

Abstract
Myxopapillary ependymomas (MPE) are considered benign (World Health Organization (WHO) grade I) neoplasms with favorable prognosis. However, malignant behavior occurs in a small subset. To our knowledge, only five anaplastic MPEs have been reported without consensus on diagnostic criteria. We retrieved 14 anaplastic MPEs from the pathology archives of six institutions. Each tumor included at least two of the following features: ≥5 mitoses per 10 high power fields, Ki‐67 labeling index (LI) ≥10%, microvascular proliferation (MVP) and spontaneous necrosis. These features were typically encountered in the foci of hypercellularity and reduced mucin. There were eight male and six female patients (age range 6–57 years, median = 16.5). Ten tumors displayed anaplasia at initial resection, and 4 were anaplastic at a second surgery for recurrence (ranging from 9 months to 14 years following initial resection). The Ki‐67 LI ranged between 8% and 40% in the anaplastic foci and <3% in the foci of classic MPE. There was documented cerebrospinal fluid (CSF) dissemination in seven cases, recurrence following an anaplastic diagnosis in three cases and bone or soft tissue invasion in two cases. One patient suffered lung metastases. Two cases evaluated by targeted next‐generation sequencing and one evaluated by fluorescence in situ hybridization (FISH) showed nonspecific chromosomal gains. We conclude that although rare, anaplastic MPE occurs in both pediatric and adult patients, similar to other ependymomas. At a minimum, closer follow‐up is recommended, given the concern for aggressive biologic potential. Further study is needed to determine WHO grading criteria and genetic indicators of tumor progression.
Keywords: anaplastic transformation, CSF dissemination, malignant neoplasm, metastasis, microvascular proliferation, myxopapillary ependymoma, necrosis, recurrence
Introduction
Myxopapillary ependymomas (MPEs) account for 9%–13% of all ependymomas, and approximately 83% of ependymomas in the region of the filum terminale 21. They predominantly arise as intradural neoplasms in the region of the conus medullaris, cauda equina and filum terminale 21, and rarely as extradural neoplasms from the filum terminale externa 9. Occasionally, they occur at other central nervous system (CNS) locations including the cervical and thoracic spinal cord, the fourth ventricle, the lateral ventricles and the brain parenchyma 21. Less commonly, they have been reported to occur in the absence of a previous or concomitant spinal cord MPE, at sites including the subcutaneous sacrococcygeal region, presacral region and mediastinum, possibly arising from ectopic ependymal rests 1, 7, 11, 12, 13, 18, 19, 23, 28, 32, 33, 34, 40.
MPEs typically have low mitotic activity and Ki‐67 labeling indices (LI), with fewer than 5 mitoses/10 high power fields (HPF), Ki‐67 LI ranging from 0% to 5.5% 27, and averaging 1.6% 29. Anaplasia within MPEs has been described as exceptional. Only five have been reported in the literature to our knowledge, most occurring within the pediatric setting with ages ranging from 7 months to 15 years 2, 4, 5, 37 and one occurring in an adult at 38 years of age 38. Two cases were intradural, two were subcutaneous sacrococcygeal lesions and one was anaplastic at the time of metastasis to an inguinal lymph node 2, 4, 5, 37, 38. Malignant features included punctate necrosis, microvascular proliferation (MVP), increased Ki‐67 LI (80%, 70%, 40% and 10% respectively) and increased mitotic activity 2, 4, 5, 37. One anaplastic sacrococcygeal MPE occurred in the setting of Schinzel–Giedion syndrome with anaplastic ependymoblastic areas 4.
Most MPEs have a favorable prognosis; however, some recur or spread within the neuraxis. Nevertheless, most such cases retain their otherwise benign (WHO grade I) histopathology. Subtotal resection, lack of initial radiation treatment and a young age at diagnosis are factors that have been associated with worse prognosis 17, 39. In some studies, pediatric patients have a less predictable outcome than their adult counterparts, even after gross total resection 21, 31, 35. Extraneural metastases from primary intradural MPEs are rare 30, as are advanced cases in which the recurrence involves the adjacent soft tissues 22, and cases that have both intradural and soft tissue involvement at initial presentation 38. In contrast, metastases in subcutaneous sacrococcygeal MPEs have been reported in up to 20% of the cases, typically to inguinal lymph nodes or the lung 11, 19, 40, and there are frequent local recurrences in postsacral and presacral MPEs 34, 36.
Materials and Methods
Fourteen patients with anaplastic MPE (10 primary, 4 diagnosed at recurrence) were encountered from six institutions, including consultations (9 in‐house cases, 5 consult cases). Anaplasia was defined as having at least two of the following features: ≥5 mitoses per 10 HPF, Ki‐67 labeling index (LI) ≥10%, MVP and spontaneous necrosis. The presence of necrosis included both palisading and non‐palisading forms; however, necrosis did not qualify as an anaplastic feature if the patient previously received radiation or chemotherapy because of the difficulties in distinguishing tumor necrosis from treatment effects. A Ki‐67 LI ≥10% was selected as it is significantly higher than the typically reported values for MPEs, which range from 0% to 5.5% 27, and average 1.6% 29.
