

SUMMARY
Aberrant chromosome numbers in cancer cells may impose distinct constraints on the emergence of drug resistance - a major factor limiting the long-term efficacy of molecularly-targeted therapeutics. However, for most anti-cancer drugs we lack analyses of drug resistance mechanisms in cells with different karyotypes. Here, we focus on GSK923295, a mitotic kinesin CENP-E inhibitor that was evaluated in clinical trials as a cancer therapeutic. We performed unbiased selections to isolate inhibitor-resistant clones in diploid and near-haploid cancer cell lines. In diploid cells we identified single-point mutations that can suppress inhibitor binding. In contrast, transcriptome analyses revealed that the C-terminus of CENP-E was disrupted in GSK923295-resistant near-haploid cells. While chemical inhibition of CENP-E is toxic to near-haploid cells, knockout of the CENPE gene does not suppress haploid cell proliferation, suggesting that deletion of the CENP-E C-terminus can confer resistance to GSK923295. Together, these findings indicate that different chromosome copy numbers in cells can alter epistatic dependencies and lead to distinct modes of chemotype-specific resistance.
Keywords: resistance, chemical inhibitor, target ID, GSK923295, HCT116, KBM7, CENPE, kinesin, ploidy, chromosome copy number
INTRODUCTION
Understanding resistance to chemical inhibitors in cells is critical for improving their efficacy as therapeutics (Daub et al., 2004). Cancer cells often harbor chromosomal translocations and abnormal ploidy (Bakhoum and Cantley, 2018; Chunduri and Storchová, 2019; Gordon et al., 2012) and these differences in karyotypes can result in unique constraints on the evolution of genetic changes (Podgornaia and Laub, 2015; Starr and Thornton, 2016). For example, variations in gene copy numbers can influence epistatic interactions between alleles that results in the evolution of distinct biochemical outputs of proteins (Siddiq et al., 2017). Therefore, the emergence of drug resistance can depend on differences in cellular karyotypes and ploidy. However, for most anti-cancer drugs we lack analyses of how differences in chromosome number impacts the chemotype-specific mechanisms of resistance that can emerge.
CENPE is a large (~316 kDa) plus-end directed microtubule motor that mediates interactions between kinetochores and microtubules and plays a key role in aligning chromosomes during mitosis (Kapoor et al., 2006; McEwen et al., 2001; Putkey et al., 2002; Wood et al., 1997). Genetic knockout or chemical inhibition of CENPE results in activation of the spindle assembly checkpoint, prolonged and death in diploid human cells (Putkey et al., 2002; Wood et al., 2010). Therefore, CENPE has been proposed as a potential target for cancer therapeutics and chemical inhibitors of CENPE have been developed (Hirayama et al., 2015; Ohashi et al., 2015; Wood et al., 2010). In particular, GSK923295 is a potent CENPE inhibitor that was tested in clinical trials as an anti-cancer agent (Chung et al., 2012; Lock et al., 2012). In vitro analyses indicate that the compound binds in an allosteric pocket proximal to the ATP-binding site in the kinesin motor domain (Wood et al., 2010). Further, GSK923295 has been proposed to block the ATPase cycle of CENPE by preventing the phosphate release and locking the motor domain in a tight microtubule-bound state (Wood et al., 2010). However, mechanisms of resistance against GSK923295 in cells are not well understood.
Analysis of resistance that is specific to a chemical inhibitor can be leveraged for determining its cellular target and validating its mechanism of action (Kapoor and Miller, 2017). We have developed an approach termed “DrugTargetSeqR” that combines transcriptome sequencing, computational discovery of drug resistance-conferring mutations, and genome editing to confirm and validate cellular targets of drugs (Kasap et al., 2014; Wacker et al., 2012). We have proposed that the “gold standard” of target validation is achieved when a resistance-conferring mutation is identified and shown to protect against the inhibitor in biochemical as well as cellular assays (Kapoor and Miller, 2017). However, we lack this level of analysis of the mechanism of action of GSK923295 in cancer cells. Furthermore, we do not know how changes in ploidy or karyotypes impact drug sensitivity or resistance.
Here, we use unbiased selections to isolate GSK923295-resistant clones in diploid or near-haploid cancer cells to investigate mechanisms of resistance. We use sequencing analyses to identify genetic changes in these cells that confer resistance to the GSK923295 inhibitor and characterize selected mutations in biochemical assays. Consistent with differential requirements of CENPE in haploid and diploid cell lines, our data suggest that cells with different ploidies can acquire distinct mechanisms of chemotype-specific drug resistance against the same chemical inhibitor.
RESULTS
Identifying and Characterizing Cells Resistant to GSK923295
To examine resistance to GSK923295 (Figure 1A) we focused on two cancer cell lines - HCT116 and KBM7. We used diploid HCT116 cells, which were employed for DrugTargetSeqR (Kasap et al., 2014; Wacker et al., 2012) as selection of inhibitor-resistant clones can be efficient due to mutations associated with impaired DNA mismatch repair pathways in these cells (Glaab and Tindall, 1997). In parallel, we used well-characterized, near-haploid mammalian KBM7 cells as they have been used to examine recessive resistance mechanisms that could be otherwise masked in diploid or polyploid cells (Elling and Penninger, 2014; Smurnyy et al., 2014).
Figure 1. Identifying GSK923295-resistant clones derived from HCT116 or KBM7 cells.
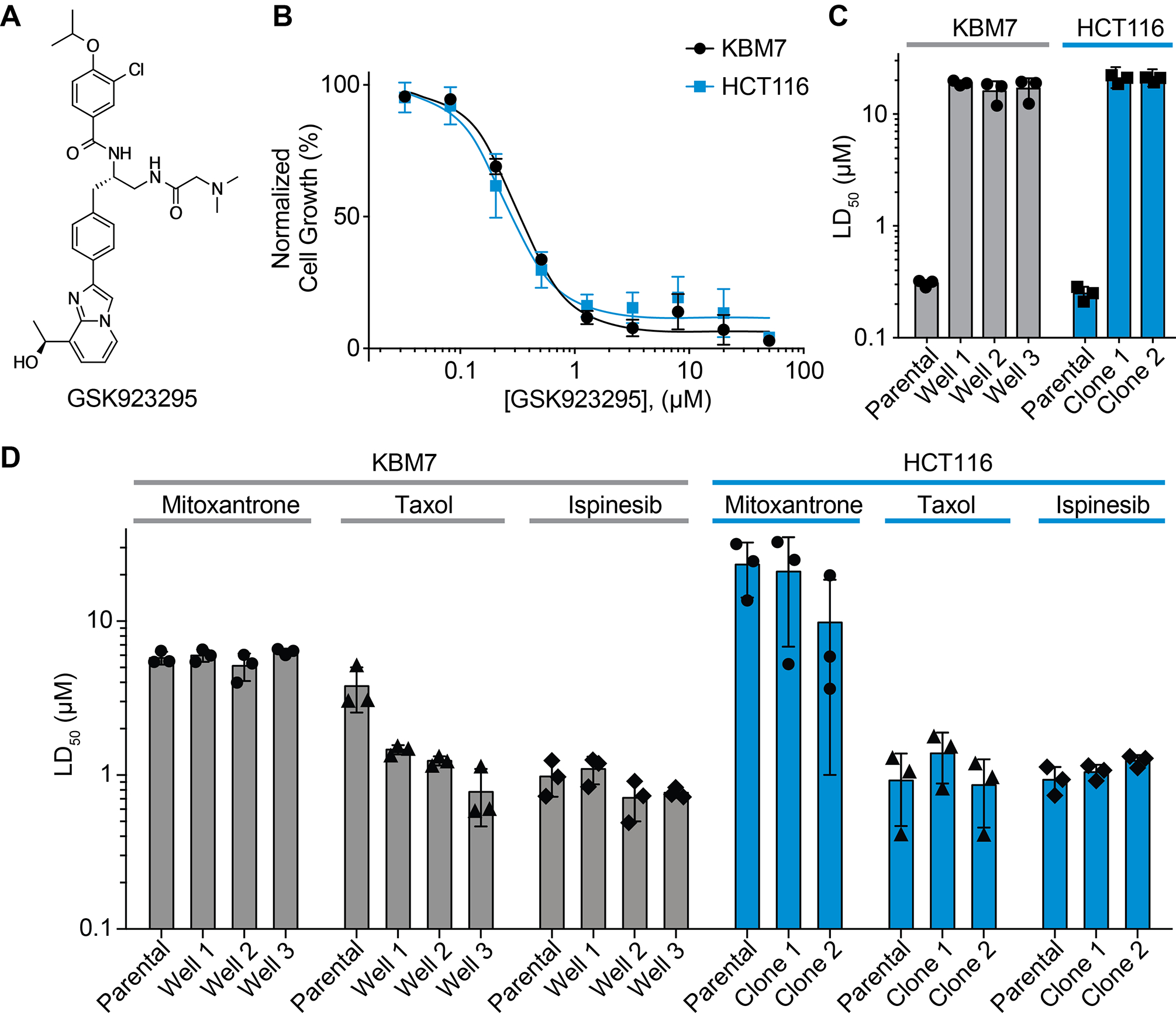
(A) Chemical structure of CENPE kinesin inhibitor (GSK923295).
