

Abstract
Purpose of Review
Macrophages play key roles in tissue homeostasis and immune surveillance, mobilizing immune activation in response to microbial invasion and promoting wound healing to repair damaged tissue. However, failure to resolve macrophage activation can lead to chronic inflammation and fibrosis, and ultimately to pathology. Activated macrophages have been implicated in the pathogenesis of systemic sclerosis (SSc), although the triggers that induce immune activation in SSc and the signaling pathways that underlie aberrant macrophage activation remain unknown.
Recent Findings
Macrophages are implicated in fibrotic activation in SSc. Targeted therapeutic interventions directed against SSc macrophages may ameliorate inflammation and fibrosis.
Summary
While current studies have begun to elucidate the role of macrophages in disease initiation and progression, further work is needed to address macrophage subset heterogeneity within and among SSc end-target tissues to determine the disparate functions mediated by these subsets and to identify additional targets for therapeutic intervention.
Keywords: Systemic sclerosis, Scleroderma, Monocytes, Macrophages, Innate immunity, Fibrosis
Introduction
Mounting evidence implicates innate immunity, specifically macrophage activation, in the pathogenesis of systemic sclerosis (SSc). Multi-tissue bioinformatics analyses indicate that activated macrophages are a driver of SSc in multiple end-target organs [1, 2, 3•], and therapeutic inhibition of macrophage recruitment is associated with clinical benefit in patients with SSc [4••]. However, the molecular mechanisms by which macrophages induce inflammatory and fibrotic activation in SSc have not been completely elucidated. This review will address recent progress in defining the immunophenotypic and functional characteristics of SSc macrophages, potential mechanisms responsible for their activation, and their potential role in the induction and progression of fibrosis in patients with SSc.
Network Evidence for an Immune-Fibrotic Axis in Systemic Sclerosis
Four distinct molecular subsets have been identified among patients with SSc: inflammatory, fibroproliferative, limited, and normal-like [5, 6]. Each of the subsets contains patients that are molecularly distinct but not clinically distinguishable, are reproducible in skin across multiple patient cohorts, and respond differently to therapy [4••, 7]. An inflammatory gene expression signature, comprised largely of genes associated with immune activation and immune response signals, can be used to subset patients with SSc who have enrichment of these inflammatory processes. Gene expression studies focusing on skin have identified significantly increased expression of innate immune pathways in the inflammatory subset, including TGF-β, IL-4, and TNF-α pathways [8], and enriched macrophage and dendritic cell (DC) gene expression signatures [1].
Mahoney et al. used consensus clustering to identify common gene expression modules and to build a gene-gene co-expression network with three gene expression cohorts of skin tissue biopsies from patients with SSc and healthy controls. Five co-expressed groups of genes were identified, representing macrophage activation, adaptive immunity, interferon response, extracellular matrix (ECM) deposition and TGF-β signaling, and cell proliferation [2]. Many interconnected genes were shared among these five clusters, with most interconnections occurring between macrophages with adaptive immunity, interferon, and ECM. Notably, the macrophage activation, adaptive immunity, and interferon clusters were comprised almost entirely of inflammatory genes from the SSc inflammatory molecular patient subset [2].
Building on these results, Hinchcliff and Toledo et al. identified a high level of CD163-positive immunohistochemistry (IHC) staining in patients with SSc in therapy-naïve skin. They demonstrated that macrophage and monocyte signatures are correlated with the inflammatory gene expression signature in skin of patients with SSc [4••].
Using integrative genomics, a conserved immune-fibrotic axis was identified across multiple end-target organs in SSc (skin, lung, esophagus, and peripheral blood) [3•]. One of the largest hubs in the lung network included genes highly associated with macrophage activation and genes that encode proteins highly expressed in lung-resident macrophages (LR-MØs). Increased expression of activated macrophage gene sets in SSc-PF (pulmonary fibrosis) lung and SSc-inflammatory skin compared with control lung and control skin, respectively, was also observed in this study [3•]. Collectively, these results implicate macrophages as drivers of fibrosis in SSc. Defining the immunophenotype of SSc macrophages will facilitate therapeutic targeting of these cells.
Beyond the M1/M2 Paradigm of SSc Macrophage Activation
To deconvolute the complexity of macrophage activation, a system of nomenclature was adopted that designated macrophages as residing at one of two ends of a spectrum of polarized states: M1 and M2. M1 macrophages, which are activated by stimuli such as IFN-γ/LPS and GM-CSF, constitute one extreme end of this spectrum and are characterized by the production of pro-inflammatory mediators, including iNOS, IL-12, IL-1β, and TNF [9]. In contrast, M2 activation is elicited by diverse stimuli, including immune complexes, glucocorticoids, IL-4/IL-13, and M-CSF, and is associated with the production of angiogenic factors, growth factors, and anti-inflammatory cytokines, including IL-10 [9].
However, there are several limitations associated with reliance on M1/M2 designations. Because macrophage activation derives from local micro-environmental signals, polarization is context-dependent and mutable. The M1/M2 paradigm is based on in vitro stimulation with single inducers of activation, while macrophages are exposed in vivo to activation with multiple stimuli and, in the case of contact-dependent activation, multiple cell types. Finally, macrophages in a disease-context may not conform to uniform activation subsets or expand clonally.
Indeed, recent evidence suggests that this is true of SSc macrophages. While early reports characterized SSc macrophage activation as M2 [10, 11] based on evaluation of CD163 and CCL18 expression, subsequent studies have shown that both circulating monocytes [12••, 13] and in vitro monocyte-derived M-CSF-differentiated macrophages [14] from patients with SSc express a mix of surface markers typically associated with M1 and M2 polarization states.