Two cases (primary tumor case #5, and recurrent tumor case #8) were evaluated by next‐generation DNA sequencing (NGS). One was evaluated by the UCSF500 Cancer Panel 10, 14, 25, 26, which assesses approximately 500 cancer‐associated genes for mutation, copy number alterations and structural variants including gene fusions. An additional tumor was evaluated by the Children's Hospital of Philadelphia Comprehensive Solid Tumor NGS Panel V2.0, which includes sequence and copy number analyses of 238 cancer genes, and fusions associated with 110 cancer genes (genes listed at https://www.testmenu.com/chop/Tests/785967). One tumor was evaluated by centromere enumerating FISH probes for chromosomes 1, 7 and 9 by the Colorado Children's Hospital Genetics Laboratory.
Results
The clinicopathologic features of the 14 patients with anaplastic MPEs, 13 of 14 documented to be initially intradural, are summarized in Table 1. There were eight male and six female patients (eight pediatric and six adults) with a median age of 14.5 years (range 6–55 years) at diagnosis of MPE, and median age of 16.5 years (range 6–57 years) at diagnosis of anaplastic MPE (anaplasia was present only at recurrence in four cases). In the majority of the cases, the initial main mass occurred in the lumbosacral region, ranging in size from 1.7 cm to 11.5 cm. One case was located at the floor of the fourth ventricle (case #11), and one case was described as a sciatic notch mass in which it could not clearly be determined if this was a primary soft tissue tumor rather than an initially intradural mass (case #10). At the time of initial surgery, there was gross total resection in eight cases, subtotal resection in five cases and in one case the extent of resection was not indicated; 11 cases received adjuvant radiation therapy after the initial surgery.
Table 1.
The clinicopathologic features for 14 cases of anaplastic myxopapillary ependymoma. Cases are ordered by age of presentation (eight pediatric cases followed by six adult cases). More than one size or location is listed for cases with recurrences, separated by commas. The term “initial” is used to indicate the initial resection, a first recurrence is denoted as 1st, a second recurrence is denoted as 2nd. Features considered anaplastic are highlighted in green. Yellow boxes indicate the presence of necrosis that occurred after radiation or chemotherapy, and therefore was not considered an anaplastic feature; this is also indicated by an asterisk. Involvement of the adjacent tissue was assessed by histologic and/or radiographic findings. In cases with a large tumor size and direct invasion of the adjacent bone or soft tissue (case #12 and #10), the integrity of the tumor capsule would be lost. In cases with bone erosion, bone remodeling, involvement of sacral rootlets, involvement of dura or an entrapped ganglion, this was not taken as a direct indication of capsule integrity. M: male, F: female, MVP: microvascular proliferation, HPF: high power fields, STR: subtotal resection, GTR: gross total resection, Res: resection in which gross total or subtotal resection was not specified, XRT: radiation therapy, pXRT: partial course of radiation therapy, Chem: chemotherapy, C: cervical, T: thoracic, L: lumbar
| Case # | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at MPE diagnosis in years, (age at anaplasia diagnosis, if later) | 6 | 7 | 10 | 10 | 11 | 12 | 13 | 16, (20) | 31, (32) | 31, (45) | 40 | 45, (57) | 50 | 55 |
| Gender | F | F | M | M | M | M | M | F | M | F | F | M | M | F |
| Location | L4‐S1 (intradural) | T12‐L3 (intradural) | S1‐S2 (intradural) | L1‐L2 (intradural, centered at conus medullaris) | filum terminale (L4‐5, S2‐3) and S1 nerve root (3 masses) (intradural) | lumbar mass, lumbar and sacral (intradural) | L1‐L2 with multiple smaller lesions (posterior fossa, C, T, and L cistern) (intradural) | L3‐L4 & L5‐S1, multiple T and L lesions (intradural) | sacral filum terminale region mass with smaller T lesions (intradural) | sciatic notch, pelvic & sciatic masses (unclear if MPE was a primary soft tissue form) | floor of 4th ventricle | T11‐L2, L1, T8‐L5 (originally an intradural mass filling the thecal sac) | L5‐S3 (intradural) | L1‐L2, L2 (intradural) |
| Size | size not specified | not specified | 4.2 cm | 4.5 cm | 3 cm mass on imaging | 3.2 cm, 1.5 and 1 cm | main mass 3.8 cm | 1.7 and 1.0 cm, multiple (up to 1.6 cm) | not specified | initial size not known, 10 cm and 5.5 cm | 5 cm | 11.5 cm, 10 cm, size of second recurrence not specified | 8.5 cm | 2.7 cm, 2.3 cm |
