

Summary
Many sarcomas contain gene fusions that can be pathogenetic mechanisms and diagnostic markers. In this article we review selected fusion sarcomas and techniques for their detection. CIC-DUX4 fusion sarcoma is a round cell tumor now considered an entity separate from Ewing sarcoma with a more aggressive clinical course, occurrence in older age, and predilection to soft tissues. It is composed of larger cells than Ewing sarcoma and often has prominent necrosis. Nuclear DUX4 expression is a promising immuno histochemical marker. BCOR-CCNB3 fusion sarcoma is cyclin B3–positive, usually occurs in bone or soft tissue of children, and may mimic a poorly differentiated synovial sarcoma. EWSR1-NFATC2 sarcoma may present in bone or soft tissue. It is typically composed of small round cells in a trabecular pattern in a myxoid matrix resembling myoepithelioma. ACTB-GLI1 fusion sarcoma may mimic a skin adnexal carcinoma, showing focal expression of epithelial markers and S100 protein. NTRK-fusion sarcomas include, in addition to infantile fibrosarcoma with ETV6-NTRK3 fusion, LMNA-NTRK1 fusion sarcoma, a low-grade spindle cell sarcoma seen in peripheral soft tissues in children and young adults. Methods to detect gene fusions include next-generation sequencing panels, anchored multiplex polymerase chain reaction systems to detect partner for a known fusion gene, and comprehensive RNA sequencing to detect virtually all gene fusions. In situ hybridization testing using probes for both fusion partners can be used as an alternative confirmation technique, especially in the absence of satisfactory RNA yield. In addition, fusion protein-related and other immunohistochemical markers can have a high specificity for fusion sarcomas.
Keywords: Fusion sarcoma, CIC-DUX4, BCOR-CCNB3, EWSR1-NFATC2, ACTB-GLI1, LMNA-NTRK1, Anchored multiplex PCR
1. Introduction
Gene fusions created by chromosomal rearrangements are associated with cancers of various types, especially sarcomas, and are often used as diagnostic markers. Among the first reported gene fusions in sarcomas was the SS18-SSX1/2 fusion resulting from the t(X;18) translocation, still believed to be specific to synovial sarcoma [1]. Other well-known gene fusions used as diagnostic markers in sarcomas include fusion of Ewing sarcoma gene (EWSR1) with FLI1 or ERG in Ewing sarcoma and PAX3 or PAX7 fusions with FOXO1 in alveolar rhabdomyosarcoma. Notably, EWSR1 gene involvement is not specific for Ewing sarcoma, but occurs in many unrelated sarcomas [2,3].
Chimeric transcripts (chimeras, or fusion transcripts) originate from the fusion of two or more different gene transcripts. Traditionally, all chimeric RNAs were thought originate from gene fusions occurring due to chromosomal rearrangements, such as deletions, inversions, or translocations [4]. More recently, some fusion RNAs have been suggested to be products of either cis-splicing between neighboring genes, or a result from RNA trans-splicing, with both phenomena having detected in some non-neoplastic tissues [5,6].
With the recent advances in next-generation sequencing, numerous new chimeric RNAs have been discovered in sarcomas. In many cases, tumors harboring these fusions have emerged as new clinicopathologic entities with characteristic histological and immunophenotypic features. In this review, we present examples of such newly described entities regularly encountered in diagnostic pathology, based on our experience, and discuss their clinical significance and strategies for their detection.
2. Classic and newer fusion search strategies
The first fusion genes, such as those found in Ewing sarcoma and synovial sarcoma, were cloned and characterized following the cytogenetic detection of chromosome rearrangements by chromosome banding and fluorescence in situ hybridization (FISH) studies. However, this classical approach is limited by the requirement of fresh tumor tissue and obtaining metaphases for chromosome analysis. Therefore, it is now largely replaced by molecular genetic techniques.
In clinical practice, an interphase FISH employing probes for fusion genes is widely used to detect rearrangements [4]. However, this technique requires a strong diagnostic hypothesis and availability of commercial probes or a capability to manufacture probes from gene libraries. FISH is often unsuitable for analyzing structural rearrangements including inversions and interstitial deletions involving closely adjacent genes.
Invention of the polymerase chain reaction (PCR) amplification revolutionized the search for fusion genes. PCR methods were developed for detection of specific fusion break-points at both DNA and RNA levels [7]. RACE (rapid amplification of cDNA ends), an RT (reverse-transcription)-PCR technique, was employed to amplify chimeric transcripts expressed by the fusion genes with only one known partner [8]. Also, gene expression profiling assays were successfully used to search for new fusion gene transcripts [9,10]. Oligo microarrays with known sarcoma fusion gene transcripts are also employed to detect exon-exon breakpoints [11,12]. Combined multiplex RT-PCR and microarray pipeline, and the GeneChip exon array screening were established to search for known, or new, fusion gene transcripts [13,14]. Additionally, digital expression profiling using NanoString Technologies nCounter Analysis System (NanoString Technologies, Seattle, WA) has proven efficient for detection of known fusion gene transcripts [5,15]. However, these methods are typically used in a research setting and not commonly applied in clinical practice.
The advent of next-generation sequencing (NGS) technologies opened a new era in the search for fusion genes and their chimeric transcripts. The whole-genome NGS of tumor DNA has been successfully used to identify new fusion genes, such as the NAB2-STAT6 fusion in solitary fibrous tumor [16]. Yet, most NGS-based studies searching for unknown fusions have employed a transcriptome sequencing (RNA-seq) [1]. The advantage of the RNA-based NGS is the ability to identify fusion gene transcript variants as well as levels of fusion gene transcript expression. However, chromosomal rearrangements that cause the replacement of regulatory sequences cannot be detected using this method. In soft tissue tumors, fusions including upstream or promoter regulatory sequences represent a relatively small fraction of all known fusions. Moreover, chromosomal rearrangements silencing the involved genes may not be detected by the NGS RNA-seq [17]. In both methods, a search for fusions require well preserved nucleic acids, which are often difficult obtain from formalin-fixed, paraffin-embedded (FFPE) tissues. Because of this and high cost, the application of whole-genome sequencing or RNA-seq into diagnostic pathology has been limited. More recently, lower-cost methods for the efficient preparation of sequencing libraries (SMARTer-Stranded Total RNA-Seq Kit v2) from small (250 pg-10 ng) inputs of partially degraded FFPE RNA has been developed and commercialized by Takara Bio USA, Inc., (Mountain View, CA).
Recently, assays have been established for fusion gene transcripts detection combining multiplex anchored PCR amplification of specific targets with next-generation sequencing [18]. These amplicon-based assays employ anchored multiplex PCR to enrich chimeric transcripts in a low input of partially degraded RNA from FFPE tissue. While conventional PCR requires knowledge of both sequences flanking the area of interest, anchored PCR allows for detection of a target when only one sequence is known. In this technique, cDNA undergoes repair, adenylation and ligation with a universal adapter and subsequent amplification with a primer for the known gene and a primer complementary to the ligated universal adapter or “anchof” [19,20]. Therefore, NGS libraries created with Anchored PCR may contain chimeric transcripts of both previously known and novel fusions. These assays, commercialized by ArcherDx, Inc, (Boulder, CO) and by NuGEN Technologies, Inc, (San Carlos, CA), are compatible with commonly used NGS platforms such as Illumina (Illumina, San Diego, CA) and Ion Torrent (Thermo Fisher Scientific, Waltham, MA), and their value in the evaluation of bone and soft tissue tumors has been well documented [21].
