

Abstract
Aims
In this exposure–response analysis, the dosing regimen for tildrakizumab, an antibody for treating moderate‐to‐severe chronic plaque psoriasis, was determined using data from 3 randomised controlled trials (P05495/NCT01225731: phase 2b, n = 355; reSURFACE 1/NCT01722331: phase 3, n = 772; reSURFACE 2/NCT01729754: phase 3, n = 1090).
Methods
A maximum drug effect (Emax) logistic‐regression exposure–efficacy model was used to describe the week 12 Psoriasis Area and Severity Index (PASI) responses with average concentration of tildrakizumab during weeks 1–12 (Cavg12) as exposure metric. The impact of covariates (e.g., body weight, region) was tested. Exposure–safety, longitudinal pharmacokinetic–pharmacodynamic and risk–benefit analyses were also conducted.
Results
At week 12, Emax was estimated at 62.2, 37.9 and 14.6% of responders for PASI75/90/100, respectively. Exposure–response curves plateaued at exposures >5 μg mL−1. Heavier subjects had a lower response rate to placebo as measured by PASI75/90/100 than lighter subjects. PASI100 placebo response was less in subjects with higher baseline PASI score and older age. Simulated week 12 PASI75 increased by ≤4% on increasing the dose from 100 to 200 mg every 12 weeks (Q12W). The pharmacokinetic–pharmacodynamic model adequately described the time course of PASI change after treatment in the entire population and in each subject. Risk–benefit profiles were favourable for the 100‐ and 200‐mg doses in different weight subgroups.
Conclusions
Patients with moderate‐to‐severe psoriasis should receive 100‐mg subcutaneous tildrakizumab Q12W. Patients with high body weight (>90 kg) may benefit from a higher dose (200‐mg Q12W).
Keywords: dermatology, pharmacodynamics, pharmacokinetic–pharmacodynamic, psoriasis, modelling and simulation
What is already known about this subject
The safety and efficacy of subcutaneous tildrakizumab, a humanised anti‐interleukin‐23 p19‐specific monoclonal antibody, in the treatment of moderate‐to‐severe chronic plaque psoriasis were demonstrated in phase 2/3 clinical trials. However, dose regimen justification for tildrakizumab is warranted.
High body mass index and weight gain are risk factors for psoriasis, and weight loss may reduce disease symptoms.
Clarity about the exposure–response relationship of tildrakizumab in chronic plaque psoriasis is needed to understand the impact of key subject characteristics, especially body weight, on this relationship.
What this study adds
In the exposure–response model, using week 12 Psoriasis Area and Severity Index responses and average concentration of tildrakizumab from weeks 1 to 12 as the exposure metric, treatment responses plateaued at doses ≥100 mg.
These results indicate that patients with moderate‐to‐severe psoriasis should receive a regimen of 100‐mg subcutaneous tildrakizumab every 12 weeks.
Patients with high body weight (>90 kg) may benefit from 200‐mg subcutaneous tildrakizumab every 12 weeks.
1. INTRODUCTION
Psoriasis is a common, chronic, immune‐mediated inflammatory disease affecting the skin and joints. 1 Plaque psoriasis is the most common type of psoriasis and has a detrimental effect on physical and mental health. 2 It is characterised by inflammatory, red, raised plaques, usually covered by grey or silvery‐white scales. 3 The plaques are often symmetrically distributed and most frequently occur on the elbows, knees, scalp, lower back and in body folds.
Available systemic treatments for moderate‐to‐severe psoriasis include tumour necrosis factor‐α inhibitors (adalimumab, etanercept and infliximab), interleukin (IL)‐17 antagonists (secukinumab, brodalumab and ixekizumab), an IL‐12 and IL‐23 antagonist (ustekinumab), and a phosphodiesterase‐4 inhibitor (apremilast).4, 5 Given the complex and chronic nature of psoriasis, the pursuit for new therapeutic agents continues.
IL‐12 and IL‐23 play important roles in chronic plaque psoriasis 6 and have a shared binding protein, p40. 7 Researchers have speculated that blocking IL‐12p40 could be an effective treatment strategy for psoriasis, acting through control of the type 1 cytokine cascade and IL‐12/IL‐23 production. 8 However, while the anti–IL‐12/23p40 agent briakinumab demonstrated robust efficacy in the treatment of moderate‐to‐severe psoriasis, it had an untoward safety profile. 9 Consequently, development of drugs shifted from targeting IL‐12/23p40 to selective targeting of the IL‐23‐specific p19 subunit, 10 leading to approval of IL‐23p19 antagonists such as guselkumab and tildrakizumab for treating moderate‐to‐severe psoriasis.
Tildrakizumab is a high‐affinity, humanised immunoglobulin G1κ (IgG1κ) monoclonal antibody that specifically binds to IL‐23p19; it does not bind to IL‐12/23p40.11, 12 Pharmacokinetics (PK), safety, bioavailability and clinical activity of tildrakizumab were evaluated among healthy subjects 12 and in subjects with moderate‐to‐severe plaque‐type psoriasis 11 in phase 1 studies. In phase 2b 13 and phase 3 14 clinical trials, subcutaneous (SC) tildrakizumab was safe and efficacious in the treatment of chronic plaque psoriasis. The relationship between exposure and response (efficacy and safety) of tildrakizumab for the treatment of chronic plaque psoriasis was evaluated using data from these trials.
The objectives of the exposure–response analyses and longitudinal PK–pharmacodynamic (PD) model were: to quantify exposure response for week 12 Psoriasis Area and Severity Index (PASI) endpoints; to examine the average concentration of tildrakizumab from weeks 1 to 12 (Cavg12) of treatment and concentration of tildrakizumab at week 12 (C12) as predictors for the exposure–response relationship and establish the clinically relevant tildrakizumab exposure range; to understand the impact of key subject characteristics, especially body weight, on the exposure–response relationship, and to compare projected PASI endpoints for 100‐ and 200‐mg fixed‐dose regimens (100‐ and 200‐mg SC tildrakizumab every 12 weeks [Q12W]) relative to alternative weight‐based regimens. In addition, the longitudinal population PK‐PD of tildrakizumab‐associated changes in PASI responses and projected PASI response (PASI responder defined by 75% [PASI75], 90% [PASI90] and 100% [PASI100] reductions in the PASI score) over time were characterised for the proposed tildrakizumab regimens in the induction and maintenance phases.
2. METHODS
2.1. Data
Pooled analyses were conducted using data from 3 randomised controlled trials: P05495 (NCT01225731: phase 2b, n = 355), 13 reSURFACE 1 (NCT01722331: phase 3, n = 772) 14 and reSURFACE 2 (NCT01729754: phase 3, n = 1090). 14 All 3 trials included subjects aged ≥18 years with predominantly plaque psoriasis for ≥6 months, a PASI score ≥12, psoriasis body surface area involvement ≥10% and a Physician's Global Assessment of moderate‐to‐severe psoriasis at baseline who were candidates for phototherapy or systemic therapy.