(B) Analyzing toxicity of GSK923295 to HCT116 and KBM7 cells (KBM7: LD50 = 0.31 ± 0.02 μM, HCT116: LD50 = 0.25 ± 0.04 μM, average ± s.d., 72 h, n=3).
(C–D) Comparisons of toxicity (LD50) of GSK923925 (C), mitoxantrone (D), taxol (D) and ispinesib (D) to parental and resistant HCT116 and KBM7 cells (72 h, n = 3). For comparison, LD50 values for parental cells in the presence of GSK9123295 are also shown (C, data from B). See also Figure S1 and Table S1.
As a first step we confirmed that GSK923295 potently inhibits proliferation of these two cell lines (HCT116: LD50 = 0.25 ± 0.04 μM; KBM7: LD50 = 0.31 ± 0.02 μM; average ± s.d.; n=3, Figure 1B and Table S1). Next, we set up unbiased selections in HCT116 cells to identify clones resistant to the GSK923295 compound. We cultured HCT116 cells in the presence of the compound (2 μM) and in approximately two weeks observed multiple resistant colonies. We isolated and characterized two resistant clones (Figure 1C). We tested these HCT116 clones and found they were not sensitive to GSK923295 at concentrations up to ~85-fold higher than the LD50 for parental cells (HCT116 clone 1: LD50 = 20.3 ± 1.1 μM; clone 2: 21.5 ± 0.7 μM, Figure 1C, S1B and Table S1).
As KBM7 cells grow in suspension, we first optimized a cell dilution-based protocol in 96-well plates in order to select GSK923295-resistant cells (Figure S1A). After ~3 weeks of selections with our cell dilution-based scheme in the presence of GSK923295 (2 μM), we isolated cells from three wells in which growth was observed. We tested these KBM7 cells and found they were also resistant to the compound (KBM7 well 1: 18.9 ± 1 μM, well 2: 16.0 ± 3.6 μM, well 3: 16.9 ± 3.9 μM, Figure 1C, S1C and Table S1).
A range of mechanisms (e.g. upregulation of drug efflux pumps) can confer resistance to chemically-unrelated cytotoxic agents in cancer cells (Gottesman et al., 2016; Robey et al., 2018). In developing and applying DrugTargetSeqR, we have found that testing resistant clones for sensitivity to chemically unrelated drugs (e.g. mitoxantrone, ispinesib, and taxol) can help identify multi-drug response-based resistance mechanisms (Kasap et al., 2014; Wacker et al., 2012). We found that compared to parental cells, GSK923295-resistant clones derived from HCT116 or KBM7 cells did not exhibit resistance to these other cytotoxic compounds (Figure 1D and S1B, C and Table S1). In fact, we note that KBM7 clones resistant to GSK923295 exhibited modestly enhanced sensitivity to the microtubule-stabilizing agent taxol (Figure 1D and S1C). Taken together, these data indicate that resistance to GSK923295 exhibited by the KBM7 and HCT116 cells we isolated is chemotype-specific and not likely due to multi-drug response mechanisms.
Examining Resistance to GSK923295 in Diploid HCT116 Cells
We have found that mutations leading to chemotype-specific resistance often emerge in the direct targets of drugs (Kapoor and Miller, 2017). Therefore, we next analyzed the sequence of the CENPE gene in the GSK923295-resistant clones isolated from HCT116 cells. In particular, we sequenced genomic DNA from the GSK923295-resistant clones and the parental cells to analyze exons 1–8, which correspond to the motor domain of CENPE kinesin. Sanger sequencing did not reveal any nonsynonymous mutations in this genomic locus isolated from parental cells, but we found a different heterozygous mutation in each of the two GSK923295-resistant clones (clone 1, chr.4, pos.: 103195988, codon change: ATG to GTG (M97V); clone 2, chr.4, pos.: 103194435, codon change: AGG to ATG (R189M), numbering from H. sapiens, genome assembly: GRCh38.p13, Figure 2A). These residues map to the predicted GSK923295-binding site in a pocket near loop 5 in the CENPE motor domain (Figure S2A).
Figure 2. Characterizing resistance to GSK923295 in diploid cells.
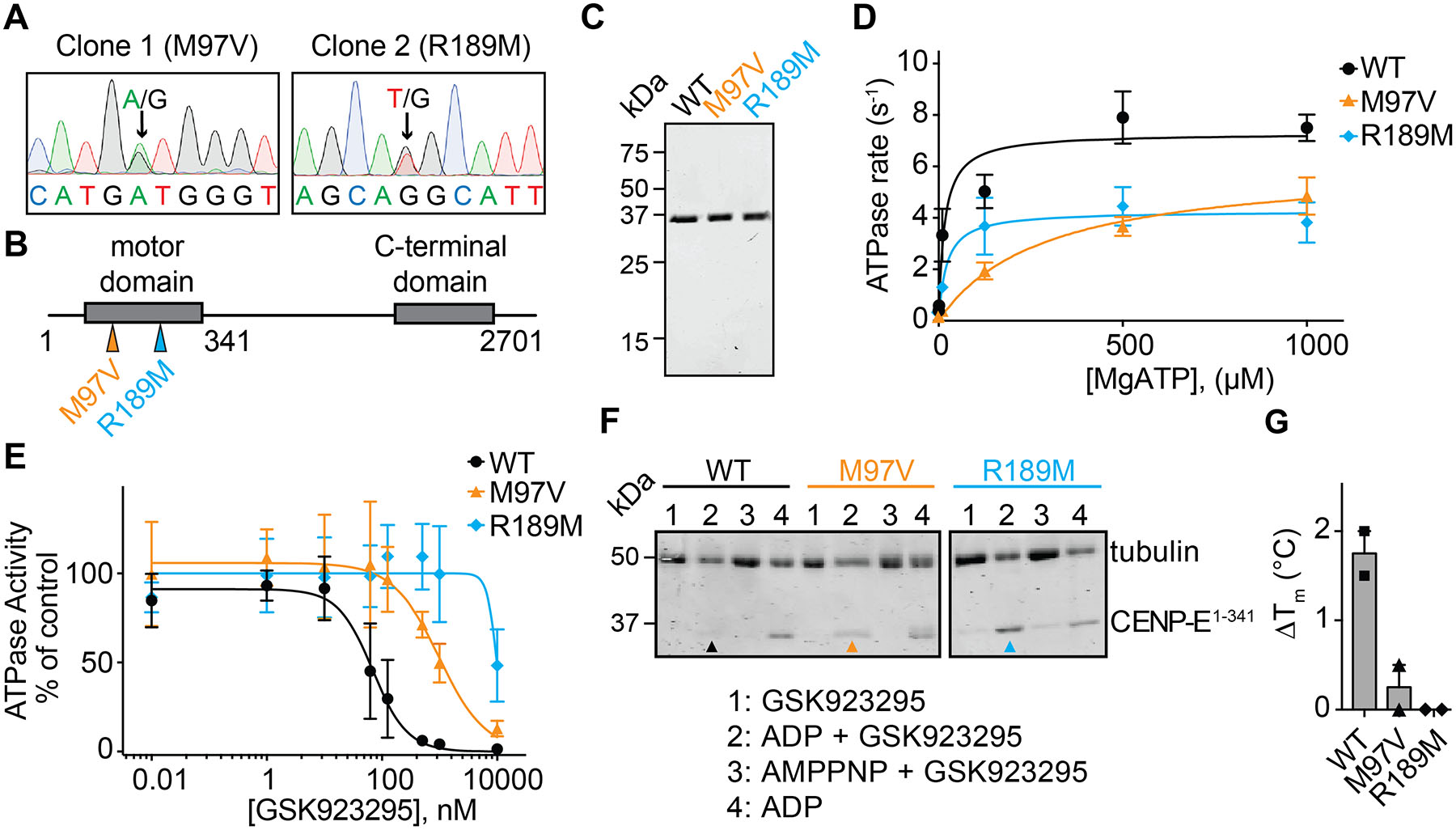
(A) Sanger sequencing traces of genomic DNA encoding CENPE kinesin domain in two GSK923295-resistant HCT116 clones. Positions of heterozygous mutations are indicated (black arrows). Sequences are shown in 5’−3’ direction of the coding strand.
(B) Schematic shows motor and C-terminal domains of the CENPE protein. Positions of identified mutations are shown (colored arrows). First and last residues of the full length CENPE protein and the site of truncation to generate the recombinant kinesin motor domain construct are also indicated (not to scale).
(C) SDS-PAGE analysis of purified wild-type (WT) and mutant (M97V and R189M) CENPE constructs (aa 1–341, Coomassie Blue).
(D) ATP-concentration dependence of the steady-state activity of wild-type and mutant CENPE motor domain constructs. Graph shows values fit to a Michaelis-Menten equation (WT: kcat = 7.5 ± 0.5 s−1, KM = 25 ± 18 μM; M97V: kcat = 6.1 ± 0.9 s−1, KM = 294 ± 58 μM; R189M: kcat = 4.3 ± 0.8 s−1, KM = 21 ± 2 μM, average ± s.d., n=3).
(E) GSK923295 concentration-dependent inhibition of microtubule-stimulated ATPase activity of recombinant CENPE kinesin motor domain constructs. Graph shows values fit to a sigmoidal dose-response equation (IC50 WT: 77 ± 60 nM, M97V: 813 ± 167 nM, R189M: n.d., average values ± s.d., 2 mM MgATP, n=3).