Increased application of fusion search techniques will likely lead to discovery of numerous novel gene fusions and improve our understanding of occurrence of known fusions. Detection of larger numbers of specific fusion tumors will broaden our understanding of the clinicopathologic correlation of various fusion tumors.
3. CIC fusion sarcomas
Although initially included in the Ewing sarcoma family, CIC-DUX4 fusion sarcoma is now considered an entity separate from Ewing sarcoma based on histologic and clinical differences, such as older patient average age and a more aggressive course than seen in Ewing sarcoma [22]. Originally identified from cytogenetic studies on tumors histologically often considered Ewing sarcoma [23,24], subsequent larger series have been identified by screening of EWSR1 gene rearrangement-negative Ewing-like sarcomas [22,25–30]. In those studies, 22% to 68% of EWSR1-FISH negative tumors had a CIC-DUX4 fusion, so that this fusion seems to be the most common one identified in EWSR1 gene rearrangement negative Ewing-like sarcomas [26,28].
Most cases contain a t(4;19)(q35;q13) corresponding to a rearrangement between the CIC and DUX4 genes, and a smaller subset contains a t(10;19)(q26;q13), which corresponds to a fusion between CIC and the paralog gene DUX4L [27]. A small number of cases (currently 2) with CIC-FOXO4 fusions corresponding to t(X;19) (q13;q13;3) translocation have been reported (discussed in the end of this section). Fluorescence in situ hybridization for CIC detects most cases, but some cases may be missed due to cryptic translocations [31].
CIC gene encodes for a morphogenetic transcription factor known from Drosophila as the morphogenetic Capicua transcription factor, a transcriptional repressor regulating kinase signaling. CIC-DUX4 fusion up-regulates ETS transcription factors of PEA3 subfamily, especially ETV4 [32,33]. Murine model of induced CIC-DUX4 fusion produced around cell sarcoma like the human one supporting pathogenetic role of this fusion [34].
Nearly 200 cases of CIC-DUX4 fusion sarcomas have been reported [22–30]. These sarcomas occur at a median age of approximately 30 years but with a wide age range from childhood to old age (6-81 years) with a mild male predominance. In contrast with Ewing sarcoma, they almost always present as soft tissue tumors occurring in the extremities, trunk wall, body cavities, and occasionally in bones or organ-based locations, such as kidney and brain. Tumors are usually large with a median size 9 to 10 cm with a grossly whitish appearance with frequent necrosis. Follow-up studies have noted poor overall survival with most patient succumbing with lung metastases in 1 to 2 years. Even small tumors have proven fatal. Median survival and responsiveness to Ewing sarcoma chemotherapy protocols is clearly inferior to typical Ewing sarcoma.
Histologically there is a round or ovoid cell tumor with occasional spindle cell change. Tumors often have extensive geographic necrosis showing a perivascular sparing pattern of cellularity (Fig. 1A). In addition to diffuse sheet-like histology, there is often myxoid matrix (Fig. 1B). The cells can have pale eosinophilic or clear cytoplasm. Nuclei are larger than in typical Ewing sarcoma, often showing prominent nucleoli (Fig. 1C).
Fig. 1.
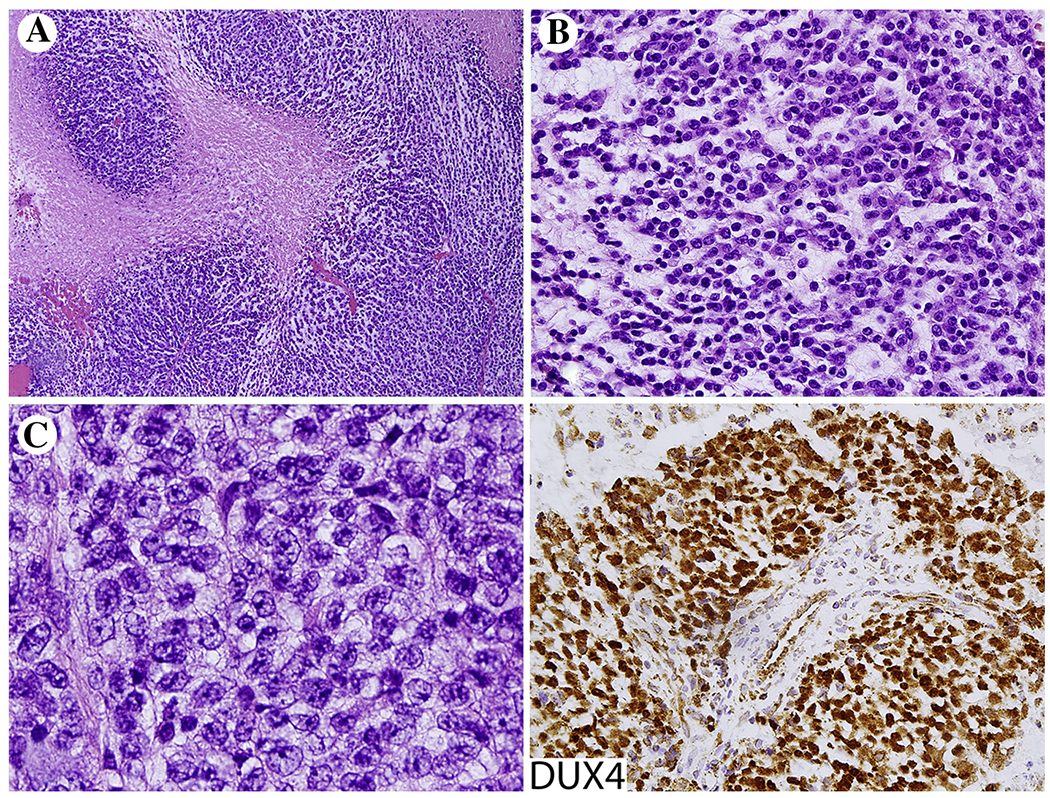
CIC-DUX4 fusion sarcoma. A, Extensive tumor necrosis is common. B and C, Tumors are composed of round cells larger than typical Ewing sarcoma cells with prominent nucleoli. Tumor cells show nuclear positivity for DUX4, which is considered a useful marker for these fusion sarcomas.
Immunohistochemically CIC-DUX fusion sarcomas are variably CD99-positive and rarely show strong membrane staining typically seen in Ewing sarcoma. However, these tumors are distinctive for nuclear expression of DUX4, which seems to be the best immunohistochemical surrogate marker for tumors carrying this gene fusion (Fig. 1) [35]. While focal keratin and rarely desmin expression has been detected, expression of calretinin, ETV4, and WT1 also seem to be distinctive although less specific features [36–38].
Two patients with CIC-FOXO4 fusion sarcomas have been reported, curiously both being intramuscular 2–3 cm tumors in the posterior neck: A 63 year-old man and a 13-year old boy [39,40]. Follow-up data of this molecular sub entity remains insufficient. Histologically these tumors may resemble classic Ewing sarcoma more than the CIC-DUX4 sarcomas (Fig. 2).
Fig. 2.