| Ki‐67 LI (data source) | 20% (initial resection) | 11% (initial resection) | 34% (initial resection) | 15% (initial resection) | 14% (initial resection) | 10%, 17% (initial resection, 1st recurrence) | 8% (initial resection) | 10% (1st recurrence) | 10% (9m re‐resection) | 40% (1st recurrence) | 20% (initial resection) | 26% (1st recurrence) | 10% (initial resection) | 20% (initial resection) |
| Mitotic Index per 10 HPF | 12 | 10 | 20 | 6 | 5 | 3, 2 | 6 | 6 | 7 | 7 | 5 | 17 | 14 | 7 |
| Necrosis | Yes | No | Yes: focal non‐palisading | No | Yes: non‐palisading | Yes: palisading (only on recurrence) | No | No | * Yes: non‐palisading and palisading | * Yes: non‐palisading and palisading | Yes: focal non‐palisading | * Yes: non‐palisading | Yes: non‐palisading and palisading | Yes: non‐palisading |
| MVP | Yes | Yes | Yes | Yes: small foci of MVP | Yes: multiple foci | Yes (initial and recurrence) | Yes | No | Yes | Yes | Yes | Yes: prominent | Yes: extensive | Yes |
| Onset of anaplastic features | initial resection | initial resection | initial resection | initial resection | initial resection | initial resection | initial resection | recurrence | present on recurrence/ second surgery at 9 months | first recurrence | initial resection | first recurrence | initial resection | initial resection |
| Treatment | GTR, proton therapy | STR, XRT | GTR, proton therapy | GTR, XRT | GTR | Initial: GTR 1st: GTR, XRT | STR, XRT, Avastin | Initial: GTR 1st: Res, XRT | Initial: STR, Chem, pXRT 1st: STR | Initial: Res, XRT 1st (pelvic mass): GTR (sciatic mass): GTR, Chem | STR, proton therapy | Initial: STR, XRT 1st: GTR 2nd: GTR, XRT | GTR, XRT | Initial: GTR, XRT 1st: unspecified |
| Recurrence/ Metastasis status | none reported within 2 yrs | stable residual disease ~1.5 yrs | none reported within 1 yr | none reproted within 4 months | none reported within 1 month | 1st: 1 yr, nothing additional reported over the following 6 yrs | stable residual disease at 4 yrs | recurrence at 4 years | lost to clinical follow‐up after 2 years, patient died at age 36, no autopsy | 1st: 14 yrs Lung metastasis: 14 yrs patient died at age 48 | none reported within 2.5 yrs | 1st: 12 yrs 2nd: 15 yrs patient died 16 yrs after diagnosis, age 60 with extensive local disease | none reported within 9 yrs | 1st: 18 months, no subsequent clinical follow‐up available |
| Documented recurrence after anaplasia | N/A | N/A | N/A | N/A | N/A | recurred once after anaplasia | N/A | N/A | N/A | one additional surgery after anaplasia was diagnosed, both masses were present at 14 yrs, surgeries were separated by 4 months | N/A | recurred once after anaplasia | N/A | recurred once after anaplasia |
| Involvement of adjacent tissue by histologic and/or radiographic findings | No | No | Yes: sacral bone erosion but not invasion, entrapped ganglion | No | No | Microscopic dural invasion, no signifigant involvement of adjacent tissues | No | No | No | Yes: invasion of adjacent soft tissues | No | first recurrence destroyed L1 vertebrae, extended into paraspinal tissues, just prior to death imaging showed a large soft tissue mass invading L2‐L4 veterbral bodies, extending into paraspianal and left psoas muscles | Yes: involvement of sacral rootlets and dura, with sacral bone remodeling (cortical thinning and destruction without invasion) | No |
| CSF dissemination | No | No | No | No | No | Yes | Yes | Yes | Yes | Yes: found 14 yrs after initial | No | Y: at time of recurrences | suspected drop metastasis | Yes |
There was CSF dissemination in 7 of 14 patients, invasion of the adjacent bone or soft tissues in 2 cases (Figure 1A, G, J; cases 12 and 10) and erosion or remodeling of the adjacent bone in another 2 cases. One tumor metastasized to the lung (Figure 1L, M; case #10) at the time of the recurrence; this patient was concurrently found to have a sciatic mass, a pelvic mass, multiple pulmonary nodules, a left‐sided pleural effusion and an enhancing pleural mass 14 years after removal of a sciatic notch MPE. Metastatic MPE was demonstrated on evaluation of pleural fluid and a pleural biopsy (Figure 1L, M). For this case, it could not be determined if the primary tumor was intradural or extradural in origin. For all the other cases, the primary tumor was confirmed by imaging (example Figure 2J, case # 4) or communication with the surgeon as being initially intradural. Though case #12 was initially intradural, it grew to extensively involve the paraspinal soft tissues (Figure 1A).
Figure 1.
