

Abstract
Novel pyridine- and pyrimidine-based allosteric inhibitors are reported that achieve PDE4D subtype selectivity through recognition of a single amino acid difference on a key regulatory domain, known as UCR2, that opens and closes over the catalytic site for cAMP hydrolysis. The design and optimization of lead compounds was based on iterative analysis of X-ray crystal structures combined with metabolite identification. Selectivity for the activated, dimeric form of PDE4D provided potent memory enhancing effects in a mouse model of novel object recognition with improved tolerability and reduced vascular toxicity over earlier PDE4 inhibitors that lack subtype selectivity. The lead compound, 28 (BPN14770), has entered mid-stage, human Phase 2 clinical trials for the treatment of Fragile X Syndrome.
>Graphical Abstract
INTRODUCTION
The phosphodiesterase-4 (PDE4) enzymes regulate the spatial and temporal patterning of signaling through the cAMP second messenger system by hydrolysis of the cyclic nucleotide.1-4 The PDE4 gene family consists of four subtypes, PDE4A-D, that are distinguished from other PDE gene families by the presence of upstream conserved regions known as UCR1 and UCR2,5-6 PDE4 enzymes can be present in cells as either dimers or monomers. UCR1 is required for dimerization while UCR2 regulates enzyme activity by opening and closing over the active site and thereby controls access of cAMP.7-8 The basal activity of dimeric isoforms is increased in response to cAMP signaling through phosphorylation of UCR1 by protein kinase A (PKA).9-12
The importance of the enzyme for normal brain function is underscored by the discovery of PDE4D mutations in an ultra-rare, neurodevelopmental disorder known as acrodysostosis type 1 with or without hormone resistance (ACRDYS2), a condition in which brachydactyly (short fingers and toes) is accompanied by intellectual disability with affected individuals showing reduction in IQ.13-17 The ACRDYS2 mutations are exclusively missense mutations of residues on the surface of the protein. In one patient, the UCR1 PKA phosphorylation site is mutated from serine to alanine, thereby preventing PKA activation in response to cAMP signaling.15, 18 A second subset of ACRDYS2 mutations alter contact residues between UCR2 and the catalytic domain, thereby affecting the closure of UCR2 across the active site.19 The remaining ACRDYS2 mutations occur at the base of the four-helix bundle needed for dimerization and appear to elevate basal enzyme activity.20
Genetic variation in PDE4D also contributes to biological variation in human cognitive ability. The PDE4D gene is a consistent hit across multiple, large-scale, genome-wide association studies (GWAS) of human cognition.21-24 Due to the large size of the gene, more than 1 million base pairs, these studies associate allelic variation over 5’ exons encoding dimeric forms of the PDE4D enzyme with human cognitive ability.25 Thus, as with the ultra-rare ACRDYS2 mutations, common genetic variants identified by GWAS also show the importance of dimeric isoforms of PDE4D for normal brain function.
The role in cognition of second messenger signaling through cAMP has been studied in model organisms. Dance, the first mutation shown to affect learning and memory in the Drosophila fruit fly, is a deletion of the single PDE4 gene present in flies.26-28 Deletion of PDE4 impairs associative memory by altering the spatial and temporal patterning of cAMP signaling.4, 29 Rutabaga, which also impairs associative memory in the fruit fly, is a mutation of the calcium-calmodulin adenylate cyclase.30 Studies in genetically engineered mice place PDE4D downstream of calcium influx through the N-methyl D-aspartate receptor (NMDA-R) with consequent activation of calcium/calmodulin adenylate cyclase in a pathway important for early- and late-stage memory formation as well as hippocampal neurogenesis and the expression of brain-derived neurotrophic factor (BDNF).31-36
Multiple studies show the benefit of PDE4 inhibition in mouse models of learning and memory, major depression, and neurodegeneration.37-42 Although several PDE4 inhibitors are used clinically, the effective dose is limited by undesirable gastro-intestinal side effects such as nausea, vomiting, and diarrhea.43-45 In rodents and other species, PDE4 inhibitors may also cause vascular inflammation, particularly in the gut, however, that toxicity has not been documented in humans.46-47 Marketed PDE4 inhibitors such as roflumilast and apremilast lack PDE4 subtype selectivity, lack selectivity for dimeric as compared to monomeric isoforms of the PDE4 enzymes, and inhibit equally the activated and basal states.
We showed previously that it is possible to design subtype-selective, PDE4D allosteric inhibitors with improved tolerability and reduced vascular toxicity.7 Subtype selective PDE4D allosteric inhibitors exploit a binding pose in which the inhibitor captures a regulatory helix from UCR2 in trans- across the opposite active site.7-8 As we describe below, such compounds are more potent against dimeric as compared to monomeric forms of PDE4D and can be optimized for potency against the activated as compared to the basal state of the PDE4D dimer. This pharmacological profile contrasts with the profiles of competitive, active-site directed PDE4 inhibitors.
RESULTS
PDE4 allosteric inhibitors utilize a common pharmacophore in which an aromatic core forms a hydrogen bond with an invariant, active site glutamine and a pair of aromatic arms capture UCR2 in trans- across the active site thereby blocking access by cAMP.7, 48-50 Within UCR2, a single amino acid difference of a phenylalanine in PDE4D (Phe271) and a tyrosine in PDE4A-C can be exploited for subtype selectivity.7 Selective PDE4B inhibitors also fit this pharmacophore,49 however, PDE4B allosteric inhibitors capture a different, C-terminal regulatory helix (CR3) in cis- across the active site.48 The initial series of PDE4D allosteric inhibitors that we described used a methoxyphenyl core in which the oxygen forms a hydrogen bond with the invariant glutamine (l)7 We speculated that the pyrimidine core of the series of PDE4B inhibitors reported by Naganuma (2)49 could be optimized to capture UCR2.
The choice of the in vitro assay format for measuring inhibition of PDE4 enzymatic activity is critical. We utilized a real-time, coupled enzyme assay in which hydrolysis of cAMP to AMP leads to the oxidation of NADH which can be measured spectroscopically.7 Many high-throughput assays of PDE4 enzymatic activity, in contrast, use a fluorescein-labeled cAMP substrate that when hydrolyzed allows detection of AMP using fluorescence polarization. However, the fluorescein label prevents closure of UCR2 and thereby alters the kinetics of PDE4 allosteric inhibitors (Supplemental Figure 1).
PDE4 enzymes are present in cells as dimers (if they contain UCR1) or as monomers (if they lack UCR1).52-53 The basal activity of dimeric forms of PDE4 is increased by phosphorylation of a serine in UCR1 by PKA.10, 12 Mutating the serine to aspartic acid mimics PKA phosphorylation and increases the activity of the enzyme. In the case of PDE4D7, we measured an 8-fold increase in hydrolytic activity due to mutation of serine129 to aspartic acid (notation: S129D). For screening, we used mutationally activated, dimeric forms of the enzymes, PDE4D7-S129D and PDE4B1-S133D. These were produced using a Baculovirus expression system in Sf9 cells and purified using C-terminal affinity tags and column chromatography.
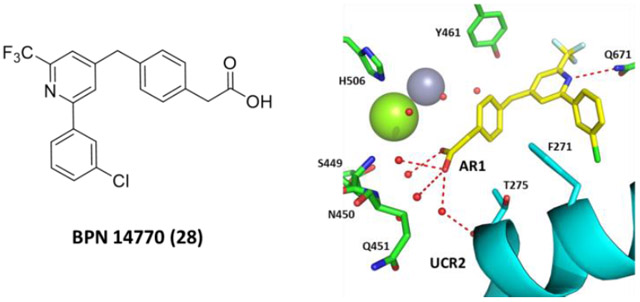
Our initial study of the structure-activity relationship (SAR) in a triazine series as in 3 confirmed that Ar1 and Ar2 could be tuned for subtype selectivity.50 Replacement of the Ar2 chlorothiophene in 2 with the 3-chlorophenyl in 1, consistently provided compounds with PDE4D selectivity through capture of UCR2 and not CR3. Exploration of Ar1 replacements achieved a 3,000-fold range in PDE4D potency across a series of amides or alcohols with the primary alcohols providing the greatest potency and selectivity (Table 1). Co-crystal structures of ethyl alcohol (5) and amide (6) derivatives show both inhibitors hydrogen bond to the invariant glutamine671 (Q671) in the active site through the triazine core. A second hydrogen bond is observed from the amine linker to a stable water molecule near the metal center and tyrosine461 (Y461) of the catalytic domain. In addition, the co-crystal structure revealed that the amide nitrogen in 6 participates in a hydrogen bond through a water network with UCR2 threonine275 (T275), while the oxygen hydrogen bonds with the catalytic domain through a water molecule that coordinates to both histidine506 (H506) and the first shell of waters surrounding the neighboring magnesium atom. The alcohol 5 lacks the interaction with UCR2 while maintaining the interaction with the catalytic domain. Phenylalanine271 (F271), which projects from UCR2 into the allosteric site, is the key residue for PDE4D selectivity. In the UCR2 of PDE4A-C, the phenylalanine is replaced by a tyrosine. When 5 or 6 occupy the allosteric site, UCR2 closes across the binding site and accommodates the phenylalanine but does not tolerate the protruding para-hydroxyl substituent of a tyrosine (Figure 2, see also Figure 7). Although potent and selective, the triazines were abandoned as they featured low or negligible solubility, poor cellular permeability, and high plasma protein binding.
Table 1:
Inhibitory activity of triazine Ar1 substitution
 |
||||||
|---|---|---|---|---|---|---|
| Compound | R | PDE4D7- S129D IC50 (nM)a |
PDE4B1- S133D IC50 (nM)b |
PDE4D7- S129(wt) IC50 (nM)c |
Ratio B/Dd |
Ratio Basal/Activatede |
| 4 | −(CH2)3-OH | 0.2 | 11 | 27 | 55 | 135 |
| 5 | −CH2CH2OH | 0.5 | 24 | 102 | 48 | 204 |
| 6 | −CH2CONH2 | 5 | 48 | 203 | 10 | 41 |
| 7 | −CH2OH | 12 | 105 | 351 | 9 | 30 |
| 8 | −CONH2 | 22 | 44 | 252 | 2 | 11 |
| 9 | −CH(Me)CO2H | 96 | 1090 | NCf | 11 | |
| 10 | −CO2H | 213 | 241 | 482 | 1 | 2 |
| 11 | −CH2CO2H | 681 | 781 | 4040 | 1 | 6 |
PDE4D7-S129D is a dimeric isoform of PDE4D that contains a UCR1 mutation (S129D) that mimics PKA phosphorylation.
PDE4B1-S133D is a dimeric isoform of PDE4B that also contains a UCR1 mutation (S133D) that mimics PKA phosphorylation.
PDE4D7-S129(wt) is the native isoform of PDE4D7 that lacks the UCR1 phosphorylation mimetic mutation.
B/D is the ratio of IC50 for PDE4B1-S133D/PDE4D7-S129D.
Basal/Activated is the ratio of IC50 for PDE4D7-S129(wt)/PDE4D7-S129D.
NC -Not calculated as IC50 >10,000 nM. See experimental section for methods and detailed synthetic procedures.
Figure 2:

PDE4D allosteric binding site. (A) view of a PDE4 dimer (PDB: 4WZI) showing the four-helix bundle and UCR2 from one subunit (colored red) positioned in trans- across the active site of the opposite subunit (colored blue).8 (B) Views of compounds 5 (PDB: 6NJI) and 6 (PDB: 6NJH) bound in the PDE4D allosteric site showing key polar contacts. Magnesium (green sphere), zinc (purple sphere), water (red spheres), UCR2 (cyan), and catalytic domain amino acid side chains (green).
Figure 7:

Comparison and selectivity of 23 and 28: (A) 23 with AR1 amide pointing up in the pocket to form water-mediated hydrogen bonds with the main-chain and side-chain of UCR2 (cyan) threonine275 (T275) and catalytic domain glutamine451 (Q451), histidine506 (H506) and waters surrounding the magnesium atom (green sphere). (B) 28 (PDB: 6NJJ) with AR1 carboxylic acid pointing down losing water-mediated hydrogen bonds with histidine506 (H506) and the side-chain of threonine275 (T275), but gaining water-mediated hydrogen bonds to serine449 (S449), asparagine450 (N450), and the main-chain of glutamine451 (Q451), while maintaining the main-chain interaction with UCR2 threonine275 (T275). (C) Modeling of tyrosine271 (Y271) in place of phenylalanine271 (Y271) onto the crystal structure of PDE4D and 28. The closest approach by phenylalanine271 to 28 is 4.4Å which is reduced with tyrosine to 3.2Å.
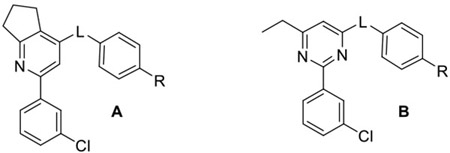
Published co-crystal structures with 2 and PDE4B showed that the methyl and ethyl arms extending from the pyrimidine core project into an invariant hydrophobic pocket known as the Q-region.7, 48 The Q-region provides an opportunity to adjust the potency, as long as groups projecting into the pocket are kept small. We explored a fused cyclopentyl group as a conservative replacement for the 6-methyl and 5-ethyl substituents of 2. Like the triazine series, the Ar2 chlorothiophene was replaced with the 3-chlorophenyl. We found that the SAR for Ar1 could be transferred to the new series with alcohols and amides showing the best PDE4D potency and selectivity (Table 2). Exploring meta- versus para- substitution (12 v 16) of the Ar1 phenyl showed that the trajectory of the distal alcohol or amide was important for potency, further supporting the conclusions drawn from the structural studies of the triazine series. However, lead compounds had low solubility with high P-glycoprotein efflux in MDCK-MDR1 cells and therefore were abandoned. SAR studies then focused on 6-subsituted pyridines or pyrimidines. A timeline plot of the different cores that were explored over the course of the lead optimization effort is shown in Figure 3.
Table 2:
Inhibitory activity of cyclopentylpyridine Ar1 substitutiona
 |
||||||
|---|---|---|---|---|---|---|
| Compound | R | PDE4D7- S129D IC50 (nM) |
PDE4B1- S133D IC50 (nM) |
PDE4D7- S129(wt) IC50 (nM) |
Ratio B/D |
Ratio Basal/Activated |
| 12 | 4-CH2CH2OH | 9 | 562 | 2111 | 60 | 226 |
| 13 | 4-(CH2)2CONH2 | 35 | 166 | 1283 | 5 | 37 |
| 14 | 4-CH2CO-NMe2 | 51 | 356 | 914 | 7 | 18 |
| 15 | 4-CH2CO-N(H)Me | 133 | 98 | 1991 | 1 | 15 |
| 16 | 3- CH2CH2OH | 226 | 1419 | 6325 | 6 | 28 |
| 17 | 4-CH2CO2H | 1165 | 1642 | NTb | 1 | - |
See table 1 for assay definitions. See experimental section for methods and detailed synthetic procedures.
NT: Not tested
Figure 3:

Timeline plot for chemical optimization of different cores and Q region substituents. Compound symbols are colored by linker type and inhibitory potency against PDE4D-S129D is indicated by the size of the symbol.
A survey of linkers using the cyclopentylpyridine core and an Ar1 amide revealed that a methylene linker was preferred over an amine linker for PDE4D potency and selectivity (Table 3). The 6-ethylpyrimidine series confirmed that trend when matched pairs of Ar1 amides and alcohols were compared. This affects PDE4 subtype selectivity as shown in Figure 4 for compounds in which Ar2 is fixed as 3-chlorophenyl and the linker is either a methylene or an amine while the core varies. In addition, compounds with a methylene linker show good proportionality between PDE4B/PDE4D and basal/activated selectivity. Apremilast, an active site inhibitor competitive with cAMP has no selectivity for PDE4B versus PDE4D, nor does it show selectivity for the activated enzyme. (R)-rolipram, a prototypical allosteric inhibitor, is 5.5-fold selective for PDE4D versus PDE4B and 21-fold selective for the activated form of PDE4D7 (Figure 4).
