

Abstract
Treatment of leukemia has become much difficult because of resistance to the existing anticancer therapies. This has thus expedited the search for alternativ therapies, and one of these is the exploitation of mesenchymal stem cells (MSCs) towards control of tumor cells. The present study investigated the effect of human umbilical cord-derived MSCs (UC-MSCs) on the proliferation of leukemic cells and gauged the transcriptomic modulation and the signaling pathways potentially affected by UC-MSCs. The inhibition of growth of leukemic tumor cell lines was assessed by proliferation assays, apoptosis and cell cycle analysis. BV173 and HL-60 cells were further analyzed using microarray gene expression profiling. The microarray results were validated by RT-qPCR and western blot assay for the corresponding expression of genes and proteins. The UC-MSCs attenuated leukemic cell viability and proliferation in a dose-dependent manner without inducing apoptosis. Cell cycle analysis revealed that the growth of tumor cells was arrested at the G0/G1 phase. The microarray results identified that HL-60 and BV173 share 35 differentially expressed genes (DEGs) (same expression direction) in the presence of UC-MSCs. In silico analysis of these selected DEGs indicated a significant influence in the cell cycle and cell cycle-related biological processes and signaling pathways. Among these, the expression of DBF4, MDM2, CCNE2, CDK6, CDKN1A, and CDKN2A was implicated in six different signaling pathways that play a pivotal role in the anti-tumorigenic activity exerted by UC-MSCs. The UC-MSCs perturbate the cell cycle process of leukemic cells via dysregulation of tumor suppressor and oncogene expression.
Keywords: leukemia, mesenchymal stem cells, cell cycle, gene expression profiling
Introduction
Leukemia is a hematopoietic malignancy, categorized into acute and chronic subtypes, of myeloid and lymphoid progenitor cells1. Over the last few decades, the treatment strategy for leukemia has been unchanged. Conventional therapies are not only associated with relapse and the acquisition of a chemotherapy resistance phenotype of leukemia cells, but also cause adverse systemic effects such as pain, anxiety, and depression which can affect the quality of life2. Research by Ayala et al. showed that the tumor microenvironment such as bone marrow stroma, and especially the hematopoietic stem cell niche, can provide a permissive environment for leukemogenesis and tumor progression3. Therefore, targeting leukemic tumor cells in the hematopoietic stem cell niche by chemotherapy, radiotherapy, and bone marrow transplantation is difficult4, hence necessitating a newer and efficient innovative approach.
Mesenchymal stem cells (MSCs) are non-hematopoietic cells which can differentiate into mesodermal lineage5. Bone marrow (BM) MSCs are the most common and well-characterized MSCs and have been extensively studied. However, the invasive and painful sample harvesting procedures, and loss of the cellular fraction in BM of aged individuals, which impairs the quantity and quality of in vitro expanded MSCs, make BM an unfavorable source for MSCs6. Therefore, alternative sources of MSCs with a multipotent phenotype and characteristics similar to BM-MSCs are actively being investigated. The Wharton’s Jelly of human umbilical cord-derived MSCs (UC-MSCs) served as an ideal substitute for BM-MSCs7, and thus the present study utilized UC-MSCs to assess the effects on the growth of leukemia cells.
The use of MSCs in clinical applications of leukemia treatment is still not conclusive, as presently only nine studies are available in the clinical trial portal of NIH, USA, when searching for leukemia and MSCs8. Most of the clinical trials employed MSCs as supportive therapy to enhance the engraftment of hematopoietic stem cells or to leverage the graft-versus-host disease that emerged due to allogeneic transplantation of hematopoietic stem cells. Although data regarding the direct use of MSCs in eradicating leukemic cells in clinical trials are not available, much laboratory evidence and numerous preclinical studies substantially support the anti-leukemic activity of MSCs. In the past, accumulated evidence has shown that MSCs are implicated in hematopoietic and non-hematopoietic cell proliferation, including leukemia cells in both in vitro and in vivo models9. For instance, several studies have shown that MSCs can support the growth and differentiation of normal hematopoietic stem cells10,11.
On the other hand, MSCs exerted a profound inhibitory activity on highly proliferating cells such as leukemic tumor cells9,12. As compared with solid tumors, there is less known regarding the function of MSCs toward hematological malignancies such as leukemia and multiple myeloma. Lee et al. stated that out of 42 clinical trial investigations deciphering the role of MSCs in tumors, only 13 precisely targeted hematologic malignancies such as leukemia, myelodysplastic syndrome, lymphoma, and multiple myeloma13. Besides the substantial evidence of anti-leukemic activity of MSCs, the underlying molecular mechanisms that govern the signaling pathways in mediating this effect are not fully understood. Therefore, the present study was designed to find new clues that may lead to the development of novel therapeutic approaches with high efficacy and fewer side effects. In the present study, the anti-tumorigenic activity of human UC-MSCs was tested on leukemic tumor cells, along with in silico analysis in which the molecular mechanisms involved in the biological processes and signaling pathways were predicted from the microarray gene expression profiling of leukemic tumor cells in the presence of UC-MSCs.
Materials and Methods
Generation and Expansion of UC-MSCs
Umbilical cord MSCs were derived from Wharton’s Jelly of the human umbilical cord (n=7). The human umbilical cords were obtained from full-term deliveries at the Britannia Women and Child Hospital, Kajang, Selangor, Malaysia, after obtaining written consent from the parents. The collection and use of human umbilical cord tissues was approved by the Ethical and Research Committee, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia. After removal of blood vessels, Wharton’s Jelly tissues were subjected to an in-house combined enzymatic-mechanical dissociation method14. The single cells obtained from Wharton’s Jelly were cultured in complete DMEM/F12 [Dulbecco’s Modified Eagle’s Medium with nutrient mixture F-12 (HAM) [1:1], (GIBCO, InvitrogenTM, Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Stem Cell Technology, Vancouver, Canada), 1% Penicillin/Streptomycin (GIBCO), 0.5% Fungizone Amphotericin (GIBCO) and 0.1% Gentamycin (GIBCO) and incubated in a humidified incubator at 37°C in a 5% CO2 atmosphere. Adherent cells from passage three onwards were employed for characterization, and upon characterization UC-MSCs up to passage seven were used in subsequent experiments.
Leukemic Tumor Cell Culture
Four different cell lines, namely K562, Jurkat, BV173, and HL-60, were used as leukemic tumor cells. These suspension cells were derived from chronic myeloid leukemia (CML) (BV173 and K562)15,16, acute promyelocytic leukemia (APL) (HL-60)17, and acute lymphoblastic leukemia (ALL) (Jurkat)18. All human leukemic cell lines were purchased from ATCC and cultured in complete RPMI 1640 (Roswell Park Memorial Institute medium) (GIBCO) supplemented with 10% heat-inactivated FBS (GIBCO) and 1% Penicillin/Streptomycin and 0.5% Fungizone Amphotericin and 0.1% Gentamycin, and incubated at 37°C in a 5% CO2 humidified cell culture incubator.
Cell Viability and Proliferation Assay
Cell viability was assessed by MTS assay CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay kit (Promega, Madison, WI, USA), while cell proliferation was measured using tritiated thymidine (3H-TdR) incorporation assay. Various ratio of tumor cells with UC-MSCs (1:0.2, 1:0.1, 1:0.02, and 1:0.01) were co-cultured with a fixed number of leukemic tumor cell lines (0.5 × 105 cells/well) in 96-well plates in complete RPMI 1640 medium for 3 days. For the viability assay, 30 µl of MTS mixed with PMS (phenazine methosulfate) was added to each well and incubated for 3–4 h. The absorbance values of tumor cells alone and tumor cells in co-culture with UC-MSCs were measured by using a microtiter plate reader (Dynex Technologies, MRX II, Chantilly, VA, USA) at 490 nm. For the tritiated thymidine (3H-TdR) assay, 3H-TdR (0.037 MBq/well [0.5 µCi/well]) (Perkin Elmer, Waltham, MA, USA) was pulsed to the cells at the final 18 h of incubation. At 72 h, cells were harvested using an automated cell harvester (Harvester 96 MACH III M, TOMTEC, Hamden, CT, USA) onto glass fiber filter mats (Perkin Elmer, Waltham, MA, USA). After drying the filter mats, scintillation fluid was added, and the DNA content was measured by thymidine incorporation using the beta counter (WALLAC Micro Beta Trilux, Perkin Elmer, Waltham, MA, USA).
