyond the traditional low molecular weight of 500 and hydrogen bond acceptors of 10 (Mullard, 2018). Besides, TPSA of all promising dioxolo-derivatives was observed in range 104.37 to 163.43 Å which was a very good indicator of the bioavailability of the investigated NLPs. In addition, the calculated %ABS was ranged between 52.62% and 72.99%, indicating that the investigated NLPs may have good cell membrane permeability and oral bioavailability.
Molecular target prediction and network analysis
Severe Acute Respiratory Syndrome diseases (C1175175) displayed 117 genes based on DisGeNET online software. Additionally, targeted genes for the identified dioxolo-derivatives as potent Mpro inhibitors were collected using online SwissTargetPrediction tools, giving 100 targets. Classification of the predicted targets for each examined dioxolo-derivative is depicted in Supporting Information Figure S2. The Venn diagram comparison analysis between Severe Acute Respiratory Syndrome diseases and predicted targeted genes was demonstrated and plotted in Figure 6.
Figure 6.

Venn diagram analysis for the four identified dioxolo-derivatives as potent SARS-CoV-2 main protease (Mpro) inhibitors and Severe Acute Respiratory Syndrome disease genes.
According to data presented in Venn diagram (Figure 6), Acute Respiratory Syndrome diseases and predicted targeted genes displayed commonly shared MAPK14 and EGFR (Figure 6). The host protein angiotensin-converting enzyme 2 (ACE2), serves as an entry receptor for SARS-CoV-2. Targeting MAPK14 would result in blocking of ACE2 production pathways, and in turn, reducing the probability of SARS-CoV-2 virus to be received and internalized by human cells (Kindrachuk et al., 2015). In addition, SARS-CoV-2 inhibition might be achieved by inhibition of MAPKs which are activated also by GFRs, such as in case of chloroquine (Hondermarck et al., 2020).
The possible targets predicted by SwissTargetPrediction for all examined dioxolo-derivatives were further analyzed using STRING PPI network and visualized by Cytoscape 3.8.0. The network topological analysis by Cytoscape demonstrated that the targets within the top 10 scores of degree were MAPK14 and EGFR for the four examined dioxolo-derivatives (Supporting Information Table S4). The STRING PPI network for the identified top 10 targets is depicted in Figure 7.
Figure 7.

The STRING PPI network for the top 10 targets identified by network analyzer for the identified dioxolo-derivatives as potent SARS-CoV-2 main protease (Mpro) inhibitors.
Conclusion
SARS-CoV-2 main protease (Mpro) is characterized to be an essential and highly potent target for the inhibition of the novel coronavirus. In this study, a total of 113,756 natural and natural like products were screened against Mpro to discover potential SARS-CoV-2 Mpro inhibitors. Filtration of MolPort database was carried out using combined molecular docking and molecular dynamics (MD) followed by molecular mechanics–generalized Born surface area (MM-GBSA) binding energy calculations. Based on docking scores and MM-GBSA binding energies, nine NLPs showed promising binding affinities > −50.0 kcal/mol with SARS-CoV-2 Mpro. Interestingly, four of the nine NLPs—namely, MolPort-004-849-765, MolPort-000-708-794, MolPort-002-513-915 and MolPort-000-702-646—are bis([1,3]dioxolo)pyran-5-carboxamide derivatives showing high affinity towards Mpro with binding energies > −56.0 kcal/mol. Post-dynamics analyses demonstrated the stability and affinity of the identified dioxolo-derivatives with Mpro. Predicted physicochemical parameters of the promising dioxolo-derivatives fit drug-likeness properties, indicating the probability of these NLPs as prospective SARS-CoV-2 drug candidates. Protein-protein interaction (PPI) showed the linked top targets genes that could have an effect on viral infection, as well as, the host. The current results establish that bis([1,3]dioxolo)pyran-5-carboxamide derivatives hold promise as inhibitors against SARS-CoV-2 Mpro and are ready for in vitro inhibition against SARS-CoV-2.
