

Abstract
Phaeochromocytomas (PCC) and paragangliomas (PGL) are rare tumours arising from the chromaffin cells of the adrenal medulla (PCC) or the paraganglia located outside the adrenal gland (PGL). However, their incidence is likely to be underestimated; around 10 % of all PCC/PGL are metastatic, with higher metastatic potential of PGLs compared to PCCs. If benign, surgery is the treatment of choice, but if metastatic, therapy is challenging. Here we review the currently existing therapy options for metastatic PCCs/PGLs including conventional chemotherapy (the original Averbuch scheme, but updated), radiopharmaceutical treatments (131I-MIBG, 90Y- and 177Lu-DOTATATE) and novel targeted therapies (anti-angiogenic tyrosine kinase inhibitors and mTORC1 inhibitors), emphasising future therapeutic approaches (HIF-2α and PARP inhibitors, temozolomide alone, metronomic temozolomide, somatostatin analogues) based on the oncogenic signalling pathways related to three different clusters comprising more than 20 well-characterised PCC/PGL susceptibility genes. We suggest that targeted combination therapies including repurposed agents may offer more effective future options worthy of exploration.
Keywords: Malignant phaeochromocytoma, therapy, oncogenic signalling
Introduction
Phaeochromocytomas (PCC) and paragangliomas (PGL) originate from neural crest-derived chromaffin cells of the adrenal medulla (PCC) or from the paraganglia located outside the adrenal gland (extra-adrenal phaeochromocytoma, PGL), and are generally highly vascular. They are frequently heritable albeit rare tumours with an incidence of 0.8 per 100,000 persons per year [1], with the highest prevalence in the fourth and fifth decades, but 10 %–20 % occur in paediatric patients [2, 3]. However, the incidence may be underestimated since over 50 % of phaeochromocytomas found at autopsy were not clinically suspected, at least in earlier studies [4]. They commonly produce catecholamines including adrenaline (epinephrine), noradrenaline (norepinephrine), or dopamine. They may produce one, two or all three different types of catecholamines, depending on the underlying somatic or germline mutation [5]. The excess of secretion can produce a whole variety of symptoms including hypertension (which may be episodic), palpitations, severe headache, sweating, and anxiety. For benign PCCs/PGLs, comprising around 90 % of phaeochromocytomas but less for PGLs, the therapy of choice is surgery following adequate blood pressure control with alpha-adrenoreceptor blockade and less often β-adrenoreceptor blockade. However, around 10 % of PCCs/PGLs are metastatic, and therapy for these tumours is currently challenging. Although there are some radiological, biochemical, histopathological (“Phaeochromocytoma of the Adrenal gland Scales Score” (PASS) > 6, Ki-67 > 2 %) and genetic clues suggesting the risk of metastases of primary PCCs/PGLs [5–9], malignant disease is currently only defined by the presence of distant metastases. Currently, the term “malignant” PCC/PGL has been replaced by “metastatic” PCC/PGL in the most recent WHO classification [10]. The localisation of the primary tumour can give some idea of the risk of metastases: around 5 %–20 % of PCCs and around 15 %–35 % of PGLs are metastatic [11–13]. Tumours that are extra-adrenal and greater than 5 cm in size are associated with a higher risk for metastatic spread [6, 14]. Due to poor differentiation and disrupted catecholamine production, metastatic PCCs/PGLs often produce high levels of dopamine and its metabolite 3–methoxytyramine [15, 16]. Moreover, high chromogranin A (CgA) levels have been linked to metastatic spread [14]. Most importantly at present, the specific genetic background is related to a high potential risk of metastasis [5]. Up to 30–40 % of these tumours show germline mutations, with an equal number showing identifiable somatic mutations in more than 20 well-characterised PCC/PGL susceptibility genes (Fig. 1), as recently reviewed [17]. These different mutations can be separated into three different clusters: pseudohypoxia-associated cluster-1 mutations, kinase signalling-associated cluster-2 mutations, and most recently Wnt-signalling linked cluster-3 mutations (Fig. 1) [5, 17]. The pseudohypoxia-associated cluster-1 mutations show a more aggressive behaviour, with the highest metastatic potential seen in SDHB carriers, compared to cluster-2 mutations. Cluster-3 mutations also seem to be associated with high metastatic potential [18]. In metastatic PCCs/PGLs, primary tumour resection can be recommended based on careful evaluation of tumour burden as well as the extent of metastatic disease. This option would help to reduce cardiovascular and other such risks from high catecholamine levels, alleviate symptoms from the tumour invading surrounding structures, or increase the entry of a radiopharmaceutical into metastatic lesions [19, 20]. In other types of neuroendocrine tumours (NETS) there is evidence that surgical resection of metastases may extend progression-free and overall survival although there is no direct evidence for PCC/PGLs apart from single case reports [21, 22] due to the rarity of disease. Therefore, surgery to remove metastatic lesions may be considered in individual cases of PCC/PGL, if at all possible. This is not to say that we would recommend removal of all metastatic lesions in all cases, but where it is possible to remove all or nearly all lesions with minimal morbidity, this should be actively considered, especially in the context of a possible curative approach, as previously suggested [23]. If such R0 resection is not possible, current therapy options rely on classic chemotherapy regimens (the Averbuch scheme [24]), but modified and updated), radiopharmaceuticals (131I-MIBG, 90Y/177Lu-DO-TATATE), as well as on molecular targeted therapies based on the activation of oncogenic signalling pathways associated with the different molecular clusters (anti-angiogenic tyrosine kinase inhibitors and mTORC1 inhibitors) including agents with potential future therapeutic possibilities (HIF-2α and PARP inhibitors, temozolomide alone, metronomic temozolomide, somatostatin analogues) [5, 25]. In spite of striking recent progress in this area, none of the therapy options mentioned has been officially approved by the U.S. Food and Drug Administration or the European Medicines Agency for metastatic PCC/PGL due to the rarity of the disease and the lack of prospective studies. Here we review the currently existing therapeutic options for metastatic PCCs/PGLs, and provide some ideas regarding upcoming promising future approaches, closely linked to new discoveries in molecular biology and pathogenesis of these tumours, especially those with underlying germline and somatic mutations.
Fig. 1.
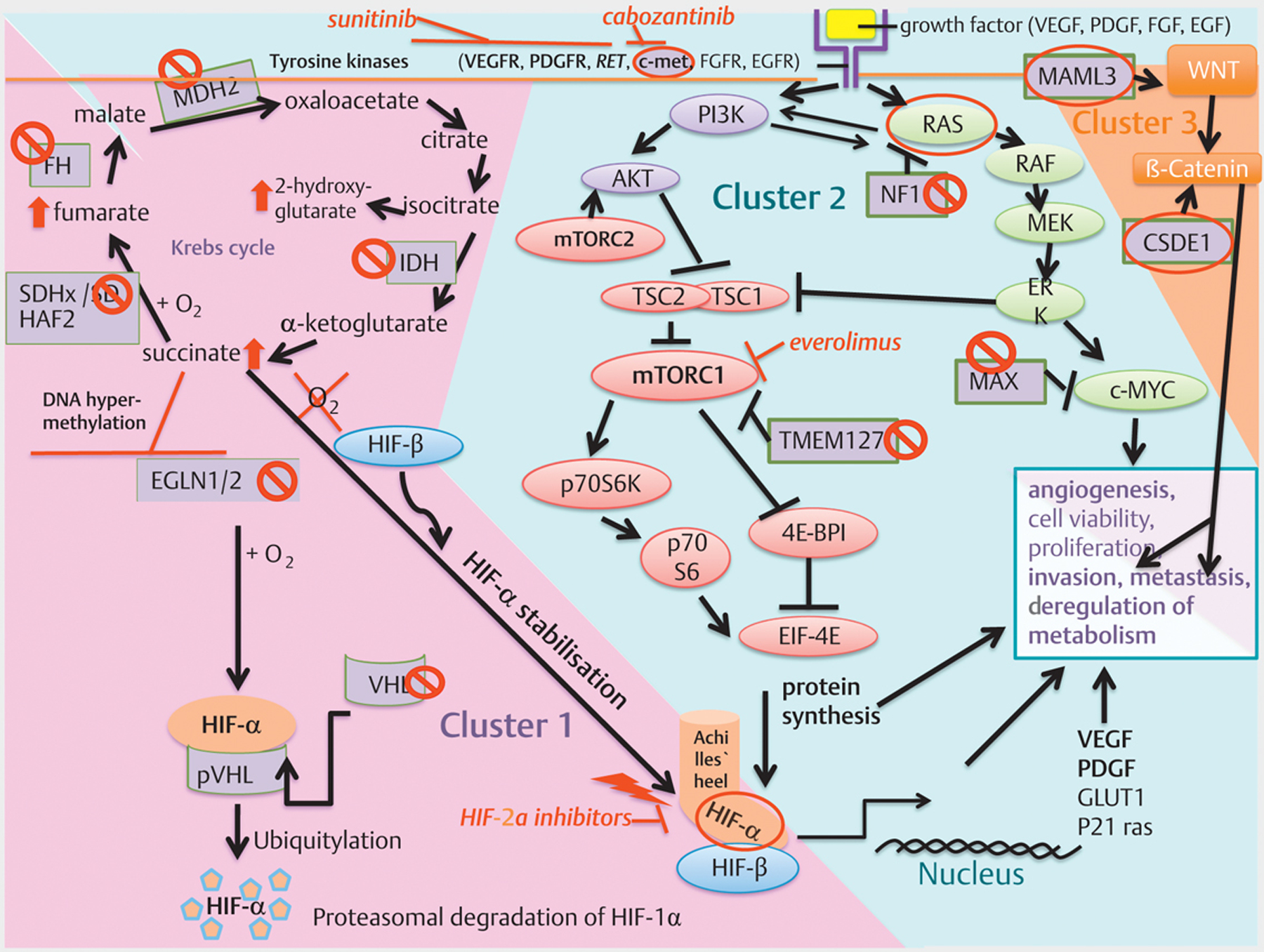