Transwell Assay
A transwell assay was employed to decipher the mode of UC-MSCs-mediated antiproliferation on leukemic tumor cells. In this experiment, UC-MSCs (0.5 × 105 cells/ml) were seeded in the upper chamber of transwell insert with a 0.1 micron pore size of the membrane (BD Falcon Cell Culture Insert, BD Bioscience, San Jose, CA, USA) in a 12-well plate for 24 h in complete D-MEM/F12 media. Upon removal of non-adherent UC-MSCs, 0.5 × 106 cells/ml of the leukemic cell line (1:0.1=Tumor cells: UC-MSCs) were added in the lower chamber and incubated for 2 days in complete RPMI 1640 medium. After 2 days, 200 µl of the leukemic tumor cell suspension (approximately 0.1 × 106) were re-plated in 96-well plates, and cells were pulsed with 3H-TdR at the final 18 h of incubation prior to being assessed for cell proliferation.
Apoptosis Assay
The apoptosis assay was performed by using the Annexin V-FITC Apoptosis Detection Kit (Merck, Darmstadt, Germany). Annexin V is a member of the Annexin family that binds to phosphatidylserine, an intracellular leaflet of the cell membrane. During the early phase of apoptosis, there is loss of integrity of the cell membrane, which leads to the translocation of phosphatidylserine from the inner surface to the outside of cells. Annexin V has high affinity toward phosphatidylserine, and can be used to target and identify apoptotic cells. However, the binding of Annexin V alone is not sufficient to differentiate apoptotic and necrotic cells. In this regards, propidium iodide (PI), a fluorescent dye that passes the disintegrated cell membrane and binds to DNA, specifically stains necrotic cells. Leukemic tumor cells (106 cells/ml) were co-cultured with UC-MSCs at 1:0.1 ration at different time points (24, 48, and 72 h). After incubation, the leukemic tumor cells were washed, re-suspended in 500 µl of isotonic binding buffer, and incubated with 5 µl Annexin V-FITC and PI for 15 min. Finally, the leukemic cells were analyzed by flow cytometer BD LSR FORTESATM CELL ANALYZER (BD Biosciences, San Jose, CA, USA), and 104 events were acquired and analyzed using Becton Dickinson FACSDiva™ software.
Cell Cycle Assay
The DNA content of leukemic tumor cells that in co-culture with UC-MSCs (1:0.1 ratio) was determined by PI dye. Leukemic tumor cells were washed and fixed in 90% ethanol at -20°C for overnight. The fixed cells were washed with 1% bovine serum albumin (BSA) (AmrescoTM) and incubated in 50 µg/ml RNAase (InvitrogenTM, Carlsbad, CA, USA) with 450 µl BSA. The cells were left at room temperature for 30 minutes in a dark environment with cell cycle staining buffer consists of 50 µg/ml PI (InvitrogenTM). The DNA content was measured by the acquisition of 104 PI-labeled cells using flow cytometer. The acquired cells were subjected to the doublet discrimination, and the data obtained were analyzed using FCS Express 5 software (De Novo Software).
RNA Extraction and Microarray Gene Expression Profiling
For the microarray experiment, 1 × 106 leukemic tumor cells (BV173 and HL-60) were either cultured alone (control group) or co-cultured with 1 × 105 UC-MSCs (test group) in a 10 cm Petri dish. Total RNA of HL-60 and BV173 cell lines was extracted using the RNeasy Plus Mini kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s instructions. The integrity and quality of the extracted RNA were evaluated by Agilent 2100 bioanalyzer (Agilent Technologies, in Molecular Genomics Pte Ltd, Singapore) and Nanodrop ND-1000 spectrophotometer (Thermo Scientific, Waltham, MA, USA), respectively. The extracted RNA was labeled with Low Input Quick Amp Labeling Kit, One-Color (Agilent Technologies, CA, USA), and then hybridized to the Agilent SurePrint G3 Human 1 Color GE 8×60 K Microarray chip (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s instruction. All microarray experiments and raw data analysis were conducted at the Microarray Core Facility (Molecular Genomics Pte. Ltd. in Singapore). Only genes with raw p-values less than 0.05 and fold change more than 2 were considered as differentially expressed genes (DEGs). Data were analyzed by using GeneSpring 13 software (Agilent Technologies) for DEG clustering followed by DAVID online bioinformatics tool (Database for Annotation, Visualization and Integrated Discovery v6.8) for gene ontology (GO) analysis and KEGG (Kyoto Encyclopaedia of Genes and Genomes) pathways analysis. The raw data were deposited in the NCBI Gene Expression Omnibus (GEO) database (accession number: GSE116677).
Quantitative Real-Time PCR (qRT-PCR)
The expressions of selected genes from the microarray results were validated by qRT-PCR. Six DEGs from BV173 and HL-60 with the same experimental design were selected based on either having the same expression directions and/or two different expression directions (MDM2, CCNE2, MYL9, TGFβI, CDKN1A, and CDKN2A) to confirm the fold difference. The extracted RNA (1 µg) was converted to cDNA using the Roche® cDNA Synthesis Kit (Roche, Basel, Switzerland) according to the manufacturer’s protocol. Primer pairs for the selected genes were designed by First Base Company (First Base, Kuala Lumpur, Malaysia) (see additional file 1 for the list of primers). The qRT-PCR reaction was run using Light Cycler 480 DNA SYBR Green I Master by Roche (Roche, Basel, Switzerland). Triplicate PCR reactions for each sample (BV173 and HL-60 cells) were conducted in 96-well PCR plates in 20 µl final reaction volume as recommended by the manufacturer’s protocol. The relative fold change in RNA expression was analyzed using the 2−ΔΔ C T method, where the average of ΔC T values for the amplicon of interest was normalized to that of two endogenous gene TATA box binding protein (TBP) and actin beta (ACTB), compared with control specimens19.
Western Blot Analysis
Western blot analysis was conducted to validate the microarray results. Tumor cells (HL-60 cells, 106 cell/ml), were cultured with UC-MSCs (1:0.1 ration) or alone in 6-well plates. After 72 h of incubation, the HL-60 cell suspensions were collected, washed, and lysed with 100 µl lysis buffer (125 mM Tris-HCL, PH 6.8, 4% SDS, 20% Glycerol, 200 mM DL-Dithiothreitol) (Promega, Madison, WI, USA). Proteins concentration was determined by Bradford reagent (Sigma-Aldrich, St. Louis, MO, USA) using a microtiter plate reader (Dynex Technologies, MRX II, Chantilly, VA, USA) at 595 nm wavelength. The samples were electrophoresed in sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto polyvinylidene fluoride (PVDF) membrane (ThermoFisher Scientific, Waltham, MA, USA); the membrane was then incubated in blocking buffer with 5% BSA (AmrescoTM, Solon, OH, USA) for 1 h at room temperature on an orbital shaker. Then, the membrane was washed and incubated with the respective primary antibodies (see additional file 2) at 4°C overnight with gentle agitation. The membranes were washed with TBS-Tween 20 (TBS/T) and incubated with horseradish peroxidase-conjugated secondary antibody (1:1000 dilution, Cell Signaling Technology, USA) for 1 h at room temperature. Finally, after washing, 150 µl of chemiluminescence (SuperSignal West Femto Maximum Sensitivity Substrate; Thermo Fisher Scientific, Rockford, IL, USA) was applied to the membrane and incubated for 5 min in darkness before viewing using the Alpha Imager (Alpha Innotech, San Leandro, CA, USA).
