

Abstract
Purpose:
Pheochromocytomas and paragangliomas (PCPG) are usually benign neuroendocrine tumors. However, PCPGs with mutations in the succinate dehydrogenase B subunit (SDHB) have a poor prognosis and frequently develop metastatic lesions. SDHB-mutated PCPGs exhibit dysregulation in oxygen metabolic pathways, including pseudohypoxia and formation of reactive oxygen species, suggesting that targeting the redox balance pathway could be a potential therapeutic approach.
Experimental Design:
We studied the genetic alterations of cluster I PCPGs compared with cluster II PCPGs, which usually present as benign tumors. By targeting the signature molecular pathway, we investigated the therapeutic effect of ascorbic acid on PCPGs using in vitro and in vivo models.
Results:
By investigating PCPG cells with low SDHB levels, we show that pseudohypoxia resulted in elevated expression of iron transport proteins, including transferrin (TF), transferrin receptor 2 (TFR2), and the divalent metal transporter 1 (SLC11A2; DMT1), leading to iron accumulation. This iron overload contributed to elevated oxidative stress. Ascorbic acid at pharmacologic concentrations disrupted redox homeostasis, inducing DNA oxidative damage and cell apoptosis in PCPG cells with low SDHB levels. Moreover, through a preclinical animal model with PCPG allografts, we demonstrated that pharmacologic ascorbic acid suppressed SDHB-low metastatic lesions and prolonged overall survival.
Conclusions:
The data here demonstrate that targeting redox homeostasis as a cancer vulnerability with pharmacologic ascorbic acid is a promising therapeutic strategy for SDHB-mutated PCPGs.
Introduction
Pheochromocytomas and paragangliomas (PCPG) are catecholamine-producing tumors, which are stratified into two major molecular subtypes: cluster I and cluster II. Cluster I PCPGs commonly exhibit abnormal activation of hypoxia signaling, particularly those carrying mutations in succinate dehydrogenase subunits (SDHx), von Hippel–Lindau (VHL), hypoxia-inducible factor 2A (HIF2A), or fumarate hydratase (FH). Cluster II PCPGs commonly show hyperactivation of protein kinase pathways, carry mutations in ret proto-oncogene (RET), neurofibromatosis type 1 (NF1), MYC-associated factor X (MAX), transmembrane protein 127 (TMEM127), or the kinesin family member 1B (KIF1B; refs. 1–3). Succinate dehydrogenase (SDH), also known as the mitochondrial respiratory complex II, comprises SDHA, SDHB, SDHC, and SDHD subunits (4). Loss-of-function mutations in SDHx lead to substantial loss of the complex II activity and result in reprogramming of cellular metabolic pathways, as indicated by the accumulation of succinate, genome-wide hypermethylation, and a pseudohypoxia phenotype (5–10). Furthermore, compromised mitochondrial complex II disrupts electron transfer to oxygen, leading to increased formation of reactive oxygen species (ROS) and redox imbalance (11, 12).
Clinically, SDHB-deficient PCPGs are one of the most common subtypes of cluster I PCPGs. In contrast to other PCPG molecular subtypes, SDHB-deficient PCPGs, especially those producing and secreting catecholamines/metanephrines, typically present an early onset and display tumor aggressiveness with development of metastases, all resulting in patient mortality (13–16). For advanced PCPGs with metastatic lesions, surgical resection coupled with radio- or chemotherapies are considered the standards of care. For chemotherapy, the most widely used regimen is a combination of cyclophosphamide, vincristine, and dacarbazine (CVD regimen; ref. 17). However, emerging evidence shows that in some patients, the CVD regimen produces only transient effects with only minimally prolonged overall patient survival (18). Therefore, therapeutic approaches with enhanced efficacy have been advocated to better handle advanced PCPGs.
The pseudohypoxic “signature” and angiogenic pathways have been proposed as targets for limiting tumor expansion in cluster I PCPGs (19). Humanized monoclonal VEGFA antibody and tyrosine kinase inhibitors showed efficacy in cluster I PCPGs and have advanced to clinical studies (20, 21). Similarly, disturbance of the NAD+/NADH metabolism in cluster I PCPGs has been reported, establishing a therapeutic vulnerability to PARP inhibitors (22). Overall, the distinctive pattern in molecular signaling and metabolism in cluster I PGPCs suggests that there may be novel therapeutic targets for this type of malignancy.
To expand the therapeutic possibilities for cluster I PCPGs, we investigated the redox homeostasis in SDHB-deficient cells. Furthermore, we hypothesized and probed the associations between metabolic deficiency and iron uptake in these cells. Finally, we evaluated the impact of pharmacologic ascorbic acid on cluster I PCPGs using in vitro assays and an animal model.
Materials and Methods
This study was approved by the Institutional Review Board of the Eunice Kennedy Shriver NICHD/NIH, and all patients gave written informed consent.
Patient samples
All patient samples, including frozen tissue and formalin-fixed, paraffin-embedded (FFPE) slides, were collected from clinically identified patients with cluster I and cluster II PCPG. Tissue preparation and dissection were performed as reported previously (22), and clinical samples used are summarized in Supplementary Table S1.
Cell lines and reagents
Mouse pheochromocytoma (MPC) cells (4/30 PRR; ref. 23) were cultured in DMEM supplemented with 10% (v/v) FBS and penicillin/streptomycin at 37°C. To establish the SDHB wild-type (SDHBWT) and SDHB-knockdown (SDHBKD) cell lines, mouse pheochromocytoma cells were transduced by lentivirus with either nontargeted or SDHB-targeted short hairpin RNA (shRNA). The specific targeting sequencing for SDHB was TGA GTA ACT TCT ACG CAC A (GE Dharmacon). The virus was added to the supernatant of the cell culture medium and cultured at 37°C, 5% CO2, for 3 days. Then, the cells were selected by puromycin (2 μg/mL). Human pheochromocytoma precursor cells (hPheo1; ref. 24) were cultured in RPMI1640 medium with 10% FBS, and SDHB was knocked down using a lentivirus with shRNA (targeting sequence: CAG AGC TGA ACA TAA TTT A; GE Dharmacon). Control hPheo1 cells were transduced with nontargeted shRNA. Knockdown efficacy was evaluated in all transformed cells by qRT-PCR and Western blot analysis. For in vivo studies, SDHB knockdown mouse pheochromocytoma cells were transduced with a retrovirus containing firefly luciferase and selected using G418. Human renal carcinoma cells University of Michigan-Renal Carcinoma-6 (UMRC6; refs. 25, 26) were cultured in DMEM supplemented with 10% (v/v) FBS and penicillin/streptomycin.
Ascorbic acid, N-acetyl-L-cysteine (NAC), and catalase (Sigma-Aldrich) were dissolved in PBS before being added to the cell medium. The final concentrations of ascorbic acid, NAC, and catalase were 1 mmol/L, 2.5 mmol/L, and 500 U/mL, respectively. The pH of the medium was adjusted to 7.4 before use. Cells were treated overnight for oxidative stress and DNA damage experiments and treated 24 hours for experiments related to cell viability.
ROS measurement
Cells were seeded in a 96-well plate and treated under various conditions. Cellular levels of H2O2 were determined using the ROS-Glo H2O2 Assay Kit (Promega). Briefly, derivatized luciferin substrate from the kit was incubated with samples, reacting directly with H2O2 to generate the luciferin precursor. After the addition of the ROS-Glo Detection Solution, which converts the luciferin precursor to luciferin, the luminescence signal was recorded by a FLUOstar Omega Plate Reader (BMG Labtech). The ROS luminescence signal was normalized to protein quantification. Mitochondrial ROS levels were estimated by MitoSOX Staining (Thermo Fisher Scientific). Briefly, cells were incubated with 5 μmol/L MitoSOX-Red for 30 minutes at 37°C. The fluorescence signal was assessed on a FACSCanto II Flow Cytometer (BD Biosciences), and fluorescence intensity was quantified using ImageJ (version 1.52K, NIH, Bethesda, MD).
Cellular labile iron pool quantification and iron assay
The labile iron pool level was evaluated using Calcein Red-Orange AM Fluorescent Dye (Thermo Fisher Scientific) combined with 2′,2′−bipyridyl (BIP), as described previously (27). Briefly, 106 cells were pelleted and resuspended in 1 mL PBS with 500 nmol/L Calcein Red-Orange AM. After incubation for 15 minutes at 37°C (5% CO2), cells were washed with PBS and resuspended in 1 mL PBS. The cells were divided into two flow cytometry tubes, and 500 μmol/L BIP was added to one tube. Cells were then incubated at room temperature for more than 15 minutes before analysis using an LSRFortessa Flow Cytometer (BD Biosciences). The labile iron pool (A.U.) = MFIBIP−MFINoBIP and the relative labile iron pool for each sample were compared with the control sample.
Iron was quantified with the Iron Assay Kit (Sigma-Aldrich). Cells or tissue specimens were collected and rapidly homogenized in cold iron assay buffer from the kit. Ferrous and total iron from samples were assessed following the manufacturer’s protocol, and absorbance was taken at 593 nm using the FLUOstar Omega plate reader.
RNAi
Small interference RNA was designed and purchased from Integrated DNA Technologies. The sequences of small interference RNA oligos used in this study are listed in Supplementary Table S2.
Antioxidant response element luciferase reporter assay
Nrf2-driven transcriptional activation was quantified as antioxidant response element (ARE) transcriptional activity using a dual-luciferase reporter system (28, 29). pGL3.47-ARE-Luc plasmid (900 ng) and 100 ng pRL-TK plasmid (Promega) were cotransfected into 8,000 cells using Lipofectamine 3000 (Thermo Fisher Scientific). The firefly (ARE activity) and Renilla (internal control) luminescence signal were assessed using a Polarstar Optima Plate Reader (BMG Labtech).
Cell viability analysis
The dose–response curve of ascorbic acid treatment was assessed with the CCK-8 Assay (Dojindo). Cells were seeded in a 96-well plate and treated with ascorbic acid for 24 hours. After replacing the culture medium with fresh medium, CCK8 was added, and the cells were incubated at 37°C for 1 hour. The absorbance at 450 nm was assessed using a FLUOstar Omega plate reader. Cells were enumerated using a Vi-CELL Cell Counter (Beckman Coulter) to evaluate cell viability after treatment.
