

Abstract
Appropriate ways to measure the similarity between single-cell RNA-sequencing (scRNA-seq) data are ubiquitous in bioinformatics, but using single clustering or classification methods to process scRNA-seq data is generally difficult. This has led to the emergence of integrated methods and tools that aim to automatically process specific problems associated with scRNA-seq data. These approaches have attracted a lot of interest in bioinformatics and related fields. In this paper, we systematically review the integrated methods and tools, highlighting the pros and cons of each approach. We not only pay particular attention to clustering and classification methods but also discuss methods that have emerged recently as powerful alternatives, including nonlinear and linear methods and descending dimension methods. Finally, we focus on clustering and classification methods for scRNA-seq data, in particular, integrated methods, and provide a comprehensive description of scRNA-seq data and download URLs.
Keywords: single-cell RNA-seq, clustering, classification, similarity metric, sequences analysis, machine learning
Introduction
Many bioinformatics problems involve DNA and protein sequence analyses or temporal series analyses. Each cell has its unique phenotype and biological function, which is reflected in the differences between different histology. Bulk RNA sequencing (RNA-seq) is based on studies on a large number of cells, and its expression level is the relative average level of a group of cells. Therefore, in the mixed cell population, the traditional bulk RNA-seq cannot analyze the critical differences of individual cells. In particular, it cannot study the complex system with ever-changing expression and the expression characteristics of genes in the system. The emergence of single-cell RNA-seq solves this problem by providing the expression profile information of single cells. Although it is impossible to obtain the complete information of each RNA expressed by each cell, with limited raw materials, gene clustering analysis/identification of gene expression patterns can reveal or discover the existence of rare cell types in the cell population.
Single-cell RNA-seq (scRNA-seq) provides precision and details. It uses optimized next-generation sequencing technologies and acquires transcriptomic information from individual cells to provide a better understanding of cell functions at genetic and cellular levels. scRNA-seq has been used to study cancer, metagenomics and regulatory and evolutionary networks [1–3]. Identification of genes that are essential for a given cell type is critical for understanding the biological characteristics of cells. Kater et al. [4] showed a clustering robustness score to solve the problem that most clustering methods are not robust to noise. By artificially adding noise, they obtained clusters of cells with biological meaningful in single-cell expression dataset. Studies have shown there is significant heterogeneity in gene expression between individual cells of the same cell type. The rapid development of molecular biology technologies has dramatically improved the ability to analyze transcriptomes; in particular, high-throughput sequencing technology and transcriptome sequencing (RNA-seq) analysis are now common experimental methods. Xie et al. [5] presented a novel biclustering algorithm for the analysis of large-scale bulk RNA-seq and scRNA-seq data.
In recent years, ‘scRNA-seq technology’ has become an essential tool for molecular biology research. Compared with traditional cell-based RNA-seq, scRNA-seq can better reflect the molecular biological processes within a particular cell population. In addition, scRNA-seq enables more precise subpopulation analysis of specific cell types and allows the detection of different responses of individual cells to the same stimulus in the same cell type [6, 7]. In many studies on gene transcription [8, 9], what is detected is the average gene expression of a population of somatic cells, tissue cells or organism cells. Although these studies have helped the progress of gene transcriptome research, traditional cell-based RNA-seq cannot clearly show the heterogeneity between the cells in an organism. scRNA-seq can provide information on individual cell transcriptomes and can be used to develop cell subsets to determine the time stage of cell differentiation and the progression of single cells. In addition to cell heterogeneity, the analysis of cell differentiation requires the clustering of scRNA-seq data. Such clustering helps in understanding potential cellular mechanisms, which can promote the discovery of new markers on specific types of cells and the recognition of tumor subtypes.
Köster et al. [10] proposed a Bayesian model for analyzing the transcript expression of single cells. Transcriptome analysis helps to predict gene expression from genotype data. However, the cells in our bodies have almost the same genotype, but the transcriptome information only reflects the activity of some genes. In addition, gene expression is heterogeneous even within similar cell types, and individual cell transcriptome is critical to elucidating stochastic biological processes. The analysis of scRNA-seq data presents some challenges. scRNA-seq provides deep scrutiny into the gene expression character of diverse cell types. The current main challenge is the noisy nature of the scRNA data. Many of the features of scRNA-seq data are zero or nearly zero, so processing noise information is of great significance. This noise makes it difficult to distinguish very similar cell types, and this is where the technology needs to be improved. Second, scRNA-seq data always have high dimensions, which increase the difficulty of the analysis to some extent.
Therefore, dimensionality reduction methods have been used to process the original data. The most common dimensionality reduction method is principal components analysis (PCA) [11], which is unsupervised and aims to find a lower dimensional representation of the data. Peng et al. [12] proposed two models, unsupervised Gene Ontology AutoEncoder (GOAE) and supervised Gene Ontology Neural Network (GONN), to reduce dimensionality. They combined scRNA-seq data and gene ontology information to extract the hidden layer information and obtain lower dimensionality representations of the data. Because of the large numbers of zeros in scRNA-seq, the classical dimensionality building method often fails. Pierson and Yau [13] proposed zero-inflated factor analysis (ZIFA), which makes drop-out events exactly zero, thereby modeling scRNA-seq data and improving the modeling precision. Although ZIFA is easy to use, it is likely to lose information; however, drop-out events can be recovered to reduce the loss.
Sequence analysis is essential in bioinformatics [14–17], and metric learning often plays an important role in this. Common metric learning algorithms for classification include information-theoretic metric learning (ITML) [18], large margin nearest neighbor (LMNN) [19] and geometric mean metric learning (GMML) [20]. ITML, which was proposed by Davis et al. in 2007 [18], uses Bregman divergence or divergence distance to measure similarity. LMNN [19] was proposed by Weinberger et al. in 2000 and is the most commonly used Mahalanobis distance metric learning method. GMML [20, 21]was proposed by Zadeh and Hosseini in 2016 [19], and the main innovation of this method is its ability to achieve the effect that the similarity distance is small and the dissimilarity distance is large using only one objective function. In scRNA-seq data, the classification accuracy of several datasets can be more than 90% and sometimes close to 100%, whereas the clustering task is more difficult. Common clustering machine-learning methods include K-means [21], expectation maximization (EM) [22] and spectral clustering [23]. K-means [21] is a classical clustering method that is easy to operate, and the number of clusters required can be set before the experiment. The K-means method is robust on a variety of datasets. Many of the newly proposed clustering methods are extensions of K-means. EM [22] is an integrated clustering method that also has been shown to be stable and reliable. Spectral clustering [23] is based on graph theory, which introduces the concept of degree and then uses K-means to cluster after steps such as eigenvalue decomposition. Common combinations of methods include T-distributed stochastic neighbor embedding (t-SNE) with K-means [24] and PCA with hierarchical clustering [25]. Jiang et al. embedded the cell similarity measurement method based on variance analysis into the hierarchical clustering framework and developed the clustering algorithm ‘Corr’ [26].
scRNA-seq technology can be used to perform gene expression studies on a variety of cells simultaneously, avoiding the need to label each cell. The results of existing scRNA-seq studies have shown that gene expression profiles between individual cells are significantly different in the same cell type, indicating only a few of the expressed genes detected in the same cell group are shared by a single cell, and most of the genes are expressed randomly in each cell. Monier et al. [27] present a tool called IRIS-EDA, which is a Shiny web server for expression data analysis. scRNA-seq analysis has incomparable advantages over traditional sequencing analysis in the fields of cancer evolution and drug resistance analysis [28, 29] in the treatment process, as well as epidermal mesenchymal transformation [30], which is extremely important in the cancer transformation process and the measurement of mutation rate in cancer cells [31–33]. Single-cell level analysis can help in understanding the complex processes of cancer occurrence, development, metastasis and recurrence. Individualized clinical treatment also will be the target and direction of scRNA-seq technology. Generally, scRNA-seq has three advantages (applications) over traditional bulk RNA-seq: (i) it can reveal complex and rare cell populations, (ii) uncover heterogenous gene regulatory relationships among cells, and (iii) track the trajectories of distinct cell lineages in development in terms of both temporal and spatial information.