Statistical Analysis
The results obtained from treated cells at different ratio of UC-MSCs were compared with those of untreated controls by Student’s t-test using GraphPad Prism (Version 7). Furthermore, statistical analysis of microarray data was performed by GeneSpring 13 software (Agilent Technologies, Santa Clara, CA, USA). Identification of genes differentially expressed in microarray analysis was carried out by Student t-test followed by Benjamini–Hochberg correction. Statistical significance was determined at p<0.05.
Results
Umbilical Cord-derived MSCs Inhibit the Leukemic Tumor Cells Proliferation Through Cell-to-Cell Contact
The suspendad leukemic tumor cells were co-cultured with the successfully isolated and fully characterized adherent MSCs from the human umbilical cord (see additional file 3) at different ratios (1:0.2, 1:0.1, 1:0.02, and 1:0.01) to determine the anti-proliferative effect of UC-MSCs. After 72 h of incubation, the viability and proliferation of leukemic tumor cells were evaluated by MTS assay and 3H-TdR incorporation, respectively. The results obtained showed that UC-MSCs reduced the viability and proliferation of the leukemic tumor cells in a dose-dependent manner. The anti-proliferative activity of UC-MSCs is profound against the BV173 cell line, and the robustness of inhibition gradually declines from HL-60, K562, and Jurkat cells onwards (Fig. 1). The BV173 and HL-60 cells recorded a significant inhibition at a ratio up to 1:0.1, and the ratio of 1:0.2 scored the highest inhibition (more than 60% inhibition). The cell ratio that yielded the optimal anti-proliferative effect (1:0.1) in the co-culture assay was adopted and employed in subsequent experiments. In order to decipher the mode of USC-MSCs-mediated antiproliferation, tumor cells were either co-cultured in direct contact with UC-MSCs, or they were physically separated by a transwell insert, where UC-MSCs were seeded onto the upper chamber of a transwell system while tumor cells were added to the lower chamber. The cells were also cultured with the UC-MSCs-conditioned medium (SN) at 1:1 ratio, v/v of culture media and MSCs-conditioned media. The most significant anti-proliferative effect was noted when UC-MSCs were in physical contact with tumor cells. A marginal inhibition of leukemic tumor cells was observed in the transwell system, which was neither significant nor equivalent to the co-culture system. Likewise, UC-MSCs-conditioned medium had also failed to prevent the proliferation of tumor cell growth (Fig. 1c).
Figure 1.

Umbilical cord-derived MSCs inhibit the proliferation of leukemic tumor cells through cell-to-cell contact. Tumor cells at 0.5 × 105 cells/well of 96-well plates were cultured in the presence or absence of UC-MSC at various ratios for 3 days. Cell viability and proliferation were assessed by MTS assay (Panel a) and 3H-TdR incorporation (Panel b) during the final 4 and 18 h of culture, respectively. The results represent the mean of at least four experiments ±SD. Panel (c) shows the tumor cells were cultured alone (Tumor cell), in direct contact with UC-MSC (Tumor+UC-MSCs), with UC-MSCs separated by Transwell (Transwell+Tumor) and in supernatant aspirated from UC-MSCs culture (SN+Tumor). Data show results from more than three independent experiments with mean ±SD. [*] statistically significant at p<0.05.
Anti-proliferative Effect of UC-MSCs Surpasses Apoptosis and Prevents the Tumor Cells from Entering into S or G2/M Phase of the Cell Cycle
The apoptosis assay was utilized to decipher the mechanism of the anti-proliferative activity of UC-MSCs on leukemic tumor cells. The apoptosis results suggested that the anti-proliferative effect of UC-MSCs on leukemic tumor cells at 1:0.1 ratio is not due to apoptosis induction. The early phase of apoptosis is indicated by positive staining of Annexin V, molecules that flip out from the internal surface of the cell membrane to the outer surface during apoptosis, and the lack of PI stain that binds to accessible DNAs. Although tumor cells displayed a residual level of early apoptosis in culture in vitro, supplementation of UC-MSCs neither escalated nor reduced the early apoptosis. However, a small decline in early apoptosis was observed in tumor cells co-cultured with UC-MSCs, but without statistical significance (Fig. 2a).
Figure 2.

Anti-proliferative effect of UC-MSCs surpasses apoptosis and prevent the tumor cells from entering into S or G2/M phase of the cell cycle. One million leukemic tumor cells were co-cultured in presence or absence of UC-MSC at a 1:01 ratio for 24, 48 and 72 h. After incubation, cells were analyzed for apoptotic and necrotic markers, Annexin V and PI, respectively, using by flow cytometry. Data showed results from three different experiments ±SD (Panel a). For cell cycle analysis, 106 cells/ml of leukemic tumor cells were co-cultured with or without of UC-MSC in 12-well plates. At day 3 the cells were collected and stained with PI and assessed by flow cytometry (Panel b).
As apoptosis does not account for the UC-MSCs-mediated anti-proliferative activity, other possible physiological activities of tumor cells that could be potentially affected by UC-MSCs were further investigated. One of the candidate mechanisms is the cell cycle progression of tumor cells. Fig. 2b indicates that DNA synthesis decreased in all leukemic tumor cell lines in the presence of UC-MSCs. Furthermore, the results showed that UC-MSCs-induced cell cycle arrest in both BV173 and HL-60 cell lines was confined to the G0/G1 phase of the cell cycle. Further analysis showed that K562 and Jurkat cells were mostly accumulated in G0/G1, albeit to a lesser degree compared with BV173 and HL-60 cells. Moreover, BV173, K562, and Jurkat cells were also prevented from entry to the G2/M phase of the cell cycle (Fig. 2b).
Microarray Results Analysis
The microarray analysis was conducted using BV173 and HL-60 cell lines as UC-MSCs profoundly inhibited these cell lines. The extracted RNA after passing the prerequisite quality and quantity checks was used for the gene profiling. The results were analyzed using GeneSpring 13 software, and DEGs were filtered by fold change of more than two along with a p-value <0.05. The microarray analysis showed approximately 563 and 3933 probes were altered at least two-fold in HL-60 and BV173 by UC-MSCs as compared with the control, respectively. It was found that, out of 563 probes, 61 were duplicated in HL-60 cells, and after subtraction, the remaining number was 502 DEGs. Further analysis revealed that 411 and 91 genes were down and up-regulated, respectively, in HL-60 cells when in culture with UC-MSCs. In parallel with this, analysis of BV173 cell line data indicated that out of 3933 probes, 914 probes were duplicated. Moreover, out of the remaining 3019 genes, 1019 and 2000 DEGs were down and up-regulated, respectively, in BV173 in the presence of UC-MSCs as compared with the control. Besides this, further analysis revealed that BV173 and HL-60 cell lines shared 276 DEGs in which 35 (see additional file 4) of 276 genes were dysregulated in the same direction with different levels of expression, while 241 DEGs were dysregulated in two different directions. Furthermore, the results highlighted that most of 35 DEGs are involved in the cell cycle, DNA damage, and cell proliferation processes.
Gene Ontology Analysis
GO was performed to identify the potentially altered biological processes in HL-60 and BV173 cell lines by UC-MSCs. For this, DEGs with 2-fold change and p-value <0.05 as cut off were used through the DAVID online bioinformatics tool. The 35 common DEGs were selected for further GO analysis. The GO analysis shows that 31 DEGs were known by DAVID bioinformatics tools and analyzed, while four genes were not annotated or had limited biological information, and these were removed from the list. Approximately 14 significant biological functions were identified based on p-value (p<0.05) (Table 1), which are essential processes in cancer development. Further analysis revealed that several DEGs are tumor suppressor genes (THY1, FSTL1, DCN) and proto-oncogenes (MDM2, PSME3, MYBL2) which were up and down-regulated in both cell lines in the presence of UC-MSCs, respectively.