(modified from [17] and [103]) Overview: Cluster-1, −2 and −3 with molecular-targeted therapeutic options: Cluster-1: The pseudohypoxic signalling cluster includes mutations in genes encoding for hypoxia-inducible factor 2 alpha (HIF2A), Krebs-Cycle enzymes such as succinate dehydrogenase subunits (SDHx [SDHA, SDHB, SDHC, SDHD]), succinate dehydrogenase complex assembly factor 2 (SDHAF2), fumarate hydratase (FH), malate dehydrogenase 2 (MDH2), and isocitrate dehydrogenase (IDH), moreover, von Hippel–Lindau tumour suppressor (VHL) and egl-9 prolyl hydroxylase 1 and 2 (EGLN1/2). SDH(A[AF2]/B/C/D), FH, MDH2 and IDH mutations impair the Krebs cycle and lead to an increase in succinate, fumarate, or 2-hydroxyglutarate. The accumulation of oncometobolites promote DNA hypermethylation and inactivate tumour suppressor genes, including egl-9 prolyl hydroxylase 1/2 (EGLN1/2). The impaired activity of EGLN1/2 leads less ubiquitination/degradation of HIF-α. The HIF-α degradation is VHL-dependent; Therefore, these mutations promote HIF-α stabilisation independent of hypoxia resulting in increased angiogenesis (VEGF/PDGF transcription amongst others), dysregulation of metabolism, migration, invasion and finally metastases. HIF-α is the common final point, the “Achilles’ heel”, of cluster-1 mutations, interconnecting cluster-1 with cluster-2 mutations. Cluster-2: The kinase signalling cluster comprises mutations in the RET proto-oncogene, NF1 tumour suppressor, H-RAS and K-RAS proto-oncogenes, TMEM127 and MAX. Receptor tyrosine kinases (amongst others RET, VEGFR, c-met) activate PI3K. PI3K activates AKT, which inhibits TSC1/2 leading to disinhibition/activation of mTORC1; mTORC1 phosphorylates and activates various proteins including p70S6K, by which p70S6 is phosphorylated. Activated p70S6 promotes cell growth, proliferation, cell survival, and leads amongst others to protein synthesis of HIF-1α, which favours angiogenesis (VEGF/PDGF transcription amongst others), invasion and metastasis under hypoxic or pseudohypoxic conditions in the case of SDHx-mutations. The RAS/RAF/ERK-pathway is also activated by tyrosine kinases (amongst others RET) and activates mTORC1. NF1 mutations lead to disinhibiton/activation of RAS. TMEM127 mutations lead to disinhibition/activation of mTORC1. The tumour suppressor MAX antagonises Myc-dependent cell survival, proliferation and angiogenesis: mutations lead to increased cell proliferation and angiogenesis. Cluster-3: The Wnt signalling cluster comprises somatic mutations in CSDE1 and the mastermind like transcriptional coactivator 3 (MAML3) fusion genes. MAML3 mutated PGLs/PCCs show hypomethylation and over-activation of Wnt signalling. CSDE1 mutations lead to over-activation of β-catenin, a taget of Wnt signalling. Over-activation of Wnt/β-catenin signalling favors tumour proliferation, invasion and metastases. ⦸Phaeochromocytoma promoting loss of function mutation of a tumour suppressor gene.  Phaeochromocytoma promoting gain of function mutation of a proto-oncogene. ⬆Increase/up-regulation in the case of cluster-1 mutations of the Krebs cycle enzymes. ⟘
Phaeochromocytoma promoting gain of function mutation of a proto-oncogene. ⬆Increase/up-regulation in the case of cluster-1 mutations of the Krebs cycle enzymes. ⟘ Inhibition. ↘Activation.
Inhibition. ↘Activation.
Oncogenic Signalling in PCC and PGL
For the successful development of novel therapeutic strategies, it is extremely important to note that now more than 20 PCC/PGL-associated germline and/or somatic mutations have been identified, and these can be divided into three main clusters (Fig. 1) [5, 17]:
1) The pseudohypoxic signalling cluster (cluster-1) is related to mutations of genes encoding for proteins that are associated with significant regulation of the hypoxia signalling pathway and therefore hypoxia-inducible factor (HIF)-α, most significantly 2α, and includes mutations in genes encoding for hypoxia-inducible factor 2α (HIF2A), Krebs cycle enzymes such as succinate dehydrogenase subunits (SDHx [SDHA, SDHB, SDHC, SDHD]), succinate dehydrogenase complex assembly factor-2 (SDHAF2), fumarate hydratase (FH), malate dehydrogenase 2 (MDH2), and isocitrate dehydrogenase 1 (IDH1); it also includes von Hippel–Lindau tumour suppressor (VHL) and egl-9 prolyl hydroxylase-1 and −2 (EGLN1/2). PCCs and PGLs resulting from cluster-1 mutations are often multiple, aggressive and metastatic, and have a poorer prognosis compared to PCCs/PGLs bearing other susceptibility gene mutations. The cluster is called pseudohypoxic since it mimicks cellular hypoxia leading to an increased dependence on glycolysis due to an impaired Krebs cycle (SDH(A[AF2]/B/C/D), FH, MDH2 and IDH mutations) with impaired oxidative phosphorylation. In this manner the oncometabolites succinate, fumarate or 2-hydroxyglutarate accumulate, and in turn promote DNA hypermethylation and thus inactivate tumour suppressor genes, including egl-9 prolyl hydroxylase 1/2 (EGLN1/2), as reviewed in [26]. The impaired activity of EGLN1/2 leads to less dihydroxylation of HIF-α and thus, less ubiquitination/degradation of HIF-α. The HIF-α degradation is VHL-dependent; therefore, these mutations promote HIF-α stabilisation and accumulation independent of hypoxia resulting in increased angiogenesis (VEGF/PDGF transcription amongst others), dysregulation of metabolism, migration, invasion and finally metastases. HIF-α is the common final point, the „Achilles’ heel“, of cluster-1 mutations, interconnecting cluster-1 with cluster-2 mutations [27], as explained below in detail, and is thus a particularly significant possible therapeutic target. Almost all tumours belonging to cluster-1 have a noradrenergic phenotype and produce noradrenaline (normetanephrine), with some also producing 3-methoxytyramine, but little or no adrenaline (metanephrine).
2) The kinase signalling cluster (cluster-2) is related to mutations of genes encoding for proteins that belong to the phosphatidylinositol-3-kinase/mammalian target of rapamycin (PI3K/mTORC1) pathway/receptor kinase signalling and comprises mutations in the rearranged-during-transfection (RET) proto-oncogene, neurofibromin 1 (NF1) tumour suppressor, H-RAS and K-RAS proto-oncogenes, transmembrane protein 127 (TMEM127) and Myc-associated factor X (MAX). Most PCCs/PGLs patients belonging to this cluster have a relatively good prognosis. Activation of PI3K/AKT and RAS/RAF/ERK signalling promotes cell growth, proliferation, cell survival, and chromatin remodelling, and is also involved in the metabolic ‚switch’ towards glycolysis and glutaminolysis in cancer cells. Receptor tyrosine kinases (amongst others RET, VEGFR, c-met) activate PI3K: PI3K activates AKT, which in turn inhibits TSC1/2 leading to disinhibition/activation of mTORC1; mTORC1 phosphorylates and activates various proteins including p70S6K, by which p70S6 is phosphorylated. Activated p70S6 promotes cell growth, proliferation, cell survival, and amongst others, leads to protein synthesis of HIF-α, especially HIF-1α, which favors angiogenesis (VEGF/PDGF transcription amongst others), invasion and metastases under hypoxic or pseudohypoxic conditions, as in the case of SDHx-mutations. The RAS/RAF/ERK-pathway is also activated by receptor tyrosine kinases (amongst others RET) and activates mTORC1. NF1 mutations lead to disinhibition of RAS and to mTORC1 activation. TMEM127 is a tumour suppressor gene inhibiting mTORC1 with mutations also leading to disinhibition/activation of mTORC1. The tumour suppressor MAX antagonises Myc-dependent cell survival, proliferation and angiogenesis: mutations lead to increased cell proliferation and angiogenesis [17, 28]. Most of the cluster-2 mutations have a typical adrenergic phenotype with adrenaline (metanephrine) together with or without noradrenaline (normetanephrine) production.