However, there are discrepancies in the characteristic SSc macrophage surface marker profiles reported in these studies, which likely derive from differences between circulating blood monocytes [12••, 13] and in vitro differentiated macrophages [14]. Soldano et al. found that patients with SSc have a significantly higher percentage of monocytes that are dual positive for M2 surface markers CD163, CD206, and CD204 and M1 surface markers CD80, CD86, TLR2, and TLR4, compared with healthy controls [12••]. In contrast, Lescoat et al. noted significant decreases in CD200R1, CD204, and CD169 surface expression in SSc patient cells compared with healthy controls. Notably, in the Lescoat et al. report, peripheral blood monocytes isolated from SSc patients or healthy control subjects were differentiated with M-CSF, a cytokine that promotes a tissue resident macrophage phenotype and is constitutively produced under homeostatic conditions [14]. While increased M-CSF production has been reported in patients with arthritis and pulmonary fibrosis [15], plasma levels of M-CSF are actually lower in SSc-ILD (interstitial lung disease) patients compared with control subjects [16], so the contribution of M-CSF to SSc macrophage polarization is unclear. It is certainly possible that local tissue levels of M-CSF are elevated in SSc, although these data are not currently available. Accordingly, further studies are needed to define the complete repertoire of local regulators of macrophage activation in SSc end-target tissues to identify optimal in vitro models.
In this regard, a recent gene expression study of in vitro–differentiated macrophages from SSc patients may provide critical clues to the signaling pathways upregulated in SSc macrophages. RNA sequencing of SSc patient-derived monocyte-differentiated macrophages (MDMs) revealed a transcriptomic signature characterized by increased expression of pathways associated with glycolysis, hypoxia, and mTOR signaling and downregulation of IFNγ response pathways. Differential expression (DE) of MDMs from patients with SSc or healthy controls found over 600 genes that showed significantly different gene expression patterns, including enrichment of genes that regulate innate immune processes, and genes previously implicated in SSc pathogenesis. Ten percent of the genes previously associated with SSc from GWAS studies overlapped with this set of DE genes, allowing for a genome-wide eQTL (expression quantitative trait loci) analysis. Expression of GSDMA was highest in SSc-MDMs and was found to be significantly cis-regulated by the SNP rs3859192 [17•]. GSDMA was identified as an SSc-susceptibility gene from a transethnic GWAS study by Terao et al [18], and GSDMA is known to be regulated by TNF and is necessary for TNF-induced apoptosis in mice [19]. However, while MDMs from patients with SSc showed a strong association with GSDMA expression, this difference was not observed in SSc patient skin samples, limiting the relevance of this finding to blood-derived SSc-MDMs only [17•].
Potential Triggers of SSc Macrophage Activation
As key innate immune regulators, macrophages recognize invading microbes using pathogen-associated molecular patterns (PAMPs) and sterile stressors, which express damage-associated molecular patterns (DAMPs). Binding of these ligands to their cognate receptors initiates a cascade of signal transduction that culminates in the production of inflammatory mediators. Although genome-wide expression studies provide evidence of macrophage activation in SSc, the trigger for this is unclear.
In this regard, studies by Farina et al. implicate EBV-mediated TLR8 signaling in the chronic induction of innate immune activation in diffuse SSc patient monocytes [20]. Consistent with these findings, aberrant expression and activation of TLR8 in plasmacytoid dendritic cells (pDCs) from patients with SSc have been shown to result in release of IFN-α and CXCL4 [21], which are upregulated in SSc. TLR activation may also result from mycobiome dysregulation in patients with SSc [22, 23], although the cell type responsible for recognition of potential fungal antigens has not been identified yet.
The persistence of fibrosis in SSc suggests disease progression may also develop because of chronic stimulation by endogenous ligands released during cell injury or death. DAMPs have been implicated in the pathogenesis of many autoimmune diseases, including systemic lupus erythematosus (SLE) and rheumatoid arthritis (reviewed in [24]), and innate immune activation by sterile stressors may also play a role in SSc [25]. Endogenous mediators of activation in SSc include the TLR4 ligand Tenascin C, an ECM glycoprotein that is expressed at elevated levels in the skin of SSc patients in the inflammatory subset [26], and potentially heat shock proteins 70 and 90, which are upregulated in SSc [27, 28] and facilitate TLR signaling [29, 30]. While most studies of DAMPs have focused on their regulation of fibroblast activation, it is possible that these or other pattern recognition ligands transduce signals that culminate in SSc macrophage activation. Given these results and the important role of macrophages in microbial clearance and T cell activation, further investigation of TLR signaling is warranted in SSc macrophages.
Regulation of Macrophage Activation in SSc
Signaling Regulators
Because increasing evidence implicates a role for macrophages in disease pathogenesis, therapeutic targeting of these cells may provide benefit for patients with SSc. Identification of the signaling pathways that regulate SSc macrophage activation will be key to determining which mediators to target. Two recent studies in mouse models of fibrosis have shown that aberrant signaling of the Wnt/β-catenin pathway results in pro-fibrotic macrophage activation and failure to resolve fibrosis [31, 32]. Wnt signaling is known to be dysregulated in patients with SSc [33], and recent clinical trial results suggest the blockade of β-catenin signaling by application of a topical inhibitor, C-82, to skin shows therapeutic promise [34•]. Although this study did not examine drug effects on immune cells, it is possible that macrophage activation was locally altered by Wnt signaling inhibition, given the plasticity of these cells.
The JAK/STAT signaling pathway [33] is another potential mediator of SSc macrophage activation, as dysregulation of this pathway has been implicated in a number of fibrotic diseases, including renal and hepatic fibrosis. For example, it has been shown that binding of IL-4 to its receptor induces STAT6 phosphorylation, resulting in macrophage skewing toward a pro-fibrotic phenotype [35, 36]. Notably, IL-4 is produced at significantly higher levels following stimulation of PBMCs from patients with SSc and lung fibrosis or digital ulcers compared with healthy control subjects [37], and elevated serum IL-4 levels have also been reported in patients with SSc [38]. Thus, increased IL-4 expression may lead to enhanced STAT6 phosphorylation and macrophage activation in SSc patients. Alternatively, STAT3, a well-established modulator of macrophage polarization (reviewed in [39]) that is known to be dysregulated in SSc fibroblasts [40–42], may also be responsible for inducing a pro-fibrotic phenotypic and functional profile in SSc macrophages.