Table 1.
The List of 14 Significant Biological Processes.
| Biological Process | Count | p-value |
|---|---|---|
| G1/S Transition of Mitotic Cell Cycle | 5 | 3.10E-05 |
| Negative Regulation of Protein Kinase Activity | 4 | 7.20E-04 |
| Cell Cycle Arrest | 4 | 2.00E-03 |
| Negative Regulation of Cell Proliferation | 5 | 5.10E-03 |
| DNA Damage Response, Signal Transduction by P53 Class Mediator Resulting in Cell Cycle Arrest | 3 | 5.50E-03 |
| Negative Regulation of Monocyte Differentiation | 2 | 1.20E-02 |
| Regulation of Cell Cycle | 3 | 2.10E-02 |
| Replicative Senescence | 2 | 2.10E-02 |
| T-Cell Receptor Signaling Pathway | 3 | 2.90E-02 |
| Cellular Response to Extracellular Stimulus | 2 | 3.20E-02 |
| Response to Corticosterone | 2 | 3.20E-02 |
| Negative Regulation of Phosphorylation | 2 | 3.50E-02 |
| Negative Regulation of Cyclin-Dependent Protein Serine/Threonine Kinase Activity | 2 | 3.90E-02 |
| Cellular Senescence | 2 | 4.00E-02 |
To analyze the involvement of DEGs in different biological functional groups, all 35 common genes between HL-60 and BV173 were annotated for their role in biological processes. Differentially expressed genes IDs were uploaded into online DAVID bioinformatics software, and functional annotation clustering analysis was performed on GOTERM_BP_FAT gene ontology (GO). Categories with a p<0.05 were considered as statistically significant. The software mapped 31 of 35 genes and highlighted 14 different biological processes in leukemic cells cultured in the presence of UC-MSCs.
Pathway Analysis
Pathway analyses were performed to identify the putative signaling pathways that were dysregulated by UC-MSCs in both BV173 and HL-60 cell lines. Signaling pathways were evaluated by the DAVID bioinformatics tools through the KEGG database to cluster the significant pathways based on p-value (p<0.05). The results showed that UC-MSCs could trigger 13 different signaling pathways in both BV173 and HL-60 cell lines (Table 2), which were mostly associated with proliferation of tumor cells. Cell cycle, p53 signaling pathway, chronic myeloid leukemia, microRNAs in cancer, PI3K-Akt signaling pathway, and pathways in cancer were found to be the most interesting functional signaling pathways in HL-60 and BV173 cell lines (Table 3).
Table 2.
The List of Dysregulated Signaling Pathways.
| Signaling Pathways | Count | p-value |
|---|---|---|
| Cell Cycle | 6 | 8.90E-06 |
| p53 Signaling Pathway | 5 | 1.70E-05 |
| Glioma | 4 | 4.90E-04 |
| Melanoma | 4 | 6.40E-04 |
| Chronic Myeloid Leukemia | 4 | 6.60E-04 |
| Viral Carcinogenesis | 5 | 1.30E-03 |
| microRNAs in Cancer | 5 | 4.40E-03 |
| Bladder Cancer | 3 | 4.40E-03 |
| PI3K-AKt Signaling Pathway | 5 | 8.70E-03 |
| Pathways in Cancer | 5 | 1.40E-02 |
| Prostate Cancer | 3 | 1.90E-02 |
| HTLV-I Infection | 4 | 2.30E-02 |
| Hepatitis B | 3 | 4.80E-02 |
Total RNA of leukemic tumor cells (106 cells) either cultured alone (control group) or co-culture with 105 UC-MSCs (test group) were extracted. After integrity and quality evaluation the microarray experiments and raw data analysis were conducted at the Microarray Core Facility. Finally the KEGG database was used to identify the putative signaling pathways through DAVID online bioinformatics tools. The results showed 13 significant signaling pathways mediated by DEGs in HL-60 and BV173 cell lines.
Table 3.
The Most Important Signaling Pathways Altered in Leukemic Tumor Cell Lines in the Presence of UC-MSCs and their Respective DEGs.
| Signaling Pathways | Gene Symbol |
|---|---|
| Cell cycle | MDM2, CCNE2, CDK6, CDKN1A, CDKN2A, DBF4 |
| p53 signaling pathway | MDM2, CCNE2, CDK6, CDKN1A, CDKN2A |
| Chronic myeloid leukemia | MDM2, CDK6, CDKN1A, CDKN2A |
| MicroRNAs in cancer | MDM2, CCNE2, CDK6, CDKN1A, CDKN2A |
| PI3K-Akt signaling pathway | MDM2, COL6A3, CCNE2, CDK6, CDKN1A |
| Pathways in cancer | MDM2, CCNE2, CDK6, CDKN1A, CDKN2A |
Further analysis using the KEGG database through the DAVID online bioinformatics tool highlighted the most interesting functional signaling pathways and their respective DEGs in HL-60 and BV173 cell lines.
Microarray Data Validation
In order to validate the microarray results, sic DEGs (MDM2, CCNE2, MYL9, TGFβI, CDKN1A, and CDKN2A) in HL-60 and BV173 cell lines were selected for additional analysis using RT-qPCR. In addition, five DEGs (CDK6, CCND1, CCNE2, CDKN1A, and CDKN2A) in the HL-60 cell line were selected, and validation was conducted using western blot analysis. The selected genes were expressed with a similar expression pattern as DEGs in the microarray analysis of HL-60 and BV173 cells (Table 4). The western blot results showed that UC-MSCs alter the protein expression as well as mRNA expression in the HL-60 cell line. Fig. 3 shows that UC-MSCs elevate the expression of CDKN1A and CDKN2A, whereas the levels of protein expression of CDK6, CCND1, and CCNE2 were decreased in the HL-60 cell line by UC-MSCs.
Table 4.
mRNA Expression of Selected Genes after Co-cultured with UC-MSCs were Validated by RT-qPCR in BV173 and HL-60 Cell Lines.
| Genes | HL-60 Cell line | BV173 Cell line | ||
|---|---|---|---|---|
| Microarray FC | RT-qPCR FC | Microarray FC | RT-qPCR FC | |
| MDM2 | -9.355383 | -21.93 | -4.118405 | -17.85 |
| CCNE2 | -5.861723 | -12.04 | -2.821386 | -2.93 |
| MYL9 | -4.603044 | -5.29 | 530.4346 | 1351.17 |
| TGFβI | -365.5724 | -858.09 | 25509.12 | 30573.62 |
| CDKN1A | 3.77062 | 4.80 | 5.14529 | 12.99 |
| CDKN2A | 2.224852 | 3.61 | 3.622785 | 3.86 |
For microarray result validation, the extracted RNA from leukemic tumor cells in presence or absence of UC-MSCs were converted to cDNA and after designing primer pairs for selected genes, the qRT-PCR reaction was run using Light Cycler 480 DNA SYBR Green I Master by Roche. Then, the relative fold change in RNA expression was analyzed using the 2−ΔΔ C T method. The RT-qPCR results show that the mRNA expression level in HL-60 and BV173 cells in the presence of UC-MSCs showed similar results with the microarray (n=3/ triplicate).
Figure 3.