P05495, reSURFACE 1 and reSURFACE 2 were 3‐part trials conducted over 72, 64 and 52 weeks, respectively. In part 1 (weeks 0–16) of P05495, subjects were randomised to receive tildrakizumab or placebo. 13 In part 2 (weeks 16–52), subjects were rerandomised based on responder status, and all subjects received active treatment. In part 3 (weeks 52–72), subjects discontinued treatment at week 52 and were followed up for 20 weeks. In part 1 (weeks 0–12) of the reSURFACE trials, subjects were randomised to receive tildrakizumab or placebo (reSURFACE 1) or tildrakizumab, placebo or etanercept (reSURFACE 2). 14 In part 2 (weeks 12–28), subjects in the placebo group were rerandomised to receive tildrakizumab. In part 3 (weeks 28–52 or 64), subjects were rerandomised at week 28 based on responder status to receive tildrakizumab or placebo until week 64 (reSURFACE 1) or week 52 (reSURFACE 2).
Data for all subjects treated with tildrakizumab or placebo with valid exposure and PD data (post‐treatment PASI results) were included in the exposure–response and PK‐PD analyses. The PK‐PD dataset included the same subjects as the exposure–response dataset and 16 additional subjects with baseline information: 15 subjects with no postbaseline data, and 1 subject with 1 unscheduled postdose visit. Subjects treated with etanercept in reSURFACE 2 14 were excluded. Only data from part 1 of the reSURFACE trials 14 were included in the exploratory exposure–safety analysis. A comparison of trial designs, detailing which data were used in which analysis, is provided in Table 1.
TABLE 1.
Comparison of the trial designs
| Study | Part 1 | Part 2 | Part 3 |
|---|---|---|---|
|
P05495 (phase 2b) N = 355 |
Weeks 0–16 tildrakizumab or placebo |
Weeks 16–52 tildrakizumab |
Weeks 52–72 follow up, treatment discontinued |
| ER, PK‐PD and safety analyses | PK‐PD analysis | PK‐PD analysis | |
|
reSURFACE 1 (phase 3) N = 772 |
Weeks 0–12 tildrakizumab or placebo |
Weeks 12–28 tildrakizumab |
Weeks 28–64 tildrakizumab or placebo |
| ER, PK‐PD and safety analyses | PK‐PD analysis | PK‐PD analysis | |
|
reSURFACE 2 (phase 3) N = 1090 |
Weeks 0–12 tildrakizumab, etanercept or placebo |
Weeks 12–28 tildrakizumab |
Weeks 28–52 tildrakizumab or placebo |
| ER, PK‐PD and safety analyses (except patients on etanercept) | PK‐PD analysis | PK‐PD analysis |
ER, exposure–response; PK‐PD, pharmacokinetic–pharmacodynamic.
2.2. Exposure–response analysis
2.2.1. Prior experience
In prior exposure–response analyses of week 16 data from P05495, 13 PASI75 could be modelled using a maximum drug effect (Emax) model. Baseline PASI score, body weight, prior biological treatments, presence of psoriatic arthritis and region (Japan vs non‐Japan) did not have significant effects (P < .001) on model parameters. The current analysis was developed further using this prior experience.
2.2.2. Exposure–response analysis: Efficacy
Two efficacy exposure metrics—Cavg12 and C12—were considered. Cavg12 was calculated using individual PK parameters obtained from a population PK model. 15 In brief, the PK of tildrakizumab was described by a 1‐compartment model with first‐order absorption and elimination kinetics. In subjects with psoriasis, tildrakizumab clearance was 0.32 L/day and volume of distribution was 10.8 L. Raw‐data concentration–time profiles declined in a monophasic manner, indicating a 1‐compartmental model. There was no evidence of target‐mediated drug disposition. Steady‐state was achieved by 16 weeks with the clinical regimen, with 1.1‐fold accumulation in maximum plasma concentration. Body weight was identified as the most significant covariate on the PK; both clearance and volume of distribution increase in subjects with higher body weight.
PASI75, PASI90 and PASI100 responses at week 12 in each of the 3 trials were used for the exposure–response analyses for efficacy. Missing PASI values were imputed using the last observation carried forward approach. An Emax logistic‐regression exposure–response model was used to describe week 12 PASI responses. The conceptual model was as follows:
where i denotes subject, Pr denotes probability and LEC50 is the logarithm of EC50; EC50 is the drug concentration required to achieve 50% of the Emax.
The best exposure metric (Cavg12 vs C12) was evaluated as a predictor of PASI response. If no clear choice was evident, Cavg12 was selected. Alternative structural models included a sigmoid Emax relationship and a linear model, although the latter was deemed unlikely based on a prior analysis of P05495 data. 13
Covariates were tested and were included in the final model if they demonstrated a significant effect. Continuous covariates were included in the exposure–response model using power equations after centring on the median covariate value or a suitable representative value. Categorical covariates, tested if ≥50 subjects belonged to each category, were incorporated into the model as index variables as the exponent of a parameter. Thus, the effect of continuous covariates was modelled as follows:
whereas the effect of categorical covariates was modelled as follows:
where θ is an exposure–response model parameter, Cov is a continuous covariate, X is an indicator variable for a categorical covariate, i is the index for subject, pop is an index denoting the typical value in the population and k cov is a coefficient describing the strength of the covariate effect on the ER parameter.
Thus, the impact of covariates, including body weight, age, sex, race, region (Japan vs non‐Japan), presence of psoriatic arthritis, prior biologic use, prior steroid use, disease duration, baseline PASI and trial, on the exposure–response model was tested using a forward addition (P < .01) and backward elimination (P < .001) approach to develop the final model. If a certain covariate did not have a significant effect in the forward step, it was not tested further.
2.2.3. Exposure–response analysis: Safety
Adverse event (AE) data of the reSURFACE trials were used for exposure–response analyses for safety. For the exploratory exposure–safety analysis, endpoint probabilities binned by concentration quartiles and stratified by dose groups were calculated. AEs of potential interest included severe infections, infections (nasopharyngitis and upper respiratory tract infections [URTIs]), malignancies, nonmelanoma skin cancer, melanoma skin cancer, confirmed extended major adverse cardiovascular event and drug‐related hypersensitivity.
2.2.4. Clinical comparability bounds
Clinical comparability bounds were determined for tildrakizumab exposure as defined by Cavg12 (which is directly linearly correlated with the area under the concentration–time curve [AUC]) relative to the median exposure of the clinical dose of 100 mg SC. The 100‐ and 200‐mg doses had similar (maximal) efficacy and (acceptable) safety across the range of tildrakizumab exposure quartiles. Therefore, the median lowest quartile (Q1) exposure of the 100‐mg dose and median highest quartile (Q4) of the 200‐mg dose were taken as clinical comparability bounds and expressed as fractions of the median exposure of the therapeutic dose (100 mg); i.e., (median Q1 exposure 100 mg)/(median exposure 100 mg) and (median Q4 exposure 200 mg)/(median exposure 100 mg). Any exposures between these ratios (multiples) of median exposure of the clinical dose of 100 mg are expected to yield similar response associated with maximal efficacy and acceptable safety.