Oxidative DNA damage ELISA assay
Oxidative DNA damage was assessed by the DNA/RNA Oxidative Damage ELISA Kit (Cayman Chemical) according to the manufacturer’s instructions. Briefly, DNA was purified from cell lysates, digested with nuclease P1, and incubated with alkaline phosphatase for 30 minutes at 37°C. Digested DNA samples were then incubated in the provided plate for 18 hours at 4°C. Next, the samples were incubated with freshly reconstituted Ellman reagent on an orbital shaker for 90 minutes. The absorbance of each well was assessed on a FLUOstar Omega plate reader at 405 nm.
IHC
IHC was performed as reported previously (22). Briefly, 8 μm FFPE tissue sections from patients or animals were deparaffinized in xylene three times and rehydrated in a series of ethanol solutions ranging from 100% to 75%. The slides were boiled in sodium citrate buffer for 20 minutes for antigen retrieval and blocked with blocking solution (Thermo Fisher Scientific) for 30 minutes at room temperature. Next, slides were probed with anti-TF, anti-TFR2 (Thermo Fisher Scientific), or anti-DMT1 (Abcam) antibodies overnight and developed with the DAB+ Substrate Chromogen System (Dako). Slides were counter-stained with hematoxylin, dehydrated, and mounted. Samples were visualized using a KEYENCE microscope with 40-fold magnification.
Alkaline comet assay
An alkaline comet assay was performed as described previously (22, 30) to analyze DNA damage levels. Briefly, cells were harvested and resuspended in molten agarose and evenly spread on precoated glass slides. Cells on the slides were lysed and subjected to electrophoresis for 30 minutes at 1 V/cm. Slides were stained with diluted SYBR Safe (Invitrogen) and visualized by fluorescence microscopy (Keyence).
DNA fragmentation assay
Genomic DNA was collected and purified with the DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer’s protocol. 500 ng DNA samples were resolved by electrophoresis in 4%–20% Novex TBE Gel (Thermo Fisher Scientific). The gel was stained with SYBR Safe DNA Gel Dye for 20 minutes and visualized in a ChemiDoc Imaging System (Bio-Rad).
Mitochondria were isolated from each sample, and a mitochondrial DNA damage assay was performed as described previously (31). The long (10.1 kb) and short (241 bp) fragments from the mitochondrial genome were amplified by semiquantitative PCR with the primers listed in Supplementary Table S2. The PCR products were resolved in a 1% agarose gel, stained with SYBR Safe DNA Gel Dye, and visualized in a ChemiDoc imaging system.
Annexin V/PI apoptosis assay
Cell apoptosis was measured using Annexin V/PI Staining (Thermo Fisher Scientific). Cells (including the culture media) were harvested and washed once with PBS and then incubated with Annexin V and PI for 20 minutes on ice. The fluorescence signal was measured on a FACSCanto II flow cytometer, and the percentage of apoptotic cells was quantified.
Caspase 3/7 activity assay
The activity of caspase 3/7 was determined using the Caspase-Glo 3/7 Assay Kit (Promega). In brief, cells were seeded in a 96-well plate and incubated with the Caspase-Glo 3/7 reagent for 30 minutes. The luminescence signal was evaluated using a Polarstar Optima plate reader.
Western blot
Cells or tissue samples were collected, and total protein was extracted in ice-cold RIPA lysis buffer supplemented with a Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Total protein (20–30 μg) was resolved using the NuPAGE 4%–12% Novex Bis-Tris Gel (Invitrogen). The proteins were transferred to PVDF membranes (Bio-Rad) and probed with primary antibodies at 4°C overnight. The membrane was then washed with PBST (PBS+0.1% Tween20), incubated with horseradish peroxidase–conjugated secondary antibodies, and visualized using a ChemiDoc imaging system. Anti–β-actin IgG (Cell Signaling Technology, 1:5,000) was used as an internal control. The primary antibodies used included anti–HIF-1α, anti–HIF-2α, and anti-DMT1 (Abcam), and anti-TF and anti-TFR2 (Thermo Fisher Scientific). All primary antibodies were diluted 1:1,000.
qRT-PCR
Cell or tissue samples were dissected, and total RNA was extracted using the RNeasy Mini Kit (Qiagen). RNA (1 μg) was reverse transcribed to cDNA using the SuperScript III First-Strand Synthesis System (Invitrogen). Primer sets for each gene are listed in Supplementary Table S2.
Chromatin immunoprecipitation assay
Cells were seeded in 150 mm Petri dishes, and a chromatin immunoprecipitation (ChIP) assay was performed using the ChIP-IT High Sensitivity Kit (Active Motif) according to the manufacturer’s protocol. Cells (1.5 × 107) were fixed, and chromatin was prepared from each sample. Thirty micrograms of the chromatin preparation were fragmented by sonication and precipitated with anti–HIF-1α antibody (Abcam). Purified ChIP DNA was analyzed by qRT-PCR to measure promoter enrichment, comparing pull-down DNA and input DNA. The sequences of the primer set used are listed in Supplementary Table S2.
Cell viability measured by trypan blue exclusion assay
A total of 1 × 105 cells were seeded in a 6-well plate. To determine viability, cells were detached with 0.05% trypsin. All the culture media and wash buffer were collected for the analysis. The measurement of cell viability was assessed by a Vi-Cell XR Cell Counter (Beckman Coulter) using a trypan blue exclusion staining protocol. The cell suspension was mixed with an equal volume of trypan blue. Fifty random images were taken to determine the live and dead cells.
Allograft animal model
Animal experiments were performed following the principles and procedures of the Eunice Kennedy Shriver NICHD animal protocol (ASP 15–028) approved by the Animal Care and Use Committee of the NIH. Eight-week-old female athymic mice (Jackson Laboratory) were injected in the tail vein with 1.5 × 106 MPC-SDHBKD-Luci or MPC-SDHBWT-Luci cells. Tumor growth was assessed on an in vivo imaging system every week. Two weeks after the tail vein injection, the luminescence signals could be detected in the liver or spleen, and then mice were randomly divided into two groups (10 mice/group for SDHBWT cells and 10 mice/group for SDHBKD cells) and treated with saline solution or ascorbic acid. Ascorbic acid was dissolved in saline solution and neutralized to pH 7.3 with sodium hydroxide. The freshly prepared ascorbic acid solution was injected intraperitoneally every day at 4 g/kg (32, 33). The behavior and body weight of animals was monitored every day. Animals were sacrificed, and metastatic lesions in the liver were harvested at the end for further evaluation.
Colony formation
SDHBWT and SDHBKD mouse pheochromocytoma cells were seeded in a 6-well plate at a density of 2,000 cells per well. Cells were treated with 0.5 mmol/L ascorbic acid for 2 weeks. Then, the cells were fixed in 4% cold formaldehyde (Sigma) and stained with 2% crystal violet (Sigma). This experiment was repeated in three replicates, and the images were quantified by ImageJ.
Statistical analysis
The statistical significance of differences between the two groups was analyzed using the Student t test. All statistical tests were two-sided. The results were presented as mean value ± SEM. A P value of less than 0.05 was considered statistically significant. The statistical tests in this study were analyzed using GraphPad Prism 7.01 (Graph-Pad Software).
Results
SDHB deficiency triggers pseudohypoxia and reprograms iron metabolism
Genetic defects in SDHx result in substantial loss of mitochondrial complex II (SDH and succinate-ubiquinone oxidoreductase activity) activity, leading to abnormal activation of the hypoxia signaling pathway and a switch to the pseudohypoxia phenotype (10). To investigate the impact of pseudohypoxia in cluster I PCPGs, we established cell lines with considerably decreased levels of SDHB using mouse pheochromocytoma and hPheo1 cells. Knockdown efficiency was confirmed by Western blotting (Fig. 1A; Supplementary Fig. S1A), and qRT-PCR (Fig. 1B). We found that decreased levels of SDHB led to substantial activation of the hypoxia signaling pathway resulting in upregulation of genes that regulate iron homeostasis, such as transferrin (TF), transferrin receptor 2 (TFR2), and solute carrier family 11 member 2 (SLC11A2, DMT1; Fig. 1C and D; Supplementary Fig. S1B). We confirmed activation of the hypoxia signaling pathway, as well as the upregulation of genes that are associated with iron metabolism in cluster I PCPG tumor specimens obtained from patients (Fig. 1E–G). To further validate the correlation between the hypoxia signaling pathway and iron homeostasis in an SDHB-low background, we performed ChIP assay. This assay revealed stronger affinity of HIF-1α to the promoter regions of TF, TFR2, and SLC11A2 in the SDHBKD cells compared with their wild-type counterparts, indicating that HIF-1α mediated transcriptional activation of genes that regulate iron transport (Fig. 1H; Supplementary Fig. S1C). Consistent with the upregulation of iron transporters, we found elevated intracellular labile iron in SDHBKD cells compared with SDHBWT cells (Fig. 1I; Supplementary Fig. S1D and S1E). Reexpression of SDHB reduced labile iron to baseline level in SDHBKD cells (Supplementary Fig. S1F). Genetic silencing of TF, HIF-1a, or HIF-2a by siRNA reduced the iron level in SDHBKD cells (Fig. 1J–L; Supplementary Fig. S1G). Besides, we measured the expression of TF and SLC11A2 under chemical hypoxic conditions. Both SDHBWT and SDHBKD cells showed strong induction of TF and SLC11A2 expression under cobalt chloride treatment (Supplementary Fig. S1H). Moreover, the expression of TF and TFR2 was measured in a VHL-low UMRC6 cell line. Results showed that the reexpression of VHL in UMRC6 cells led to reduced expression of TF and TFR2 and reduced labile iron pools (Supplementary Fig. S1I and S1J). These findings suggest that activation of the hypoxia (pseudo-hypoxia) signaling pathway in SDHBKD cells resulted in TF-dependent ferrous iron overload.
Figure 1.