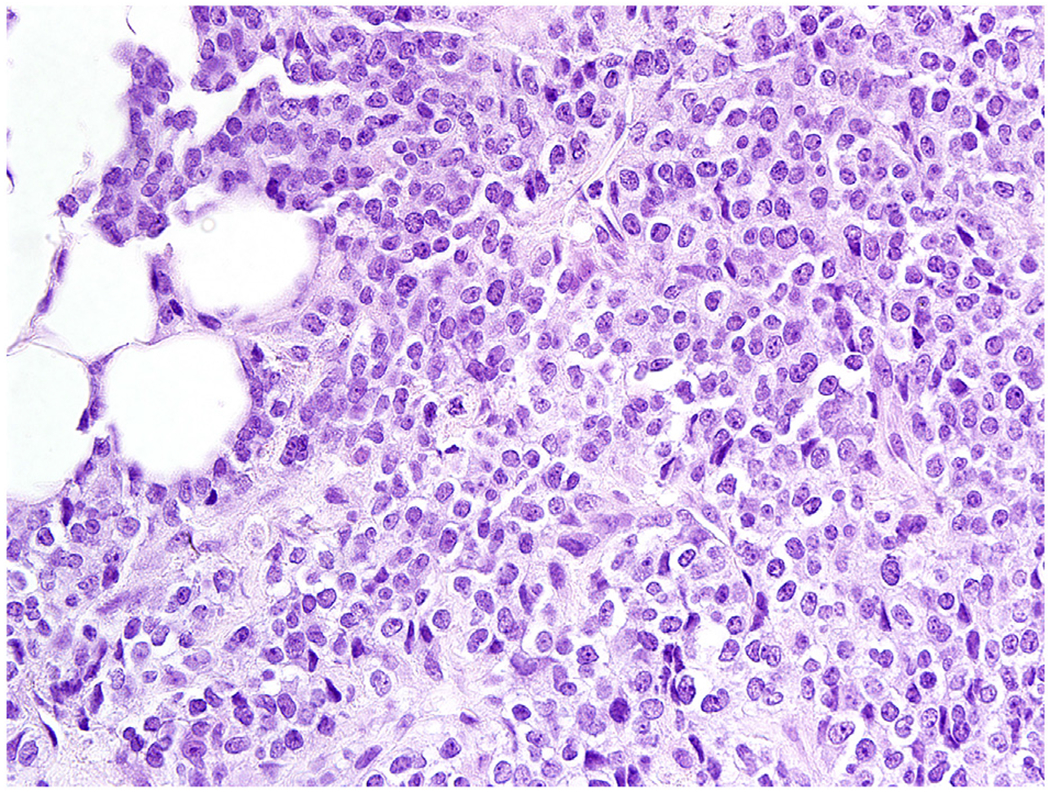
CIC-FOXO1 fusion sarcoma resembles a classic Ewing sarcoma and is composed of uniform small round cells.
4. BCOR-CCNB3 fusion sarcoma
This tumor is defined as a spindle, oval, or round cell sarcoma containing a gene fusion involving BCOR (BCL6 corepressor) and CCNB3 (cyclin B3) genes. At the chromosomal level, there is a paracentric inversion in the X chromosome involving these two genes. The fusion causes overexpression of cyclin B3 protein [41].
BCOR-CCNB3 fusion sarcomas occur from early childhood to middle age (2-44 years), with the median age of 15 years. The reported series have shown a marked male predominance, 9:1 with approximately an equal number of cases reported in soft tissue and bone [41–49]. The soft tissue examples have occurred in the extremities (40%), trunk wall (33%), abdominal cavity (17%), and head and neck (<10%), whereas in bone 40% they have arisen in long bones, pelvis (33%), and small bones (27%), with an apparent predilection for the calcaneus. Isolated cases have also been reported in the kidney, where this tumor can resemble clear cell sarcoma of the kidney. In fact, these tumors have also genetic overlap as clear cell sarcoma of the kidney can have the same fusion, although it more often contains an internal tandem duplication of the BCOR gene [49–52]. Tumor size has varied from 1.5 to 27 cm (median size, 10 cm). Tumors have been typically treated with Ewing sarcoma protocols, with similar treatment responses.
Histologically BCOR-CCNB3 fusion sarcoma is highly cellular sarcoma composed of spindle or ovoid cells with a prominent delicate capillary network (Fig. 3). The tumors composed of uniform, ovoid to spindled cells can resemble poorly differentiated synovial sarcoma, malignant peripheral nerve sheath tumor, or an undifferentiated sarcoma. Pleomorphic evolution has been seen in recurrent tumors [41–49].
Fig. 3.
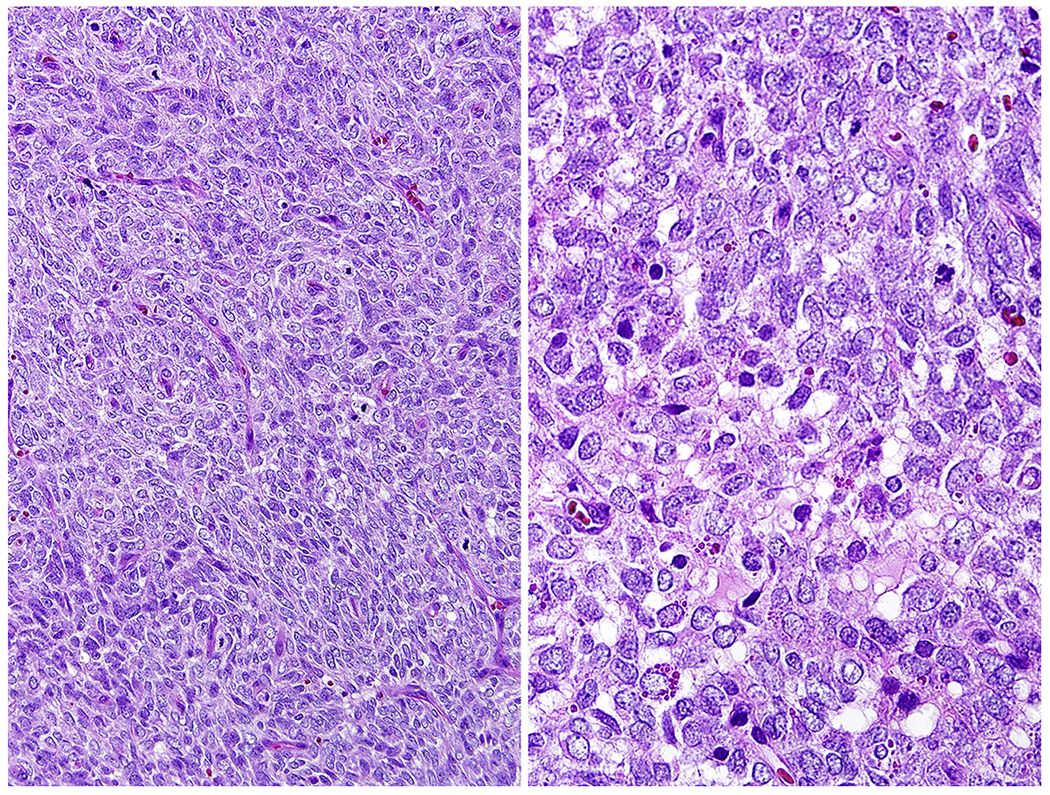
A BCOR-CCNB3 fusion sarcoma is composed of spindled cells resembling a poorly differentiated synovial sarcoma.
Immunohistochemistry for cyclin B3, the protein product of the CCNB3 gene, is positive in these fusion tumors, in contrast to classical Ewing sarcoma [45,53,54]. BCOR has also been suggested as a diagnostic marker due to overexpression of this gene; however, this protein is widely expressed in different tissues [55]. BCOR overexpression has also been reported in aggressive, newly described endometrial sarcomas containing other BCOR gene fusions [56].