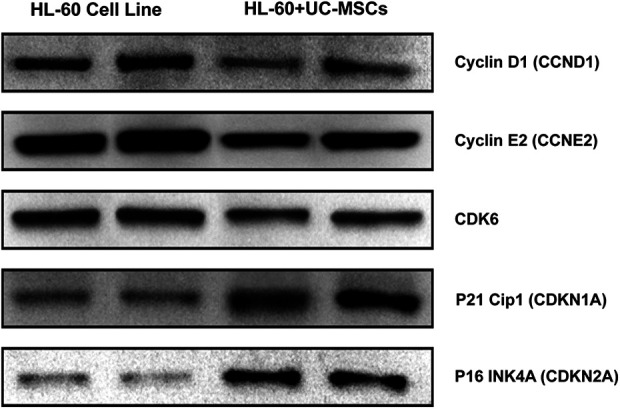
Protein expression validation of selected genes after co-culture with UC-MSCs. One million leukemic tumor cells were culture in presence or absence of UC-MSC at 1:01 ratio for 72 h. After 3 days cell were lysed and proteins were loaded onto SDS-PAGE gel and separating by electrophoresis. Then proteins were transferred onto PVDF membrane and incubated with primary antibodies and then secondary antibody for overnight and 1 h, respectively. All western blots were performed with two independent biological samples and repeated at least thrice. The representative data are shown.
Discussion
To date, it has been found that MSCs exert an anti-proliferative effect against various tumor cells ranging from hematopoietic or non-hematopoietic origins9,12,20. However, the mechanism and biomolecules that execute the UC-MSCs-induced anti-tumorigenic effect have not yet been fully elucidated. The current study shows that UC-MSCs inhibit various leukemic tumor cell proliferation mechanisms, and to our knowledge, the present study deciphers for the first time that UC-MSCs dysregulate the expression of 35 DEGs such as PSME3, NDN, IQSEC2, and PCBP4 in leukemic cells when co-cultured.
The present study showed that UC-MSCs possess a dose-dependent anti-tumorigenic effect through direct cell-to-cell contact against hematopoietic tumor cell lines of various origins such as CML, APL, and ALL. However, the observed anti-proliferative activity of UC-MSCs on leukemic tumor cell lines are non-specific, albeit different degrees of inhibition were noted on different types of tumor cells. The most robust inhibition of proliferation was recorded in BV173 and HL-60 cell lines as compared with the other tumor cell lines tested. Identification of the susceptibility of a particular type of leukemic tumor cell to UC-MSCs-induced inhibition is still elusive. However, the molecular mechanism which governs these biological processes in different tumor cells might contribute to this discrepancy. In this regard, the potential factors that cause these discrepancies may reside in the ability of UC-MSCs to affect an array of specific genes that differ between the BV173 and HL60 cells. The microarray results showed that UC-MSCs contributed 3019 DEGs in BV173, compared with 502 DEGs in the HL-60 cell line. These differences imply that the impact of UC-MSCs on the BV173 cell line is more prominent than on HL-60. Furthermore, with more DEGs observed in BV173, the probability of having key biological processes and signaling pathways altered by the UC-MSCs is higher in BV173 as compared with HL-60, hence projecting an increased susceptibility to UC-MSCs-induced inhibition.
The data from cell division and apoptosis highlighted that UC-MSCs-induced inhibition mainly corresponds to alteration in the cell cycle mechanism and interruption of protein and DNA synthesis surpassing apoptosis. Moreover, the results showed that UC-MSCs predominantly arrest the cell cycle progression of leukemic tumor cells in the G0/G1 phase of the cell cycle, and thus fewer cells proceeded to the subsequent S and G2/M phases. This suggests that treatment with UC-MSCs leads to potent growth inhibition of HL-60 and BV173 cells without being subject to apoptosis. The observed disruption of cell cycle progression in the tumor cells was consistent with the outcome of the microarray analysis. The expression levels of genes encoding cell cycle regulators such as CDK6, CCNE2, CDKN1A, CDKN2A, DBF4, MDM2, PSME3, and G0S2 were dysregulated, and consistent results were obtained from validation studies of CCNE2, CDKN1A, and CDKN2A both at the mRNA and protein levels. Thus, it could be deduced that UC-MSCs-mediated inhibition of leukemic cells occurs at the gene expression level with CCNE2, CDKN1A, and CDKN2A; these could be potential target genes. Other studies also have shown that MSCs from adipose and BM tissue can disrupt the cell cycle progression of solid tumor cells21,22. Despite considerable progress by different research groups toward the identification of the cell cycle mechanism, the reasons for the retardation or cell cycle arrest induced by UC-MSCs have yet to be fully understood.
In line with in vitro experiments, the microarray assay was designed to assess the molecular interplay and the respective signaling pathways that may be associated with the anti-tumorigenic activity of UC-MSCs on leukemic tumor cells (HL-60 and BV173). The GO analysis using the DAVID online bioinformatics software indicated that UC-MSCs mainly target the cell cycle and cell proliferation mechanism in leukemic tumor cells. Likewise, the GO analysis showed that UC-MSCs had affected biological processes such as cellular and replicative senescence, acting as an anti-tumor mechanism23, and negative regulation of protein kinase activity and negative regulation of phosphorylation, which regulates protein function and activity in different cellular processes such as cell cycle and cell growth24. Furthermore, DAVID analysis using the KEGG database also revealed that 13 signaling pathways were dysregulated by UC-MSCs in HL-60 and BV173 cells, most of which are cancer-related signaling pathways. Cell cycle and p53 signaling pathways are the most significantly dysregulated signaling pathways in leukemic tumor cells. Consequently, the microarray results showed that the expression of DBF4, MDM2, CCNE2, and CDK6 were down-regulated, while the level of CDKN1A and CDKN2A expression was elevated, which is relevant to the cell cycle signaling pathway.
In addition, the DAVID analysis showed that the expression of MDM2, CCNE2, CDK6, CDKN1A, and CDKN2A was dysregulated. These genes are part of the p53 signaling pathway, chronic myeloid leukemia, microRNAs in cancer, PI3K-Akt signaling pathway, and pathways in cancer signaling pathways. These genes play a pivotal role in the anti-tumorigenic activity of UC-MSCs in the proliferation of leukemic tumor cells. Except for DBF4, the other DEGs in cell cycle signaling pathways affect p53, microRNAs in cancer, and pathways in cancer signaling pathways as well. Dumbbell former 4 (DBF4), CDC7 regulatory subunit, can play a critical role in cell proliferation, DNA replication, G1/S transition and progression of S phase25. For instance, Nambiar et al. showed the reduction of cell proliferation and viability in NHM cells, a human cutaneous melanoma cell, upon siRNA-mediated depletion of DBF426. Besides, the present study showed that the UC-MSCs also decreased the expression level of MDM2 (mouse double minute-2) in BV173 and HL-60 cell lines not only in microarray analysis data, but further confirmed by RT-qPCR in both leukemic cell lines. MDM2 is the negative regulator of unstable tumor suppressor p53 activity which can act as a proto-oncogene in the cells. It has been shown that MDM2 inhibition can play a role as an anti-tumor mechanism by stabilizing p53, and subsequently p53 can induce cell cycle arrest through the induction of CDKN1A (p21) at the G1 phase of the cell cycle27,28. In parallel, CDK6 and CCNE2 are additional vital genes in cell cycle regulation, which were found to be down-regulated by UC-MSCs in leukemic cells. In normal circumstances, CDK6 can drive cells from G1 to S phase of the cell cycle, and it has been reported that CDK6 is frequently expressed at a higher level in leukemia and lymphoma29. Interestingly, the present study showed that UC-MSCs decreased the mRNA level of CDK6 in both tumor cell lines, and also confirmed the expression at the protein level by reduction of CDK6 protein level in HL-60 cells. Furthermore, other studies also revealed that miR-34a, miR-26a, and miR-138 significantly diminished the expression of CCNE2 and CDK6 in tumor cells and arrested the cells at the G1/S transition30,31.