Dysregulation of TGF-β signaling may also underlie aberrant macrophage activation in SSc. TGF-β is a key mediator of fibrosis in SSc [43] and has been shown to induce macrophage activation in fibrotic disease [44]. Macrophages secrete TGF-β during wound healing and efferocytosis [45]. Given the pro-fibrotic phenotype of SSc macrophages, it is also possible that aberrant autocrine production of TGF-β may influence macrophage activation in SSc. Intriguingly, a recent study suggests efferocytosis is defective in SSc blood-derived macrophages [46], which may result in altered cytokine production. In addition, an open clinical trial with fresolimumab, a neutralizing anti-TGF-β antibody, led to clinical improvement in early diffuse patients with SSc [47], supporting a role for TGF-β in disease pathogenesis. While the analysis of clinical endpoints in this study was limited to measures of fibrosis, it is possible that macrophage activation was also altered, thus accounting in part for the demonstrated clinical benefit. However, given the complexity of macrophage polarization and the exquisite sensitivity of these cells to local micro-environmental cues (both soluble and contact-dependent), it is likely that SSc macrophage activation derives from regulation by multiple signaling pathways and that the use of combination therapies will be necessary to effectively combat disease progression.
Monocyte Epigenetics
Trained immunity is the concept that the innate immune system has memory and adaptive characteristics, and epigenetic reprogramming has been shown to play a critical role in maintaining transcriptional memory in innate immune training and tolerance [48]. In the context of chronic autoimmune and autoinflammatory disorders, there is an epigenetic transcriptional reprogramming toward increased monocyte and macrophage activation, where changes and dysregulation of chromatin architecture, epigenetic enzymes, and DNA methylation all play a role in disease pathogenesis [49]. In Ciechomska et al., the addition of a histone methylation inhibitor (DZNep) and a TLR-8 agonist to a monocyte and fibroblast co-culture system induced the upregulation of collagen and α-SMA genes in fibroblasts compared with untreated cells. These data suggest that histone demethylation and chromatin dysregulation in monocytes contribute to the transdifferentiation of fibroblasts [50]. In a more recent study, chromatin immunoprecipitation followed by sequencing (ChIP-seq) of monocytes from patients with SSc and healthy controls demonstrated that SSc monocytes have altered global chromatin marks. RNA-seq analysis showed that 381 genes were differentially expressed proportional to the chromatin mark alterations near their transcriptional start sites. These genes were associated with immune, interferon, and antiviral pathways, with binding sites for IRF and STAT transcription factors at their promoters [51••].
Consistent with these results, a recent study by Ramos et al. demonstrated robust enrichment of DNAse I hypersensitive sites (DHS) specific to myeloid cells in diffuse cutaneous SSc (dcSSc) patients [52•]. This work, which assessed differential DNA methylation in whole blood of twins discordant for SSc, used regulatory annotation to predict disease-relevant cell types and showed that methylated cytosines in regulatory regions for myeloid cells are enriched in dcSSc patients [52•].
Collectively, these data support the hypothesis that there is an inherent dysregulation in monocytes from patients who develop SSc, and aberrant epigenetic reprogramming may be critical in the initiation and development of this disease.
Therapeutic Implications
SSc treatments in current clinical use are broad-acting immunosuppressors and inhibitors of proliferation directed mainly against mediators of adaptive immunity, yet none of these therapies specifically target macrophages. Despite this, a number of clinical trials conducted with SSc patients have documented effects on macrophage gene and surface marker expression. The effects of pharmacological interventions on the immunophenotypic profile of SSc macrophages are summarized here.
Fresolimumab
Fresolimumab is a high affinity human monoclonal anti-TGFβ antibody that neutralizes all three isoforms of TGFβ [47, 53, 54]. Phase 1 and 2 clinical trials demonstrated that fresolimumab is well-tolerated in patients with primary resistant glomerulosclerosis, which is a progressive fibrosis of the glomerulus in the kidneys, and has modest, though not significant, effects on fibrosis [53, 54]. In a trial with SSc patients, fresolimumab improved skin fibrosis and significantly decreased the expression of TGFβ-regulated genes, including THBS1, SERPINE1, CTGF, and COL10A1. Significantly, the expression of macrophage/monocyte-associated genes CD163 and MS4A4A in SSc patient skin was also attenuated by fresolimumab treatment [47].
Mycophenolate Mofetil
A study of mycophenolate mofetil (MMF) in SSc demonstrated that patients most likely to derive clinical benefit from drug treatment were in the inflammatory molecular subset, suggesting MMF modulates immune cell activation [7]. In a follow-up gene expression analysis with a larger patient cohort, MMF treatment was associated with decreased inflammatory gene expression scores, downregulated mRNA and protein levels of the macrophage chemo-attractant CCL2, inhibited monocyte migration, and reduced numbers of macrophages in SSc patient skin. This longitudinal inflammatory signature pattern correlated significantly with monocyte, macrophage, and dendritic cell (DC) gene expression signatures. Notably, these effects were abrogated with cessation of MMF use. These combined data suggest that these myeloid cell types are highly correlated with the inflammatory molecular signature in the skin and may regulate skin fibrosis in SSc [4].
Pirfenidone
Pirfenidone (PFD), which is approved for the treatment of idiopathic pulmonary fibrosis (IPF)/interstitial lung disease (ILD), attenuates lung fibrosis by inhibiting the expression of inflammatory cytokines, chemokines, growth factors, and pro-collagens. PFD dose-dependently decreased the secretion of TNF, IL-1β, IL-10, GM-CSF, IP-10, and MIP-1β by alveolar macrophages isolated from patients with IPF/ILD [55], and suppressed M2 alveolar macrophage polarization, but had no effect on the expression of M1 macrophage markers [56]. In the cGVHD (chronic graft-versus-host disease) mouse model of fibrosis, PFD restored pulmonary function and reversed IPF symptoms, coincident with decreases in macrophage infiltration and TGF-β production. PFD also significantly reduced macrophage cell numbers in skin and in vitro CCL2-directed macrophage chemotaxis [57••].