5. EWSR1-NFATC2 fusion sarcoma
This gene fusion corresponds to translocation t(20;22) (q13.2;q12.2) involving 2 genes encoding transcription factors, Ewing sarcoma protein and calcineurin-dependent nuclear factor of activated T-cells 2 (NFATC2 protein, also known as NFAT1) [57]. This transcription factor has homology to the NF-kappa B proteins and is involved in T-cell differentiation and transcriptional activation of cytokine genes. It also participates in osteoclast differentiation, cardiac valve morphogenesis, and regulates myosin heavy chain gene expression in skeletal muscle [58].
The frequency of occurrence of EWSR1-NFATC2 fusion sarcoma is not precisely known, but 1 of 24 (4%) of tumors histologically compatible with bone fibrosarcoma was found to have this fusion [59], and there were 2 cases with this fusion among 32 Ewing sarcomas (6%) in another study [60]. Only 5 cases of EWSR1-NFATC2 fusion sarcoma have been reported in detail. Histologically it is often characterized as an Ewing sarcoma–like tumor. Those tumors have occurred in male patients of ages 16–39 years (median, 25 years). All but one was a bone tumor involving long bones, 3 in the femur and 1 in the humerus. The tumor is clinically aggressive with potential for both local recurrence and metastasis [56,61]. Tumors with FUS-NFATC2 fusion have also been recently reported [62].
EWSR1-NFATC2 fusion sarcomas are small round cell tumors, often with corded patterns and myxoid matrix mimicking myoepithelioma of soft tissue (Fig. 4). When occurring in a bone, the tumor can resemble a poorly differentiated osteosarcoma by marked reactive sclerotic bone formation.
Fig. 4.
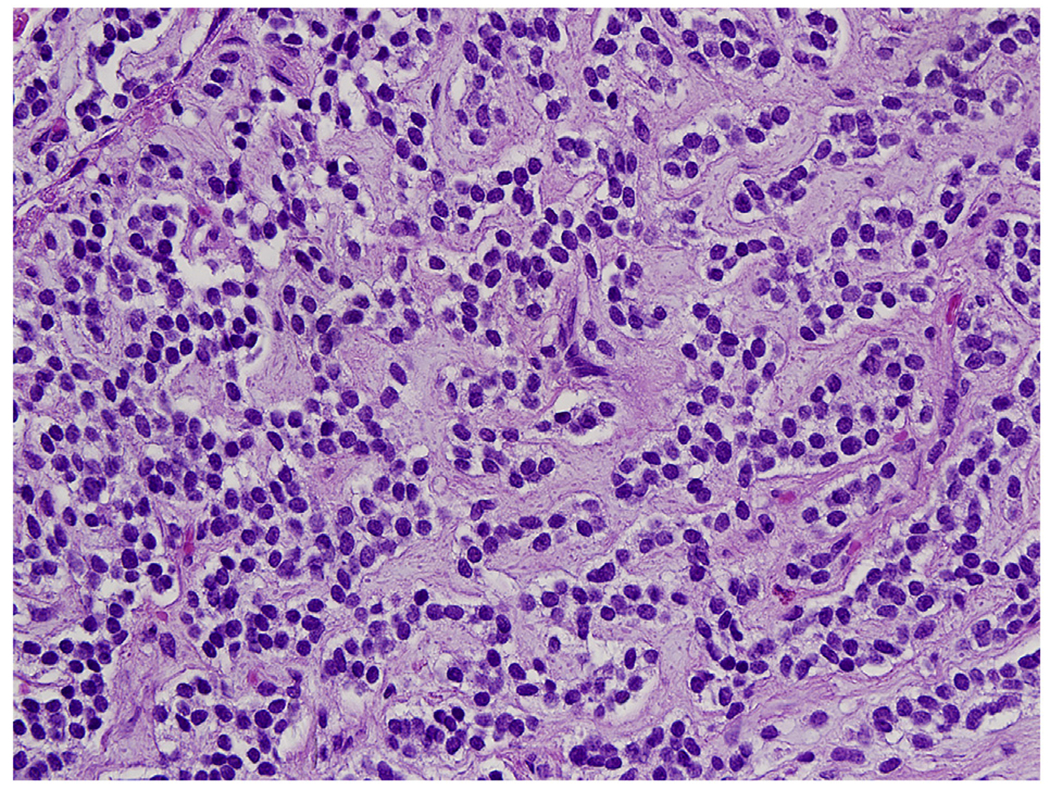
EWSR1-NFATC2 fusion sarcoma has often a corded cellular pattern with myxoid matrix between the cellular cords composed of uniform round cells.
Immunohistochemically the tumors are usually positive for vimentin and CD99 showing membrane expression for the latter as seen in typical Ewing sarcoma, while the reported tumors have been negative for desmin, EMA, keratins, SMA, and S100 protein [56,61]. Weak FLI1 and ERG expression has been noted [4]. With its transcriptome differing from Ewing sarcoma, CIC-DUX4 fusion sarcoma and BCOR-CCNB3 fusion sarcoma, EWSR1-NFATC2 fusion sarcoma should be considered a separate tumor entity [63].
6. GLI1 fusion sarcomas
GLI1 gene encodes glioma-associated oncogene 1 which is involved in the sonic hedgehog signaling pathway. It is a morphogenetic transcription factor with zinc finger elements known to be important in the development of the nervous system. Recently, 3 different fusions involving this gene were reported in tumors mostly occurring in soft tissues. There were 4 tumors with fusions involving the beta-actin gene: ACTB-GLI1 and 1 each of fusions involving patched 1 gene: PTCH1-GLI1, and a third fusion with the MALAT1 gene encoding metastasis associated lung adenocarcinoma transcript [64]. The latter fusion was previously detected in plexiform fibromyxoma of the stomach [65] and gastroblastoma, a rare biphasic epithelial-mesenchymal neoplasm of stomach [66]. In the latter reports, immunohistochemical nuclear and cytoplasmic positivity for GLI1 was detected suggesting its value as surrogate immunohistochemical marker for GLI1 fusion tumors.
ACTB-GLI1 fusion tumors occur in various soft tissue locations and have a predilection for female adults <40 years of age. Histologically it is an epithelioid neoplasm with a nested or trabecular pattern differing from Ewing sarcoma family tumors. Mitotic activity has been reported as low, <5/10 HPFs, and Ki67-labeling as 5% [64]. In our experience, these tumors can histologically mimic mixed tumor/myoepithelioma of soft tissue when composed of epithelioid nests with myxoid matrix (Fig. 5A and B). Immunohistochemically these tumors are often positive for S100 protein (Fig. 5) and sometimes focally for keratins. However, in contrast with myoepitheliomas, they are negative for SOX10 [64]. Like Ewing sarcomas, they can have membrane positivity for CD99 (Fig. 5).
Fig. 5.