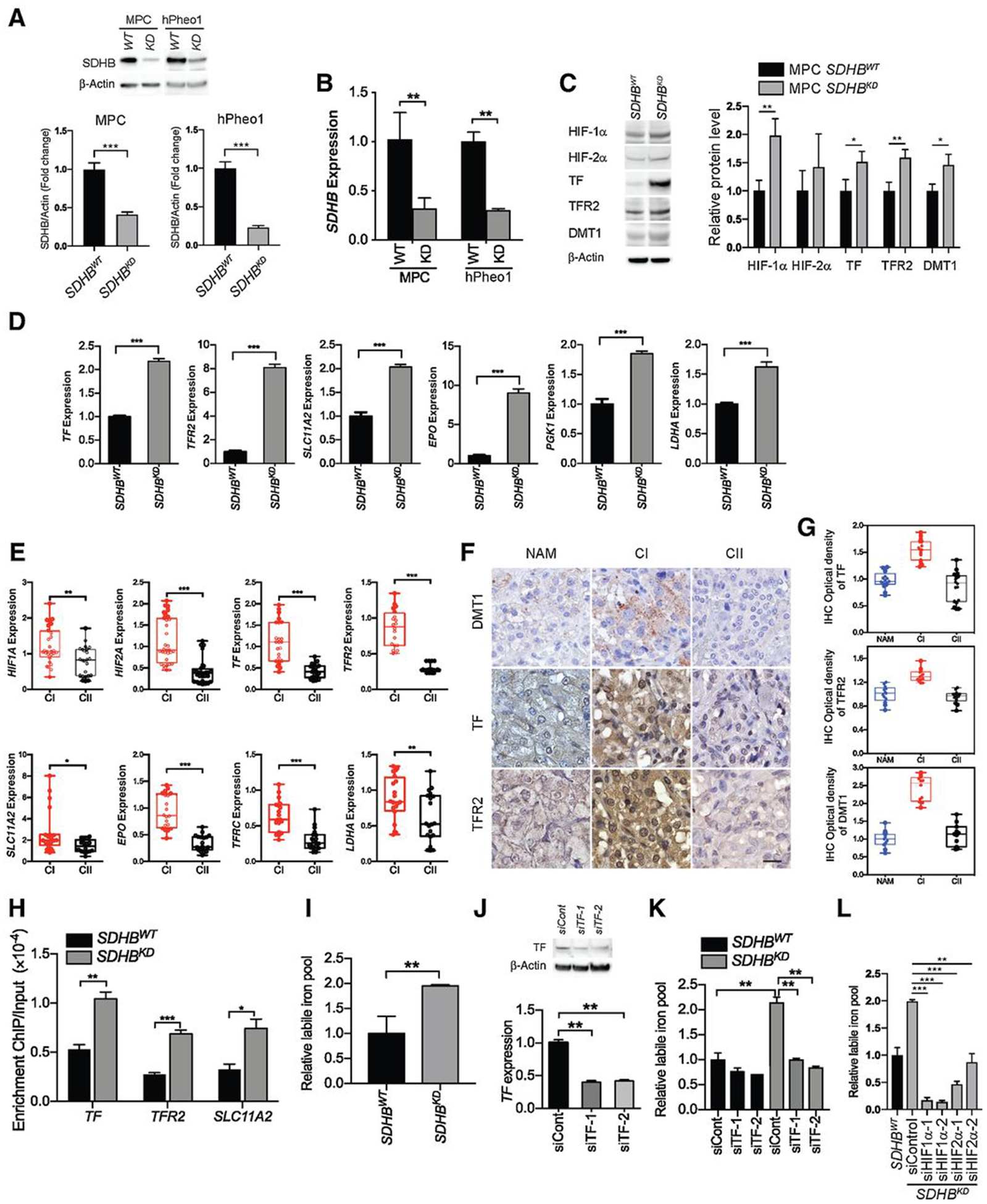
SDHB deficiency induces pseudohypoxia and reprograms iron homeostasis. A, SDHB knockdown efficiency in mouse pheochromocytoma (MPC) and hPheo1 cell lines was measured by Western blot analysis (top) and quantification (bottom, n = 3). β-actin was used as a loading control. B, SDHB knockdown efficiency in MPC and hPheo1 cell lines was measured by qRT-PCR (n = 4). C, HIF-1α, HIF-2α, TF, TFR2, and DMT1 proteins in SDHBWT and SDHBKD MPC cells were measured by Western blot analysis (left) and quantification (right, n = 3). β-actin was used as a loading control. D, Levels of TF, TFR2, SLC11A2 (DMT1), EPO, PGK1, and LDHA mRNA were measured by qRT-PCR. Data normalized to SDHBWT. E, Levels of HIF1A, HIF2A, TF, TFR2, SLC11A2, EPO, TFRC, and LDHA mRNA in cluster I and cluster II PCPG frozen tissues were measured by qRT-PCR. CI, cluster I (n = 5–8); CII, cluster II (n = 4–7). F, Expression of DMT1, TF, and TFR2 in cluster I (n = 4) and cluster II (n = 5) PCPGs was measured by IHC staining obtained from cluster I and cluster II PCPG FFPE tissues. Scale bar, 50 μm. CI, cluster I; CII, cluster II; NAM, normal adrenal medulla. G, Integrated optical density quantification of the IHC staining shown in (F). Cluster I (CI), n = 4; cluster II (CII), n = 5. H, Promoter affinity of HIF-1α assay in SDHBWT and SDHBKD hPheo1 cells was measured by a ChIP PCR assay. I, The level of the labile iron pools in SDHBWT and SDHBKD mouse pheochromocytoma cells was measured by a cellular labile iron pool quantification assay. J, Knockdown efficiency of siRNA targeting TF in mouse pheochromocytoma cells was measured by Western blot analysis (top) and qRT-PCR (bottom). K, The level of the labile iron pools in SDHBWT and SDHBKD mouse pheochromocytoma cells after TF knockdown was measured by a cellular labile iron pool quantification assay. L, The level of the labile iron pools in SDHBKD mouse pheochromocytoma cells after HIF1A and HIF2A knockdown was measured by a cellular labile iron pool quantification assay. All significance in differences were determined by t test [*, P < 0.05; **, P < 0.01; ***, P < 0.001 (±SEM)].
Iron overload compromises redox balance in SDHBKD cells
Previous findings indicated that abnormal iron metabolism results in oxidative stress in a variety of malignancies, including SDHB-deficient ones (34, 35). Using the ROS-Glo assay, we showed that a decrease of SDHB correlated with a significant elevation of oxidative stress (Fig. 2A; Supplementary Fig. S1K). A MitoSOX assay confirmed increased ROS in the mitochondria of SDHBKD cells (Fig. 2B; Supplementary Fig. S1L). Moreover, the AREluciferase reporter assay revealed that SDHBKD cells exhibited higher ARE-dependent transcriptional activity, indicating increased oxidative stress (Fig. 2C). Iron overload plays a crucial role in deleterious consequences of elevated ROS through the Fenton reaction, which converts H2O2 into hydroxyl radicals via an iron-dependent mechanism (36). We confirmed that the iron overload in SDHBKD cells was correlated with elevated oxidative stress, as the ROS levels were suppressed in the presence of the exogenous iron chelator deferoxamine. In SDHBKD cells, ROS was suppressed by deferoxamine, consistent with the notion that an elevated iron pool in these cells mediates ROS formation (Fig. 2D). Similarly, RNAi-targeting TF lowered the level of ROS, suggesting that the internalization of iron could promote the enhancement of oxidative stress (Fig. 2E). Moreover, incubation of mouse pheochromocytoma cells in high iron media with 500 μmol/L ferric chloride or 500 μmol/L ferrous chloride for 18 hours resulted in increased levels of ROS and higher cytotoxicity, confirming the association between iron overload and disrupted redox homeostasis (Fig. 2F and G; Supplementary Fig. S1M).
Figure 2.
