

Abstract
Aim
Preclinical evidence suggests that oxidized macrophage migration inhibitory factor (oxMIF) may be involved in carcinogenesis. This phase 1 study (NCT01765790) assessed the safety, tolerability, pharmacokinetics and antitumour activity of imalumab, an oxMIF inhibitor, in patients with advanced cancer using ‘3 + 3’ dose escalation.
Methods
In Schedule 1, patients with solid tumours received doses from 1 to 50 mg/kg IV every 2 weeks. In Schedule 2, patients with metastatic colorectal adenocarcinoma, non‐small‐cell lung, or ovarian cancer received weekly doses of 10 or 25 mg/kg IV (1 cycle = 28 days). Treatment continued until disease progression, unacceptable toxicity, dose‐limiting toxicity, or withdrawal of consent.
Results
Fifty of 68 enrolled patients received imalumab. The most common treatment‐related adverse events (TRAEs) included fatigue (10%) and vomiting (6%); four grade 3 serious TRAEs (two patients) occurred. The dose‐limiting toxicity was allergic alveolitis (one patient, 50 mg/kg every 2 weeks). The maximum tolerated and biologically active doses were 37.5 mg/kg every 2 weeks and 10 mg/kg weekly, respectively. Of 39 assessed patients, 13 had stable disease (≥4 months in 8 patients).
Conclusions
Imalumab had a maximum tolerated dose of 37.5 mg/kg every 2 weeks in patients with advanced solid tumours, with a biologically active dose of 10 mg/kg weekly. Further investigation will help define the role of oxMIF as a cancer treatment target.
Keywords: clinical trials, pharmacokinetics, phase I
What is already known about this subject
The oxidized form of macrophage migration inhibitory factor (oxMIF) appears to be associated with sites of inflammation and solid tumours.
Preclinical studies of imalumab, a monoclonal anti‐oxMIF antibody, showed promising antitumor activity in vitro and in vivo.
What this study adds
In patients with advanced solid tumours, the dose‐limiting toxicity was grade 3 allergic alveolitis (one patient, 50 mg/kg every 2 weeks) and the maximum tolerated dose was 37.5 mg/kg every 2 weeks or 25 mg/kg weekly.
Stable disease was achieved by 26% of the patient population.
1. INTRODUCTION
Macrophage migration inhibitory factor (MIF) is a pleiotropic, pro‐inflammatory cytokine that plays a key role in regulating innate immunity, especially macrophage functionality and differentiation. 1 , 2 It is widely and constitutively expressed by immune and nonimmune cells, and is also present in the circulation of healthy individuals. 3 , 4 MIF has been implicated in the pathogenesis of several inflammatory diseases 3 , 5 and tumour growth promotion. 3 , 6 Compared with healthy people, patients with cancer can have higher levels of circulating MIF and overexpression of MIF in tumour tissue, which have been associated with high tumour burden and grade, increased metastasis risk and poor prognosis. 7 , 8 , 9 , 10 , 11 , 12 Neutralizing anti‐MIF antibodies inhibited tumour growth and angiogenesis in animal models of B cell lymphoma, 13 prostate cancer 14 and colon cancer. 15
MIF occurs in two conformational isoforms: oxidized MIF (oxMIF) and reduced MIF (redMIF). 5 RedMIF is predominantly detected in plasma from healthy individuals, 5 whereas oxMIF is detected in patients with acute and chronic inflammatory diseases or solid tumours. 5 , 16 MIF oxidation has been reported by several groups, with modifications occurring at different sites resulting in functional implications including loss of tautomerase activity (inhibiting pro‐inflammatory activities of MIF), interference with CD74 binding (preventing signalling and co‐activation of CD44), association with disease, regulation of activated B and T cells, increased cardioprotective properties, and decreased activation of ERK1/2 and AKT signalling. 17 OxMIF is specifically expressed in tumour types including colorectal carcinoma (CRC), non‐small cell lung cancer (NSCLC) and ovarian cancer. 16
Imalumab (BAX69) is a recombinant, fully human, monoclonal antibody that binds specifically to oxMIF. 4 It is specific for a β‐sheet structure within MIF including a highly conserved catalytic motif (57Cys‐Ala‐Leu‐Cys60) of the thiol protein oxidoreductase, which is linked to the biologic function of MIF. 4 , 18 , 19 In preclinical studies, imalumab and two similar, specific anti‐oxMIF antibodies were shown to interfere with key signalling pathways involved in cell survival and proliferation. 16 , 20 These antibodies inhibited MIF‐induced phosphorylation of ERK1/2 and AKT, promoted apoptosis through activation of caspase‐3 in prostate cancer cells and led to inhibition of PC3 xenografts in a dose‐dependent manner. 20 Additionally, they sensitized prostate and ovarian cancer cell lines to cytotoxic drugs. 16
Here, we report results from the first‐in‐human phase I study to assess the safety, dose‐limiting toxicity (DLT), maximum tolerated dose (MTD), pharmacokinetics, pharmacodynamics and antitumour activity of imalumab in patients with advanced solid tumours.
2. METHODS
2.1. Patient population
Patients were included if they were aged ≥18 years; had an anticipated life expectancy of >3 months at screening; histologically confirmed malignant solid tumour (Schedule 1) or histologically or cytologically confirmed metastatic CRC (mCRC), NSCLC (mNSCLC), or ovarian cancer (Schedule 2); had refractory tumours, or had progressed on, were considered medically unsuitable for, or refused standard of care treatment; had measurable or evaluable disease, defined by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1; had an Eastern Cooperative Oncology Group (ECOG) performance status ≤1; had adequate haematological, renal, and liver function; and (for Schedule 2) had a tumour amenable to biopsy and willingness to undergo a biopsy before and at least once after treatment.
Key exclusion criteria were known brain tumours or central nervous system metastases; uncontrolled hypertension or clinically significant cardiovascular disease; any antitumour therapy within 4 weeks (6 weeks for nitrosoureas, mitomycin C) before starting imalumab; and previous treatment‐related toxicity not recovered to <grade 2 (according to National Cancer Institute Common Terminology Criteria for Adverse Events [NCI CTCAE] v4.03).
Patients provided informed consent before entering into the study (ClinicalTrials.gov identifier: NCT01765790). The study was conducted in accordance with the International Conference on Harmonization Guideline for Good Clinical Practice E6 (April 1996), Title 21 of the US Code of Federal Regulations, the European Clinical Trial Directive (2001/20/EC and 2005/28/EC), and applicable national and local regulatory requirements. The study was approved by the relevant ethics committees.
2.2. Study design and treatment
This was a multicentre, open‐label, dose‐escalation phase I study evaluating two schedules of imalumab using a classic ‘3 + 3’ design in Schedule 1 (Supporting Information Figure S1). The study initiated with Schedule 1 to assess the safety, pharmacokinetic and pharmacodynamic endpoints in patients with malignant solid tumours receiving the study drug every 2 weeks (Q2W). Schedule 2 started after Schedule 1 completion to evaluate a weekly (QW) dose escalation in patients with mCRC, mNSCLC and metastatic ovarian cancer refractory to or failing standard treatments (platinum‐free interval and platinum resistance were not considered for metastatic ovarian cancer). This schedule was designed based on results of early pharmacokinetic analyses in Schedule 1, which indicated that imalumab had a shorter than expected half‐life (t 1/2). The typical t 1/2 of Immunoglobulin G (IgG)‐derived molecules such as imalumab is 2‐3 weeks. 21
In Schedule 1, imalumab was administered intravenously every 2 weeks (days 1 and 15) at escalating planned dose levels of 1, 3, 10, 25, 37.5 and 50 mg/kg body weight. The starting dose of 1 mg/kg represents a 25‐fold safety factor versus the repeated dose no‐observed‐adverse‐effect‐level of 25 mg/kg in cynomolgus monkeys from nonclinical studies. In Schedule 2, imalumab was administered intravenously QW (days 1, 8, 15 and 22) at planned dose levels of 10, 25 and 37.5 mg/kg body weight. In Schedule 2, an expansion cohort (Supporting Information Figure S1) was planned to enrol up to 24 additional patients with mCRC, mNSCLC and metastatic ovarian cancer (eight or fewer patients per tumour type) treated at or below the MTD for this schedule. Cycles were repeated every 28 days until disease progression, unacceptable toxicity, DLT or consent withdrawal.