(F) SDS-PAGE analysis of soluble supernatant fractions from a microtubule co-sedimentation assay. CENPE motor domains in the presence of GSK923925 and ADP are indicated with colored arrows (WT: black, M97V: orange, R189M: cyan). Source gels are shown in Figure S2C.
(G) Change in the heat-induced unfolding (∆Tm) of the recombinant CENP-E motor domain constructs in the presence of GSK923295 (50 µM) in comparison to vehicle control (DMSO, 1%; mean ± range, n = 2).
To characterize the mutations identified in CENPE, we designed recombinant constructs of its motor domain. We purified wild-type and two mutant constructs (M97V and R189M) using a multistep protocol that included affinity pulldown, ion exchange, and size exclusion chromatography (aa 1–341, Figure 2B, C and S2B) and tested ATPase activity. These analyses revealed that the rates of ATP hydrolysis of the mutant alleles differ less than two-fold in comparison with the wild-type construct (WT: kcat = 7.5 ± 0.5 s−1; M97V: kcat = 6.1 ± 0.9 s−1; R189M: kcat = 4.3 ± 0.8 s−1, Figure 2D). The ATP concentration required for half maximal enzyme activity (KM) of the R189M allele is also similar to the wild-type protein (WT: KM = 25 ± 18 μM; R189M: KM = 21 ± 2 μM). In contrast, the KM for ATP of the M97V mutant allele is increased ~12-fold in comparison to the wild-type protein (M97V: KM = 294 ± 58 μM). However, as intracellular concentration of ATP is in the mM range, these CENPE mutant alleles are likely functional in cells. These data are also consistent with our cell biological analyses (see below).
Next, we examined if GSK923295 inhibits the microtubule-stimulated ATPase activity of these constructs. Consistent with a previous report (Wood et al., 2010), the ATPase activity of the wild-type construct was potently inhibited by the GSK923295 compound (IC50 = 77 ± 60 nM, Figure 2E). In contrast, the mutant constructs were inhibited by GSK923295 with substantially lower potency (M97V: IC50 = 813 ± 167 nM, R189M: IC50 value was not determined as complete inhibition was not observed at the highest dose tested).
The binding of kinesin motor domains to microtubules is regulated by nucleotide hydrolysis, with strong binding occurring in the AMPPNP-state and weaker binding in the ADP-state (Moyer et al., 1998; Wood et al., 2010). The GSK923295 inhibitor has been proposed to stabilize the kinesin motor in a strongly-bound microtubule state even in the presence of ADP (Wood et al., 2010). Consistent with these data, we found that in a co-sedimentation assay with microtubules in the presence of ADP (1mM), the wild-type and mutant CENPE motor constructs could be detected in the supernatant, but in the presence of AMPPNP (1 mM) were bound to microtubules (Figure 2F and S2C). In addition, the wild-type construct co-sedimented with microtubules in the presence of the GSK923295 inhibitor (20 μM) and ADP (1 mM). In contrast, the mutant motor domain constructs (M97V and R189M) remained in the soluble fraction in the presence of GSK923295 (20 μM) and ADP (1 mM). To further characterize binding of GSK923295 to the recombinant CENPE constructs, we used differential scanning fluorimetry. These analyses revealed an increase in the melting temperature of the wild-type construct in the presence of GSK923295 (ΔTm ~1.5°C, 50 μM GSK923295, Figure 2G). The potency of GSK923295 in this assay matches the lower potency of CENPE inhibition in ATPase assays in the absence of microtubules (Wood et al., 2010). Importantly, we did not observe any changes in the melting temperature of the mutant constructs at the highest doses tested (50 μM GSK923295, Figure 2G). Taken together, these data suggest that single-point mutations in the CENPE motor domain confer resistance by preventing GSK923295 binding.
Characterizing HCT116 Cells Resistant to GSK923295
Chemical inhibition or genetic knockout of CENPE in cells promotes accumulation of chromosomes associated with the spindle poles (so-called “pole-stuck” chromosomes) (Wood et al., 2010). Therefore, we next focused on examining the mitotic phenotype in parental and GSK923295-resistant HCT116 cells using confocal fluorescence microscopy. In parental cells treated with a vehicle control (DMSO), we observed that chromosomes can congress to the equator (Figure 3A and E). Our analyses of GSK923295-resistant clones 1 and 2 revealed that these cells can also proceed through mitosis without substantial detectable aberrant chromosome alignment (Figure 3C and E). This is consistent with the presence of functional CENPE protein in the GSK923295-resistant cells (Kapoor et al., 2006; McEwen et al., 2001; Putkey et al., 2002; Wood et al., 1997).
Figure 3. Immunofluorescence analysis of parental and GSK923295-resistant diploid cells.
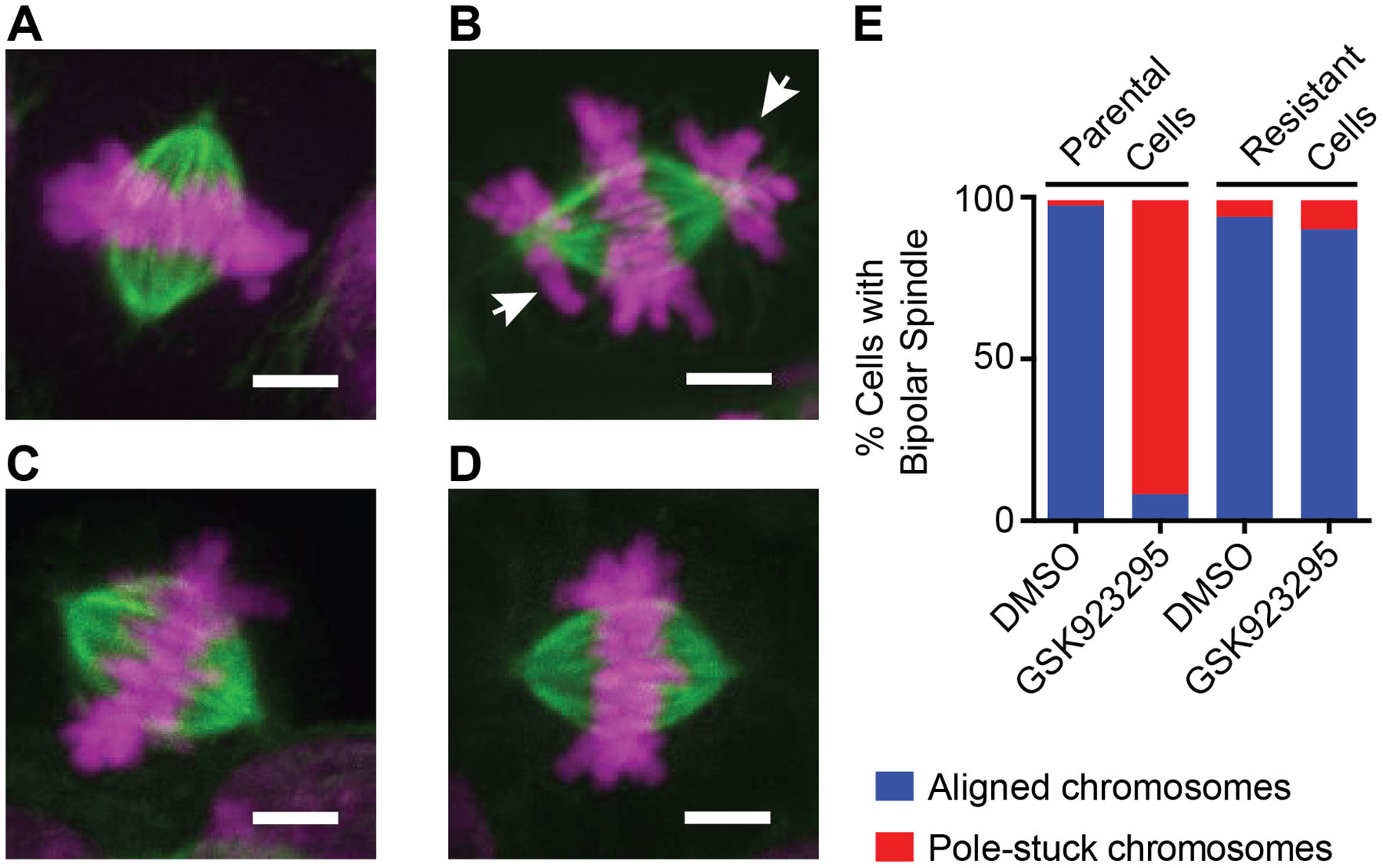
(A-D) Representative immunofluorescence images of mitotic spindles in parental and GSK923295-resistant HCT116 cells. Maximum intensity projections of DNA (magenta) and tubulin (green) are shown. Parental cells treated with vehicle control (A) or GSK923295 (B), and GSK923295-resistant cells (clone 2) treated with vehicle control (C) or GSK923295 (D) are shown. Pole-stuck chromosomes are indicated with white arrows. Scale bars, 4 μm.