Table 3:
Inhibitory activity of linker modified analogs
 |
||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | Series | L | R | PDE4D7- S129D IC50 (nM) |
PDE4B1- S133D IC50 (nM) |
PDE4D7- S129(wt) IC50 (nM) |
Ratio B/D |
Ratio Basal/Activated |
| 18 | A | CH2 | −CH2CONH2 | 8.6 | 137 | 810 | 16 | 94 |
| 19 | A | NH | −CH2CONH2 | 42.1 | 136 | 1665 | 3 | 40 |
| 20 | A | SO2 | −CH2CONH2 | 475 | 3830 | NTb | 8 | - |
| 21 | A | N(Me) | −CH2CONH2 | 733 | 3287 | NTb | 4 | - |
| 22 | B | NH | −CH2CH2OH | 1.1 | 57 | 149 | 52 | 135 |
| 23 | B | CH2 | −CH2CONH2 | 1.7 | 225 | 313 | 132 | 184 |
| 24 | B | O | −CH2CH2OH | 1.8 | 58 | 251 | 32 | 139 |
| 25 | B | CH2 | −CH2CH2OH | 2 | 584 | 534 | 292 | 267 |
| 26 | B | NH | −CH2CONH2 | 2.6 | 43 | 146 | 17 | 56 |
| 27 | B | O | −CH2CONH2 | 35 | 953 | 2431 | 27 | 69 |
See table 1 for assay definitions. See experimental section for methods and detailed synthetic procedures.
NT: Not tested
Figure 4:

Effect of amine and methylene linker replacements on PDE4 selectivity across multiple cores. The ratios of selectivity for PDE4B3-S133D/PDE4D7-S129D are plotted versus PDE4D7-S129(wt)/PDE4D7-S129D. The dotted line is the best-fit in the methylene linker series (Bravais Pearson test, r=0.677) for direct proportionality between selectivity for PDE4B-S133D versus PDE4D7(wt). Symbols: ● methylene linker, ● amine linker, ★ - apremilast, ▲ - rolipram; The relative size of the symbol indicates potency against PDE4D7-S129D. Units are in nM.
Compounds with the methylene linker tend to achieve greater PDE4D selectivity than those with the amine linker. Comparing PDE4D co-crystal structures of compounds with a methylene linker with those containing an amine linker shows little movement of the core which is fixed in place by the hydrogen bond to the invariant active site glutamine671 (Q671) and the phenylalanine-isoleucine clamp.51 Also, both 6 and 23 bind similarly to UCR2 threonine275 (T275). However, the methylene linker in 23, unlike the amine linker in 6, is unable to hydrogen bond with the water molecule coordinated between catalytic domain residues tyrosine461 (Y461), aspartic acid620 (D620) and a water molecule near the zinc atom (Figure 5). This has the effect of reducing binding affinity for the catalytic domain while maintaining UCR2 interactions, effectively improving selectivity for the PDE4D subtype.
Figure 5:

Comparison of methylene (compound 23 - magenta) and amine (compound 6 – yellow) linkers. (A) Only residues from the compound 6 co-crystal structure are shown for simplicity as no major changes are observed in the side-chain rotamers. Waters associated with 6 (red) and 23 (blue) are shown with hydrogen bonds for 6 (black) and 23 (red). UCR2 is in cyan and the zinc atom is a gray sphere. (B) Overlay of 6 (yellow) and 23 (magenta) with 2Fo-Fc electron density contoured at 1σ (blue mesh) and Fo-Fc electron density contoured at 3σ (green/red mesh). Waters associated with 6 (red) and 23 (sand) are shown with the hydrogen bond unique to 6 highlighted in red.
Based on the genetic analysis of ACRDYS2 mutations, our therapeutic hypothesis was that inhibition of the activated, dimeric forms of PDE4D would be associated with cognitive benefit. We noted that as compounds achieved greater PDE4D subtype selectivity, selectivity for the activated form of the PDE4D dimer also increased (Figure 4). This is most striking for the compounds with a methylene linker where there is a good correlation between PDE4B/PDE4D selectivity and basal/activated selectivity. In contrast to the allosteric inhibitors, PDE4 inhibitors such as apremilast (30) that bind in the active site competitively with cAMP have no PDE4 subtype selectivity due to the absolute amino acid sequence conservation of the active site,51 nor are they selective for the activated versus basal forms of the PDE4 enzymes (Figure 4). (R)-rolipram, a prototypical allosteric inhibitor that is highly efficacious in many preclinical models, but also highly emetic, has 5.5 fold selectivity for PDE4D versus PDE4B and 21 fold selectivity for activated PDE4D versus the basal enzyme.54 We postulated that as we improved PDE4D subtype selectivity, as well as the window between inhibition of the activated and basal forms of the PDE4D dimer, we would improve tolerability and reduce toxicity.
To profile in vivo pharmacology, compounds were screened for improvement of long term memory in mice using novel object recognition after a 24-hour delay and then counter screened for emetic potential (Figure 6). As mice are unable to vomit, a pharmacologic construct of PDE4 inhibitor-induced emesis was used to assess potential tolerability, the shortening of the duration of anesthesia induced by the combination of ketamine and xylazine.55-56 The anesthesia duration test provides an estimate of the potential therapeutic window between efficacy and tolerability.
Figure 6:
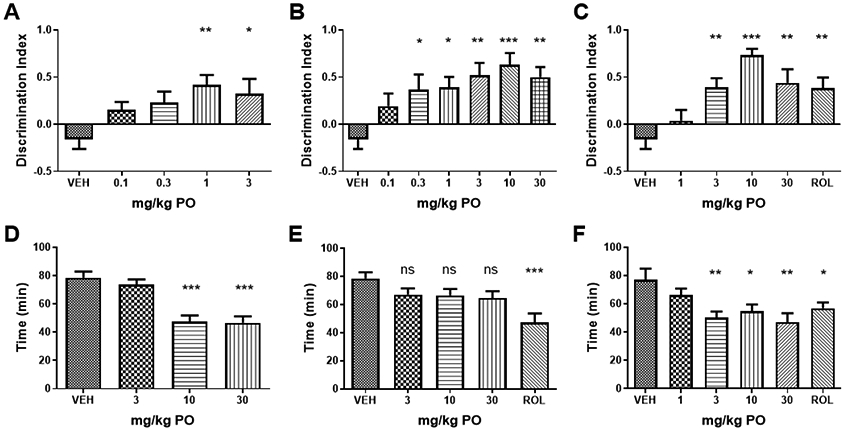
Mouse NOR and anesthesia duration test. (A) 23 significantly improved novel object discrimination in comparison to vehicle (VEH; Two-ANOVA F(4,28) = 3.96, p<0.001) at doses of 1 and 3 mg/kg. (B) 28 significantly improved novel object discrimination (F(6,42) = 4.18, p<0.005) at doses above 0.3 mg/kg PO. (C) 29 significantly improved novel object discrimination (F(5,35) = 8.26, p<0.005) at doses above 3 mg/kg PO as did rolipram (ROL) at a dose of 0.1 mg/kg SC. (D) 23 significantly reduced the duration of anesthesia induced by ketamine and xylazine, a pharmacological construct of emetic potential (F(3.27) = 20.9, p<0.001) at doses of 10 and 30 mg/kg PO. (E) 28 had no significant effect on anesthesia duration at doses of 3, 10 and 30 mg/kg PO (F(4.36) = 1.08), while (F) 29 significantly reduced anesthesia duration at doses of 3 mg/kg PO and higher (F(4,28) = 4.7, p<0.005). Rolipram significantly reduced anesthesia duration at 0.1 mg/kg IP. P-values shown are: ns, not significant; * p<0.01; ** p<0.05, and ***p<0.001 (Dunnett’s multiple comparison test).
Compound 23 (Table 3), an early lead, was moderately stable in human, dog, mouse, rat and monkey liver microsomes (54-90% parent remaining at 30 min), with good permeability and low efflux in MDR1-MDCK cells (A-B = 24.1 x 10−6 cm/sec; B-A=22 x 10−6 cm/sec ; Efflux Ratio = 0.9). Aqueous kinetic solubility was low (< 1.6 μM at pH 7), while solubility increased (100 μM) in fasted state simulated intestinal fluid (FaSSIF). Protein binding in plasma from humans, dogs, rats and mice was >99.5 −99.8%, although there was no shift in the IC50 for PDE4D7-S129D inhibition in the presence of bovine serum albumin (BSA) at 1 mg/mL. 23 was a moderate cytochrome P450 inhibitor (CYP3A4 IC50 = 5.9 μM, 2C19 = 29 μM; 2D6 >100 μM; 2C9 > 100 μM). Evaluation of the compound in mice showed good oral bioavailability when dosed in 5% DMSO, 5% Solutol, 90% PBS (F% = 53%), extended absorption over 4-8 hours, and excellent distribution to brain (brain/plasma ratio = 2.7 based on AUC). However, half-lives in plasma and brain were short (t ½ = 0.46 hr and 0.44 hr, respectively). 23 improved discrimination of the novel object at oral doses of 1 and 3 mg/kg, while it reduced anesthesia duration at doses of 10 and 30 mg/kg. Thus, the potential therapeutic window was estimated as 10-fold. Based on these promising data, we focused chemical optimization on improving solubility and metabolic stability.
To identify sites of metabolism, compound 23 was incubated with rat or human hepatocytes and the products were characterized (Table 4). Hepatocyte metabolism resulted in the hydrolysis of the amide to a carboxylic acid and mono-oxidation of the 6-ethyl side chain with further glucuronidation. Hydrolysis to the acid was more extensive in rat (29.1%) than in human hepatocytes (<1%), while both species produced mono-oxygenated and glucuronidated products. We replaced the amide with the acid to yield 28 and introduced a 6-CF3 on the pyridine core to eliminate the possibility of side-chain oxidation. This also might balance chemical properties and could improve solubility. As an alternative strategy to reduce hydrolysis of the amide, we replaced the amide with an ethanolamide as in 29 with the intention that the N-(2-hydroxyethyl)acetamide group would reduce hydrolysis of the amide while also improving solubility. Surprisingly, both compounds were potent and selective PDE4D allosteric inhibitors (Table 5). Our previous SAR had indicated that Ar1 carboxylic acid analogs were neither potent nor selective for PDE4D in the triazine or cyclopentylpyridine series (Table 1 and Table 2).
Table 4:
In vitro metabolism of compound 23 by human and rat hepatocytes.a
 | |||||||
|---|---|---|---|---|---|---|---|
| Metabolites | |||||||
| M1 | M2 | M3 | M4 | M5 | M6 | Parent | |
| [M+H]+, m/z | 558 | 558 | 558 | 382 | 382 | 367 | 366 |
| Δ MW | +192 | +192 | +192 | +16 | +16 | +1 | - |
| Δ RT, min | −4.7 | −4.4 | −4.0 | −3.0 | −2.6 | +2.2 | - |
| Proposed Reactionb | moxy+ gluc | moxy+ gluc | moxy+ gluc | moxy | moxy | hydrolysis | - |
| Hepatocytes | |||||||
| Human | <1% | 1.4% | - | 2.5% | 1.0% | <1% | 93.2% |
| Rat | <1% | - | <1% | 1.5% | 1.5% | 29.1% | 66.8% |
percentage of metabolite present is after 4 hour incubation with hepatocytes.
Abbreviations: moxy - monooxygenation; gluc -glucuronidation.
Table 5.
PDE4 inhibitory activity of 28 and 29 compared to apremilast (30).
 |
|||||||
|---|---|---|---|---|---|---|---|
| Compound | Parameter | PDE4D7- S129D dimer activated |
PDE4D7- S129(wt) dimer basal |
PDE4B1- S133D dimer activated |
PDE4D3- S54D dimer activated |
PDE4D2 monomer |
PDE4D-cat monomer |
| 28 | IC50 nM | 7.8 ± 1.8 | 1018 ± 239 | 2013 ± 256 | 7.4 ± 2.0 | 127 ± 1.2 | 6561 ± 10 |
| Hill Slope | 0.55± 0.12 | 0.58 ± 0.11 | 0.92 ± 0.21 | 0.68 ± 0.14 | 0.60± 0.16 | 1.2 ± 0.10 | |
| Imax%c | 90.2 ± 4.8a | * | * | 89 ±2.3 b | * | * | |
| Count | 15 | 9 | 6 | 5 | 4 | 3 | |
| 29 | IC50 nM d | 0.8 ± 1.6 | 885 ± 2.2 | 128 ± 1.3 | - | - | - |
| Hill Slope d | 0.66± 0.03 | 0.50 ± 0.1 | 0.37 ± 0.04 | - | - | - | |
| Imax%c, d | 88 ± 3.6 | * | * | - | - | ||
| Countd | 2 | 2 | 2 | - | - | - | |
| 30 (apremilast) | IC50 nM | 26 ± 1.3 | 32 ± 1.6 | 24 ± 1.2 | 23 ± 1.2 | 35 ± 1.3 | 21 ± 1.2 |
| Hill Slope | 1.0 ± 0.14 | 1.2 ± 0.3 | 1.0 ± 0.05 | 1.1 ± 0.14 | 1.1 ± 0.09 | 1.1 ± 0.11 | |
| Imax % | 97 ± 3.5 | 96 ± 2.4 | 100 ± 0 | 98 ± 2.0 | 99 ± 1.0 | 98 ± 1.8 | |
| Count | 8 | 6 | 2 | 5 | 4 | 3 | |
Two-Way ANOVA (F(1.29) = 27.3, p <0.001; Sidak’s multiple comparisons test p= 0.0012
Two-Way ANOVA (F(1,29) = 27.3, p < 0.001; Sidak’s multiple comparisons test, p= 0.0019
Imax could not be calculated due to incomplete inhibition curve
- not tested. Values tabulated are mean ± SD and Count -the number of times that the compound was tested.
The crystal structure of 28 shows binding of its core and Ar2 similar to 23, with differences in how the AR1 group engages the UCR2 and catalytic domain (Figure 7). 28 was stable in human, rat, mouse and dog liver microsomes (88-100% remaining after 30 min incubation) with improved kinetic solubility (95 μM at pH 7.4). Protein binding was 99.5 – 99.8% in plasma from humans, rats, mice and dogs, although there was no shift in PDE4D inhibitory potency in the presence of BSA at 1 mg/mL. 28 had low potential for cytochrome P450 inhibition (CYP3A4 IC50 = 15 μM, 2C19 = 43 μM; 2D6 >100 μM; 2C9 > 51 μM). MDCK-MDR1 cellular permeability was high with low efflux (A-B = 53 x 10−6 cm/sec; B-A = x 10−6 cm/sec; Efflux Ratio = 1.3) despite the presence of the carboxylic acid. 28 lacked off target activity against PDE1-11 (BPS Bioscience), a panel of G-protein coupled receptors, ion channels and transporters (Cerep), or the human ERG cardiac channel with IC50 in all assays >10 μM. The compound was not mutagenic in bacteria. Evaluation of 28 in C57Bl6 mice showed high bioavailability (F% = 82%) and prolonged half-life (10-12 hr), although brain distribution was moderate (brain/plasma ratio = 0.45). 28 provided cognitive benefit in the mouse NOR at doses above 0.3 mg/kg PO (plasma Cmax = 153 ± 29 ng/ml, mean ± SD), while no change in anesthesia duration was observed at doses up to 30 mg/kg (plasma Cmax = 11,475 ± 3,570 ng/ml) (Figure 6).
Compound 29 showed reduced metabolic stability in liver microsomes in contrast to 23 (e.g., 36% remaining in mouse liver microsomes after 30 min incubation) with low kinetic solubility at pH 7.4 (29 μM), had increased plasma protein binding (99.6% - 99.8%), and was a weak CYP3A4 inhibitor (IC50 = 3.1 μM). 29 improved novel object discrimination in mice at a dose of 3 mg/kg, but also reduced the duration of anesthesia at the same dose (Figure 6). Therefore, the compound was abandoned.