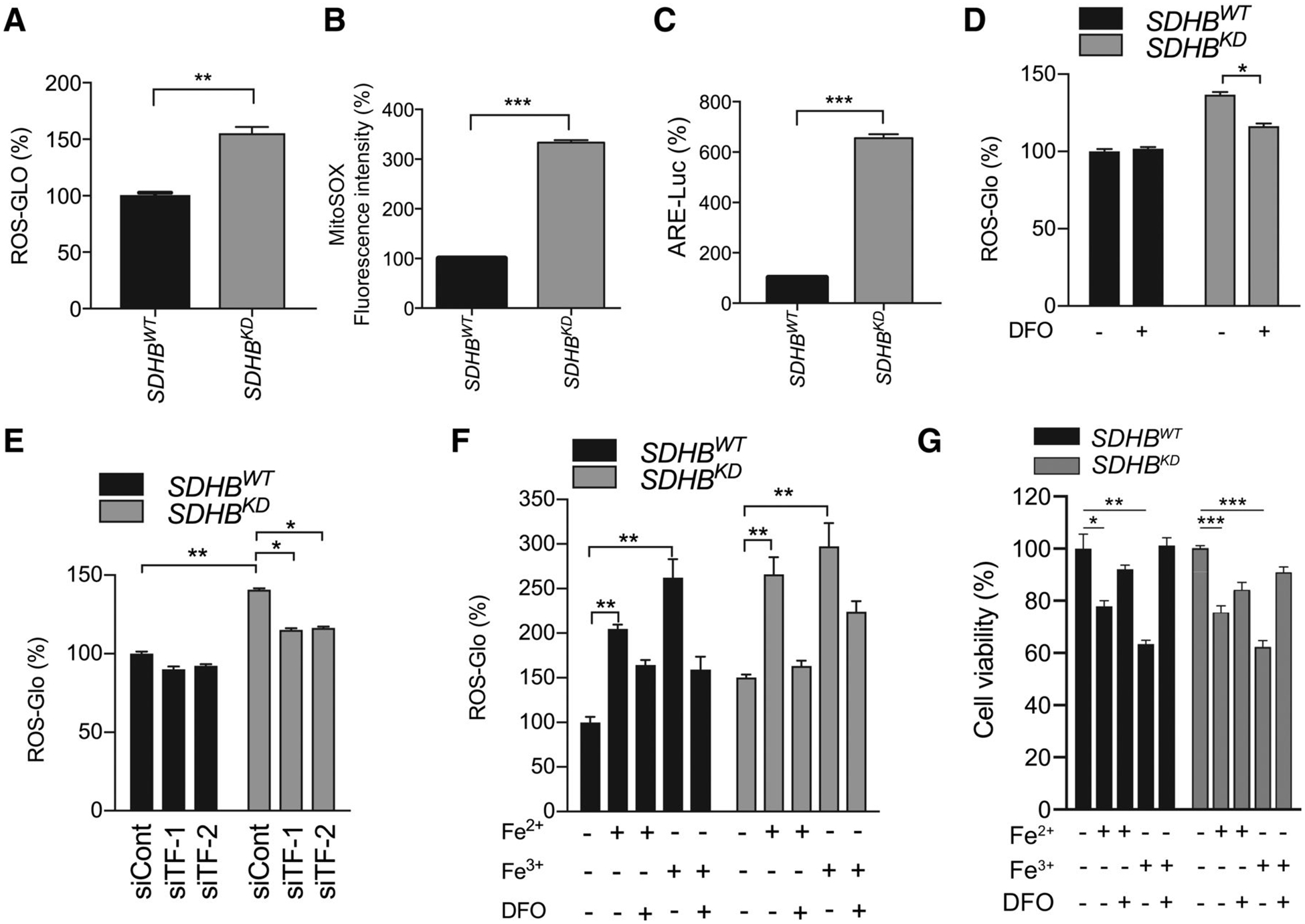
Iron overload leads to high ROS stress in SDHB-deficient PCPG cells. A, Cellular ROS levels in SDHBWT and SDHBKD mouse pheochromocytoma cells were measured by the ROS-Glo H2O2 assay. B, The MitoSOX-Red signal in SDHBWT and SDHBKD mouse pheochromocytoma cells was measured by flow cytometry, and fluorescence intensity was quantified. C, The ARE transcriptional activity in SDHBWT and SDHBKD mouse pheochromocytoma cells was measured by a luciferase reporter assay. D, Cellular ROS levels in SDHBWT and SDHBKD mouse pheochromocytoma cells treated with deferoxamine (DFO) were measured by the ROS-Glo H2O2 assay. Deferoxamine was used as an iron chelator. E, Cellular ROS levels in SDHBWT and SDHBKD mouse pheochromocytoma cells with siRNA-silenced TF was measured by the ROS-Glo H2O2 assay. F, Cellular ROS levels in SDHBWT and SDHBKD mouse pheochromocytoma cells treated with exogenous Fe2+, Fe3+, and deferoxamine were measured by the ROS-Glo H2O2 assay. G, Cell viabilities in SDHBWT and SDHBKD mouse pheochromocytoma cells treated with exogenous Fe2+, Fe3+, and deferoxamine were measured by trypan blue exclusion assay. All significance in differences was determined by t test [*, P < 0.05; **, P < 0.01; ***, P < 0.001 (±SEM)].
Ascorbic acid induces enhanced cytotoxicity in SDHB-deficient cells
Pharmacologic concentrations of ascorbic acid (>1 mmol/L) result in cytotoxicity in cancer cells via the generation of H2O2 (32, 37). We hypothesized that pharmacologic concentrations of ascorbic acid could serve as an antitumor agent for SDHB-mutant PCPGs via synergism with the iron overload found in SDHBKD cells. Findings were that, in SDHBKD cells, ascorbic acid further enhanced ROS levels, which were reversed by NAC or catalase (Fig. 3A). Accumulation of ROS was also reduced by the reexpression of wild-type SDHB (Supplementary Fig. S1N). Consistently, both the ARE luciferase reporter assay and the MitoSOX Red assay showed increased ROS after ascorbic acid treatment in both cell lines, and higher ROS levels in SDHBKD compared with SDHBWT cells (Fig. 3B–D; Supplementary Fig. S1K and S1O). The reduction of ROS levels upon catalase addition indicates that at least some effects of pharmacologic ascorbic acid require the formation of extracellular hydrogen peroxide (37–39). To further probe whether iron metabolism underlies redox imbalance, we supplemented SDHBWT and SDHBKD cells with exogenous iron and ascorbic acid and found that iron enhanced ROS with ascorbic acid (Fig. 3E). Moreover, we found elevated labile iron in both SDHBWT and SDHBKD cells after ascorbic acid treatment, although labile iron in SDHBKD cells was higher than in wild-type cells, and knockdown of TF reduced labile iron (Supplementary Fig. S2A). Deferoxamine treatment significantly reduced the elevation of labile iron (Supplementary Fig. S2B). However, deferoxamine did not reverse ascorbate-induced oxidative DNA damage (Supplementary Fig. S2C and S2D), suggesting additional mechanisms of pharmacologic ascorbate action distinct from those related to iron-associated metabolism.
Figure 3.
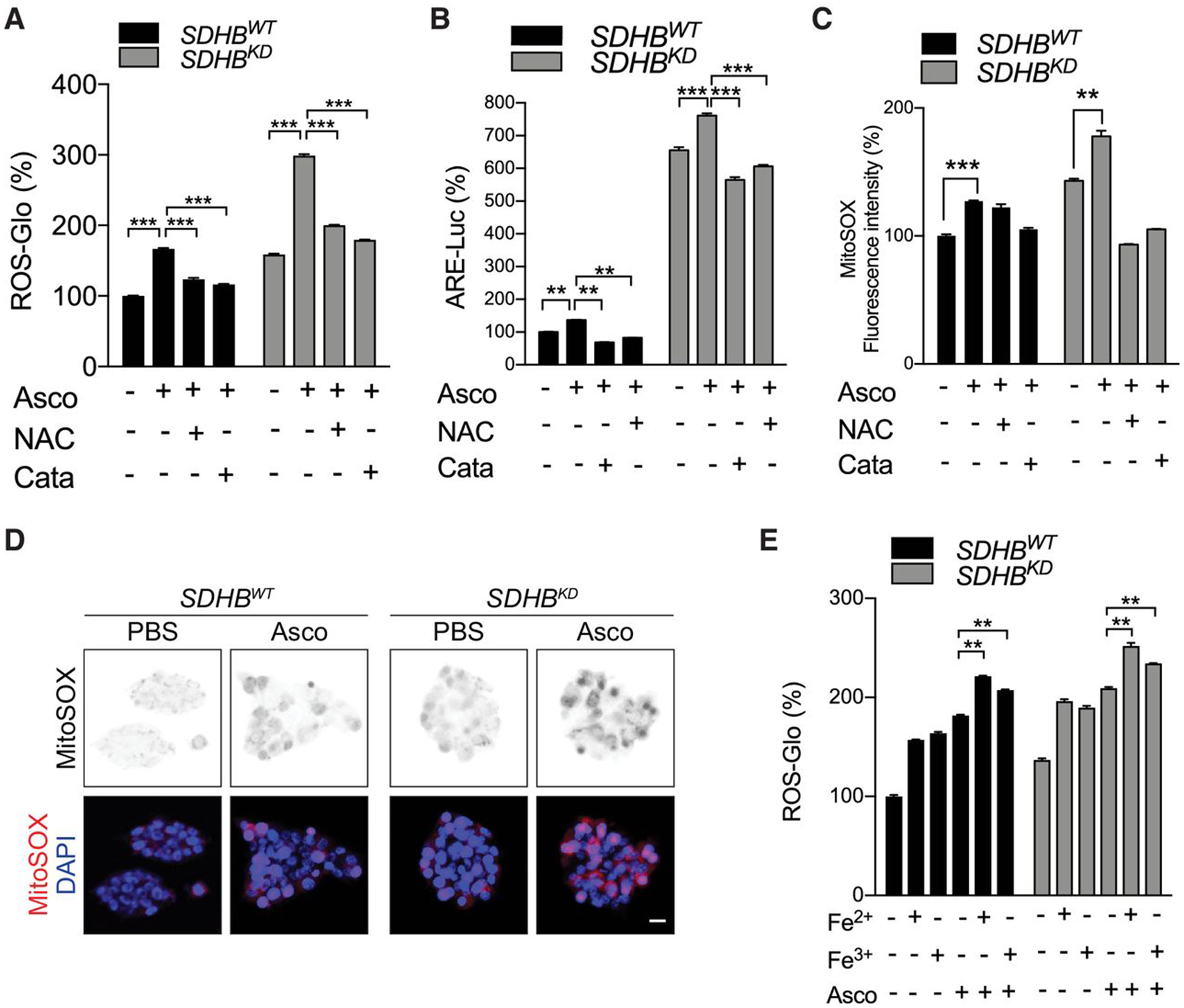
Pharmacologic ascorbic acid induces ROS overload in SDHB-low cells. A, Cellular ROS levels in SDHBWT and SDHBKD mouse pheochromocytoma cells under ascorbic acid treatment were measured by the ROS-Glo H2O2 assay. Exogenous ROS scavengers restored ROS levels. Asco, ascorbic acid; Cata, catalase. Ascorbic acid, catalase, and NAC concentrations in this and other panels were 1 mmol/L, 500 U/mL, and 2.5 mmol/L, respectively. Cells were treated with NAC or catalase for 18 hours. B, The ARE luciferase activity in the presence of ascorbic acid was measured in SDHBWT and SDHBKD mouse pheochromocytoma cells by a reporter assay. Exogenous ROS scavengers restored ARE activity to the normal level. C, The MitoSOX-Red signal was measured in SDHBWT and SDHBKD mouse pheochromocytoma cells under ascorbic acid treatment by flow cytometry. Exogenous ROS scavengers restored the MitoSOX-Red fluorescence signal to normal levels. D, ROS levels were measured in SDHBWT and SDHBKD mouse pheochromocytoma cells under ascorbic acid treatment by MitoSOX-Red staining. Scale bar, 10 μm. E, Cellular ROS levels under exogenous Fe2+, Fe3+, and ascorbic acid treatment were measured in SDHBWT and SDHBKD mouse pheochromocytoma cells by the ROS-Glo H2O2 assay. All significance in differences was determined by t test [**, P < 0.01; ***, P < 0.001 (±SEM)].