2.3. PK‐PD analyses
Prior experience of developing an indirect‐effect PK‐PD model using data from P05495 13 demonstrated a good fit with a model similar to that developed by Salinger et al. 16 Other indirect response models17, 18 were also considered but did not perform as well. Thus, the longitudinal PASI reduction data were described by an indirect‐effect longitudinal PK‐PD model (Figure 1) that used the concentration‐related suppression of plaque formation with a placebo effect on plaque degradation, representing the healing process. Transit compartments were used to describe the delay of PASI response to the treatment. Covariates with significant effects in the exposure–response analysis were also tested in the PK‐PD analysis. The goodness‐of‐fit was assessed by standard diagnostic residual plots. Details of the PK‐PD model are provided in the Appendix.
FIGURE 1.
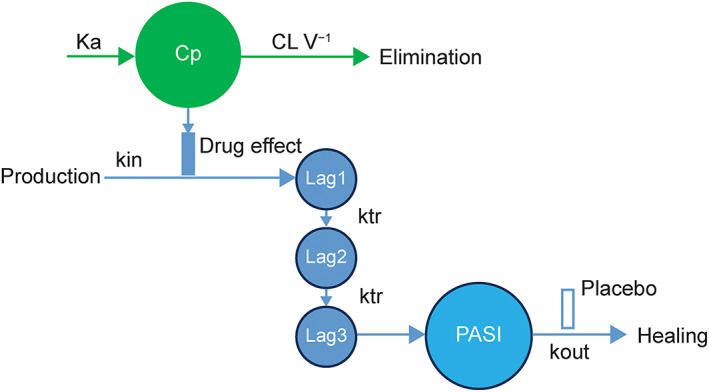
Longitudinal pharmacokinetic–pharmacodynamic model. CL V−1, clearance over volume; Cp, predicted concentration–time course from the population pharmacokinetic model using post hoc parameters; ka, absorption rate constant; kin, zero‐order constant for production of the response; kout, first‐order rate constant for loss of the response; ktr, transit rate constant; Lag1–3, PASI transit compartments 1–3; PASI, Psoriasis Area and Severity Index
2.4. Applications
2.4.1. Simulations
Simulations were used to characterise the PASI response based on population parameters from the PK‐PD/exposure–response models and sampled covariates from the phase 2/3 clinical trial populations. This approach incorporated PK‐PD/exposure–response model variability and parameter uncertainty.
Exposure–response simulations were used to assess advantages, if any, of the weight‐based tildrakizumab regimens (≤90 kg or >90 kg) over 100‐ and 200‐mg fixed‐dose regimens in terms of week 12 PASI response in the general population and different subject subsets. Simulations included the PK fixed‐effect parameters, PK parameter interindividual variability, subject subset‐specific covariates and uncertainties in the exposure–response model parameters. These simulations were performed by simulating 10,000 trials with 10,000 subjects per arm. The impact of body weight, dose regimens (25, 100, 200 and 400 mg Q12W) and region (Japan vs non‐Japan) on week 12 PASI response was assessed.
In the PK‐PD model simulations, tildrakizumab regimens were compared in different subject subgroups, throughout induction and maintenance. Each simulation included 400 subjects and 100 trial replicates; each trial replicate included fixed‐effect model uncertainty. PASI 75/90/100 response rates at weeks 12, 28 and 52 were summarised (median, 5th percentile and 95th percentile).
2.4.2. Integrated risk–benefit analysis
In an integrated risk–benefit analysis, the most frequently occurring AEs relative to the PASI75 responder rate at week 28 for the 100‐ or 200‐mg tildrakizumab doses were compared in different weight subgroups. The percentage of subjects with infections/infestations was paired with the corresponding PASI75 response rate and plotted in a horizontal bar chart to show the risk–benefit of alternate dosing regimen options from parts 1 and 2 of the reSURFACE trials.
2.5. Software
Exposure–response models were developed in R version 3.2.3 (www.r-project.org). The longitudinal PK‐PD model was developed in the nonlinear mixed effects software NONMEM version 7.2 (ICON Development Solutions, Ellicott City, MD, USA). Simulations were performed in R.
2.6. Ethics
All concerned Independent Ethics Committees (IECs), Ethical Review Committees (ERCs), or Institutional Review Boards (IRBs) reviewed and approved the protocols and applicable amendments. These studies conformed to recognised standards. All subjects signed written informed consent. The studies followed the principles of good clinical practice. All drugs/molecular targets conform to the IUPHAR/BPS Guide to PHARMACOLOGY nomenclature classification. 19 Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
3. RESULTS
3.1. Exposure–efficacy response
Both exposure metrics were equally predictive for PASI response, supporting interchangeability of exposure metrics. However, Cavg12 was chosen based on the theoretical grounds that Cavg12, but not C12, reflects the full exposure of the dosing interval during which continuous target engagement may be important to achieve PASI response.
Thus, the exposure–response base model developed to assess week 12 PASI response was as follows:
Model parameters for week 12 PASI response are summarised in Table 2. Emax was estimated at 62.2% for the PASI75 response, at 37.9% for the PASI90 response and at 14.6% for the PASI100 response, and EC50 estimates were 0.36, 0.46 and 0.55 μg mL−1, respectively (Table 2). For a typical 90‐kg subject, the placebo response rate was 5.42% for PASI75. Lower placebo response was observed in heavier subjects and higher placebo response was observed in subjects with lower body weight. Treatment response plateaued at tildrakizumab doses ≥100 mg Q12W. Exposure–response curves flattened markedly after exposure of >5 μg mL−1 (Figure 2).
TABLE 2.
Estimate (90% confidence interval) for exposure–response model parameters (based on week 12 PASI response)
| Model parameter | PASI75 | PASI90 | PASI100 a |
|---|---|---|---|
| Placebo response at 90 kg (%) | 5.42 (3.74, 7.72) | 1.65 (0.86, 3.09) | 0.45 (0.15, 1.51) |
| Emax (%) | 62.16 (58.73, 65.41) | 37.89 (34.04, 41.73) | 14.63 (11.23, 17.93) |
| EC50 (μg mL−1) | 0.36 (0.22, 0.61) | 0.46 (0.25, 0.86) | 0.55 (0.21, 1.37) |
| Placebo response at 56 kg b (%) | 8.40 (5.84, 11.94) | 3.15 (1.67, 5.77) | 0.73 (0.24, 2.51) |
| Placebo response at 131 kg b (%) | 3.77 (2.48, 5.56) | 0.97 (0.49, 1.89) | 0.30 (0.09, 1.04) |
EC50, drug concentration needed to achieve 50% of the maximum drug effect; Emax, maximum drug effect; PASI, Psoriasis Area and Severity Index.