(E) Bipolar spindles were counted and manually classified as aligned or containing pole-stuck chromosome(s). Spindles in parental cells treated with DMSO: aligned: 98%, pole-stuck: 2%; parental cells treated with GSK923295: aligned: 8%, pole-stuck: 92%; spindles in resistant cells (clones 1 and 2) treated with DMSO: aligned, 95%; pole-stuck, 5%; GSK923295: aligned, 91%; pole-stuck, 9%. See also Figure S3.
Further, our analyses of parental cells treated with GSK923295 revealed an increase in misaligned, pole-stuck chromosomes in mitosis (Figure 3B and E) that match the phenotype of CENPE depletion or knock-out (Weaver et al., 2003; Wood et al., 1997, 2010). In contrast, we observed that GSK923295-resistant clones treated with the inhibitor do not accumulate polar chromosomes and proceed through mitosis similar to cells treated with vehicle control (Figure 3D and E). Together, these analyses suggest that the GSK923295-resistant HCT116 cells express functional CENP-E that cannot bind the inhibitor, as the wildtype allele would be inhibited by the compound. Although further studies of cells with engineered CENPE alleles will be needed to examine if the point mutations we identified in the motor domain are sufficient to confer resistance in cells, we believe this is likely based on our application of DrugTargetSeqR to other drugs, including another kinesin inhibitor (Kasap et al., 2014).
Examining Resistance to GSK923295 in Haploid KBM7 Cells
To examine mechanisms of resistance against the GSK923295 inhibitor in KBM7 cells we first focused on the motor domain of CENPE. However, Sanger sequencing of genomic DNA in this locus did not reveal any nonsynonymous variants in KBM7 cells resistant to GKS923295; therefore we performed transcriptome sequencing of the three GSK923295-resistant cell populations and the parental cells. We employed a pipeline using available open-source software for alignment of the reads to the human genome, gene expression and exon coverage analyses (Figure S4A). These analyses of single nucleotide variations and short insertions and/or deletions (indels) in the KBM7 cells also did not reveal non-synonymous mutations in the kinesin motor domain of the CENPE gene.
We next focused on gene expression analyses to compare changes in RNA expression between the parental and GSK923295-resistant cells (Figure S4B). First, we examined the expression of MDR efflux transporter P-glycoprotein (also known as Pgp, MDR1 or ABCB1), as overexpression of this pump was reported to confer resistance to GSK923295 (Tcherniuk and Oleinikov, 2015). We observed almost no reads mapping to the ABCB1 gene in parental and GSK923295-resistant cells, suggesting low or no expression of the protein (Figure S4C), consistent with the observed chemotype-specific resistance (Figure 1C, D)
Next, we performed a more extensive transcriptome-wide analysis that revealed several genes with substantially altered expression profiles, including CENPE, in GSK923295-resistant clones in comparison to the parental cells (Figure S4B, C and Table S2). In particular, exon coverage analyses using the DexSeq algorithm (Anders et al., 2012) revealed that the expression of exons corresponding to the N-terminal kinesin domain of CENPE was not substantially altered in the parental and GSK923295-resistant cells. But interestingly, we found essentially no coverage for exons corresponding to the C-terminal portion of the CENPE gene in the resistant cells (exons 25–48, Figure 4A). We note that the residual coverage of the C-terminal exons in cells from well 1 (Figure 4A - red lines) likely represents contamination of this well with RNA from parental cells due to incomplete separation in our dilution-based selection protocol. As the coverage of multiple adjacent genes (BDH2, SLC9B1, SLC9B2, CISD2, Figure S4C and Table S2) was also nearly zero, we suspected a possible genomic deletion.
Figure 4. Characterizing resistance to GSK923295 in KBM7 cells.
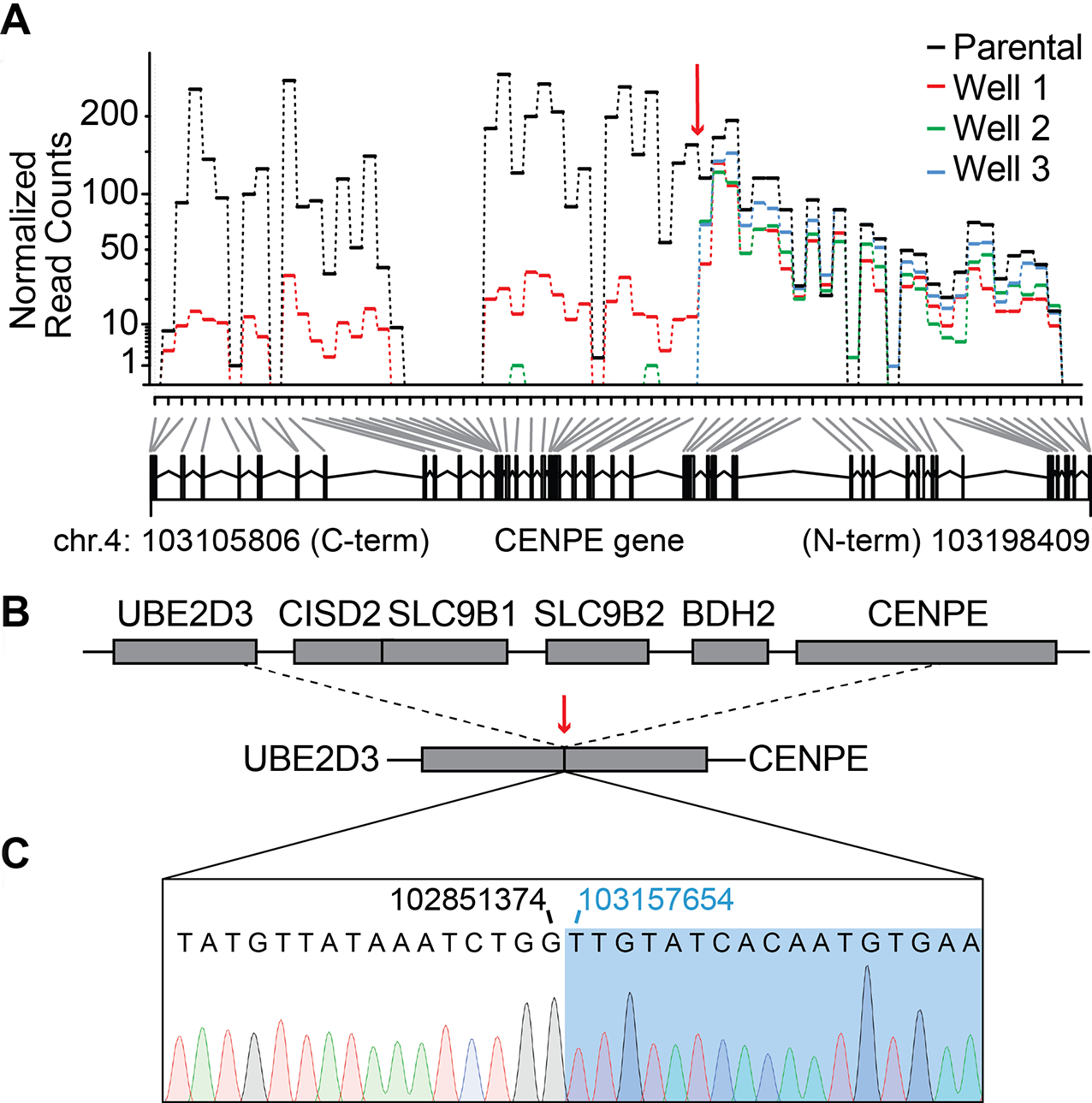
(A) Exon usage over CENPE gene in parental and GSK923295-resistant KBM7 cells. Normalized read counts for each sample are shown (top panel). Deletion site in the CENPE gene is indicated (red arrow). Counting bins generated by the DexSeq algorithm are also shown (bottom panel, gray and black lines). First and last base pairs of the CENPE gene on chromosome 4 are indicated.
(B) Schematics show UBE2D3-CENPE genomic locus in wild-type (top) and GSK923295-resistant KBM7 cells (bottom). Deletion/fusion site is indicated (dashed lines and red arrow).
(C) Sanger sequencing trace of the deletion/fusion site in GSK923295-resistant KBM7 cells. Positions of the last base pair of the UBE2D3 gene (black) and the first base pair of the CENPE gene (blue) at the deletion/fusion site are indicated.
We next used PCR analyses of the CENPE genomic locus and identified the site of the deletion event (Figure S4D, E). Sanger sequencing confirmed a ~300kb deletion of genomic DNA that led to a fusion between intronic regions in UBE2D3 and CENPE genes (Figure 4B, C). The deletion includes exons 25–48 of CENPE, spans the four adjacent genes, and extends into the first intron of the UBE2D3 gene. We posit that resistance to GSK923295 in these clones is likely CENPE-specific, as CRISPR/Cas9-based deletion of CENPE gene in haploid mammalian cells can confer resistance to GSK923295 (Raaijmakers et al., 2018). Taken together, these data suggest that loss of the C-terminal domain of CENPE, which is needed for kinetochore targeting, is likely equivalent to the genetic knockout may protect haploid cells against GSK923295-induced toxicity (Raaijmakers et al., 2018).