To further evaluate whether ascorbic acid is a therapeutic candidate for the treatment of cancers with mutated SDHB, we characterized concentration-dependent cytotoxicity. SDHB deficiency predisposed cells to be sensitive to ascorbic acid–induced cytotoxicity, with IC50 = 1.859 mmol/L (36 pmol/cell) for SDHBWT cells and IC50 = 0.330 mmol/L (6 pmol/cell) for SDHBKD cells (Fig. 4A). Furthermore, we found that ascorbic acid enhanced oxidative damage in SDHBKD cells, as shown by 8-hydroxydeoxyguanosine (8-OH-dG) ELISA and mitochondrial DNA oxidative damage assays (Fig. 4B and C; Supplementary Fig. S2E). Accordingly, DNA electrophoresis and the comet assay revealed increased DNA fragmentation in SDHBKD cells treated with ascorbic acid compared with SDHBWT cells (Fig. 4D–F; Supplementary Fig. S2F). The DNA fragmentation was prevented with catalase, consistent with the formation of hydrogen peroxide as a prerequisite for and initiator of subsequent DNA damage. The ascorbic acid–induced DNA damage was associated with the onset of the apoptotic cascade, as evidenced by the caspase-3/7, Annexin V/PI cell apoptosis assay, and CCK8 cell viability assay (Fig. 4G–J). Furthermore, the clonogenic assay showed that ascorbic acid significantly reduced colony formation of SDHBKD mouse pheochromocytoma cells (Fig. 4K). Moreover, the trypan blue exclusion assay showed reduced cell number of SDHBKD cells compared with SDHBWT counterparts (Fig. 4L), while reexpression of SDHB reversed ascorbic acid–induced cytotoxicity (Supplementary Fig. S2G). ROS scavengers, such as NAC or catalase, largely reversed cytotoxicity induced by ascorbic acid, again suggesting that the genotoxicity is ROS dependent.
Figure 4.
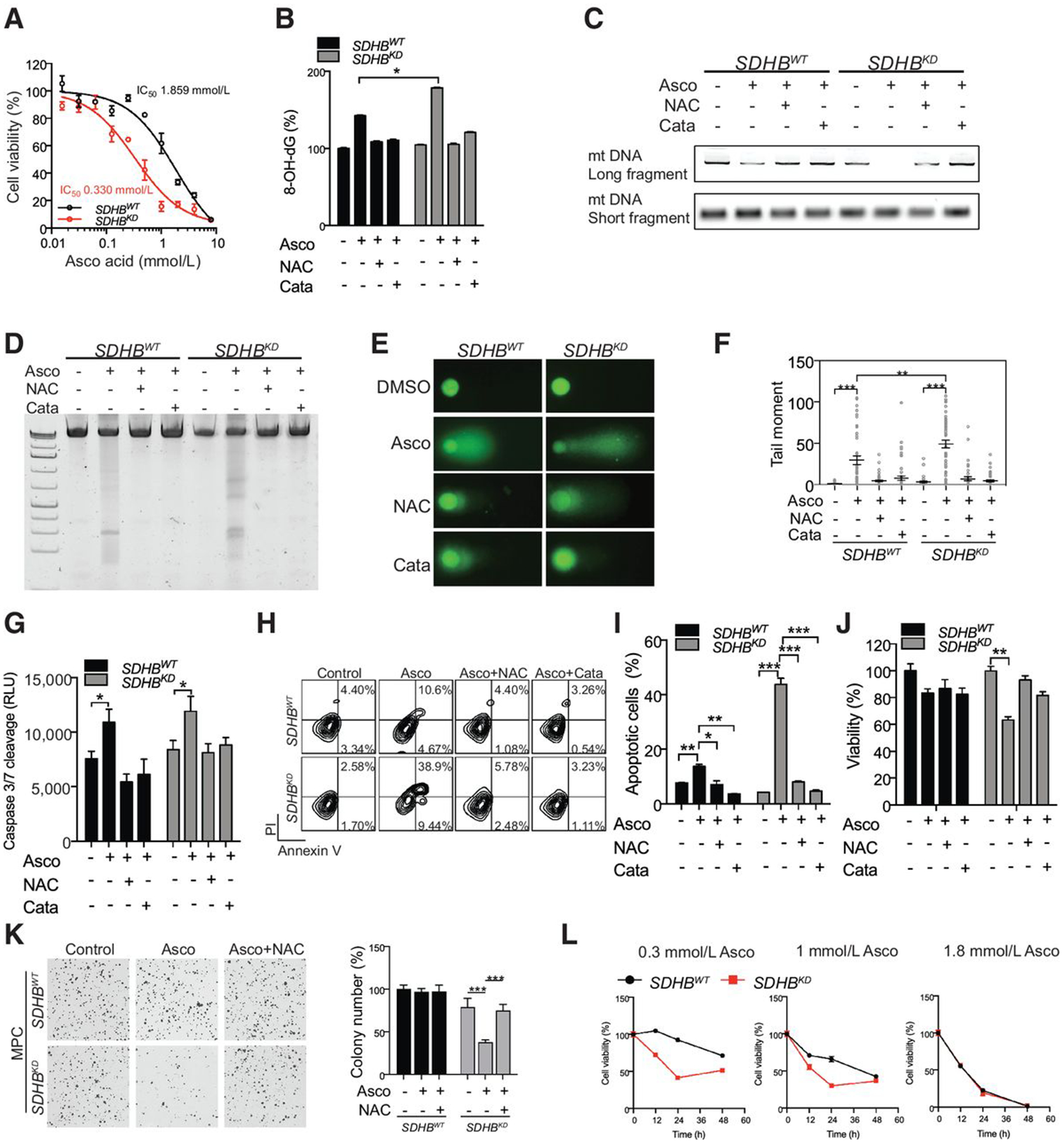
Ascorbic (Asco) acid leads to more oxidative DNA damage and cellular injury in oxidative stress–vulnerable SDHB-low PCPG cells. A, Dose–response curves of SDHBWT and SDHBKD mouse pheochromocytoma (MPC) cells to ascorbic acid treatment were measured by the CCK8 assay. The cells were treated with ascorbic acid for 24 hours. Data were fit for nonlinear regression. B, 8-OH-dG levels in SDHBWT and SDHBKD mouse pheochromocytoma cells after ascorbic acid treatment were measured by an oxidative DNA damage ELISA assay. NAC or catalase (Cata) was supplemented for 18 hours. C, Mitochondrial DNA (mt DNA) PCR assay was performed to measure the amplification of long and short fragments from mitochondrial DNA in SDHBWT and SDHBKD mouse pheochromocytoma cells after ascorbic acid treatment. D, DNA fragmentation in SDHBWT and SDHBKD mouse pheochromocytoma cells after ascorbic acid treatment was measured by electrophoresis. ROS scavengers restored the DNA fragmentation induced by ascorbic acid. E, DNA fragmentation (Comet tail) in SDHBWT and SDHBKD mouse pheochromocytoma cells after ascorbic acid treatment was measured by comet assay. ROS scavengers restored the DNA fragmentation induced by ascorbic acid. F, Quantification of comet assay shown in (E) by measuring tail moment. G, Caspase 3/7 activity in SDHBWT and SDHBKD mouse pheochromocytoma cells after ascorbic acid treatment was measured by a caspase 3/7–Glo assay. H, Apoptosis in SDHBWT and SDHBKD mouse pheochromocytoma cells after ascorbic acid treatment was measured by Annexin V/PI staining and analyzed by flow cytometry. I, Quantification of the fraction of apoptotic cells in (H). J, Cell viability in SDHBWT and SDHBKD mouse pheochromocytoma cells after ascorbic acid treatment was measured by the CCK8 assay. ROS scavengers rescued the cell viability of SDHBKD cells. K, Colony formation assay in SDHBWT and SDHBKD mouse pheochromocytoma cells after ascorbic acid treatment (left) and quantification of colony number (right). L, Trypan blue exclusion cell counting assay in 100,000 SDHBWT and SDHBKD mouse pheochromocytoma cells after three doses of ascorbic acid treatment. 0.3 mmol/L (6 pmol/cell), 1 mmol/L (20 pmol/cell), and 1.8 mmol/L (36 pmol/cell). All significance in differences was determined by t test [*, P < 0.05; **, P < 0.01; ***, P < 0.001 (±SEM)].
Ascorbic acid suppresses SDHBKD metastatic lesions and improves disease outcome
To explore the effects of pharmacologic ascorbic acid administration on SDHB interference in vivo, we established metastatic pheochromocytoma tumors by injecting SDHBWT or SDHBKD mouse pheochromocytoma cells into athymic nude mice (22). Bioluminescence imaging revealed that pharmacologic ascorbic acid (4 g/kg/day) strongly delayed the expansion of the metastatic lesions in the SDHBKD group. Four weeks after treatment, tumor growth was suppressed by approximately 80% in mice with SDHBKD allografts after receiving ascorbic acid (Fig. 5A and B; Supplementary Fig. S3). Importantly, ascorbic acid improved disease outcome of the SDHBKD allograft–bearing mice, with median survival increased by 21.8% (median survival: saline, t = 27.5 days; ascorbic acid, t = 33.5 days; P = 0.0014; Fig. 5C). For further validation, we assessed oxidative damage by measurement of 8-OH-dG in metastatic lesions dissected from the liver. In SDHBKD allograft tissues, increased oxidative damage was found in hepatic lesions from the ascorbic acid–treated mice compared with the saline group (Fig. 5D). Moreover, the histologic analysis showed higher Ki67 expression in the saline group and increased γH2A.X expression in the ascorbic acid treatment group (Fig. 5E and F), indicating elevated DNA damage and cytotoxicity. In the SDHBWT group, ascorbic acid treatment alone did not significantly inhibit tumor growth nor prolong survival. (Fig. 5A–C; Supplementary Fig. S6). No noticeable weight loss or other side effects were observed in the experimental animals.
Figure 5.