The microarray results showed that UC-MSCs not only elevate the mRNA expression levels of CDKN1A and CDKN2A, cyclin-dependent kinase inhibitors, in both leukemic cell lines, but also increase the protein levels of p21 (CDKN1A) and p16 (CDKN2A) in the HL-60 cell line. Cyclin-dependent kinase inhibitors, p21cip and p16INK4a, can block cell cycle progression through inhibiting the CDK4 and CDK6 complex or monomeric CDK4 and CDK632,33. Several studies have shown that up-regulation of CDKN1A and CDKN2A arrests cell cycle process in G1 phase, and the loss of CDKN1A and CDKN2A is associated with cancer progression34,35. In line with this, the present study showed that UC-MSCs could induce the expression of THY1 (Thy-1 cell surface antigen) and reduce the expression level of PSME3 (proteasome activator subunit 3), ARID5B (AT-rich interaction domain 5B), and MYBL2 (MYB proto-oncogene like 2), which mediate essential roles in the genesis and progression of tumor cells36 in both leukemic cell lines. It was shown that up-regulation of THY1 (as a tumor suppressor gene) and down-regulation of PSME3, ARID5B (by up-regulating p21), and MYBL2 could arrest the tumor cell in the G0/G1 phase of the cell cycle37–41.
It was noted that UC-MSCs down-regulated the expression of COL6A3 (Collagen Type VI Alpha 3 Chain) that may act as an oncogene42, and enhanced the expression of DEGs in both leukemic cell lines in our study. There are DEGs which serve as tumor suppressor genes such as NDN (Necdin, MAGE Family Member) and PCBP4 [Poly(RC) Binding Protein 4]43 that inhibit the cell cycle progression and suppress lung cancer cells in the G2/M phase of the cell cycle44. In addition, PTPRC (Protein Tyrosine Phosphatase, Receptor Type C)45 and DCN (Decorin) which up-regulates p2146,47; FSTL1 (Follistatin Like 1)48 and G0S2 (G0/G1 Switch 2), which promotes quiescence in hematopoietic stem cells49,50; H19 (H19, Imprinted Maternally Expressed Transcript)51 and DKK-1 (Dickkopf WNT Signaling Pathway Inhibitor 1)52 are important tumor suppressor genes that support our notion.
The increases in CD2453 and APOC154 are essential for tumor proliferation, while the microarray results showed that UC-MSCs decreased the mRNA of CD24 and APOC1 in HL-60 and BV173 cell lines. For instance, Takano et al. have shown that the inhibition of APOC1 expression inhibited pancreatic cancer proliferation55. Likewise, the microarray results showed that UC-MSCs could down-regulate the expression level of C10orf10, EMP1, and ARHGAP26, and up-regulate the mRNA level of PHACTR1, HNMT, LY6K, SARDH, and FOSL1 in HL-60 and BV173 cell lines; however, their functional attributes in the inhibition of leukemic tumor cells remain to be clarified.
Finally, the microarray results highlighted several DEGs, the expression of which, to our knowledge, has not been previously reported in leukemia in the presence of UC-MSCs. These DEGs include KIF9, ZBTB46, IQSEC2, XLOC_006291, XLOC_014512, and SPTB. For instance, Demonchy et al. claimed that the KIF9 protein is required for trypanosome motility56, or based on sequence similarity, they have claimed that ZBTB46 might function as a transcriptional repressor for B-lymphocyte-induced maturation protein 1 (Blimp1) where it could block the proliferation of T and B lymphocytes57,58. Therefore, more experiments are needed to decipher the role of these genes in leukemic tumor cells when cultured in the presence of UC-MSCs.
The present study was conducted to investigate the therapeutic potential of UC-MSCs in the treatment of leukemia, as well as to identify the mechanisms involved. Collectively, this study demonstrated that the anti-proliferative activity of UC-MSCs on different tumor cells from the leukemic origin is mediated by cell-to-cell contact, predominantly by the accumulation of tumor cells in the G0/G1 phase of the cell cycle without inducing apoptosis. The in silico analysis predicted that UC-MSCs exerts its anti-tumorigenic activity through 35 DEGs in both HL-60 and BV173 cell lines. These genes may play a pivotal role in cell cycle regulation and tumor suppressor gene function, or may function as oncogenes through dysregulation of several signaling pathways. Although these genes have previously been predicted to serve as cell cycle regulatory genes, tumor suppressor or oncogenes, most of them have not been reported to mediate tumor cell suppression by UC-MSCs.
Conclusion
The data provided in this report may bring newer insight into therapeutic clinical trials using UC-MSCs for the treatment of leukemia. However, in vivo model studies are required in order to precisely elucidate the effect of microenvironment on the proliferation of leukemic tumor cells whereby the role of MSCs may become significant.
Supplemental Material
Supplemental Material, Additional_file_1 for Human Mesenchymal Stem Cells-mediated Transcriptomic Regulation of Leukemic Cells in Delivering Anti-tumorigenic Effects by Vahid Hosseinpour Sarmadi, Salma Ahmadloo, Mohadese Hashem Boroojerdi, Cini Mathew John, Satar Jabbar Rahi al-Graitte, Hamza Lawal, Maryam Maqbool, Ling King Hwa and Rajesh Ramasamy in Cell Transplantation
Supplemental Material, Additional_file_2 for Human Mesenchymal Stem Cells-mediated Transcriptomic Regulation of Leukemic Cells in Delivering Anti-tumorigenic Effects by Vahid Hosseinpour Sarmadi, Salma Ahmadloo, Mohadese Hashem Boroojerdi, Cini Mathew John, Satar Jabbar Rahi al-Graitte, Hamza Lawal, Maryam Maqbool, Ling King Hwa and Rajesh Ramasamy in Cell Transplantation
Supplemental Material, Additional_file_3a for Human Mesenchymal Stem Cells-mediated Transcriptomic Regulation of Leukemic Cells in Delivering Anti-tumorigenic Effects by Vahid Hosseinpour Sarmadi, Salma Ahmadloo, Mohadese Hashem Boroojerdi, Cini Mathew John, Satar Jabbar Rahi al-Graitte, Hamza Lawal, Maryam Maqbool, Ling King Hwa and Rajesh Ramasamy in Cell Transplantation
Supplemental Material, Additional_File_4 for Human Mesenchymal Stem Cells-mediated Transcriptomic Regulation of Leukemic Cells in Delivering Anti-tumorigenic Effects by Vahid Hosseinpour Sarmadi, Salma Ahmadloo, Mohadese Hashem Boroojerdi, Cini Mathew John, Satar Jabbar Rahi al-Graitte, Hamza Lawal, Maryam Maqbool, Ling King Hwa and Rajesh Ramasamy in Cell Transplantation
Footnotes
Ethical Approval: The collection and use of human umbilical cord tissues were approved by the Ethical and Research Committee, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia (reference number: UPM/FPSK/PADS/T7-MJKEtikaPer/F01[Lect_Sept(08)14]).
Statement of Human and Animal Rights: This article does not contain any studies with human or animal subjects.
Statement of Informed Consent: Written and signed informed consent was obtained from a legally authorized representative (parents).
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Research University Grant Scheme (RUGS), 04-02-12-1757RU and Putra Grant-Putra Graduate Initiative (IPS), GP-PS/2014/9432500, Universiti Putra Malaysia. The publication fee is funded by Research Management Center, Universiti Putra Malaysia.