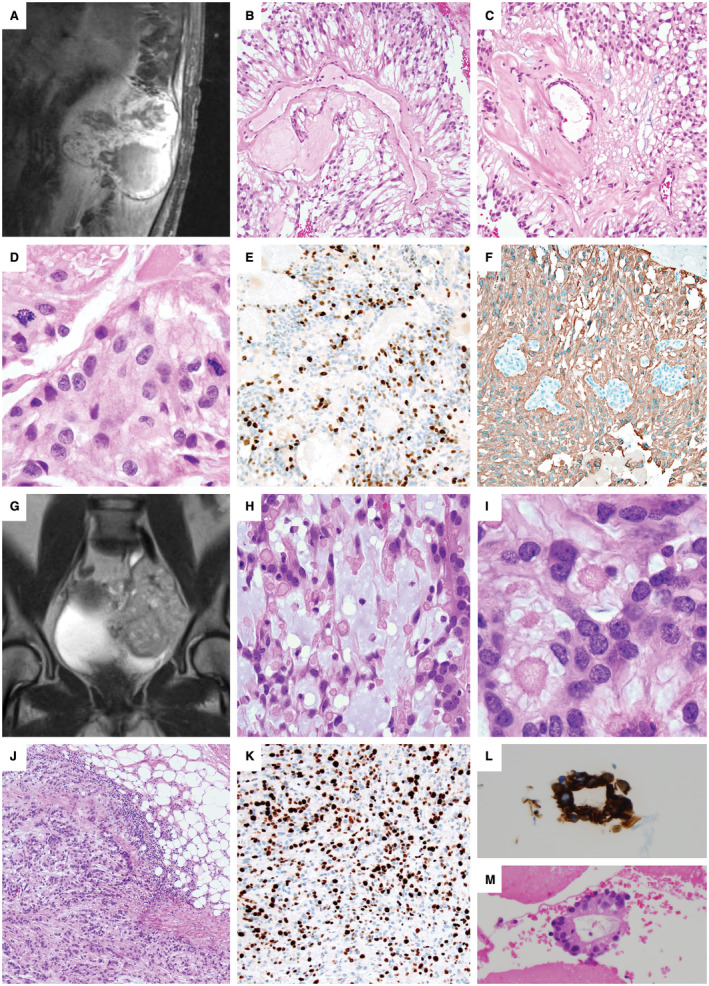
Anaplastic myxopapillary ependymomas with aggressive clinical features. A–F. Case #12: A 45‐year‐old man underwent subtotal resection and radiation of an 11.5 cm T11‐L2 MPE. (A). Recurrence detected by MRI 12 years later showed destruction of the L1 vertebrae with invasion into the paraspinal soft tissues. Areas of classic MPE histology were present (B,C), but were accompanied by anaplastic features, including increased mitotic activity of 17 mitoses per 10 HPF (D), Ki‐67 LI up to 26% and prominent microvascular proliferation (MVP). There were foci of non‐palisading necrosis in this patient that had previously received radiation therapy. A representative area of increased Ki‐67 staining is shown (E), and foci of MVP are seen among GFAP‐positive neoplastic cells (F). G–M. Case #10: A 10‐cm pelvic recurrence on MR imaging (G) was found in a 45‐year‐old woman 14 years after resection and radiation of a sciatic notch MPE. Careful review of the medical record could not establish if the original mass was intradural or extradural in origin, or if it arose as an extraspinal primary soft tissue MPE. The recurrence contained areas of classic MPE with increased mucin and collagen balloons H–I. The mass was invasive into the adjacent soft tissues (J), with a Ki‐67 LI up to 40% (K), a mitotic index of 7/10 HPF, MVP and palisading necrosis. As the patient had received radiation therapy, the palisading necrosis was not considered an anaplastic feature. The patient was concurrently found to have multiple pulmonary nodules, a large left‐sided pleural effusion and an enhancing pleural‐based mass. Evaluation of the pleural fluid, shown here with GFAP immunostaining (L), and a pleural biopsy (M) demonstrated metastatic MPE. At the time of the pelvic recurrence, a sacral mass was also present, which was resected 4 months later in a separate surgery.
Figure 2.
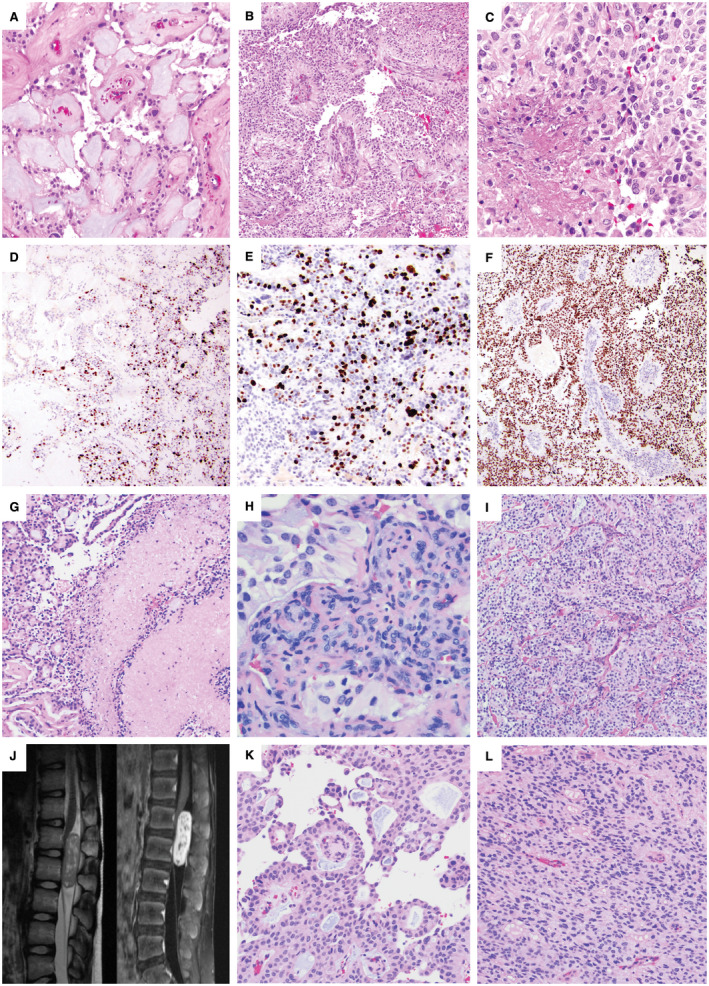
Adult and pediatric intradural anaplastic myxopapillary ependymomas. A–F. Case #14: A 55‐year‐old woman with a 2.7‐cm L1‐L2 mass and CSF dissemination with masses involving the cauda equina at the time of diagnosis underwent gross total resection and radiation therapy. The initial resection included areas of classic MPE (A) intermixed with anaplastic areas demonstrating hypercellularity and reduced mucin (B). The mitotic index reached 7 mitoses per 10 HPF, and there was both non‐palisading necrosis (C) and microvascular proliferation (MVP). A sharp transition between the classic and anaplastic components was present, reflected both by the histology and the variation in Ki‐67 (D), which was 20% in the anaplastic component (E) and <1% in conventional areas (D); left side). There was diffuse nuclear p53 positivity within the areas of anaplastic transformation (F). Recurrence 18 months later as a 2.3‐cm L2 mass also showed anaplastic features. G–I. Case #9: A 31‐year‐old man with an intradural sacral mass and smaller thoracic tumor deposits underwent subtotal resection, chemotherapy and a partial course of radiation. Re‐resection of the sacral MPE 9 months later showed both palisading and non‐palisading necrosis (G), which was not considered an anaplastic feature because of prior chemoradiation therapy. Anaplastic features included a Ki‐67 LI of 10%, a mitotic index of 7/10 HPF and MVP (H). Many areas of the tumor showed hypercellularity with decreased mucin (I). The patient was lost to clinical follow‐up and died at 36 years of age; no autopsy was performed. J–L. Case #4: A 10‐year‐old boy presented with an intradural extramedullary L1‐L2 enhancing mass centered at the conus medullaris by spinal MRI (J). Portions of the tumor showed classic MPE histology with papillary architecture and a myxoid stroma (K), while anaplastic regions were hypercellular with reduced mucin (L). Anaplastic features included a Ki‐67 LI of up to 15%, 6 mitoses per 10 high power fields and small foci of MVP.