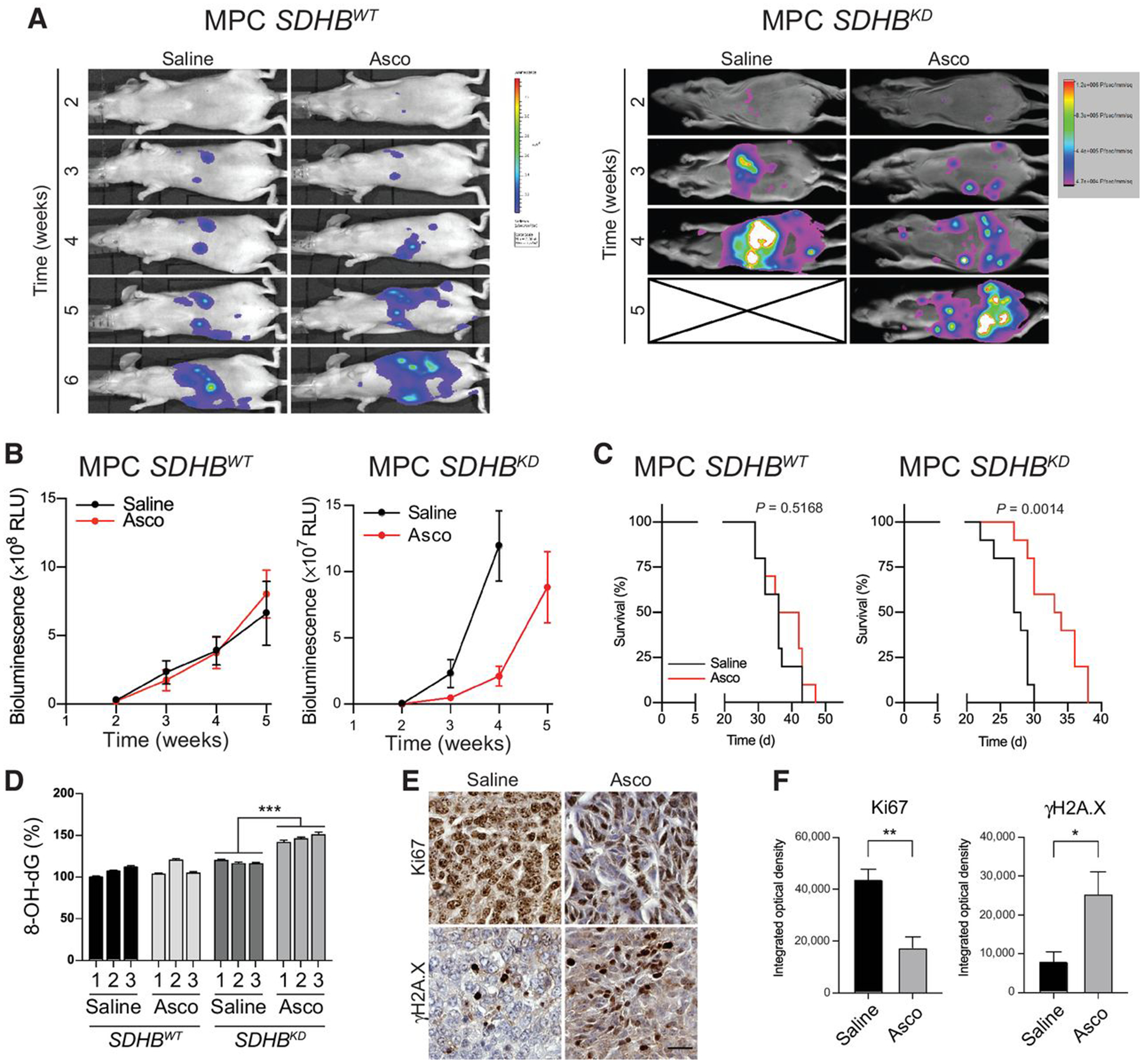
Ascorbic acid delays SDHB-mutated allograft in vivo. A, Luciferase imaging showing representative SDHBWT (n = 10 for each group) and SDHBKD (n = 10 for each group) hepatic lesions in vivo under ascorbic acid treatment. The saline-treated group is shown as the control. Asco, ascorbic acid. B, Quantification of tumor volume shown in (A). C, Kaplan–Meier analysis showing overall survival of tumor-bearing animals under ascorbic acid treatment (SDHBWT, P = 0.5168; SDHBKD, P = 0.0014). D, Levels of oxidative DNA damage in ascorbic acid–treated hepatic lesions from animal models were measured by an 8-OH-dG ELISA assay. Allograft tissues (n = 3 for each group) were analyzed. All significance in differences was determined by one-way ANOVA; ***, P < 0.001 (±SEM). E, Expression of Ki67 and γH2A.X in metastatic lesions under ascorbic acid treatment were measured by an IHC assay (n = 4 for each group). Scale bar, 50 μm. F, Quantification of IHC staining in (E) on the basis of integrated optical density. Significance in differences was determined by t test [*, P < 0.05; ** , P < 0.01 (±SEM)]. MPC, mouse pheochromocytoma.
Discussion
In this study, we demonstrated that cluster I PCPGs with SDHB deficiency exhibited increased expression of iron transport genes, which were induced by activation of the transcriptional factor HIF-α. Increased intracellular iron transport contributed to an increase in the labile iron pool and elevated ROS burden in SDHBKD cells. Pharmacologic levels of ascorbic acid aggravated the oxidative burden of SDHB-deficient cluster I PCPGs, leading to genetic instability and apoptotic cell death. Our study highlights the importance of the labile iron pool and ROS balance in cluster I PCPGs and reveals that targeting the intrinsic redox status could be a potential therapeutic strategy for these tumors (Fig. 6).
Figure 6.
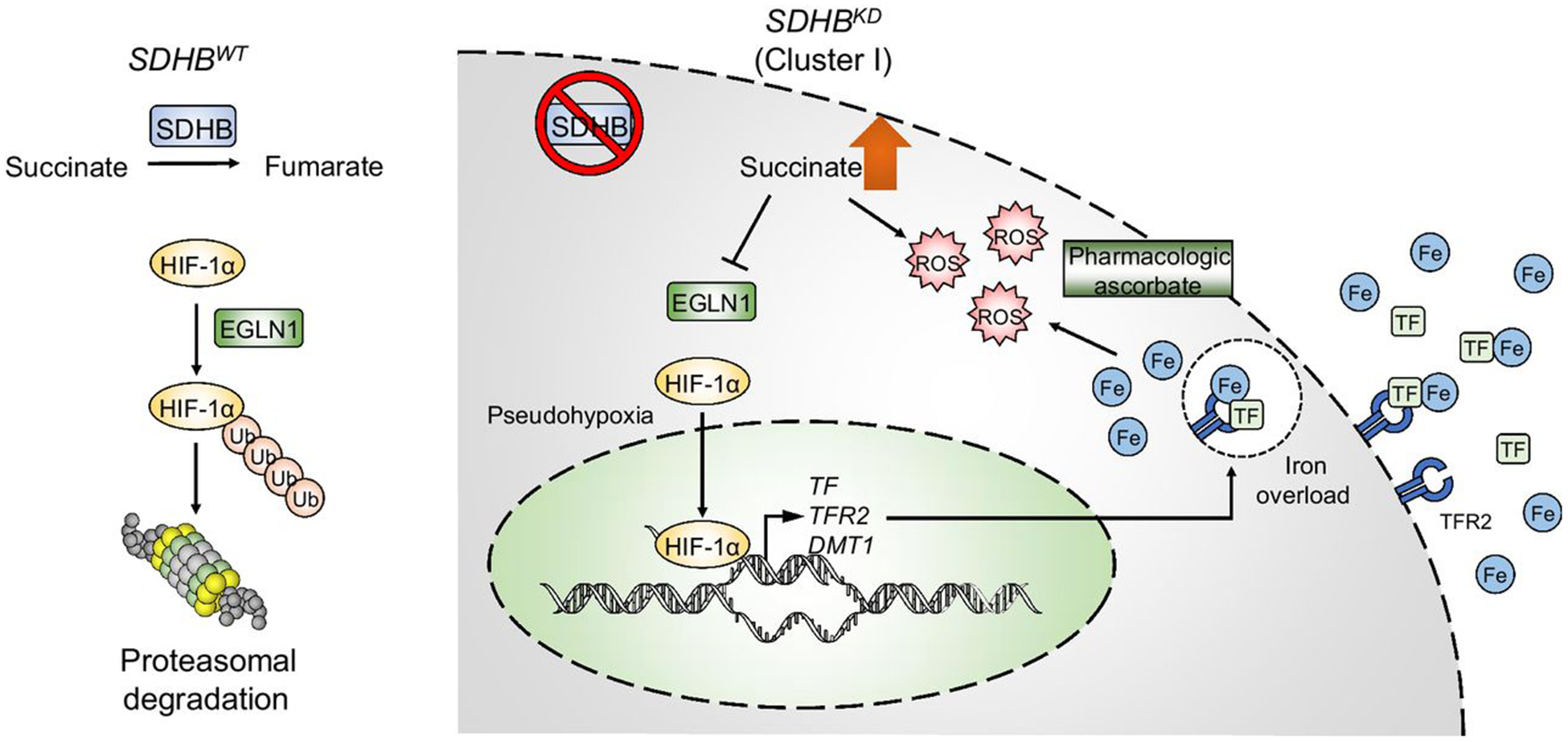
Schematic of the molecular mechanism of the ascorbic acid anticancer effect. SDHB-low PCPGs exhibit an imbalance in intracellular iron levels due to abnormal activation of the HIF signaling pathway, which further leads to increased vulnerability to oxidative stress. Targeting of the ROS synthetic pathway with pharmacologic doses of ascorbic acid could be a valuable therapeutic strategy for cluster I PCPGs with complex II deficiencies.
Pseudohypoxia triggers iron overload in SDHBKD cells
Transcriptomic profiling revealed that cluster I PCPGs exhibit signatures of hypoxia-related gene expression, suggesting activation of HIF-α and increased expression of its downstream genes that regulate angiogenesis, tumor growth, and energy metabolism (40–42). Here, we demonstrate that SDHBKD cells recapitulate the pseudohypoxia phenotype observed in human tumors, evidenced by the upregulation of hypoxia-related genes, such as TF, TFR2, SLC11A2, EPO, PGK1, and LDHA. Importantly, TF, TFR2, and DMT1 are critical components of cellular iron uptake, and the upregulation of these proteins indicates an increased level of iron in SDHBKD cells. It has been reported that as a major iron-binding and -transporting protein, increased level of TF could lead to ferric iron elevation, which could induce ferroptosis, an iron-dependent type of cell death (43). Conversely, a missense mutation at a splice junction of SLC11A2 that impaired iron uptake in erythroid cells and led to iron deficiency–related anemia was discovered in a patient (44). Another study described the overexpression of the DMT1 and TFR proteins in human colonic carcinoma, resulting in increased intracellular iron by means of increased intracellular iron import and reduced iron efflux (45). Besides pseudohypoxia-induced iron uptake, SDHB deficiency results in loss of mitochondrial Fe-S cluster, which contributes to the labile iron elevation and vulnerability to oxidative damage (46). Our data further demonstrate that increased expression of iron transport proteins is a critical component of iron overload–induced by pseudohypoxia in SDHB-disrupted tumor cells.