Supplementary Material
Funding Statement
The computational work was completed with resources supported by the Science and Technology Development Fund, STDF, Egypt, Grants No. 5480 & 7972.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Al-Khafaji, K., Al-Duhaidahawi, D., & Taskin Tok, T. (2020). Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. Journal of Biomolecular Structure and Dynamics, 1–9. 10.1080/07391102.2020.1764392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayly, C. I., Cieplak, P., Cornell, W., & Kollman, P. A. (1993). A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges - The RESP model. The Journal of Physical Chemistry, 97(40), 10269–10280. 10.1021/j100142a004 [DOI] [Google Scholar]
- Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., & Haak, J. R. (1984). Molecular-dynamics with coupling to an external bath. The Journal of Chemical Physics, 81(8), 3684–3690. 10.1063/1.448118 [DOI] [Google Scholar]
- Boopathi, S., Poma, A. B., & Kolandaivel, P. (2020). Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. Journal of Biomolecular Structure and Dynamics, 1–10. 10.1080/07391102.2020.1758788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, D. A., Betz, R. M., Cerutti, D. S., Cheatham, T. E., Darden, T. A., Duke, R. E., Giese, T. J., Gohlke, H., Goetz, A. W., Homeyer, N., Izadi, S., Janowski, P., Kaus, J., Kovalenko, A., Lee, T. S., LeGrand, S., Li, P., Lin, C., Luchko, T., … Kollman, P. A. (2016). AMBER 2016. University of California. [Google Scholar]
- Clark, A. M. (1996). Natural products as a resource for new drugs. Pharmaceutical Research, 13(8), 1133–1144. 10.1023/a:1016091631721 [DOI] [PubMed] [Google Scholar]
- Cragg, G. M., Newman, D. J., & Snader, K. M. (1997). Natural products in drug discovery and development. Journal of Natural Products, 60(1), 52–60. 10.1021/np9604893 [DOI] [PubMed] [Google Scholar]
- Cucinotta, D., & Vanelli, M. (2020). WHO declares COVID-19: A pandemic. Acta Bio-Medica: Atenei Parmensis, 91(1), 157–160. 10.23750/abm.v91i1.9397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden, T., York, D., & Pedersen, L. (1993). Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. The Journal of Chemical Physics, 98(12), 10089–10092. 10.1063/1.464397 [DOI] [Google Scholar]
- De Vivo, M., Masetti, M., Bottegoni, G., & Cavalli, A. (2016). Role of molecular dynamics and related methods in drug discovery. Journal of Medicinal Chemistry, 59(9), 4035–4061. 10.1021/acs.jmedchem.5b01684 [DOI] [PubMed] [Google Scholar]
- Forli, S., Huey, R., Pique, M. E., Sanner, M. F., Goodsell, D. S., & Olson, A. J. (2016). Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nature Protocols, 11(5), 905–919. 10.1038/nprot.2016.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G. A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., … Fox, D. J. (2009). Gaussian 09 (version revision E01). Gaussian Inc. [Google Scholar]
- Gasteiger, J., & Marsili, M. (1980). Iterative partial equalization of orbital electronegativity - A rapid access to atomic charges. Tetrahedron, 36(22), 3219–3228. 10.1016/0040-4020(80)80168-2 [DOI] [Google Scholar]
- Gentile, D., Patamia, V., Scala, A., Sciortino, M. T., Piperno, A., & Rescifina, A. (2020). Putative inhibitors of SARS-CoV-2 main protease from a library of marine natural products: A virtual screening and molecular modeling study. Marine Drugs, 18(4), 225–233. 10.3390/md18040225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Paz, L., Lossada, C., Moncayo, L., Romero, F., Paz, J., Vera, J., Pérez, A., San-Blas, E., & Alvarado, Y. (2020). Theoretical molecular docking study of the structural disruption of the viral 3CL-protease of COVID19 induced by binding of capsaicin, piperine and curcumin, part 1: A comparative study with chloroquine and hydrochloroquine two antimalaric drugs. 10.21203/rs.3.rs-21206/v1 [DOI]