PASI100 model parameters were centred to a baseline PASI score of 18. Subjects with higher baseline PASI responses had lower PASI100 placebo responses. Note that no baseline effect was detected for PASI75 and PASI90; therefore, responses were considered valid across all baseline PASI scores and did not require centring.
Body weight levels (56 kg and 131 kg) represent the 5th and 95th percentiles, respectively.
FIGURE 2.
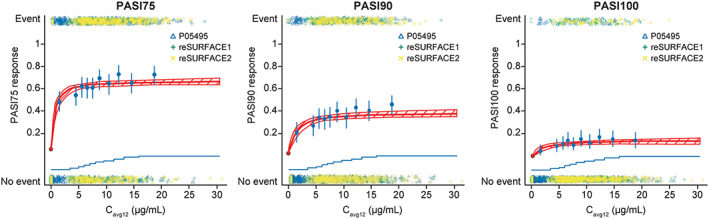
Week 12 PASI exposure–response relationships. Cavg12, average concentrations from weeks 1 to 12; CI, confidence interval; PASI, Psoriasis Area and Severity Index. Blue points are PASI response probabilities (error bars: 95% exact CIs) corresponding to average concentrations (Cavg12), stratified by concentration decile. The blue lines (stair‐steps) represent the cumulative distribution of pharmacokinetic values and the location of each decile. The multicolour points represent individual subject response with (PASI response = yes; top) and without (PASI response = no; bottom) an event. The red line represents the model fit, with shading indicating 95% CIs
Baseline body weight on placebo response was the only significant (predefined significance level of P < .001) covariate in the week 12 PASI75 and PASI90 models. Baseline PASI on placebo response was the only significant covariate in the PASI100 response model. Because body weight was a significant covariate for both PASI75 and PASI90, it was also included in the PASI100 model, although its impact on the model was marginally significant (P = .0031). Age also had a marginally significant impact on the placebo response of PASI100 (P = .0262) and was also included in the final model.
Exposure–efficacy response simulations showed the magnitude of negative correlation between body weight and placebo response for the week 12 PASI responses (Table 3). PASI100 placebo response was less in subjects with higher baseline PASI scores and older age. Among all subpopulations (by body weight and region), modelled PASI75 increased by ≤4% when the tildrakizumab dose was increased from 100 to 200 mg Q12W (Table 3). The proportion of PASI75 responders increased only by ≤2% in the 400‐mg Q12W group compared with the 200‐mg tildrakizumab Q12W group. In contrast, when the dose was reduced from 100 to 25 mg Q12W, modelled PASI75 decreased by 10, 16 and 16% in subjects weighing ≤90, >120 and > 135 kg, respectively.
TABLE 3.
Week 12 PASI response by tildrakizumab dose and by weight and region subgroups (exposure–response simulations)
| Response | Subgroup | 0 mg | 25 mg | 100 mg | 200 mg | 400 mg | Difference between 200‐ and 100‐mg groups | Difference between 400‐ and 200‐ mg groups |
|---|---|---|---|---|---|---|---|---|
| PASI75 | ≤90 kg | 7 | 58 | 68 | 70 | 71 | 2 | 1 |
| >90 kg | 5 | 44 | 57 | 60 | 62 | 3 | 2 | |
| >120 kg | 4 | 34 | 50 | 53 | 55 | 4 | 2 | |
| >135 kg | 3 | 31 | 47 | 51 | 53 | 4 | 2 | |
| >150 kg | 3 | 28 | 44 | 48 | 51 | 4 | 2 | |
| Non‐Japan | 6 | 51 | 63 | 65 | 67 | 2 | 1 | |
| Japan | 7 | 59 | 69 | 71 | 72 | 2 | 1 | |
| PASI90 | ≤90 kg | 2 | 29 | 41 | 44 | 45 | 3 | 2 |
| >90 kg | 1 | 17 | 27 | 30 | 32 | 3 | 2 | |
| >120 kg | 1 | 10 | 19 | 22 | 24 | 3 | 2 | |
| >135 kg | 1 | 9 | 17 | 20 | 22 | 3 | 2 | |
| >150 kg | 1 | 7 | 15 | 18 | 20 | 3 | 2 | |
| Non‐Japan | 2 | 23 | 35 | 37 | 39 | 3 | 2 | |
| Japan | 3 | 31 | 43 | 46 | 47 | 3 | 2 | |
| PASI100 | ≤90 kg | 1 | 9 | 14 | 16 | 17 | 2 | 1 |
| >90 kg | 0 | 5 | 9 | 11 | 12 | 2 | 1 | |
| >120 kg | 0 | 3 | 7 | 8 | 9 | 1 | 1 | |
| >135 kg | 0 | 3 | 6 | 7 | 8 | 1 | 1 | |
| >150 kg | 0 | 2 | 5 | 6 | 7 | 1 | 1 | |
| Non‐Japan | 1 | 7 | 12 | 14 | 15 | 2 | 1 | |
| Japan | 1 | 8 | 13 | 15 | 16 | 2 | 1 |
Data are presented as % of subjects.
PASI, Psoriasis Area and Severity Index.
Japanese subjects had better PASI response than non‐Japanese subjects (Table 3).
No discernible trend of PASI responses by exposure (Cavg12) quartiles was evident (Table 4). Subjects in the corresponding quartiles of the 100‐ and 200‐mg groups had similar PASI response rates.
TABLE 4.