The primary objective was to determine the safety and tolerability of imalumab, including the MTD and immunogenicity, in patients with malignant solid tumours (Schedule 1) and in patients with mCRC, mNSCLC or metastatic ovarian cancer (Schedule 2). Secondary objectives were to determine the pharmacokinetics of imalumab, explore pharmacodynamic markers indicative of antitumour and anti‐MIF activity, explore tissue penetration and target binding of imalumab in tumour tissue, and to assess the antitumour activity of imalumab, if any.
2.3. Safety assessments
NCI CTCAE v4.03 criteria were used to assess the severity of adverse effects (AEs) and their causal relationship to imalumab. DLT was defined as any treatment‐emergent ≥grade 3 nonhematologic toxicity, grade 4 hematologic toxicity, grade 3 febrile neutropenia, or grade 3 thrombocytopenia associated with bleeding. MTD was defined as the highest dose level examined at which less than one‐third of evaluable patients experienced a DLT during the first treatment cycle.
2.4. Assessments of immunoglobulin against imalumab
Plasma samples obtained at baseline and after exposure were tested for the presence of total immunoglobulin against imalumab (binding antibodies) using an internally validated homogeneous bridging electrochemiluminescence assay (IPM Biotech GmbH [now part of Bioagilytix Europe GmbH] Hamburg, Germany). Samples with confirmed positive antibodies were analysed for the presence of neutralizing antibodies against imalumab using an internally validated competitive ligand binding assay (IPM Biotech GmbH).
2.5. Pharmacokinetics and pharmacodynamics
Blood samples to determine the pharmacokinetics of imalumab and the measurement of circulating oxMIF (oxMIF ± bound imalumab) and circulating total MIF levels were collected in Schedule 1 at baseline; in cycle 1, pre and post infusion on day 1 and at 1, 4, 8, 24, 72 and 168 hours after infusion, as well as pre infusion on day 15; in cycle 2, pre infusion on day 1, pre and post infusion on day 15, and at 1, 4, 8, 24, 72 and 168 hours after infusion; pre infusion on day 1 of all subsequent cycles; and at the end of treatment.
In Schedule 2, blood samples were collected at baseline; in cycle 1, pre infusion on days 1, 8, 15 and 22, as well as immediately post infusion on day 1 and at 1, 4, 8, 24 and 72 hours after this infusion; in cycle 2, pre infusion on days 1, 8, 15 and 22, and post infusion on day 15, and at 1, 4, 8, 24 and 72 hours after this infusion; pre infusion on day 1 of all subsequent cycles; and at the end of treatment.
Imalumab concentration was determined by a specific anti‐MIF enzyme‐linked immunosorbent assay. Briefly, imalumab was captured by precoated MIF and detected after blocking and washing steps by a rabbit antihuman IgG peroxidase conjugate. Imalumab was quantified against an internal reference standard.
Details regarding precision, accuracy and sensitivity of the assays used in both schedules are included in the Supporting Information data S1.
Pharmacodynamic markers indicative of imalumab antitumour and anti‐MIF activities were explored by assessing tumour response, tissue penetration and target occupancy. Imalumab tissue penetration and target occupancy were investigated in patients from Schedule 2 by analysing levels of circulating oxMIF, circulating total MIF and imalumab in tumour tissue from metastatic lesions obtained pre and post treatment. Biopsies were embedded in optimal cutting temperature compound and frozen, transferred to and distributed by Quest Diagnostics (Madison, New Jersey) and cryosectioned by BioQuant (Heidelberg, Germany). Separate, consecutive tissue sections from tumour biopsies were stained for imalumab and oxMIF by immunohistochemistry at Baxalta Innovations GmbH (a Takeda company, Vienna, Austria). Details of pathological review and digital image analyses are summarized in the Supporting Information data S1.
Tumour measurement was assessed at baseline, on cycle 2, day 28, at the end of every subsequent even‐numbered cycle thereafter, and at the end of the study, with tumour response assessed by investigators using RECIST v1.1.
2.6. Statistical analysis
The enrolled analysis set included all enrolled patients, the safety population comprised all patients who received ≥1 dose of imalumab and the full analysis dataset included all patients who received ≥1 dose of imalumab and had pharmacodynamic data at baseline and at least one time point after treatment. The pharmacokinetic analysis dataset included all patients who received at least one scheduled dose of imalumab and provided one or more evaluable post‐dose concentration (ie, concentration above the lower limit of quantitation of the assay).
Best overall response (assessed using RECIST v1.1) was summarized by treatment group. Plasma concentrations of imalumab, oxMIF and total MIF and pharmacokinetic parameters were summarized using descriptive statistics. Individual subject concentration‐time data were graphically presented by treatment group on linear and semilogarithmic scales.
All data processing, analyses and summaries used SAS® software package (Version 9.3 or higher). Pharmacokinetic parameters were derived using noncompartmental methods with Phoenix® WinNonlin® Version 6.4 (Certara L.P., Princeton, NJ, USA) or SAS® Version 9.2, or higher (SAS Institute, Inc., Cary, NC, USA).
3. RESULTS
3.1. Patients and treatment
From June 2012 to August 2016, 68 patients were enrolled; 50 patients received at least one dose of imalumab and were included in the safety, full and pharmacokinetics analysis sets (Figure 1). Reasons for discontinuation of enrolled patients prior to treatment included screening failure, withdrawal by physician, worsening performance status (clinical progression) and one death. Patient demographics are presented in Table 1. The median age was 61.5 years (range 40‐87), 54% (n = 27) of patients were female. Most patients were Caucasian (90%, n = 45). Primary diagnoses included CRC (50%, n = 25), ovarian carcinoma (20%, n = 10) and NSCLC (14%, n = 7). Patients had been heavily pretreated with chemotherapy, with 84% (n = 42) of patients having received three or more prior regimens. In addition, 34% (n = 17) of patients had received one and 10% (n = 5) of patients had received two prior radiotherapy regimens.
FIGURE 1.
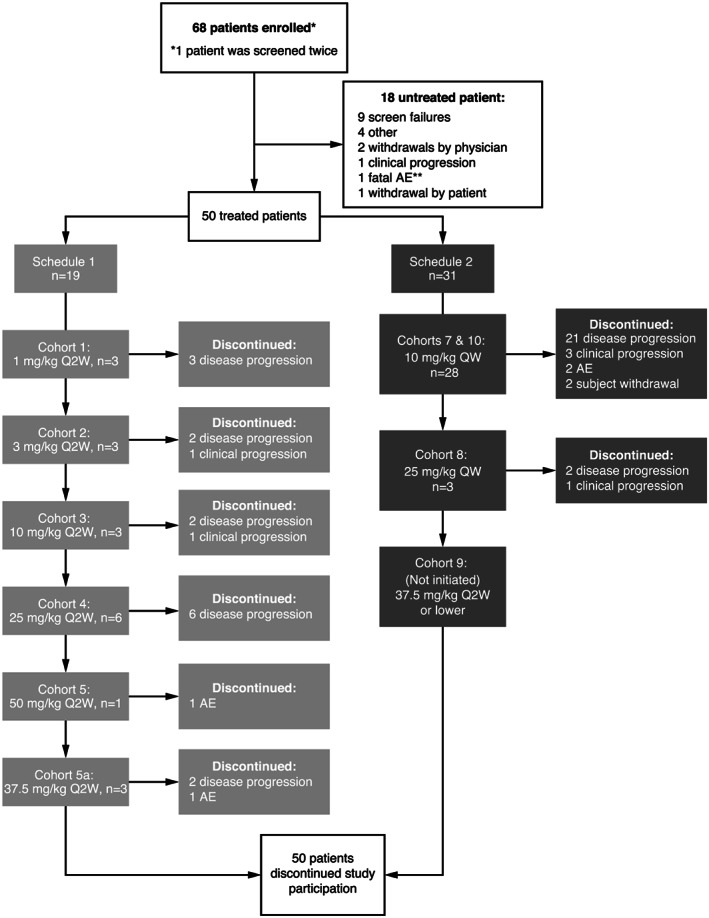
Disposition of patients in both schedules and all cohorts of the phase 1 imalumab study. Enrolled patients were those who had signed an informed consent form. AE, adverse event; Q2W, every two weeks; QW, every week
TABLE 1.