Given these results, a phase II trial was undertaken to study the safety and tolerability of PFD in patients with scleroderma-associated interstitial lung disease (LOTUSS). Data from this study demonstrated that PFD has an acceptable tolerability profile that is not adversely affected by concomitant treatment with MMF [58•]. Drug effects on macrophage activation were not evaluated in this trial, but based on earlier reports in IPF, it is possible that SSc macrophage polarization and/or recruitment are altered through PFD modulation of signaling pathway activation. A recent report demonstrated that lung tissues from patients with SSc-ILD have increased the activation of the hedgehog (Hh) signaling pathway, which was inhibited by PFD [59]. Notably, Hh ligands mobilize myeloid cell migration [60] and enhance macrophage activation [61]. In addition to these studies, there are several active clinical trials that are evaluating the efficacy of PFD in SSc-related manifestations, one completed, two recruiting, and one not yet recruiting. This includes the Scleroderma Lung Study III, which will evaluate the efficacy of combining PFD with MMF in the treatment of SSc-ILD (NCT03221257).
Nintedanib
Nintedanib (NTD), a small-molecule tyrosine kinase inhibitor, is another recently approved therapy for IPF and ILD that targets angiogenic factor receptors, including PDGF, FGF, and VEGF [62••]. NTD has been shown to reduce macrophage activation and lessen vascular and fibrotic symptoms in the Fra-2 mouse model of SSc [62••, 63]. In vitro reports indicate that NTD significantly reduces M2 macrophage markers, including CD11b, CD206, CD200R, and CD209, but does not suppress membrane surface expression of M1 markers, CD40, CD80, CD83, and CD86 [64]. Based on preclinical evidence, the FDA granted NTD fast-track status for SSc-ILD patients, and phase III (SENSCIS) [65] clinical trial results should provide significant information about the clinical course of SSc-ILD and NTD-mediated effects on immune activation and fibrosis.
Tocilizumab
IL-6 contributes to the pathogenesis of SSc through direct effects on fibrosis and inflammation, and blockade of IL-6 receptor binding with the humanized monoclonal antibody tocilizumab results in improvements in both skin sclerosis and SSc-associated polyarthritis in SSc patients [66, 67]. These promising results led to the faSScinate study, which was the first randomized, placebo-controlled, phase II trial of tocilizumab for the treatment of SSc. Although tocilizumab-treated SSc patients did not demonstrate statistically significant improvement in skin thickness, clinically relevant increases in lung function were observed. Expression of genes associated with macrophage activation, including CCL18, was reduced by tocilizumab treatment [68•], suggesting IL-6 plays a role in the regulation of SSc macrophage activation. IL-6/STAT3 signaling is a well-established regulator of macrophage polarization in a variety of pathologies, including cancer (reviewed in [69]), although this signaling pathway has not been shown to direct the activation of SSc macrophages.
As greater insight into the molecular mechanisms that regulate disease pathogenesis and progression emerges, targeted therapies that interrupt these mediators and signaling pathways can be rigorously tested in clinical trials. Because of the clinical heterogeneity associated with SSc, it may be optimal to consider molecular subsetting of SSc patients to tailor the appropriate intervention to the patient population most likely to derive clinical benefit. In this regard, macrophage-targeted therapies may be most useful for treating SSc patients in the inflammatory molecular subset.
Conclusion
Given the role imputed to macrophages in the regulation of fibrosis in SSc, therapeutic interventions to modulate SSc macrophage activation may have significant clinical benefit (Fig. 1). However, translational opportunities must be developed in the context of emerging knowledge of the functional phenotype of SSc macrophage subsets. Characterization of these subsets will likely reveal new targets for precision therapy. Much of the confusion about the SSc macrophage immunophenotype stems from bulk purification methods and reliance on a handful of surface markers to define polarization. It is entirely possible, indeed likely, that SSc end-target organs contain multiple subsets of differentially polarized macrophages that mediate different functions in disease pathogenesis. Comparative transcriptomic analyses and single-cell RNA-sequencing approaches may address SSc macrophage complexity by providing unbiased classification of cells. Characterizing SSc macrophages based on their functional and transcriptional profiles will help eliminate polarization ambiguity and will inform studies to determine the function of these cells in SSc pathology.
Fig. 1.