PASI response and AEs by exposure quartile from part 1 of the reSURFACE trials
| Placebo | 100 mg Q1 | 100 mg Q2 | 100 mg Q3 | 100 mg Q4 | 200 mg Q1 | 200 mg Q2 | 200 mg Q3 | 200 mg Q4 | |
|---|---|---|---|---|---|---|---|---|---|
| Efficacy (at week 12) | |||||||||
| n | 350 | 154 | 154 | 152 | 152 | 153 | 153 | 155 | 155 |
| Cavg12 (μg mL−1) | 4.4 (2.1, 5.2) | 5.7 (5.2, 6.4) | 7.0 (6.4, 7.8) | 8.7 (7.8, 16.1) | 8.7 (4.2, 10.4) | 11.5 (10.4, 12.5) | 14.1 (12.5, 15.5) | 17.3 (15.5, 30.6) | |
| PASI75 | 18 (5.9) | 90 (58.4) | 96 (62.3) | 90 (59.2) | 112 (73.7) | 81 (52.9) | 105 (68.6) | 100 (64.5) | 116 (74.8%) |
| PASI90 | 5 (1.6) | 46 (29.9) | 59 (38.3) | 47 (30.9) | 75 (49.3) | 32 (20.9) | 58 (37.9) | 62 (40) | 73 (47.1%) |
| PASI100 | 1 (0.3) | 14 (9.1) | 22 (14.3) | 16 (10.5) | 29 (19.1) | 10 (6.5) | 26 (17) | 21 (13.5) | 24 (15.5%) |
| Safety | |||||||||
| n | 310 | 154 | 154 | 154 | 154 | 156 | 155 | 155 | 156 |
| Cavg12, (μg mL−1) | 0 | 4.4 (2.1, 5.2) | 5.7 (5.2, 6.4) | 7.0 (6.4, 7.8) | 8.7 (7.8, 16.1) | 8.7 (4.2, 10.4) | 11.5 (10.4, 12.5) | 14.1 (12.5, 15.5) | 17.3 (15.5, 30.6) |
| Any AE | 160 (51.6) | 76 (49.4) | 71 (46.1) | 62 (40.3) | 73 (47.4) | 72 (46.2) | 70 (45.2) | 68 (43.9) | 74 (47.4) |
| Infection | 63 (20.3) | 33 (21.4) | 33 (21.4) | 27 (17.5) | 36 (23.4) | 35 (22.4) | 30 (19.4) | 32 (20.6) | 30 (19.2) |
| Severe infection | 1 (0.3) | 1 (0.6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (0.6) | 1 (0.6) | 0 (0) |
| URTI | 30 (9.7) | 19 (12.3) | 18 (11.7) | 17 (11) | 22 (14.3) | 19 (12.2) | 18 (11.6) | 20 (12.9) | 16 (10.3) |
| Malignancies | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (0.6) | 0 (0) | 0 (0) | 1 (0.6) | 0 (0) |
| Nonmelanoma skin cancer | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (0.6) | 0 (0) | 0 (0) | 1 (0.6) | 0 (0) |
| Melanoma skin cancer | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Confirmed extended MACE | 0 (0) | 0 (0) | 1 (0.6) | 0 (0) | 0 (0) | 1 (0.6) | 0 (0) | 0 (0) | 0 (0) |
| Drug‐related hypersensitivity | 1 (0.3) | 0 (0) | 0 (0) | 0 (0) | 1 (0.6) | 1 (0.6) | 0 (0) | 0 (0) | 0 (0) |
PASI response and AEs are presented as n (%) and Cavg12 is reported as median (range).
AE, adverse event; Cavg12, average concentration from weeks 1 to 12; MACE, major adverse cardiovascular event; PASI, Psoriasis Area and Severity Index; Q, quartile; URTI, upper respiratory tract infection.
3.2. Exposure–safety response
The number of AEs observed and AE rates in the 100‐ and 200‐mg tildrakizumab groups were generally similar to those in the placebo groups in part 1 of the reSURFACE trials, 14 although the rate of URTIs was slightly higher in the tildrakizumab groups than in the placebo group (Table 4). No trend in AEs by exposure quartiles during weeks 0 to 12 was observed despite a wide Cavg12 range (Table 4).
3.3. Clinical comparability bounds
Based on the observed lack of dependence on exposure in safety and PASI response (Table 4), the clinical comparability bounds for Cavg12 expressed as ratios were defined by the median of the lowest exposure quartile for the 100‐mg group (4.4 μg mL−1) and the median of the highest exposure quartile for the 200‐mg group (17.3 μg mL−1) relative to the median exposure of the 100‐mg dose (6.4 μg mL−1). Thus, the clinical comparability bounds were 0.7–2.7. Overall, 86.9 and 84.8% of subjects in the analysis had Cavg12 concentrations within the clinical comparability bounds at the 100‐ and 200‐mg dose, respectively.
3.4. Longitudinal PK‐PD model
In the longitudinal PK‐PD model, the maximum placebo response was estimated at 19%, and Emax was set to 100% reduction from baseline in the PASI response (Table 5). The drug concentration needed to achieve EC50 was 0.25 μg mL−1, similar to the EC50 from the exposure–efficacy modelling. Goodness‐of‐fit plots for the final model are shown in Figure 3.
TABLE 5.
Longitudinal pharmacokinetic–pharmacodynamic model parameters
| Parameter | Variable | Estimate | RSE | Between‐subject variability |
|---|---|---|---|---|
| Placebo response (PASI reduction from baseline) | Pmax | 19% | 7.0% | 199% |
| Maximum tildrakizumab effect (PASI reduction from baseline) | Emax | 100% | ‐ | ‐ |
| Potency (μg mL−1) | EC50 | 0.25 | 4.8% | 183% |
| Healing time (wk) | t1/2 kout | 4.2 | 2.6% | 86% |
| Drug effect onset (wk) | 3 × t1/2 ktr | 1.7 | 4.0% | 98% |
| Placebo response at 131 kg (%) a | ‐ | 23% | ‐ | ‐ |
| Placebo response at 56 kg (%) a | ‐ | 14% | ‐ | ‐ |
EC50, drug concentration needed to achieve 50% of the maximum drug effect; Emax, maximum drug effect; kout, first‐order rate constant for the loss of the response; ktr, transit rate constant; PASI, Psoriasis Area and Severity Index; Pmax, maximum placebo response; RSE, relative standard error; t1/2, half‐life.
Body weight levels (56 and 131 kg) represent the 5th and 95th percentiles, respectively.
FIGURE 3.
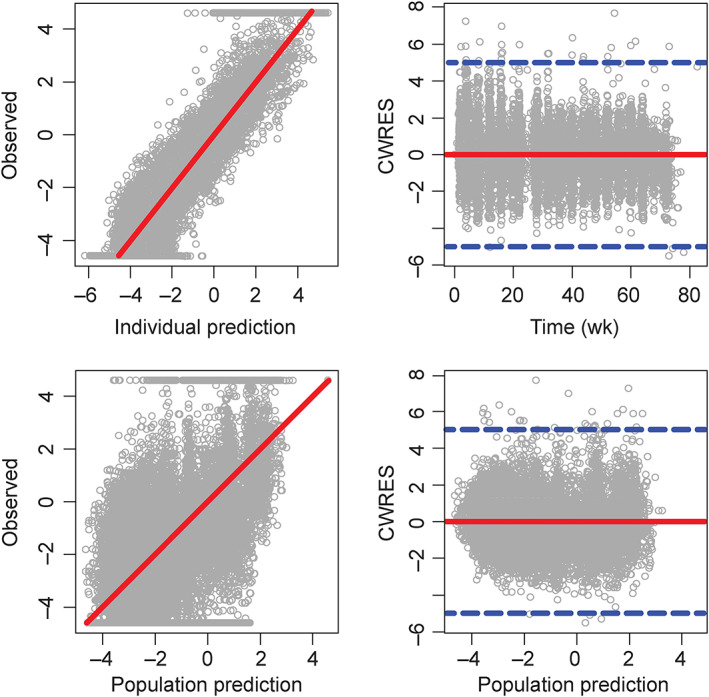
Goodness‐of‐fit plots for the final pharmacokinetic–pharmacodynamic model. Circles are individual observed values; solid red lines are either lines of unity (left panels) or zero horizon lines (right panels). Blue dashed lines at the right panels indicate conditional weighted residuals (CWRES) equal to −5 and + 5. Observed, individual, and population prediction Psoriasis Area and Severity Index (PASI) scores shown in the plots were transformed as PASI/(100 − PASI)
This model adequately described the time course of PASI change after treatment in the entire population and in each subject. More importantly, the model described PASI75 and PASI90 responder probabilities without bias across dose regimens. Across all time points between 12 and 72 weeks, 11.5% of the simulated probabilities were below the 90% confidence interval (CI) of actual responder probabilities and 12.8% were above. However, the model underpredicted the PASI100 response rates.