All cases showed typical histological features of MPE such as patchy papillary architecture, hyalinized blood vessels surrounded by cuboidal to columnar glial cells with an intermediate layer of myxoid material, mucinous microcysts and spiculated eosinophilic “collagen balloons” (Figure 1B, C, H, I and 2A, K). Anaplasia typically occurred in foci with hypercellularity and reduced mucin (Figure 2B, I and L).
Representative images depicting anaplastic features in the tumors, including necrosis (Figure 2C), MVP (Figure 2H) and elevated Ki‐67 LI (Figure 1E, K and 2E), are shown from cases 12 (Figure 1A–F), 10 (Figure 1G–M), 14 (Figure 2A–F), 9 (Figure 2G–I) and 4 (Figure 2J–L). Palisading and non‐palisading necrosis were also seen in patients after chemotherapy or radiation therapy (Figure 2G), which was not counted as an anaplastic feature since the necrosis may have been at least partially because of prior therapy. Some tumors showed a distinct transition between the classic and anaplastic components, reflected in both histological features and variations in the Ki‐67 LI. While the Ki‐67 LI ranged from 8% to 40% within the highest areas, it was typically <3% in the foci of classic MPE. This was well demonstrated in case #14 (Figure 2A–F), in which the Ki‐67 LI was 20% in the anaplastic component and <1% in the areas with classic morphology, with a visible transition zone (Figure 2D–E). This case also showed diffuse nuclear p53 positivity within areas of anaplastic transformation (Figure 2F).
Six cases demonstrated four anaplastic features, five cases had three, two cases had two and one case progressed from two to three anaplastic features at recurrence (case #6). The anaplastic MPEs in adult patients demonstrated 3–4 anaplastic criteria for our study, while pediatric cases were more variable, with cases showing between 2 and 4 anaplastic criteria. Mitotic activity and Ki‐67 LI were comparable between pediatric and adult cases (median mitotic index of 6/10 HPF in pediatric cases and 7/10 HPF in adult cases; median Ki‐67 LI of 14% in pediatric cases and 20% in adult cases). For all the cases, the maximal mitotic index ranged from 2 to 20/10 HPF (median 7/10 HPF), with a median Ki‐67 LI of 15%.
The presence of anaplasia within pediatric cases was for the most part not solely a consequence of increased proliferation, as most pediatric tumors had MVP or necrosis. Among eight pediatric cases, only one (case #8) qualified for anaplasia based on increased proliferative activity (6 mitoses/10 HPF and Ki‐67 LI of 10%) in the absence of other anaplastic features; another pediatric case (case #4) contained only small foci of MVP in addition to an elevated Ki‐67 LI and mitotic index (ie., manifested three criteria). No primitive or embryonal component was identified in any of the cases; in particular, the pediatric and adult cases had similar histologic features.
Ten cases displayed anaplasia at initial resection before the administration of any radiation therapy, while 4 were anaplastic at the time of a second surgery for recurrence 9 months to 14 years subsequently. Of the four cases that developed anaplasia at the time of second surgery/recurrence (cases #8, 9, 10 and 12 in Table 1), three had received radiation therapy after the initial resection, including two that were subtotally resected and one in which the extent of resection was not indicated. While radiation therapy, and in one case chemotherapy, may have contributed to the presence of necrosis, each of these cases was considered anaplastic regardless, as they contained MVP with an elevated mitotic index and Ki‐67 LI. There were three cases with documented recurrence after a diagnosis of anaplasia (cases #6, 12 and 14 in Table 1). Two of these recurred despite radiation therapy; one was a subtotal resection, and one was gross totally resected. The third case of recurrence after a diagnosis of anaplasia occurred after gross total resection without adjuvant therapy.