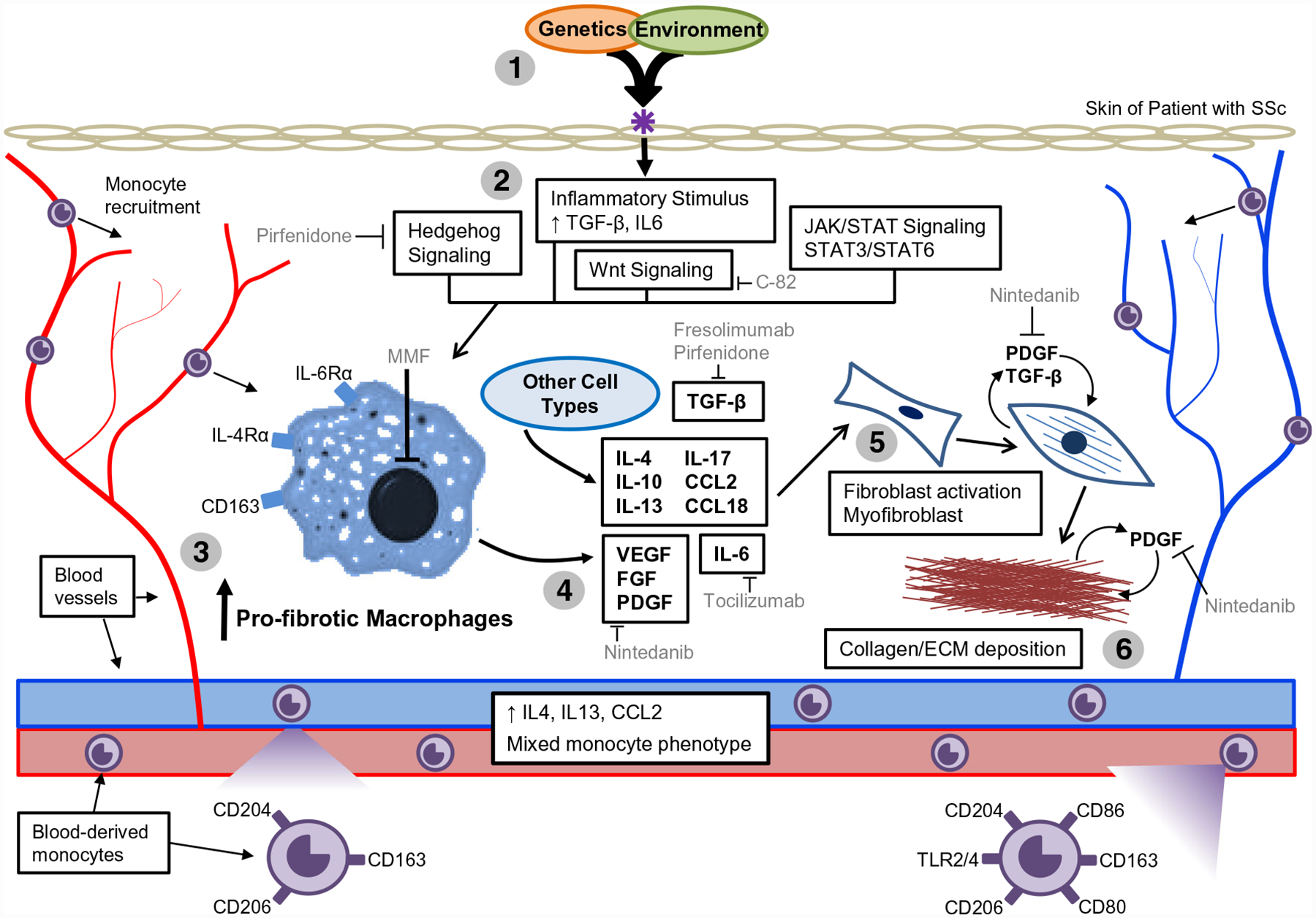
Model of SSc pathogenesis, highlighting monocyte/macrophage involvement and therapeutic interventions. (1) Genetic susceptibility combined with an environmental insult provides a signal to initiate SSc pathogenesis. (2) This triggers innate immune and inflammatory activation, downstream Wnt, JAK/STAT, and Hedgehog signaling activation, leading to the increased expression of fibrotic mediators, including TGF-β and IL-6. (3) Monocytes are recruited to the end-target tissue, resulting in pro-fibrotic macrophage activation. (4) Activated macrophages release pro-fibrotic cytokines, growth factors, and inflammatory mediators, resulting (5) in the differentiation of fibroblasts from adipocytes, fibroblast activation, and activation/recruitment of myofibroblasts. Persistent activation and inflammation cause sustained fibrosis, which is exacerbated by TGF-β-mediated stimulation of pro-fibrotic PDGF production. (6) Continued proliferation leads to collagen deposition, extracellular matrix production, and chronic fibrosis. Therapies that modulate fibrosis and macrophage activation include as follows: pirfenidone (PFD), an anti-fibrotic therapy that inhibits hedgehog signaling and TGF-β; C-82, a topical inhibitor of β-catenin signaling; mycophenolate mofetil (MMF), which inhibits lymphocyte proliferation; fresolimumab, an anti-TGF-β antibody that inhibits all three isoforms of TGF-β; and nintedanib (NTD), a small-molecule tyrosine kinase inhibitor that targets angiogenic factor receptors
Funding
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases grants R56-AR0639835 and R03-AR068097 (PAP) and by a grant from the Scleroderma Foundation (PAP). DMT received support from the National Institutes of Health p50 Specialized Center Grant, Research Diversity Supplement (3P50AR060780 - 07W1) and the John H. Copenhaver, Jr. and William H. Thomas, MD 1952 Junior Fellowship from Dartmouth Graduate Studies.
Abbreviations
- SSc
Systemic sclerosis
- ECM
Extracellular matrix
- VEGF
Vascular endothelial growth factor
- pDCs
Plasmacytoid dendritic cells
- MMF
Mycophenolate mofetil
- PFD
Pirfenidone
- IPF
Idiopathic pulmonary fibrosis
- NTD
Nintedanib
- PDGF
Platelet-derived growth factor
- FGF
Fibroblast growth factor
Footnotes
Conflict of Interest Ms. Toledo reports grants from NIH and from Dartmouth Graduate Studies, during the conduct of the study.
Dr. Pioli reports grants from Celdara Medical, LLC, grants from NIH/NIAMS, and grants from Scleroderma Foundation, during the conduct of the study; in addition, Dr. Pioli has a patent Cellular Based Therapies Targeting Disease-Associated Molecular Mediators of Fibrotic, Inflammatory, and Autoimmune Conditions pending.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Assassi S, Swindell WR, Wu M, Tan FD, Khanna D, Furst DE, et al. Dissecting the heterogeneity of skin gene expression patterns in systemic sclerosis. Arthritis Rheum. 2015;67:3016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahoney JM, Taroni J, Martyanov V, Wood TA, Greene CS, Pioli PA, et al. Systems level analysis of systemic sclerosis shows a network of immune and profibrotic pathways connected with genetic polymorphisms. PLoS Comput Biol. 2015;11:e1004005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.•.Taroni JN, Greene CS, Martyanov V, Wood TA, Christmann RB, Farber HW, et al. A novel multi-network approach reveals tissue-specific cellular modulators of fibrosis in systemic sclerosis. Genome Med. 2017;9:27. [DOI] [PMC free article] [PubMed] [Google Scholar]; A meta-analysis combining multiple gene expression datasets from different SSc-affected tissues, identifying a cluster of genes associated with alternatively activated macrophages.