Patient demographics and baseline characteristics
| Characteristic | All patients (n = 50) a | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Median age, years (range) | 61.5 (40–87) | ||||||||
| Gender, n (%) | |||||||||
| Male | 23 (46) | ||||||||
| Female | 27 (54) | ||||||||
| Race, n (%) | |||||||||
| Caucasian | 45 (90) | ||||||||
| African American | 4 (8) | ||||||||
| Multiple | 1 (2) | ||||||||
| Ethnicity, n (%) | |||||||||
| Hispanic/Latino | 12 (24) | ||||||||
| Not Hispanic/Latino | 38 (76) | ||||||||
| Median weight, kg (range) | 73.75 (45.9–158.6) | ||||||||
| Median BMI, kg/m2 (range) | 25.82 (17.4–49.0) | ||||||||
| Schedule 1 | Schedule 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 mg/kg Q2W (n = 3) | 3 mg/kg Q2W (n = 3) | 10 mg/kg Q2W (n = 3) | 25 mg/kg Q2W (n = 6) | 37.5 mg/kg Q2W (n = 3) | 50 mg/kg Q2W (n = 1) | 10 mg/kg QW (n = 28) | 25 mg/kg QW (n = 3) | All patients (N = 50) | |
| ECOG performance status, n (%) | |||||||||
| 0 | 2 (67) | 3 (100) | 0 | 3 (50) | 2 (67) | 1 (100) | 11 (39) | 2 (67) | 24 (48) |
| 1 | 1 (33) | 0 | 3 (100) | 3 (50) | 1 (33) | 0 | 17 (61) | 1 (33) | 26 (52) |
| Primary diagnosis, n (%) | |||||||||
| CRC | 2 (67) | 0 | 1 (33) | 1 (17) | 3 (100) | 1 (100) | 14 (50) | 3 (100) | 25 (50) |
| NSCLC | 1 (33) | 0 | 0 | 0 | 0 | 0 | 6 (21) | 0 | 7 (14) |
| Ovarian carcinoma | 0 | 1 (33) | 0 | 1 (17) | 0 | 0 | 8 (29) | 0 | 10 (20) |
| Other | 0 | 2 (67) | 2 (67) | 4 (67) | 0 | 0 | 0 | 0 | 8 (16) |
| Number of prior chemotherapy regimens, n (%) | |||||||||
| <3 | 0 | 0 | 0 | 0 | 1 (33) | 1 (100.0) | 6 (21) | 0 | 8 (16) |
| ≥3 | 3 (100) | 3 (100) | 3 (100) | 6 (100) | 2 (67) | 0 | 22 (79) | 3 (100) | 42 (84) |
| Number of prior radiotherapy treatments, n (%) | |||||||||
| 1 | 1 (33) | 0 | 2 (67) | 2 (33) | 1 (33) | 1 (100) | 9 (32) | 1 (33) | 17 (34) |
| 2 | 0 | 1 (33) | 0 | 0 | 0 | 0 | 4 (14) | 0 | 5 (10) |
Abbreviations: BMI, body mass index; CRC, colorectal carcinoma; ECOG, Eastern Cooperative Oncology Group; NSCLC, non‐small cell lung cancer; Q2W, every 2 weeks; QW, weekly.
Due to the small treatment groups of this study, patient baseline demographic data that contain multiple indirect identifiers have been grouped to preserve patient anonymity.
Nineteen patients (38%) in Schedule 1 received imalumab at doses from 1 to 50 mg/kg Q2W and 31 patients in Schedule 2 (62%, including 22 patients in the expansion cohort) received imalumab 10 or 25 mg/kg QW. Overall, the median number of imalumab treatment cycles received was 2 (range 1‐17).
Of 68 patients enrolled in the study, all 50 patients who received one or more treatment discontinued treatment because of disease progression according to RECIST v1.1 criteria (76%, n = 38), clinical disease progression (12%, n = 6), an AE (8%, n = 4) or consent withdrawal (4%, n = 2) (Figure 1).
3.2. Exposure and dose interruptions
Overall, 9 (18%) of the 50 patients who received one or more dose of treatment were exposed to imalumab for <1 month, 33 (66%) from 1 to <4 months, six (12%) from 4 to <7 months and two (4%) patients for ≥10 months.
A total of 41 patients (82%) had no imalumab dose reductions during the study. Of the nine patients (18%) who required one or more dose reduction, six had one reduction and three had more than one dose reduction.
3.3. Safety
Forty‐six (92%) patients who received at least one dose of imalumab had treatment‐emergent AEs (TEAEs), the majority of which (84%) were grade 1 or 2 (Supporting Information Tables S1 and S2). Immunologically relevant TEAEs in at least one patient included three (11%) patients with rash and two (7%) with pruritus in the 10 mg/kg QW group. Fifteen (30%) patients had AEs that were considered related to imalumab, with the most common being fatigue (10%) and vomiting (6%) (Table 2). All treatment‐related AEs were grade 1 or 2, except for four grade 3 events that occurred in two (4%) patients (all serious adverse events (SAEs)). One of these treatment‐related SAEs was grade 3 allergic alveolitis resulting in pulmonary haemorrhage occurring during cycle 1 in a patient receiving 50 mg/kg Q2W. This Caucasian male patient was hospitalized and recovered, receiving therapeutic doses of prednisone for several weeks. The patient had been receiving concomitant therapy including albuterol sulfate, amlodipine besylate, diphenhydramine and fluconazole, and his relevant medical history comprised anxiety‐exacerbated asthma, Barrett's oesophagus, hypertension, seasonal allergies and shortness of breath. The three remaining SAEs (grade 3 vomiting, constipation and nausea, Table 2), in one patient treated with 10 mg/kg QW, were considered to be possibly treatment related; all SAEs resolved without dose modification.
TABLE 2.