We compared and contrasted the kinetics of PDE4 enzyme inhibition for 28 as compared to apremilast (30) across a panel of PDE4 enzymes (Figure 8). 28 was equipotent against two dimeric forms of PDE4D containing activating mutations that mimic UCR1 phosphorylation by PKA, PDE4D7-S129D and PDE4D5-S54D (Table 5). The two isoforms differ in the length of the N-terminal sequence preceding UCR1, but are identical in sequence thereafter. 28 is 14-fold less potent against a monomeric form of PDE4D, PDE4D2, which contains a truncated UCR2 that retains the portion of UCR2 needed for allosteric inhibitor binding. 28 is 137-fold selective for the activated PDE4D7 enzyme as compared to the basal enzyme, PDE4D7-S129(wt). Potency and selectivity against PDE4B1-S133D is roughly similar (225 fold), while deletion of UCR2 as in the PDE4D catalytic domain construct (PDE4D-cat) greatly reduces inhibitory potency. For comparison, apremilast (30) has no selectivity for PDE4 subtypes, inhibits equally dimeric and monomeric isoforms of PDE4D, and does not require the presence of UCR2. The steric bulk of the phthalate moiety in apremilast projects towards solvent when the compound is bound in the PDE4 active site and likely prevents closure of UCR2 (Supplemental Figure 2).
Figure 8:
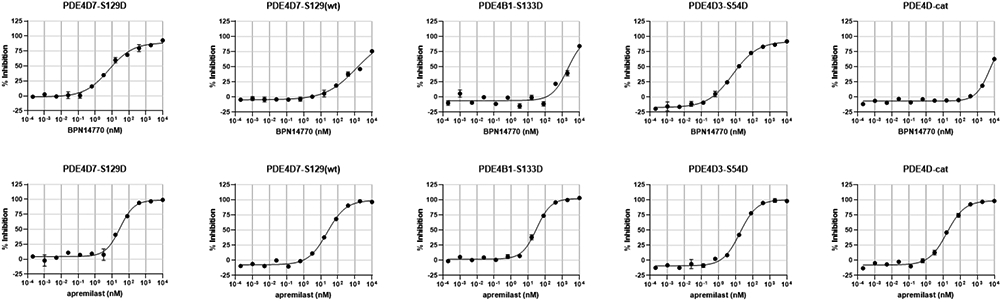
Kinetics of PDE4 enzyme inhibition contrasting a PDE4D subtype selective allosteric inhibitor (28, top row) with apremilast (30), an active site-directed PDE4 inhibitor competitive with cAMP (bottom row). The low Hill slope of the allosteric inhibitor against the activated, dimeric forms of PDE4D indicates negative cooperativity between the two active sites in the PDE4 dimer. Negative cooperativity is reduced (Hill slope tends towards 1) when UCR2 is deleted as in the truncated PDE4D-cat enzyme. Apremilast inhibits equally well all PDE4 subtypes independent of their activation state and does not require UCR2 for binding. Compounds were tested in duplicate wells using a coupled enzyme assay. The Z’ quality factor for the in vitro inhibition assay was >0.6. Representative assays are shown plotting the well mean ± SD (note: the variance between duplicate wells may be less than the size of the symbols).
We showed previously that PDE4D allosteric inhibitors behave kinetically as partial inhibitors with Imax ~90%.7 This kinetic behavior is seen with 28 (Figure 8, Table 5). Imax is ~90% against the two dimeric, activated forms of PDE4D tested as compared to apremilast (30) for which Imax =100%. This difference in kinetic behavior between 28 and 30 is statistically significant (Two-Way ANOVA, F(1,29) = 27.3, p <0.001). We showed previously that the two active sites of the PDE4D dimer exhibit negative cooperativity such that binding of inhibitor to the first site reduces the affinity for the second site (Hill slope < l).7 Binding of the allosteric inhibitor in the first site also reduces the catalytic activity (Kcat) of the second site, so rather than Imax = 50% (1st site closed, 2nd site open and fully active), Imax = 90% due to the decrease in Kcat of the 2nd site (1st site closed, 2nd site open but only partially active, see also Burgin et al. 20107). The kinetic behavior of 28 is altered when UCR2 is removed as in the truncated PDE4D catalytic domain (PDE4D-cat). Negative cooperativity is lost, 28 binding is competitive with cAMP, and the Hill slope for the inhibition curve tends towards to 1 (Figure 8).
Evaluation of 28 pharmacokinetics in rats and dogs showed high oral bioavailability (F% 80-100%). Safety pharmacology studies showed no adverse CNS or pulmonary effects in rats up to a maximum oral dose of 60 mg/kg, and no effect on dog cardiovascular function up to a maximum oral dose of 100 mg/kg. Dose range finding toxicology studies demonstrated inappetence and weight loss in rats at an oral dose of 100 mg/kg and emesis, tremor and weight loss in dogs at 100 mg/kg. Toxicological studies of 28 and 90 days duration in rats and dogs did not reveal vascular toxicity nor any other microscopic findings in contrast to PDE4 inhibitors that lack subtype selectivity. 28 did not cause emesis in ferrets at oral doses up to 30 mg/kg, nor did it cause emesis in cynomolgus monkeys at oral doses up to 50 mg/kg. Based on this profile, 28 was advanced into human clinical trials.
Compounds were synthesized as depicted in Scheme 1. 1,3,5-Triazines 4-11 were assembled as previously described.49 Pyrimidines 22-27 and 29 were assembled from the amidine 30 by cyclization with an appropriate beta-keto ester and chlorination with POCl3 to yield 31, then displacement of the pyrimidine chloride to obtain the desired Ar1 products. Pyridines 12-21 and 28 were synthesized by a regioselective Suzuki reaction of 32 to yield 33. Regioselective substitution of the pyridine chloride yielded the desired Ar1 products 12-21. Regioselective Suzuki reaction of 34 gave 35, which was then subjected to a second Suzuki reaction with a benzyl boronate which afforded 28.
Scheme 1:
Synthetic schemesa
a Reagents and Conditions: a) EtCOCH2CO2Et, NaOMe; b) POCl3; c) H2NAr1, heat; d) BrCH2Ar1, Pd(dppf)Cl2; e) HOAr1, K2CO3; f) 3-Cl PhB(OH)2, Pd(Ph3P)4, K3PO4; g) 1) HSAr1, Et3N; 2) m-CPBA;
DISCUSSION
We have shown that it is possible to discover potent, subtype selective, allosteric inhibitors of PDE4D. These exploit a key selectivity residue in the UCR2 regulatory domain, a phenylalanine in PDE4D versus a tyrosine in PDE4B. This key selectivity residue is biologically significant as it affects KM for substrate. PDE4D is more active at lower cAMP concentration with a KM of 1.5 μM as compared to 7.7 μM for PDE4B, while reciprocal exchange of the phenylalanine and the tyrosine between the two PDE4 subtypes reverses the difference in KM.7 Modeling studies suggest that the tyrosine hydroxyl in PDE4B may form a hydrogen bond with the cAMP ribose and that this is absent when a phenylalanine is present on UCR2 as in PDE4D. Based on these data, we offer the working hypothesis that the two active sites in the PDE4 dimer communicate by rotation around the four-helix bundle such that UCR2 alternately opens and closes in trans- across the opposite active site during the catalytic cycle. Capture of cAMP by the closure of UCR2 facilitates hydrolysis of the cyclic phosphodiester bond and subsequent release of AMP when UCR2 re-opens.
PDE4 allosteric inhibitors capture the dimer in a non-productive conformation in which one active site is closed by UCR2 while the other is open.7 The Kcat of the open active site is reduced as the corresponding UCR2 is prevented from closing over the cAMP substrate. Compounds with high affinity for both the catalytic domain and UCR2 are able to fully inhibit the dimeric enzyme by binding simultaneously in both active sites. Binding in the closed active site is due to completion of a hydrophobic surface that allows capping by UCR2, while binding in the open active site is competitive with substrate and does not require UCR2. Reducing affinity for the catalytic domain, as when the linker is a methylene rather than an amine, yields compounds with improved potency and selectivity for PDE4D as well as for the activated state of the enzyme. Allosteric inhibitors bind strongly in the closed active site, but due to reduced affinity for the open active site, behave as partial inhibitors with Imax = 90%. In this model, the N-terminus of UCR1 likely regulates rotation around the four-helix bundle. We hypothesize that this holds the dimer in a non-productive conformation which thereby reduces the basal activity of the enzyme. Correspondingly, phosphorylation by PKA releases inhibition by UCR1, allows rotation around the four-helix bundle, and permits full enzyme activity. Based on the genetic analysis of ACRDYS2 mutations, our therapeutic hypothesis is that inhibition of the activated, dimeric forms of PDE4D will be associated with cognitive benefit as this will augment cAMP signaling while maintaining the spatial and temporal patterning of information at the synapse.
One of the earliest biochemical findings in patients with Fragile X Syndrome (FXS) was decreased production of cAMP at baseline and in response to signaling through adenylate cyclase.57-59 FXS is an X chromosome-linked, genetic disorder associated with cognitive impairment, reduced IQ, and autistic behavior.60 Subsequent cloning of the FMR1 gene responsible for FXS allowed the creation of genetically exact models of the disorder in the Drosophila fruit fly and in mice.61-63 In both species, deletion of the FMR1 gene causes a decrease in baseline and stimulated levels of cAMP production.64-66 This causes defects in associative learning and neurodevelopment in the fly model and global changes in behavior in the mouse model. The FMR1 knockout mice show anxiety-related behaviors (increased open field activity), impaired social interaction with wild-type mice, and impairment of natural behaviors such as nest building and marble burying.62, 67 In patients, the FMR1 gene deletion prevents the maturation of excitatory synapses in cortex and hippocampus.68-69 Excitatory synapses occur on dendritic spines. These remain thin and elongate in patients with FXS and that defect also is seen in the mouse FMR1 knockout model. We show separately that 28 normalizes the behavior of adult FMR1 knockout mice and promotes the maturation of dendritic spines on layer 2/3 pyramidal cells, a type of neuron in cortex that expresses PDE4D.67 Knockdown of PDE4D in normal mice improves maturation of dendritic spines on cortical pyramidal neurons.70
In separate studies we have shown that 28 modulates signaling through cAMP leading to activation of PKA and phosphorylation of the CREB transcription factor.54 A challenge in exploring PDE4D pharmacology in mice is the absence of the key phenylalanine residue in UCR2 that is exploited for PDE4D subtype selectivity. Instead, this is a tyrosine as in PDE4A-C.7 The core amino acid sequence of PDE4D from UCR1 through the end of the catalytic domain is absolutely conserved across mammalian species except for the phenylalanine in UCR2 which is unique to primate.7 That single amino acid difference may be an important biological adaption in primates that allows the enzyme to function at lower cAMP concentrations, an adaption in primates that may enhance dendritic compartmentalization in cortical neurons.71
To better understand PDE4D pharmacology in mice, we humanized the mouse PDE4D gene by replacing the single codon encoding the key UCR2 tyrosine with the corresponding codon for phenylalanine.54 The single amino change in the humanized mouse enzyme lowers 28 IC50 from 133 ± 18 nM to 2.9 ± 0.3 nM. Correspondingly, 28 shows increased potency in humanized PDE4D mice as compared to wild-type mice. 28 increases brain cAMP, increases phosphorylation of CREB, augments the late phase of hippocampal long-term potentiation (LTP), improves short and long term memory, and increases production of brain-derived neurotrophic factor (BDNF) in hippocampus.54
CONCLUSION
Modification of this fundamental pathway of cognition has been the target of extensive research over multiple decades, but has not been evaluated clinically because earlier compounds lacked PDE4D subtype selectivity or could not allow for dosing within the therapeutic range. Evaluation of 28 (BPN14770) will provide the first proof-of-concept study for this therapeutic mechanism in humans and, if successful, will facilitate the development of a novel molecule for the treatment of Fragile X Syndrome and other brain disorders.
EXPERIMENTAL SECTION
General synthetic procedures.
Unless otherwise noted, all purchased reagents were used as received without further purification. 1H NMR spectra were recorded on a Bruker Avance III 400 MHz or Bruker Avance III 500 MHz spectrometer. 1H NMR spectra were reported in parts per million (ppm) downfield from tetramethylsilane. In reported spectral data, the format (δ) chemical shift (multiplicity, J values in Hz, integration) was used with the following abbreviations: s = singlet, br s = broad singlet, d = doublet, t = triplet, q = quartet, m = multiplet. Mass spectroscopy data were obtained on a Micromass LCT. Compound HPLC retention times using the following HPLC methods: Method 1: Agilent 1100 series, X-Bridge C18 column, 5-100% ACN/water (0.07% H3PO4) (3.5 min), 100% ACN (0.07% H3PO4) (0.25 min), 100% - 5% ACN/water (0.07% H3PO4)(1.25 min) and 5% ACN/water (0.07% H3PO4) (0.25 min), flow rate: 2 mL/min. Method 2: Varian Star, Gemini-NX C18 Column, 55% ACN (0.025% TFA)/water (3 min), 55-90% ACN (0.025% TFA)/water (18 min), 90% ACN (0.025% TFA)/water (2 min), 90-2% ACN (0.025% TFA)/water (2 min). Method 3: Varian Star, Gemini-NX C18 Column, 2-90% ACN (0.025% TFA)/water (18 min), 90% ACN (0.025% TFA)/water (2 min), 90-2% ACN (0.025% TFA)/water (2 min), flow rate: 1.0 mL/min. Method 4: Varian Star, Phenomenex Luna C18(2) Column, 10-95% ACN/water (0. 1% TFA) (10 min), 95% ACN/water (0.1% TFA) (6 min), flow rate: 1.0 mL/min. Method 5: Varian Star, Phenomenex Luna C18(2) Column, 5-95% ACN/water (0. 1% TFA) (15 min), 95% ACN/water (0.1% TFA) (5 min), flow rate: 0.75 mL/min. Method 6: Varian Star, Phenomenex Luna C18(2) Column, 10-90% ACN/water (0.025% TFA) (10 min), 90% ACN/water (0.025% TFA) (10 min), flow rate: 1.0 mL/min. Unless otherwise stated compounds are ≥ 95% determined by HPLC or elemental analysis.
General Procedure for the Synthesis of Compoound 4-8.
To a suspension of 2-chloro-4-(3-chlorophenyl)-6-ethyl-1,3,5-triazine (1 eq) and appropriate aniline (1.5-2.0 eq) were suspended in HOAc and heated to 100 °C until the reaction was complete by HPLC. The cooled reaction mixture was poured into water and the precipitate collected and washed with water to obtain the desired product.
3-(4-((4-(3-Chlorophenyl)-6-ethyl-1,3,5-triazin-2-yl)amino)phenyl)propan-1-ol (4).
1H NMR (DMSO-d6, 500 MHz) δ 10.19 (s, 1H), 8.41–8.31 (m, 2H), 7.71–7.67 (m, 3H), 7.60 (t, J= 8.0 Hz, 1H), 7.24–7.16 (m, 2H), 4.45 (t, J= 5.5 Hz, 1H), 3.43 (q, J= 5.5 Hz, 2H), 2.77 (q, J = 7.5 Hz, 2H), 2.60 (t, J= 7.5 Hz, 2H), 2.51–2.49 (m, 2H), 1.75–1.69 (m, 2H), 1.32 (t, J = 7.5 Hz, 3H); APCI MS m/z 369.2 [M + H]+. HPLC Method 2: retention time: 12.85 min.
2-(4-(4-(3-Chlorophenyl)-6-ethyl-1,3,5-triazin-2-ylamino)phenyl)ethanol (5).
1H NMR (CDCl3, 360 MHz) δ 8.46 (s, 1H), 8.35 (d, J = 7.6 Hz, 1H), 7.66 (d, J = 8.4 Hz, 2H), 7.50 (m, 1H), 7.43 (dd, J = 7.6 and 7.8 Hz, 1H), 7.28 (d, J = 8.4 Hz, 2H), 3.89 (m, 2H), 2.89 (m, 4H), 1.40 (t, J = 7.5 Hz, 3H). TOF MS ES+ m/z 355.1 [M + H]+. HPLC Method 1: retention time: 3.23 min.