- 4.••.Hinchcliff M, Toledo DM, Taroni JN, Wood TA, Franks JM, Ball MS, et al. Mycophenolate mofetil treatment of systemic sclerosis reduces myeloid cell numbers and attenuates the inflammatory gene signature in skin. J Invest Dermatol. 2018;138:1301–10 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study focuses on the molecular effects of long-term MMF therapy on the macrophages in SSc patient skin.
- 5.Milano A, Pendergrass SA, Sargent JL, George LK, McCalmont TH, Connolly MK, et al. Molecular subsets in the gene expression signatures of scleroderma skin. PLoS One. 2008;3:e2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pendergrass SA, Lemaire R, Francis IP, Mahoney JM, Lafyatis R, Whitfield ML. Intrinsic gene expression subsets of diffuse cutaneous systemic sclerosis are stable in serial skin biopsies. J Invest Dermatol. 2012;132:1363–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hinchcliff M, Huang CC, Wood TA, Matthew Mahoney J, Martyanov V, Bhattacharyya S, et al. Molecular signatures in skin associated with clinical improvement during mycophenolate treatment in systemic sclerosis. J Invest Dermatol. 2013;133:1979–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson ME, Pioli PA, Whitfield ML. Gene expression profiling offers insights into the role of innate immune signaling in SSc. Semin Immunopathol. 2015;37:501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86. [DOI] [PubMed] [Google Scholar]
- 10.Christmann RB, Sampaio-Barros P, Stifano G, Borges CL, de Carvalho CR, Kairalla R, et al. Association of Interferon- and transforming growth factor beta-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheum. 2014;66:714–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higashi-Kuwata N, Jinnin M, Makino T, Fukushima S, Inoue Y, Muchemwa FC, et al. Characterization of monocyte/macrophage subsets in the skin and peripheral blood derived from patients with systemic sclerosis. Arthritis Res Ther. 2010;12:R128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.••.Soldano S, Trombetta AC, Contini P, Tomatis V, Ruaro B, Brizzolara R, et al. Increase in circulating cells coexpressing M1 and M2 macrophage surface markers in patients with systemic sclerosis. Ann Rheum Dis. 2018;77:1842–+ [DOI] [PubMed] [Google Scholar]; This study finds that circulating monocytes from the blood of patients with SSc have a mixed M1/M2 phenotype.
- 13.Trombetta AC, Soldano S, Contini P, Tomatis V, Ruaro B, Paolino S, et al. A circulating cell population showing both M1 and M2 monocyte/macrophage surface markers characterizes systemic sclerosis patients with lung involvement. Respir Res. 2018;19:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lescoat A, Ballerie A, Jouneau S, Fardel O, Vernhet L, Jego P, Lecureur V. 2018. M1/M2 polarisation state of M-CSF blood-derived macrophages in systemic sclerosis. Ann Rheum Dis, annrheumdis-2018–214333. [DOI] [PubMed] [Google Scholar]
- 15.Ushach I, Zlotnik A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J Leukoc Biol. 2016;100:481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, et al. Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Investig. 2010;90:812–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.•.Moreno-Moral A, Bagnati M, Koturan S, Ko JH, Fonseca C, Harmston N, et al. Changes in macrophage transcriptome associate with systemic sclerosis and mediate GSDMA contribution to disease risk. Ann Rheum Dis. 2018;77:596–601 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study focuses on the gene expression of monocyte-derived macrophages from patients with SSc and healthy controls, and performs an eQTL analysis with previous GWAS findings and reports that GSDMA gene upregulation was cis-regulated by SNP rs3859192.
- 18.Terao C, Kawaguchi T, Dieude P, Varga J, Kuwana M, Hudson M, et al. Transethnic meta-analysis identifies GSDMA and PRDM1 as susceptibility genes to systemic sclerosis. Ann Rheum Dis. 2017;76:1150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lei M, Bai X, Yang T, Lai X, Qiu W, Yang L, et al. Gsdma3 is a new factor needed for TNF-alpha-mediated apoptosis signal pathway in mouse skin keratinocytes. Histochem Cell Biol. 2012;138:385–96. [DOI] [PubMed] [Google Scholar]
- 20.Farina A, Peruzzi G, Lacconi V, Lenna S, Quarta S, Rosato E, et al. Epstein-Barr virus lytic infection promotes activation of Toll-like receptor 8 innate immune response in systemic sclerosis monocytes. Arthritis Res Ther. 2017;19:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ah Kioon MD, Tripodo C, Fernandez D, Kirou KA, Spiera RF, Crow MK, et al. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci Transl Med. 2018;10: eaam8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arron ST, Dimon MT, Li Z, Johnson ME, Wood TA, Feeney L, et al. High Rhodotorula sequences in skin transcriptome of patients with diffuse systemic sclerosis. J Invest Dermatol. 2014;134:2138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson ME, Franks JM, Cai G, Mehta BK, Wood TA, Archambault K, et al. Microbiome dysbiosis is associated with disease duration and increased inflammatory gene expression in systemic sclerosis skin. Arthritis Res Ther. 2019;21:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Reilly S Pound the alarm: danger signals in rheumatic diseases. Clin Sci (Lond). 2015;128:297–305. [DOI] [PubMed] [Google Scholar]
- 25.O’Reilly S, van Laar JM. Targeting the TLR4-MD2 axis in systemic sclerosis. Nat Rev Rheumatol. 2018;14:564–6. [DOI] [PubMed] [Google Scholar]
- 26.Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, et al. Tenascin-C drives persistence of organ fibrosis. Nat Commun. 2016;7:11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogawa F, Shimizu K, Hara T, Muroi E, Hasegawa M, Takehara K, et al. Serum levels of heat shock protein 70, a biomarker of cellular stress, are elevated in patients with systemic sclerosis: association with fibrosis and vascular damage. Clin Exp Rheumatol. 2008;26:659–62. [PubMed] [Google Scholar]