This review is organized as follows. In ‘the challenges ofscRNA-seq’, the challenges of scRNA-seq are discussed. In ‘methods for dimensionality reduction and clustering analysistools’, we review dimensionality reduction methods and describe some of the advanced clustering tools and trends, including SC3, an R package for clustering and single-cell regulatory network inference and clustering (SCENIC). In ‘performances of the methods on scRNA-seq datasets’, we describe scRNA-seq datasets and provide the download URLs. In ‘discussion and conclusions’, we discuss the current limitations of some of the methods based on the existing literature.
The challenges of scRNA-seq
The study of a single cell is irreplaceable for the exploration of the mystery of biology and also faces significant challenges. Gene transcription is not stable and continuous but a sporadic active transcription. For a fixed cell, transcription is in a constantly changing state [34–36]. Due to the technical limitations of single-cell transcription level measurement, it is difficult to detect low-level gene expression, and most intelligent detection methods can detect about 10–20% of the actual mRNA molecules [37, 38]. Since there are very few single-cell materials available, an amplification step is generally required to generate a larger amount. However, due to amplification is nonlinear, the proportion of cDNA in cells is not balanced, and the amplification is biased, so some markers cannot be amplified.
scRNA-seq has great limitations in obtaining information. The main limitation is caused by biological noise during gene expression [34, 36]. A significant feature of scRNA-seq data is a large number of zero-inflated counts due to dropout or transient gene expression, which may mislead downstream analyses. The read count is connected with gene-specific expression level, while the nuisance variables are difficult to estimate. The commonly used method of inter-sample normalization is trimmed mean of M values (TMM) and differential expression analysis for sequence count data (DESeq) [39–41]. Both methods eliminated some genes based on a weighted average or median of samples, but both methods performed poorly when a large number of zero counts were counted. In addition to normalization, confounding factors such as biological variables and technical noise also influence the observed read counts.
In 1992, Eberwine et al. [42] used in vitro transcriptional amplification to study acutely dissociated cells in restricted areas of rat brain and analyzed gene expression characteristics in single living neurons. However, the number of gene detection and detection flux were low. With the continuous development of detection technology, Tang et al. have combined single-cell RNA with high-throughput sequencing for the 1st time [43], significantly increasing the detection flux. However, due to the limitation of single-cell isolation technology, not all laboratories can successfully complete single-cell sequencing experiments.
Although many researchers began to study scRNA-seq and there were some experimental methods that were easy to start using, only a very small number of single cells were labeled, and individual cells could not be enriched, and the computational channels for processing original data were limited, which caused difficulties in sequencing single cells. Sequencing has the risks of low coverage, low mappability, high duplicate rate and high error rate. Some companies have developed tools to process scRNA raw data but only in the early stages.
The workflow for scRNA-seq is summarized in Figure 1. Single-cell data are high-dimensional and contain a lot of noise, so the raw single-cell data generally are processed for dimensionality reduction and denoising, before being classified for analysis, including clustering analysis, cell type identification, sorting and other operations.
Figure 1.
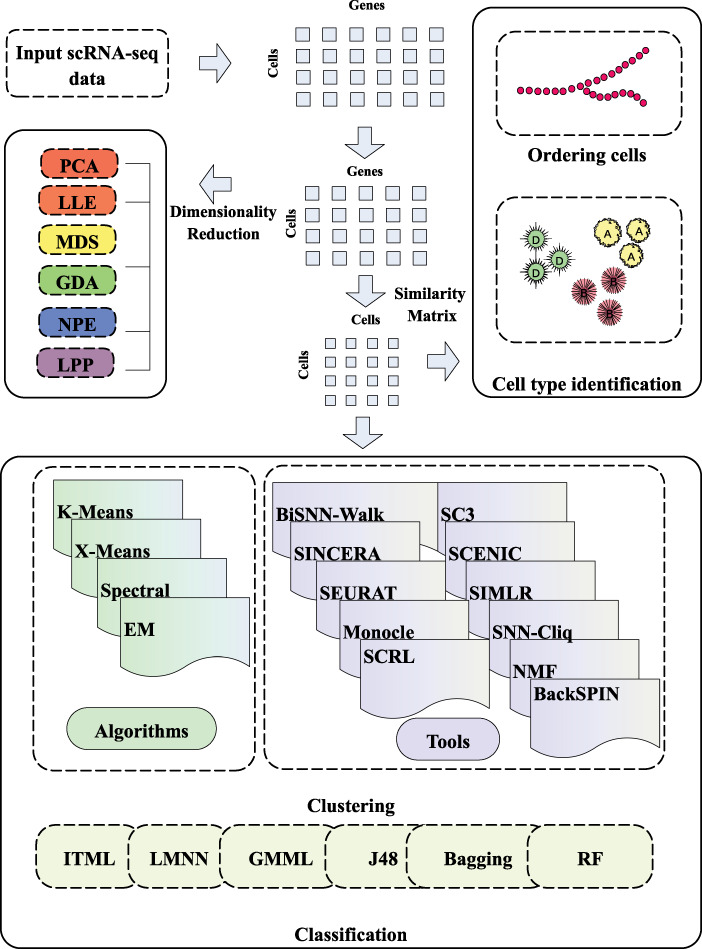
The workflow of scRNA-seq data and the pipeline of scRNA-seq data application. The 1st stage is dataset processing. Cells–genes matrix was obtained by processing the original data with the effective dimensionality reduction method. The 2nd stage is to calculate the similarity matrix. The similarity matrix can be used for cell sequencing, cell type recognition and other applications. In addition, machine learning can be used to cluster or classify single-cell data.
Methods for dimensionality reduction and clustering analysis tools
Clustering plays an essential role in single-cell analysis. Given the high dimensionality of single-cell data, many approaches combine classic clustering and dimension reduction. Effective dimensionality reduction methods are critical because most scRNA-seq data are large and noisy, with the characteristics of a small number of samples but a large number of dimensions. Dimensionality reduction is usually carried out after counting normalization to avoid the curse of dimensionality.
Dimensionality reduction methods
Principal components analysis
PCA [11] is a commonly used unsupervised dimensionality reduction method [44]. PCA assumes that the data are normally distributed, diagonalizes the covariance matrix of the original matrix and the resulting covariance matrix is a set of new variables of the diagonal matrix. The orthogonal transformation is used to transform a set of potential linear correlation variables into linear independent variables, which means that linear dimensionality reduction is realized. One of the main problems with linear dimensionality reduction algorithms is that when they concentrate dissimilar data points in a lower dimensional region, the data points are far apart. By projecting cells into two-dimensional space, PCA can easily visualize samples and improve the interpretation ability. An extended version of PCA, pcaReduce [25], creates the relationship between the data patterns and cell type. pcaReduce is a hierarchical clustering combining PCA, k-means and iteration. It starts with a large number of clusters, and pcaReduce iteratively combines similar clusters. After each combination, the component of the smallest variance in the data is deleted.
T-distributed stochastic neighbor embedding
To represent high-dimensional data on the low-dimensional and nonlinear manifold, we also need to show similar data points together, which is not what the linear dimension-reduction algorithms can do. t-SNE [45] is a nonlinear dimensionality reduction method, and it converts the distance of high-dimensional space of points into the probability of similarity of points and maintains the sum of the difference of conditional probabilities between a pair of points in high-dimensional space and low-dimensional space to be the minimum. At the same time, the long tail of t-distribution is used to solve the overlapping problem when the high-dimensional data is mapped to the low-dimensional data. t-SNE algorithm defines the soft boundary between the local and global structure of data, which can make the points scattered locally and aggregated globally, and take care of the points at close range and far range at the same time. t-SNE has been used recently to reduce the dimensionality of scRNA-seq data [46, 47].