DISCUSSION
Here we report unbiased selections in diploid and near-haploid cultured cancer cells and identify mechanisms that confer resistance to GSK923295, a chemical inhibitor of the motor protein CENPE. We find that diploid HCT116 cells resistant to GSK923295 carry heterozygous, single-point mutations in the kinesin motor domain. We characterize these CENPE alleles using in vitro biochemical assays and show that the mutations block inhibitor binding. In contrast to diploid cells, drug-resistant haploid KBM7 cells do not carry mutations in the motor domain but have lost the kinetochore-targeting C-terminal domain of CENPE. Together, our results suggest that distinct mechanisms of resistance to GSK923295 can emerge in cancer cells with different karyotypes.
The presence of GSK923295 leads to tight binding of CENPE’s motor domain to the microtubule tracks in dividing cells. This likely prevents chromosome alignment at the start of cell division and leads to activation of the spindle assembly checkpoint (Putkey et al., 2002; Wood et al., 2010). In the case of haploid cells, the loss of the CENPE motor domain can be tolerated, as these cells grow in the absence of a robust spindle-assembly checkpoint (Raaijmakers et al., 2018). However, when the wild-type CENP-E motor protein is present in haploid cells, GSK923295 likely traps chromosomes bound to microtubules and thereby induces extensive chromosome mis-segregation and toxicity. Interestingly, it has been found that many human tumors and cancer cells can have compromised spindle-assembly checkpoints (Weaver and Cleveland, 2005), and loss of CENP-E may be a strategy to overcome GSK923295-induced toxicity. In contrast, in diploid or polyploid cells with robust checkpoints, the resistance mechanisms must prevent the binding of the inhibitor to CENP-E through mutations that are functionally silent. Further studies will be needed to examine if distinct mechanisms of resistance can arise in cells with different ploidies and karyotypes for other drugs.
Single-point mutations identified in this study (M97V and R189M) map to the kinesin motor domain of CENPE and support the predicted binding model of the CENPE-GSK923295 inhibitor complex (Wood et al., 2010). M97 and R189 residues likely form critical interactions with the GSK923295 inhibitor as mutations of these residues, which emerged in our unbiased selections, prevent binding of the compound. Analyses of resistance-conferring mutations can not only help establish the direct physiological targets of chemical inhibitors but can also be useful during inhibitor development (Cupido et al., 2019; Pisa et al., 2019). The single-point mutations in the CENPE motor domain we identified could therefore be valuable for the design and optimization of new potent and specific CENPE inhibitors that can bind the protein in different poses or at distinct sites to overcome resistance.
Evolution of protein functions, such as ligand binding, depends on available trajectories in sequence space. These trajectories are constrained by epistasis within proteins as well as genetic interactions across different loci (Lehner, 2011; Starr and Thornton, 2016). These constraints can be altered in cancer cells that have aberrant gene and chromosome copy numbers and thereby lead to the emergence of distinct drug-resistance mechanisms. Our findings suggest that studying resistance mechanisms to drugs in cells with different karyotypes could be useful for designing treatments and improving the long-term efficacy of targeted anticancer therapeutics.
STAR Methods
LEAD CONTACT AND RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tarun M Kapoor (kapoor@rockefeller.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Escherichia coli BL21 Rosetta™ (DE3) pLysS cells used for protein expression in this study were grown in LB media supplemented with 34 mg/L chloramphenicol and 50 mg/L kanamycin (for details see STAR Methods - Protein Expression and Purification). HCT116 cells were obtained from ATCC (#CCL-247). KBM7 cells were a kind gift from Dr. Kivanc Birsoy (Rockefeller University).
METHOD DETAILS
Mammalian cell culture
HCT116 cells were maintained in McCoy’s medium supplemented with 10% Fetal Bovine Serum (FBS), 2 mM L-glutamine and 100 U/ml penicillin/streptomycin. KBM7 cells were grown in IMDM media supplemented with 10% FBS, 2 mM L-glutamine and 100 U/ml penicillin/streptomycin. All mammalian cells were incubated at 37°C and 5% CO2.
Selection of resistant clones
Drug-resistant clones in HCT116 cells were isolated using approaches similar to those we have previously described (Wacker et al., 2012). Briefly, ~1.106 cells were plated on a 10 cm culture dish in media supplemented with GSK923295 (2 μM, 0.1 – 0.5% DMSO was used as a vehicle). Medium with compound was exchanged every 2–3 days for ~2 weeks. Most cells died but a few resistant colonies emerged on the plate. Surviving colonies were isolated by ring cloning, transferred to a new plate and expanded in media containing the drug (2 μM GSK923295 and 0.1 – 0.5% DMSO).
KBM7 cell populations resistant to GSK923295 were isolated by a cell dilution-based protocol in 96-well plates. Briefly, cells were grown in media supplemented with GSK923295 (2 μM, 0.1 – 0.5% DMSO was used as a vehicle). After 2–3 days cells were diluted ~3-fold into fresh media containing the drug (2 μM) and this process was repeated for ~3 weeks. All cells died in most wells on the plate but in a few wells, we observed cell growth. Cells from these wells were transferred to 10-cm dishes and expanded in media with the drug (GSK923295, 2 μM) before cell proliferation, RNAseq and immunofluorescence analysis (see below).
Cell proliferation assays
To quantify growth of HCT116 cells in the presence of drugs, cells were plated (1000–2000 cells per well) in clear flat-bottom, 96-well plates and treated with 8–9 doses of a serial dilution of the desired compound (0.5% DMSO was used as a vehicle in these assays). Growth of KBM7 cells was quantified using similar procedures (10000–20000 cells per well). 0.5% DMSO and 0.1% SDS were used as controls. After 3 days, cell proliferation was determined using an Alamar Blue assay (O’brien et al., 2000). Briefly, 50 μl of Alamar Blue stock solution (sterile-filtered solution of 0.5 M resazurin sodium salt in PBS) was added to each well and the plates were incubated at 37°C and 5% CO2 until the ratio of fluorescent readouts between positive and negative control wells reached 5–10-fold difference. Fluorescence readout was determined using a Synergy NEO Microplate Reader (excitation: 550nm, emission: 590nm).
Immunofluorescence
HCT116 cells were plated on round glass coverslips (Fisher Scientific) in six-well dishes 24 h before fixation. Cells were exposed to DMSO (vehicle control, 0.1%) or GSK923295 (200 nM), at 37 °C and 5% CO2 in complete medium for 4 h and then fixed for 10 min at 37 °C in fix solution (4% formaldehyde in 10 mM MES, 138 mM KCl, 2 mM EGTA, 3 mM MgCl2, 0.32 M sucrose, 10% FBS and 0.01% saponin, pH 6.1). Coverslips were washed three times with PBS, treated for 30 min in blocking buffer (10% FBS, 0.01% saponin, 0.01% NaN3 in PBS) and incubated for 1 h at room temperature with FITC-conjugated mouse anti-tubulin monoclonal antibody (1:2000 dilution in blocking buffer). Coverslips were washed three times in PBS, and DNA was stained with Hoechst 33342 or JF646-Hoechst conjugate (Legant et al., 2016) (1:10,000 in PBS). Coverslips were mounted in 0.5% n-propyl gallate in 20 mM Tris-HCl, pH 8.0, with 90% glycerol and sealed with nail polish. A Nikon TE2000 confocal microscope (Morrell Instruments) equipped with 100x objective (Plan Apo, 1.45 NA), Yokogawa CSU10 confocal head, and EMCCD Photometrics Cascade II 512B camera (Roper Scientific) was used for all fluorescent visualizations. NIS-Elements software was used for acquisition of images. Fiji software was used to generate maximum intensity projections of acquired images and to adjust brightness and contrast (Schindelin et al. 2012).
Dose response analysis and LD50 calculations
For each experiment, technical replicates were averaged, and percent cellular growth was calculated by normalizing the background-corrected fluorescence of wells with compounds to wells with vehicle control (0.5% DMSO). Background fluorescence was calculated as the mean of fluorescent readouts of wells treated with SDS (0.1%). To determine the LD50 values, percent cellular growth was plotted against concentration of compound and the data were fit using a sigmoidal dose-response curve (Equation 1) using Prism software. The values from at least three independent experiments were averaged and standard deviations were calculated.
| Equation 1 |
In this equation x denotes concentration of the compound and h is the Hill coefficient.
RNAseq and gene expression analysis
Parental and GSK923295-resistant KBM7 cells (~2.107) were washed once with 1X PBS and lysed using a High-Pure RNA extraction kit followed by RNA isolation according to the manufacturer’s protocol. RNA-Seq libraries were prepared at the Rockefeller University Genomics Core Facility with the Illumina TruSeq RNA Library prep kit and sequenced using the NextSeq500 system. Raw sequence reads were aligned to a human genome template (GRCh38, version 25 (Ensembl 85) from GENCODE, 2016-07-15) with the STAR aligner software (Dobin et al., 2013). Read groups were assigned and PCR duplicates were removed using Picard (http://broadinstitute.github.io/picard/). Aligned reads assigned to exons were counted using featureCounts (Liao et al., 2014). Normalized counts were determined as follows: first, sample normalization factors were calculated by dividing the total number of counts in each of the resistant clone samples by the total number of counts in the parental sample. Normalized read counts were then calculated by dividing the read count value for each gene in a sample by the sample normalization factor. Fold change values were calculated as follows. First, the values of normalized read counts in all samples were augmented by 1. The fold change values were then calculated by dividing the counts value for each gene in the sample by the corresponding count value in the parental sample. Differential exon usage was determined by the DexSeq algorithm in RStudio by using the default settings (Anders et al., 2012). Numbering in Figure 4 corresponds to H. sapiens, genome version: GRCh38.p13.
