

Abstract
Aims
Branebrutinib (BMS‐986195) is a potent, highly selective, oral, small‐molecule, covalent inhibitor of Bruton's tyrosine kinase (BTK). This study evaluated safety, pharmacokinetics and pharmacodynamics of branebrutinib in healthy participants.
Methods
This double‐blind, placebo‐controlled, single‐ and multiple‐ascending dose (SAD; MAD) Phase I study (NCT02705989) enrolled participants into 3 parts: SAD, MAD and JMAD (MAD in first‐generation Japanese participants). In each part, participants were randomised 3:1 to receive branebrutinib (SAD: 0.3–30 mg; [J]MAD: 0.3–10 mg) or placebo. Participants in the MAD parts received branebrutinib daily for 14 days and were followed for 14 days postdosing. Safety was assessed by monitoring, laboratory and physical examinations, vital signs, and recording adverse events (AEs). Pharmacodynamics were assessed with a mass spectrometry assay that measured drug‐occupied and free BTK.
Results
The SAD, MAD and JMAD parts of the study included 40, 32 and 24 participants. Branebrutinib was well tolerated and AEs were mild/moderate, except for 1 serious AE that led to discontinuation. Branebrutinib was rapidly absorbed, with maximum plasma concentration occurring within 1 hour and a half‐life of 1.2—1.7 hours, dropping to undetectable levels within 24 hours. BTK occupancy was rapid, with 100% occupancy reached after a single 10‐mg dose. BTK occupancy decayed predictably over time (mean half‐life in MAD panels: 115–154 hours), such that pharmacodynamic effects were maintained after branebrutinib plasma levels fell below the lower limit of quantification.
Conclusion
Rapid and high occupancy of BTK and the lack of notable safety findings support further clinical development of branebrutinib.
Keywords: clinical trials, drug safety, pharmacodynamics, pharmacokinetics, Phase I
What is already known about this subject
Bruton's tyrosine kinase (BTK) inhibitors have shown promise for the treatment of immune‐mediated and inflammatory diseases, as well as B‐cell malignancies.
Branebrutinib (BMS‐986195), a covalent inhibitor of BTK, has demonstrated robust in vivo efficacy in animal models of immune‐mediated diseases.
What this study adds
Branebrutinib rapidly inactivated BTK following exposure to oral doses ≤30 mg in healthy participants and was well tolerated.
A novel assay provided high‐resolution data describing pharmacodynamics, BTK‐occupancy and BTK turnover in vivo.
1. INTRODUCTION
Bruton's tyrosine kinase (BTK) is a member of the Tec family of nonreceptor tyrosine kinases. 1 It is expressed in haematopoietic cells, but selectively downregulated in T cells and terminally differentiated plasma cells. 2 BTK is required for signal transduction via the B‐cell receptor and Fc receptors (i.e. FcγRIIa, FcγRIIIa and FcεRI) and regulates the development and functions of B lymphocytes. 3 In addition, BTK is required for myeloid cell activation via the Fc receptors. 4
The pathology of rheumatoid arthritis (RA) and other immune‐mediated diseases is associated with several BTK‐regulated pathways. 5 , 6 , 7 In murine B cells, monocytes, macrophages, dendritic cells, natural killer cells, platelets and granulocytes, BTK is involved in regulation of proinflammatory signalling pathways related to development of collagen‐induced arthritis via the FcγRIIa and FcγRIIIa receptors. 4 , 8 , 9 , 10 In addition, BTK regulates signalling pathways involved in RA pathology via the immunoglobulin E receptor with high FcεRI affinity found on mast cells, basophils and eosinophils. 11 , 12 , 13 The differentiation of osteoclasts, which mediate bone destruction in RA, is also regulated by BTK through the receptor and activator of nuclear factor‐ΚB (RANK) and RANK ligand (RANK‐L) pair. 14 , 15 Given the key role played by BTK in these signalling pathways, a BTK inhibitor could be a rational treatment strategy for immune‐mediated diseases such as RA, Sjögren's syndrome and systemic lupus erythematosus.
The properties of the BTK protein make it an excellent target for covalent drug inhibitors. The reactive residue, cysteine 481 (Cys481), is located within the active site. Furthermore, the long half‐life (t1/2) of BTK (BTK t1/2; previously estimated at 48–72 hours in human B cells) 16 could potentially extend the pharmacodynamic (PD) effect of a covalent inhibitor, as well as reduce the dosage or dosing frequency required for target inhibition. Lower drug dosages should limit the likelihood and severity of drug–drug interactions, as well as the likelihood of adverse events (AEs) due to off‐target activities.