PK‐PD simulations illustrated PASI responses over time for alternative regimens and in different subject subgroups. Treatment with the 200‐mg dose Q12W throughout the trial period resulted in increased week 52 PASI75, PASI90 and PASI100 responses of 7, 10 and 8%, respectively, as compared with the 100‐mg dose. The benefit of increasing the dose to 200 mg in partial responders at 28 weeks was also investigated: PASI75 response rates increased by 15% at week 52 among subjects with dose increase, defined as subjects who received 100 mg tildrakizumab and had a partial response at 28 weeks but subsequently received 200 mg tildrakizumab instead of continuing the 100‐mg dose. However, at a population level, compared with subjects who received 100 mg tildrakizumab throughout the simulation, PASI75, PASI90 and PASI100 responses only increased by 2, 1 and 0% at week 52, respectively, after a dose increase in partial responders at 28 weeks to the 200‐mg dose (Table 6), due to the fact that partial responders only constituted a small portion of the overall population.
TABLE 6.
Impact of dose increase to 200 mg in partial responders at week 28
| Response | Time (wk) | 100 mg | 200 mg | 100/200 mg dose | Difference between100 mg and 100/200 mg |
|---|---|---|---|---|---|
| PASI75 | 12 | 57.1 | 60.1 | 56.4 | −0.7 |
| PASI75 | 28 | 73.5 | 79.8 | 72.9 | −0.6 |
| PASI75 | 52 | 77.8 | 84.4 | 79.6 | 1.8 |
| PASI90 | 12 | 35.4 | 38.9 | 34.9 | −0.5 |
| PASI90 | 28 | 50.2 | 58.3 | 49.8 | −0.4 |
| PASI90 | 52 | 57.6 | 67.5 | 58.7 | 1.1 |
| PASI100 | 12 | 9.0 | 11.5 | 8.8 | −0.2 |
| PASI100 | 28 | 12.3 | 17.8 | 12.2 | −0.1 |
| PASI100 | 52 | 16.0 | 23.5 | 15.7 | −0.3 |
PASI, Psoriasis Area and Severity Index. Data are presented as % of subjects.
Expected percentage of subjects with PASI response, by regimen, with mean differences between the 100‐mg and 100‐/200‐mg regimens. The 100‐mg regimen means that all responders and partial responders received 100 mg over weeks 0–52, and week 28 nonresponders were discontinued and counted as nonresponders thereafter. The 200‐mg regimen has a similar description as the 100‐mg regimen, except with a different dose. The 100‐/200‐mg regimen means that all subjects started off with 100 mg, partial responders were switched to 200 mg at 28 weeks and nonresponders were discontinued and counted as nonresponders thereafter. No difference between 100 mg vs 100/200 mg is expected at weeks 12 and 28; however, results are presented for completeness. Times selected at 12, 28 and 52 weeks to reflect landmark time points in the phase 3 trials.
Subjects with body weight > 90 kg receiving 200 mg tildrakizumab had similar PASI75, PASI90 and PASI100 responses as those with body weight ≤ 90 kg receiving 100 mg tildrakizumab. However, among subjects with very high body weight (>120 kg), despite receiving 200 mg tildrakizumab, fewer were PASI75, PASI90 and PASI100 responders than in the subgroup with body weight ≤ 90 kg receiving 100 mg tildrakizumab. PASI response rates up to 52 weeks were marginally higher (<3%) in Japanese subjects than in the overall subject population, whereas the rates were slightly lower (<5%) in diabetic subjects than in all subjects with psoriasis (data not shown). Response rates for diabetic subjects with psoriasis were similar to those of the subject subpopulation >90 kg.
3.5. Integrated risk–benefit analysis
Safety events were generally similar between the 100‐ and 200‐mg groups in parts 1 and 2 of the reSURFACE trials, even in those with high body weight. However, in Part 3, subjects from the 100‐mg group who were switched to the 200‐mg dose at week 28 had 55% infections/infestations, while partial responders who continued on the 100‐mg dose had 30% infections/infestations. This difference also persisted in subjects with body weight > 90 kg or > 120 kg.
The risk of adverse events in the system organ class of infections and infestations in parts 1 and 2 of the clinical trials relative to the PASI75 responder rate at week 28 for subjects ≤90 kg receiving 100 mg tildrakizumab was similar to that for subjects >90 kg receiving 200 mg tildrakizumab (Figure 4).
FIGURE 4.
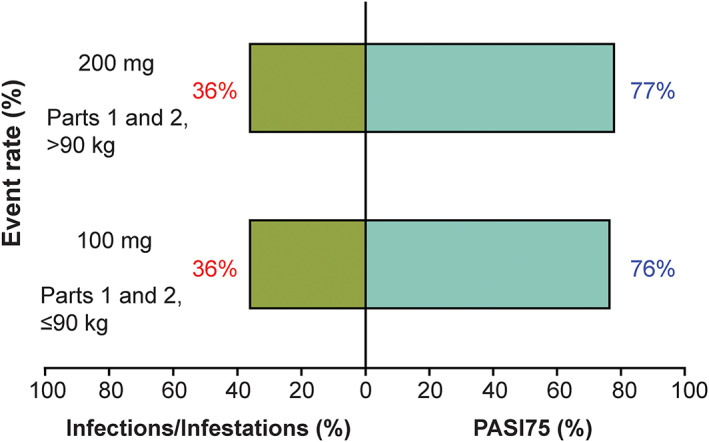
Risk–benefit analysis of the week 28 PASI75 response. PASI, Psoriasis Area and Severity Index
4. DISCUSSION
4.1. Exposure–response analyses
Both the 100‐ and 200‐mg SC tildrakizumab regimens were deemed clinically viable. In the exposure–response model, which described week 12 PASI75, PASI90 and PASI100 responses using Cavg12 as the prespecified exposure metric, treatment responses plateaued at tildrakizumab doses ≥100 mg, indicating that patients with moderate‐to‐severe psoriasis should receive a regimen of 100 mg SC tildrakizumab at weeks 0 and 4 and subsequently every 12 weeks.