HIF-α, the interconnection between cluster-1 and cluster-2: As extensively reviewed in [26], it is not completely clear whether HIF-1α or HIF-2α promote tumorigenesis in cluster-1 PCCs and PGLs and both HIFs seem to be involved; nevertheless, HIF-2α seems to be predominantly overexpressed in cluster-1 mutations and is considered as an important player in the aggressive behaviour of these tumours. For example, overexpression of HIF-2α has been described in patients with PCCs and PGLs with SDHB and SDHD mutations [29, 30]. On the other hand, another study [31, 32] found more frequent HIF-2α overexpression in VHL-mutated PCCs and PGLs, whereas in SDH-related tumours nuclear HIF-1α staining was more prominent, although these findings in a single study are controversial. In the studies of Favier et al. [33], HIF-2α mRNA overexpression was detected in both VHL- and SDH-mutated PCCs and PGLs, as compared to cluster 2 (NF1- and RET-mutated) PCCs and PGLs. A study by Koh et al. [34] also indicated a leading role for HIF-2α in tumour development and progression in cluster-1 tumours. Moreover, somatic and germline gain-of-function mutations have been identified in the HIF2A gene in patients with multiple or recurrent PCCs and PGLs and polycythaemia, with a metastatic potential of around 30–40 % [35–40]. These mutations disrupt HIF-2α prolyl hydroxylation, as well as the binding of mutated HIF-2α to pVHL, resulting in HIF-2α accumulation.
The cluster-2 mutations in RET and NF-1 promote nuclear accumulation of HIF-1α via Ras/RAF/MEK/ERK over-activation [41]. ERK directly phosphorylates HIF-1α and induces transcription of VEGF, a key regulator of angiogenesis [42, 43]. RET and NF1 mutations, moreover, lead to over-activation of PI3K signalling and, thus, induce HIF-1α mRNA expression and transcription via overexpression of NF-κB subunits. Activation of PI3K also may lead to mTORC2 activation; this in turn induces HIF-2α expression [44]. HIF-2α in turn may also activate mTORC1 [45]. In tumours caused by TMEM127 and MAX mutations, HIF-1α levels seem to be increased due to mTORC1 activation [46, 47]. MAX acts as a tumour suppressor, and mutations in MAX disinhibit/activate c-Myc signalling [28, 48]. HIF-1α is also one of the transcription targets of c-Myc[49]. Somatic H-RAS mutations were found in PCCs and PGLs, causing the activation of the Ras/RAF/ERK signalling pathway [50]. Ras/RAF/ERK pathway activation leads to an increase in HIF-1α signalling and to the transcription of HIF target genes. Therefore, both HIF-1α and HIF-2α are common checkpoints/gatekeepers of cluster-1 and cluster-2 mutated PCC/PGLs and, thus, interconnect both clusters as a potential common druggable target. However, cluster-1 mutations are predominantly associated with HIF-2α accumulation which is also associated with more aggressive tumour behaviour in PCC/PGL and other tumours [18, 34, 51], while cluster-2 mutations are predominantly linked to HIF-1α accumulation (although both clusters may involve both types of HIFs, which have been found to be overexpressed in most human cancers and have been considered as an essential checkpoint (gatekeeper) of tumorigenesis, as reviewed in [26, 27]). Nevertheless, we essentially do not know why cluster-1 tumours have a much worse prognosis. Several studies are underway now to decipher the molecular biology of these tumours in even more detail.
3) The Wnt signalling cluster (cluster-3) comprises somatic mutations in Cold Shock Domain-containing E1 gene (CSDE1) and the ‚mastermind-like’ transcriptional coactivator 3 (MAML3) fusion genes. MAML3-mutated PGLs/PCCs show hypomethylation and over-activation of Wnt and Hedgehog signalling. These tumours strongly express the neuroendocrine tumour marker CgA. CSDE1 mutations lead to over-activation of β-catenin, a target of Wnt signalling: over-activation of Wnt/β-catenin signalling favours tumour proliferation, invasion and metastases. The catecholamine phenotype of the recently discovered cluster-3 mutations [18] is not known as yet. In a recent study, cluster-3 mutations, with MAML3 being the most common, were associated with high Ki-67, aggressive behaviour and the early occurence of distant metastases [18].
The association of the three different clusters with effects on different oncogenic signalling pathways related to a different risk of metastatic PCC/PGL emphasises the importance of genetic testing in all PCC/PGL patients [52].
Very recently, a gain of function mutation in the DNA methyltransferase DNMT3A has been identified as a potential PCC/PGL susceptibility gene leading to hypermethylation and inactivation of tumour suppressor genes, similar to the increased oncometabolites associated with cluster-1 mutations [53]. Currently, it is unknown whether this gene may play a primary or secondary role in the pathogenesis of these tumours.
Chemotherapy: Classic Chemotherapy Updated
Chemotherapeutic agents inhibit the cell cycle at different phases. Besides other sporadically-used chemotherapy protocols including cisplatin, 5-fluorouracil, methotrexate, ifosfamide and streptozotocin which show low evidence to support their clinical use [54, 55], the best studied chemotherapy protocol for advanced PCC/PGL combines cyclophosphamide, vincristine, and dacarbazine (CVD) according to the Averbuch scheme (cyclophosphamide 750 mg/m2, vincristine 1.4 mg/m2, and dacarbazine 600 mg/m2 on day 1 and dacarbazine 600 mg/m2 on day 2) [56]. However, all studies are retrospective and prognostic factors indicating metastatic behaviour such as size and location of the primary tumour, size, location and the timing of metastases, progression prior to chemotherapy, and SDHB mutation status, are frequently missing [55]. A meta-analysis of the largest studies on CVD showed a partial response concerning tumour size in 37 % of patients [57]. In two of the studies included in this meta-analysis in PCC/PGL patients, the median progression-free survival (PFS) of CVD-treated patients was 20 months and 40 months, respectively. The other studies included in the meta-analysis did not report PFS [57]. However, these studies also included some patients with slow-growing tumours and minimal or no progression prior to study entrance. One of the retrospective studies on CVD which only included patients with progression prior to chemotherapy found radiographic and clinical evidence of a response in 33 % of patients [58]. This was the only retrospective study showing an apparent survival benefit from CVD therapy, although while responders had a median overall survival (OS) of 6.4 years versus 3.7 years in non-responders, this was not statistically significant (p = 0.095). However, in a multivariable analysis adjusting for tumour size at the time of diagnosis, median OS was significantly longer among those who received CVD (p = 0.05; hazard ratio = 0.22; 95 % confidence interval = 0.05–1.0) [58]. There is evidence that patients, especially those with rapidly-growing SDHB-related PCCs/PGLs with radiographic progression over a short period of time (< 6 months), may benefit from CVD therapy [57, 59–61]. Accordingly, a recent study has shown promising results for prolonged CVD chemotherapy in 12 patients harbouring an SDHB mutation with a complete response in two of 12 patients (16.7 %) and a partial response in 8 of 12 (66.7 %) patients [62]. Therefore, a total of 83 % of SDHB mutation carriers responded to prolonged CVD therapy. All patients showed tumour reduction (12–100 % by Response Evaluation Criteria in Solid Tumours [RECIST]). A median of 20.5 cycles (range 4–41) was administered. PFS and OS were 930 and 1190 days, respectively. Thus, prolonged CVD therapy resulted in continued tumour reduction, and the authors suggested that CVD chemotherapy be considered part of the initial management in patients with metastatic SDHB-related PCC/PGL [62]. This study again provides strong evidence for the benefits of genetic testing in order to identify the best possible treatment decision. However, the side effects of vincristine may include peripheral sensory and autonomic neuropathy [63] while rare cases of leukaemia and myelodysplastic syndrome have been observed [58]. Whether adjuvant treatment with 4–6 cycles of CVD after surgery in patients with positive predictors of metastatic potential could improve PFS and OS has not yet been studied [55].
Recently, monotherapy with temozolomide, the oral precursor for dacarbazine, has shown a 50 % response rate in SDHB carriers, 33 % partial responses and 47 % stable disease over the whole study population, and thus may be considered for tumour stabilisation as a maintenance regime subsequent to CVD chemotherapy in SDHB carriers [64]. Interestingly, 80 % of these responders showed low tumour levels of O6-methylguanine-DNA methyltransferase (MGMT) [64]. Furthermore, a correlation between SDHB-mutated tumours and hypermethylation of the MGMT promoter region was observed [64]. The increase in the oncometabolites succinate, fumarate, or 2-hydroxyglutarate in SDHB cluster-1-mutated PCCs/PGLs leads to DNA hypermethylation [17] associated with hypermethylation of the MGMT promoter region leading to down-regulation of MGMT expression by epigenetic silencing (Fig. 1) [65, 66]. Temozolomide is a DNA alkylating substance leading to DNA adduction, double-strand breaks and apoptosis. The only enzyme capable of repairing the temozolomide-induced adducts is MGMT, which is irreversibly inactivated during the repair process [65, 67]. Down-regulation of MGMT expression makes tumours with mutations in Krebs cycle enzymes such as SDHB very sensitive to temozolomide. Due to frequent hypermethylation of the MGMT promoter region in SDHB-mutated tumors, these SDHB-mutated tumors show lower MGMT expression leading to a higher susceptibility to temozolomide. Therefore, the first step is the genetic testing of the PCC/PGL patients for an SDHB mutation which most likely increases sensitivity to temozolomide. Since the postulated mechanism of tumourigenesis is similar in all SDHx mutated tumours with a pseudohypoxia-associated increase in succinate and DNA hypermethylation (Fig. 1)[1], it is likely that temozolomide would also show efficacy in patients with SDHA/C/D mutations, although this has to be explored in further studies. Additional measurement of MGMT expression in PCC/PGLs might be beneficial for assessment of the tumor sensitivity to temozolomide but this still needs to be systematically investigated. At present, we would not advise its routine use. The long-term tolerability of temozolomide monotherapy may be better as compared to maintenance therapy with CVD. Maintenance with dacarbazine or temozolomide alone after 6–9 cycles of CVD may be reasonable for patients who initially responded to CVD (partial radiographic response or disease stabilisation) [55, 64]. If temozolomide monotherapy is not effective or not tolerated at standard doses, a metronomic scheme with long-term low-dose temozolomide (75 mg/m2/d with a schedule of 3 weeks on treatment followed by 1 week off treatment) in combination with high-dose lanreotide autogel (120 mg s.c. every 14 days) might stabilise PCCs/PGLs with low MGMT levels and MGMT hypermethylation, as recently published for two patients [68]. This study again emphasised the benefit of genetic testing in all patients in order to provide the best individualised treatment approach depending on specific oncogenic signalling, although the patient numbers are small.