2-(4-(4-(3-Chlorophenyl)-6-ethyl-1,3,5-triazin-2-ylamino)phenyl)acetamide (6).
1H NMR (CDCl3, 360 MHz) δ 8.44 (s, 1H), 8.35 (d, J = 7.7 Hz, 1H), 7.71 (d, J = 8.4 Hz, 2H), 7.50 (m, 1H), 7.42 (dd, J = 7.7 and 8.0 Hz, 1H), 7.32 (d, J = 8.4 Hz, 2H), 5.40 (br s, 2H), 3.59 (s, 2H), 2.84 (q, J = 7.5 Hz, 2H), 1.39 (t, J = 7.5 Hz, 3H). TOF MS ES+ m/z 368.1 [M + H]+. HPLC Method 1: retention time: 2.89 min.
(4-((4-(3-Chlorophenyl)-6-ethyl-1,3,5-triazin-yl)amino)phenyl)methanol (7).
1H NMR (DMSO-d6, 500 MHz) δ 10.24 (s, 1H), 8.42–8.31 (m, 2H), 7.75–7.68 (m, 3H), 7.61 (t, J = 7.5 Hz, 1H), 7.36–7.28 (m, 2H), 5.11 (t, J = 5.5 Hz, 1H), 4.48 (d, J = 5.5 Hz, 2H), 2.78 (q, J = 7.5 Hz, 2H), 1.32 (t, J = 7.5 Hz, 3H); APCI MS m/z 341.1 [M + H]+. HPLC Method 2: retention time: 10.79 min.
4-(4-(3-chlorophenyl)-6-ethyl-1,3,5-triazin-2-ylamino)benzamide (8).
1H NMR (DMSO-d6, 360 MHz,) δ 10.53 (s, 1H), 8.35 (m, 2H), 7.89 (br s, 4H), 7.70 (d, J = 7.7 Hz, 2H), 7.61 (dd, J = 7.8 and 8.0 Hz, 1H), 7.26 (br s, 2H), 2.82 (q, J = 7.5 Hz, 2H), 1.32 (t, J = 7.5 Hz, 3H). TOF MS ES+ m/z 354.1 [M + H]+. HPLC Method 1: retention time: 2.95 min.
General Procedure for the Synthesis of Compoound 9-11.
A suspension of 2-chloro-4-(3- chlorophenyl)-6-ethyl-1,3,5-triazine (1 eq) and the appropriate aniline (2.0 eq) in HOAc was heated to 100 °C until the reaction was complete by HPLC. The cooled reaction mixture was diluted with saturated NaHCO3 and EtOAc. The organic phase was dried over Na2SCO4, filtered and concentrated. The crude material was purified by chromatography to obtain desired product.
2-(4-(4-(3-Chlorophenyl)-6-ethyl-1,3,5-triazin-2-ylamino)phenyl)propanoic acid (9).
1H NMR (DMSO-d6, 360 MHz,) δ 12.26 (s, 1H), 10.28 (s, 1H), 8.32 (m, 2H), 7.73 (d, J = 8.4 Hz, 2H), 7.68 (d, J = 7.7 Hz, 1H), 7.60 (dd, J = 7.7 and 7.9 Hz, 1H), 7.27 (d, J = 8.4 Hz, 2H), 3.64 (q, J = 7.1 Hz, 1H), 2.76 (q, J = 7.5 Hz, 2H), 1.35 (d, J = 7.1 Hz, 3H), 1.30 (t, J = 7.5 Hz, 3H). TOF MS ES+ m/z 353.3 [M + H]+. HPLC Method 1: retention time: 3.34 min.
4-((4-(3-Chlorophenyl)-6-ethyl-1,3,5-triazin-2-yl)amino)benzoic acid (10).
1H NMR (DMSO-d6, 360 MHz,) δ 10.65 (s, 1H), 8.36 (m, 2H), 7.95 (br s, 4H), 7.70 (d, J = 7.4 Hz, 1H), 7.61 (dd, J = 7.7 and 8.2 Hz, 1H), 2.81 (q, J = 7.5 Hz, 2H), 1.33 (t, J = 7.6 Hz, 3H). TOF MS ES+ m/z 355.1 [M + H]+. HPLC Method 1: retention time: 3.30 min.
2-(4-((4-(3-Chlorophenyl)-6-ethyl-1,3,5-triazin-2-yl)amino)phenyl)acetic acid (11).
1H NMR (DMSO-d6, 360 MHz,) δ 10.29 (s, 1H), 8.32 (m, 2H), 7.58~7.73 (m, 4H), 7.24 (d, J = 7.4 Hz, 2H), 3.53 (s, 2H), 2.76~2.80 (q, J = 7.5 Hz, 2H), 1.30 (t, J = 7.5 Hz, 3H). TOF MS ES+ m/z 369.1 [M + H]+. HPLC Method 1: retention time: 3.19 min.
2-[4-[[2-(3-Chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl]amino]phenyl]ethanol (12).
An 18-mL test tube was charged with 33 (30 mg, 0.1 mmol) and methyl 2-(4-aminophenyl)acetate (33 mg, 0.2 mmol) and the resulting mixture heated at 150 °C under Ar for 1 hour. After cooling to rt, the mixture was partitioned between NaHCC3 (10 ml) and DCM (10 ml). The organic phase was concentrated and the crude material purified by chromatography (DCM/MeOH, 0 to 3%) to give methyl 2-[4-[[2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl]amino]phenyl]acetate (31 mg, 77% yield).
LAH (20 mg, 0.53 mmol, 10 eq) was added portion-wise to a 0 °C solution of methyl 2-[4-[[2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl]amino]phenyl]acetate (20.7 mg, 0.053 mmol) in THF (2 ml). When the addition was complete, the reaction was stirred under Ar at rt overnight. The reaction was quenched with 2N HCl (aq.) (2 ml) and saturated NaHCO3. The mixture was extracted with EtOAc (3 x 5 ml), and the organic layers combined and concentrated. The crude material was purified by chromatography using (DCM/EtOAc, 0 to 30%) to give compound 12 (3.5 mg, 18% yield). 1H NMR (CDCl3, 360 MHz) δ 7.84 (m, 2H), 7.41 (m, 3H), 7.21 (m, 3H), 7.16 (s, 1H), 5.95 (br s, 1H), 3.90 (m, 2H), 3.18 (m, 2H), 2.87 (m, 4H), 2.24 (m, 2H). TOF MS ES+ m/z 331.1 [M + H]+. HPLC Method 1: retention time: 1.86 min.
3-(4-((2-(3-Chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)amino)phenyl)propanamide hydrochloride (13).
A mixture of 33 and 3-(4-aminophenyl)propanamide (0.066 g, 0.40 mmol) in NMP (3 mL) was heated under microwave conditions at 120 °C for 3 hrs, then at 140 °C for 2.5 hrs. The reaction was diluted with EtOAc, washed with water and brine, then dried with Na2SO4, and concentrated. The crude material was purified by silica gel chromatography (hexanes/EtOAc, 0 to 100%, then DCM/MeOH, 0 to 5%). The resulting solids were further purified by preparative HPLC to afford compound 13 (0.042 g, 54%) as a white solid. 1H NMR (DMSO-d6, 500 MHz) δ 14.09 (br s, 1H), 9.82 (s, 1H), 7.90-7.89 (m, 1H), 7.71-7.58 (m, 3H), 7.33-7.27 (m, 5H), 6.98 (s, 1H), 6.77 (br s, 1H), 3.18-3.15 (m, 2H), 2.93-2.80 (m, 4H), 2.40-2.34 (m, 2H), 2.27-2.21 (m, 2H); ESI MS m/z 392.0 [M + H]+. HPLC Method 1: retention time: 1.81 min.
General Procedure for the Synthesis of Compoound 14 and 15.
The appropriate aniline (3 eq) and 33 (1 eq) were heated for 3 hrs at 150 °C. Toluene was added to the reaction, which was then heated for an additional 7 hours. After this time, the mixture was purified by silica gel chromatography eluting with DCM and MeOH and the resulting solid converted to the HCl salt.
2-(4-((2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)amino)phenyl)-N,N-dimethylacetamide hydrochloride (14).
1H NMR (DMSO-d6, 500 MHz) δ 14.2 (br s, 1H), 9.88 (s, 1H), 7.91-7.90 (m, 1H), 7.72-7.66 (m, 2H), 7.61-7.58 (m, 1H), 7.37-7.32 (m, 1H), 7.01 (s, 1H), 3.73 (s, 2H), 3.17 (t, J = 7.7 Hz, 2H), 3.04 (s, 3H), 2.93 (t, J = 7.2 Hz, 2H), 2.84 (s, 3H), 2.27-2.21 (m, 2H); ESI+ MS m/z 406.0 [M + H]+. HPLC Method 3: retention time: 11.83 min.
2-(4-((2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)amino)phenyl)-N-methylacetamide hydrochloride (15).
1H HNMR (DMSO-d6, 500 MHz) δ 14.07 (br s, 1H), 9.82 (s, 1H), 8.04-8.03 (m, 1H), 7.90-7.89 (m, 1H), 7.71-7.58 (m, 3H), 7.38-7.34 (m, 4H), 7.01 (s, 1H), 3.44 (s, 2H), 3.18-3.14 (m, 2H), 2.93 (t, J = 13 Hz, 2H), 2.59 (d, J = 4.6 Hz, 3H), 2.27-2.21 (m, 2H); ESI MS m/z 392.0 [M + H]+. HPLC Method 3: retention time: 11.29 min.
2-(3-((2-(3-Chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)amino)phenyl)ethanol hydrochloride (16).
Methyl 2-(3-aminophenyl)acetate (0.130 g, 0.79 mmol) and 33 (0.160 g, 0.53 mmol) in dioxane were treated with Pd(OAc)2 (5.8 mg, 0.026 mmol), rac-BINAP (24 mg, 0.040 mmol), and CS2CO3 (430 mg, 1.33 mmol) and, under Ar, was heated until 33 was consumed. The reaction was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SC4, filtered, and concentrated. The residue was purified by chromatography (hexanes/EtOAc, 0 to 100%) to afford the product which then converted to the hydrochloride to afford methyl 2-(3-((2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)amino)phenyl)acetate hydrochloride (0.190 g, 91%) as a white solid. 1H NMR (DMSO-d6, 500 MHz) δ 14.04 (s, 1H), 9.81 (s, 1H), 7.92 (t, J = 1.8 Hz, 1H), 7.76–7.71 (m, 1H), 7.69–7.64 (m, 1H), 7.60 (t, J = 7.9 Hz, 1H), 7.44 (t, J = 7.9 Hz, 1H), 7.38–7.34 (m, 1H), 7.33–7.28 (m, 1H), 7.23–7.17 (m, 1H), 7.09 (s, 1H), 3.77 (s, 2H), 3.62 (s, 3H), 3.16 (t, J = 7.5 Hz, 2H), 2.93 (t, J = 7.5 Hz, 2H), 2.24 (quin, J = 7.5 Hz, 2H); APCI MS m/z 393 [M + H]+.
To a solution of methyl 2-(3-((2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)amino)phenyl)acetate (0.080 g, 0.20 mmol) in THF at 0 °C was added, BH3•DMS (2.0 M, 0.30 mL, 0.60 mmol). The reaction was warmed to rt and stirred until starting material was consumed by HPLC. The reaction was quenched with 0.5 M HCl, diluted with saturated NaHCO3 and extracted with EtOAc. The organic layer was dried over Na2SO4, filtered and concentrated. The crude material was purified by chromatography (hexanes/EtOAc, 0 to 100%) and the isolated material treated with HCl in MeOH to give compound 16 (0.064 g, 80%) as a light-yellow solid. 1H NMR (DMSO-d6, 500 MHz) δ 14.08(s, 1H), 9.81 (s, 1H), 7.90 (t, J = 1.8 Hz, 1H), 7.76–7.68 (m, 1H), 7.67–7.63 (m, 1H), 7.59 (t, J = 7.8 Hz, 1H), 7.40 (t, J = 7.8 Hz, 1H), 7.30–7.27 (m, 1H), 7.26–7.23 (m, 1H), 7.20–7.16 (m, 1H), 7.09 (s, 1H), 3.65 (t, J = 6.5 Hz, 2H), 3.16 (t, J = 7.5 Hz, 2H), 2.92 (t, J = 7.5 Hz, 2H), 2.77 (t, J = 6.5 Hz, 2H), 2.24 (quin, J = 7.5 Hz, 2H); APCI MS m/z 365.2 [M + H]+. HPLC Method 3: retention time: 11.92 min.
2-[4-[[2-(3-Chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl]amino]phenyl]acetic acid (17).
Methyl 2-[4-[[2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl]amino]phenyl]acetate (31 mg, 0.77 mmol) and LiOH (48 mg, 1.14 mmol) in THF/water (2:1, 1.5 mL) were stirred at rt overnight. 2N HCl (0.6 ml) was added and the volatiles removed. The crude product was purified by chromatography (DCM/MeOH, 2 to 5%) to give compound 17 (26 mg, HCl salt, 81% yield). 1H NMR (DMSO-d6, 360 MHz) δ 14.23 (br s, 1H), 12.39 (br s, 1H), 9.84 (br s, 1H), 7.91 (s, 1H), 7.70 (d, J = 7.70 Hz, 1H), 7.63 (d, J = 8.40, 1H), 7.57 (dd, J = 7.70, 8.40 Hz, 1H), 7.36 (br s, 4H), 7.02 (s, 1H), 3.60 (s, 2H), 3.11 (t, J = 7.90 Hz, 2H), 2.90 (t, J = 7.40 Hz, 2H), 2.20 (m, 2H). TOF MS ES+ m/z 378.9 [M + H]+. HPLC Method 1: retention time: 2.01 min.
2-(4-((2-(3-Chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)methyl)phenyl)acetamide hydrochloride (18).
A 10-mL sealed tube was charged with 33 (0.100 g, 0.33 mol), T-l (0.096 g, 0.33 mmol), Pd(dppf)Cl2 (0.027 g, 0.033 mmol), and powdered Na2CO3 (0.141 g, 1.33 mmol). Dioxane (3 mL) and water (1.5 mL) were added and the mixture stirred under Ar at 90 °C for 3 days until 33 was consumed. The reaction was cooled and purified by chromatography (hexanes/EtOAc, 0 to 100%) to afford methyl 2-(4-((2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)methyl)phenyl)acetate (0.036 g, 48%) as an off-white solid. 1H NMR (CDCl3, 500 MHz) δ 7.94–7.91 (m. 1H), 7.77 (dt, J = 7.0. 2.0 Hz. 1H). 7.36–7.30 (m. 2H), 7.24 (s, 1H), 7.21 (d, J = 8.0 Hz, 2H), 7.13 (d, J = 8.0 Hz, 2H), 3.95 (s, 2H), 3.69 (s, 3H), 3.60 (s, 2H), 3.08 (t, J = 8.0 Hz, 2H), 2.86 (t, J = 7.5 Hz, 2H), 2.18–2.09 (m, 2H); APCI MS m/z 392 [M+H]+.
A 10-mL vial was charged with methyl 2-(4-((2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)methyl)phenyl)acetate (0.036 g, 0.09 mmol) and NH4Cl (0.015 g, 0.27 mmol). To this was added MeOH (2 mL) followed by 7N NH3 / MeOH (4 mL, 27.6 mmol). The vial was sealed and heated at 100 °C for 48 hrs. The reaction was concentrated, purified by chromatography (hexanes/EtOAc (0 to 100%). The isolated material was treated with HCl in MeOH to afford the compound 18 (0.037 g, 99%) as a white solid, mp 178-180 °C. 1H NMR (DMSO-d6, 500 MHz) δ 8.11–8.07 (m, 1H), 8.00–7.93 (m, 1H), 7.81 (s, 1H), 7.57–7.47 (m, 2H), 7.43 (br s, 1H), 7.25–7.34 (m, 4H), 6.84 (br s, 1H), 4.60 (br s, 1H), 4.00 (s, 2H), 3.31 (s, 2H), 3.00 (t, J = 7.5 Hz, 2H), 2.87 (t, J = 7.5 Hz, 2H), 2.07 (q, J = 7.5 Hz, 2H); APCI MS m/z 377.4 [M+H]+. HPLC Method 3: retention time: 12.20 min.