Overall, of the 14 patients within the series, 6 patients had disease complications. There were four patients who underwent a second surgery for treatment of recurrent MPE, two patients had three surgeries, in one case there was extraneural metastatic disease and three patients died (one with extensive local disease at the time of death). Six patients thus far have had favorable outcomes (four pediatric and two adult) with either no reported recurrence, or stable residual disease with clinical follow‐up times ranging from 1 to 9 years (median follow‐up of approximately 2 years). All patients with disease complications also had CSF dissemination, while most patients with more favorable outcomes did not have CSF dissemination. For two patients, significant clinical follow‐up times were not yet available.
For the two cases that underwent NGS (primary tumor case #5, and recurrent tumor case #8), no pathogenic mutations, variants with strong evidence of clinical significance, amplifications, deletions or structural rearrangements were demonstrated among the cancer‐associated genes that were evaluated. Both NGS tests included the evaluation of NF2, and the UCSF500 Cancer Panel included the coverage of RELA and YAP1. Similar to classic MPE, the main finding was aneuploidy, with gains of numerous whole chromosomes. In one case, there were relative gains of chromosomes 1, 2, 4, 5, 7, 8, 9, 11, 12, 15, 16, 17, 18, 19, 20, 21 and X, with copy‐neutral loss of heterozygosity of chromosome 10. The second case demonstrated gains of chromosomes 4, 5, 7, 9, 16, 17, 18, 19, 20 and X. Neither case demonstrated deletions of chromosome 6q or 22q, which are frequently observed in spinal cord subependymomas and ependymomas, respectively 6, 24. The case evaluated by FISH for chromosomes 1, 7 and 9 demonstrated three to four copies of each chromosome.
Discussion
Though histologic anaplasia in myxopapillary ependymomas is exceptional, we were able to retrieve 14 patients from the files of six institutions for this study of clinicopathologic features. We defined anaplasia based on the histopathologic findings that are distinctly uncommon in MPE overall, but are similar to the criteria currently used to define anaplasia in classic ependymomas. Although our study findings are based on small case numbers, the chosen criteria were nevertheless associated with aggressive clinical features in 6 of the 14 cases, such as bone/soft tissue invasion, extraneural metastasis, multiple surgeries and tumor recurrence. As such, the recognition of MPEs with aggressive features will lead to the study of additional cases and further refinement of grading criteria.
Additionally, there was CSF dissemination in 7 of the 14 cases, 3 of the 8 pediatric cases (37%) and 4 of the 6 adult cases (66%). In a study of 183 patients (mean age at diagnosis of 35.5 ± 15.8) with classic MPE, distant spinal or brain metastases indicating CSF dissemination were observed in 9.3% and 6% of the patients, respectively, with local recurrence in 26% of the patients 39. Other studies have reported higher rates of CSF dissemination for MPE ranging from 35% to 57% 8, 15, particularly within pediatric cases of MPE and including pediatric dissemination at initial presentation in 14%–58% of the cases 8, 16. However, the higher percentages are usually reported within studies of smaller patient numbers.
While extraneural metastatic disease to the lung was present in one anaplastic case, careful review of the medical record could not establish if the primary MPE for this particular case was initially intradural in origin or not. As subcutaneous sacrococcygeal myxopapillary ependymomas more frequently have metastatic disease including lung metastases, it cannot be excluded that the metastases in this case are related to an extraspinal origin rather than anaplastic histology. Though extraspinal MPE and intradural MPE may have inherent biological differences, the metastatic potential of extraspinal myxopapillary ependymomas is thought to be attributable to increased access to the lymphovascular structures 19. Rare cases of initially intradural MPE with subsequent direct extension into the adjacent tissues could achieve comparable lymphovascular exposure.
Although the genetic alterations that drive MPEs have not been identified to date, copy number analyses have demonstrated chromosomal instability with frequent chromosomal gains, and methylation profiling clearly distinguishes myxopapillary ependymomas from other molecular subtypes of ependymoma 24. Gene expression profiling also highlights their unique biology, with MPEs showing a Warburg phenotype 20 and increased gene expression of HOXB13 compared to non‐myxopapillary ependymomas 3. The observed aneuploidy in our anaplastic MPEs is concordant with studies of MPE in general. Unfortunately, candidate genetic drivers of anaplastic transformation were not identified within the limited available data from two anaplastic cases evaluated on a NGS cancer panel. Whether this is because of targets not being represented on our panels or other mechanisms is not clear based on the limited data to date. Of interest, case #14 demonstrated strong and diffuse nuclear p53 immunostaining within the anaplastic component, raising the question of TP53 mutation, but this case was unfortunately not available for genetic testing. Nonetheless, TP53 mutations were not present in the two cases within our series studied by NGS.
We conclude that histologic anaplastic transformation of MPE can occur in both pediatric and adult patients. Though our series consists of small numbers, several of our cases were associated with either disease recurrence, local invasion, CSF dissemination or metastatic disease, which is at least suggestive of a more aggressive biologic potential than classic MPE. Therefore, at a minimum, closer clinical observation is recommended. Additional studies are needed to further refine the proposed grading criteria, and to identify the genetic biomarkers of MPE tumorigenesis and progression/anaplastic transformation.
Compliance with Ethical Standards
The authors declare no conflict of interest. All procedures performed in studies involving human participants were in accordance with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Presented in abstract format at the United States and Canadian Academy of Pathology meeting in Vancouver, British Columbia, Canada, March 17–23, 2018.