Radiopharmaceuticals: 131I-metaiodbenzylguanidin (MIBG) and Peptide Receptor Radionuclide Therapy (PRRT)
MIBG is a noradrenaline analogue that is taken up by the chromaffin cells of the sympathomedullary system which can be specifically targeted by the radiopharmaceutical 131I-MIBG. 131I-MIBG therapy is currently the most studied treatment option in metastatic PCC/PGL, including even (albeit small) prospective studies [69–73]. It is still the recommended first-line treatment for 123I-MIBG positive patients with slow-growing metastases [5], although this situation may be changing. Treatment regimens can be classified into two basic strategies: fractionated multiple low-dose treatments, or a limited number of high-dose treatments. Both high-dose regimens and low-dose regimens have shown efficacy [74]. In a large meta-analysis of 17 studies on 131I-MIBG published between 1984 and 2012 including 243 PCC/PGL patients (follow-up durations 24 to 62 months), 3 % of patients showed a complete response, 27 % of patients showed a partial response, while 52 % of patients exhibited stable disease [75]. In two of the studies included in this meta-analysis on 131I-MIBG treatment in PCC/PGL patients, the mean PFS of 131I-MIBG-treated patients was 23.1 and 28.5 months, respectively. Two other studies reported a 5-year survival rate of 45 % (with a median OS of 4.7 years) and 64 %, respectively, and one study reported a median OS of 42 months. The other studies included in the meta-analysis did not report PFS or OS [75]. In another retrospective study, the median PFS of MIBG-treated PCC patients (n = 6, none with SDHB mutations) was 20.6 months with a median OS of 41.2 months; the median PFS of 131I-MIBG-treated PGL patients was 14.4 months (n = 5, one with an SDHB mutation) with a median OS of 22.8 months [76]. Clearly, the robustness and duration of these responses depends on a host of factors, probably most importantly on the genetic background. For patients with bone metastases a poorer response to 131I-MIBG has been reported compared to patients with metastases limited to soft tissue [71]. After low-dose treatment, adverse effects are generally mild including anorexia, nausea, vomiting, mild leukopenia, thrombocytopenia, and gonadal failure; however, severe bone marrow aplasia has been reported sporadically [71]. After high-dose treatment, complete (3/12) and partial responses (7/12) have been reported, but the adverse effects on the bone marrow including grade 3 thrombocytopenia and grade 3 and grade 4 neutropenia were more common [77]. Acute myeloid leukaemia and myelodysplastic syndrome have also been observed after several infusions of high-dose treatments [69]. A new preparation of ultratrace 131I-MIBG with high specific activity (radiolabelled MIBG without the addition of a carrier) produced on the Ultratrace® platform, may increase tumour tissue uptake and treatment efficacy with less side effects [78]. Ultratrace iobenguan-131I is being evaluated in a phase II trial (NCT00874614) that has reported a preliminary radiological partial response rate and stable disease in more than 90 % of patients (n = 34) [79]. The estimated study completion date is February 2021. In March 2018 a certfication or an extension request to delay submission of results was submitted. The study results should first be awaited before Ultratrace Iodine treatment might be recommended. However, the increasing use of PRRT will probably render such data otiose as newer radioisotopes are used.
Since most PCCs/PGLs strongly express somatostatin receptor subtype 2 (SSTR2) [80, 81], PRRT has also been investigated in some small studies [82–85], and was reported to be superior to 131I-MIBG therapy in PCC/PGL patients (n = 22) regarding PFS and response to treatment according to a very recently published retrospective study [76]. In this case, a radiocative ligand, 90Y or more usually 177Lu, is combined with octreotide via a chelating agent. In PGL patients the OS was also significantly higher after PRRT compared to 131I-MIBG therapy [76]. The recent prospective NETTER-1 trial in midgut neuroendocrine tumours has led to the approval of PRRT for the treatment of midgut and pancreatic neuroendocrine tumours in many countries [86], and initiation of an analogous prospective phase II clinical trial with PCC/PGL patients seems reasonable. Nevertheless, more data regarding different hereditary and sporadic metastatic PCCs/PGLs is needed for more accurate conclusions regarding the efficacy of this type of radiotherapy.
Conventional External Beam Radiation Therapy (cEBRT)
CEBRT is the most frequently used treatment in patients with PCC/PGL bone metastases; in a retrospective study with 24 PCC/PGL patients treated with cERBT, more than 80 % showed symptomatic and imaging improvement [87], and in a very recently published study with 41 PCC/PGL patients treated with cEBRT, 81 % of all lesions showed local control at 5 years while 94 % of patients showed symptomatic improvement [88]. A prospective randomised control trial published in 2005 showed that patients with spinal cord compression due to metastases of any cancer benefited most from first-line decompressive surgery followed by cERBT, compared to cEBRT alone [89]. Moreover, spinal stereotactic radiosurgery (SSRS) (cyberknife), which allows the delivery of a high dose of radiation directly to the lesion, decreasing toxic effects on adjacent tissue, may also be a therapeutic option for PCC/PGL bone metastases [90–92].
Molecular Targeted Therapies: Receptor Tyrosine Kinase Inhibitors, mTORC1 Inhibitors, HIF-2α Antagonist, SSTR2 analogues
The receptor tyrosine kinase inhibitors previously considered as potential therapeutic targets for PGL/PCC include sunitinib, cabozantinib, axitinib and pazopanib, and all have anti-angiogenic potential. Sunitinib is the most studied tyrosine kinase inhibitor in PGL/PCC with anti-angiogenic potential due to inhibition of vascular endothelial growth factors-1 and −2 receptors (VEGFR1/2), platelet-derived growth factor-β receptor (PDGFR) and RET, amongst others. Accordingly, sunitinib is an interesting therapeutic agent for pseudohypoxia associated cluster-1 mutations (amongst others SDHx) leading to HIF-α stabilisation and increased angiogenesis, but also for the kinase signalling associated cluster-2 mutations including RET and NF1 and converging with the pseudohypoxia pathway into HIF-α with increased angiogenesis (Fig. 1). Sunitinib has already been approved by the U.S. Food and Drug Administration and the European Medicines Agency for renal cell carcinomas, pancreatic neuroendocrine tumours and gastrointestinal stromal tumours. At the moment, sunitinib is being investigated in the first prospective, randomised, placebo-controlled clinical phase II trial in PCC and PGL (FIRST-MAPPP, NCT01371202). At the time the current manuscript was written, the trial had almost completed recruitment (n = 74). The current largest published retrospective trial investigating sunitinib (dose: 37.5 mg or 50 mg) in PCC and PGL included 17 patients [93]: 3 of the 17 patients could not be evaluated for tumour response to sunitinib since they suffered from early toxicities and medication was stopped. Side effects of sunitinib include hypertension, diarrhoea, mucositis, hand-food syndrome and fatigue. Of the remaining 14 patients, 3 (21.4 %) showed partial responses (PR), 5 (35.7 %) had stable disease (SD), while the other six patients (43 %) had progressive disease (PD). Therefore, a total of 57 % of patients evaluated showed clinical and radiographic benefit from sunitinib, indicating that sunitinib might be an effective treatment option. The median OS of sunitinib-treated patients was 26.7 months. Although the median PFS after initiating sunitinib was only 4.1 months (95 % confidence interval = 1.4–11.0), the PFS of the responders to sunitinib was much longer compared to non-responders: the three partial responses to sunitinib lasted for 11, 12 and 4.5 months, respectively. Two cases with stable disease had no progression until the end of the observation period of 36 months (one of these patients was under additional therapy with the mTORC1 inhibitor rapamycin for 1.5 years until the end of the observation period), the other three patients with stable disease showed a PFS of 27, 8 and 6 months, respectively. The time to progression of non-responders to sunitinib was 0.4–4 months, which strongly decreased the median PFS of the whole study population [93]. Current unpublished data from the NIH suggest that such therapy may be particularly effective for SDHB-related tumours, although again in many patients rapid relapse may occur after a brief ‚honeymoon’ period. It is likely that in the future specific molecular features may be identified to indicate the type of patient who will show a prolonged response to such agents. Four of the 5 patients with SD were SDHB mutation carriers and one of 3 patients with PR was an SDHB carrier. Five of the 6 patients with PD suffered from apparently sporadic PCCs and one of them was an SDHB mutation carrier. Thus, most patients with a clinical benefit were carriers of SDHB mutations. It should also be noted that several SDHB patients with metastatic PCC/PGL who initially had a good response then progressed rapidly (K. Pacak, personal experience). Therefore, caution should be taken when sunitinib is considered for the therapy of these particular patients. For one of the SD-HB-mutated patients with SD, the sunitinib dose was decreased to 25 mg due to fatigue and 18 months after initiation of sunitinb therapy the mTORC1 inhibitor rapamycin (4 mg) was added: this patient showed persistent SD and overall lower glucose uptake in 18F-FDG PET/CT 36 months after initiation of sunitinib therapy and 18 months after first application of the mTORC1 inhibitor rapamycin, and had experienced no disease progression at the time of the writing of that study [93]. Although single treatment with the mTORC1 inhibitor everolimus was not effective in PCC and PGL in a small clinical study [94], nevertheless, this implies that combination treatments might be more effective at lower doses with less toxicity, compared to higher dose treatment with each drug separately. A study (NCT00655655) with the mTORC1 inhibitor everolimus plus the EGFR-1 inhibitor vatalanib is ongoing. Consistently, our in vitro and in vivo data on PCC/PGL [95–98] showed additive effects of the mTORC1 inhibitor everolimus plus the statin lovastatin (which leads to AKT and ERK inhibition), or additive effects of a dual PI3K/mTORC1/2 inhibitor plus lovastatin; moreover, we could show synergism for lovastatin plus 13-cis retinoic acid at low therapeutically-relevant doses. Re-purposing older well-tolerated drugs such as statins or 13-cis retinoic acid for novel therapeutic purposes seems to be a novel approach worthy of further research, particularly in combination treatments.