Treatment‐emergent, treatment‐related adverse events
| AE, n (%) | Schedule 1 | Schedule 2 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 mg/kg Q2W (n = 3) | 3 mg/kg Q2W (n = 3) | 10 mg/kg Q2W (n = 3) | 25 mg/kg Q2W (n = 6) | 37.5 mg/kg Q2W (n = 3) | 50 mg/kg Q2W (n = 1) a | 10 mg/kg QW (n = 28) | 25 mg/kg QW (n = 3) | All patients (N = 50) | ||||||||||
| Grades | All | 3‐5 | All | 3‐5 | All | 3‐5 | All | 3‐5 | All | 3‐5 | All | 3‐5 | All | 3‐5 | All | 3‐5 | All | 3‐5 |
| Any b | 3 (100) | 0 | 2 (67) | 0 | 1 (33) | 0 | 1 (17) | 0 | 1 (33) | 0 | 1 (100) | 1 (100) a | 6 (21) | 1 (4) a | 0 | 0 | 15 (30) | 2 (4) a |
| Fatigue | 2 (67) | 0 | 0 | 0 | 1 (33) | 0 | 0 | 0 | 1 (33) | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 5 (10) | 0 |
| Vomiting | 1 (33) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (7) | 1 (4) a | 0 | 0 | 3 (6) | 1 (2) a |
| Constipation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (7) | 1 (4) a | 0 | 0 | 2 (4) | 1 (2) a |
| Dysgeusia | 1 (33) | 0 | 1 (33) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (4) | 0 |
| Nausea | 1 (33) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4) | 1 (4) a | 0 | 0 | 2 (4) | 1 (2) a |
| Rash | 1 (33) | 0 | 1 (33) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (4) | 0 |
| Allergic alveolitis | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (100)c | 1 (100)a,c | 0 | 0 | 0 | 0 | 1 (2) | 1 (2) a |
| Arthralgia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 1 (2) | 0 |
| Chills | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (100) | 0 | 0 | 0 | 0 | 0 | 1 (2) | 0 |
| Decreased appetite | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 1 (2) | 0 |
| Diarrhoea | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 1 (2) | 0 |
| Oedema peripheral | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (33) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2) | 0 |
| Headache | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (100) | 0 | 0 | 0 | 0 | 0 | 1 (2) | 0 |
| Infusion‐related reaction | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (100) | 0 | 0 | 0 | 0 | 0 | 1 (2) | 0 |
| Mental impairment | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2) | 0 |
| Mucosal inflammation | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (100) | 0 | 0 | 0 | 0 | 0 | 1 (2) | 0 |
| Night sweats | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 1 (2) | 0 |
| Proctalgia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 1 (2) | 0 |
| Pruritus | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (100) | 0 | 0 | 0 | 0 | 0 | 1 (2) | 0 |
| Pyrexia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (4) | 0 | 0 | 0 | 1 (2) | 0 |
| Urticaria | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (100) | 0 | 0 | 0 | 0 | 0 | 1 (2) | 0 |
Abbreviations: AE, adverse event; DLT, dose‐limiting toxicity; G, grade; Q2W, every 2 weeks; QW, weekly.
AE rated grade 3; there were no treatment‐related grade 4 or grade 5 AEs.
This row does not represent the total of individual AEs, as each patient may have undergone more than one AE.
Grade 3 allergic alveolitis was considered to be a DLT in the patient treated at 50 mg/kg Q2W.
The SAE of grade 3 allergic alveolitis resulting in pulmonary haemorrhage in the first patient treated at the 50 mg/kg Q2W dose level was considered a DLT (ie, a grade 3 nonhaematologic toxicity during the first cycle of treatment). Diagnosis was made clinically via exclusion. Computed tomographic angiography was used to exclude pulmonary embolism and demonstrated the presence of ground‐glass opacities in the lungs, suggesting the presence of multifocal bronchopneumonia. There was no evidence of pulmonary infection. Allergy was suspected as symptoms resolved following treatment with prednisone. Due to the clinical significance of this serious adverse event, treatment at this dose level was suspended. Subsequently, the dose of 37.5 mg/kg every 2 weeks was chosen to be tested and it was found to be the MTD for Schedule 1. In Schedule 2, the MTD was not reached and imalumab was well tolerated up to the highest dose tested (25 mg/kg QW).
Five on‐treatment deaths occurred during the study; none were considered related to imalumab. All five deaths were due to disease progression (three in the 10 mg/kg QW cohort, one in the 10 mg/kg Q2W cohort and one in the 37.5 mg/kg cohort).
3.4. Pharmacokinetics
Area under the curve (AUC) and maximum concentration (C max) increased with increasing doses of imalumab (Figure 2). Pharmacokinetic parameters for the most commonly administered dose, 10 mg/kg QW, are shown in Table 3. The t 1/2 of imalumab ranged from 56‐150 hours (day 1, cycle 1) to 73‐176 hours (day 15, cycle 2). The median C max increased from 213.0 mg/L (range 122.0‐621.0 mg/L) at day 1, cycle 1, to 321.5 mg/L (range 169.0‐476.0 mg/L) at day 15, cycle 2. The concentration‐time profiles for the Q2W and QW dosing schedules are shown in Figure 3A,B.
FIGURE 2.
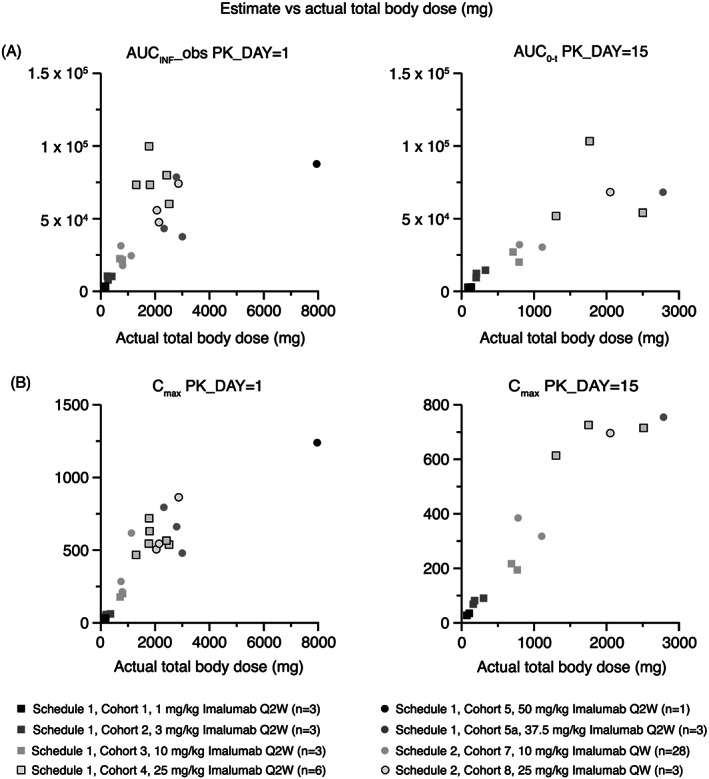
Area under the curve and maximum plasma concentration of imalumab given every two weeks (schedule 1) and once weekly (schedule 2). Data plotted are individual patient pharmacokinetic parameter values (estimate) versus actual total body dose of imalumab. AUCINF_obs, area under the plasma concentration‐time curve from time zero (pre‐dose) extrapolated to infinity (mg*h/L); AUC(0‐t), area under the plasma concentration‐time curve from time zero (pre‐dose) to time ‘t’ (338 hours post‐dose for schedule 1 and 168 hours post‐dose for schedule 2) (mg*h/L); Cmax, maximum plasma concentration (mg/L); PK_DAY = 1, cycle 1 day 1); PK‐DAY = 15, cycle 2 day 15. Q2W, every two weeks; QW, every week. The legend shows the treatment schedule, treatment cohort, and imalumab dose level for each data point
TABLE 3.
Pharmacokinetic parameters for patients treated with 10 mg/kg weekly
| Time point | Statistic | AUC(0‐inf) (mg.h/L) | AUC(0‐tau) (mg.h/L) | C max (mg/L) | C min (mg/L) | t ½ (h) | MRT(0‐inf) (h) | CL (L/h) | V dss (L) |
|---|---|---|---|---|---|---|---|---|---|
| Cycle 1, day 1 | n | 27 | 27 | 28 | ‐ | 27 | 27 | 27 | 27 |
| Mean | 19910 | 14490 | 231.11 | ‐ | 87.00 | 124.8 | 0.04058 | 4.837 | |
| SD | 6826 | 4437 | 93.82 | ‐ | 20.06 | 29.16 | 0.01259 | 1.045 | |
| CV% | 34.3 | 30.6 | 40.6 | ‐ | 23.1 | 23.4 | 31.0 | 21.6 | |
| Minimum | 9930 | 8650 | 122.0 | ‐ | 56.0 | 79.3 | 0.0208 | 3.12 | |
| Median | 18420 | 13510 | 213.00 | ‐ | 85.60 | 122.5 | 0.03930 | 4.772 | |
| Maximum | 36400 | 24100 | 621.0 | ‐ | 150 | 214 | 0.0770 | 6.97 | |
| Geometric mean | 18870 | 13890 | 218.33 | ‐ | 84.97 | 121.8 | 0.03883 | 4.727 | |
| Geometric CV% | 34.0 | 30.0 | 33.2 | ‐ | 22.4 | 22.8 | 30.9 | 22.4 | |
| Cycle 2, day 15 | n | ‐ | 18 | 18 | 18 | 18 | ‐ | 18 | 18 |
| Mean | ‐ | 26320 | 313.83 | 83.78 | 118.3 | ‐ | 0.03152 | 5.122 | |
| SD | ‐ | 9925 | 76.70 | 39.15 | 32.32 | ‐ | 0.01050 | 1.599 | |
| CV% | ‐ | 37.7 | 24.4 | 46.7 | 27.3 | ‐ | 33.3 | 31.2 | |
| Minimum | ‐ | 13500 | 169.0 | 33.9 | 72.7 | ‐ | 0.0182 | 2.93 | |
| Median | ‐ | 26250 | 321.50 | 83.05 | 120.3 | ‐ | 0.02929 | 5.028 | |
| Maximum | ‐ | 50500 | 476.0 | 156.0 | 176 | ‐ | 0.0568 | 8.33 | |
| Geometric mean | ‐ | 24600 | 304.41 | 74.77 | 113.8 | ‐ | 0.02998 | 4.892 | |
| Geometric CV% | ‐ | 39.6 | 26.5 | 53.9 | 29.5 | ‐ | 33.3 | 32.2 |
Abbreviations: AUC(0‐inf), area under the curve from time 0 to infinity; AUC(0‐tau), area under the curve to the end of the dosing period; CL, clearance; C max, maximum observed concentration; C min, minimum observed concentration; CV, coefficient of variation; MRT(0‐inf), mean residence time from time 0 to infinity; t ½, half‐life; SD, standard deviation; V dss, volume of distribution at steady state.