- 28.Tomcik M, Zerr P, Pitkowski J, Palumbo-Zerr K, Avouac J, Distler O, et al. Heat shock protein 90 (Hsp90) inhibition targets canonical TGF-beta signalling to prevent fibrosis. Ann Rheum Dis. 2014;73: 1215–22. [DOI] [PubMed] [Google Scholar]
- 29.Saito K, Kukita K, Kutomi G, Okuya K, Asanuma H, Tabeya T, et al. Heat shock protein 90 associates with Toll-like receptors 7/9 and mediates self-nucleic acid recognition in SLE. Eur J Immunol. 2015;45:2028–41. [DOI] [PubMed] [Google Scholar]
- 30.Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–12. [DOI] [PubMed] [Google Scholar]
- 31.Feng Y, Ren J, Gui Y, Wei W, Shu B, Lu Q, et al. Wnt/beta-catenin-promoted macrophage alternative activation contributes to kidney fibrosis. J Am Soc Nephrol. 2018;29:182–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sennello JA, Misharin AV, Flozak AS, Berdnikovs S, Cheresh P, Varga J, et al. Lrp5/beta-catenin signaling controls lung macrophage differentiation and inhibits resolution of fibrosis. Am J Respir Cell Mol Biol. 2017;56:191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei J, Fang F, Lam AP, Sargent JL, Hamburg E, Hinchcliff ME, et al. Wnt/beta-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. 2012;64:2734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.•.Lafyatis R, Mantero JC, Gordon J, Kishore N, Carns M, Dittrich H, et al. Inhibition of beta-catenin signaling in the skin rescues cutaneous adipogenesis in systemic sclerosis: a randomized, double-blind, placebo-controlled trial of C-82. J Investig Dermatol. 2017;137:2473–83 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that the inhibition of β-catenin signaling with a topical inhibitor showed therapeutic effects to SSc-affected skin.
- 35.Tao B, Jin W, Xu JQ, Liang ZY, Yao JL, Zhang Y, et al. Myeloid-specific disruption of tyrosine phosphatase Shp2 promotes alternative activation of macrophages and predisposes mice to pulmonary fibrosis. J Immunol. 2014;193:2801–11. [DOI] [PubMed] [Google Scholar]
- 36.Weng SY, Wang XY, Vijayan S, Tang YL, Kim YO, Padberg K, et al. IL-4 receptor alpha signaling through macrophages differentially regulates liver fibrosis progression and reversal. Ebiomedicine. 2018;29:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dantas AT, de Almeida AR, Sampaio MCPD, Cordeiro MF, de Oliveira PSS, Mariz HD, et al. Different profile of cytokine production in patients with systemic sclerosis and association with clinical manifestations. Immunol Lett. 2018;198:12–6. [DOI] [PubMed] [Google Scholar]
- 38.Needleman BW, Wigley FM, Stair RW. Interleukin-1, interleukin-2, interleukin-4, interleukin-6, tumor-necrosis-factor-alpha, and interferon-gamma levels in sera from patients with scleroderma. Arthritis Rheum. 1992;35:67–72. [DOI] [PubMed] [Google Scholar]
- 39.Rebe C, Vegran F, Berger H, Ghiringhelli F. STAT3 activation: a key factor in tumor immunoescape. JAKSTAT. 2013;2:e23010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chakraborty D, Sumova B, Mallano T, Chen CW, Distler A, Bergmann C, et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun. 2017;8:1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Papaioannou I, Xu S, Denton CP, Abraham DJ, Ponticos M. STAT3 controls COL1A2 enhancer activation cooperatively with JunB, regulates type I collagen synthesis posttranscriptionally, and is essential for lung myofibroblast differentiation. Mol Biol Cell. 2018;29:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pedroza M, To S, Assassi S, Wu M, Tweardy D, Agarwal SK. Role of STAT3 in skin fibrosis and transforming growth factor beta signalling. Rheumatology (Oxford). 2018;57:1838–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morris E, Chrobak I, Bujor A, Hant F, Mummery C, Ten Dijke P, et al. Endoglin promotes TGF-beta/Smad1 signaling in scleroderma fibroblasts. J Cell Physiol. 2011;226:3340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nacu N, Luzina IG, Highsmith K, Lockatell V, Pochetuhen K, Cooper ZA, et al. Macrophages produce TGF-beta-induced (beta-ig-h3) following ingestion of apoptotic cells and regulate MMP14 levels and collagen turnover in fibroblasts. J Immunol. 2008;180:5036–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ballerie A, Lescoat A, Augagneur Y, Lelong M, Morzadec C, Cazalets C, et al. Efferocytosis capacities of blood monocyte-derived macrophages in systemic sclerosis. Immunol Cell Biol. 2019;97:340–7. [DOI] [PubMed] [Google Scholar]
- 47.Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest. 2015;125:2795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van der Heijden C, Noz MP, Joosten LAB, Netea MG, Riksen NP, Keating ST. Epigenetics and trained immunity. Antioxid Redox Signal. 2018;29:1023–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arts RJW, Joosten LAB, Netea MG. The potential role of trained immunity in autoimmune and autoinflammatory disorders. Front Immunol. 2018;9:298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ciechomska M, O’Reilly S, Przyborski S, Oakley F, Bogunia-Kubik K, van Laar JM. Histone demethylation and toll-like receptor 8-dependent cross-talk in monocytes promotes transdifferentiation of fibroblasts in systemic sclerosis via Fra-2. Arthritis Rheum. 2016;68:1493–504. [DOI] [PubMed] [Google Scholar]
- 51.••.van der Kroef M, Castellucci M, Mokry M, Cossu M, Garonzi M, Bossini-Castillo LM, et al. Histone modifications underlie monocyte dysregulation in patients with systemic sclerosis, underlining the treatment potential of epigenetic targeting. Ann Rheum Dis. 2019;78:529–38 [DOI] [PubMed] [Google Scholar]; This study identified altered global methylation marks in monocytes from patients with SSc when compared with healthy control monocytes.