Another promising tyrosine kinase inhibitor might be the c-met inhibitor cabozantinib. In renal cell carcinoma patients it was more effective than sunitinib in terms of PFS and objective response [99, 100]. Patients with metastatic prostate cancer with bone metastases experienced pain relief, increased haemoglobin and decreased bone turnover in response to cabozantinib [101]. Preliminary results of a phase II study (NCT 02302833) assessing cabozantinib in 11 patients with PCC/PGL have shown a tumour size decrease and disease stabilisation in most patients with a PFS of 11.2 months, without serious adverse events [55]. The initial dose of cabozantinib was 60 mg daily and the dose was titrated down on the basis of tolerability. However, two phase II studies with the tyrosine kinase inhibitors pazopanib (6 patients) (NCT01340794) [102] and axitinib (9 patients) (NCT01967576) did not show any clear benefit in PCC/PGL. Due to gastrointestinal and serious cardiovascular adverse events (Takotsubo cardiomyopathy) of several patients, the pazopanib trial was terminated [102] and the axitinib trial is being closed. All tyrosine kinase inhibitors mentioned increase the risk of severe hypertension and require adequate blood pressure monitoring.
HIF-α is the „Achilles heel“ of the cluster-1 PCC/PGL susceptibility gene mutations (detailed under „Oncogenic signalling“, Fig. 1) and, moreover, interconnects cluster-1 with cluster-2 kinase signalling (also details under „Oncogenic signalling) [26, 27, 35, 103]. As described in detail above and reviewed in [26, 27], all the pseudohypoxia-associated cluster-1 PCC/PGL susceptibility mutations predominantly lead to HIF-2α stabilisation and accumulation as a consequence of increases in succinate and other oncometabolites. Therefore, the HIF-2α inhibitor PT2399, which has already shown efficacy in renal cell carcinoma mouse models and was superior to sunitinib [104, 105], might be a promising novel therapy option for PCC/PGL. It deserves further investigation in PCC/PGL in vitro and in vivo, especially in cluster-1 models and eventually in patients carrying SDHx or other Krebs cycle-associated mutations [27].
Long-acting SSTR2 analogues have already been approved for tumour growth control in midgut neuroendocrine tumours (PROMID trial) and pancreatic neuroendocrine tumours with a Ki-67 < 10 % (CLARINET trial) [106, 107]. Due to strong SSTR2 expression of PCC/PGL [81], treatment with long-acting SSTR2 analogues deserves evaluation in a clinical phase II trial with PCC/PGL patients.
The current ongoing clinical trials in PCC/PGL investigating molecular targeted therapy are reviewed in [25].
PARP inhibitors – Novel Targeted Therapy to Improve Old Fashioned Chemotherapy
As mentioned above, chemotherapeutic agents such as temozolomide lead to DNA damage and tumour cell apoptosis. Poly(ADP-ribose) polymerase (PARP) is a highly conserved enzyme in eukaryotes which repairs DNA breaks and stabilises DNA replication [108], similar to MGMT. PARP-deficient mice were very sensitive to DNA damaging agents [109]. The PARP DNA repair system is very active in PCCs/PGLs with pseudohypoxia-associated cluster-1 mutations through increased intracellular NAD + levels secondary to enhanced catalytic activity of NADH dehydrogenase. This leads to chemoresistance in cluster-1 (SDHB) mutation carriers. PARP inhibitors, which have been demonstrated to potentiate DNA damaging effects of chemotherapeutic agents [110, 111], could be a promising target for metastatic PCCs/PGLs with cluster-1 mutations such as SDHB. In a very recent study in an allograft mouse model of SDHB-knockdown PCC/PGLs, therapy with the PARP inhibitor olaparib sensitised PCC/PGL cells to temozolomide, suppressed metastatic allograft lesions and improved overall survival [112]. The NAD + /PARP pathway might be a crucial target in SDHB-mutated PCC/PGL. Combination therapy with olaparib and temozolomide could become a very promising approach for cluster-1 metastatic tumours.
Immunotherapy
PCC/PGL of the cluster-1 group grow under pseudohypoxic conditions and this potentially prevents them from being recognised by the immune system. The binding of programmed death-ligand 1 (PD-L1) to its receptor programmed cell death protein 1 (PD-1) on T cells delivers a signal that prevents T-cell-receptor-mediated activation of interleukin-2 production and T cell proliferation, and this may promote tumour growth. Pembrolizumab, an antibody that inhibits PD-1, is currently being investigated in a phase II clinical trial (NCT02721732) for the treatment of metastatic PCC/PGLs [20]. The study is still recruiting at the time of writing and, therefore, results are pending. It is clearly important to understand the effects of the molecular targeted agents on the immune system such that they do not have potentially antagonistic effects [113]. It will be also important to find out whether metastatic PCC/PGL, especially those that are very aggressive and are associated with an SDHB mutation, present with PD-1 and PD-L1 since our preliminary results did not detect either one in these tumours (K. Pacak, personal observation). Immunotherapy will be an important area of future research and novel therapeutic options in these patients, but should be investigated in experimental and other models before translation to patients with metastatic PCC/PGL.
Conclusions
Fig. 2 shows a flow chart comprising current and potential future therapeutic options for PCC/PGL. If curative surgery is not possible, CVD chemotherapy should be considered as first-line therapy in patients with metastatic rapidly growing SDHB-related PCC/PGL; this may be followed by temozolomide monotherapy. Metronomic temozolomide scheme might be beneficial in patients with pseudohypoxia-associated cluster-1 mutations and low MGMT tumour levels. 131I-MIBG therapy is currently recommended as first-line therapy for slowly-growing metastatic PCCs/PGLs. Preliminary data from a phase II clinical study with ultratrace iobenguan 131I with high specific activity look promising and may improve I131-MIBG uptake and activity, and reduce side effects. However, it seems likley that PRRT based on SSTR2 expression of PCC/PGL may be superior to 131I-MIBG therapy and needs to be evaluated in prospective clinical trials. CEBRT or spinal stereotactic radiosurgery are therapy options for bone metastases. Molecular targeted therapies with anti-angiogenic tyrosine kinase inhibitors such as sunitinib and cabozantinib are being studied in prospective phase II clinical trials, and appear to be promising agents for patients with pseudohypoxia-associated cluster-1 and kinase signalling-associated cluster-2 mutations; mTORC1 inhibitor monotherapy seems to show little activity, but combination treatments look encouraging. Novel molecular targeted therapies such as HIF-2α inhibitors, focusing on a convergent target of many mutations, as well as PARP inhibitors interrupting the repair pathway in cluster-1 mutated PCCs/PGLs, seem to have the potential to open up the road for future therapy approaches. A prospective phase II clinical study on immunotherapy is ongoing. Cell line and in vivo studies suggest that molecular targeted combination treatments, possibly with currently available repurposed agents, may show the way to more effective treatment in the future. Finally, combined therapies using either radiotherapy or chemotherapy together with immunotherapy are on the horizon in many metastatic cancers, and such studies should also include metastatic PCC/PGL patients.
Fig. 2.