FIGURE 3.
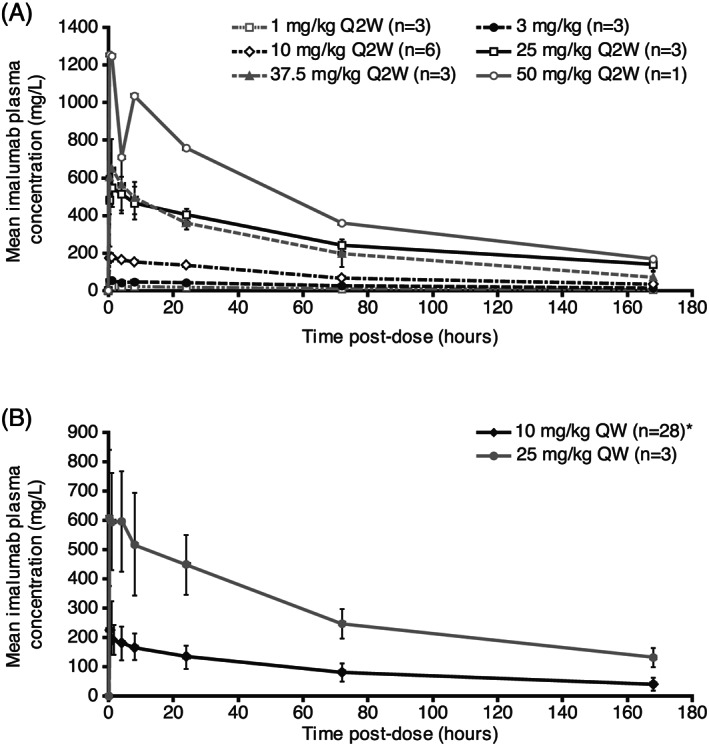
Concentration‐time curves of imalumab in schedule 1 (doses every two weeks, A) and schedule 2 (doses every week, B). QW, weekly; Q2W, every two weeks. Error bars represent standard deviation
3.5. Immunogenicity
Anti‐imalumab antibody data were available for 48 treated patients at baseline and 27 patients at study completion. Seven patients showed detectable anti‐imalumab antibodies at one or more visits; they were present at baseline in five patients (one patient each receiving 1 mg/kg and 25 mg/kg Q2W and three patients treated with 10 mg/kg QW) with no subsequent increase after exposure to imalumab. Pre‐existing antibodies against imalumab were neutralizing at all time points in one patient, only after exposure to imalumab in three patients and there was no neutralizing activity in the last patient. Two (4%) of 50 patients, both treated with 10 mg/kg QW, developed de novo anti‐imalumab binding and neutralizing antibodies at days 58 and 184, respectively.
3.6. Tumour response
All 50 treated patients were evaluated for response based on RECIST v1.1 criteria; the response was known in 39 patients and unknown in 11. The best overall response was stable disease (SD) in 13 patients (26.0% of total treated patients, 33.3% of those with known response), and progressive disease in 26 patients (52% of total treated patients, 66.7% of those with known response). Three patients with SD were treated at 3 mg/kg Q2W, three at 10 mg/kg Q2W, four at 10 mg/kg QW and one each at 1 mg/kg Q2W, 25 mg/kg Q2W and 25 mg/kg QW. Of the 13 patients with SD, eight had SD lasting for more than 4 months. There were no complete or partial objective tumour responses or evidence of a dose response (Supporting Information Figure S2 and Supporting Information Table S3). The tumour types of patients with SD of greater than 4 months included NSCLC (n = 2), ovarian cancer (n = 2), CRC (n = 1); oesophageal cancer (n = 1) and cancer of the parotid gland (n = 1). Median time on treatment for those with SD of greater than 4 months was 5.44 months (range 4.37‐15.64). One patient with ovarian cancer (granulosa cell tumour) received 10 mg/kg QW imalumab for 17 cycles.
3.7. Pharmacodynamics
3.7.1. Levels of circulating oxMIF and total MIF
Across all patients in the study, the median level of circulating oxMIF remained at 4.75 ng/mL, although the range varied from 4.75‐15.98 at baseline (n = 50) to 4.75‐9.59 at cycle 4 (n = 10) and 4.75‐4.75 at study completion (n = 2) (Table 4). The median level of total plasma MIF (oxMIF plus redMIF) in all patients also remained at 4.75 ng/mL, with a range of 4.75‐27.78 (n = 50) at baseline, 4.75‐10.57 (n = 10) at cycle 4 and 4.75‐4.75 (n = 2) at study completion (Table 4). There were no notable changes from baseline in plasma levels of circulating oxMIF and total MIF.
TABLE 4.
Concentrations of circulating oxMIF and total plasma MIF in the safety population
| Patients (n) | Median circulating oxMIF (range), ng/mL | Median total MIF (range), ng/mL | |
|---|---|---|---|
| Baseline | 50 | 4.75 (4.75–15.98) | 4.75 (4.75–27.78) |
| Cycle 2 | 41 | 4.75 (4.75–13.01) | 4.75 (4.75–11.94) |
| Cycle 3 | 13 | 4.75 (4.75–9.33) | 4.75 (4.75–11.00) |
| Cycle 4 | 10 | 4.75 (4.75–9.59) | 4.75 (4.75–10.57) |
| Cycle 5 | 6 | 4.75 (4.75–6.80) | 4.75 (4.75–10.02) |
| Cycle 6 | 7 | 4.75 (4.75–22.73) | 4.75 (4.75–24.74) |
| Cycle 7 | 3 | 4.75 (4.75–4.75) | 4.75 (4.75–4.75) |
| Cycle 8 | 1 | 4.75 (4.75–4.75) | 4.75 (4.75–4.75) |
| Cycle 9 | 2 | 4.75 (4.75–4.75) | 4.75 (4.75–4.75) |
| Cycle 10 | 2 | 4.75 (4.75–4.75) | 4.75 (4.75–4.75) |
| Cycle 11 | 2 | 4.75 (4.75–4.75) | 4.75 (4.75–4.75) |
| Cycle 12 | 2 | 4.75 (4.75–4.75) | 4.75 (4.75–4.75) |
| Cycle 13 | 2 | 4.75 (4.75–4.75) | 4.75 (4.75–4.75) |
| Cycle 14 | 1 | 4.75 (4.75–4.75) | 4.75 (4.75–4.75) |
| Cycle 15 | 1 | 4.75 (4.75–4.75) | 4.75 (4.75–4.75) |
| Cycle 16 | 1 | 4.75 (4.75–4.75) | 4.75 (4.75–4.75) |
| Cycle 17 | 1 | 4.75 (4.75–4.75) | 4.75 (4.75–4.75) |
| Completion | 2 | 4.75 (4.75–4.75) | 4.75 (4.75–4.75) |
Baseline measurement is pre‐cycle 1 dose. All other measurements are pre‐dose. MIF, macrophage migration inhibitory factor; oxMIF, oxidized macrophage migration inhibitory factor.