2-[4-[[2-(3-Chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl]amino]phenyl]acetamide hydrochloride (19).
An 18-mL test tube was charged with 33 (30 mg, 0.1 mmol) and 2-(4-aminophenyl)acetonitrile (30 mg, 0.23 mmol), then heated at 150 °C under Ar for 1 hr. After cooling to rt, the reaction was diluted with NaHCO3 (10 ml) and DCM (10 ml). The organic layer was concentrated and the crude material purified by chromatography using DCM followed by EtOAc to give 2-[4-[[2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl]amino]phenyl]acetonitrile (16 mg, 44% yield). 1H NMR (CDCl3, 360 MHz) δ 7.84 (s, 1H), 7.68 (m, 1H), 7.32 (m, 4H), 7.21 (m, 2H), 7.15 (s, 1H), 5.82 (br s, 1H), 3.75 (s, 2H), 3.09 (t, J = 7.50 Hz, 2H), 2.83 (t, J = 7.20 Hz, 2H), 2.21 (m, 2H).
Con H2SO4 (0.5 ml) was added at 0 °C to an 18 mL vial containing 2-[4-[[2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl]amino]phenyl]acetonitrile (12 mg, 0.033 mmol) and stirred at rt for 6 hrs.. The reaction was slowly poured onto 0 °C Na2CO3. The resulting precipitate was collected and washed with water, then dried. The solid was treated with 4N HCl in dioxane and the volatile material was removed to give compound 19. (2.6 mg, 19% yield). 1H NMR (CDCl3, 360 MHz) δ 7.83 (s, 1H), 7.69 (m, 1H), 7.18 ~7.30 (m, 7H), 5.77 (br s, 1H), 5.44 (br s, 2H), 3.58 (s, 2H), 3.08 (t, J = 7.70 Hz, 2H), 2.83 (t, J = 7.10 Hz, 2H), 2.21 (m, 2H). ESI+ MS m/z 378.2 [M + H]+. HPLC Method 3: retention time: 11.37 min.
2-(4-((2-(3-Chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)sulfonyl)phenyl)acetamide (20).
A mixture of 33 (0.120 g, 0.40 mmol), methyl 2-(4-mercaptophenyl)acetate (0.100 g, 0.55 mmol), and Et3N (0.145 g, 1.3 mmol) in DMF was heated at 110 °C for 24 hrs. The reaction was concentrated, diluted with EtOAc (100 mL), washed with 10% aqueous LiCl (2 × 100 mL), dried over Na2SO4, filtered, and concentrated. The residue was purified by chromatography (hexane/EtOAc, 0 to 50%) to afford methyl 2-(4-((2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)thio)phenyl)acetate (0.164 g, 100%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 7.78-7.77 (m, 1H), 7.61–7.46 (m, 3H), 7.37 (d, J = 8.2 Hz, 2H), 7.30–7.28 (m, 3H), 6.89 (s, 1H), 3.73 (s, 3H), 3.70 (s, 2H), 3.09 (t, J = 7.7 Hz, 2H), 2.90 (t, J = 7.4 Hz, 2H), 2.24–2.05 (m, 4H); APCI MS m/z 410 [M + H]+.
A solution of methyl 2-(4-((2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)thio)phenyl)acetate (0.164 g, 0.40 mmol) and m-CPBA (0.269 g, 1.2 mmol) in DCM (15 mL) was stirred at rt for 2 hrs. The reaction was diluted with DCM (100 mL), washed with water (100 mL) and saturated NaHCO3 (100 mL), dried over MgSO4, filtered, and concentrated. The crude material was purified by chromatography (hexane/EtOAc, 0 to 50%) to afford methyl 2-(4-((2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)sulfonyl)phenyl)acetate (0.109 g, 62%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 8.09–8.0 (m, 2H), 7.97–7.85 (m, 3H), 7.57–7.39 (m, 4H), 3.75–3.68 (m, 5H), 3.18–3.07 (m, 4H), 2.22–2.05 (m, 2H); APCI MS m/z 442 [M + H]+.
A suspension of methyl 2-(4-((2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)sulfonyl)phenyl)acetate (0.109 g, 0.25 mmol) in 7N NH3/MeOH was heated at 100 °C in a sealed tube. After 24 hours, the mixture was concentrated and purified by chromatography (DCM/(89:9:1, DCM/MeOH/con NH4OH), 0 to 50%) to afford compound 20 (0.055 g, 53%) as a white solid. 1H NMR (DMSO-d6, 500 MHz) δ 8.20–8.18 (m, 2H), 8.10–8.08 (m, 1H), 8.03 (d, J = 8.4 Hz, 2H), 7.56–7.51 (m, 5H), 6.98 (s, 1H), 3.50 (s, 2H), 3.12 (t, J = 7.6 Hz, 2H), 3.03 (t, J = 7.8 Hz, 2H), 2.13–2.08 (m, 2H); APCI MS m/z 427.3 [M + H]+. HPLC Method 3: retention time: 18.03 min.
2-(4-((2-(3-Chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridin-4-yl)(methyl)amino)phenyl) acetamide (21).
A mixture of 33 (0.060 g, 0.20 mmol) and 2-(4-(methylamino)phenyl)acetamide (0.066 g, 0.40 mmol) in NMP (3 mL) was microwaved for 3 hrs at 140 °C. The reaction was diluted with EtOAc, washed with water and brine, then dried with Na2SO4, and concentrated. The crude material was purified by silica gel chromatography (hexanes/EtOAc, 0 to 100%, then DCM/MeOH, 0 to 5%). The resulting solids were further purified by preparative HPLC to afford the title compound 21 (0.012 g, 15%) as a white solid. 1H NMR (DMSO-d6, 500 MHz) δ 14.18 (br s, 1H), 8.10 (s, 1H), 7.94 (d, J = 7.5 Hz, 1H), 7.70-7.66 (m, 2H), 7.55 (br s, 1H), 7.37-7.27 (m, 5H), 6.92 (br s, 1H), 3.57 (s, 3H), 3.01 (t, J = 7.1 Hz, 2H), 1.96-1.85 (m, 4H); ESI MS m/z 392.1 [M + H]+. HPLC Method 4: retention time: 8.80 min.
2-(4-((2-(3-Chlorophenyl)-6-ethylpyrimidin-4-yl)amino)phenyl)ethanol (22).
A mixture of 31 (0.150 g, 0.59 mmol), 2-(4-aminophenyl)ethanol (0.134 g, 0.89 mmol), and 4M HCl in dioxane (0.222 mL, 0.89 mmol) in EtOH was heated for 2.5 h at 85 °C. The mixture was poured onto NaHCO3 at 0 °C and extracted with EtOAc. The combined organic layers were dried with Na2SO4 and concentrated. The crude residue was purified by silica gel chromatography (DCM/MeOH, 0 to 5%) to afford compound 22 (0.145 g, 70%) as a yellow solid. 1H NMR (DMSO-d6, 500 MHz) δ 9.53 (s, 1H), 8.33-8.28 (m, 2H), 7.62-7.55 (m, 4H), 7.22 (d, J = 8.4 Hz, 2H), 6.58 (s, 1H), 4.62 (t, J = 5.2 Hz, 1H), 3.63-3.59 (m, 2H), 2.72-2.64 (m, 4H), 1.26 (t, J = 7.6 Hz, 3H); ESI MS m/z 354.0 [M + H]+. HPLC Method 5: retention time: 10.85 min.
Preparation of 2-(4-((2-(3-Chlorophenyl)-6-ethylpyrimidin-4-yl)methyl)phenyl)acetamide (23).
A mixture of 31 (870 mg, 3.44 mol), T-l (1.00 g, 3.44 mmol), Pd(dppf)Cl2 (280 mg, 0.34 mmol), and powdered Na2CO3 (1.09 g, 10.3 mmol) in dioxane (16 mL) and water (8 mL) was heated at 90 °C under Ar for 2 hrs . The reaction was cooled to rt, then filtered through Celite and the pad washed well with EtOAc. The filtrate was washed with brine (3 x 25 mL), dried over Na2SO4, filtered and concentrated. The residue (1.8 g) was purified by chromatography (hexanes/DCM, 0 to 100%, then hexanes/EtOAc (0 to 24%) to afford methyl 2-(4-((2-(3-chlorophenyl)-6-ethylpyrimidin-4-yl)methyl)phenyl)acetate (0.65 g, 50% yield) as a colorless oil. 1H NMR (CDCl3, 500 MHz) δ 8.49–8.47 (m, 1H), 8.37 (dt, J = 7.5, 1.5 Hz, 1H), 7.44–7.38 (m, 2H), 7.31–7.27 (m, 2H), 7.27–7.23 (m, 2H, overlaps with CDCl3), 6.85 (s, 1H), 4.10 (s, 2H), 3.69 (s, 3H), 3.61 (s, 2H), 2.76 (q, J = 7.5 Hz, 2H), 1.31 (t, J = 7.5 Hz, 3H).
In a20-mLvial, methyl 2-(4-((2-(3-chlorophenyl)-6-ethylpyrimidin-4-yl)methyl)phenyl)acetate (113 mg, 0.30 mmol) and NH4Cl (48 mg, 0.89 mmol) were suspended in MeOH (3 mL) and treated with 7N NH3/MeOH (7.5 mL, 53 mmol). The vial was sealed and stirred at 100 °C for 43 hours. The crude material was purified by chromatography (DCM/MeOH, 0 to 5%; then hexanes/EtOAc, 0 to 100%) to give compound 23 (37 mg, 35% yield) as a white solid. 1H NMR (DMSO-d6, 500 MHz) δ 8.38–8.33 (m, 2H), 7.61–7.53 (m, 2H), 7.41 (br s, 1H), 7.29 (d, J = 8.0 Hz, 2H), 7.25 (s, 1H), 7.20 (d, J = 8.0 Hz, 2H), 6.82 (br s, 1H), 4.09 (s, 2H), 3.32 (s, 2H), 2.77 (q, J = 7.5 Hz, 2H), 1.26 (t, J = 7.5 Hz, 3H). APCI MS m/z 366.3 [M + H]+. HPLC Method 3: retention time: 19.54 min.
2-(4-((2-(3-Chlorophenyl)-6-ethylpyrimidin-4-yl)oxy)phenyl)ethanol (24).
A solution of 31 (0.200 g, 0.79 mmol), methyl 2-(4-hydroxyphenyl)acetate (0.131 g, 0.79 mmol) and K2CO3 (0.546 g, 3.95 mmol) in ACN (5 mL) was heated to 85 °C for 7 hours. The reaction mixture was diluted with water and extracted with EtOAc. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by chromatography (hexanes/DCM, 0 to 100%) to afford methyl 2-(4-((2-(3-chlorophenyl)-6-ethylpyrimidin-4-yl)oxy)phenyl)acetate (0.130 g, 65%). 1H NMR (CD3OD, 500 MHz) δ 8.21 (t, J = 1.8 Hz, 1H), 8.16–8.12 (m, 1H), 7.46–7.35 (m, 4H), 7.21-7.17 (m, 2H), 6.77(s, 1H), 3.73 (s, 2H), 3.72 (s, 3H), 2.83 (q, J = 7.6 Hz, 2H), 1.35 (t, J = 6.4 Hz, 3H); APCI MS m/z 383 [M + H]+.
To a solution of methyl 2-(4-((2-(3-chlorophenyl)-6-ethylpyrimidin-4-yl)oxy)phenyl)acetate (0.075 g, 0.20 mmol) in THF (1.3 mL) at 0 °C was added BH3•SMe2 (0.031 g, 0.39 mmol) and the mixture stirred at rt overnight. The reaction was quenched with 0.5M HCl, diluted with NaHCO3 and extracted with EtOAc. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by chromatography (hexanes/DCM, 0 to 100%) to afford compound 24 as a white solid (0.052 g, 69%). 1H NMR (DMSO-d6, 500 MHz) δ 8.17–8.10 (m, 2H), 7.59–7.54 (m, 1H),7.51 (t, J = 7.9 Hz, 1H), 7.37-7.32 (m, 2H), 7.22–7.17 (m, 2H), 6.88 (s, 1H), 4.67 (t, J = 5.2 Hz, 1H), 3.68–3.62 (m, 2H), 2.79 (q, J = 7.3 Hz, 4H), 1.28 (t, J = 7.6 Hz, 3H); APCI MS m/z 355.1 [M + H]+. HPLC Method 4: retention time: 14.83 min.
2-(4-((2-(3-Chlorophenyl)-6-ethylpyrimidin-4-yl)methyl)phenyl)ethanol (25).
A solution of 31 (52 mg, 0.13 mmol) and THF (3 mL) at 0 °C was treated with a solution of 1M LAH in THF (0.31 mL, 0.31 mmol) then slowly warmed to rt. After 4.5 hours, the mixture was quenched with MeOH, diluted with water (10 mL), and acidified with 2N HCl to pH ~3. It was extracted with EtOAc (3 x 12 mL) and the combined organic layers washed with saturated brine (5 mL), dried over Na2SO4, filtered, and concentrated. The crude material was purified by chromatography (hexanes/EtOAc, 0 to 50%) to afford compoound 25 (17 mg, 36% yield) as a colorless oil. 1H NMR (DMSO-d6, 500 MHz) δ 8.38–8.33 (m, 2H), 7.61–7.53 (m, 2H), 7.28–7.25 (m, 3H), 7.16 (d, J = 8.0 Hz, 2H), 4.58 (t, J = 5.5 Hz, 1H), 4.08 (s, 2H), 3.60–3.53 (m, 2H), 2.77 (q, J = 7.5 Hz, 2H), 2.67 (t, J = 7.0 Hz, 2H), 1.26 (t, J = 7.5 Hz, 3H). ESI MS m/z 353.3 [M + H]+. HPLC Method 3: retention time: 21.65 min.
2-(4-((2-(3-Chlorophenyl)-6-ethylpyrimidin-4-yl)amino)phenyl)acetamide (26).
A mixture of 31 (0.150 g, 0.59 mmol) and 2-(4-aminophenyl)acetamide (0.107 g, 0.71 mmol) and 4M HCl in dioxane (2 drops) in HOAc (3 mL) was heated for 2 hrs at 108 °C. The mixture was cooled to rt, neutralized with saturated NaHCO3, extracted with EtOAc, dried over Na2SO4, filtered, and concentrated. The residue was purified by chromatography (DCM/MeOH, 0 to 5%) to afford the title compound 26 (0.135 g, 62%) as a white solid. 1H NMR (DMSO-d6, 500 MHz) δ 9.57 (s, 1H), 8.33-8.29 (m, 2H), 7.64 (d, J = 8.4 Hz, 2H), 7.58-7.54 (m, 2H), 7.43 (br s, 1H), 7.27 (d, J = 8.5 Hz, 2H), 6.86 (br s, 1H), 6.59 (s, 1H), 3.35 (s, 2H), 2.67 (q, J = 7.6 Hz, 2H), 1.26 (t, J = 7.6 Hz, 3H); ESI MS m/z 367.0 [M + H]+. HPLC Method 5: retention time: 10.14 min.
2-(4-((2-(3-Chlorophenyl)-6-ethylpyrimidin-4-yl)oxy)phenyl)acetamide (27).
A solution of 31 (0.200 g, 0.79 mmol), methyl 2-(4-hydroxyphenyl)acetate (0.131 g, 0.79 mmol) and K2CO3, (0.546 g, 3.95 mmol) in ACN (5 mL) was heated to 85 °C for 7 hours. The reaction mixture was diluted with water and extracted with EtOAc. The combined organic layers were dried over Na2SO4, filtered, and concentrated. The residue was purified by chromatography (hexanes/DCM, 0 to 100%) to afford methyl 2-(4-((2-(3-chlorophenyl)-6-ethylpyrimidin-4-yl)oxy)phenyl)acetate (0.130 g, 65%). 1H NMR (CD3OD, 500 MHz) δ 8.21 (t, J = 1.8 Hz, 1H), 8.16–8.12 (m, 1H), 7.46–7.35 (m, 4H), 7.21-7.17 (m, 2H), 6.77(s, 1H), 3.73 (s, 2H), 3.72 (s, 3H), 2.83 (q, J = 7.6 Hz, 2H), 1.35 (t, J = 6.4 Hz, 3H); APCI MS m/z 383 [M + H]+.