Results of the exposure–efficacy response simulations that included body weight indicated that psoriatic patients with body weight >90 kg may benefit from a higher dose of tildrakizumab (200 mg; 2 100‐mg SC injections) at weeks 0 and 4 and subsequently every 12 weeks. In the underlying population PK model providing post hoc parameters for the current analysis, 15 high body weight was associated with decreased tildrakizumab exposure. Specifically, extremes in body weight (range: 40.6–222.2 kg) were positively correlated to a −53 to +163% change in clearance and to a −43 to +107% change in volume of distribution compared with subjects with median body weight (85 kg). In addition to the PK effect, body weight also impacted placebo PASI responses, which has important clinical implications given the association between obesity and psoriasis. Indeed, higher body mass index 20 and weight gain 20 are risk factors for psoriasis, and weight loss may reduce disease symptoms.21, 22
In a previous study, the PK of tildrakizumab was similar in Japanese, Caucasian and Chinese subjects matched by age, sex, height, body weight and body mass index. 23 Furthermore, in the present study, the Japan region had no impact on the PASI response as a covariate. However, marginally higher PASI responses were observed in Japanese subjects because of body weight differences, which affected tildrakizumab PK and placebo response. Similarly, body weight differences may also result in lower response in diabetic subjects.
Overall, tildrakizumab was well tolerated. Numbers and rates of AEs were generally comparable between the tildrakizumab and placebo groups, except for slightly more URTIs (~2%) in the tildrakizumab groups. No consistent trend was observed in AEs between 0 and 12 weeks of treatment across exposure quartiles, starting from the lowest exposure quartile of the 100‐mg group to the highest exposure quartile of the 200‐mg group.
4.2. Longitudinal PK‐PD analyses
An indirect‐effect PK‐PD model with drug suppression of plaque formation and placebo‐induced healing rate was used to characterise longitudinal PASI reduction over 72 weeks. The EC50 was estimated at 0.25 μg mL−1, similar to the exposure–response model. Administering the 200‐ vs 100‐mg dose throughout the trial period increased the week 52 PASI75 response rates by 7% and PASI90 response rates by 10%. Thus, 200 mg did provide an advantage over 100 mg in certain patients. Among partial responders that switched from the 100‐mg to the 200‐mg dose, the PASI75 response rates were increased by 15% at week 52. However, the net impact on the overall population was an increase of 2% because the fraction of partial responders was relatively small. Starting subjects with high body weight (>90 kg) on 200 mg tildrakizumab corrected the PASI response to match subjects with lower body weight (≤90 kg) who received 100 mg tildrakizumab; however, this scenario did not hold true for subjects with very high body weight (>120 or >150 kg).
Japanese psoriasis subjects had marginally better response rates (<3%) and diabetic psoriasis subjects had marginally lower response rates (<5%) than the overall population, presumably due to body weight differences. It therefore appears that Japanese subjects and diabetic subjects are not markedly different from the reference population.
4.3. Risk–benefit profile
Overall, subjects ≤90 kg who received the 100‐mg dose and subjects >90 kg who received the 200‐mg dose had similar risk–benefit profiles over a 28‐week treatment period, suggesting that patients with moderate‐to‐severe psoriasis with body weight ≤90 kg should receive a 100‐mg SC Q12W tildrakizumab regimen (1 100‐mg injection), whereas those >90 kg may benefit from a dose of 200 mg (2 100‐mg injections).
4.4. Strengths and limitations
The large sample size, robust methodology of primary phase 2 and 3 trials and rigorous analysis as per the predecided statistical analysis plan are the strengths of this study. Nevertheless, it must be noted that the exposure–response model was based on data at 12 weeks, a relatively short period of treatment, during which all efficacy or safety responses may not be evident. Integrated risk–benefit analysis evaluated the risk vs benefits of the treatment at 28 weeks. The PASI100 probabilities were generally underpredicted in the PK‐PD model. Despite this limitation, the PASI PK‐PD model brings into play all dose changes, washout profiles, the entire 0–72‐week PASI time course and the continuum of PASI responses and provides a robust evaluation of the overall profile of tildrakizumab that can be used to make inferences for dose recommendations.
The simulations from the PK‐PD model were based on an induction and maintenance treatment phase. However, subject drop‐out was not taken into account, and therefore the simulations are not true trial simulations. Nonetheless, the simulation‐based dose recommendation is valid and appropriate because dropout rates were low and similar across dose groups.
5. CONCLUSIONS
High body weight (>90 kg) was correlated with decreased tildrakizumab exposure and decreased PASI response. A 100‐mg SC Q12W dose regimen was found to be efficacious for subjects with moderate‐to‐severe psoriasis and a body weight ≤90 kg, whereas a 200‐mg SC dose may be beneficial for subjects with high body weight (>90 kg). Results of the integrated risk–benefit analysis support the use of a tildrakizumab SC regimen of 100 mg (one 100‐mg injection) Q12W vs 200 mg (two 100‐mg injections) Q12W in patients with moderate‐to‐severe psoriasis with a body weight ≤90 kg vs >90 kg, respectively.
COMPETING INTERESTS
L.W. is an employee of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. P.J., T.K., R.W. and H.L. are consultants paid for by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. They also consult for numerous other pharmaceutical companies.
CONTRIBUTORS
P.J. developed the population PK model (provided post hoc PK parameters) and coauthored and reviewed the manuscript. T.K. provided scientific supervision and interpretation of results and coauthored and reviewed the manuscript. L.W. provided scientific supervision and interpretation of results and reviewed the manuscript. R.W. developed the PK‐PD model, provided scientific supervision and interpretation of results and co‐authored and reviewed the manuscript. H.L. developed the exposure–response model and coauthored and reviewed the manuscript.
ACKNOWLEDGEMENTS
Medical writing and/or editorial assistance was provided by Vidula Bhole, MD, MHSc, and Maribeth Bogush, PhD, of Cactus Communications. This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
APPENDIX 1.
Details of the pharmacokinetic–pharmacodynamic model
where i denotes subject, Pmax is the maximum placebo response and j denotes time. The plaque formation rate is represented by kin. PASI is the fraction of baseline, which ranges from 0 to 1. The plaque degradation rate is represented by kout. The variables PTR1, PTR2 and PTR3 reference 3 sequential PASI transit compartments, each with transit rate ktr. The drug effect is a standard Emax model, where Cpij is the prediction from the population pharmacokinetic model using post hoc parameters. The placebo effect is an initial increase in healing. The initial conditions of the above differential equations are as follows:
The formation rate kin was set equal to the degradation rate kout, to put the system into steady‐state at baseline.
Subject‐specific parameters (Pmax, EC50, kout and ktr) were modelled as log‐normally distributed with a diagonal covariance matrix. No variability was put on Emax; it was not possible to estimate subject‐specific variability on both Pmax and Emax.
The residual error model was as follows:
Where PASIm refers to the measured PASI value, and the residual error (εij) is normally distributed with mean 0. Both PASIm and PASI were truncated to be between 0.01 and 0.99, to enable use of the logit‐transform, which was needed to describe the skewed PASI distribution, with frequent data measurements near 0.