3.7.2. Tissue penetration and target occupancy
Posttreatment (cycle 2, day 1) tissue suitable for calculating target occupancy was available from 15 patients in the 10 mg/kg QW cohort and one patient in the 25 mg/kg QW cohort. All tumour biopsies showed satisfactory tissue penetration (Figure 4A) and the 10 mg/kg dose of imalumab resulted in a target occupancy of >100% in 10/15 (67%) evaluable patients by the end of the first treatment cycle (Figure 4B). No accumulation of imalumab was observed in adjacent liver tissue with normal morphology in patients treated with 10 mg/kg QW, although accumulation was seen in one patient treated with 25 mg/kg QW.
FIGURE 4.
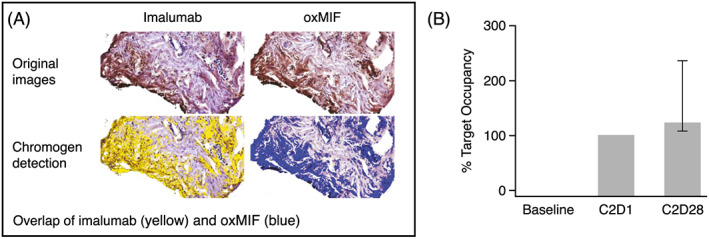
Tissue penetration (a) and target occupancy (B) showing imalumab binding to oxMIF in mCRC tumour tissue. (A) Example tissue penetration illustrated by stained regions: Consecutive slides were stained for imalumab and oxMIF. Target occupancy was calculated based on digital image analysis. (B) Target occupancy in a patient with mCRC: Median target occupancy in total tumour tissue (tumor + stroma) reached 102% after the first treatment cycle and increased to 119% after the second cycle. Patient was treated with 10 mg/kg imalumab, QW. C2D1, cycle 2, day 1; C2D28, cycle 2, day 28; mCRC, metastatic colorectal cancer; oxMIF, oxidized macrophage migration inhibitory factor; QW, weekly
4. DISCUSSION
Imalumab is an inhibitor of oxMIF, whose significance as a tumour‐related conformational isoform of MIF has only been reported relatively recently. 16 Although cancer immunotherapy was initiated in the 1980s, it only reached widespread recognition as a breakthrough treatment in 2013; 22 we initiated our first‐in‐human anti‐oxMIF antibody study in 2012.
The limited assessment in this phase 1 study did not reveal any safety concern with imalumab in patients with advanced solid tumours at doses up to 37.5 mg/kg Q2W. The DLT was allergic alveolitis, an interstitial inflammation of the distal lung involving alveolar macrophage activation and antibody‐dependent inflammation. 23 No data regarding antibody titre were collected for this patient. Immunologically related AEs are usually not considered to be dose‐related or DLTs due to their idiosyncratic nature and the DLT of allergic alveolitis was observed in a patient with a history of shortness of breath, asthma and allergic conditions. However, it has been shown to be related to biologic therapies in previous studies. 24 , 25 As MIF promotes the release of cytokines, including tumour necrosis factor α, there is the possibility of similar complications with imalumab, such as interstitial lung disease/noninfectious pulmonary complications.
The assessed pharmacokinetic parameters suggest a potentially subproportional increase of systematic exposure with increasing total dose. However, due to the large range of doses tested and small sample sizes in Schedule 1 dose cohorts and the 25 mg/kg QW dose cohort in Schedule 2, the results should be interpreted with caution.
Despite a heavily pretreated patient population, disease stabilized in approximately a quarter of patients, with durable SD after treatment with imalumab for more than 4 months in eight patients (16% of treated patients), and over 15 months for one patient with ovarian cancer. When evaluating tumour types that had prolonged stable disease, patients with immune‐responsive tumours such as NSCLC, ovarian and oesophageal appear to benefit, although the molecular profiles of these tumours are unavailable. This study chose to focus expansion in patients with mCRC based on preclinical data, 9 , 15 , 16 although limited clinical activity was seen with imalumab in these tumours. In addition, the microsatellite status of the patients with mCRC is unknown, which would be relevant considering the activity of immunotherapy in high‐level microsatellite instability mCRC tumors. 26 , 27
MIF appears to promote tumour growth in nonclinical studies of breast cancer, 28 glioblastoma 29 and childhood rhabdomyosarcoma 30 models, and pancreatic cancer, 31 , 32 CRC and osteosarcoma 33 , 34 cell lines. Conversely, MIF downregulation appears to drive anti‐angiogenic therapy resistance in glioblastoma xenograft models, 35 and MIF appears to have tumour‐suppressive activity in murine skin. 36 The varying biological consequences of MIF oxidation at different sites point to more than one form of ‘oxMIF’. The impact of MIF and its associated pathways are therefore complex and the effect of anti‐oxMIF treatment may require additional exploration with chemotherapy, as anti‐oxMIF antibodies have been found to sensitize human prostate and ovarian cancer cell lines to cytotoxic drugs. 16
Levels of circulating oxMIF and total MIF (oxMIF plus redMIF) remained steady during treatment with imalumab, and the corresponding median values of oxMIF and total MIF suggested that there was a lack of redMIF present. We are unclear on any direct effect from the blockade of oxMIF with imalumab and its impact on pathway regulation. A previous report of median oxMIF levels in patients with various cancers ranged from 0.0 to 3.5 ng/mL, although a considerable interpatient variation was observed, similar to findings in this study, and we observed higher median levels of 4.75 ng/mL in evaluable patients. 16 Also aligned with our results, plasma levels of total MIF have been shown to correlate with oxMIF in oxMIF‐positive patients; significantly lower levels of oxMIF versus total MIF are found in healthy tissue. 16 Anti‐oxMIF antibodies have been found to induce neither complement‐dependent nor antibody‐dependent cell lysis/toxicity and it cannot be ruled out that they may interfere with pro‐angiogenic properties of MIF, 16 therefore despite the target binding shown in this study, the mechanism of tumour‐related cell death is still unclear.
A biologically active dose of imalumab was determined as 10 mg/kg QW based on its safety, pharmacokinetic and pharmacodynamic profile, penetration of tumour tissue and high oxMIF target occupancy. There was an insufficient number of samples from higher doses to confirm a dose‐response relationship.
A phase I/IIa imalumab monotherapy study in patients with malignant ascites of ovarian cancer was initiated in 2015 but was terminated prematurely for logistic reasons due to poor design and patient enrolment. A phase IIa combination study investigating imalumab plus 5‐fluorouracil/leucovorin or panitumumab versus standard of care in patients with mCRC was initiated in 2015 but was terminated prematurely based on an overall benefit‐risk assessment by the data safety monitoring board after a review of the available safety and efficacy data. Although no clinical studies evaluating imalumab in advanced solid tumours are currently planned, future clinical development would require integrative biomarkers in order to predict response.