Methyl 2-(4-((2-(3-chlorophenyl)-6-ethylpyrimidin-4-yl)oxy)phenyl)acetate (0.075 g, 0.20 mmol) and 7 M NH3/MeOH (3 mL) were heated at 100 °C for 72 hours. The reaction was cooled, evaporated, and purified by column chromatography (DCM/MeOH, 0 to 5%) to afford compound 27 (0.048 g, 64%) as a white solid. 1H NMR (DMSO-d6, 500 MHz) δ 8.16–8.13 (m, 1H), 8.14–8.11 (m, 1H), 7.59–7.55 (m, 1H), 7.51 (t, J = 7.9 Hz, 2H), 7.39–7.36 (m, 2H), 7.24–7.21 (m, 2H), 6.92 (s, 1H), 6.90 (s, 1H), 3.44 (s, 2H), 2.80 (q, J = 7.6 Hz, 2H), 1.28 (t, J = 7.6 Hz, 3H); APCI MS m/z 368 [M + H]+. HPLC Method 4: retention time: 13.32 min.
Preparation of 2-(4-((2-(3-chlorophenyl)-6-(trifluoromethyl)pyridin-4-yl)methyl)phenyl)acetic acid (28).
In a 250 mL round bottomed flask, 35 (1.66 g, 5.67 mmol), T-l (1.89 g, 6.52 mmol), Na2CO3 (2.92 g, 27.5 mmol), Pd(dppf)Cl2•DCM (460 mg, 0.56 mmol) in dioxane (30 mL) and H2O (15 mL) were purged with Ar for 20 min, then sealed and stirred at 90 °C for 4 hours. The reaction was concentrated to dryness and residue chromatographed (hexanes/DCM, 2 to 50%) to afford methyl 2-(4-((2-(3-chlorophenyl)-6-(trifluoromethyl)pyridin-4-yl)methyl)phenyl)acetate (1.76 g, 74%) as a white solid: mp 72–74 °C. 1H NMR (CDCl3, 500 MHz) δ 8.03–8.00 (m, 1H), 7.91–7.86 (m, 1H), 7.68 (s, 1H), 7.46–7.44 (m, 1H), 7.42–7.37 (m, 2H), 7.29–7.26 (m, 2H), 7.18–7.15 (m, 2H), 4.09 (s, 2H), 3.70 (s, 3H), 3.62 (s, 2H); APCI MS m/z 420 [M+H]+.
Methyl 2-(4-((2-(3-chlorophenyl)-6-(trifluoromethyl)pyridin-4-yl)methyl)phenyl)acetate (1.75 g, 4.18 mmol) was dissolved in MeOH/THF/H2O (1:1:1, 45 mL), treated with LiOH•H20 (1.0 g, 24 mmol) and stirred at rt overnight. The reaction was concentrated, diluted with H2O (100 mL) and the pH adjusted to 3 with 2N HCl. The solid was collected and dried to yield 28 (1.58 g, 90%) as a white solid, mp: 124–126 °C. 1H NMR (CDCl3, 500 MHz) δ 8.03–8.00 (m, 1H), 7.91–7.85 (m, 1H), 7.68 (s, 1H), 7.46–7.44 (m, 1H), 7.42–7.37 (m, 2H), 7.30–7.26 (m, 2H), 7.19–7.15 (m, 2H), 4.09 (s, 2H), 3.65 (s, 2H); APCI MS m/z 406 [M+H]+. Elemental analysis for C21H15ClF3NO2: Calculated: C (62.16%), H (3.73%), Cl (8.74%), F (14.05%), N (3.45%) Found: C (62.20%), H (3.55%), Cl (8.77%), F (14.43%), N (3.32%).
2-(4-((2-(3-Chlorophenyl)-6-ethylpyrimidin-4-yl)methyl)phenyl)-N-(2-hydroxyethyl)acetamide (29).
Methyl 2-(4-((2-(3-chlorophenyl)-6-ethylpyrimidin-4-yl)methyl)phenyl)acetate (64 mg, 0.17 mmol, 1.0 eq) and LiOH•IrhO (21 mg, 0.50 mmol, 3.0 eq) were stirred in THF (3 mL) and water (3 mL) at rt until the starting material was consumed. The reaction was diluted with water (10 mL) and acidified with 2N HCl. It was extracted with EtOAc, which was washed with brine (5 mL), dried over Na2SO4, filtered, and concentrated. The residue was purified by chromatography (DCM/MeOH, 0 to 10%) to afford 2-(4-((2-(3-chlorophenyl)-6-ethylpyrimidin-4-yl)methyl)phenyl)acetic acid (40 mg, 66% yield) as a white solid. 1H NMR (DMSO-d6, 500 MHz) δ 12.31 (br s, 1H), 8.40–8.33 (m, 2H), 7.62–7.53 (m, 2H), 7.3l(d, J = 8.0 Hz, 2H), 7.26 (s, 1H), 7.21(d, J = 8.0 Hz, 2H),4.10 (s, 2H), 3.51 (s, 2H), 2.78 (q, J = 7.5 Hz, 2H), 1.26 (t, J = 7.5 Hz, 3H).
A solution of 2-(4-((2-(3-chlorophenyl)-6-ethylpyrimidin-4-yl)methyl)phenyl)acetic acid (0.22 g, 0.60 mmol) in DCM (15 ml) at 0 °C was treated with oxalyl chloride (0.25 mL, 3.00 mmol) followed by DMF (1 drop). After 4 hrs, the volatile material was removed to afford crude 2-(4-((2-(3-chlorophenyl)-6-ethylpyrimidin-4-yl)methyl)phenyl)acetyl chloride as a red oil. This was dissolved in DCM (8 mL), cooled to 0 °C, then treated with ethanolamine (0.060 mL, 1.00 mmol) and DIEA (0.17 mL, 1.00 mmol). After 3 hrs, the reaction mixture was diluted with DCM (50 mL) and water (5 mL), then the pH was adjusted to 5 with 2N HCl. The organic layer was washed with brine (5 mL), dried over Na2SO4, filtered, and concentrated. The crude material was purified by chromatography (hexanes/EtOAc, 0 to 100%) followed by preparative HPLC (water/acetonitrile with 0.05% TFA) to afford compound 29 (0.036 g, 45%) as a white solid, mp 136–138 °C. 1H NMR (DMSO-d6, 500 MHz) δ 8.37–8.31 (m, 2H), 8.02–7.96 (m, 1H), 7.59 (dt, J = 6.5, 2.0 Hz, 1H), 7.55 (t, J = 8.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 2H), 7.25 (s, 1H), 7.20 (d, J = 8.0 Hz, 2H), 4.63 (t, J = 5.0 Hz, 1H), 4.09 (s, 2H), 3.40–3.35 (m, 4H), 3.90 (q, J = 6.0 Hz, 2H), 2.77 (q, J = 7.5 Hz, 2H), 1.26 (t, J = 7.5 Hz, 3H); APCI MS m/z 410.6 [M+H]+. HPLC Method 6: retention time: 17.42 min.
4-Chloro-2-(3-chlorophenyl)-6-ethyl pyrimidine (31).
In a 1-L round bottomed flask 3-chlorobenzimidamide HCl 30 (23.9 g, 154 mmol, 1 eq), ethyl 3-oxopentanoate (27.8 g, 193 mmol, 1.25 eq), and ethanol (500 mL) were treated with NaOMe (10.0 g, 185 mmol, 1.20 eq) and heated to reflux for 20 hrs. The resulting mixture was cooled to rt and concentrated to ~50 mL The slurry was cautiously treated with 2N HCl (219 mL), filtered and the solid washed with H2O and ether to afford crude 2-(3-chlorophenyl)-6-ethylpyrimidin-4(3H)-one (9.3 g, 39% yield). The crude solid was treated with POCl3 (50 mL, 536 mmol, 13.4 eq) at 0 °C and stirred at 100 °C for 5hr. After cooling to rt, the mixture was added slowly to cold aq NaHCO3. The mixture was extracted with EtOAc and the organics dried with Na2SO4, and concentrated. The organic extract was dried over Na2SO4, filtered and concentrated under reduced pressure. Purification by silica gel chromatography using DCM/EtOAc as eluent afforded 31 (9.3 g, 99% yield) as a white solid. 1H NMR (CD3OD, 500 MHz) δ 8.32 (s, 1H), 8.28 (d, J = 7.5 Hz, 1H), 7.50–7.46 (m, 1H), 7.45–7.40 (m, 1H), 7.27 (s, 1H), 2.83 (q, J = 7.5 Hz, 2H), 1.34 (t, J = 7.5 Hz, 3H).
Synthesis of 4-Chloro-2-(3-hlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridine hydrochloride (33).
An 18-mL vial was charged with 2,4-dichloro-6,7-dihydro-5H-cyclopenta[b]pyridine HCl salt (32) (225 mg, 1 mmol), (3-chlorophenyl)boronic acid (165 mg, 1.06 mmol), Pd(Ph3P)4 (84 mg, 0.07 mmol), and K2CO3 (500 mg, 3.6 mmol). Toluene (6 ml), EtOH (2 ml) and water (2.5 ml) were added and the reaction was stirred under Ar at 90 °C until 32 was consumed. The reaction was cooled to rt resulting in a biphasic system. The organic phase was concentrated, and the crude material purified via chromatography using hexanes/DCM (9:1 then 8:1) to afford 4-chloro-2-(3-chlorophenyl)-6,7-dihydro-5H-cyclopenta[b]pyridine, which was converted into its corresponding HCl salt by treating with 4N HCl in dioxane (228 mg, 76% yield).
Preparation of 4-chloro-2-(3-chlorophenyl)-6-(trifluoromethyl)pyridine (35).
In a 500-mL round bottomed flask, (3-chlorophenyl)boronic acid (1.59 g, 10.2 mmol), Pd(Ph3P)4 (0.608 g, 0.52 mmol) and K2CO3 (3.9 g, 27.8 mmol) in toluene (40 ml), EtOH (10 ml) and water (20 ml) were purged with Ar for 30 min. 34 (2.00 g, 9.26 mmol) was added and purging continued for 10 min. The reaction was sealed and stirred under Ar at 90 °C for 4 h. After cooling to rt, the reaction mixture was diluted with EtOAc (100 mL) and hexanes (100 mL). The aqueous layer was separated, and the organic layer washed with brine (2 × 25 mL), dried over Na2SO4, filtered, and concentrated. The residue was purified by chromatography (hexanes) to afford 35 (1.82 g, 67%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 8.06–8.04 (m, 1H), 7.92 (dt, J = 7.0, 2.0 Hz, 1H), 7.89 (d, J = 1.5 Hz, 1H), 7.64 (d, J = 1.5 Hz, 1H), 7.48–7.42 (m, 2H).
Methyl 2-(4-((4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)methyl)phenyl)acetate (T-1).
Methyl 2-(4- (bromomethyl)phenyl)acetate (3.45 g, 14.2 mmol), pinacol diborane (4.32 g, 17.0 mmol), Pd(PPh3)4 (1.64 g, 1.42 mmol), and K2CO3 (5.88 g, 42.6 mmol) in dioxane (80 mL) were stirred under Ar at 80 °C for 23 hours. The reaction was cooled and diluted with EtOAc (200 mL) before it was filtered through Celite. The filtrate was washed with brine (3 x 25 mL), dried over Na2SO4, filtered, and concentrated. The crude material was purified by chromatography (hexanes/EtOAc, 0 to 100%) to afford methyl 2-(4-((4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)methyl)phenyl)acetate (2.88 g, 70% yield) as a colorless semisolid. 1H NIVIR (CDCl3, 500 MHz) δ 7.14 (s, 4H), 3.67 (s, 3H), 3.57 (s, 2H), 2.27 (s, 2H), 1.23 (s, 12H).
Protein Expression and Purification.
PDE4D residue numbering was based on the reference PDE4D7 isoform, NCBI Reference Sequence: NP_001159371.1. Human PDE4D7 contained a mutation of S129D to mimic activation by cAMP-dependent protein kinase A (PKA), PDE4D3 contained the corresponding mutation S54D, and PDE4B1 contained the mutation S133D. All PDE4 proteins contained the mutations S579A and S581A to remove the potential for ERK-dependent phosphorylation. Methods used to generate synthetic genes for human PDE4 subtypes and isoforms were as described previously.7 Synthetic genes were engineered with carboxyl- or amino-terminal hexahistidine tags for Baculovirus-infected insect cell expression and purification.
PDE4 Enzyme Assay Protocol.
Kinetic assay of cAMP hydrolysis by purified PDE4 was measured by coupling the formation of the PDE4 reaction product, 5′-adenosine monophosphate to the oxidation of reduced nicotinamide adenine dinucleotide (NADH) by the use of three coupling enzymes (yeast myokinase, pyruvate kinase and lactate dehydrogenase), which allowed fluorescent determination of reaction rates.7 Assays were performed in 96-well plates in a total volume of 200 μl/well. Compounds were dissolved in dimethylsulfoxide (DMSO) and added to plates in a volume of 10 μl followed by addition of 165 μl of assay mix. Plates were preincubated at 25 °C for 5-10 min and the reactions were initiated by the addition of 25 μl of cAMP followed by thorough mixing. Reaction rates were measured by monitoring the decrease in fluorescence using excitation at 355 nm and emission at 460 nm for a period of 10 min in a fluorescence plate reader. Initial rates (slopes) were determined from linear portions of the progress curves. Final concentrations of assay components were as follows: 50 mM Tris, pH 8, 10 mM MgCl2, 50 mM KCl, 2% DMSO, 5 mM tris(2-carboxyethyl)phosphine (TCEP), 0.4 mM phosphenolpyruvate (PEP), 0.01 mM NADH, 0.04 mM adenosine triphosphate (ATP), 0.004 mM cAMP, 7.5 units myokinase from yeast, 1.6 units pyruvate kinase, 2 units lactate dehydrogenase, and ~0.5 nM PDE4D7-S129D and PDE4B1-S133D to yield an initial rate of approximately −0.7 RFU/s. Less active PDE4 enzymes were used at concentrations up to 4 nM. All data were normalized relative to control wells lacking cAMP and are presented as percent inhibition. An inhibitory concentration 50% (IC50) value was calculated by fitting a sigmoidal dose response curve. Z′ quality factors were routinely >0.6 for the assay.
PDE4 Enzyme Crystallization and Structure Determination.
PDE4D with an N-terminal UCR2 fusion7 in 10mM HEPES, pH7.5, 100mM NaCl, 1mM dithiothreitol, 0.1mM ZnCl2, and 0.1mM MgCl2, was crystallized at 5-10 mg/ml in the presence of 0.5mM compound (5, 6 or 28). Crystals were produced in a few days by sitting drop vapor diffusion at 16°C using a 1:1 mixture of protein:ligand to crystallization condition containing 12.5% w/v PEG1,000, 12.5% w/v PEG3,350, 12.5% w/v MPD, 0.03M NPS (sodium nitrate, disodium hydrogen phosphate, ammonium sulfate), and 0.1mM MES/Imidazole, pH6.5. Crystals were harvested, flash frozen and shipped to the listed synchrotron for screening and data collection. Data for 5 and 6 were collected at the Advanced Proton Source on Beamline 21-ID-F equipped with a MAR Mosaic 300 and 225 CCD X-ray detector, respectively. Data for 28 was collected at the Advanced Light Source on Beamline 5.0.3 equipped with an ADSC Q315R CCD X-ray detector. Diffraction data were reduced and scaled with XDS and XSCALE.72 Structures were solved by molecular replacement with previously solved PDE4D structures and were refined using iterative cycles of restrained refinement with Refmac573 and model-building using COOT,74 both part of the CCP4 suite of programs. 75-76 Structures were validated by Molprobity77 ahead of deposition in the Protein Data Bank.78-79 Diffraction data and refinement statistics are listed in Supplemental Table 1.