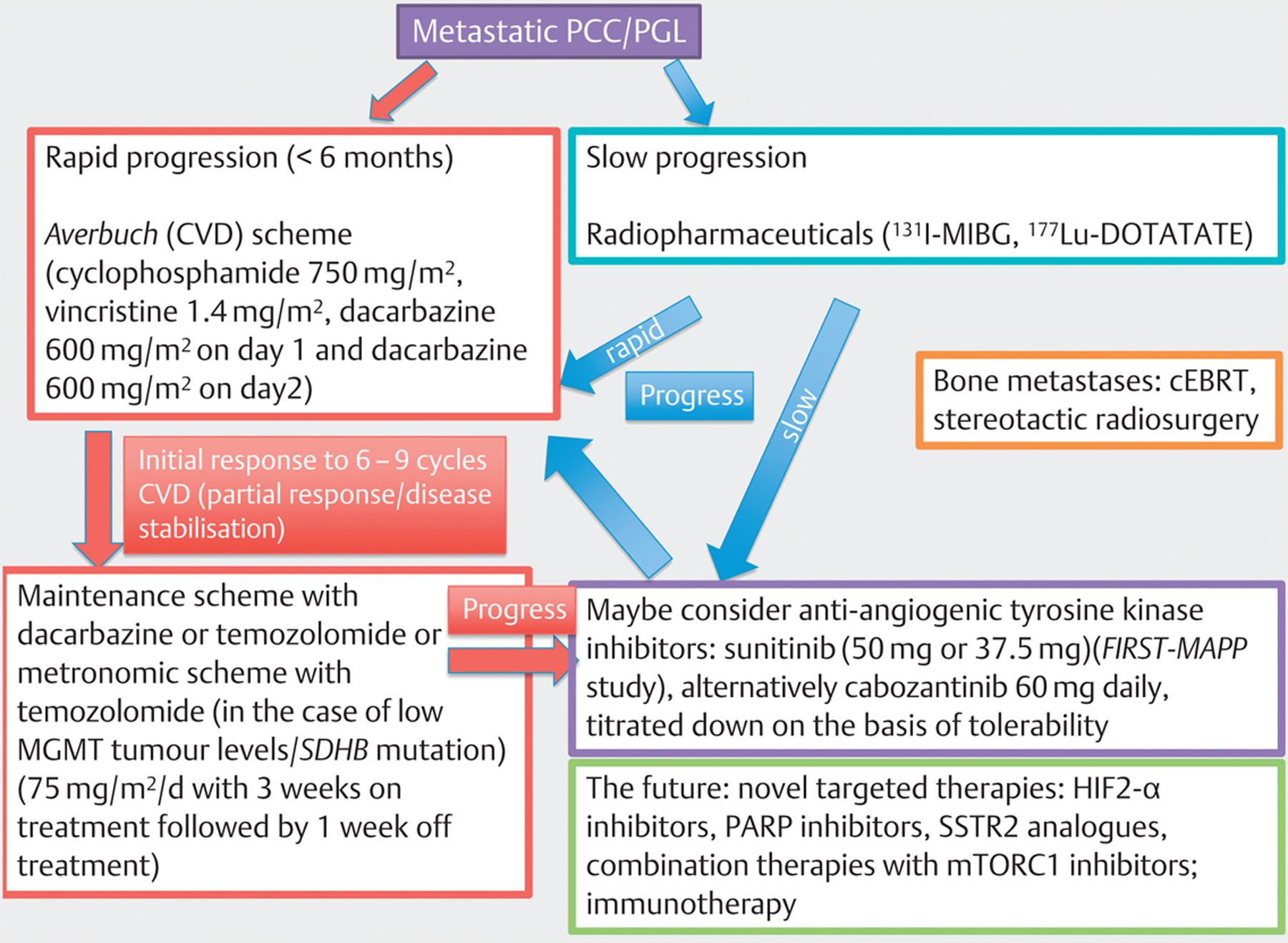
Current and potential future therapeutic options for PCC/PGL
Acknowledgement
We acknowledge the support of Deutsche Forschungsgemeinschaft (DFG) within the CRC/Transregio 205/1 “The Adrenal: Central Relay in Health and Disease“.
Footnotes
Conflict of Interest
No conflict of interest has been declared by the authors.
References
- [1].Beard CM, Sheps SG, Kurland LT et al. Occurrence of pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin Proc 1983; 58: 802–804 [PubMed] [Google Scholar]
- [2].Barontini M, Levin G, Sanso G. Characteristics of pheochromocytoma in a 4- to 20-year-old population. Ann NY Acad Sci 2006; 1073: 30–37 [DOI] [PubMed] [Google Scholar]
- [3].Beltsevich DG, Kuznetsov NS, Kazaryan AM et al. Pheochromocytoma surgery: Epidemiologic peculiarities in children. World J Surg 2004; 28: 592–596 [DOI] [PubMed] [Google Scholar]
- [4].Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc 1981; 56: 354–360 [PubMed] [Google Scholar]
- [5].Crona J, Taieb D, Pacak K. New Perspectives on pheochromocytoma and paraganglioma: Toward a molecular classification. Endocr Rev 2017; 38: 489–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Korevaar TI, Grossman AB. Pheochromocytomas and paragangliomas: Assessment of malignant potential. Endocrine 2011; 40: 354–365 [DOI] [PubMed] [Google Scholar]
- [7].Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: A clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol 2002; 26: 551–566 [DOI] [PubMed] [Google Scholar]
- [8].Strong VE, Kennedy T, Al-Ahmadie H et al. Prognostic indicators of malignancy in adrenal pheochromocytomas: Clinical, histopathologic, and cell cycle/apoptosis gene expression analysis. Surgery 2008; 143: 759–768 [DOI] [PubMed] [Google Scholar]
- [9].Liu TH, Chen YJ, Wu SF et al. [Distinction between benign and malignant pheochromocytomas]. Zhonghua Bing Li Xue Za Zhi 2004; 33: 198–202 [PubMed] [Google Scholar]
- [10].Lam AK. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocr Pathol. 2017: 28: 213–227 [DOI] [PubMed] [Google Scholar]
- [11].O’Riordain DS, Young WF Jr., Grant CS et al. Clinical spectrum and outcome of functional extraadrenal paraganglioma. World J Surg 1996; 20: 916–921 discussion 922 [DOI] [PubMed] [Google Scholar]
- [12].Amar L, Bertherat J, Baudin E et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 2005; 23: 8812–8818 [DOI] [PubMed] [Google Scholar]
- [13].King KS, Prodanov T, Kantorovich V et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol 2011; 29: 4137–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Grossman A, Pacak K, Sawka A et al. Biochemical diagnosis and localization of pheochromocytoma: can we reach a consensus? Ann NY Acad Sci 2006; 1073: 332–347 [DOI] [PubMed] [Google Scholar]
- [15].Eisenhofer G, Lenders JW, Siegert G et al. Plasma methoxytyramine: A novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer 2012; 48: 1739–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Parenti G, Zampetti B, Rapizzi E et al. Updated and new perspectives on diagnosis, prognosis, and therapy of malignant pheochromocytoma/paraganglioma. J Oncol 2012; 2012: 872713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jochmanova I, Pacak K. Genomic Landscape of pheochromocytoma and paraganglioma. Trends Cancer 2018; 4: 6–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fishbein L, Leshchiner I, Walter V et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell 2017; 31: 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Roman-Gonzalez A, Zhou S, Ayala-Ramirez M et al. Impact of surgical resection of the primary tumor on overall survival in patients with metastatic pheochromocytoma or sympathetic paraganglioma. Ann Surg 2017. doi: 10.1097/SLA.0000000000002195 [DOI] [PubMed] [Google Scholar]
- [20].Roman-Gonzalez A, Jimenez C. Malignant pheochromocytoma-paraganglioma: Pathogenesis, TNM staging, and current clinical trials. Curr Opin Endocrinol Diabetes Obes 2017; 24: 174–183 [DOI] [PubMed] [Google Scholar]
- [21].Wei S, Wu D, Yue J. Surgical resection of multiple liver metastasis of functional malignant pheochromocytoma: A case report and literature review. J Cancer Res Ther 2013; 9 (Suppl:): S183–S185 [DOI] [PubMed] [Google Scholar]
- [22].Arnas-Leon C, Sanchez V, Santana Suarez AD et al. Complete remission in metastatic pheochromocytoma treated with extensive surgery. Cureus 2016; 8: e447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ellis RJ, Patel D, Prodanov T et al. Response after surgical resection of metastatic pheochromocytoma and paraganglioma: can postoperative biochemical remission be predicted? J Am Coll Surg 2013; 217: 489–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Averbuch SD, Steakley CS, Young RC et al. Malignant pheochromocytoma: Effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med 1988; 109: 267–273 [DOI] [PubMed] [Google Scholar]
- [25].Angelousi A, Dimitriadis GK, Zografos G et al. Molecular targeted therapies in adrenal, pituitary and parathyroid malignancies. Endocr Relat Cancer 2017; 24: R239–R259 [DOI] [PubMed] [Google Scholar]
- [26].Jochmanova I, Yang C, Zhuang Z et al. Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Cancer Inst 2013; 105: 1270–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tella SH, Taieb D, Pacak K. HIF-2alpha: Achilles’ heel of pseudohypoxic subtype paraganglioma and other related conditions. Eur J Cancer 2017; 86: 1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nature Genetics 2011; 43: 663–667 [DOI] [PubMed] [Google Scholar]
- [29].Gimenez-Roqueplo AP, Favier J, Rustin P et al. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J Clin Endocrinol Metab 2002; 87: 4771–4774 [DOI] [PubMed] [Google Scholar]
- [30].Gimenez-Roqueplo AP, Favier J, Rustin P et al. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet 2001; 69: 1186–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pollard PJ, El-Bahrawy M, Poulsom R et al. Expression of HIF-1alpha, HIF-2alpha (EPAS1), and their target genes in paraganglioma and pheochromocytoma with VHL and SDH mutations. J Clin Endocrinol Metab 2006; 91: 4593–4598 [DOI] [PubMed] [Google Scholar]
- [32].Pollard PJ, Briere JJ, Alam NA et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 2005; 14: 2231–2239 [DOI] [PubMed] [Google Scholar]
- [33].Favier J, Briere JJ, Burnichon N et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One 2009; 4: e7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Koh MY, Lemos R Jr., Liu X et al. The hypoxia-associated factor switches cells from HIF-1alpha- to HIF-2alpha-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res 2011; 71: 4015–4027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhuang Z, Yang C, Lorenzo F et al. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med 2012; 367: 922–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lorenzo FR, Yang C, Ng Tang Fui M et al. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med (Berl) 2013; 91: 507–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pacak K, Jochmanova I, Prodanov T et al. New syndrome of paraganglioma and somatostatinoma associated with polycythemia. J Clin Oncol 2013; 31: 1690–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Taieb D, Yang C, Delenne B et al. First report of bilateral pheochromocytoma in the clinical spectrum of HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab 2013; 98: E908–E913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Comino-Mendez I, de Cubas AA, Bernal C et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum Mol Genet 2013; 22: 2169–2176 [DOI] [PubMed] [Google Scholar]
- [40].Toledo RA, Qin Y, Srikantan S et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr Relat Cancer 2013; 20: 349–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mylonis I, Chachami G, Samiotaki M et al. Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J Biol Chem 2006; 281: 33095–33106 [DOI] [PubMed] [Google Scholar]