References
- 1. Akpolat N, Bozlak N, Kazez A, Köseoğullari AA (2003) Sacrococcygeal extraspinal ependymoma: a case report. Turk J Pediatr 45:276–279. [PubMed] [Google Scholar]
- 2. Awaya H, Kaneko M, Amatya VJ, Takeshima Y, Oka S, Inai K (2003) Myxopapillary ependymoma with anaplastic features. Pathol Int 53:700–703. [DOI] [PubMed] [Google Scholar]
- 3. Barton VN, Donson AM, Kleinschmidt‐DeMasters BK, Birks DK, Handler MH, Foreman NK (2010) Unique molecular characteristics of pediatric myxopapillary ependymoma. Brain Pathol 20:560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beschorner R, Wehrmann M, Ernemann U, Bonin M, Horber V, Oehl‐Jaschkowitz B et al (2007) Extradural ependymal tumor with myxopapillary and ependymoblastic differentiation in a case of Schinzel‐Giedion syndrome. Acta Neuropathol 113:339–346. [DOI] [PubMed] [Google Scholar]
- 5. Chakraborti S, Kini H, Pai KG, Upadhyaya V (2012) Sacrococcygeal myxopapillary ependymoma with anaplastic ependymoma component in an infant. J Pediatr Neurosci 7:218–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ebert C, von Haken M, Meyer‐Puttlitz B, Wiestler OD, Reifenberger G, Pietsch T, von Deimling A (1999) Molecular genetic analysis of ependymal tumors. NF2 mutations and chromosome 22q loss occur preferentially in intramedullary spinal ependymomas. Am J Pathol 155:627–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Estrozi B, Queiroga E, Bacchi CE, Faria Soares de Almeida V, Lucas de Carvalho J, Lageman GM et al (2006) Myxopapillary ependymoma of the posterior mediastinum. Ann Diagn Pathol 10:283–287. [DOI] [PubMed] [Google Scholar]
- 8. Fassett DR, Pingree J, Kestle JR (2005) The high incidence of tumor dissemination in myxopapillary ependymoma in pediatric patients. Report of five cases and review of the literature. J Neurosurg 102(Suppl. 1):59–64. [DOI] [PubMed] [Google Scholar]
- 9. Fourney DR, Fuller GN, Gokaslan ZL (2000) Intraspinal extradural myxopapillary ependymoma of the sacrum arising from the filum terminale externa. Case report. J Neurosurg 93(Suppl. 2):322–326. [DOI] [PubMed] [Google Scholar]
- 10. Goode B, Mondal G, Hyun M, Ruiz DG, Lin YH, Van Ziffle J et al (2018) A recurrent kinase domain mutation in PRKCA defines chordoid glioma of the third ventricle. Nat Commun 9:810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Helwig EB, Stern JB (1984) Subcutaneous sacrococcygeal myxopapillary ependymoma. A clinicopathologic study of 32 cases. Am J Clin Pathol 81:156–161. [DOI] [PubMed] [Google Scholar]
- 12. Idowu MO, Rosenblum MK, Wei XJ, Edgar MA, Soslow RA (2008) Ependymomas of the central nervous system and adult extra‐axial ependymomas are morphologically and immunohistochemically distinct–a comparative study with assessment of ovarian carcinomas for expression of glial fibrillary acidic protein. Am J Surg Pathol 32:710–718. [DOI] [PubMed] [Google Scholar]
- 13. Johnson JM, Jessurun J, Leonard A (1999) Sacrococcygeal ependymoma: case report and review of the literature. J Pediatr Surg 34:1405–1407. [DOI] [PubMed] [Google Scholar]
- 14. Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C et al (2017) Targeted next‐generation sequencing of pediatric neuro‐oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro‐Oncol 19:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kraetzig T, McLaughlin L, Bilsky MH, Laufer I (2018) Metastases of spinal myxopapillary ependymoma: unique characteristics and clinical management. J Neurosurg Spine 28:201–208. [DOI] [PubMed] [Google Scholar]
- 16. Kukreja S, Ambekar S, Sin AH, Nanda A (2014) Cumulative survival analysis of patients with spinal myxopapillary ependymomas in the first 2 decades of life. J Neurosurg Pediatr 13:400–407. [DOI] [PubMed] [Google Scholar]
- 17. Kukreja S, Ambekar S, Sharma M, Sin AH, Nanda A (2015) Outcome predictors in the management of spinal myxopapillary ependymoma: an integrative survival analysis. World Neurosurg 83:852–859. [DOI] [PubMed] [Google Scholar]
- 18. Lynch J, Kelly N, Fitzpatrick B, Regan P (2002) A sacrococcygeal extraspinal ependymoma in a 67‐year‐old man: a case report and review of the literature. Br J Plast Surg 55:80–82. [DOI] [PubMed] [Google Scholar]
- 19. Ma YT, Ramachandra P, Spooner D (2006) Case report: primary subcutaneous sacrococcygeal ependymoma: a case report and review of the literature. Br J Radiol 79:445–447. [DOI] [PubMed] [Google Scholar]