Animal Experiments.
Male ICR mice weighing 25–30 g were used (Harlan, Indianapolis, IN) for all of the experiments. All behavioral tests were performed during 9:30 am–16:30 pm and in accordance with the “NIH Guide for the Care and Use of Laboratory Animals” (revised 2011) and were approved by the Institutional Animal Care and Use Committee of the University at Buffalo. Compounds were dosed by per os gavage (PO) in a vehicle of 5% DMSO, 5% Solutol in phosphate-buffered saline (PBS). Rolipram was dosed at 1 mg/kg by intraperitoneal injection.
Novel Object Recognition.
The novel object recognition test was performed in a Plexiglas open field box (L30 cm, W50 cm, H40 cm) as described elsewhere.80-81 Briefly, the task procedure consists of three phases: habituation phase on day 1 for 10 min, training (T1) phase on day 2 for 5 min, and testing (T2) phase on day 3 for 5 min. The duration each animal spent exploring the objects was recorded. Time spent exploring the identical objects in T1 was recorded as a1 and a2; time spent exploring the familiar and the novel objects in T2 was recorded as ‘a’ and ‘b’, respectively. The following variables were calculated: e1 = a1 + a2, e2 = a + b, the relative Discrimination Index d2 = (b – a)/e2.
Anesthesia Duration Test.
Mice were dosed PO with the PDE4 inhibitor or vehicle or IP with rolipram; 30 min or 15 min later (for PO and IP doses, respectively), mice were injected IP with a mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg). The duration of anesthesia was determined as the time between the loss and return of the righting reflex and was used as an endpoint to measure the duration of anesthesia.
ADME.
Assays of kinetic solubility, liver microsome stability, CYP inhibition using a fluorescent substrate and cellular permeability in MDCK-MDR1 cells were performed by Albany Molecular Research Inc. using standard protocols. Metabolite identification was performed by incubation with cryopreserved hepatocytes (0.5 x 106 cells/mL in suspension) from male Wistar rats and human for 4 hours at 37°C. At the end of the incubations, samples of the incubation medium were analyzed by LC-UV/MS/MS analysis. Counter assays were performed against PDE1-10 (BPS Biosciences), G-protein coupled receptors, ion channels and transporters (Cerep), and human cardiac ERG channel (Eurofins Panlabs).
Supplementary Material
Figure 1:
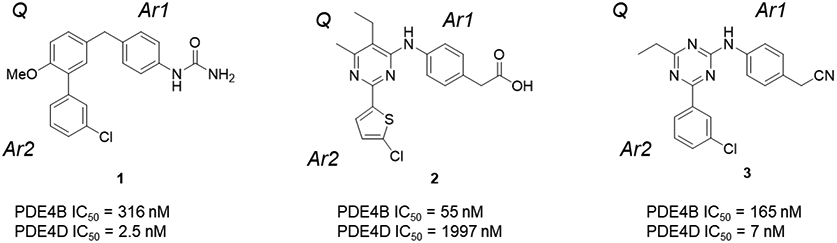
(1) Burgin et al. compound D1596877; (2) Naganuma et al. compound 3349; (3) Hagen et al. compound 8.50 The aliphatic substituents on the core project into a pocket of the active site known as the Q region.51 A pair of aromatic arms projects from the core. These have been labeled Ar1 and Ar2 as in Burgin et al.7
ACKNOWLEDGEMENTS
This work was supported by a research grant from the National Institutes of Health Blueprint Neurotherapeutics Program through the National Institute of Neurological Disorders and Stroke and the National Institute on Aging (grant number NS078034) to M.E.G. with a sub-award to Y.X. and independently by Tetra Discovery Partners, Inc. The authors wish to thank Dr. Chuck Cywin, Dr. Enrique Michelotti, and Anirudh Chowdhary for their contributions to the project.
ABBREVIATIONS USED
- MeOH
(methanol)
- DCM
(dichloromethane)
- EtOAc
(ethyl acetate)
- EtOH
(ethanol)
- NMP
(N-Methyl-2-pyrrolidinone)
- ACN
(acetonitrile)
- THF
(tetrahydrofuran)
- DMF
(N,N-dimethylformamide)
- AcOH
(acetic acid)
- HCl
(hydrochloric acid)
- TFA
(trifluoroacetic acid)
- NaHCO3
(sodium bicarbonate)
- Na2CO3
(sodium carbonate)
- K2CO3
(potassium carbonate)
- Na2SO4
(sodium sulfate)
- MgSO4
(magnesium sulfate)
- NH4Cl
(ammonium chloride)
- LiCl
(lithium chloride)
- NaCl
(sodium chloride)
- NH4OH
(ammonium hydroxide)
- LAH
(lithium aluminum hydride)
- POCl3
(phosphoryl chloride)
- LiOH•H2O
(lithium hydroxide monohydrate)
- KC1
(potassium chloride)
- DIEA
(diisopropylethylamine)
- Pd(PPh3)4
(tetrakis(triphenylphosphine)palladium (0))
- Pd(dppf)Cl2
([1,1’- Bis(diphenylphosphino)ferrocene]dichloropalladium (II))
- BH 3 •DMS
(borane dimethyl sulfide complex)
- TFA
(trifluoroacetic acid)
- H3PO4
(phosphoric acid)
- cone H2SO4
(concentrated sulfuric acid)
- Ar
(Argon)
- ACRDYS2
(acrodysostosis type 1 with or without hormone resistance)
- UCR
(upstream conserved regions)
- GWAS
(genome-wide association studies)
- NMDA-R
(N-methyl D-aspartate receptor)
- BDNF
(brain-derived neurotrophic factor)
- PDE4D7-S129D
(dimeric isoform of PDE4D that contains a UCR1 mutation (S129D) that mimics PKA phosphorylation)
- PDE4B1-S133D
(dimeric isoform of PDE4B that also contains a UCR1 mutation (S133D) that mimics PKA phosphorylation)
- PDE4D7-S129(wt)
(native isoform of PDE4D7 that lacks the UCR1 phosphorylation mimetic mutation)
- FaSSIF
(fasted state simulated intestinal fluid)
Footnotes
COMPETING INTEREST STATEMENT
M.E.G, R.N., X.M. and J.A.S are employees of Tetra Discovery Partners, Inc. which has a financial interest in BPN14770.
ACCESSION CODES
Protein Structure Database: Compound 5, PDB ID: 6NJI; compound 6, PDB ID:6NJH; compound 28, PDB ID: 6NJJ. Authors will release the atomic coordinates and experimental data upon article publication.
SUPPORTING INFORMATION
(1) Molecular String formula (CSV file) (2) X-ray diffraction data and refinement statistics (3) comparison of 28 in vitro potency against PDE4D7-S129D using the coupled enzyme assay of cAMP hydrolysis versus the BPS Bioscience PDE Assay Kit Catalog (3) binding pose of apremilast in the PDE4 active site.
REFERENCES
- 1.Matthiesen K; Nielsen J, Cyclic AMP control measured in two compartments in HEK293 cells: phosphodiesterase K(M) is more important than phosphodiesterase localization. PLoS One 2011, 6 (9), e24392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Houslay MD, Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci 2010, 35 (2), 91–100. [DOI] [PubMed] [Google Scholar]
- 3.Blackman BE; Horner K; Heidmann J; Wang D; Richter W; Rich TC; Conti M, PDE4D and PDE4B function in distinct subcellular compartments in mouse embryonic fibroblasts. J Biol Chem 2011, 286 (14), 12590–12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gervasi N; Tchenio P; Preat T, PKA dynamics in a Drosophila learning center: coincidence detection by rutabaga adenylyl cyclase and spatial regulation by dunce phosphodiesterase. Neuron 2010, 65(4), 516–529. [DOI] [PubMed] [Google Scholar]
- 5.Bolger G; Michaeli T; Martins T; St John T; Steiner B; Rodgers L; Riggs M; Wigler M; Ferguson K, A family of human phosphodiesterases homologous to the dunce learning and memory gene product of Drosophila melanogaster are potential targets for antidepressant drugs. Mol Cell Biol 1993, 13 (10), 6558–6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Francis SH; Blount MA; Corbin JD, Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev 2011, 91 (2), 651–690. [DOI] [PubMed] [Google Scholar]
- 7.Burgin AB; Magnusson OT; Singh J; Bjornsson JM; Thorsteinsdottir M; Hrafnsdottir S; Hagen T; Witte P; Staker BL; Kiselyov AS; Stewart LJ; Gurney ME, Design of phosphodiesterase type 4D (PDE4D) allosteric modulators for cognition with improved safety. Nature Biotechnology 2010, 28 (1), 63–70. [DOI] [PubMed] [Google Scholar]
- 8.Cedervall P; Aulabaugh A; Geoghegan KF; McLellan TJ; Pandit J, Engineered stabilization and structural analysis of the autoinhibited conformation of PDE4. Proc Natl Acad Sci U S A 2015, 112 (12), E1414–E1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alvarez R; Sette C; Yang D; Eglen RM; Wilhelm R; Shelton ER; Conti M, Activation and selective inhibition of a cyclic AMP-specific phosphodiesterase, PDE-4D3. Mol Pharmacol 1995, 48 (4), 616–622. [PubMed] [Google Scholar]
- 10.Sette C; Conti M, Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. J Biol Chem 1996, 271 (28), 16526–16534. [DOI] [PubMed] [Google Scholar]
- 11.Byrne AM; Elliott C; Hoffmann R; Baillie GS, The activity of cAMP-phosphodiesterase 4D7 (PDE4D7) is regulated by protein kinase A-dependent phosphorylation within its unique N-terminus. FEBS Lett 2015, 589 (6), 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffmann R; Wilkinson IR; McCallum JF; Engels P; Houslay MD, cAMP-specific phosphodiesterase HSPDE4D3 mutants which mimic activation and changes in rolipram inhibition triggered by protein kinase A phosphorylation of Ser-54: generation of a molecular model. Biochem J 1998, 333 (Pt 1), 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linglart A; Fryssira H; Hiort O; Holterhus PM; Perez de Nanclares G; Argente J; Heinrichs C; Kuechler A; Mantovani G; Leheup B; Wicart P; Chassot V; Schmidt D; Rubio-Cabezas O; Richter-Unruh A; Berrade S; Pereda A; Boros E; Munoz-Calvo MT; Castori M; Gunes Y; Bertrand G; Bougneres P; Clauser E; Silve C, PRKAR1A and PDE4D mutations cause acrodysostosis but two distinct syndromes with or without GPCR-signaling hormone resistance. J Clin Endocrinol Metab 2012, 97 (12), E2328–E2338. [DOI] [PubMed] [Google Scholar]
- 14.Lee H; Graham JM Jr.; Rimoin DL; Lachman RS; Krejci P; Tompson SW; Nelson SF; Krakow D; Cohn DH, Exome sequencing identifies PDE4D mutations in acrodysostosis. Am J Hum Genet 2012, 90 (4), 746–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michot C; Le Goff C; Goldenberg A; Abhyankar A; Klein C; Kinning E; Guerrot AM; Flahaut P; Duncombe A; Baujat G; Lyonnet S; Thalassinos C; Nitschke P; Casanova JL; Le Merrer M; Munnich A; Cormier-Daire V, Exome sequencing identifies PDE4D mutations as another cause of acrodysostosis. Am J Hum Genet 2012, 90 (4), 740–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lynch DC; Dyment DA; Huang L; Nikkel SM; Lacombe D; Campeau PM; Lee B; Bacino CA; Michaud JL; Bernier FP; Consortium FC; Parboosingh JS; Innes AM, Identification of novel mutations confirms Pde4d as a major gene causing acrodysostosis. Hum Mutat 2013, 34 (1), 97–102. [DOI] [PubMed] [Google Scholar]
- 17.Lindstrand A; Grigelioniene G; Nilsson D; Pettersson M; Hofmeister W; Anderlid BM; Kant SG; Ruivenkamp CA; Gustavsson P; Valta H; Geiberger S; Topa A; Lagerstedt-Robinson K; Taylan F; Wincent J; Laurell T; Pekkinen M; Nordenskjold M; Makitie O; Nordgren A, Different mutations in PDE4D associated with developmental disorders with mirror phenotypes. J Med Genet 2014, 51 (1), 45–54. [DOI] [PubMed] [Google Scholar]
- 18.Hoppmann J; Gesing J; Silve C; Leroy C; Bertsche A; Hirsch FW; Kiess W; Pfaffle R; Schuster V, Phenotypic variability in a family with acrodysostosis type 2 caused by a novel PDE4D mutation affecting the serine target of protein kinase-A phosphorylation. J Clin Res Pediatr Endocrinol 2017, 9 (4), 360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurney ME; D'Amato EC; Burgin AB, Phosphodiesterase-4 (PDE4) molecular pharmacology and Alzheimer's Disease. Neurotherapeutics 2015, 12 (1), 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Briet C; Pereda A; Le Stunff C; Motte E; de Dios Garcia-Diaz J; de Nanclares GP; Dumaz N; Silve C, Mutations causing acrodysostosis-2 facilitate activation of phosphodiesterase 4D3. Hum Mol Genet 2017, 26 (20), 3883–3894. [DOI] [PubMed] [Google Scholar]
- 21.Lam M; Trampush JW; Yu J; Knowles E; Davies G; Liewald DC; Starr JM; Djurovic S; Melle I; Sundet K; Christoforou A; Reinvang I; DeRosse P; Lundervold AJ; Steen VM; Espeseth T; Raikkonen K; Widen E; Palotie A; Eriksson JG; Giegling I; Konte B; Roussos P; Giakoumaki S; Burdick KE; Payton A; Ollier W; Chiba-Falek O; Attix DK; Need AC; Cirulli ET; Voineskos AN; Stefanis NC; Avramopoulos D; Hatzimanolis A; Arking DE; Smyrnis N; Bilder RM; Freimer NA; Cannon TD; London E; Poldrack RA; Sabb FW; Congdon E; Conley ED; Scult MA; Dickinson D; Straub RE; Donohoe G; Morris D; Corvin A; Gill M; Hariri AR; Weinberger DR; Pendleton N; Bitsios P; Rujescu D; Lahti J; Le Hellard S; Keller MC; Andreassen OA; Deary IJ; Glahn DC; Malhotra AK; Lencz T, Large-scale cognitive GWAS meta-analysis reveals tissue-specific neural expression and potential nootropic drug targets. Cell Rep 2017, 21 (9), 2597–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trampush JW; Yang MLZ; Yu J; Knowles E; Davies G; Liewald DC; Starr JM; Djurovic S; Melle I; Sundet K; Christoforou A; Reinvang I; DeRosse P; Lundervold AJ; Steen VM; Espeseth T; Raikkonen K; Widen E; Palotie A; Eriksson JG; Giegling I; Konte B; Roussos P; Giakoumaki S; Burdick KE; Payton A; Ollier W; Horan M; Chiba-Falek O; Attix DK; Need AC; Cirulli ET; Voineskos AN; Stefanis NC; Avramopoulos D; Hatzimanolis A; Arking DE; Smyrnis N; Bilder RM; Freimer NA; Cannon TD; London E; Poldrack RA; Sabb FW; Congdon E; Conley ED; Scult MA; Dickinson D; Straub RE; Donohoe G; Morris D; Corvin A; Gill M; Hariri AR; Weinberger DR; Pendleton N; Bitsios P; Rujescu D; Lahti J; Le Hellard S; Keller MC; Andreassen OA; Deary IJ; Glahn DC; Malhotra AK; Lencz T, GWAS meta-analysis reveals novel loci and genetic correlates for general cognitive function: a report from the COGENT consortium. Mol Psychiatry 2017, 22 (11), 1651–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savage JE; Jansen PR; Stringer S; Watanabe K; Bryois J; de Leeuw CA; Nagel M; Awasthi S; Barr PB; Coleman JRI; Grasby KL; Hammerschlag AR; Kaminski JA; Karlsson R; Krapohl E; Lam M; Nygaard M; Reynolds CA; Trampush JW; Young H; Zabaneh D; Hagg S; Hansell NK; Karlsson IK; Linnarsson S; Montgomery GW; Munoz-Manchado AB; Quinlan EB; Schumann F; Skene NG; Webb BT; White T; Arking DE; Avramopoulos D; Bilder RM; Bitsios P; Burdick KE; Cannon TD; Chiba-Falek O; Christoforou A; Cirulli ET; Congdon E; Corvin A; Davies G; Deary IJ; DeRosse P; Dickinson D; Djurovic S; Donohoe G; Conley ED; Eriksson JG; Espeseth T; Freimer NA; Giakoumaki S; Giegling I; Gill M; Glahn DC; Hariri AR; Hatzimanolis A; Keller MC; Knowles E; Koltai D; Konte B; Lahti J; Le Hellard S; Lencz