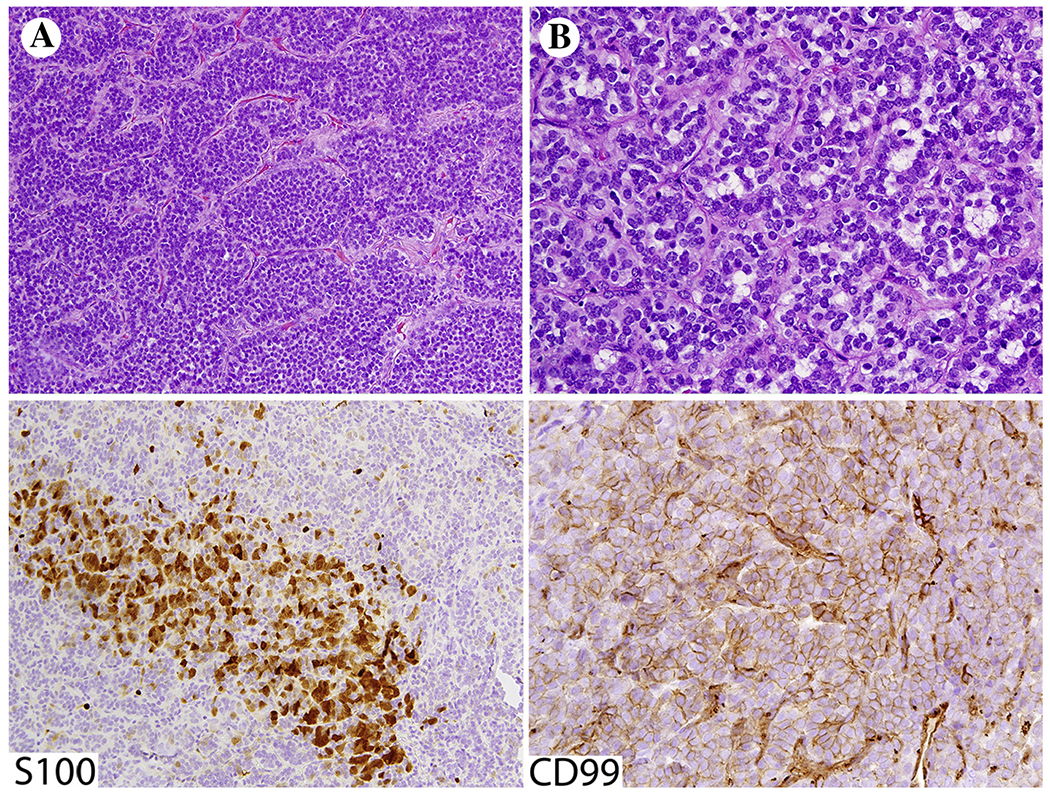
A and B, ACTB-GLI1 fusion sarcoma shows histologic resemblance to skin adnexal tumors such as myoepithelioma. Tumor cells show variable positivity for S100 protein and CD99.
The ACTB-GLI1 fusion tumors were originally identified by cytogenetic studies showing the corresponding t(7;12) (p22;q13) translocation and subsequently demonstrated to harbor the ACTB-GLI1 fusion. These tumors were designated pericytomas based on histological perivascular patterns and variable smooth muscle actin expression. Three of those tumors originated in the tongue, and one each in the calf and stomach [67]. One example was subsequently reported in the talus bone [68]. Clinical follow-up of ACTB-GLI1 fusion tumors have shown only regional lymph node metastases with no evidence for distant metastases, but follow-up data is scant necessitating caution in prognostication [64,67,68].
7. NTRK1/2/3 gene-involving fusions
Neurotrophic tyrosine kinases (NTRK1, NTRK2, and NTRK3), also known as tropomyosin receptor kinases and high-affinity nerve growth factor receptors, are a family of receptor tyrosine kinases involved in the development, differentiation, and metabolism of neural and other tissues. Their activation may lead to activation of at least MAP kinase and PIK3CA downstream pathways. The clinical significance in recognizing NTRK gene fusions lies in the utilization of NTRK inhibitors as a targeted oncologic therapy [69–71].
ETV6-NTRK3 fusions were originally reported in infantile fibrosarcoma and cellular mesoblastic nephroma as recurrent genetic events [72,73]. A similar fusion was subsequently described in secretory carcinoma of the breast [74], and secretory carcinoma analog tumors in the salivary glands [75]. Here we discuss newly described sarcomas with NTRK fusions.
7.1. LMNA-NTRK1 fusion sarcoma with a spindle cell pattern
This fusion corresponds to t(1;1) (q22;q23) intrachromosomal inversion translocation. It involves genes LMNA encoding nuclear envelope protein lamin A/C and NTRK1 encoding neurotrophic receptor tyrosine kinase 1 in the MAP kinase signaling pathway. The fusion protein contains an active tyrosine kinase domain of the NTRK1 protein. Tumors with fusions TPM3-NTRK1 may have similar histologic and clinicopathologic features [76–79].
LMNA-NTRK1 fusion sarcoma with a spindle cell pattern occurs in children from early infancy and young adults in a wide variety of peripheral soft tissue locations. This tumor has significant potential for local recurrence, but so far, no metastases have been rare. In tumors that are inoperable or metastasize, NTRK inhibitor therapy could be applicable. Histologically these tumors are infiltrative, non-pleomorphic spindle cell sarcomas with low-grade morphology showing mitotic rate <5/10 HPFs (Fig. 6A–C).
Fig 6.
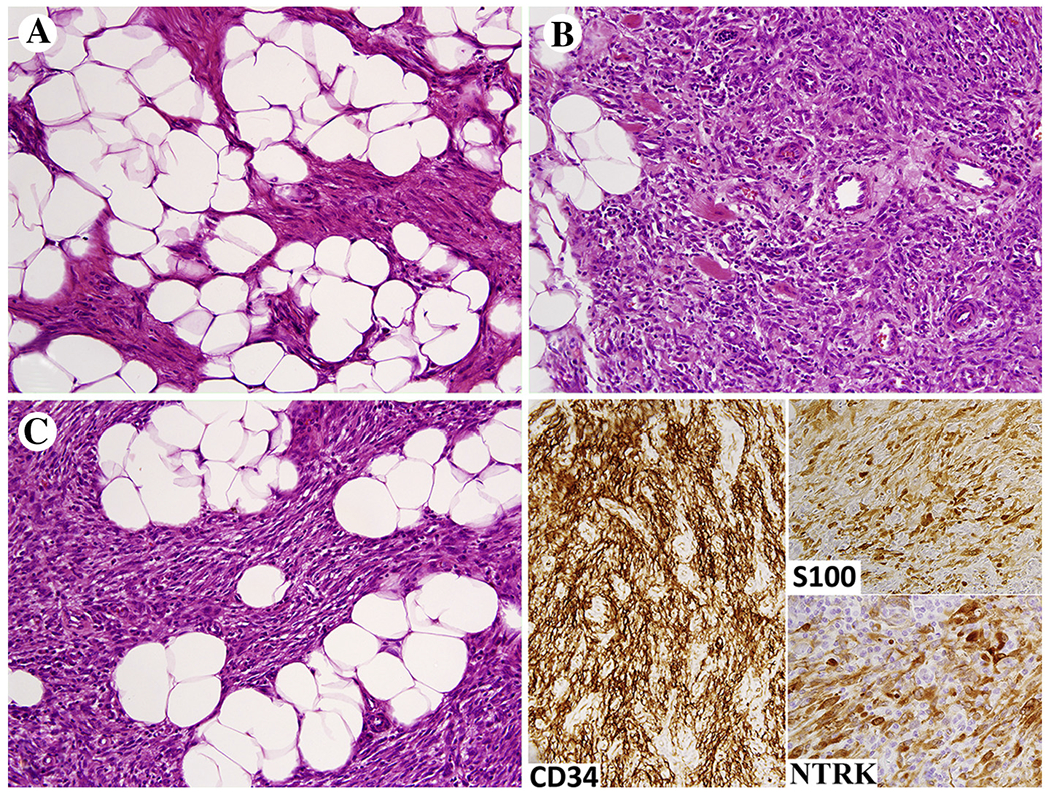
A-C, LMNA-NTRK1 fusion sarcoma shown here grows permeatively in subcutaneous fat and is composed of uniform spindled to ovoid cells with low mitotic activity. The tumor cells are immunohistochemically positive for S100 protein and often CD34. Immunoreactivity for NTRK is a consistent although not totally specific feature.