ORCID iD: Rajesh Ramasamy  https://orcid.org/0000-0003-4227-0458
https://orcid.org/0000-0003-4227-0458
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Colby-Graham MF, Chordas C. The childhood leukemias. J Pediatr Nurs. 2003;18(2):87–95. [DOI] [PubMed] [Google Scholar]
- 2. Musarezaie A, Khaledi F, Esfahani HN, Ghaleghasemi TM. Factors affecting quality of life and fatigue in patients with leukemia under chemotherapy. J Educ Health Promot. 2014;3:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ayala F, Dewar R, Kieran M, Kalluri R. Contribution of bone microenvironment to leukemogenesis and leukemia progression. Leukemia. 2009;23(12):2233–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kurtova AV, Balakrishnan K, Chen R, Ding W, Schnabl S, Quiroga MP, Sivina M, Wierda WG, Estrov Z, Keating MJ, Shehata M, et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood. 2009;114(20):4441–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, Du J, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418(6893):41–49. [DOI] [PubMed] [Google Scholar]
- 6. Rao MS, Mattson MP. Stem cells and aging: expanding the possibilities. Mech Ageing Dev. 2001;122(7):713–734. [DOI] [PubMed] [Google Scholar]
- 7. Nagamura-Inoue T, He H. Umbilical cord-derived mesenchymal stem cells: their advantages and potential clinical utility. World J Stem Cells. 2014;6(2):195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. U.S. National Library of Medicine. Search of: Leukemia | mesenchymal stem cells-ClinicalTrials.gov. 2019. DOI: 10.1177/0963689719885077 Available from <https://clinicaltrials.gov/ct2/results?cond=Leukemia&term=&type=&rslt=&age_v=&gndr=&intr=mesenchymal+stem+cells+&titles=&outc=&spons=&lead=&id=&cntry=&state=&city=&dist=&locn=&strd_s=&strd_e=&prcd_s=&prcd_e=&sfpd_s=&sfpd_e=&lupd_s=&lupd_e=&sort=> (accessed June 21, 2019).
- 9. Ramasamy R, Lam EW, Soeiro I, Tisato V, Bonnet D, Dazzi F. Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: impact on in vivo tumor growth. Leukemia. 2007;21(2):304–310. [DOI] [PubMed] [Google Scholar]
- 10. Keating A. Mesenchymal stromal cells: new directions. Cell Stem Cell. 2012;10(6):709–716. [DOI] [PubMed] [Google Scholar]
- 11. Dazzi F, Ramasamy R, Glennie S, Jones SP, Roberts I. The role of mesenchymal stem cells in haemopoiesis. Blood Rev. 2006;20(3):161–171. [DOI] [PubMed] [Google Scholar]
- 12. Sarmadi VH, Tong CK, Vidyadaran S, Abdullah M, Seow HF, Ramasamy R. Mesenchymal stem cells inhibit proliferation of lymphoid origin haematopoietic tumour cells by inducing cell cycle arrest. Med J Malaysia. 2010;65(3):209–214. [PubMed] [Google Scholar]
- 13. Lee MW, Ryu S, Kim DS, Lee JW, Sung KW, Koo HH, Yoo KH. Mesenchymal stem cells in suppression or progression of hematologic malignancy: current status and challenges. Leukemia. 2019;33(3):597–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tong CK, Vellasamy S, Tan BC, Abdullah M, Vidyadaran S, Seow HF, Ramasamy R. Generation of mesenchymal stem cell from human umbilical cord tissue using a combination enzymatic and mechanical disassociation method. Cell Biol Int. 2011;35(3):221–226. [DOI] [PubMed] [Google Scholar]
- 15. Pegoraro L, Matera L, Ritz J, Levis A, Palumbo A, Biagini G. Establishment of a Ph1-positive human cell line (BV173). J Natl Cancer Inst. 1983;70(3):447–453. [PubMed] [Google Scholar]
- 16. Lozzio CB. Cytotoxicity of a factor isolated from human spleen. Natl Cancer Inst. 1973;50(2):535–538. [DOI] [PubMed] [Google Scholar]
- 17. Collins SJ, Ruscetti FW, Gallagher RE, Gallo RC. Terminal differentiation of human promyelocytic leukemia cells induced by dimethyl sulfoxide and other polar compounds. Proc Natl Acad Sci U S A. 1978;75(5):2458–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schneider U, Schwenk HU, Bornkamm G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int J Cancer. 1977;19(5):621–626. [DOI] [PubMed] [Google Scholar]
- 19. Nygard AB, Jorgensen CB, Cirera S, Fredholm M. Selection of reference genes for gene expression studies in pig tissues using SYBR green qPCR. BMC Mol Biol. 2007;8:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sarmadi VH, Heng FS, Ramasamy R. The effect of human mesenchymal stem cells on tumour cell proliferation. Med J Malaysia. 2008;63(Suppl A):63–64. [PubMed] [Google Scholar]
- 21. Ahn JO, Coh YR, Lee HW, Shin IS, Kang SK, Youn HY. Human adipose tissue-derived mesenchymal stem cells inhibit melanoma growth in vitro and in vivo. Anticancer Res. 2015;35(1):159–168. [PubMed] [Google Scholar]
- 22. Tian LL, Yue W, Zhu F, Li S, Li W. Human mesenchymal stem cells play a dual role on tumor cell growth in vitro and in vivo. J Cell Physiol. 2011;226(7):1860–1867. [DOI] [PubMed] [Google Scholar]
- 23. Smith SK, Kipling D. The role of replicative senescence in cancer and human ageing: utility (or otherwise) of murine models. Cytogenet Genome Res. 2004;105(2–4):455–463. [DOI] [PubMed] [Google Scholar]
- 24. Jin L, Chun J, Pan C, Alesi GN, Li D, Magliocca KR, Kang Y, Chen ZG, Shin DM, Khuri FR, Fan J, et al. Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis. Oncogene. 2017. 36(27):3797–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matthews LA, Guarne A. Dbf4: the whole is greater than the sum of its parts. Cell Cycle. 2013;12(8):1180–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nambiar S, Mirmohammadsadegh A, Hassan M, Mota R, Marini A, Alaoui A, Tannapfel A, Hegemann JH, Hengge UR. Identification and functional characterization of ASK/Dbf4, a novel cell survival gene in cutaneous melanoma with prognostic relevance. Carcinogenesis. 2007;28(12):2501–2510. [DOI] [PubMed] [Google Scholar]
- 27. Dickens MP, Fitzgerald R, Fischer PM. Small-molecule inhibitors of MDM2 as new anticancer therapeutics. Semin Cancer Biol. 2010;20(1):10–18. [DOI] [PubMed] [Google Scholar]
- 28. Deb SP. Cell cycle regulatory functions of the human oncoprotein MDM2. Mol Cancer Res. 2003;1(14):1009–1016. [PubMed] [Google Scholar]
- 29. Kollmann K, Heller G, Schneckenleithner C, Warsch W, Scheicher R, Ott RG, Schafer M, Fajmann S, Schlederer M, Schiefer AI, Reichart U, et al. A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell. 2016;30(2):359–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Erdmann K, Kaulke K, Rieger C, Salomo K, Wirth MP, Fuessel S. MiR-26a and miR-138 block the G1/S transition by targeting the cell cycle regulating network in prostate cancer cells. J Cancer Res Clin Oncol. 2016;142(11):2249–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun F, Fu H, Liu Q, Tie Y, Zhu J, Xing R, Sun Z, Zheng X. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 2008;582(10):1564–1568. [DOI] [PubMed] [Google Scholar]
- 32. Canepa ET, Scassa ME, Ceruti JM, Marazita MC, Carcagno AL, Sirkin PF, Ogara MF. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life. 2007;59(7):419–426. [DOI] [PubMed] [Google Scholar]
- 33. Vidal A, Koff A. Cell-cycle inhibitors: three families united by a common cause. Gene. 2000;247(1-2):1–15. [DOI] [PubMed] [Google Scholar]
- 34. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Romagosa C, Simonetti S, Lopez-Vicente L, Mazo A, Lleonart ME, Castellvi J, Ramon y Cajal S. p16(Ink4a) overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. Oncogene. 2011;30(18):2087–2097. [DOI] [PubMed] [Google Scholar]