Zero-inflated factor analysis
A large number of dropout events in single-cell RNA data make most dimension-reduction algorithms fail to work. ZIFA [13] is a linear dimensionality reduction method for scRNA-seq data, which is completed on the basis of modification by probabilistic PCA/factor analysis (PPCA/FA). In the absence of dropout event, it is equivalent to PPCA/FA. ZIFA regards 0 in data as normal data and models it. So, drop-out events in the data are assumed to result in zero counts and are set as precisely 0 rather than approximately 0. However, these drop-out events could be recreated by detecting technology or environmental effects, so this assumption could lead to loss of information and decreased accuracy.
Neural networks
Neural networks can continuously extract the main features through a hidden layer to achieve the effect of dimensionality reduction. For example, denoising self-coding has been used widely to reconstruct data from higher to lower dimensions [48, 49]. Lin et al. [46] proposed a supervised method for scRNA-seq dimensionality reduction based on a neural network. This method uses protein–protein and protein–DNA interaction data to learn the neural network of the structure and parameters in the model. Lin et al.’s method promotes the development of supervised models and has been shown to perform best in unsupervised models.
Some of the open dimensionality reduction methods are listed in Table 1. These methods each have their characteristics, so the most appropriate dimensionality reduction method should be chosen according to the characteristics of the target dataset. In 2007, van der Maaten published a MATLAB Toolbox for Dimensionality Reduction that contains implementations of many methods for dimensionality reduction [50].
Table 1.
Summary of dimensionality reduction methods
| Dimensionality reduction method | Reference | Year | Usage or download URL | |
|---|---|---|---|---|
| 1 | PCA | [11] | 1987 | MATLAB, Python, R etc. all have a free package |
| 3 | FA | [51] | 1980 | http://personality-project.org/r |
| 4 | Classical multidimensional scaling | [52] | 1978 | https://www.statmethods.net/advstats/mds.html |
| 5 | Sammon mapping | [53] | 1969 | MATLAB, Python, R, etc. all have a free package |
| 6 | Linear discriminant analysis | / | 1936 | MATLAB, Python, R, etc. all have a free package |
| 9 | Local linear embedding (LLE) | [55] | 2000 | https://cs.nyu.edu/∼roweis/lle/code.html |
| 10 | Laplacian eigenmaps | [56] | 2003 | MATLAB, Python, R, etc. all have a free package |
| 11 | Hessian LLE | [57] | 2003 | https://cs.nyu.edu/∼roweis/code.html |
| 12 | Local tangent space alignment | [58] | 2004 | https://manifoldlearningjl.readthedocs.io/en/latest/ltsa.html |
| 18 | Generalized discriminant analysis | [59] | 2000 | https://github.com/mhaghighat/gda |
| 20 | Neighborhood preserving embedding | [60] | 2005 | http://www.cad.zju.edu.cn/home/dengcai/Data/code/NPE.m |
| 21 | Locality preserving projection | [61] | 2005 | http://www.cad.zju.edu.cn/home/dengcai/Data/code/LPP.m |
| 22 | t-distributed stochastic neighbor embedding | [45] | 2008 | https://lvdmaaten.github.io/tsne/ |
| 23 | LMNN | [19] | 2005 | http://kilian.cs.cornell.edu/code/lmnn/lmnn.html |
Classic clustering methods
Clustering methods aim to group data objects into multiple classes or clusters so that objects in the same cluster are similar and different from objects in different clusters [62]. Clustering can be based on partitioning or layering, where partitioning divides objects into different clusters, and layering classifies objects into levels. Clustering based on distance clusters similar objects close to each other. Clustering based on a probability distribution model finds a set of objects in a group of objects that conform to a specific distribution model. The objects are not necessarily the closest or most similar, but they perfectly fit the model described by the probability distribution. Most clustering methods need prior knowledge on a number of clusters, and the quality of clustering needs to be improved. Classic clustering methods such as K-means [21], X-means [63], spectral clustering [23] and EM [22] can be used in single-cell clustering directly. K-means [21] is an unsupervised machine-learning technique that operates on a complete dataset without the need for a special training dataset. X-means [63] is an extended version of K-means by an improve-structure part where Euclidean distance is used to calculate the distance between each use case, and other distance functions are used to calculate the distance between any two use cases. Spectral clustering [23] is an evolutionary algorithm from graph theory. The main idea is to consider all the data as points in space that can be connected by edges. EM [22] was proposed by Arthur et al. in 1977. The EM algorithm assigns a probability distribution to each instance, which indicates the probability of it belonging to each of the clusters.
Popular clustering analysis tools
Clustering is another effective method to detect cell types. Poisson and error models can be used to count data and explain technical noise in various sources of noise in single-cell data. scRNA-seq data analysis can help in studying the heterogeneity and evolution of cancer cells. Except for a few early methods, most of the currently available integrated methods achieve state-of-the-art performances on some problems. Although the analyses of scRNA-seq data are complex and procedures may vary depending on the purpose, many mature tools have been developed to integrate two or more functions that greatly simplify the independent operations.
The challenges of clustering in scRNA-seq research are mainly reflected in an unclear number of single-cell clusters, unfixed cell types and poor scalability. In the past few years, the number of cells in scRNA-seq experiments has grown by several orders of magnitude. Although researchers have developed a variety of tools, they are not user-friendly because they use different programming languages and require different input data formats. In this section, we describe 11 of the most advanced scRNA-seq tools currently available. A summary of these methods and tools, the download URLs and other useful information are given in Table 2.
Table 2.
Summary of advanced tools
| Tools | Language | Method | Download | Cite | |
|---|---|---|---|---|---|
| 1 | SC3 | R | Cluster | https://github.com/hemberg-lab/sc3 | 10.1101/036558; 10.1038/nmeth.4236 |
| 2 | SCENIC | R/Python | Cluster | https://github.com/aertslab/SCENIC | 10.1101/144501; 10.1038/nmeth.4463 |
| 3 | BackSPIN | Python | Bicluster | https://github.com/linnarsson-lab/BackSPIN | 10.1126/science.aaa1934 |
| 4 | BiSNN-Walk | / | Bicluster | / | 10.1089/cmb.2017.0049 |
| 5 | SNN-Cliq | MATLAB/Python | Cluster | http://bioinfo.uncc.edu/SNNCliq | 10.1093/bioinformatics/btv088 |
| 6 | NMF | Python | Cluster | https://github.com/ccshao/nimfa | 10.1093/bioinformatics/btw607 |
| 7 | SIMLR | MATLAB | ML+cluster | https://github.com/BatzoglouLabSU/SIMLR | 10.1038/nmeth.4207 |
| 8 | SINCERA | R | Cluster | https://github.com/xu-lab/SINCERA | 10.1371/journal.pcbi.1004575; 10.1007/978-1-4939-7710-9_15 |
| 9 | SEURAT | R | Cluster | https://github.com/satijalab/seurat | 10.1038/nbt.3192; 10.1101/164889 |
| 10 | Monocle | R | Cluster | https://github.com/cole-trapnell-lab/monocle-release | 10.1038/nbt.2859; 10.1038/nmeth.4150; 10.1101/110668; 10.1038/nmeth.4402 |
| 11 | SCRL | C++ | ML+cluster |
https://github.com/SuntreeLi/
SCRL |
10.1093/nar/gkx750 |
SC3, an R package for clustering
SC3 was proposed by Kiselev et al. [64] in 2017. It is an interactive R package that uses a parallelization approach to avoid the need for user-specified parameters. SC3 was verified experimentally on 12 scRNA-seq datasets. SC3 constrained parameter values via a pipeline and was found to be superior to five other tested methods in terms of accuracy and stability. Because SC3 has a long run time, Kiselev et al. proposed randomly selecting subsets and constructing clusters based on the random matrix theory. They found that the estimated value was consistent with the number of original clusters suggested by them. SC3 is based on PCA and spectral dimensionality reductions, and it utilizes k-means and additionally performs the consensus clustering.
Single-cell regulatory network inference and clustering
SCENIC [64, 65]was proposed by Aibar et al. in 2017 [63] who used it to identify stable cell states in tumor and brain scRNA-seq data based on the activity of the gene regulatory networks in each cell. The authors proposed two complementary methods to handle the large dimensions of single-cell data: (i) small sample extraction to infer the gene regulatory network and (ii) gradient enhancement instead of the random forest (RF) to achieve a more efficient solution. They demonstrated that single-cell data were suitable for gene regulation and that genomic regulatory codes can be used to guide the identification of transcription factors and cell states.