Immunohistochemically, the tumors are often positive for S100 protein, which could raise the differential diagnosis of low-grade MPNST (Fig. 6). However, these tumors do not have true neuroectodermal differentiation and are SOX10-negative. The tumors are typically positive for CD34 (Fig. 6) and may also be positive for SMA while negative for desmin. A pan-NTRK antibody recognizing NTRKs 1–3 has been reported useful in screening NTRK gene-rearranged sarcomas [80,81]. This antibody typically strongly labels the membranes/cytoplasm (Fig. 6). However, potential positivity for NTRK antibody in other sarcomas, such as rhabdomyosarcoma, and physiological positivity in some endothelial and skeletal muscle cells, should be considered in the differential.
Some low-grade LMNA-NTRK1 fusion sarcomas have been reported with myopericytoma-like features [77]. The same fusion has also been detected in unrelated malignancies of various lineages, such as uterine spindle cell sarcoma, generalized eruptive histiocytosis, and colonic adenocarcinoma [82–84].
7.2. ETV6-NTRK3 fusions in sarcomas other than infantile fibrosarcoma
ETV6-NTRK3 fusions have been recently in KIT/PDGRA and other known GIST driver mutation-negative (wild-type) gastrointestinal stromal tumors. These tumors occurred in middle-aged men in the small intestine and rectum [85,86]. These tumors were high-risk GISTs with one of them metastatic to liver and other containing high mitotic activity. One of the tumors had epithelioid cytology.
Recognition of an ETV6-NTRK3 fusion GIST ultimately requires genomic sequencing, but application of pan NTRK immunohistochemistry could also be useful [80].
Disclosures and acknowledgements:
The authors report no conflicts of interest. This work was supported by NIH’s intramural research program. The views expressed in this article are those of the authors and do not reflect the official policy of the Department of Army/Navy/Air Force, Department of Defense, or U.S. Government.
References
- [1].Clark J, Rocques PJ, Crew AJ, et al. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet 1994;7:502–8. [DOI] [PubMed] [Google Scholar]
- [2].de Alava E Ewing sarcoma, an update on molecular pathology with therapeutic implications. Surg Pathol Clin 2017;10:575–85. [DOI] [PubMed] [Google Scholar]
- [3].Arnold MA, Barr FG. Molecular diagnostics in the management of rhabdomyosarcoma. Expert Rev Mol Diagn 2017;17:189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mertens F, Tayebwa J. Evolving techniques for gene fusion detection in soft tissue tumours. Histopathology 2014;64:151–62. [DOI] [PubMed] [Google Scholar]
- [5].Liew M, Mao R, Wittwer CT, et al. Detection of chromosomal translocations in formalin-fixed paraffin-embedded (FFPE) leukemic specimens by digital expression profiling. Int J Lab Hematol 2015;37:690–8. [DOI] [PubMed] [Google Scholar]
- [6].Chwalenia K, Facemire L, Li H. Chimeric RNAs in cancer and normal physiology. Wiley Interdiscip Rev RNA 2017;6:1427. [DOI] [PubMed] [Google Scholar]
- [7].Pfeifer JD. Molecular genetic testing in surgical pathology. Philadelphia: Lippincott Williams & Wilkins; 2006. [Google Scholar]
- [8].Yeku O, Frohman MA. Rapid amplification of cDNA ends (RACE). Methods Mol Biol 2011;703:107–22. [DOI] [PubMed] [Google Scholar]
- [9].Beck AH, West RB, van de Rijn M. Gene expression profiling for the investigation of soft tissue sarcoma pathogenesis and the identification of diagnostic, prognostic, and predictive biomarkers. Virchows Arch 2010;456: 141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang L, Motoi T, Khanin R, et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 2012;51:127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Løvf M, Thomassen GO, Mertens F, et al. Assessment of fusion gene status in sarcomas using a custom made fusion gene microarray. PLoS One 2013;8:e70649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Skotheim RI, Thomassen GO, Eken M, et al. A universal assay for detection of oncogenic fusion transcripts by oligo microarray analysis. Mol Cancer 2009;8:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Xiong FF, Li BS, Zhang CX, et al. A pipeline with multiplex reverse transcription polymerase chain reaction and microarray for screening of chromosomal translocations in leukemia. Biomed Res Int 2013;2013:135086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wada Y, Matsuura M, Sugawara M, et al. Development of detection method for novel fusion gene using GeneChip exon array. J Clin Bioinforma 2014;4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Akhter A, Mughal MK, Elyamany G, et al. Multiplexed automated digital quantification of fusion transcripts: comparative study with fluorescent in-situ hybridization (FISH) technique in acute leukemia patients. Diagn Pathol 2016;11:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Robinson DR, Wu YM, Kalyana-Sundaram S, et al. Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing. Nat Genet 2013;45:180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Agerstam H, Lilljebjörn H, Lassen C, et al. Fusion gene-mediated truncation of RUNX1 as a potential mechanism underlying disease progression in the 8p11 myeloproliferative syndrome. Genes Chromosomes Cancer 2007;46:635–43. [DOI] [PubMed] [Google Scholar]
- [18].Scolnick JA, Dimon M, Wang IC, et al. An efficient method for identifying gene fusions by targeted RNA sequencing from fresh frozen and FFPE samples. PLoS One 2015;10:e0128916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Loh EY, Elliott JF, Cwirla S, et al. Polymerase chain reaction with single-sided specificity: analysis of T cell receptor delta chain. Science 1989; 243:217–20. [DOI] [PubMed] [Google Scholar]
- [20].Zheng Z, Liebers M, Zhelyazkova B, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nat Med 2014;20:1479–84. [DOI] [PubMed] [Google Scholar]
- [21].Szurian K, Kashofer K, Liegl-Atzwanger B. Role of next-generation sequencing as a diagnostic tool for the evaluation of bone and soft-tissue tumors. Pathobiology 2017;84:323–38. [DOI] [PubMed] [Google Scholar]
- [22].Antonescu CR, Owosho AA, Zhang L, et al. Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am J Surg Pathol 2017;41:941–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Richkind KE, Romansky SG, Finklestein JZ. T(4;19)(q35;q13.1): a recurrent change in primitive mesenchymal tumors? Cancer Genet Cytogenet 1996;87:71–4. [DOI] [PubMed] [Google Scholar]
- [24].Somers GR, Shago M, Zielenska M, et al. Primary subcutaneous primitive neuroectodermal tumor with aggressive behavior and an unusual karyotype: case report. Pediatr Dev Pathol 2004;7:538–45. [DOI] [PubMed] [Google Scholar]