Iron overload: the sword of Damocles for SDHB-deficient malignancies
As an important part of DNA synthesis, cellular respiration, and redox balance, iron plays a crucial role in cell metabolism and proliferation (47). However, because of the harmful effect of an overload of “free” iron, the labile iron pool is usually maintained at the lowest sufficient level by tight regulation of the expression of proteins that are associated with iron transport and homeostasis (34). High levels in the labile iron pool are often observed during malignant transformation, together with the disruption of cellular redox homeostasis (27). Redox-active iron converts hydrogen peroxide largely to the highly toxic hydroxyl radical via the Fenton/Haber–Weiss reaction cycles (Fe2+ + H2O2 → Fe3+ + HO• + OH−; Fe3+ + H2O2 → Fe2+ + HOO• + H+; ref. 48). Thus, elevated labile iron pool in SDHBKD cells could prime cancer cells for ascorbic acid–induced oxidative damage. In this study, we found high levels of cellular and mitochondrial ROS in SDHBKD cells and depletion of iron by using deferoxamine-reduced oxidative stress in SDHBKD cells, suggesting that ROS accumulation was due to the presence of the intracellular labile iron pool. Genetic silencing of TF reduced the labile iron pool as well as the ROS level indicating a close association between low levels of SDHB, iron overload, and oxidative stress, consistent with reports that showed a correlation between ROS accumulation and SDH dysfunction (49, 50). One study, based on prokaryotic SDH, suggested that electron transport chain dysfunction induced by SDH deficiency was associated with ROS formation (51). Moreover, blocking interception of electrons generated at SDHA by displacement of their acceptor, coenzyme Q, from the membrane part of cluster II by vitamin E succinate and, even more so, its mitochondria-targeted variant caused the massive formation of ROS with ensuing apoptosis induction (52). Our findings show that the increased labile iron pool induced by pseudohypoxia in SDHBKD cells contributes to ROS generation.
Besides iron overload and oxidative cellular damage, other ascorbic acid–associated molecular mechanisms could be relevant for tumor suppression. SDH depletion leads to the accumulation of succinate, which serves as a competitive inhibitor of DNA/histone demethylases (53). Inhibition of the demethylation reactions eventually leads to a genome-wide hypermethylation phenotype in these cancer cells (54). Conversely, ascorbic acid has been shown to remodel the epigenome by enhancing histone demethylases and ten-eleven translocation (TET) protein (55, 56). Using TET as an example, ascorbic acid reduces Fe3+ to Fe2+, which then fuels the demethylation steps from 5-methylcytosine to 5-hydroxymethylcytosine (5hmC), 5hmC to 5-fluorocytosine (5fC), and 5fC to 5-carboxylcytosine (39, 57). Thus, it is possible that the presence of high concentrations of ascorbic acid partially rectifies the hypermethylation phenotype, and therefore limits the aggressive phenotypes during cancer growth, here particularly SDHB-PCPG. Investigation of epigenome reprogramming in SDH-depleted cancers will be of importance to elucidate the molecular mechanisms of ascorbic acid treatment.
Ascorbic acid as a potential therapy for cancer
Ascorbic acid has been shown to act as an anticancer agent for certain advanced tumors, such as pancreatic and ovarian cancer (58, 59). A single-arm pilot trial also revealed that about half of the patients with refractory acute myeloid leukemia or myelodysplastic syndromes exhibited a clinical response to ascorbic acid administration (60). Studies on several types of tumor cells with increased steady-state ROS levels, due to oxidative metabolism defects, and with an increased labile iron pool (61) revealed that pharmacologic doses of ascorbic acid could be used as a potential therapeutic agent by targeting oxidative metabolic pathways (27). It has been suggested that iron-enriched tumors, such as non-Hodgkin lymphomas and renal cell carcinomas, are also good candidates for ascorbic acid trials (38). In this study, ascorbic acid increased ROS to higher levels in SDHBKD cells that had increased levels of iron, indicating ROS-mediated toxicity of ascorbic acid for SDHB PCPGs.
While ascorbic acid at pharmacologic concentrations has few side effects in healthy organs or tissues, it is more toxic to tumor cells with higher prooxidative status (27). Two clinical trial datasets have determined that intravenous administration of ascorbic acid was well tolerated and showed efficacy in advanced and metastatic diseases (33, 59). In healthy tissues, ROS and labile iron pool levels are maintained at a basal level, increased ROS induced by the labile iron pool plus pharmacologic ascorbic acid are rapidly neutralized by elevated removal of hydrogen peroxide or by normal cellular repair mechanisms (39). However, for tumors with higher labile iron pool and ROS levels, it is difficult to eliminate the severely increased ROS induced by ascorbic acid (27).
SDHB-deficient PCPGs are examples of tumor types sensitive to pharmacologic doses of ascorbic acid. Ascorbic acid induces more oxidative DNA damage and efficiently promotes apoptosis in SDHBKD cells, which exhibit a higher ROS stress burden than their SDHBWT counterparts with lower ROS and labile iron pool levels (Fig. 4). Furthermore, ROS scavengers, such as NAC and catalase, reverse the genotoxicity and cytotoxicity in these cells, strongly indicating the central role of ROS in ascorbic acid–induced cell death and of an increased labile iron pool as a critical modulator of elevated ROS generation. Aberrant iron metabolism combined with redox disbalance induced by ascorbic acid, could be a promising therapeutic target for malignancies, especially those with “fragile” redox homeostasis. This approach is particularly appealing, given the safety and low adverse effects of pharmacologic doses of ascorbic acid. Nevertheless, our study shows that the SDHBKD allograft growth was delayed but not eliminated by pharmacologic ascorbic acid. One of our recent findings showed that the activity of nuclear factor erythroid 2–related factor 2 (NRF2) is significantly upregulated in SDH-depleted cells. In accordance with this, many antioxidant genes, such as HMOX1, GCLC, GCLM, and SLC7A11, are highly presented in this disease subtype (62). It is possible that in SDH-depleted cancers, the intrinsic antioxidant pathway is upregulated, which partially counteracts ascorbic acid–induced oxidative damage. Combination treatment with an NRF2/antioxidant pathway inhibitor may be useful to synergize with ascorbic acid and achieve a better disease outcome. SDHB-depleted tumors are generally resistant to standard chemotherapy. Ascorbate here was used as a single treatment agent, both for wild-type and mutant tumors in athymic mice. Other cell and animal studies with highly aggressive and treatment-resistant cancer cell types, that is, pancreatic, ovarian, and glioblastoma, show that ascorbate as a single-treatment agent has some but not full efficacy (32, 63). A combination of pharmacologic ascorbate with other agents has often reported being synergistic (37, 63), including a recent report using combination treatment with pharmacologic ascorbate plus anti–PD-1agent (64).
In summary, we demonstrate that cluster I SDHB-low PCPG cell lines and SDHB-deficient tumors feature an increased labile iron pool induced by the HIF signaling pathway, leading to higher levels of ROS. Disruption of this redox state by ascorbic acid may cause an overwhelming ROS burden, which presents a potential strategy toward this tumor type treatment. The difficult-to-treat PCPGs are typified by a frequency of nonsense SDHB mutations showing a proof of concept for this approach as well as candidates for clinical trials.
Supplementary Material
Translational Relevance.
Readership should be interested in this article because it demonstrates that SDHB-low cluster I pheochromocytoma and paraganglioma (PCPG) cells exhibited pseudohypoxia, which reprogrammed iron homeostasis and consequently resulted in high reactive oxygen species generation. This effect was enhanced by pharmacologic concentrations of ascorbic acid, presenting a new approach for treating these types of cancers. Ascorbic acid is currently a treatment agent in several clinical trials for acute myeloid leukemia, metastatic prostate and colon cancer, advanced non–small cell lung cancer, and glioblastoma. Because pharmacologic ascorbic acid has an excellent clinical safety profile, it is likely that ascorbic acid can be a valuable tool for the treatment of PCPGs as well, for which few therapeutic options currently exist.