- Gordon, J. C., Myers, J. B., Folta, T., Shoja, V., Heath, L. S., & Onufriev, A. (2005). H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Research, 33(Web Server issue), W368–W371. 10.1093/nar/gki464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey, A. L. (2008). Natural products in drug discovery. Drug Discovery Today, 13(19–20), 894–901. 10.1016/j.drudis.2008.07.004 [DOI] [PubMed] [Google Scholar]
- Hawkins, P. C. D., Skillman, A. G., Warren, G. L., Ellingson, B. A., & Stahl, M. T. (2010). Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. Journal of Chemical Information and Modeling, 50(4), 572–584. 10.1021/ci100031x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heberle, H., Meirelles, G. V., da Silva, F. R., Telles, G. P., & Minghim, R. (2015). InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinformatics, 16(1), 169. 10.1186/s12859-015-0611-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondermarck, H., Bartlett, N. W., & Nurcombe, V. (2020). The role of growth factor receptors in viral infections: An opportunity for drug repurposing against emerging viral diseases such as COVID-19? FASEB Bioadvances, 2(5), 296–303. 10.1096/fba.2020-00015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakalian, A., Jack, D. B., & Bayly, C. I. (2002). Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. Journal of Computational Chemistry, 23(16), 1623–1641. 10.1002/jcc.10128 [DOI] [PubMed] [Google Scholar]
- Jin, Z., Du, X., Xu, Y., Deng, Y., Liu, M., Zhao, Y., Zhang, B., Li, X., Zhang, L., Peng, C., Duan, Y., Yu, J., Wang, L., Yang, K., Liu, F., Jiang, R., Yang, X., You, T., Liu, X., … Yang, H. (2020). Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature, 582(7811), 289–293. 10.1038/s41586-020-2223-y [DOI] [PubMed] [Google Scholar]
- Kerrigan, J. E. (2013). Molecular dynamics simulations in drug design. In Kortagere S. (Ed.), In silico models for drug discovery (pp. 95–113). Humana Press. [Google Scholar]
- Kindrachuk, J., Ork, B., Hart, B. J., Mazur, S., Holbrook, M. R., Frieman, M. B., Traynor, D., Johnson, R. F., Dyall, J., Kuhn, J. H., Olinger, G. G., Hensley, L. E., & Jahrling, P. B. (2015). Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for middle east respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrobial Agents and Chemotherapy, 59(2), 1088–1099. 10.1128/AAC.03659-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurapati, K. R. V., Atluri, V. S., Samikkannu, T., Garcia, G., & Nair, M. P. N. (2015). Natural products as anti-HIV agents and role in HIV-associated neurocognitive disorders (HAND): A brief overview. Frontiers in Microbiology, 6, 1444–1457. 10.3389/fmicb.2015.01444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R., Ma, X., Song, Y., Zhang, Y., Xiong, W., Li, L., & Zhou, L. (2019). Anti-colorectal cancer targets of resveratrol and biological molecular mechanism: Analyses of network pharmacology, human and experimental data. Journal of Cellular Biochemistry, 120(7), 11265–11273. 10.1002/jcb.28404 [DOI] [PubMed] [Google Scholar]
- Liu, C., Zhou, Q., Li, Y., Garner, L. V., Watkins, S. P., Carter, L. J., Smoot, J., Gregg, A. C., Daniels, A. D., Jervey, S., & Albaiu, D. (2020). Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Central Science, 6(3), 315–331. 10.1021/acscentsci.0c00272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, R., Zhao, X., Li, J., Niu, P., Yang, B., Wu, H., Wang, W., Song, H., Huang, B., Zhu, N., Bi, Y., Ma, X., Zhan, F., Wang, L., Hu, T., Zhou, H., Hu, Z., Zhou, W., Zhao, L., … Tan, W. (2020). Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet (London, England), 395(10224), 565–574. 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., & Simmerling, C. (2015). ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation, 11(8), 3696–3713. 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashour, N. H., Lin, G. I., & Frishman, W. H. (1998). Herbal medicine for the treatment of cardiovascular disease: Clinical considerations. Archives of Internal Medicine, 158(20), 2225–2234. 10.1001/archinte.158.20.2225 [DOI] [PubMed] [Google Scholar]