SEURAT
Satija et al. [66] proposed SEURAT, a toolbox for spatial cell localization. SEURAT combines scRNA-seq data within situ RNA patterns to predict spatial cell localization. The toolkit was applied to infer the spatial location of a complete transcriptome and correctly located unusual subpopulations. The reliability of SEURAT was verified using the RNA-seq data of 851 single cells from Danio rerio embryos. SEURAT’s test dataset is Pollan [67], which includes the following four cell types: ‘NPC’, ‘GW16’, ‘GW21’ and ‘GW21+3’. The toolkit’s expression matrix includes the number of genes, the number of cells and the number of genes in each cell, as well as the number of cells in which each gene is expressed. In addition, users can look for genes that fluctuate significantly and then use those genes rather than all of them for subsequent analysis to reduce the amount of computation. Users can look at the cell population with the toolkit, and then you can look for markers for each subpopulation.
Single-cell interpretation via multi-kernel learning
In 2017, Wang et al. [68] proposed single-cell interpretation via multi-kernel learning (SIMLR), a kernel-based similarity learning method, for dimensionality reduction of scRNA-seq data. SIMLR can also be applied to large-scale datasets. They conducted single-kernel comparisons on four datasets without weight terms and showed that adding weight terms significantly enhanced SIMLR performance. Further experimental studies conducted by Zhang et al. [69] verified the robustness of SIMLR for drop-out events in single-cell data and its application to the imputed data by low-rank to have better performance than the general clustering algorithm.
SINCERA
Guo et al. [70, 71] proposed SINCERA, a pipeline for scRNA-seq profiling analysis. SINCERA can identify cell types, gene signatures and can determine key nodes. Analysis of mouse lung cells using the SINCERA pipeline distinguished the main cell types of the fetal lung. Guo et al. subsequently introduced logistic regression models that predict gene sequences, providing a valuable tool for analyzing scRNA-seq data. SINCERA is based on hierarchical clustering, by which data is converted to z-score before clustering, and the number k for clustering is determined by finding the 1st singleton in the hierarchy.
Shared nearest neighbor (SNN-Cliq)
Xu et al. [72] developed SNN-Cliq in 2005 for grouping cells of the same type. scRNA-seq data usually have tens of thousands of dimensions, and only a few of the thousands of genes are significantly expressed in different types of cells, which make the clustering problem difficult. SNN-Cliq combined with an SNN similarity metric can automatically determine the number of clusters, especially in high-dimensional single-cell data, which is a great advantage.
Nonnegative matrix factorization
In 2016, Shao et al. [73] proposed nonnegative matrix factorization (NMF) to identify subgroups in scRNA-seq datasets. Identifying cell types from single-cell data is an unsupervised problem. Although PCA is used widely, single-cell data are generally too noisy. The 1st few principal components extracted from PCA can explain only a small part of the differences, and cell subgroups are not easy to distinguish through the projection of the 1st several dimensions. The NMF approach is different from PCA because its feature superposition constraint is nonnegative. NMF was designed specially to detect single parts, which helps to detect the natural groupings of individual cells and functional cell subsets.
Monocle
To study cell differentiation, the expression profiles of individual cells are required. Monocle was developed by Trapnell et al. [74] as an unsupervised algorithm for analyzing single-cell gene expression data to reveal the expression sequence of key regulatory factors and the interactions associated with differentiation. The authors used the Monocle algorithm to study mouse myoblasts and found eight transcription factors that had not been considered previously. scRNA-seq data collected at different time points can help to reveal key events in differentiation. Monocle requires users to prepare phenotype data and feature data required by Monocle objects as well as the expression matrix, and the expression matrix is counted. This tool not only contains the general functions of a single-cell toolkit, such as quality control, difference analysis etc. In the monocle package, it is interesting to note that dimensionality reduction must be followed by clustering in order to be visualized. In addition, Monocle develops the function to infer the development trajectory, which becomes the highlight of this tool.
BackSPIN
Zeisel et al. [75] developed BackSPIN in 2015 and tested it on the adult nervous system, which is highly complex and has many cell types that are challenging to identify. scRNA-seq data were used to classify mammalian cortical cells. BackSPIN detected different types of cells based on molecule clustering and showed that transcription factors formed a complex hierarchical regulatory code, revealing the diversity of brain cell types and their transcriptomes.
BiSNN-Walk
Shi and Huang [76] proposed BiSNN-Walk, an iterative biclustering method based on SNN-Cliq [72]. BiSNN-Walk differs from SNN-Cliq in that it returns a sorted list as a reliable indicator of a cluster. In addition, BiSNN-Walk uses a metric method based on entropy to select the starting point of clustering, and its clustering ability was tested on three scRNA-seq datasets.
Single-cell representation learning
Single-cell representation learning (SCRL) [47] is a nonlinear dimensionality reduction method based on machine learning and clustering that was developed by Li et al. in 2017. To process drop-out events of single-cell RNA data, SCRL uses biological knowledge such as high-throughput RNA sequencing and adopts a network-embedding method to express a more abundant and low-dimensional expression of scRNA-seq data.
We have provided the download URLs and references for 14 other popular scRNA-seq analysis tools in Table 3, so interested readers can find them easily.
Table 3.
Summary of other popular analytical tools
| Tools | Download | Tools | Download |
|---|---|---|---|
| SAMtools | https://github.com/samtools/samtools | SCDE | https://github.com/hms-dbmi/scde |
| SART | https://github.com/alexdobin/STAR | GeneQC | http://bmbl.sdstate.edu/GeneQC/home.html |
| MAST | https://github.com/RGLab/MAST | IRIS-EDA | http://bmbl.sdstate.edu/IRIS/ |
| Kallisto | https://github.com/pachterlab/kallisto | QUBIC2 | https://github.com/maqin2001/qubic2 |
| BPSC | https://github.com/nghiavtr/BPSC | CellRanger | https://github.com/10XGenomics/cellranger |
| salmon | https://github.com/COMBINE-lab/salmon | Scater | https://github.com/davismcc/scater |
| UML-tools | https://github.com/CGATOxford/UMI-tools | SAVER | https://github.com/mohuangx/SAVER |
Performances of the methods on scRNA-seq datasets
Transcriptome datasets
The 12 datasets summarized in Table 4 are named after the primary provider of the published dataset. The 1st six datasets are benchwork labeled and are considered as the gold standard; the other six datasets are computationally labeled and are considered as silver standard. Yan’s dataset consists of the transcriptomes of human oocytes and early embryonic cells at seven key stages of development, using two to three embryos per stage. Deng’s dataset includes the transcriptomes of single cells isolated from mouse embryos at different pre-implantation stages. Treutlein’s dataset contains transcriptome data from single distal lung epithelium cells. Zeisel’s dataset contains 19 972 genes from 3005 cells and was used to study specialized cell types in mouse cortex and hippocampus.
Table 4.