The goodness‐of‐fit was assessed by standard diagnostic residual plots:
Histograms of interindividual variability (ETA) estimates with mean 0 and variance ω2
Pairwise plots of individual ETA estimates
Box‐plots of ETAs vs dose and trial
Population and individual predictions vs observations
Weighted residuals vs time and population predictions
Kerbusch T, Li H, Wada R, Jauslin PM, Wenning L. Exposure–response characterisation of tildrakizumab in chronic plaque psoriasis: Pooled analysis of 3 randomised controlled trials. Br J Clin Pharmacol. 2020;86:1795–1806. 10.1111/bcp.14280
This work is based on a pooled analysis of data from 3 randomised controlled trials: P05495 (NCT01225731: phase 2b, n = 355), 13 reSURFACE 1 (NCT01722331: phase 3, n = 772) 14 and reSURFACE 2 (NCT01729754: phase 3, n = 1090). 14 Primary results of these 3 trials have already been published. Dr Kerbusch, the corresponding author, has led the pooled analysis, and he can be considered as the PI for this paper.
DATA AVAILABILITY STATEMENT
Merck & Co., Inc.'s data sharing policy, including restrictions, is available at http://engagezone.merck.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the EngageZone site or via email to dataaccess@merck.com.
REFERENCES
- 1. Parisi R, Symmons DP, Griffiths CE, Ashcroft DM, on behalf of the Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) project team . Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol. 2013;133(2):377‐385. [DOI] [PubMed] [Google Scholar]
- 2. Lewis‐Beck C, Abouzaid S, Xie L, Baser O, Kim E. Analysis of the relationship between psoriasis symptom severity and quality of life, work productivity, and activity impairment among patients with moderate‐to‐severe psoriasis using structural equation modeling. Patient Prefer Adherence. 2013;7:199‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Langley RG, Krueger GG, Griffiths CE. Psoriasis: epidemiology, clinical features, and quality of life. Ann Rheum Dis. 2005;64(Suppl 2):ii18‐ii23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feldman SR, Goffe B, Rice G, et al. The challenge of managing psoriasis: unmet medical needs and stakeholder perspectives. Am Health Drug Benefits. 2016;9(9):504‐513. [PMC free article] [PubMed] [Google Scholar]
- 5. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. 2017;140(3):645‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schurich A, Raine C, Morris V, Ciurtin C. The role of IL‐12/23 in T cell‐related chronic inflammation: implications of immunodeficiency and therapeutic blockade. Rheumatology (Oxford). 2018;57:246‐254. [DOI] [PubMed] [Google Scholar]
- 7. Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL‐12p40 to form a cytokine, IL‐23, with biological activities similar as well as distinct from IL‐12. Immunity. 2000;13(5):715‐725. [DOI] [PubMed] [Google Scholar]
- 8. Toichi E, Torres G, McCormick TS, et al. An anti‐IL‐12p40 antibody down‐regulates type 1 cytokines, chemokines, and IL‐12/IL‐23 in psoriasis. J Immunol. 2006;177(7):4917‐4926. [DOI] [PubMed] [Google Scholar]
- 9. Gordon KB, Langley RG, Gottlieb AB, et al. A phase III, randomized, controlled trial of the fully human IL‐12/23 mAb briakinumab in moderate‐to‐severe psoriasis. J Invest Dermatol. 2012;132(2):304‐314. [DOI] [PubMed] [Google Scholar]
- 10. Puig L. The role of IL 23 in the treatment of psoriasis. Expert Rev Clin Immunol. 2017;13(6):525‐534. [DOI] [PubMed] [Google Scholar]
- 11. Kopp T, Riedl E, Bangert C, et al. Clinical improvement in psoriasis with specific targeting of interleukin‐23. Nature. 2015;521(7551):222‐226. [DOI] [PubMed] [Google Scholar]
- 12. Khalilieh S, Hodsman P, Xu C, Tzontcheva A, Glasgow S, Montgomery D. Pharmacokinetics of tildrakizumab (MK‐3222), an anti‐IL‐23 monoclonal antibody, after intravenous or subcutaneous administration in healthy subjects. Basic Clin Pharmacol Toxicol. 2018;123(3):294‐300. [DOI] [PubMed] [Google Scholar]
- 13. Papp K, Thaçi D, Reich K, et al. Tildrakizumab (MK‐3222), an anti‐interleukin‐23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo‐controlled trial. Br J Dermatol. 2015;173(4):930‐939. [DOI] [PubMed] [Google Scholar]
- 14. Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390(10091):276‐288. [DOI] [PubMed] [Google Scholar]
- 15. Jauslin P, Kulkarni P, Wada R, et al. Population pharmacokinetic modeling of tildrakizumab (MK‐3222), an anti‐interleukin‐23‐p19 monoclonal antibody, in healthy volunteers and subjects with psoriasis. American Academy of Dermatology Conference San Diego, California 2018. p. Poster Abstract #6721. [DOI] [PubMed]
- 16. Salinger DH, Endres CJ, Martin DA, Gibbs MA. A semi‐mechanistic model to characterize the pharmacokinetics and pharmacodynamics of brodalumab in healthy volunteers and subjects with psoriasis in a first‐in‐human single ascending dose study. Clin Pharmacol Drug Dev. 2014;3(4):276‐283. [DOI] [PubMed] [Google Scholar]
- 17. Tham LS, Tang CC, Choi SL, Satterwhite JH, Cameron GS, Banerjee S. Population exposure‐response model to support dosing evaluation of ixekizumab in patients with chronic plaque psoriasis. J Clin Pharmacol. 2014;54(10):1117‐1124. [DOI] [PubMed] [Google Scholar]
- 18. Zhou H, Hu C, Zhu Y, et al. Population‐based exposure‐efficacy modeling of ustekinumab in patients with moderate to severe plaque psoriasis. J Clin Pharmacol. 2010;50(3):257‐267. [DOI] [PubMed] [Google Scholar]
- 19. Alexander SPH, Kelly E, Marrion NV, et al. The concise guide to PHARMACOLOGY 2017/18: overview. Br J Pharmacol. 2017;174:S1‐S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Setty AR, Curhan G, Choi HK. Obesity, waist circumference, weight change, and the risk of psoriasis in women: Nurses' Health Study II. Arch Intern Med. 2007;167(15):1670‐1675. [DOI] [PubMed] [Google Scholar]
- 21. Romero‐Talamás H, Aminian A, Corcelles R, Fernandez AP, Schauer PR, Brethauer S. Psoriasis improvement after bariatric surgery. Surg Obes Relat Dis. 2014;10(6):1155‐1159. [DOI] [PubMed] [Google Scholar]
- 22. Hossler EW, Wood GC, Still CD, Mowad CM, Maroon MS. The effect of weight loss surgery on the severity of psoriasis. Br J Dermatol. 2013;168(3):660‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zandvliet A, Glasgow S, Horowitz A, et al. Tildrakizumab, a novel anti‐IL‐23 monoclonal antibody, is unaffected by ethnic variability in Caucasian, Chinese, and Japanese subjects. Int J Clin Pharmacol Ther. 2015;53(02):139‐146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Merck & Co., Inc.'s data sharing policy, including restrictions, is available at http://engagezone.merck.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the EngageZone site or via email to dataaccess@merck.com.