Genomic PCR analysis
Genomic DNA for PCR analysis and Sanger sequencing was isolated from parental cells and drug-resistant clones using DNeasy Blood & Tissue Kit (Qiagen).
Primers used for identifying M97V mutation in CENPE motor domain (PCR product spanning exons 3 and 4):
ATAGTTGATCATCCTACATTCTGAC
GATAAAAGTGAATCTATGCTATGCC
Primers used for identifying R189M mutation in CENPE motor domain (PCR product spanning exons 6 to 8):
TCCTACAGAATTATGGTTCCATTTGC
CCTCCCACATTTAGTTTAGTTAACAG
Primers for PCR amplification of the deletion/fusion site on chromosome 4 in KBM7 cells:
GATGACCTAGCAACTACACAGTCGA
GTTACATTTGGGGAACAGTGACAGG
Primers for Sanger sequencing of the amplified deletion/fusion site:
GAAGTTTCCAGGAATTTGCATATGG
CTAGCTATCTTTGTGACTTG
All primers are shown in 5’ to 3’ direction and were ordered from IDT (https://www.idtdna.com/).
Vectors for Recombinant Protein Expression
Constructs for expression of CENPE motor domain were generated as described previously (Garcia-Saez et al., 2004). Briefly, the coding sequence for CENPE motor domain (aa 1–341) was PCR amplified (CloneAmp HiFi, Clontech) from pcDNA5-FRT-TO-Myc-LAP-Cenp-E plasmid and cloned into pET28 vector using NcoI and XhoI sites to generate C-terminal His6-fusion construct (XhoI site introduces leucine and glutamate residues (LE) between the CENPE motor domain (aa 1–341) and the C-terminal hexahistidine tag). M97V and R189M mutations were introduced by PCR using primers containing the mutant sequences. The coding sequences for all constructs were verified by Sanger sequencing. pcDNA5-FRT-TO-Myc-LAP-Cenp-E vector (Kim et al., 2010) was a kind gift from Dr. Ekaterina Grishchuk (University of Pennsylvania) and Dr Don Cleveland (Ludwig Institute for Cancer Research, UCSD). Sequence alignments were performed using the ClustalW algorithm in MacVector software (MacVector, Inc).
Protein Expression and Purification
Human CENPE motor domain (aa 1–341) wild-type and mutant constructs (M97V and R189M) were expressed and purified as follows. Briefly, E.coli Rosetta cells were grown in Miller’s LB medium to O.D.600 ~0.8 at 30°C, chilled to 18°C and the protein was expressed overnight (14–16 h, 0.5 mM IPTG). The cells were resuspended in lysis buffer (50 mM NaH2PO4 pH = 8.0, 250 mM NaCl, 20 mM imidazole, 1 mM MgCl2, 1 mM β-mercaptoethanol, 0.2 mM ATP, 1 tablet of Roche cOmplete protease inhibitor cocktail per 50 ml of buffer), and lysed by sonication or using Emulsiflex-C5 homogenizer (Avestin, 5–10 cycles at 10–15 kPsi). The lysate was spun at 38,000 rpm for 40 min at 4°C in a Ti-45 rotor using Beckman Coulter Optima LE-80K ultracentrifuge. The clarified supernatant was incubated with Ni-NTA agarose resin for ~45 min. The beads were washed with ~150 ml of lysis buffer and the protein was eluted by a gradient of elution buffer (25 mM PIPES.NaOH pH = 6.8, 400 mM imidazole, 250 mM NaCl, 1 mM MgCl2, 0.2 mM ATP, 1 mM β-mercaptoethanol). Eluted protein was dialyzed into low salt buffer (25 mM PIPES.NaOH pH = 6.6, 20 mM NaCl, 2 mM MgCl2, 1 mM β-mercaptoethanol, 0.2 mM ATP), loaded onto HiTrap™ SP column (GE Healthcare) and eluted with a gradient of high salt buffer (25 mM PIPES.NaOH pH = 6.6, 500 mM NaCl, 2 mM MgCl2, 1 mM β-mercaptoethanol, 0.2 mM ATP, 1 mM EGTA). The protein was further purified over a HiLoad 16/60 Superdex 200 or Superdex 200 Increase 10/300 GL columns (GE Healthcare) in size exclusion buffer (25 mM PIPES-KOH pH = 6.8, 250 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.2 mM ATP, 1 mM β-mercaptoethanol) and the desired fractions were dialyzed into storage buffer (25 mM PIPES-KOH pH = 6.8, 250 mM KCl, 2 mM MgCl2, 1 mM EGTA, 50 μM ATP, 1 mM DTT, 30 % sucrose) and flash frozen in liquid nitrogen.
Microtubule co-sedimentation assays
Taxol-stabilized microtubules were polymerized from purified pre-cleared bovine tubulin as described (Kapitein et al., 2008). Purified CENPE motor domain constructs were incubated with microtubules in BRB80 buffer and spun in TLA 120.1 rotor (Beckman Coulter) at 90000 rpm for 10 mins at 30°C. Pelleted material was resuspended in 1x BRB80 buffer (80 mM PIPES.KOH, 1 mM MgCl2, mM EGTA, pH = 6.8) supplemented with desired nucleotide and/or GSK923295 and incubated for 25 min at room temperature. The solution was then transferred into centrifuge tubes with a cushion of 100 μl of BRB80 with 40% glycerol and subjected to sedimentation in TLA 120.1 rotor (Beckman Coulter) at 90000 rpm for 10 mins at 30°C. The soluble supernatant and the pellet were analyzed by SDS-PAGE and the gels imaged on LI-COR Odyssey scanner.
Analyses of ATPase Activity
Steady-state ATPase activity of CENPE motor domain constructs was determined using the NADH-coupled assay in the presence of 6 μM taxol-stabilized microtubules. Briefly, the time course of fluorescence decrease corresponding to the consumption of NADH was measured using a Synergy NEO Microplate Reader (λex = 340 nm, 440 nm emission filter). Fluorescence values were plotted against time and fit by linear regression to obtain a rate of fluorescence decrease. The ATPase rate was calculated from rates of fluorescence decrease using ADP calibration curve. Percent inhibition of the ATPase activity was calculated by normalizing the rate of fluorescence decrease in the presence of compounds to DMSO control. GSK923295 compound stocks were made using DMSO. Final conditions for CENPE motor domain wild type and mutants (30–40 nM): 1x BRB80 (80 mM PIPES.KOH, 1 mM MgCl2, mM EGTA, pH = 6.8), 1 mM TCEP, 0.1 mg/ml BSA, 175–200 μM NADH, 1 mM phosphoenolpyruvate, 15–30 U/ml lactate dehydrogenase, 15–30 U/ml pyruvate kinase, 2 mM MgATP, 6 μM microtubules. 0.5–1% DMSO was used as a vehicle control. Taxol-stabilized, bovine microtubules were prepared as described previously (Kapitein et al., 2008).
Compound IC50 Calculation
For each experiment, values of percent inhibition of ATPase activity were calculated, plotted against concentration of compound, and the data were fit using a sigmoidal dose-response curve (Equation 2) using Prism software to determine the IC50. IC50 values from at least three independent experiments were averaged and standard deviations were calculated.
| Equation 2 |
In this equation x denotes concentration of the compound and h is the Hill coefficient.
Determining enzyme activity parameters
To obtain enzyme activity parameters of the CENPE motor domain constructs, we fitted steady state ATPase rates obtained at increasing concentrations of the substrate (MgATP) to the Michaelis-Menten equation (3):
| Equation 3 |
In this equation Vmax denotes the maximum ATPase rate, x denotes ATP concentration and KM is the ATP concentration required for half maximal enzyme rate and has units of concentration (e.g. mM). Catalytic turnover number (kcat) was calculated by dividing the Vmax value by the concentration of enzyme in the assay.
Differential Scanning Fluorimetry
These experiments were carried out on a C1000 Touch Thermal cycler CFX-96 instrument (GE Healthcare) using 96-well plates (Hard-shell® HSP9665 Bio-Rad). CENPE motor domain recombinant constructs (WT, M97V and R189M; aa 1–341) were diluted to a final concentration of 7–10 μM in 1x BRB80 supplemented with 1 mM TCEP and mixed with GSK923295 (50 μM) or DMSO (1%). SYPRO® Orange (Sigma S5692, excitation 490 nm, emission 590 nm) was used at 1:500 to 1:1000 dilution. The temperature was linearly increased with a step of 0.5°C for 55 minutes, from 25°C to 95°C and fluorescence readings were taken at each interval. Melting temperatures were determined as the minimum value of the first derivative of the fluorescence vs temperature curves.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters including the number of replicates, mean ± standard deviation (s.d.) or range are reported in the respective figure legends.