- [42].Richard DE, Berra E, Gothie E et al. p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem 1999; 274: 32631–32637 [DOI] [PubMed] [Google Scholar]
- [43].Lim JH, Lee ES, You HJ et al. Ras-dependent induction of HIF-1alpha785 via the Raf/MEK/ERK pathway: a novel mechanism of Ras-mediated tumor promotion. Oncogene 2004; 23: 9427–9431 [DOI] [PubMed] [Google Scholar]
- [44].Mohlin S, Hamidian A, von Stedingk K et al. PI3K-mTORC2 but not PI3K-mTORC1 regulates transcription of HIF2A/EPAS1 and vascularization in neuroblastoma. Cancer Res 2015; 75: 4617–4628 [DOI] [PubMed] [Google Scholar]
- [45].Elorza A, Soro-Arnaiz I, Melendez-Rodriguez F et al. HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol Cell 2012; 48: 681–691 [DOI] [PubMed] [Google Scholar]
- [46].Brugarolas J, Lei K, Hurley RL et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 2004; 18: 2893–2904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Land SC, Tee AR. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem 2007; 282: 20534–20543 [DOI] [PubMed] [Google Scholar]
- [48].Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res 2012; 44: 328–333 [DOI] [PubMed] [Google Scholar]
- [49].Fukuda R, Hirota K, Fan F et al. Insulin-like growth factor 1 induces hypoxia-inducible factor 1-mediated vascular endothelial growth factor expression, which is dependent on MAP kinase and phosphatidylinositol 3-kinase signaling in colon cancer cells. J Biol Chem 2002; 277: 38205–38211 [DOI] [PubMed] [Google Scholar]
- [50].Crona J, Delgado Verdugo A, Maharjan R et al. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J Clin Endocrinol Metab 2013; 98: E1266–E1271 [DOI] [PubMed] [Google Scholar]
- [51].Kim WY, Perera S, Zhou B et al. HIF2alpha cooperates with RAS to promote lung tumorigenesis in mice. J Clin Invest 2009; 119: 2160–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sbardella E, Cranston T, Isidori AM et al. Routine genetic screening with a multi-gene panel in patients with pheochromocytomas. Endocrine 2018; 59: 175–182 [DOI] [PubMed] [Google Scholar]
- [53].Remacha L, Curras-Freixes M, Torres-Ruiz R et al. Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genet Med 2018, doi: 10.1038/s41436-018-0003-y [DOI] [PubMed] [Google Scholar]
- [54].Grogan RH, Mitmaker EJ, Duh QY. Changing paradigms in the treatment of malignant pheochromocytoma. Cancer Control 2011; 18: 104–112 [DOI] [PubMed] [Google Scholar]
- [55].Jimenez P, Tatsui C, Jessop A et al. Treatment for Malignant Pheochromocytomas and Paragangliomas: 5 Years of Progress. Curr Oncol Rep 2017; 19: 83. [DOI] [PubMed] [Google Scholar]
- [56].Keiser HR, Goldstein DS, Wade JL et al. Treatment of malignant pheochromocytoma with combination chemotherapy. Hypertension 1985; 7 (3 Pt 2): I18–I24 [DOI] [PubMed] [Google Scholar]
- [57].Niemeijer ND, Alblas G, van Hulsteijn LT et al. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis. Clin Endocrinol (Oxf) 2014; 81: 642–651 [DOI] [PubMed] [Google Scholar]
- [58].Ayala-Ramirez M, Feng L, Habra MA et al. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer 2012; 118: 2804–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jimenez C, Rohren E, Habra MA et al. Current and future treatments for malignant pheochromocytoma and sympathetic paraganglioma. Curr Oncol Rep 2013; 15: 356–37123674235 [Google Scholar]
- [60].Plouin PF, Fitzgerald P, Rich T et al. Metastatic pheochromocytoma and paraganglioma: focus on therapeutics. Horm Metab Res 2012; 44: 390–399 [DOI] [PubMed] [Google Scholar]
- [61].Huang H, Abraham J, Hung E et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer 2008; 113: 2020–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Jawed I, Velarde M, Darr R et al. Continued tumor reduction of metastatic pheochromocytoma/paraganglioma harboring succinate dehydrogenase subunit b mutations with cyclical chemotherapy. Cell Mol Neurobiol 2018. doi: 10.1007/s10571-018-0579-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tay CG, Lee VWM, Ong LC et al. Vincristine-induced peripheral neuropathy in survivors of childhood acute lymphoblastic leukaemia. Pediatr Blood Cancer 2017; 64: [DOI] [PubMed] [Google Scholar]
- [64].Hadoux J, Favier J, Scoazec JY et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int J Cancer 2014; 135: 2711–2720 [DOI] [PubMed] [Google Scholar]
- [65].Pegg AE, Dolan ME, Moschel RC. Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase. Prog Nucleic Acid Res Mol Biol 1995; 51: 167–223 [DOI] [PubMed] [Google Scholar]
- [66].Hegi ME, Liu L, Herman JG et al. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol 2008; 26: 4189–4199 [DOI] [PubMed] [Google Scholar]
- [67].Bignami M, O’Driscoll M, Aquilina G et al. Unmasking a killer: DNA O(6)-methylguanine and the cytotoxicity of methylating agents. Mutat Res 2000; 462: 71–82 [DOI] [PubMed] [Google Scholar]
- [68].Tena I, Gupta G, Tajahuerce M et al. Successful second-line metronomic temozolomide in metastatic paraganglioma: Case reports and review of the literature. Clin Med Insights Oncol 2018; 12: 1179554918763367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Gonias S, Goldsby R, Matthay KK et al. Phase II study of high-dose [131I]metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J Clin Oncol 2009; 27: 4162–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Krempf M, Lumbroso J, Mornex R et al. Treatment of malignant pheochromocytoma with [131I]metaiodobenzylguanidine: a French multicenter study. J Nucl Biol Med 1991; 35: 284–287 [PubMed] [Google Scholar]
- [71].Loh KC, Fitzgerald PA, Matthay KK et al. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): a comprehensive review of 116 reported patients. J Endocrinol Invest 1997; 20: 648–658 [DOI] [PubMed] [Google Scholar]
- [72].Mukherjee JJ, Kaltsas GA, Islam N et al. Treatment of metastatic carcinoid tumours, phaeochromocytoma, paraganglioma and medullary carcinoma of the thyroid with (131)I-meta-iodobenzyl-guanidine [(131)I-mIBG]. Clin Endocrinol (Oxf) 2001; 55: 47–60 [DOI] [PubMed] [Google Scholar]
- [73].Sze WC, Grossman AB, Goddard I et al. Sequelae and survivorship in patients treated with (131)I-MIBG therapy. Br J Cancer 2013; 109: 565–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Basu S, Abhyankar A, Jatale P. The current place and indications of 131I-metaiodobenzylguanidine therapy in the era of peptide receptor radionuclide therapy: Determinants to consider for evolving the best practice and envisioning a personalized approach. Nucl Med Commun 2015; 36: 1–7 [DOI] [PubMed] [Google Scholar]
- [75].van Hulsteijn LT, Niemeijer ND, Dekkers OM et al. (131)I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: systematic review and meta-analysis. Clin Endocrinol (Oxf) 2014; 80: 487–501 [DOI] [PubMed] [Google Scholar]
- [76].Nastos K, Cheung VTF, Toumpanakis C et al. Peptide receptor radionuclide treatment and (131)I-MIBG in the management of patients with metastatic/progressive phaeochromocytomas and paragangliomas. J Surg Oncol 2017; 115: 425–434 [DOI] [PubMed] [Google Scholar]
- [77].Rose B, Matthay KK, Price D et al. High-dose 131I-metaiodobenzyl-guanidine therapy for 12 patients with malignant pheochromocytoma. Cancer 2003; 98: 239–248 [DOI] [PubMed] [Google Scholar]
- [78].Barrett JA, Joyal JL, Hillier SM et al. Comparison of high-specific-activity ultratrace 123/131I-MIBG and carrier-added 123/131I-MIBG on efficacy, pharmacokinetics, and tissue distribution. Cancer Biother Radiopharm 2010; 25: 299–308 [DOI] [PubMed] [Google Scholar]