- 36. Ren F, Wang L, Shen X, Xiao X, Liu Z, Wei P, Wang Y, Qi P, Shen C, Sheng W, Du X. MYBL2 is an independent prognostic marker that has tumor-promoting functions in colorectal cancer. Am J Cancer Res. 2015;5(4):1542–1552. [PMC free article] [PubMed] [Google Scholar]
- 37. Xiong S, Zheng Y, Jiang P, Liu R, Liu X, Qian J, Gu J, Chang L, Ge D, Chu Y. PA28gamma emerges as a novel functional target of tumour suppressor microRNA-7 in non-small-cell lung cancer. Br J Cancer. 2014;110(2):353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sanchez N, Gallagher M, Lao N, Gallagher C, Clarke C, Doolan P, Aherne S, Blanco A, Meleady P, Clynes M, Barron N. MiR-7 triggers cell cycle arrest at the G1/S transition by targeting multiple genes including Skp2 and Psme3. PLoS One. 2013;8(6):e65671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang JJ, Xu H, Bhojwani D, Moriyama T, Qian M, Yang X, Seibel NL, Relling MV, Carroll WL, Pui C-H. ARID5B regulates leukemia sensitivity to antimetabolites in children with acute lymphoblastic leukemia via effects on cell cycle progression. Blood. 2014;124(21):791–791.24755410 [Google Scholar]
- 40. Papetti M, Augenlicht LH. MYBL2, a link between proliferation and differentiation in maturing colon epithelial cells. J Cell Physiol. 2011;226(3):785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lung HL, Cheung AKL, Cheng Y, Kwong FM, Lo PHY, Law EWL, Chua D, Zabarovsky ER, Wang N, Tsao SW. Functional characterization of THY1 as a tumor suppressor gene with antiinvasive activity in nasopharyngeal carcinoma. Int J Cancer. 2010;127(2):304–312. [DOI] [PubMed] [Google Scholar]
- 42. Xie X, Liu X, Zhang Q, Yu J. Overexpression of collagen VI alpha3 in gastric cancer. Oncol Lett. 2014;7(5):1537–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yan W, Scoumanne A, Jung Y-S, Xu E, Zhang J, Zhang Y, Ren C, Sun P, Chen X. Mice deficient in poly (C)-binding protein 4 are susceptible to spontaneous tumors through increased expression of ZFP871 that targets p53 for degradation. Genes Dev. 2016;30(5):522–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Castano Z, Vergara-Irigaray N, Pajares MJ, Montuenga LM, Pio R. Expression of alpha CP-4 inhibits cell cycle progression and suppresses tumorigenicity of lung cancer cells. Int J Cancer. 2008;122(7):1512–1520. [DOI] [PubMed] [Google Scholar]
- 45. Porcu M, Kleppe M, Gianfelici V, Geerdens E, De Keersmaecker K, Tartaglia M, Foa R, Soulier J, Cauwelier B, Uyttebroeck A, Macintyre E, et al. Mutation of the receptor tyrosine phosphatase PTPRC (CD45) in T-cell acute lymphoblastic leukemia. Blood. 2012;119(19):4476–4479. [DOI] [PubMed] [Google Scholar]
- 46. Xu Y, Xia Q, Rao Q, Shi S, Shi Q, Ma H, Lu Z, Chen H, Zhou X. DCN deficiency promotes renal cell carcinoma growth and metastasis through downregulation of P21 and E-cadherin. Tumour Biol. 2016;37(4):5171–5183. [DOI] [PubMed] [Google Scholar]
- 47. Jarvinen TA, Prince S. Decorin: a growth factor antagonist for tumor growth inhibition. Biomed Res Int. 2015;2015:654765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhou X, Xiao X, Huang T, Du C, Wang S, Mo Y, Ma N, Murata M, Li B, Wen W, Huang G, et al. Epigenetic inactivation of follistatin-like 1 mediates tumor immune evasion in nasopharyngeal carcinoma. Oncotarget. 2016;7(13):16433–16444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yamada T, Park CS, Burns A, Nakada D, Lacorazza HD. The cytosolic protein G0S2 maintains quiescence in hematopoietic stem cells. PLoS One 2012;7(5):e38280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yim CY, Sekula DJ, Hever-Jardine MP, Liu X, Warzecha JM, Tam J, Freemantle SJ, Dmitrovsky E, Spinella MJ. G0S2 suppresses oncogenic transformation by repressing a MYC-regulated transcriptional program. Cancer Res. 2016;76(5):1204–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yoshimizu T, Miroglio A, Ripoche MA, Gabory A, Vernucci M, Riccio A, Colnot S, Godard C, Terris B, Jammes H, Dandolo L. The H19 locus acts in vivo as a tumor suppressor. Proc Natl Acad Sci U S A. 2008;105(34):12417–12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhu Y, Sun Z, Han Q, Liao L, Wang J, Bian C, Li J, Yan X, Liu Y, Shao C, Zhao RC. Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia. 2009;23(5):925–933. [DOI] [PubMed] [Google Scholar]
- 53. Huang L, Lv W, Zhao X. CD24 as a molecular marker in ovarian cancer: a literature review. Cancer Trans Med. 2016;2(1):29–32. [Google Scholar]
- 54. Ko HL, Wang YS, Fong WL, Chi MS, Chi KH, Kao SJ. Apolipoprotein C1 (APOC1) as a novel diagnostic and prognostic biomarker for lung cancer: a marker phase I trial. Thorac Cancer. 2014;5(6):500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Takano S, Yoshitomi H, Togawa A, Sogawa K, Shida T, Kimura F, Shimizu H, Tomonaga T, Nomura F, Miyazaki M. Apolipoprotein C-1 maintains cell survival by preventing from apoptosis in pancreatic cancer cells. Oncogene. 2008;27(20):2810–2822. [DOI] [PubMed] [Google Scholar]
- 56. Demonchy R, Blisnick T, Deprez C, Toutirais G, Loussert C, Marande W, Grellier P, Bastin P, Kohl L. Kinesin 9 family members perform separate functions in the trypanosome flagellum. J Cell Biol. 2009;187(5):615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Martins G, Calame K. Regulation and functions of Blimp-1 in T and B lymphocytes. Annu Rev Immunol. 2008;26:133–169. [DOI] [PubMed] [Google Scholar]
- 58. Martins GA, Cimmino L, Liao J, Magnusdottir E, Calame K. Blimp-1 directly represses Il2 and the Il2 activator Fos, attenuating T cell proliferation and survival. J Exp Med. 2008;205(9):1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material, Additional_file_1 for Human Mesenchymal Stem Cells-mediated Transcriptomic Regulation of Leukemic Cells in Delivering Anti-tumorigenic Effects by Vahid Hosseinpour Sarmadi, Salma Ahmadloo, Mohadese Hashem Boroojerdi, Cini Mathew John, Satar Jabbar Rahi al-Graitte, Hamza Lawal, Maryam Maqbool, Ling King Hwa and Rajesh Ramasamy in Cell Transplantation
Supplemental Material, Additional_file_2 for Human Mesenchymal Stem Cells-mediated Transcriptomic Regulation of Leukemic Cells in Delivering Anti-tumorigenic Effects by Vahid Hosseinpour Sarmadi, Salma Ahmadloo, Mohadese Hashem Boroojerdi, Cini Mathew John, Satar Jabbar Rahi al-Graitte, Hamza Lawal, Maryam Maqbool, Ling King Hwa and Rajesh Ramasamy in Cell Transplantation
Supplemental Material, Additional_file_3a for Human Mesenchymal Stem Cells-mediated Transcriptomic Regulation of Leukemic Cells in Delivering Anti-tumorigenic Effects by Vahid Hosseinpour Sarmadi, Salma Ahmadloo, Mohadese Hashem Boroojerdi, Cini Mathew John, Satar Jabbar Rahi al-Graitte, Hamza Lawal, Maryam Maqbool, Ling King Hwa and Rajesh Ramasamy in Cell Transplantation
Supplemental Material, Additional_File_4 for Human Mesenchymal Stem Cells-mediated Transcriptomic Regulation of Leukemic Cells in Delivering Anti-tumorigenic Effects by Vahid Hosseinpour Sarmadi, Salma Ahmadloo, Mohadese Hashem Boroojerdi, Cini Mathew John, Satar Jabbar Rahi al-Graitte, Hamza Lawal, Maryam Maqbool, Ling King Hwa and Rajesh Ramasamy in Cell Transplantation