- [25].Yoshimoto M, Graham C, Chilton-MacNeill S, et al. Detailed cytogenetic and array analysis of pediatric primitive sarcomas reveals a recurrent CIC-DUX4 fusion gene event. Cancer Genet Cytogenet 2009;195:1–11. [DOI] [PubMed] [Google Scholar]
- [26].Graham C, Chilton-MacNeill S, Zielenska M, et al. The CIC-DUX4 fusion transcript is present in a subgroup of pediatric primitive round cell sarcomas. Hum Pathol 2012;43:180–9. [DOI] [PubMed] [Google Scholar]
- [27].Italiano A, Sung YS, Zhang L, et al. High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosomes Cancer 2012;51:207–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Choi EY, Thomas DG, McHugh JB, et al. Undifferentiated small round cell sarcoma with t(4;19)(q35;q13.1) CIC-DUX4 fusion: a novel highly aggressive soft tissue tumor with distinctive histopathology. Am J Surg Pathol 2013;37:1379–86. [DOI] [PubMed] [Google Scholar]
- [29].Yoshida A, Goto K, Kodaira M, et al. CIC-rearranged sarcomas: a study of 20 cases and comparisons with Ewing sarcomas. Am J Surg Pathol 2016;40:313–23. [DOI] [PubMed] [Google Scholar]
- [30].Gambarotti M, Benini S, Gamberi G, et al. CIC-DUX4 fusion-positive round-cell sarcomas of soft tissue and bone: a single-institution morphological and molecular analysis of seven cases. Histopathology 2016;69: 624–34. [DOI] [PubMed] [Google Scholar]
- [31].Yoshida A, Arai Y, Kobayashi E, et al. CIC break-apart fluorescence in-situ hybridization misses a subset of CIC-DUX4 sarcomas: a clinicopathological and molecular study. Histopathology 2017;71:461–9. [DOI] [PubMed] [Google Scholar]
- [32].Kawamura-Saito M, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35; q13)translocation. Hum Mol Genet 2006;15:2125–37. [DOI] [PubMed] [Google Scholar]
- [33].Tanaka M, Yoshimoto T, Nakamura T. A double-edged sword: The world according to Capicua in cancer. Cancer Sci 2017;108:2319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yoshimoto T, Tanaka M, Homme M, et al. CIC-DUX4 induces small round cell sarcomas distinct from Ewing sarcoma. Cancer Res 2017; 77:2927–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Siegele B, Roberts J, Black JO, et al. DUX4 immunohistochemistry is a highly sensitive and specific marker for CIC-DUX4 fusion-positive round cell tumor. Am J Surg Pathol 2017;41:423–9. [DOI] [PubMed] [Google Scholar]
- [36].Specht K, Sung YS, Zhang L, et al. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer 2014;53:622–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ito M, Ishikawa M, Kitajima M, et al. A case report of CIC-rearranged undifferentiated small round cell sarcoma in the cerebrum. Diagn Cytopathol 2016;44:828–32. [DOI] [PubMed] [Google Scholar]
- [38].Tsukamoto Y, Futani H, Yoshiya S, et al. Primary undifferentiated small round cell sarcoma of the deep abdominal wall with a novel variant of t(10;19) CIC-DUX4 gene fusion. Pathol Res Pract 2017;213:1315–21. [DOI] [PubMed] [Google Scholar]
- [39].Sugita S, Arai Y, Tonooka A, et al. A novel CIC-FOXO4 gene fusion in undifferentiated small round cell sarcoma: a genetically distinct variant of Ewing-like sarcoma. Am J Surg Pathol 2014;38:1571–6. [DOI] [PubMed] [Google Scholar]
- [40].Solomon DA, Brohl AS, Khan J, et al. Clinicopathologic features of a second patient with Ewing-like sarcoma harboring CIC-FOXO4 gene fusion. Am J Surg Pathol 2014;38:1724–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Gen 2012;44:461–6. [DOI] [PubMed] [Google Scholar]
- [42].Cohen-Gogo S, Cellier C, Coindre JM, et al. Ewing-like sarcomas with BCOR-CCNB3 fusion transcript: a clinical, radiological and pathological retrospective study from the Société Française des cancers de L’Enfant. Pediatr Blood Cancer 2014;61:2191–8. [DOI] [PubMed] [Google Scholar]
- [43].Puls F, Niblett A, Marland G, et al. BCOR-CCNB3 (Ewing-like) sarcoma: a clinicopathologic analysis of 10 cases, in comparison with conventional Ewing sarcoma. Am J Surg Pathol 2014;38:1307–18. [DOI] [PubMed] [Google Scholar]
- [44].Peters TL, Kumar V, Polikepahad S, et al. BCOR-CCNB3 fusions are frequent in undifferentiated sarcomas of male children. Mod Pathol 2015;28:575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Shibayama T, Okamoto T, Nakashima Y, et al. Screening of BCOR-CCNB3 sarcoma using immunohistochemistry for CCNB3: a clinicopathological report of three pediatric cases. Pathol Int 2015;65: 410–4. [DOI] [PubMed] [Google Scholar]
- [46].Li WS, Liao IC, Wen MC, Lan HH, Yu SC, Huang HY. BCOR-CCNB3-positive soft tissue sarcoma with round-cell and spindle-cell histology: a series of four cases highlighting the pitfall of mimicking poorly differentiated synovial sarcoma. Histopathology 2016;69:792–801. [DOI] [PubMed] [Google Scholar]
- [47].Matsuyama A, Shiba E, Umekita Y, et al. Clinicopathologic diversity of undifferentiated sarcoma with BCOR-CCNB3 fusion: analysis of 11 cases with a reappraisal of the utility of immunohistochemistry for BCOR and CCNB3. Am J Surg Pathol 2017;41:1713–21. [DOI] [PubMed] [Google Scholar]
- [48].Kao YC, Owosho AA, Sung YS, et al. BCOR-CCNB3 fusion positive sarcomas: a clinicopathologic and molecular analysis of 36 cases with comparison to morphologic spectrum and clinical behavior of other round cell sarcomas. Am J Surg Pathol 2018;42:604–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Argani P, Kao YC, Zhang L, et al. Primary renal sarcomas with BCOR-CCNB3 gene fusion: a report of 2 cases showing histologic overlap with clear cell sarcoma of kidney, suggesting further link between BCOR-related sarcomas of the kidney and soft tissues. Am J Surg Pathol 2017;41:1702–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wong MK, Ng CCY, Kuick CH, et al. Clear cell sarcomas of the kidney are characterised by BCOR gene abnormalities, including exon 15 internal tandem duplications and BCOR-CCNB3 gene fusion. Histopathology 2018;72:320–9. [DOI] [PubMed] [Google Scholar]
- [51].Gooskens SL, Gadd S, van den Heuvel-Eibrink MM, Perlman EJ. BCOR internal tandem duplications in clear cell sarcoma of the kidney. Genes Chromosomes Cancer 2016;55:549–50. [DOI] [PubMed] [Google Scholar]
- [52].Astolfi A, Melchionda F, Perotti D, et al. Whole transcriptome sequencing identifies BCOR internal tandem duplication as a common feature of clear cell sarcoma of the kidney. Oncotarget 2015;6:40934–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ludwig K, Alaggio R, Zin A, et al. BCOR-CCNB3 undifferentiated sarcoma—does immunohistochemistry help in the identification? Pediatr Dev Pathol 2017;20:321–9. [DOI] [PubMed] [Google Scholar]
- [54].Yamada Y, Kuda M, Kohashi K, et al. Histological and immunohistochemical characteristics of undifferentiated small round cell sarcomas associated with CIC-DUX4 and BCOR-CCNB3 fusion genes. Virchows Arch 2017;470:373–80. [DOI] [PubMed] [Google Scholar]