Acknowledgments
This research was supported by the Intramural Research Program of the Center for Cancer Research, NCI, the Eunice Kennedy Shriver National Institute of Child Health and Human Development, and the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases. J. Neuzil and K. Hadrava Vanova were supported by the Czech Science Foundation (18-10832S).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
H.K. Ghayee has received royalties from the University of Texas Southwestern Medical Center. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res 2012;44:328–33. [DOI] [PubMed] [Google Scholar]
- 2.Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell 2017;31:181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer 2014;14:108–19. [DOI] [PubMed] [Google Scholar]
- 4.Bezawork-Geleta A, Rohlena J, Dong L, Pacak K, Neuzil J. Mitochondrial complex II: at the crossroads. Trends Biochem Sci 2017;42:312–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013;23:739–52. [DOI] [PubMed] [Google Scholar]
- 6.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005;7:77–85. [DOI] [PubMed] [Google Scholar]
- 7.Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG Jr. Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 2002;1:237–46. [DOI] [PubMed] [Google Scholar]
- 8.Favier J, Briere JJ, Burnichon N, Riviere J, Vescovo L, Benit P, et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One 2009;4:e7094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kluckova K, Tennant DA. Metabolic implications of hypoxia and pseudohypoxia in pheochromocytoma and paraganglioma. Cell Tissue Res 2018;372:367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Favier J, Gimenez-Roqueplo AP. Pheochromocytomas: the (pseudo)-hypoxia hypothesis. Best Pract Res Clin Endocrinol Metab 2010;24:957–68. [DOI] [PubMed] [Google Scholar]
- 11.Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol 2008;28: 718–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korge P, John SA, Calmettes G, Weiss JN. Reactive oxygen species production induced by pore opening in cardiac mitochondria: the role of complex II. J Biol Chem 2017;292:9896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amar L, Baudin E, Burnichon N, Peyrard S, Silvera S, Bertherat J, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab 2007;92: 3822–8. [DOI] [PubMed] [Google Scholar]
- 14.Crona J, Lamarca A, Ghosal S, Welin S, Skogseid B, Pacak K. Genotype-phenotype correlations in pheochromocytoma and paraganglioma. Endocr Relat Cancer 2019;26:539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turkova H, Prodanov T, Maly M, Martucci V, Adams K, Widimsky J Jr, et al. Characteristics and outcomes of metastatic SDHB and sporadic pheochromocytoma/paraganglioma: an National Institutes of Health Study. Endocr Pract 2016;22:302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hescot S, Curras-Freixes M, Deutschbein T, van Berkel A, Vezzosi D, Amar L, et al. Prognosis of malignant pheochromocytoma and paraganglioma (MAPP-Prono Study): a European Network for the Study of Adrenal Tumors Retrospective Study. J Clin Endocrinol Metab 2019;104:2367–74. [DOI] [PubMed] [Google Scholar]
- 17.Fishbein L, Ben-Maimon S, Keefe S, Cengel K, Pryma DA, Loaiza-Bonilla A, et al. SDHB mutation carriers with malignant pheochromocytoma respond better to CVD. Endocr Relat Cancer 2017;24:L51–5. [DOI] [PubMed] [Google Scholar]
- 18.Huang H, Abraham J, Hung E, Averbuch S, Merino M, Steinberg SM, et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer 2008;113:2020–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nolting S, Grossman A, Pacak K. Metastatic phaeochromocytoma: spinning towards more promising treatment options. Exp Clin Endocrinol Diabetes 2019; 127:117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hahn NM, Reckova M, Cheng L, Baldridge LA, Cummings OW, Sweeney CJ. Patient with malignant paraganglioma responding to the multikinase inhibitor sunitinib malate. J Clin Oncol 2009;27:460–3. [DOI] [PubMed] [Google Scholar]
- 21.Jimenez C, Cabanillas ME, Santarpia L, Jonasch E, Kyle KL, Lano EA, et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel-Lindau disease: targeting angiogenic factors in pheochromocytoma and other von Hippel-Lindau disease-related tumors. J Clin Endocrinol Metab 2009;94:386–91. [DOI] [PubMed] [Google Scholar]
- 22.Pang Y, Lu Y, Caisova V, Liu Y, Bullova P, Huynh TT, et al. Targeting NAD (+)/PARP DNA repair pathway as a novel therapeutic approach to SDHB-mutated cluster I pheochromocytoma and paraganglioma. Clin Cancer Res 2018; 24:3423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Powers JF, Evinger MJ, Tsokas P, Bedri S, Alroy J, Shahsavari M, et al. Pheochromocytoma cell lines from heterozygous neurofibromatosis knockout mice. Cell Tissue Res 2000;302:309–20. [DOI] [PubMed] [Google Scholar]
- 24.Ghayee HK, Bhagwandin VJ, Stastny V, Click A, Ding LH, Mizrachi D, et al. Progenitor cell line (hPheo1) derived from a human pheochromocytoma tumor. PLoS One 2013;8:e65624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grossman HB, Wedemeyer G, Ren LQ. Human renal carcinoma: characterization of five new cell lines. J Surg Oncol 1985;28:237–44. [DOI] [PubMed] [Google Scholar]
- 26.Zhuang Z, Frerich JM, Huntoon K, Yang C, Merrill MJ, Abdullaev Z, et al. Tumor derived vasculogenesis in von Hippel-Lindau disease-associated tumors. Sci Rep 2014;4:4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schoenfeld JD, Sibenaller ZA, Mapuskar KA, Wagner BA, Cramer-Morales KL, Furqan M, et al. O2(−) and H2O2-mediated disruption of Fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell 2017;31:487–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y, Lu Y, Celiku O, Li A, Wu Q, Zhou Y, et al. Targeting IDH1-mutated malignancies with NRF2 blockade. J Natl Cancer Inst 2019;111:1033–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang X, Fu X, Liu Y, Yu D, Cai SJ, Yang C. Blockade of glutathione metabolism in IDH1-mutated glioma. Mol Cancer Ther 2020;19:221–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu Y, Liu Y, Yang C. Evaluating in vitro DNA damage using comet assay. J Vis Exp 2017:56450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito H, Fujita K, Tagawa K, Chen X, Homma H, Sasabe T, et al. HMGB1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice. EMBO Mol Med 2015;7:78–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A 2008;105:11105–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffer LJ, Levine M, Assouline S, Melnychuk D, Padayatty SJ, Rosadiuk K, et al. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann Oncol 2008;19:1969–74. [DOI] [PubMed] [Google Scholar]
- 34.Kruszewski M Labile iron pool: the main determinant of cellular response to oxidative stress. Mutat Res 2003;531:81–92. [DOI] [PubMed] [Google Scholar]
- 35.Saxena N, Maio N, Crooks DR, Ricketts CJ, Yang Y, Wei MH, et al. SDHB-deficient cancers: the role of mutations that impair iron sulfur cluster delivery. J Natl Cancer Inst 2016;108:djv287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas C, Mackey MM, Diaz AA, Cox DP. Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress: implications for diseases associated with iron accumulation. Redox Rep 2009; 14:102–8. [DOI] [PubMed] [Google Scholar]
- 37.Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A 2005;102:13604–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levine M, Violet PC. Data triumph at C. Cancer Cell 2017;31:467–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shenoy N, Creagan E, Witzig T, Levine M. Ascorbic acid in cancer treatment: let the phoenix fly. Cancer Cell 2018;34:700–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003;3: 721–32. [DOI] [PubMed] [Google Scholar]
- 41.Ivan M, Kaelin WG Jr. The EGLN-HIF O2-sensing system: multiple inputs and feedbacks. Mol Cell 2017;66:772–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med 2011;365: 537–47. [DOI] [PubMed] [Google Scholar]
- 43.Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis 2016;7:e2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mims MP, Guan Y, Pospisilova D, Priwitzerova M, Indrak K, Ponka P, et al. Identification of a human mutation of DMT1 in a patient with microcytic anemia and iron overload. Blood 2005;105:1337–42. [DOI] [PubMed] [Google Scholar]
- 45.Brookes MJ, Hughes S, Turner FE, Reynolds G, Sharma N, Ismail T, et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut 2006;55:1449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lill R, Hoffmann B, Molik S, Pierik AJ, Rietzschel N, Stehling O, et al. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim Biophys Acta 2012;1823:1491–508. [DOI] [PubMed] [Google Scholar]
- 47.Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol 2014;10:9–17. [DOI] [PubMed] [Google Scholar]
- 48.Lawen A, Lane DJ. Mammalian iron homeostasis in health and disease: uptake, storage, transport, and molecular mechanisms of action. Antioxid Redox Signal 2013;18:2473–507. [DOI] [PubMed] [Google Scholar]
- 49.Ishii T, Yasuda K, Akatsuka A, Hino O, Hartman PS, Ishii N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res 2005;65:203–9. [PubMed] [Google Scholar]
- 50.Szeto SS, Reinke SN, Sykes BD, Lemire BD. Ubiquinone-binding site mutations in the Saccharomyces cerevisia succinate dehydrogenase generate superoxide and lead to the accumulation of succinate. J Biol Chem 2007;282:27518–26. [DOI] [PubMed] [Google Scholar]
- 51.Yankovskaya V, Horsefield R, Tornroth S, Luna-Chavez C, Miyoshi H, Leger C, et al. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 2003;299:700–4. [DOI] [PubMed] [Google Scholar]
- 52.Dong LF, Low P, Dyason JC, Wang XF, Prochazka L, Witting PK, et al. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 2008;27:4324–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sciacovelli M, Frezza C. Oncometabolites: unconventional triggers of oncogenic signalling cascades. Free Radic Biol Med 2016;100:175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pang Y, Liu Y, Pacak K, Yang C. Pheochromocytomas and paragangliomas: from genetic diversity to targeted therapies. Cancers 2019;11:pii: E436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang M, Pollard PJ. Succinate: a new epigenetic hacker. Cancer Cell 2013;23: 709–11. [DOI] [PubMed] [Google Scholar]
- 56.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 2012; 26:1326–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Young JI, Zuchner S, Wang G. Regulation of the epigenome by vitamin C. Annu Rev Nutr 2015;35:545–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Drisko JA, Chapman J, Hunter VJ. The use of antioxidants with first-line chemotherapy in two cases of ovarian cancer. J Am Coll Nutr 2003;22:118–23. [DOI] [PubMed] [Google Scholar]
- 59.Monti DA, Mitchell E, Bazzan AJ, Littman S, Zabrecky G, Yeo CJ, et al. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS One 2012;7:e29794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park CH, Kimler BF, Yi SY, Park SH, Kim K, Jung CW, et al. Depletion of L-ascorbic acid alternating with its supplementation in the treatment of patients with acute myeloid leukemia or myelodysplastic syndromes. Eur J Haematol 2009;83:108–18. [DOI] [PubMed] [Google Scholar]
- 61.Caltagirone A, Weiss G, Pantopoulos K. Modulation of cellular iron metabolism by hydrogen peroxide. Effects of H2O2 on the expression and function of iron-responsive element-containing mRNAs in B6 fibroblasts. J Biol Chem 2001;276: 19738–45. [DOI] [PubMed] [Google Scholar]
- 62.Liu Y, Pang Y, Caisova V, Ding J, Yu D, Zhou Y, et al. Targeting NRF2-governed glutathione synthesis for SDHB-mutated pheochromocytoma and paraganglioma. Cancers 2020;12:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Padayatty SJ, Sun AY, Chen Q, Espey MG, Drisko J, Levine M. Vitamin C: intravenous use by complementary and alternative medicine practitioners and adverse effects. PLoS One 2010;5:e11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luchtel RA, Bhagat T, Pradhan K, Jacobs WR Jr, Levine M, Verma A, et al. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc Natl Acad Sci U S A 2020;117:1666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.