- 20. Mack SC, Agnihotri S, Bertrand KC, Wang X, Shih DJ, Witt H et al (2015) Spinal myxopapillary ependymomas demonstrate a Warburg phenotype. Clin Cancer Res 21:3750–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McLendon R, Schiffer D, Rosenblum M, Wiestler O (2016) Myxopapillary ependymoma. In: WHO Classification. Louis D, Ohgaki H, Wiestler O, Cavenii W (eds), pp. 104–105. WHO: Lyon. [Google Scholar]
- 22. Metcalf JS, Balentine JD, Maize JC (1984) Myxopapillary ependymoma of the cauda equina with extension to the skin of the sacrococcygeal region. Am J Dermatopathol 6:249–257. [DOI] [PubMed] [Google Scholar]
- 23. Morantz RA, Kepes JJ, Batnitzky S, Masterson BJ (1979) Extraspinal ependymomas. Report of three cases. J Neurosurg 51:383–391. [DOI] [PubMed] [Google Scholar]
- 24. Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F et al (2015) Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27:728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pekmezci M, Stevers M, Phillips JJ, Van Ziffle J, Bastian BC, Tsankova NM et al (2018) Multinodular and vacuolating neuronal tumor of the cerebrum is a clonal neoplasm defined by genetic alterations that activate the MAP kinase signaling pathway. Acta Neuropathol 135:485–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pekmezci M, Villanueva‐Meyer JE, Goode B, Van Ziffle J, Onodera C, Grenert JP et al (2018) The genetic landscape of ganglioglioma. Acta Neuropathol Commun 6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prayson RA (1997) Myxopapillary ependymomas: a clinicopathologic study of 14 cases including MIB‐1 and p53 immunoreactivity. Mod Pathol 10:304–310. [PubMed] [Google Scholar]
- 28. Pulitzer DR, Martin PC, Collins PC, Ralph DR (1988) Subcutaneous sacrococcygeal (“myxopapillary”) ependymal rests. Am J Surg Pathol 12:672–677. [DOI] [PubMed] [Google Scholar]
- 29. Rezai AR, Woo HH, Lee M, Cohen H, Zagzag D, Epstein FJ (1996) Disseminated ependymomas of the central nervous system. J Neurosurg 85:618–624. [DOI] [PubMed] [Google Scholar]
- 30. Rickert CH, Kedziora O, Gullotta F (1999) Ependymoma of the cauda equina. Acta Neurochir (Wien) 141:781–782. [DOI] [PubMed] [Google Scholar]
- 31. Safaee M, Oh MC, Mummaneni PV, Weinstein PR, Ames CP, Chou D et al (2014) Surgical outcomes in spinal cord ependymomas and the importance of extent of resection in children and young adults. J Neurosurg Pediatr 13:393–399. [DOI] [PubMed] [Google Scholar]
- 32. Satti M, Firoze M, Malaker K, Hussain M, Maniyar I (2005) Mediastinal myxopapillary ependymoma primary or late metastases of paracoccygeal ependymoma: a case report. Ann Diagn Pathol 9:215–218. [DOI] [PubMed] [Google Scholar]
- 33. Schiavello E, Biassoni V, Antonelli M, Modena P, Cesaro S, Pierani P, Gandola L (2018) Pediatric extraspinal sacrococcygeal ependymoma (ESE): an Italian AIEOP experience of six cases and literature review. Childs Nerv Syst 34:1291–1298. [DOI] [PubMed] [Google Scholar]
- 34. Shelekhova KV, Egorenkov VV, Kheinstein VA, Konstantinova AM, Iyevleva A, Imyanitov EN et al (2018) Myxopapillary ependymoma of lumbar soft tissue: a case report with gene expression evaluation. Int J Surg Pathol 26:364–369. [DOI] [PubMed] [Google Scholar]
- 35. Stephen JH, Sievert AJ, Madsen PJ, Judkins AR, Resnick AC, Storm PB et al (2012) Spinal cord ependymomas and myxopapillary ependymomas in the first 2 decades of life: a clinicopathological and immunohistochemical characterization of 19 cases. J Neurosurg Pediatr 9:646–653. [DOI] [PubMed] [Google Scholar]
- 36. Timmerman W, Bubrick MP (1984) Presacral and postsacral extraspinal ependymoma. Report of a case and review of the literature. Dis Colon Rectum 27:114–119. [DOI] [PubMed] [Google Scholar]
- 37. Trivedi D, Xiong Z (2017) Anaplastic myxopapillary ependymoma in an infant: case report and literature review. Intractable Rare Dis Res 6:128–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vega‐Orozco R, Rembao‐Bojórquez D, Salmerón‐Mercado M, García‐Marquez A, Tena‐Suck ML (2011) Inguinal lymph nodal metastasis of myxopapillary ependymoma confirmed by fine‐needle aspiration cytology, biopsy, and immunohistochemistry: case report. Diagn Cytopathol 39:689–693. [DOI] [PubMed] [Google Scholar]
- 39. Weber DC, Wang Y, Miller R, Villà S, Zaucha R, Pica A et al (2015) Long‐term outcome of patients with spinal myxopapillary ependymoma: treatment results from the MD Anderson Cancer Center and institutions from the Rare Cancer Network. Neuro Oncol 17:588–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yust Katz S, Cachia D, Kamiya‐Matsuoka C, Olar A, Theeler B, Penas Prado M et al (2018) Ependymomas arising outside of the central nervous system: a case series and literature review. J Clin Neurosci 47:202–207. [DOI] [PubMed] [Google Scholar]