- [55].Kao YC, Sung YS, Zhang L, et al. BCOR overexpression is a highly sensitive marker in round cell sarcomas with BCOR genetic abnormalities. Am J Surg Pathol 2016;40:1670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Chiang S, Lee CH, Stewart CJR, et al. BCOR is a robust diagnostic immunohistochemical marker of genetically diverse high-grade endometrial stromal sarcoma, including tumors exhibiting variant morphology. Mod Pathol 2017;30:1251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Szuhai K, Jczenga MI, de Jong D, et al. The NFATc2 gene is involved in a novel cloned translocation in a Ewing sarcoma variant that couples its function in immunology to oncology. Clin Cancer Res 2009;15: 2259–68. [DOI] [PubMed] [Google Scholar]
- [58].Hogan PG, Chen L, Nardone J, et al. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 2003;17:2205–32. [DOI] [PubMed] [Google Scholar]
- [59].Romeo S, Bovée JV, Kroon HM, et al. Malignant fibrous histiocytoma and fibrosarcoma of bone: a re-assessment in the light of currently employed morphological, immunohistochemical and molecular approaches. Virchows Arch 2012;461:561–70. [DOI] [PubMed] [Google Scholar]
- [60].Wang WL, Patel NR, Caragea M, et al. Expression of ERG, an Ets family transcription factor, identifies ERG-rearranged Ewing sarcoma. Mod Pathol 2012;25:1378–83. [DOI] [PubMed] [Google Scholar]
- [61].Sadri N, Barroeta J, Pack SD, et al. Malignant round cell tumor of bone with EWSR1-NFATC2 gene fusion. Virchows Arch 2014;465:233–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Watson S, Perrin V, Guillemot D, et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas. J Pathol 2018;245:29–40. [DOI] [PubMed] [Google Scholar]
- [63].Baldauf MC, Orth MF, Dallmayer M, et al. Robust diagnosis of Ewing sarcoma by immunohistochemical detection of super-enhancer-driven EWSR1-ETS targets. Oncotarget 2017;9:1587–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Antonescu CR, Agaram NP, Sung Y-S, et al. A distinct malignant epithelioid neoplasm with gli1 gene rearrangements, frequent s100 protein expression, and metastatic potential expanding the spectrum of pathologic entities with ACTB/MALAT1/PTCH1-GLI1 fusions. Am J Surg Pathol 2018;42:553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Spans L, Fletcher CD, Antonescu CR, et al. Recurrent MALAT1-GLI1 oncogenic fusion and GLI1 up-regulation define a subset of plexiform fibromyxoma. J Pathol 2016;239:335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Graham RP, Nair AA, Davila JI, et al. Gastroblastoma harbors a recurrent somatic MALAT1-GLI1 fusion gene. Mod Pathol 2017;30:1443–52. [DOI] [PubMed] [Google Scholar]
- [67].Dahlen A, Fletcher CD, Mertens F, et al. Activation of the GLI oncogene through fusion with the beta-actin gene (ACTB) in a group of distinctive pericytic neoplasms: pericytoma with t(7;12). Am J Pathol 2004;164:1645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bridge JA, Sanders K, Huang D, et al. Pericytoma with t(7;12) and ACTB-GLI1 fusion arising in bone. Hum Pathol 2012;43:1524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lange AM, Lo HW. Inhibiting TRK proteins in clinical cancer therapy. Cancers (Basel) 2018;10(4):e105 10.3390/cancers10040105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wong V, Pavlick D, Brennan T, et al. Evaluation of a congenital infantile fibrosarcoma by comprehensive genomic profiling reveals an LMNA-NTRK1 gene fusion responsive to crizotinib. J Natl Cancer Inst 2015; 108:djv307. [DOI] [PubMed] [Google Scholar]
- [71].Doebele RC, Davis LE, Vaishnavi A, et al. An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov 2015;5:1049–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Knezevich SR, McFadden DE, Tao W, et al. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet 1998;18:184–7. [DOI] [PubMed] [Google Scholar]
- [73].Rubin BP, Chen CJ, Morgan TW, et al. Congenital mesoblastic nephroma t(12;15) is associated with ETV6-NTRK3 gene fusion: cytogenetic and molecular relationship to congenital infantile brosarcoma. Am J Pathol 1998;153:1451–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Tognon C, Knezevich SR, Huntsman D, et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell 2002;2:367–76. [DOI] [PubMed] [Google Scholar]
- [75].Skálová A, Vanecek T, Sima R, et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: a hitherto undescribed salivary gland tumor entity. Am J Surg Pathol 2010; 34:599–608. [DOI] [PubMed] [Google Scholar]
- [76].Haller F, Knopf J, Ackermann A, et al. Paediatric and adult soft tissue sarcomas with NTRK1 gene fusions: a subset of spindle cell sarcomas unified by a prominent myopericytic/haemangiopericytic pattern. J Pathol 2016;23:700–10. [DOI] [PubMed] [Google Scholar]
- [77].Agaram NP, Zhang L, Sung YS, et al. Recurrent NTRK1 gene fusions define a novel subset of locally aggressive Lipofibromatosis-like neural tumors. Am J Surg Pathol 2016;40:1407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kohsaka S, Saito T, Akaike K, et al. Pediatric soft tissue tumor of the upper arm with LMNA-NTRK1 fusion. Hum Pathol 2018;72:167–73. [DOI] [PubMed] [Google Scholar]
- [79].Davis JL, Lockwood CM, Albert CM, et al. Infantile NTRK-associated mesenchymal tumors. Pediatr Dev Pathol 2018;21:68–78. [DOI] [PubMed] [Google Scholar]
- [80].Hechtman JF, Benayed R, Hyman DM, et al. Pan-Trk immunohistochemistry is an efficient and reliable screen for the detection of NTRK fusions. Am J Surg Pathol 2017;41:1547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Rudzinski ER, Lockwood CM, Stohr BA, et al. Pan-Trk immunohistochemistry identifies NTRK rearrangements in pediatric mesenchymal tumors. Am J Surg Pathol 2018;42:927–35. [DOI] [PubMed] [Google Scholar]
- [82].Chiang S, Cotzia P, Hyman DM, et al. NTRK fusions define a novel uterine sarcoma subtype with features of fibrosarcoma. Am J Surg Pathol 2018;42:791–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Pinney SS, Jahan-Tigh RR, Chon S. Generalized eruptive histiocytosis associated with a novel fusion in LMNA-NTRK1. Dermatol Online J 2016;22(8):13030/qt07d3f2xk. [PubMed] [Google Scholar]
- [84].Sartore-Bianchi A, Ardini E, Bosotti R, et al. Sensitivity to entrectinib associated with a novel LMNA-NTRK1 gene fusion in metastatic colorectal cancer. J Natl Cancer Inst 2016;108(1):djv306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Shi E, Chmielecki J, Tang CM, et al. FGFR1 and NTRK3 actionable alterations in “wild-type” gastrointestinal stromal tumors. J Transl Med 2016;14:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Brenca M, Rossi S, Polano M, et al. Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J Pathol 2016;238: 543–9. [DOI] [PubMed] [Google Scholar]