Summary of scRNA-seq datasets
| Dataset | # of genes | # of cells | # of clusters | Cells in each cluster | Standard | Cell resource | Recommended methods | |
|---|---|---|---|---|---|---|---|---|
| 1 | Biase’s [77] | 25 737 | 49 | 3 | 9 + 20 + 20 | Bench | Two and four-cell Mouse embryo |
SC3, pcaReduce and SINCERA |
| 2 | Yan’s [78] | 20 214 | 124 | 7 | 3 + 3 + 6 + 12 + 20 + 16 + 30 |
Bench | Human preimplantation embryos and embryonic stem cells | pcaReduce |
| 3 | Goolam’s [79] | 41 480 | 124 | 5 | 6 + 64 + 42 + 6 + 6 | Bench | Four-cell mouse embryos | pcaReduce |
| 4 | Deng’s [80] | 22 457 | 268 | 10 | 50 + 14 + 37 + 8 + 43 + 10 + 30 + 12 + 60 + 4 |
Bench | Mouse preimplantation embryos | SC3 and pcaReduce |
| 5 | Pollen’s [67] | 23 730 | 301 | 11 | 22 + 17 + 11 + 37 + 31 + 54 + 24 + 40 + 24 + 15 + 25 |
Bench | Human | SC3, SIMLR and pcaReduce |
| 6 | Kolodziejczyk’s [81] |
38 653 | 704 | 3 | 295 + 159 + 250 | Bench | Mouse embryonic stem cell | SC3, SINCERA and SEURAT |
| 7 | Treutlein’s [82] | 23 271 | 80 | 5 | 41 + 14 + 12 + 11 + 3 | Computational | Human lung epithelium | SC3 |
| 8 | Ting’s [83] | 29 018 | 149 | 7 | 24 + 41 + 11 + 34 + 12 + 12 + 15 |
Computational | Human pancreatic circulating tumor cells | SC3 |
| 9 | Patel’s [84] | 5948 | 430 | 5 | 118 + 94 + 75 + 73 + 70 | Computational | Human glioblastomas | SC3, tSNE+kmeans and SINCERA |
| 10 | Usoskin’s [85] | 25 334 | 622 | 11 | 125 + 233 + 26 + 48 + 12 + 17 + 32 + 64 + 22 + 31 + 12 |
Computational | Human neuron | SC3 |
| 11 | Klein’s [86] | 24 175 | 2717 | 4 | 933 + 303 + 683 + 798 | Computational | Human embryonic stem cells | SC3, tSNE+kmeans and SINCERA |
| 12 | Zeisel’s [75] | 19 972 | 3005 | 9 | 290 + 390 + 948 + 820 + 98 + 175 + 198 + 26 + 60 |
Computational | Mouse cortex | SC3 |
Dataset size
We considered a small dataset as one with RNA-seq data of less than 100 cells, a large dataset had more than 1000 cells and a medium-size dataset was in the middle. As shown in Table 4, Biase’s and Treutlein’s datasets were classified as small, and Klein’s and Zeisel’s datasets were large with 2717 and 3005 cells, respectively. The numbers of genes in these 12 datasets were extremely large. Except for Patel’s dataset, which contained 5948 genes, the other 11 datasets contained from 19 972 to 41 480 genes. These scRNA-seq datasets are large and contain a lot of expression data.
Performances on raw scRNA-seq datasets
To better understand the performance of each method on scRNA-seq data, we conducted classification and clustering experiments on the raw datasets. The experimental results are particularly important because they can be used to analyze and judge whether the data preprocessing steps and algorithm improvements are effective.
We used the raw scRNA-seq data of the 12 datasets without any preprocessing with four widely used machine-learning classification methods, including KNN, RF, J48 and bagging. KNN is the most commonly used classification method, which determines the class of samples to be classified by the class of adjacent k samples. J48 is a decision tree-based algorithm. RF and bagging are integrated machine-learning algorithms. These methods are free and efficient in Weka software. The results are shown in Figure 2. The methods’ classification performance was measured with accuracy.
Figure 2.
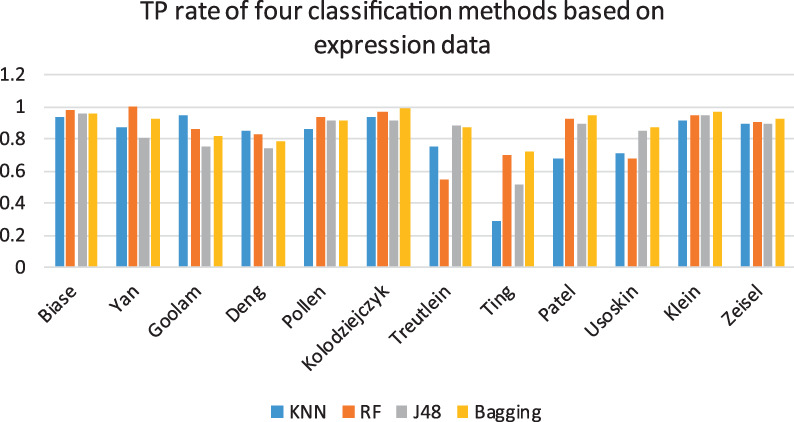
True Positive (TP) rate of four classification methods based on expression data. The blue, orange, gray and yellow bars represent the classification performance of KNN, RF, J48 and bagging, respectively.
As shown in Figure 2, In general, although the four methods showed differences in the results for the 12 datasets, the classification of expression data showed accuracies that could reach over 80%. Overall, bagging was the most stable achieving good classification accuracy on all 12 datasets, which may be explained by its integrated classification mechanism. For the six gold datasets, RF was better than the other three methods. Ting’s dataset showed the worst results among all the datasets and methods, possibly because the dataset contains too much noise, which affected the ability of the algorithms to accurately classify the expression data. Thus, for complex datasets, machine learning still has room for improvement.
Unsupervised clustering is currently the core part of the scRNA-seq analysis. It does not require researchers to make any input to the known expressed genes and can directly cluster similar cells by an algorithm. Because many subsequent analyses of single cells are based on clustering, the results of clustering have a great impact on the final conclusion. We performed experiments on the 12 datasets with three clustering methods. The results are shown in Figure 3. Greedy [87] is a hierarchical clustering algorithm adapted to large networks, which can detect high modularity partitions quickly and without limit to the number of nodes. Overall, the spectral method was more stable on all 12 datasets, which may be because of its integrated classification mechanism. We need to input the number of clustering on used clustering methods except for X-means and greedy. The default parameters of the algorithm were used in all experiments. In the 2nd picture in Figure 3, it can be easily seen that in the overall experiment results, spectral and greedy were significantly better than EM. EM showed the worst result because EM is unable to recognize expression data when there is a lot of noise.
Figure 3.
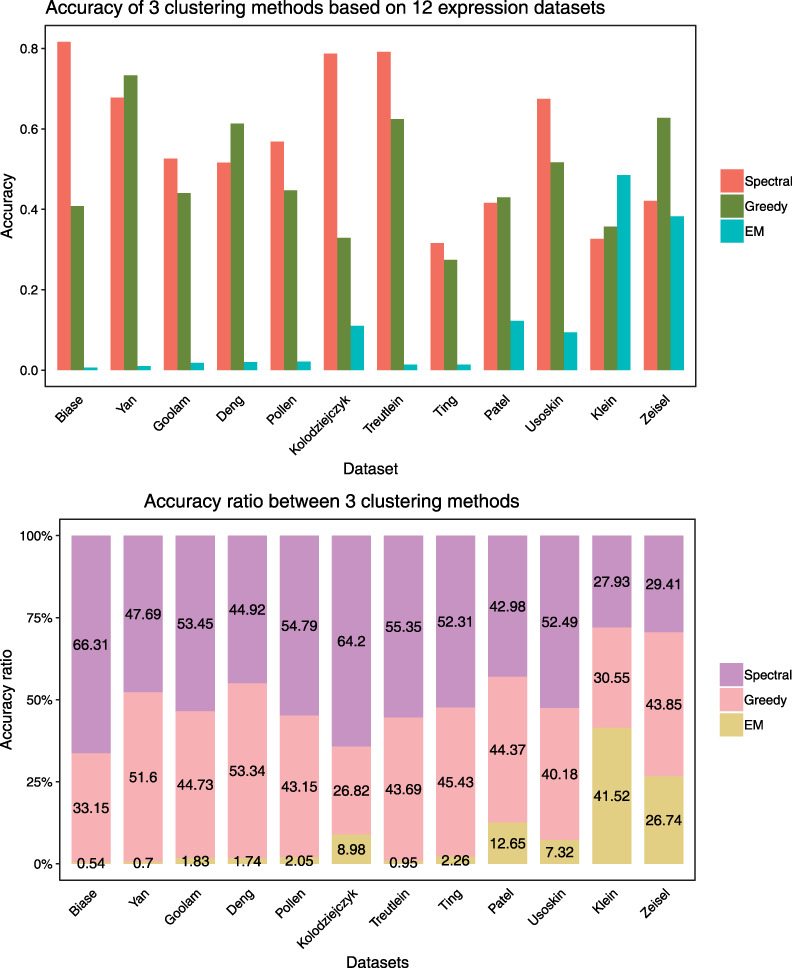
Accuracy of 3 clustering methods based on 12 expression datasets. The figure above shows the accuracy of the spectral, greedy and EM clustering on the dataset, while the figure below intuitively shows the percentage of each of the three methods in clustering performance accuracy.