- 52.•.Ramos PS, Zimmerman KD, Haddad S, Langefeld CD, Medsger TA Jr, Feghali-Bostwick CA. Integrative analysis of DNA methylation in discordant twins unveils distinct architectures of systemic sclerosis subsets. Clin Epigenetics. 2019;11:58. [DOI] [PMC free article] [PubMed] [Google Scholar]; Recent study that found an enrichment of methylated cytosines in regulatory regions in myeloid cells.
- 53.Trachtman H, Fervenza FC, Gipson DS, Heering P, Jayne DR, Peters H, et al. A phase 1, single-dose study of fresolimumab, an anti-TGF-beta antibody, in treatment-resistant primary focal segmental glomerulosclerosis. Kidney Int. 2011;79:1236–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vincenti F, Fervenza FC, Campbell KN, Diaz M, Gesualdo L, Nelson P, et al. A phase 2, double-blind, placebo-controlled, randomized study of fresolimumab in patients with steroid-resistant primary focal segmental glomerulosclerosis. Kidney Int Rep. 2017;2:800–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He X, Theegarten D, Guzman J, Costabel U, Bonella F. Effect of pirfenidone (PFD) on cytokine/chemokine release from alveolar macrophages (AMs) in interstitial lung diseases (ILD): preliminary results. Eur Respir J. 2013;42:P2334. [Google Scholar]
- 56.Toda M, Mizuguchi S, Minamiyama Y, Yamamoto-Oka H, Aota T, Kubo S, et al. Pirfenidone suppresses polarization to M2 phenotype macrophages and the fibrogenic activity of rat lung fibroblasts. J Clin Biochem Nutr. 2018;63:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.••.Du J, Paz K, Flynn R, Vulic A, Robinson TM, Lineburg KE, et al. Pirfenidone ameliorates murine chronic GVHD through inhibition of macrophage infiltration and TGF-beta production. Blood. 2017;129:2570–80 [DOI] [PMC free article] [PubMed] [Google Scholar]; This study finds that in the cGVHD mouse model of fibrosis and SSc, pirfenidone decreased macrophage infiltration, reduced macrophage chemotaxis toward CCL2, and ameliorated symptoms of fibrosis.
- 58.•.Khanna D, Albera C, Fischer A, Khalidi N, Raghu G, Chung L, et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: the LOTUSS trial. J Rheumatol. 2016;43:1672–9 [DOI] [PubMed] [Google Scholar]; Phase II trial to study the safety and tolerability of pirfenidone in SSc patients with interstitial lung disease.
- 59.Xiao H, Zhang GF, Liao XP, Li XJ, Zhang J, Lin H, et al. Anti-fibrotic effects of pirfenidone by interference with the hedgehog signalling pathway in patients with systemic sclerosis-associated interstitial lung disease. Int J Rheum Dis. 2018;21:477–86. [DOI] [PubMed] [Google Scholar]
- 60.Seki E HEDGEHOG signal in hepatocytes mediates macrophage recruitment: a new mechanism and potential therapeutic target for fatty liver disease. Hepatology. 2016;63:1071–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pereira TA, Xie G, Choi SS, Syn WK, Voieta I, Lu J, et al. Macrophage-derived hedgehog ligands promotes fibrogenic and angiogenic responses in human schistosomiasis mansoni. Liver Int. 2013;33:149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.••.Huang J, Maier C, Zhang Y, Soare A, Dees C, Beyer C, et al. Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis. Ann Rheum Dis. 2017;76:1941–8 [DOI] [PubMed] [Google Scholar]; This study shows that nintedanib reduces macrophage activation and ameliorates symptoms of fibrosis in the Fra-2 mouse model of SSc.
- 63.Maurer B, Distler JH, Distler O. The Fra-2 transgenic mouse model of systemic sclerosis. Vasc Pharmacol. 2013;58:194–201. [DOI] [PubMed] [Google Scholar]
- 64.Bellamri N, Morzadec C, Lecureur V, Joannes A, Wollin L, Jouneau S, et al. Effects of Nintedanib on the M1 and M2a polarization of human macrophages. Eur Respir J. 2018;52:PA5250. [Google Scholar]
- 65.Distler O, Brown KK, Distler JHW, Assassi S, Maher TM, Cottin V, et al. Design of a randomised, placebo-controlled clinical trial of nintedanib in patients with systemic sclerosis-associated interstitial lung disease (SENSCIS). Clin Exp Rheumatol. 2017;35(Suppl 106):75–81. [PubMed] [Google Scholar]
- 66.Elhai M, Meunier M, Matucci-Cerinic M, Maurer B, Riemekasten G, Leturcq T, et al. Outcomes of patients with systemic sclerosis-associated polyarthritis and myopathy treated with tocilizumab or abatacept: a EUSTAR observational study. Ann Rheum Dis. 2013;72:1217–20. [DOI] [PubMed] [Google Scholar]
- 67.Shima Y, Kuwahara Y, Murota H, Kitaba S, Kawai M, Hirano T, et al. The skin of patients with systemic sclerosis softened during the treatment with anti-IL-6 receptor antibody tocilizumab. Rheumatology (Oxford). 2010;49:2408–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.•.Khanna D, Denton CP, Lin CJF, van Laar JM, Frech TM, Anderson ME, et al. Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: results from the open-label period of a phase II randomised controlled trial (faSScinate). Ann Rheum Dis. 2018;77:212–20 [DOI] [PMC free article] [PubMed] [Google Scholar]; Phase II study of tocilizumab treatment for SSc, finding that expression of macrophage activation markers, like CCL18, was reduced after treatment, and clinical increases in lung function were observed.
- 69.Mauer J, Denson JL, Bruning JC. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015;36:92–101. [DOI] [PubMed] [Google Scholar]