BTK inhibitors have shown promise in multiple disease areas, including immune‐mediated and inflammatory diseases 17 and cancer. 18 Studies of BTK inhibitors in rheumatic diseases demonstrated their ability to reduce disease development in lupus 19 and RA 20 in animal models. Similarly, interventional trials in human participants demonstrated the robust clinical activity of BTK inhibitors in B‐cell malignancies, especially chronic lymphocytic leukaemia, mantle cell lymphoma and Waldenstrom's macroglobulinaemia. 18
Branebrutinib (BMS‐986195) is a potent, highly selective, small‐molecule, covalent inhibitor of BTK that can be administered orally. It covalently modifies a cysteine residue in the active site, resulting in rapid inactivation of BTK. 21 Branebrutinib has demonstrated >5000‐fold selectivity for BTK over 240 other kinases, with only 4 related Tec family kinases demonstrating selectivity of <5000‐fold. 21 Robust in vivo efficacy of branebrutinib was demonstrated in murine models of collagen‐ and collagen antibody–induced arthritis, protecting against clinically evident disease, histological joint damage and bone mineral density loss. 21 In both models, maximal efficacy was observed at doses ≥0.5 mg kg−1 administered orally once daily (QD), which achieved ≥90% inactivation of BTK in vivo. 21 Potent efficacy across a range of measures (e.g. reduction in proteinuria) was also observed in a mouse model of lupus nephritis, with robust inhibition of BTK activity at doses as low as 0.2 mg kg−1. 21
Considering the efficacy observed in animal models of rheumatic disease, we conducted this first‐in‐human study to assess the safety and tolerability, pharmacokinetics (PK), BTK receptor occupancy and PD of branebrutinib in healthy participants.
2. METHODS
2.1. Study design
This single‐centre, randomised, double‐blind, placebo‐controlled, single‐ascending dose (SAD) and multiple‐ascending dose (MAD), Phase I study (NCT02705989) was performed in Melbourne, Victoria, Australia between August 2016 and August 2017.
The study was conducted in accordance with Good Clinical Practice as defined by the International Conference on Harmonisation and in line with the ethical principles of the Declaration of Helsinki, European Union Directive 2001/20/EC and the US Code of Federal Regulations, Title 21, Part 50. The protocol, amendments and participant‐informed consent were approved by Independent Ethics Committee at the study site prior to study initiation, and all participants provided written informed consent before beginning any study procedures. The experimental protocols and design were in compliance with the British Journal of Clinical Pharmacology guidelines.
2.2. Study population
Healthy participants were recruited into the 3 study parts. Participants aged 18–55 years with a body mass of >50 kg and a body mass index of 18–32 kg m−2, inclusive, who were healthy (as determined by no clinically significant deviation from normal in medical history, physical examination, electrocardiogram [ECG] and clinical laboratory evaluations), were included in both the SAD and MAD parts. The third part of the study included an additional cohort of healthy first‐generation Japanese participants (permitted body mass index 18–30 kg m−2, inclusive, with confirmed paternal and maternal ancestry and whose residency outside of Japan did not exceed 10 years) in a MAD study (JMAD) in order to explore the effect of Japanese race on PK, PD and safety. Women of childbearing potential were required to have a negative serum or urine pregnancy test within 24 hours prior to the start of treatment and were required to use an effective method of birth control during the study. Participants with known or suspected autoimmune disorder, major surgery within 4 weeks of study drug administration, or significant acute or chronic medical illness or any other illness, condition or significant laboratory anomalies that the investigator felt may put the participant at unacceptable risk were excluded. All participants were screened within 28 days prior to study drug or placebo administration to evaluate their eligibility.
2.3. Dose rationale
The initial dose selection was made to achieve a low level of BTK occupancy based on several considerations, including regulatory requirements for safety relative to animal toxicology, in vitro characterisation of BTK inactivation rates, estimated BTK t1/2, preclinical drug stability assays, PK/PD modelling and data from nonhuman primate studies. 21 The maximum recommended starting dose based on the no‐observed‐AE level and a 10‐times safety margin was 19 mg. However, the predicted BTK occupancy at 19 mg was >90% (predicted therapeutic level); thus, 0.3 mg and 1 mg were selected for the first and second dose levels, respectively, to achieve a low level of initial activity. In the SAD study, the maximum projected dose was 45 mg, based on predicted exposure, area under the curve (AUC) and BTK occupancy. However, 30 mg was ultimately selected as the highest dose based on PK/PD analysis. After the first cohort of patients in the SAD study received branebrutinib (0.3 mg), the decision to proceed to the next higher dose level was based on available PK and PD data; dose escalation did not occur until the safety of the preceding dose panel was confirmed. The desired range of target occupancy in the MAD study was 50–98% at steady state, based on predicted pharmacologically effective doses in preclinical animal models that achieved >90% BTK occupancy. 21 Therefore, doses from 0.3–10 mg were tested.