To some extent, these diversities are also reflected in the effectiveness of the algorithm, that is, some methods are better for certain types of data. Because of the complexity of clustering problems, it is unlikely that one method is superior to all other methods.
Performances of seven clustering methods and toolboxes on the 12 datasets
Cell classification results vary because of different parameter combinations and different dataset sizes, making it difficult and time-consuming to find the optimized parameters for the best result. Hence, methods that integrate machine learning to provide a profound and easy-to-operate pipeline have been developed. At present, many clustering methods have been developed for single-cell data, but these methods show very different characteristics in model assumptions and clustering. Therefore, there is an urgent need for large-scale comparison and evaluation of these methods. To this end, we compared several clustering methods and tools, evaluated their clustering capabilities and conducted analysis to explore their effects on clustering cell types and detecting differentially expressed genes in the context of real data.
To evaluate the performance of clustering methods, we tested the clustering methods SC3, pcaReduce and tSNE+kmeans on 12 published datasets. The analysis refers to adjusted Rand index (ARI) due to its wide adoption in the field. In order to test the stability of the method, we repeated the experiment 100 times with fixed parameters. The clustering performance shows that SC3 has the best robustness, while pcaReduce and tSNE+kmeans still have worse performance. Figure 4 shows that most of the ARI values of SC3 are concentrated and located in the upper half of the graph, indicating that SC3 plays a stable role and has the best effect. The ARI values of tSNE+kmeans and pcaReduce are widely distributed.
Figure 4.
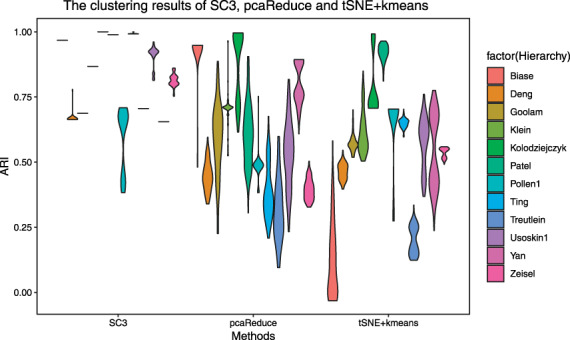
The clustering results of SC3, pcaReduce and tSNE+kmeans. We use 12 colors to represent different datasets, labeled on the right side of the picture. The column of the box in the picture represents the ARI distribution. The wider the horizontal direction is, the more clustering results are distributed in the value of ARI.
ARI of the three methods in the Pollan dataset is almost all between 0.25 and 0.6, where SC3 results are more concentrated at the endpoints of the interval, and pcaReduce values are more concentrated between 0.25 and 0.4. In comparison, tSNE+kmeans results are better, most of which are around 0.6. This shows that tSNE+kmeans is effective for dimensionality reduction of the dataset of Pollan. Based on the Biase dataset, SC3 results were stable above 0.9, and most ARI of pcaReduce were distributed between 0.8 and 0.9. On the contrary, more than half of tSNE+kmeans were below 0.1, which indicated that tSNE+kmeans was completely invalid for dimensionality reduction of Baise dataset. Usually, no dimensionality reduction method or clustering is suitable for all datasets. We can observe the characteristics of statistical datasets and find the effective dimensionality reduction method corresponding to the dataset.
Then we conducted an experiment on all datasets with the default parameters of the toolbox and recorded its experimental results as shown in Figure 5, which showed the clustering performances on five toolboxes: SC3, SNN-Cliq, SINCERA, SEURAT and pcaReduce. First, we analyze the experimental results based on the dataset Pollen and find that the results of the five clustering algorithms are very close, from which we can draw a conclusion that Pollen is universal to the algorithms. In terms of algorithmic comparison, SC3 produced the best performances, beating the other four clustering methods tested. SC3 worked best on most datasets, and its powerful integration features made it a stable toolbox. However, the SEURAT algorithm performed poorly with most datasets, and it is the most unstable method. SEURAT is a toolbox for spatial cell localization, and it may be that the noise in the single-cell RNA dataset affects the performance of the algorithm.
Figure 5.
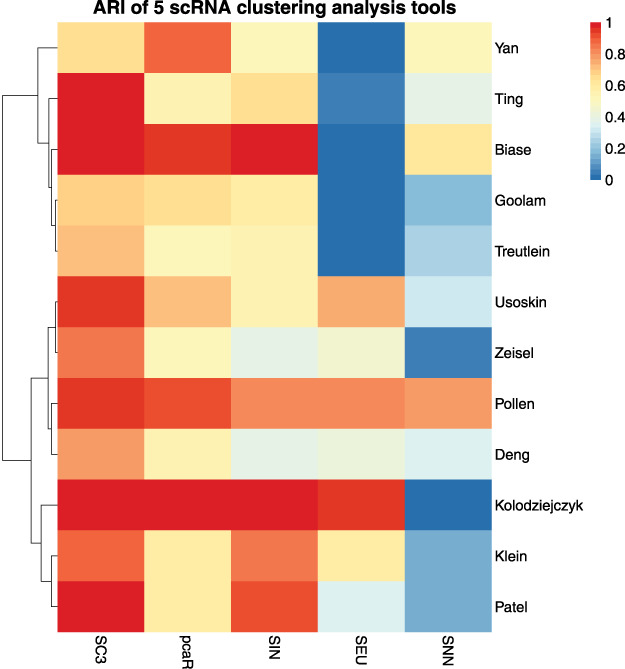
ARI of five scRNA clustering analysis tools. We use abbreviations SNN, SIN, SEU and pcaR to represent the clustering tool SNN-Cliq, SINCERA, SEURAT and pcaReduce, respectively. The closer to red means the better clustering effect, and the closer to blue means the worse clustering effect.
Discussion and conclusions
scRNA-seq provides deep scrutiny into the gene expression character of diverse cell types. The main challenge now is the noisy nature of the single-cell RNA data. This noise makes it difficult to distinguish very similar cell types, and this is where the technology needs to be improved. In this review, we covered several aspects of scRNA-seq applications, from quantitative analysis and characterization of cell types through clustering and classification to gene regulatory network reconstruction and cell-state identification. Using 12 published sets of single-cell RNA data, we showed the classification and clustering performance of each algorithm. We have listed the classical machine-learning methods, summarized the currently generally accepted toolkits and conducted experiments on the single-cell RNA dataset. At present, single-cell RNA classification has been relatively mature, but there is still a lot of room for improvement in clustering. scRNA-seq technologies allow researchers to uncover new and potentially unexpected biological discoveries compared with traditional profiling methods. scRNA-seq has also been applied to identify subclones from the transcriptomes of neoplastic cells, and the technique holds enormous potential for both basic biology and clinical applications.
Experimental studies emphasize that there is no one way to perform the best in all situations. Some of the shortcomings of these approaches, such as scalability, robustness and in some cases unavailability, need to be addressed in studies. Several recommendations have been made based on the above experiments and analysis, with details showcased in Table 4. Specifically, for the Pollen’s dataset, we recommend the algorithm with the fastest convergence (SIMLR) as it showed little difference in clustering performance among various algorithms. SIMLR is a kernel-based similarity learning method, based on dimensionality reduction of scRNA-seq data, and can be typically applied to relative large-scale datasets. For the Biase’s dataset, SC3, pcaReduce and SINCERA all achieved a better effect than the other algorithms. For the Goolam’s dataset, we recommend using pcaReduce for dimensionality reduction and clustering due to its superior prediction performance, while for the Klein’s dataset, we recommend using the tSNE+kmeans or SC3 method. From the perspective of the algorithm, it is easy to see, according to Figures 4 and 5, that SC3 is stable in most of the benchmark datasets and scales well with the most datasets in terms of their sizes and variating dimensionality. SC3, pcaReduce and SINCERA are more robust on almost datasets than the other tools in this review. For single cell datasets created from heterogeneous sources, multi-modal and multi-view learning can be introduced to combine all the gene expression data from a single cell so that the data can be complementary to each other, to make better use of the data for analysis. Further research into individual cells will contribute to the field of personalized medicine with a deeper understanding of the underlying processes of various developmental physiological and disease systems.