DATA AND CODE AVAILABILITY
Transcriptome sequencing analyses were performed using publicly available software (STAR aligner (Dobin et al., 2013), Picard (http://broadinstitute.github.io/picard/), featureCounts (Liao et al., 2014), SAMtools (Li et al., 2009), Varscan2 (Koboldt et al., 2013), DexSeq (Anders et al., 2012) and Rstudio (version 1.1.423). R packages were obtained from Bioconductor (https://www.bioconductor.org/). RNA sequencing data sets related to Figure 4 have been submitted to NCBI SRA (Sequence Read Archive) and can be found under BioProjectID: PRJNA561192.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial strains | ||
| Escherichia coli BL21 Rosetta™ (DE3) pLysS | Merck | Cat#70954 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Kanamycin sulfate | Sigma-Aldrich | Cat#K1377–25G |
| Chloramphenicol | Fisher Scientific | Cat#BP904–100 |
| Ni-NTA agarose resin | Qiagen | Cat#30210 |
| 1,4-dithiotreitol (DTT) | GoldBio | Cat#DTT25 |
| β-mercaptoethanol | Sigma-Aldrich | Cat#M6250 |
| β-Nicotinamide adenine dinucleotide, reduced disodium salt (NADH) | Sigma-Aldrich | Cat#N7410 |
| Adenosine 5′-triphosphate disodium salt hydrate | Sigma-Aldrich | Cat#A6419 |
| Bovine Serum Albumin, fraction V | Bioworld | Cat#22070008 |
| Dimethylsulfoxide (DMSO) | Sigma-Aldrich | Cat#276855 |
| L-Glutathione reduced | Sigma-Aldrich | Cat#G6529 |
| Imidazole | Sigma-Aldrich | Cat#I2399 |
| Sodium phosphate dibasic anhydrous | Fisher Scientific | Cat#S374500 |
| Sodium phosphate monobasic monohydrate | Fisher Scientific | Cat#S369500 |
| PIPES | Sigma-Aldrich | Cat#P6757 |
| MES hydrate | Sigma-Aldrich | Cat#M8250 |
| Tris base | Fisher Scientific | Cat#BP152–1 |
| Potassium chloride | Fisher Scientific | Cat#P2173 |
| Sodium chloride | Fisher Scientific | Cat#S2173 |
| Potassium hydroxide | Fisher Scientific | Cat#P250500 |
| Sodium hydroxide | Fisher Scientific | Cat#S374500 |
| Magnesium chloride hexahydrate | Fisher Scientific | Cat#M33500 |
| EGTA | Sigma-Aldrich | Cat#E3889 |
| Sucrose | Sigma-Aldrich | Cat#S0389 |
| Saponin | Sigma-Aldrich | Cat#S4521 |
| Paraformaldehyde | EMS | Cat#15710 |
| Sodium azide | Sigma-Aldrich | Cat#71290 |
| D-Lactic Dehydrogenase (LDH) | Sigma-Aldrich | Cat#L3888 |
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma-Aldrich | Cat#P7626 |
| Phospho(enol)pyruvic acid monopotassium salt (PEP) | Sigma-Aldrich | Cat#P7127 |
| Pyruvate Kinase from rabbit muscle (PK) | Sigma-Aldrich | Cat#P9136 |
| Roche cOmplete, EDTA-free Protease Inhibitor | Sigma-Aldrich | Cat#11873580001 |
| Tris(2-carboxyethyl)phosphine HCl (TCEP) | GoldBio | Cat#TCEP10 |
| Triton™ X-100 | Sigma-Aldrich | Cat#X100–100ML |
| Sodium dodecyl sulfate (SDS) | Sigma-Aldrich | Cat#L4509 |
| Miller’s LB medium | Formedium | Cat#LMM105 |
| Isopropyl-beta-D-thiogalactoside (IPTG) | GoldBio | Cat#I2481 |
| McCoy’s 5A medium | ATCC | Cat#30–2007 |
| IMDM medium | Thermo-Fisher | Cat#12440053 |
| Penicillin/Streptomycin (10000 U/ml) | Thermo-Fisher | Cat#15140122 |
| Fetal Bovine Serum (Heat Inactivated) | Sigma-Aldrich | Cat#F4135 |
| Glutamine (200 mM) | Thermo-Fisher | Cat#25030081 |
| Resazurin sodium salt | Sigma-Aldrich | Cat#R7017 |
| 1x DPBS | Thermo-Fisher | Cat#14190144 |
| CloneAmp HiFi PCR Premix | Clontech | Cat#639298 |
| High-Pure RNA isolation kit | Roche | Cat#11828665001 |
| TruSeq® Stranded Total RNA Library Prep | Illumina | Cat#20020596 |
| SYPRO® Orange | Sigma-Aldrich | Cat#S5692 |
| n-propyl gallate | Sigma-Aldrich | Cat#02370 |
| Hoechst 33342 | Thermo-Fisher | Cat#62249 |
| JF646-Hoechst | Legant et al., 2016 | NA |
| Anti-α-tubulin-FITC antibody, mouse monoclonal (clone DM1A) | Sigma-Aldrich | Cat#F2168 |
| Recombinant DNA | ||
| pcDNA5-FRT-TO-Myc-LAP-Cenp-E | Gift from Grishchuk and Cleveland labs | Kim et al., 2010 |
| pET28_Hs_CENPE (aa1–341) | This work | NA |
| Oligonucleotides (5’ to 3’) | ||
| ATAGTTGATCATCCTACATTCTGAC | This work | primer for Sanger sequencing of CENPE exons 3 and 4 |
| GATAAAAGTGAATCTATGCTATGCC | This work | primer for Sanger sequencing of CENPE exons 3 and 4 |
| TCCTACAGAATTATGGTTCCATTTGC | This work | primer for Sanger sequencing of CENPE exons 6 to 8 |
| CCTCCCACATTTAGTTTAGTTAACAG | This work | primer for Sanger sequencing of CENPE exons 6 to 8 |
| GATGACCTAGCAACTACACAGTCGA | This work | primer for PCR amplification of CENPE/UBE2D3 truncation |
| GTTACATTTGGGGAACAGTGACAGG | This work | primer for PCR amplification of CENPE/UBE2D3 truncation |
| GAAGTTTCCAGGAATTTGCATATGG | This work | primer for Sanger sequencing of CENPE/UBE2D3 truncation |
| CTAGCTATCTTTGTGACTTG | This work | primer for Sanger sequencing of CENPE/UBE2D3 truncation |
| Deposited data | ||
| CENPE motor domain, human | Garcia-Saez et al., 2004 | PDB:1T5C |
| Human genome (GRCh38), version 25 (Ensembl 85) | GENCODE | gencode-help@sanger.ec.uk |
| RNAseq: KBM7 parental cells and GSK923295-resistant KBM7 clones 1, 2, 3 | This work | BioProjectID: PRJNA561192 |
| Experimental Models: Cell Lines | ||
| HCT116 | ATCC | Cat#CCL-247 |
| KBM7 | Wang et al., 2015 | NA |
| Software and Algorithms | ||
| GraphPad Prism version 6.0 | GraphPad Software | https://www.graphpad.com/ |
| MacVector (version 17.0.4) | MacVector, Inc | https://macvector.com |
| STAR aligner | Dobin A et al., 2013 | https://github.com/alexdobin/STAR |
| Picard | Broad Institute | http://broadinstitute.github.io/picard/ |
| DexSeq | Anders et al., 2012 | https://bioconductor.org/packages/release/bioc/html/DEXSeq.html |
| Rstudio | Version 1.2.1335 | https://www.rstudio.com/ |
| featureCounts | Liao et al., 2014 | http://subread.sourceforge.net/ |
| SamTools | Li et al., 2009 | http://www.htslib.org/ |
| VarScan | Koboldt et al., 2013 | https://github.com/dkoboldt/varscan |
| Fiji | Schindelin et al. 2012 | https://imagej.net/Fiji |
SIGNIFICANCE.
The emergence of resistance is a key factor limiting the long-term efficacy of molecularly-targeted anticancer drugs. Therefore, understanding the molecular basis of drug resistance is critical for developing effective therapeutics. In particular, the specific mutations or other changes (e.g. truncations) of the target protein that confer resistance can guide chemical inhibitor design and testing. Additionally, the evolution of genetic changes in the target protein can be constrained by aberrant karyotypes and ploidies of cancer cells. However, we do not fully understand how differences in chromosome number impact the emergence of resistance to many anti-cancer drugs. Here, we focused on a potent chemical inhibitor of mitotic kinesin CENP-E GSK923295, which was evaluated in clinical trials as an anticancer agent. We analyzed resistance to the compound in diploid and near-haploid cancer cell lines and found that in diploid cells, single-point mutations in the motor domain of CENPE can block inhibitor binding in biochemical assays. In contrast, drug-resistant near-haploid cells do not carry mutations in the motor domain but have a genetic deletion that removes CENPE’s kinetochore-targeting C-terminal domain. Together, our data show that cells with different chromosome copy numbers can acquire chemotype-specific resistance by distinct mechanisms.
ACKNOWLEDGEMENT
T.M.K. is grateful to the NIH/NIGMS for funding (R01 GM98579 and R35 GM130234-01). R.P. was supported by the Tri-Institutional PhD Program in Chemical Biology and the Rockefeller University Graduate Program. D.Y.Z.P. was supported by the Rockefeller University Summer Undergraduate Research Fellowship. We thank Dr. Ekaterina Grishchuk (UPenn) and Dr. Don Cleveland (UCSD) for providing the pcDNA5-FRT-TO-Myc-LAP-Cenp-E plasmid. We also thank Dr. Luke Lavis (HHMI Janelia Research Campus) for providing the JF646-Hoechst dye.