In conclusion, imalumab had a maximum tolerated dose of 37.5 mg/kg Q2W in patients with advanced solid tumours, with a biologically active dose of 10 mg/kg QW. Further clinical investigation is warranted to assess the role of oxMIF as a therapeutic target in humans based on preclinical evidence suggesting that it may play a role in carcinogenesis and cancer‐associated inflammation. The modest antitumor activity observed in the present study may be enhanced by combining oxMIF‐targeted treatments with other anticancer agents, with the caveat that there may be additional toxicity with addition of these agents. 37
COMPETING INTERESTS
D.M. received research support from Merck and Oncolytics, and advisory board and/or speaker honoraria from Amgen, BMS, Bayer, Eisai, EMD Serono, Exelixis and Genentech. He is currently employed by the Robert H Lurie Comprehensive Cancer Center, Northwestern University, Chicago, USA. F.dJ. was an employee of Shire International GmbH, a member of the Takeda group of companies, Zug, Switzerland at the time of the study, was a stockholder of Shire, and is currently an employee of Tesaro, a GSK company. D.V. is an employee of Baxalta Innovations GmbH, a member of the Takeda group of companies, Vienna, Austria, and a stockholder in Takeda Pharmaceutical Company Limited. A.Y. is an employee of Baxalta US Inc., a member of the Takeda group of companies, Cambridge, MA, USA, and stockholder in Takeda Pharmaceutical Company limited. J.C.S. has received travel support and honoraria from Celgene, advisory board honoraria from Novartis, Puma, Tempus, research support from Genentech, and research funding from Pfizer and Celgene. R.K.R. is an employee of Merck Research Laboratories. J.S.S. is part of the Institute for Drug Development, Cancer Therapy and Research Center at University of Texas Health Science Center San Antonio; San Antonio, TX, and has received grant funding for their Cancer Center Support Grant P30CA054174. A.M.T. has received research funding from IMMATICS, OBI Pharma, Tempus, Parker Institute for Cancer Immunotherapy, EMD Serono; Baxalta; Foundation Medicine; Bayer; Boston Biomedical; Placon Therapeutics; Karus Therapeutics; and Tvardi. She had a consulting or Advisory Role for Roche, Covance, Genentech and Tempus.
CONTRIBUTORS
A.M.T., D.M., D.V., J.C.S. and R.R. designed the research. A.M.T., A.Y., D.M., J.C.S., J.S., M.P. and N.H. performed the research. A.M.T., A.Y., D.M., D.V., F.dJ., J.C.S., J.S., M.P. and N.H. analysed the data. A.M.T., A.Y., D.M., D.V., F.dJ., J.C.S., J.S., L.H., M.P., N.H. and R.R. wrote the manuscript. A.M.T. and L.H. provided patient care, evaluations and accrual. A.Y. provided support with literature and references. A.M.T., D.M., D.V. and J.S. provided administrative, technical and material support. A.M.T., D.M., M.P. and J.C.S. provided study supervision.
Supporting information
Table S1. Summary of adverse events
Table S2 Treatment‐emergent adverse events of any cause occurring in at least two patients in any one cohort
Table S3 Summary of best overall tumour response and overall response rate by treatment cohort
Figure S1. Study design. Cohort 9 was initially also planned for Schedule 2 to investigate imalumab at 37.5 mg/kg QW. This cohort was not initiated and is not presented here. Q2W, every 2 weeks; QW, every week
Figure S2. Waterfall plot of best percentage change in target lesion dimensions from baseline/best response (n = 38). Data were not available for 12 patients in cohorts 4 (n = 1), 5 (n = 1) and 10 (n = 10); one each of these patients in cohort 10 had best response listed as progressive disease (PD) and stable disease (SD), respectively. Best response was unknown for three patients with change from baseline data available. mCRC, metastatic colorectal carcinoma; NSCLC, non‐small cell lung cancer; Q2W, every 2 weeks; QW, weekly; SCLC, small cell lung cancer
Data S1: Details of assay parameters, pathological review and digital image analysis
Supporting info item
ACKNOWLEDGEMENTS
R.J. Kerschbaumer, M. Thiele and A. Schinagl were employees of Baxalta Innovations GmbH, a Takeda company, Vienna, Austria at the time that the study was conducted and were responsible for planning, analysing and evaluating biomarker and target occupancy studies. G.C. Manzur was an employee of Baxalta US Inc., a Takeda company, Cambridge, MA, USA, at the time of the study, and was involved in the clinical development of imalumab. M. Yoon of Baxalta US Inc., a Takeda company, Cambridge, MA, USA, performed the statistical analyses, was involved in the preparation of the clinical study report and reviewed the manuscript. A. Adefuye of Baxalta US Inc., a Takeda company, Cambridge, MA, USA, was involved with acquisition, analysis and interpretation of the safety data included and reviewed the manuscript. S. Sheth, P. Douillard, F. Scheiflinger, M. Azadeh and H. Glantschnig are employees of Baxalta Innovations GmbH, a Takeda company, Vienna, Austria, and reviewed the manuscript or provided additional information on the study background and methods. Medical writing support was provided by Laura McMahon of Physicians World Europe GmbH (Mannheim, Germany) and was funded by Baxalta US Inc. (a member of the Takeda group of companies, Lexington, MA, USA).
The study was sponsored by Baxalta US Inc. (a member of the Takeda group of companies, Lexington, MA, USA) and Baxalta Innovations GmbH, a Takeda company, Vienna, Austria. Support was also provided by a Cancer Center Support Grant (Institute for Drug Development, Cancer Therapy and Research Center at University of Texas Health Science Center San Antonio; San Antonio, TX) P30CA054174 and a grant from the National Institutes of Health/National Cancer Institute to the University of Texas MD Anderson Cancer Center, award number P30 CA016672.
Mahalingam D, Patel MR, Sachdev JC, et al. Phase I study of imalumab (BAX69), a fully human recombinant antioxidized macrophage migration inhibitory factor antibody in advanced solid tumours. Br J Clin Pharmacol. 2020;86:1836–1848. 10.1111/bcp.14289
The authors confirm that the principal investigators for this paper are Devalingam Mahalingam and Apostolia Maria Tsimberidou.
Devalingam Mahalingam and Apostolia Maria Tsimberidou contributed equally to the study.
DATA AVAILABILITY STATEMENT
The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participant data supporting the results, will be available 3 months after the submission to researchers who provide a methodologically sound proposal. The data will be provided after de‐identification, in compliance with applicable privacy laws, data protection and requirements for consent and anonymization.