Key Points
The paper reviewed machine-learning approaches for clustering and classification based on the characteristics of single-cell RNA-sequencing (scRNA-seq). Efficient methods and tools for dimensionality reduction were concluded in detail.
Various tools applied in scRNA-seq were explained clearly, and we highlighted the pros and cons of each approach, which could help the readers to select proper tools to distinguish tasks.
We provided a comprehensive description of scRNA-seq data and downloaded URLs. And the performances of the methods on scRNA-seq datasets were showed in the paper.
The paper stated clearly recommendations after performed various methods and tools on a series of scRNA-seq datasets. The corresponding summary can be found in Table 4 and the discussion section at the end of this article.
Supplementary Material
Ren Qi is a doctoral student at the School of Computer Science and Technology, College of Intelligence and Computing, Tianjin University, Tianjin, China. Her research interests include machine learning, metric learning and bioinformatics.
Anjun Ma is a doctoral student at the Ohio State University, USA. His research topic mainly focuses on the single-cell gene transcription regulation analysis, including biclustering algorithm development, regulon identification and functional pathway elucidation.
Qin Ma is the director of the Bioinformatics and Mathematical Biosciences Laboratory and an associate professor at the Department of Biomedical Informatics, College of Medicine, The Ohio State University. His email is qin.ma@osumc.edu.
Quan Zou is a professor at the University of Electronic Science and Technology of China. He is a senior member of Institute of Electrical and Electronics Engineers (IEEE) and Association for Computing Machinery (ACM). He won the Clarivate Analytics Highly Cited Researchers in 2018. He majors in bioinformatics, machine learning and algorithms. His email is zouquan@nclab.net.
Funding
National Key R&D Program of China (2018YFC0910405); Natural Science Foundation of China (61771331); R01 award #1R01GM131399-01 from the National Institute of General Medical Sciences of the National Institutes of Health, and the content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1. Xu Y, Zhou X. Applications of single-cell sequencing for multiomics. Methods Mol Biol 2018;1754:327–74. [DOI] [PubMed] [Google Scholar]
- 2. Yang J, Gruenewald S, Wan X-F. Quartet-net: a quartet-based method to reconstruct phylogenetic networks. Mol Biol Evol 2013;30(5):1206–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yang JL, Grunewald S, Xu YF, et al. Quartet-based methods to reconstruct phylogenetic networks. BMC Syst Biol 2014;8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kanter I, Dalerba P, Kalisky T, et al. A cluster robustness score for identifying cell subpopulations in single cell gene expression datasets from heterogeneous tissues and tumors. Bioinformatics 2019;35(6):962–71. [DOI] [PubMed] [Google Scholar]
- 5. Xie J, Ma A, Zhang Y, et al. QUBIC2: a novel biclustering algorithm for large-scale bulk RNA-sequencing and single-cell RNA-sequencing data analysis. 2018;409961. doi: 10.1101/409961. [DOI]
- 6. Marinov GK, Williams BA, McCue K, et al. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res 2014;24(3):496–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Conesa A, Madrigal P, Tarazona S, et al. A survey of best practices for RNA-seq data analysis. Genome Biol 2016;17(13):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pan G, Tang J, Guo F. Analysis of co-associated transcription factors via ordered adjacency differences on motif distribution. Sci Rep 2017;7:43597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang J, Huang T, Petralia F, et al. Synchronized age-related gene expression changes across multiple tissues in human and the link to complex diseases. Sci Rep 2015;5:15145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johannes K, Myles B, Shirley LX. A Bayesian model for single cell transcript expression analysis on MERFISH data. Bioinformatics 2019;35(6):995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wold S, Esbensen K, Geladi PJC, et al. Principal component analysis. Chemometr Intell Lab Syst 1987;2(1):37–52. [Google Scholar]
- 12. Peng J, Wang X, Shang XJ. Combining gene ontology with deep neural networks to enhance the clustering of single cell RNA-seq data. 2018;437020. doi: 10.1101/437020. [DOI] [PMC free article] [PubMed]
- 13. Pierson E, Yau C. ZIFA: dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol 2015;16(1):241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wei L, Ran S, Bing W, et al. Integration of deep feature representations and handcrafted features to improve the prediction of N6-methyladenosine sites. Neurocomputing 2019;324:3–9. [Google Scholar]
- 15. Su R, Wu H, Xu B, et al. Developing a multi-dose computational model for drug-induced hepatotoxicity prediction based on toxicogenomics data. IEEE/ACM Trans Comput Biol Bioinform 2018. doi: 10.1109/TCBB.2018.2858756. [DOI] [PubMed] [Google Scholar]
- 16. Wei L, Xing P, Zeng J, et al. Improved prediction of protein–protein interactions using novel negative samples, features, and an ensemble classifier. Artif Intell Med 2017;83:67–74. [DOI] [PubMed] [Google Scholar]
- 17. Yang J, Zhang L. Run probabilities of seed-like patterns and identifying good transition seeds. J Comput Biol 2008;15(10):1295–313. [DOI] [PubMed] [Google Scholar]
- 18. Davis JV, Kulis B, Jain P, et al. Information-theoretic metric learning. In: Icml 07: International Conference on Machine Learning, 2007. Corvalis, Oregon, USA. [Google Scholar]
- 19. Weinberger KQ, Blitzer J, Saul LK. Distance metric learning for large margin nearest neighbor classification. In: NIPS. Vancouver, British Columbia, Canada, 2005. pp. 1473–80. [Google Scholar]
- 20. Zadeh PH, Hosseini R, Sra S. Geometric mean metric learning. In ICML. New York City, NY, USA, 2016, pp. 2464–71. [Google Scholar]
- 21. Hartigan JA, Wong MA. Algorithm AS 136: a K-means clustering algorithm. J R Stat Soc Ser C Appl Stat 1979;28(1):100–8. [Google Scholar]
- 22. Dempster AP, Laird NM, Rubin DBJJRSS. Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc Series B Stat Methodol 1977;39(1):1–38. [Google Scholar]
- 23. Ng AY, Jordan MI, Weiss Y. On spectral clustering: analysis and an algorithm. In: Proceedings of the 14th International Conference on Neural Information Processing Systems: Natural and Synthetic, 2001. Kitakyushu, Japan. [Google Scholar]
- 24. Grun D, Lyubimova A, Kester L, et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 2015;525(7568):251. [DOI] [PubMed] [Google Scholar]
- 25. Žurauskienė J, Yau C. pcaReduce: hierarchical clustering of single cell transcriptional profiles. BMC Bioinformatics 2016;17(1):140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang H, Sohn LL, Huang H, et al. Single cell clustering based on cell-pair differentiability correlation and variance analysis. Bioinformatics 2018;34(21):3684–94. [DOI] [PubMed] [Google Scholar]
- 27. Monier B, McDermaid A, Wang C, et al. IRIS-EDA: an integrated RNA-Seq interpretation system for gene expression data analysis. PLoS Comput Biol 2019;15(2):e1006792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Navin NE. Tumor evolution in response to chemotherapy: phenotype versus genotype. Cell Rep 2014;6(3):417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu X, Yang J, Zhang Y, et al. A systematic study on drug-response associated genes using baseline gene expressions of the Cancer Cell Line Encyclopedia. Sci Rep 2016;6:22811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Almendro V, Cheng Y-K, Randles A, et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep 2014;6(3):514–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chenghang Z, Sijia L, Chapman AR, et al. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012;338(6114):1622–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J, Fan HC, Behr B, et al. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell 2012;150(2):402–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Y, Waters J, Leung ML, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014;512(7513):155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ross IL, Browne CM, Hume DA, et al. Transcription of individual genes in eukaryotic cells occurs randomly and infrequently. Immunol Cell Biol 1994;72(2):177–85. [DOI] [PubMed] [Google Scholar]
- 35. Ozbudak EM, Mukund T, Iren K, et al. Regulation of noise in the expression of a single gene. Nat Genet 2002;31(1):69–73. [DOI] [PubMed] [Google Scholar]
- 36. Raj A, Bogaard P, Rifkin SA, et al. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 2008;5(10):877–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Islam S, Zeisel A, Joost S, et al. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods 2014;11(2):163. [DOI] [PubMed] [Google Scholar]
- 38. Svensson V, Natarajan KN, Ly LH, et al. Power analysis of single-cell RNA-sequencing experiments. Nat Methods 2016;14(4):381–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robinson MD, Oshlack AJGB. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 2010;11(3):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li J, Witten DM, Johnstone IM, et al. Normalization, testing, and false discovery rate estimation for RNA-sequencing data. Biostatistics 2012;13(3):523–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eberwine J, Yeh H, Miyashiro K, et al. Analysis of gene expression in single live neurons. Proc Natl Acad Sci U S A 1992;89(7):3010–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tang F, Barbacioru C, Wang Y, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 2009;6(5):377–U86. [DOI] [PubMed] [Google Scholar]
- 44. Zeng X, Lin W, Guo M, et al. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput Biol 2017;13(6):e1005420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maaten L, Hinton G. Visualizing data using t-SNE. J Mach Learn Res 2008;9:2579–605. [Google Scholar]
- 46. Lin C, Jain S, Kim H, et al. Using neural networks for reducing the dimensions of single-cell RNA-Seq data. Nucleic Acids Res 2017;45(17):e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li X, Chen W, Chen Y, et al. Network embedding-based representation learning for single cell RNA-seq data. Nucleic Acids Res 2017;45(19):e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zeng X, Zhang X, Zou Q. Integrative approaches for predicting microRNA function and prioritizing disease-related microRNA using biological interaction networks. Brief Bioinform 2016;17(2):193–203. [DOI] [PubMed] [Google Scholar]
- 49. Zou Q, Li J, Song L, et al. Similarity computation strategies in the microRNA-disease network: a survey. Brief Funct Genomics 2016;15(1):55–64. [DOI] [PubMed] [Google Scholar]
- 50. Maaten L. An introduction to dimensionality reduction using Matlab. Maastricht: Maastricht University, 2007.