T; Liewald DC; London E; Lundervold AJ; Malhotra AK; Melle I; Morris D; Need AC; Ollier W; Palotie A; Payton A; Pendleton N; Poldrack RA; Raikkonen K; Reinvang I; Roussos P; Rujescu D; Sabb FW; Scult MA; Smeland OB; Smyrnis N; Starr JM; Steen VM; Stefanis NC; Straub RE; Sundet K; Tiemeier F; Voineskos AN; Weinberger DR; Widen E; Yu J; Abecasis G; Andreassen OA; Breen G; Christiansen L; Debrabant B; Dick DM; Heinz A; Hjerling-Leffler J; Ikram MA; Kendler KS; Martin NG; Medland SE; Pedersen NL; Plomin R; Polderman TJC; Ripke S; van der Sluis S; Sullivan PF; Vrieze SI; Wright MJ; Posthuma D, Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat Genet 2018, 50 (7), 912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee JJ; Wedow R; Okbay A; Kong E; Maghzian O; Zacher M; Nguyen-Viet TA; Bowers P; Sidorenko J; Karlsson Linner R; Fontana MA; Kundu T; Lee C; Li H; Li R; Royer R; Timshel PN; Walters RK; Willoughby EA; Yengo L; and Me Research T; Cogent; Social Science Genetic Association, C.; Alver M; Bao Y; Clark DW; Day FR; Furlotte NA; Joshi PK; Kemper KE; Kleinman A; Langenberg C; Magi R; Trampush JW; Verma SS; Wu Y; Lam M; Zhao JH; Zheng Z; Boardman JD; Campbell; Freese J; Harris KM; Hayward C; Herd P; Kumari M; Lencz T; Luan J; Malhotra AK; Metspalu A; Milani L; Ong KK; Perry JRB; Porteous DJ; Ritchie MD; Smart MC; Smith BH; Tung JY; Wareham NJ; Wilson; Beauchamp JP; Conley DC; Esko T; Lehrer SF; Magnusson PKE; Oskarsson S; Pers TH; Robinson MR; Thom K; Watson C; Chabris CF; Meyer MN; Laibson DI; Yang J; Johannesson M; Koellinger PD; Turley P; Visscher PM; Benjamin DJ; Cesarini D, Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet 2018, 50 (8), 1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gurney ME, Genetic association of phosphodiesterases with human cognitive performance. Front Mol Neurosci 2019, 12, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Byers D; Davis RL; Kiger JA Jr., Defect in cyclic AMP phosphodiesterase due to the dunce mutation of learning in Drosophila melanogaster. Nature 1981, 289 (5793), 79–81. [DOI] [PubMed] [Google Scholar]
- 27.Dudai Y; Jan YN; Byers D; Quinn WG; Benzer S, dunce, a mutant of Drosophila deficient in learning. Proc Natl Acad Sci US A 1976, 73 (5), 1684–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen CN; Denome S; Davis RL, Molecular analysis of cDNA clones and the corresponding genomic coding sequences of the Drosophila dunce+ gene, the structural gene for cAMP phosphodiesterase. Proc Natl Acad Sci U S A 1986, 83 (24), 9313–9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomchik SM; Davis RL, Dynamics of learning-related cAMP signaling and stimulus integration in the Drosophila olfactory pathway. Neuron 2009, 64 (4), 510–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levin LR; Han PL; Hwang PM; Feinstein PG; Davis RL; Reed RR, The Drosophila learning and memory gene rutabaga encodes a Ca2+/Calmodulin-responsive adenylyl cyclase. Cell 1992, 68 (3), 479–489. [DOI] [PubMed] [Google Scholar]
- 31.Milner B; Squire LR; Kandel ER, Cognitive neuroscience and the study of memory. Neuron 1998, 20 (3), 445–468. [DOI] [PubMed] [Google Scholar]
- 32.Bourtchuladze R; Frenguelli B; Blendy J; Cioffi D; Schutz G; Silva AJ, Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 1994, 79 (1), 59–68. [DOI] [PubMed] [Google Scholar]
- 33.Nibuya M; Nestler EJ; Duman RS, Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci 1996, 16 (7), 2365–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakagawa S; Kim JE; Lee R; Malberg JE; Chen J; Steffen C; Zhang YJ; Nestler EJ; Duman RS, Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J Neurosci 2002, 22 (9), 3673–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frey U; Huang YY; Kandel ER, Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science 1993, 260 (5114), 1661–1664. [DOI] [PubMed] [Google Scholar]
- 36.Wong ST; Athos J; Figueroa XA; Pineda VV; Schaefer ML; Chavkin CC; Muglia LJ; Storm DR, Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long-term memory and late phase LTP. Neuron 1999, 23 (4), 787–798. [DOI] [PubMed] [Google Scholar]
- 37.Wachtel H, Potential antidepressant activity of rolipram and other selective cyclic adenosine 3',5'-monophosphate phosphodiesterase inhibitors. Neuropharmacology 1983, 22 (3), 267–272. [DOI] [PubMed] [Google Scholar]
- 38.Barad M; Bourtchouladze R; Winder DG; Golan H; Kandel E, Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc Natl Acad Sci U S A 1998, 95 (25), 15020–15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vitolo OV; Sant'Angelo A; Costanzo V; Battaglia F; Arancio O; Shelanski M, Amyloid beta - peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci U S A 2002, 99 (20), 13217–13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gong B; Vitolo OV; Trinchese F; Liu S; Shelanski M; Arancio O, Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest 2004, 114 (11), 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith DL; Pozueta J; Gong B; Arancio O; Shelanski M, Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc Natl Acad Sci U S A 2009, 106 (39), 16877–16882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Myeku N; Clelland CL; Emrani S; Kukushkin NV; Yu WH; Goldberg AL; Duff KE, Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med 2016, 22 (1), 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deeks ED, Apremilast: A review in psoriasis and psoriatic Arthritis. Drugs 2015, 75 (12), 1393–1403. [DOI] [PubMed] [Google Scholar]
- 44.Spina D, PDE4 inhibitors: current status. Br J Pharmacol 2008, 155 (3), 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garnock-Jones KP, Roflumilast: A review in COPD. Drugs 2015, 75 (14), 1645–1656. [DOI] [PubMed] [Google Scholar]
- 46.Losco PE; Evans EW; Barat SA; Blackshear PE; Reyderman L; Fine JS; Bober LA; Anthes JC; Mirro EJ; Cuss FM, The toxicity of SCH 351591, a novel phosphodiesterase-4 inhibitor, in Cynomolgus monkeys. Toxicol Pathol 2004, 32 (3), 295–308. [DOI] [PubMed] [Google Scholar]
- 47.Dagues N; Pawlowski V; Sobry C; Hanton G; Borde F; Soler S; Freslon JL; Chevalier S, Investigation of the molecular mechanisms preceding PDE4 inhibitor-induced vasculopathy in rats: tissue inhibitor of metalloproteinase 1, a potential predictive biomarker. Toxicol Sci 2007, 100 (1), 238–247. [DOI] [PubMed] [Google Scholar]
- 48.Fox D 3rd; Burgin AB; Gurney ME, Structural basis for the design of selective phosphodiesterase 4B inhibitors. Cell Signal 2014, 26 (3), 657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naganuma K; Omura A; Maekawara N; Saitoh M; Ohkawa N; Kubota T; Nagumo H; Kodama T; Takemura M; Ohtsuka Y; Nakamura J; Tsujita R; Kawasaki K; Yokoi H; Kawanishi M, Discovery of selective PDE4B inhibitors. Bioorg Med Chem Lett 2009, 19 (12), 3174–3176. [DOI] [PubMed] [Google Scholar]
- 50.Hagen TJ; Mo X; Burgin AB; Fox D 3rd; Zhang Z; Gurney ME, Discovery of triazines as selective PDE4B versus PDE4D inhibitors. Bioorg Med Chem Lett 2014, 24 (16), 4031–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Card GL; England BP; Suzuki Y; Fong D; Powell B; Lee B; Luu C; Tabrizizad M; Gillette S; Ibrahim PN; Artis DR; Bollag G; Milburn MV; Kim SH; Schlessinger J; Zhang KY, Structural basis for the activity of drugs that inhibit phosphodiesterases. Structure 2004, 12 (12), 2233–2247. [DOI] [PubMed] [Google Scholar]
- 52.Richter W; Conti M, Dimerization of the type 4 cAMP-specific phosphodiesterases is mediated by the upstream conserved regions (UCRs). J Biol Chem 2002, 277 (43), 40212–40221. [DOI] [PubMed] [Google Scholar]
- 53.Bender AT; Beavo JA, Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 2006, 58 (3), 488–520. [DOI] [PubMed] [Google Scholar]
- 54.Zhang C; Xu Y; Chowdhary A; Fox D 3rd; Gurney ME; Zhang HT; Auerbach BD; Salvi RJ; Yang M; Li G; O'Donnell JM, Memory enhancing effects of BPN14770, an allosteric inhibitor of phosphodiesterase-4D, in wild-type and humanized mice. Neuropsychopharmacology 2018, 43(11), 2299–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robichaud A; Savoie C; Stamatiou PB; Tattersall FD; Chan CC, PDE4 inhibitors induce emesis in ferrets via a noradrenergic pathway. Neuropharmacology 2001, 40 (2), 262–269. [DOI] [PubMed] [Google Scholar]
- 56.Robichaud A; Stamatiou PB; Jin SL; Lachance N; MacDonald D; Laliberte F; Liu S; Huang Z; Conti M; Chan CC, Deletion of phosphodiesterase 4D in mice shortens alpha(2)-adrenoceptor-mediated anesthesia, a behavioral correlate of emesis. J Clin Invest 2002, 110 (7), 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berry-Kravis E; Huttenlocher PR, Cyclic AMP metabolism in fragile X syndrome. Ann Neurol 1992, 31 (1), 22–26. [DOI] [PubMed] [Google Scholar]
- 58.Berry-Kravis E; Sklena P, Demonstration of abnormal cyclic AMP production in platelets from patients with fragile X syndrome. Am J Med Genet 1993, 45 (1), 81–87. [DOI] [PubMed] [Google Scholar]
- 59.Berry-Kravis E; Hicar M; Ciurlionis R, Reduced cyclic AMP production in fragile X syndrome: cytogenetic and molecular correlations. Pediatr Res 1995, 38 (5), 638–643. [DOI] [PubMed] [Google Scholar]
- 60.Hagerman RJ; Berry-Kravis E; Kaufmann WE; Ono MY; Tartaglia N; Lachiewicz A; Kronk R; Delahunty C; Hessl D; Visootsak J; Picker J; Gane L; Tranfaglia M, Advances in the treatment of fragile X syndrome. Pediatrics 2009, 123 (1), 378–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morales J; Hiesinger PR; Schroeder AJ; Kume K; Verstreken P; Jackson FR; Nelson DL; Hassan BA, Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron 2002, 34 (6), 961–972. [DOI] [PubMed] [Google Scholar]
- 62.Fmr1 knockout mice: a model to study fragile X mental retardation. The Dutch-Belgian Fragile X Consortium. Cell 1994, 78 (1), 23–33. [PubMed] [Google Scholar]
- 63.Pieretti M; Zhang FP; Fu YH; Warren ST; Oostra BA; Caskey CT; Nelson DL, Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 1991, 66 (4), 817–822. [DOI] [PubMed] [Google Scholar]
- 64.Kelley DJ; Davidson RJ; Elliott JL; Lahvis GP; Yin JC; Bhattacharyya A, The cyclic AMP cascade is altered in the fragile X nervous system. PLoS One 2007, 2 (9), e931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Choi CH; Schoenfeld BP; Weisz ED; Bell AJ; Chambers DB; Hinchey J; Choi RJ; Hinchey P; Kollaros M; Gertner MJ; Ferrick NJ; Terlizzi AM; Yohn N; Koenigsberg E; Liebelt DA; Zukin RS; Woo NH; Tranfaglia MR; Louneva N; Arnold SE; Siegel SJ; Bolduc FV; McDonald TV; Jongens TA; McBride SM, PDE-4 inhibition rescues aberrant synaptic plasticity in Drosophila and mouse models of fragile X syndrome. J Neurosci 2015, 35 (1), 396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanellopoulos AK; Semelidou O; Kotini AG; Anezaki M; Skoulakis EM, Learning and memory deficits consequent to reduction of the fragile X mental retardation protein result from metabotropic glutamate receptor-mediated inhibition of cAMP signaling in Drosophila. J Neurosci 2012, 32 (38), 13111–13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gurney ME; Cogram P; Deacon RM; Rex C; Tranfaglia M, Multiple behavior phenotypes of the Fragile-X syndrome mouse model respond to chronic inhibition of phosphodiesterase-4D (PDE4D). Sci Rep 2017, 7 (1), 14653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Comery TA; Harris JB; Willems PJ; Oostra BA; Irwin SA; Weiler IJ; Greenough WT, Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci U S A 1997, 94 (10), 5401–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Irwin SA; Galvez R; Greenough WT, Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 2000, 10 (10), 1038–1044. [DOI] [PubMed] [Google Scholar]
- 70.Baumgartel K; Green A; Hornberger D; Lapira J; Rex C; Wheeler DG; Peters M, PDE4D regulates spine plasticity and memory in the retrosplenial cortex. Sci Rep 2018, 8 (1), 3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beaulieu-Laroche L; Toloza EHS; van der Goes MS; Lafourcade M; Barnagian D; Williams ZM; Eskandar EN; Frosch MP; Cash SS; Harnett MT, Enhanced dendritic compartmentalization in human cortical neurons. Cell 2018, 175 (3), 643–651 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kabsch W, Xds. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 2), 125–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Murshudov GN; Vagin AA; Dodson EJ, Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 1997, 53 (Pt 3), 240–255. [DOI] [PubMed] [Google Scholar]
- 74.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS, Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 2011, 67 (Pt 4), 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Collaborative Computational Project, N., The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 1994, 50 (Pt 5), 760–763. [DOI] [PubMed] [Google Scholar]
- 77.Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 1), 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE, The Protein Data Bank. Nucleic Acids Res 2000, 28 (1), 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Berman H; Henrick K; Nakamura H, Announcing the worldwide Protein Data Bank. Nat Struct Biol 2003, 10 (12), 980. [DOI] [PubMed] [Google Scholar]
- 80.Kamei H; Nagai T; Nakano H; Togan Y; Takayanagi M; Takahashi K; Kobayashi K; Yoshida S; Maeda K; Takuma K; Nabeshima T; Yamada K, Repeated methamphetamine treatment impairs recognition memory through a failure of novelty-induced ERK1/2 activation in the prefrontal cortex of mice. Biol Psychiatry 2006, 59 (1), 75–84. [DOI] [PubMed] [Google Scholar]
- 81.Li YF; Cheng YF; Huang Y; Conti M; Wilson SP; O'Donnell JM; Zhang HT, Phosphodiesterase-4D knock-out and RNA interference-mediated knock-down enhance memory and increase hippocampal neurogenesis via increased cAMP signaling. The Journal of Neuroscience 2011, 31 (1), 172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.