- Massova, I., & Kollman, P. A. (2000). Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspectives in Drug Discovery and Design, 18(1), 113–135. 10.1023/A:1008763014207 [DOI] [Google Scholar]
- Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., & Olson, A. J. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30(16), 2785–2791. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard, A. (2018). Re-assessing the rule of 5, two decades on. Nature Reviews. Drug Discovery, 17(11), 777. 10.1038/nrd.2018.197 [DOI] [PubMed] [Google Scholar]
- OMEGA ( Version 2.5.1.4). (2013). Santa Fe, NM: OpenEye Scientific Software. [Google Scholar]
- Quinn, R. J., Carroll, A. R., Pham, N. B., Baron, P., Palframan, M. E., Suraweera, L., Pierens, G. K., & Muresan, S. (2008). Developing a drug-like natural product library . Journal of Natural Products, 71(3), 464–468. 10.1021/np070526y [DOI] [PubMed] [Google Scholar]
- Roux, B., & Simonson, T. (1999). Implicit solvent models. Biophysical Chemistry, 78(1–2), 1–20. 10.1016/S0301-4622(98)00226-9 [DOI] [PubMed] [Google Scholar]
- Sanders, J. M., Monogue, M. L., Jodlowski, T. Z., & Cutrell, J. B. (2020). Pharmacologic treatments for coronavirus disease 2019 (COVID-19): A review. JAMA, 323(18), 1824–1836. 10.1001/jama.2020.6019 [DOI] [PubMed] [Google Scholar]
- Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., Amin, N., Schwikowski, B., & Ideker, T. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SZYBKI (Version 1.9.0.3). (2016). Santa Fe, NM: OpenEye Scientific Software. [Google Scholar]
- Thomas, G. L., & Johannes, C. W. (2011). Natural product-like synthetic libraries. Current Opinion in Chemical Biology, 15(4), 516–522. 10.1016/j.cbpa.2011.05.022 [DOI] [PubMed] [Google Scholar]
- Thorlund, K., Dron, L., Park, J., Hsu, G., Forrest, J. I., & Mills, E. J. (2020). A real-time dashboard of clinical trials for COVID-19. The Lancet Digital Health, 2(6), e286–e287. 10.1016/S2589-7500(20)30086-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul Qamar, T., Alqahtani, S. M., Alamri, M. A., & Chen, L.-L. (2020). Structural basis of SARS-CoV-2 3CL(pro) and anti-COVID-19 drug discovery from medicinal plants. Journal of Pharmaceutical Analysis. 10.1016/j.jpha.2020.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., & Case, D. A. (2004). Development and testing of a general amber force field. Journal of Computational Chemistry, 25(9), 1157–1174. 10.1002/jcc.20035 [DOI] [PubMed] [Google Scholar]
- Wells, T. N. (2011). Natural products as starting points for future anti-malarial therapies: Going back to our roots? Malaria Journal, 10 Suppl 1(1), S3. 10.1186/1475-2875-10-S1-S3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. H., Abraham, M. H., Le, J., Hersey, A., Luscombe, C. N., Beck, G., Sherborne, B., & Cooper, I. (2002). Rate-limited steps of human oral absorption and QSAR studies. Pharmaceutical Research, 19(10), 1446–1457. 10.1023/A:1020444330011 [DOI] [PubMed] [Google Scholar]
- Zhu, N., Zhang, D., Wang, W., Li, X., Yang, B., Song, J., Zhao, X., Huang, B., Shi, W., Lu, R., Niu, P., Zhan, F., Ma, X., Wang, D., Xu, W., Wu, G., Gao, G. F., Tan, W., & China Novel Coronavirus Investigating and Research Team. (2020). A Novel Coronavirus from patients with pneumonia in China, 2019 . New England Journal of Medicine, 382(8), 727–733. 10.1056/NEJMoa2001017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.