- 51. Chatfield C, Collins AJ. Factor analysis In: Introduction to Multivariate Analysis. NY, US: Springer, 1980. [Google Scholar]
- 52. Kruskal JB, Wish M. Multidimensional Scaling. Thousand Oaks, CA USA: Sage Publications, 1978. [Google Scholar]
- 53. Sammon JW., Jr A Nonlinear mapping for data structure analysis. IEEE Trans Comput 1969. [Google Scholar]
- 54. Fisher RA. The use of multiple measurements in taxonomic problems. Ann Hum Genet 2012;7(2):179–88. [Google Scholar]
- 55. Roweis ST, Saul LK. Nonlinear dimensionality reduction by locally linear embedding. Science 2000;290(5500):2323–6. [DOI] [PubMed] [Google Scholar]
- 56. Belkin M, Niyogi P. Laplacian Eigenmaps for Dimensionality Reduction and Data Representation. Cambridge, MA: MIT Press, 2003. [Google Scholar]
- 57. Donoho DL, C G. Hessian eigenmaps: locally linear embedding techniques for high-dimensional data. Proc Natl Acad Sci U S A 2003;100(10):5591–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang Z, Zha H. Principal manifolds and nonlinear dimensionality reduction via tangent space alignment. Siam J Sci Comput 2004;8(4):406–24. [Google Scholar]
- 59. Baudat G, Anouar F. Generalized discriminant analysis using a kernel approach. Neural Comput 2000;12(10):2385–404. [DOI] [PubMed] [Google Scholar]
- 60. He X, Deng C, Yan S, et al. Neighborhood preserving embedding. In: Tenth IEEE International Conference on Computer Vision, 2005. Beijing, China. [Google Scholar]
- 61. He X, Niyogi P. Locality preserving projections. In: NIPS. Vancouver, British Columbia, Canada, 2003. [Google Scholar]
- 62. Xu Y, Guo M, Liu X, et al. Identify bilayer modules via pseudo-3D clustering: applications to miRNA-gene bilayer networks. Nucleic Acids Res 2016;44(20):e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ishioka T. Extended k-means with an efficient estimation of the number of clusters. In: Seventeenth International Conference on Machine Learning, 2000. Stanford, CA, USA. [Google Scholar]
- 64. Kiselev VY, Kirschner K, Schaub MT, et al. SC3: consensus clustering of single-cell RNA-seq data. Nat Methods 2017;14(5):483–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Aibar S, Gonzálezblas CB, Moerman T, et al. SCENIC: single-cell regulatory network inference and clustering. Cell 2017;14(11):1083–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rahul S, Farrell JA, David G, et al. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 2015;33(5):495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pollen AA, Nowakowski TJ, Shuga J, et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol 2014;32(10):1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang B, Zhu J, Pierson E, et al. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat Methods 2017;14(4):414. [DOI] [PubMed] [Google Scholar]
- 69. Lihua Z, Shihua CB. Comparison of computational methods for imputing single-cell RNA-sequencing data. IEEE/ACM Trans Comput Biol Bioinform 2018;1. doi: 10.1109/TCBB.2018.2848633. [DOI] [PubMed] [Google Scholar]
- 70. Guo M, Xu Y. Single-cell Transcriptome analysis using SINCERA pipeline. Methods Mol Biol 2018;1751:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Guo M, Wang H, Potter SS, et al. SINCERA: a pipeline for single-cell RNA-Seq profiling analysis. PLoS Comput Biol 2015;11(11):e1004575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xu C, Su Z. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics 2015;31(12):1974–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shao C, Höfer TJB. Robust classification of single-cell transcriptome data by nonnegative matrix factorization. Bioinformatics 2016;33(2):btw607. [DOI] [PubMed] [Google Scholar]
- 74. Trapnell C, Cacchiarelli D, Grimsby J, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014;32(4):381–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zeisel A, Munoz-Manchado AB, Codeluppi S, et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 2015;347(6226):1138–42. [DOI] [PubMed] [Google Scholar]
- 76. Shi F, Huang H. Identifying cell subpopulations and their genetic drivers from single-cell RNA-Seq data using a biclustering approach. J Comput Biol 2017;24(7):663–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Blase FH, Cao X, Zhong S. Cell fate inclination within 2-cell and 4-cell mouse embryos revealed by single-cell RNA sequencing. Genome Res 2014;24(11):1787–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yan L, Yang M, Guo H, et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol 2013;20(9):1131–9. [DOI] [PubMed] [Google Scholar]
- 79. Goolam M, Scialdone A, Graham SJL, et al. Heterogeneity in Oct4 and Sox2 targets biases cell fate in 4-cell mouse embryos. Cell 2016;165(1):61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Deng Q, Ramskold D, Reinius B, et al. Single-cell RNA-Seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 2014;343(6167):193–6. [DOI] [PubMed] [Google Scholar]
- 81. Kolodziejczyk AA, Kim JK, Tsang JCH, et al. Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 2015;17(4):471–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Treutlein B, Brownfield DG, Wu AR, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014;509(7500):371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ting DT, Wittner BS, Ligorio M, et al. Single-cell RNA sequencing identifies extracellular matrix gene expression by pancreatic circulating tumor cells. Cell Rep 2014;8(6):1905–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014;344(6190):1396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Usoskin D, Furlan A, Islam S, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 2015;18(1):145. [DOI] [PubMed] [Google Scholar]
- 86. Klein AM, Mazutis L, Akartuna I, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015;161(5):1187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Blondel VD, Guillaume JL, Lambiotte R, et al. Fast unfolding of community hierarchies in large networks. J Stat Mech 2008; abs/0803.0476. doi: 10.1088/1742-5468/2008/10/P10008. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.