Footnotes
DECLARATION OF INTEREST
The authors declare no competing financial interests.
REFERENCES
- Anders S, Reyes A, and Huber W (2012). Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, and Cantley LC (2018). The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 174, 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chunduri NK, and Storchová Z (2019). The diverse consequences of aneuploidy. Nat. Cell Biol 21, 54–62. [DOI] [PubMed] [Google Scholar]
- Chung V, Heath EI, Schelman WR, Johnson BM, Kirby LC, Lynch KM, Botbyl JD, Lampkin TA, and Holen KD (2012). First-time-in-human study of GSK923295, a novel antimitotic inhibitor of centromere-associated protein E (CENP-E), in patients with refractory cancer. Cancer Chemother. Pharmacol 69, 733–741. [DOI] [PubMed] [Google Scholar]
- Cupido T, Pisa R, Kelley ME, and Kapoor TM (2019). Designing a chemical inhibitor for the AAA protein spastin using active site mutations. Nat. Chem. Biol 15, 444–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daub H, Specht K, and Ullrich A (2004). Strategies to overcome resistance to targeted protein kinase inhibitors. Nat. Rev. Drug Discov 3, 1001–1010. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elling U, and Penninger JM (2014). Genome wide functional genetics in haploid cells. FEBS Lett. 588, 2415–2421. [DOI] [PubMed] [Google Scholar]
- Garcia-Saez I, Yen T, Wade RH, and Kozielski F (2004). Crystal structure of the motor domain of the human kinetochore protein CENP-E. J. Mol. Biol 340, 1107–1116. [DOI] [PubMed] [Google Scholar]
- Glaab WE, and Tindall KR (1997). Mutation rate at the hprt locus in human cancer cell lines with specific mismatch repair-gene defects. Carcinogenesis 18, 1–8. [DOI] [PubMed] [Google Scholar]
- Gordon DJ, Resio B, and Pellman D (2012). Causes and consequences of aneuploidy in cancer. Nat. Rev. Genet 13, 189–203. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Lavi O, Hall MD, and Gillet J-P (2016). Toward a Better Understanding of the Complexity of Cancer Drug Resistance. Annu. Rev. Pharmacol. Toxicol 56, 85–102. [DOI] [PubMed] [Google Scholar]
- Hirayama T, Okaniwa M, Banno H, Kakei H, Ohashi A, Iwai K, Ohori M, Mori K, Gotou M, Kawamoto T, et al. (2015). Synthetic studies on centromere-associated protein-E (CENP-E) inhibitors: 2. Application of electrostatic potential map (EPM) and structure-based modeling to imidazo [1, 2-a] pyridine derivatives as anti-tumor agents. J. Med. Chem 58, 8036–8053. [DOI] [PubMed] [Google Scholar]
- Kapitein LC, Kwok BH, Weinger JS, Schmidt CF, Kapoor TM, and Peterman EJG (2008). Microtubule cross-linking triggers the directional motility of kinesin-5. J. Cell Biol 182, 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor TM, and Miller RM (2017). Leveraging Chemotype-Specific Resistance for Drug Target Identification and Chemical Biology. Trends Pharmacol. Sci 38, 1100–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor TM, Lampson MA, Hergert P, Cameron L, Cimini D, Salmon ED, McEwen BF, and Khodjakov A (2006). Chromosomes can congress to the metaphase plate before biorientation. Science 311, 388–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasap C, Elemento O, and Kapoor TM (2014). DrugTargetSeqR: a genomics- and CRISPR-Cas9-based method to analyze drug targets. Nat. Chem. Biol 10, 626–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Holland AJ, Lan W, and Cleveland DW (2010). Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of CENP-E. Cell 142, 444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Larson DE, and Wilson RK (2013). Using VarScan 2 for Germline Variant Calling and Somatic Mutation Detection. Current Protocols in Bioinformatics 15.4.1–15.4.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legant WR, Shao L, Grimm JB, Brown TA, Milkie DE, Avants BB, Lavis LD, and Betzig E (2016). High-density three-dimensional localization microscopy across large volumes. Nat. Methods 13, 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner B (2011). Molecular mechanisms of epistasis within and between genes. Trends Genet. 27, 323–331. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lock RB, Carol H, Morton CL, Keir ST, Reynolds CP, Kang MH, Maris JM, Wozniak AW, Gorlick R, Kolb EA, et al. (2012). Initial testing of the CENP-E inhibitor GSK923295A by the pediatric preclinical testing program. Pediatr. Blood Cancer 58, 916–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BF, Chan GK, Zubrowski B, Savoian MS, Sauer MT, and Yen TJ (2001). CENP-E is essential for reliable bioriented spindle attachment, but chromosome alignment can be achieved via redundant mechanisms in mammalian cells. Mol. Biol. Cell 12, 2776–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer ML, Gilbert SP, and Johnson KA (1998). Pathway of ATP hydrolysis by monomeric and dimeric kinesin. Biochemistry 37, 800–813. [DOI] [PubMed] [Google Scholar]
- O’brien J, Wilson I, Orton T, and Pognan F (2000). Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem 267, 5421–5426. [DOI] [PubMed] [Google Scholar]
- Ohashi A, Ohori M, Iwai K, Nambu T, Miyamoto M, Kawamoto T, and Okaniwa M (2015). A Novel Time-Dependent CENP-E Inhibitor with Potent Antitumor Activity. PLoS One 10, e0144675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisa R, Cupido T, Steinman JB, Jones NH, and Kapoor TM (2019). Analyzing Resistance to Design Selective Chemical Inhibitors for AAA Proteins. Cell Chem Biol 26, 1263–1273.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podgornaia AI, and Laub MT (2015). Pervasive degeneracy and epistasis in a protein-protein interface. Science 347, 673–677. [DOI] [PubMed] [Google Scholar]
- Putkey FR, Cramer T, Morphew MK, Silk AD, Johnson RS, McIntosh JR, and Cleveland DW (2002). Unstable kinetochore-microtubule capture and chromosomal instability following deletion of CENP-E. Dev. Cell 3, 351–365. [DOI] [PubMed] [Google Scholar]
- Raaijmakers JA, van Heesbeen RGHP, Blomen VA, Janssen LME, van Diemen F, Brummelkamp TR, and Medema RH (2018). BUB1 Is Essential for the Viability of Human Cells in which the Spindle Assembly Checkpoint Is Compromised. Cell Rep. 22, 1424–1438. [DOI] [PubMed] [Google Scholar]
- Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, and Gottesman MM (2018). Revisiting the role of ABC transporters in multidrug-resistant cancer. Nature Reviews Cancer 18, 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiq MA, Hochberg GK, and Thornton JW (2017). Evolution of protein specificity: insights from ancestral protein reconstruction. Curr. Opin. Struct. Biol 47, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smurnyy Y, Cai M, Wu H, McWhinnie E, Tallarico JA, Yang Y, and Feng Y (2014). DNA sequencing and CRISPR-Cas9 gene editing for target validation in mammalian cells. Nat. Chem. Biol 10, 623–625. [DOI] [PubMed] [Google Scholar]
- Starr TN, and Thornton JW (2016). Epistasis in protein evolution. Protein Sci 25, 1204–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherniuk SO, and Oleinikov AV (2015). Pgp efflux pump decreases the cytostatic effect of CENP-E inhibitor GSK923295. Cancer Lett. 361, 97–103. [DOI] [PubMed] [Google Scholar]
- Wacker SA, Houghtaling BR, Elemento O, and Kapoor TM (2012). Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat. Chem. Biol 8, 235–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BAA, and Cleveland DW (2005). Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell 8, 7–12. [DOI] [PubMed] [Google Scholar]
- Weaver BAA, Bonday ZQ, Putkey FR, Kops GJPL, Silk AD, and Cleveland DW (2003). Centromere-associated protein-E is essential for the mammalian mitotic checkpoint to prevent aneuploidy due to single chromosome loss. J. Cell Biol 162, 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KW, Sakowicz R, Goldstein LSB, and Cleveland DW (1997). CENP-E Is a Plus End–Directed Kinetochore Motor Required for Metaphase Chromosome Alignment. Cell 91, 357–366. [DOI] [PubMed] [Google Scholar]
- Wood KW, Lad L, Luo L, Qian X, Knight SD, Nevins N, Brejc K, Sutton D, Gilmartin AG, Chua PR, et al. (2010). Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc. Natl. Acad. Sci. U. S. A 107, 5839–5844. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Transcriptome sequencing analyses were performed using publicly available software (STAR aligner (Dobin et al., 2013), Picard (http://broadinstitute.github.io/picard/), featureCounts (Liao et al., 2014), SAMtools (Li et al., 2009), Varscan2 (Koboldt et al., 2013), DexSeq (Anders et al., 2012) and Rstudio (version 1.1.423). R packages were obtained from Bioconductor (https://www.bioconductor.org/). RNA sequencing data sets related to Figure 4 have been submitted to NCBI SRA (Sequence Read Archive) and can be found under BioProjectID: PRJNA561192.