- [79].Jimenez CPD, Sullivan DC, Schwarz JK et al. Long term follow-up of a pivotal phase 2 study of Ultratrace® Iobenguane I-131 (AZEDRA TM) in patients with malignant relapsed/refractory pheochromocytoma (pheo)/paraganglioma (para). In: Adrenal Tumors: Clinical Implications of the Recent Molecular and Genetic Findings;. Endocrine Society’s 97th Annual Meeting and Expo; 2015 [Google Scholar]
- [80].Van Essen M, Krenning EP, De Jong M et al. Peptide receptor radionuclide therapy with radiolabelled somatostatin analogues in patients with somatostatin receptor positive tumours. Acta Oncol 2007; 46: 723–734 [DOI] [PubMed] [Google Scholar]
- [81].Ziegler CG, Brown JW, Schally AV et al. Expression of neuropeptide hormone receptors in human adrenal tumors and cell lines: Antiproliferative effects of peptide analogues. Proc Natl Acad Sci USA 2009; 106: 15879–15884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].van Essen M, Krenning EP, Kooij PP et al. Effects of therapy with [177Lu-DOTA0, Tyr3]octreotate in patients with paraganglioma, meningioma, small cell lung carcinoma, and melanoma. J Nucl Med 2006; 47: 1599–1606 [PubMed] [Google Scholar]
- [83].Zovato S, Kumanova A, Dematte S et al. Peptide receptor radionuclide therapy (PRRT) with 177Lu-DOTATATE in individuals with neck or mediastinal paraganglioma (PGL). Horm Metab Res 2012; 44: 411–414 [DOI] [PubMed] [Google Scholar]
- [84].Forrer F, Riedweg I, Maecke HR et al. Radiolabeled DOTATOC in patients with advanced paraganglioma and pheochromocytoma. Q J Nucl Med Mol Imaging 2008; 52: 334–340 [PubMed] [Google Scholar]
- [85].Kong G, Grozinsky-Glasberg S, Hofman MS et al. Efficacy of peptide receptor radionuclide therapy for functional metastatic paraganglioma and pheochromocytoma. J Clin Endocrinol Metab 2017; 102: 3278–3287 [DOI] [PubMed] [Google Scholar]
- [86].Strosberg J, El-Haddad G, Wolin E et al. Phase 3 trial of (177) lu-dotatate for midgut neuroendocrine tumors. N Engl J Med 2017; 376: 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Vogel J, Atanacio AS, Prodanov T et al. External beam radiation therapy in treatment of malignant pheochromocytoma and paraganglioma. Front Oncol 2014; 4: 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Breen W, Bancos I, Young WF Jr. et al. External beam radiation therapy for advanced/unresectable malignant paraganglioma and pheochromocytoma. Adv Radiat Oncol 2018; 3: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Patchell RA, Tibbs PA, Regine WF et al. Direct decompressive surgical resection in the treatment of spinal cord compression caused by metastatic cancer: A randomised trial. Lancet 2005; 366: 643–648 [DOI] [PubMed] [Google Scholar]
- [90].Sahgal A, Larson DA, Chang EL. Stereotactic body radiosurgery for spinal metastases: a critical review. Int J Radiat Oncol Biol Phys 2008; 71: 652–665 [DOI] [PubMed] [Google Scholar]
- [91].Wang XS, Rhines LD, Shiu AS et al. Stereotactic body radiation therapy for management of spinal metastases in patients without spinal cord compression: a phase 1–2 trial. Lancet Oncol 2012; 13: 395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Yamada Y, Bilsky MH, Lovelock DM et al. High-dose, single-fraction image-guided intensity-modulated radiotherapy for metastatic spinal lesions. Int J Radiat Oncol Biol Phys 2008; 71: 484–490 [DOI] [PubMed] [Google Scholar]
- [93].Ayala-Ramirez M, Chougnet CN, Habra MA et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J Clin Endocrinol Metab 2012; 97: 4040–4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Druce MR, Kaltsas GA, Fraenkel M et al. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: experience with the mTOR inhibitor everolimus (RAD001). Hormone and Metabolic Research 2009; 41: 697–702 [DOI] [PubMed] [Google Scholar]
- [95].Nölting S, Maurer J, Spottl G et al. Additive anti-tumor effects of lovastatin and everolimus in vitro through simultaneous inhibition of signaling pathways. PLoS One 2015; 10: e0143830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Nölting S, Garcia E, Alusi G et al. Combined blockade of signalling pathways shows marked anti-tumour potential in phaeochromocytoma cell lines. Journal of Molecular Endocrinology 2012; 49: 79–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Nölting S, Giubellino A, Tayem Y et al. Combination of 13-cis retinoic acid and lovastatin: Marked anti-tumor potential in vivo in a pheochromocytoma allograft model in female athymic nude mice. Endocrinology 2014. doi: 10.1210/en.2014-1027:en20141027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Giubellino A, Bullova P, Nolting S et al. Combined inhibition of mTORC1 and mTORC2 signaling pathways is a promising therapeutic option in inhibiting pheochromocytoma tumor growth: In vitro and in vivo studies in female athymic nude mice. Endocrinology 2013; 154: 646–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Choueiri TK, Halabi S, Sanford BL et al. Cabozantinib versus sunitinib as initial targeted therapy for patients with metastatic renal cell carcinoma of poor or intermediate risk: The Alliance A031203 CABOSUN Trial. J Clin Oncol 2017; 35: 591–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Choueiri TK, Hessel C, Halabi S et al. Cabozantinib versus sunitinib as initial therapy for metastatic renal cell carcinoma of intermediate or poor risk (Alliance A031203 CABOSUN randomised trial): Progression-free survival by independent review and overall survival update. Eur J Cancer 2018; 94: 115–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Smith M, De Bono J, Sternberg C et al. Phase III Study of cabozantinib in previously treated metastatic castration-resistant prostate cancer: COMET-1. J Clin Oncol 2016; 34: 3005–3013 [DOI] [PubMed] [Google Scholar]
- [102].Jasim S, Suman VJ, Jimenez C et al. Phase II trial of pazopanib in advanced/progressive malignant pheochromocytoma and paraganglioma. Endocrine 2017; 57: 220–225 [DOI] [PubMed] [Google Scholar]
- [103].Nölting S, Grossman AB. Signaling pathways in pheochromocytomas and paragangliomas: prospects for future therapies. Endocr Pathol 2012; 23: 21–33 [DOI] [PubMed] [Google Scholar]
- [104].Cho H, Du X, Rizzi JP et al. On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 2016; 539: 107–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Chen W, Hill H, Christie A et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016; 539: 112–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Caplin ME, Pavel M, Cwikla JB et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med 2014; 371: 224–233 [DOI] [PubMed] [Google Scholar]
- [107].Rinke A, Muller HH, Schade-Brittinger C et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol 2009; 27: 4656–4663 [DOI] [PubMed] [Google Scholar]
- [108].Ray Chaudhuri A, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 2017; 18: 610–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].de Murcia JM, Niedergang C, Trucco C et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A 1997; 94: 7303–7307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Ben-Hur E, Utsumi H, Elkind MM. Inhibitors of poly (ADP-ribose) synthesis enhance radiation response by differentially affecting repair of potentially lethal versus sublethal damage. Br J Cancer Suppl 1984; 6: 39–42 [PMC free article] [PubMed] [Google Scholar]
- [111].Schlicker A, Peschke P, Burkle A et al. 4-Amino-1,8-naphthalimide: A novel inhibitor of poly(ADP-ribose) polymerase and radiation sensitizer. Int J Radiat Biol 1999; 75: 91–100 [DOI] [PubMed] [Google Scholar]
- [112].Pang Y, Lu Y, Caisova V et al. Targeting NAD + /PARP DNA repair pathway as a novel therapeutic approach to SDHB-mutated cluster I pheochromocytoma and paraganglioma. Clin Cancer Res 2018. doi: 10.1158/1078-0432.CCR-17-3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Allegrezza MJ, Conejo-Garcia JR. Targeted therapy and immunosuppression in the tumor microenvironment. Trends Cancer 2017; 3: 19–27 [DOI] [PubMed] [Google Scholar]