REFERENCES
- 1. Mitchell RA, Liao H, Chesney J, et al. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A. 2002;99(1):345‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3(10):791‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Conroy H, Mawhinney L, Donnelly SC. Inflammation and cancer: macrophage migration inhibitory factor (MIF) – the potential missing link. QJM. 2010;103(11):831‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kerschbaumer RJ, Rieger M, Volkel D, et al. Neutralization of macrophage migration inhibitory factor (MIF) by fully human antibodies correlates with their specificity for the beta‐sheet structure of MIF. J Biol Chem. 2012;287(10):7446‐7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thiele M, Kerschbaumer RJ, Tam FW, et al. Selective targeting of a disease‐related conformational isoform of macrophage migration inhibitory factor ameliorates inflammatory conditions. J Immunol. 2015;195(5):2343‐2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kindt N, Journe F, Laurent G, Saussez S. Involvement of macrophage migration inhibitory factor in cancer and novel therapeutic targets. Oncol Lett. 2016;12(4):2247‐2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meyer‐Siegler KL, Iczkowski KA, Vera PL. Further evidence for increased macrophage migration inhibitory factor expression in prostate cancer. BMC Cancer. 2005;5:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ren Y, Law S, Huang X, et al. Macrophage migration inhibitory factor stimulates angiogenic factor expression and correlates with differentiation and lymph node status in patients with esophageal squamous cell carcinoma. Ann Surg. 2005;242(1):55‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. He XX, Chen K, Yang J, et al. Macrophage migration inhibitory factor promotes colorectal cancer. Mol Med. 2009;15(1‐2):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tomiyasu M, Yoshino I, Suemitsu R, Okamoto T, Sugimachi K. Quantification of macrophage migration inhibitory factor mRNA expression in non‐small cell lung cancer tissues and its clinical significance. Clin Cancer Res. 2002;8(12):3755‐3760. [PubMed] [Google Scholar]
- 11. Krockenberger M, Kranke P, Hausler S, et al. Macrophage migration‐inhibitory factor levels in serum of patients with ovarian cancer correlates with poor prognosis. Anticancer Res. 2012;32(12):5233‐5238. [PubMed] [Google Scholar]
- 12. Funamizu N, Hu C, Lacy C, et al. Macrophage migration inhibitory factor induces epithelial to mesenchymal transition, enhances tumor aggressiveness and predicts clinical outcome in resected pancreatic ductal adenocarcinoma. Int J Cancer. 2013;132(4):785‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chesney J, Metz C, Bacher M, Peng T, Meinhardt A, Bucala R. An essential role for macrophage migration inhibitory factor (MIF) in angiogenesis and the growth of a murine lymphoma. Mol Med. 1999;5(3):181‐191. [PMC free article] [PubMed] [Google Scholar]
- 14. Meyer‐Siegler KL, Iczkowski KA, Leng L, Bucala R, Vera PL. Inhibition of macrophage migration inhibitory factor or its receptor (CD74) attenuates growth and invasion of DU‐145 prostate cancer cells. J Immunol. 2006;177(12):8730‐8739. [DOI] [PubMed] [Google Scholar]
- 15. Nishihira J, Ishibashi T, Fukushima T, Sun B, Sato Y, Todo S. Macrophage migration inhibitory factor (MIF): its potential role in tumor growth and tumor‐associated angiogenesis. Ann N Y Acad Sci. 2003;995:171‐182. [DOI] [PubMed] [Google Scholar]
- 16. Schinagl A, Thiele M, Douillard P, et al. Oxidized macrophage migration inhibitory factor is a potential new tissue marker and drug target in cancer. Oncotarget. 2016;7(45):73486‐73496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schindler L, Dickerhof N, Hampton MB, Bernhagen J. Post‐translational regulation of macrophage migration inhibitory factor: basis for functional fine‐tuning. Redox Biol. 2018;15:135‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kleemann R, Kapurniotu A, Frank RW, et al. Disulfide analysis reveals a role for macrophage migration inhibitory factor (MIF) as thiol‐protein oxidoreductase. J Mol Biol. 1998;280(1):85‐102. [DOI] [PubMed] [Google Scholar]
- 19. Nguyen MT, Beck J, Lue H, et al. A 16‐residue peptide fragment of macrophage migration inhibitory factor, MIF‐(50‐65), exhibits redox activity and has MIF‐like biological functions. J Biol Chem. 2003;278(36):33654‐33671. [DOI] [PubMed] [Google Scholar]
- 20. Hussain F, Freissmuth M, Volkel D, et al. Human anti‐macrophage migration inhibitory factor antibodies inhibit growth of human prostate cancer cells in vitro and in vivo. Mol Cancer Ther. 2013;12(7):1223‐1234. [DOI] [PubMed] [Google Scholar]
- 21. Mankarious S, Lee M, Fischer S, et al. The half‐lives of IgG subclasses and specific antibodies in patients with primary immunodeficiency who are receiving intravenously administered immunoglobulin. J Lab Clin Med. 1988;112(5):634‐640. [PubMed] [Google Scholar]
- 22. Couzin‐Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342(6165):1432‐1433. [DOI] [PubMed] [Google Scholar]
- 23. McSharry C, Anderson K, Bourke SJ, Boyd G. Takes your breath away – the immunology of allergic alveolitis. Clin Exp Immunol. 2002;128(1):3‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Perez‐Alvarez R, Perez‐de‐Lis M, Diaz‐Lagares C, et al. Interstitial lung disease induced or exacerbated by TNF‐targeted therapies: analysis of 122 cases. Semin Arthritis Rheum. 2011;41(2):256‐264. [DOI] [PubMed] [Google Scholar]
- 25. Hadjinicolaou A, Nisar M, Bhagat S, Parfrey H, Chilvers E, Ostor A. Non‐infectious pulmonary complications of newer biological agents for rheumatic diseases – a systematic literature review. Rheumatology. 2011;50(12):2297‐2305. [DOI] [PubMed] [Google Scholar]
- 26. Gryfe R, Kim H, Hsieh ETK, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med. 2000;342(2):69‐77. [DOI] [PubMed] [Google Scholar]
- 27. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372(26):2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Balogh KN, Templeton DJ, Cross JV. Macrophage migration inhibitory factor protects cancer cells from immunogenic cell death and impairs anti‐tumor immune responses. PLoS ONE. 2018;13(6):e0197702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Otvos B, Silver DJ, Mulkearns‐Hubert EE, et al. Cancer stem cell‐secreted macrophage migration inhibitory factor stimulates myeloid derived suppressor cell function and facilitates glioblastoma immune evasion. Stem Cells. 2016;34(8):2026‐2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johler SM, Fuchs J, Seitz G, Armeanu‐Ebinger S. Macrophage migration inhibitory factor (MIF) is induced by cytotoxic drugs and is involved in immune escape and migration in childhood rhabdomyosarcoma. Cancer Immunol Immunother. 2016;65(12):1465‐1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guo D, Guo J, Yao J, et al. D‐dopachrome tautomerase is over‐expressed in pancreatic ductal adenocarcinoma and acts cooperatively with macrophage migration inhibitory factor to promote cancer growth. Int J Cancer. 2016;139(9):2056‐2067. [DOI] [PubMed] [Google Scholar]
- 32. Yang S, He P, Wang J, et al. A novel MIF signaling pathway drives the malignant character of pancreatic cancer by targeting NR3C2. Cancer Res. 2016;76(13):3838‐3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu LH, Xia HH, Ma WQ, et al. Macrophage migration inhibitory factor siRNA inhibits hepatic metastases of colorectal cancer cells. Front Biosci (Landmark Ed). 2017;22:1365‐1378. [DOI] [PubMed] [Google Scholar]
- 34. Liu W, Liu SY, He YB, et al. MiR‐451 suppresses proliferation, migration and promotes apoptosis of the human osteosarcoma by targeting macrophage migration inhibitory factor. Biomed Pharmacother. 2017;87:621‐627. [DOI] [PubMed] [Google Scholar]
- 35. Castro BA, Flanigan P, Jahangiri A, et al. Macrophage migration inhibitory factor downregulation: a novel mechanism of resistance to anti‐angiogenic therapy. Oncogene. 2017;36(26):3749‐3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brocks T, Fedorchenko O, Schliermann N, et al. Macrophage migration inhibitory factor protects from nonmelanoma epidermal tumors by regulating the number of antigen‐presenting cells in skin. FASEB j. 2017;31(2):526‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lilenbaum RC, Herndon JE II, List MA, et al. Single‐agent versus combination chemotherapy in advanced non‐small‐cell lung cancer: the cancer and leukemia group B (study 9730). J Clin Oncol. 2005;23(1):190‐196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of adverse events
Table S2 Treatment‐emergent adverse events of any cause occurring in at least two patients in any one cohort
Table S3 Summary of best overall tumour response and overall response rate by treatment cohort
Figure S1. Study design. Cohort 9 was initially also planned for Schedule 2 to investigate imalumab at 37.5 mg/kg QW. This cohort was not initiated and is not presented here. Q2W, every 2 weeks; QW, every week
Figure S2. Waterfall plot of best percentage change in target lesion dimensions from baseline/best response (n = 38). Data were not available for 12 patients in cohorts 4 (n = 1), 5 (n = 1) and 10 (n = 10); one each of these patients in cohort 10 had best response listed as progressive disease (PD) and stable disease (SD), respectively. Best response was unknown for three patients with change from baseline data available. mCRC, metastatic colorectal carcinoma; NSCLC, non‐small cell lung cancer; Q2W, every 2 weeks; QW, weekly; SCLC, small cell lung cancer
Data S1: Details of assay parameters, pathological review and digital image analysis
Supporting info item
Data Availability Statement
The datasets, including the redacted study protocol, redacted statistical analysis plan, and individual participant data supporting the results, will be available 3 months after the submission to researchers who provide a methodologically sound proposal. The data will be provided after de‐identification, in compliance with applicable privacy laws, data protection and requirements for consent and anonymization.