2.4. Study conduct
In the SAD part, 40 participants were randomised 3:1 to receive a single dose of branebrutinib (0.3, 1, 3, 10 or 30 mg; n = 6 for each) or placebo (n = 10) in ascending dose panels. In the MAD part, 32 participants were randomised 3:1 to receive branebrutinib (0.3, 1, 3 or 10 mg; n = 6 for each) QD for 14 days or placebo (n = 8) similarly. In the JMAD part, 24 participants were randomised 3:1 to receive branebrutinib (0.3, 3 or 10 mg; n = 6 for each) QD or placebo (n = 6). Information on blinding and randomisation can be found in the supporting information (online). Although the number of participants for each dose group was not determined based on statistical power considerations, a sample size of 6 participants treated with branebrutinib provides an 80.7% probability (using nQuery Advisor 7.0) of observing at least 1 occurrence of any AE with a 24% incidence in the population from which the sample was drawn. Dose escalation only occurred following review of the PD and safety data from the previous dose panel by the sponsor's study physician and principal investigator.
2.5. Safety assessment
The safety of branebrutinib was assessed until study discharge and then at follow‐up visits on Day 14 (SAD) and Day 28 (MAD). Safety assessments included recording of AEs, physical examinations, vital sign measurements, 12‐lead ECG and clinical laboratory evaluations. Specific monitoring of pancreatic function, including assessments of amylase, lipase and fasting glucose levels, was also implemented. Reports of serious AEs were collected until 30 days after the last dose.
2.6. PK assessment
The PK of branebrutinib were characterised by AUC to time infinity, AUC to the end of the dosing period (AUCtau), maximum serum concentration (Cmax), time to reach maximum concentration and t1/2 in both parts. BMT‐250433 is a compound that rapidly forms a covalent bond to Cys481 of BTK. Excess BMT‐250433 was added as a quencher to PK samples to quench any free BTK and stop target mediated consumption of branebrutinib. Plasma concentrations of branebrutinib were quantified using a validated liquid chromatography with tandem mass spectrometry (LC–MS/MS) assay. PK assessment of samples in the SAD part was conducted using an AB SCIEX Triple Quad 5500 mass spectrometer in positive TurboIonSpray ionisation with multiple reaction monitoring (MRM) mode. The within‐run and between‐run coefficients of variation were ≤4.7 and ≤3.3%, respectively; the lower limit of quantification (LLOQ) was 0.100 ng mL−1. PK samples from the MAD part were also analysed by AB SCIEX API 5000 mass spectrometer in positive TurboIonSpray MRM mode. The within‐run and between‐run coefficients of variation were ≤4.5 and ≤3.7%, respectively; the LLOQ was 0.100 ng mL−1.
Samples for PK analysis were collected predose (t = 0) and at 0.5, 1, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 60 and 72 hours postdose in the SAD part of the study. In the MAD part, samples were collected predose (t = 0) on Days 1, 2, 3, 4, 5, 8, 10 and 14, and at 0.5, 1, 2, 3, 4, 6, 8, 12 and 16 hours postdose on Days 1 and 14, and at 24, 36, 48, 60 and 72 hours after the last dose on Day 14.
2.7. PD assessment
The PD of branebrutinib were characterised by BTK occupancy (the proportion of BTK covalently bound by branebrutinib to Cys481 relative to the amount of total BTK). To obtain robust, high‐resolution data, a novel immunocapture LC–MS/MS–based assay was developed. 22 A lysis buffer cocktail was prepared by mixing 15 mL of 10× lysis buffer, 1.0 mL of protease inhibitor and 80 μL of 0.5 mg mL−1 BMT‐250433 (quencher) in dimethyl sulfoxide. The solution was then diluted to a total volume of 75 mL with deionised water. The quencher was added in excess and it reacted more rapidly than branebrutinib with BTK Cys481; thus, the quencher stopped consumption of unreacted branebrutinib, preserving the PK of branebrutinib and the in vivo levels of BTK occupied by branebrutinib in the collected samples. BTK reacted with the quencher is termed quencher‐bound BTK (QBBTK). Blood samples were collected in 4 mL of acid citrate dextrose‐A (ACD‐A) in a Vacutainer tube and mixed well. Immediately after sample collection, an aliquot of 3.5 mL ACD‐A blood was transferred into a 15‐mL centrifuge tube containing 7.0 mL of the preprepared lysis buffer cocktail. After vortex mixing, the samples in 15‐mL centrifuge tubes were shaken with a reciprocal shaker for 1 hour at room temperature. Three aliquots of 3 mL blood lysate were prepared for each time point and stored at or below −70°C immediately after collection until analysis. During subsequent analysis, both drug‐bound BTK (DBBTK) and QBBTK were enriched by automated bead‐based immunocapture from 3 mL of blood lysate using a biotinylated rabbit anti‐BTK monoclonal antibody prior to on‐bead trypsin digestion and LC–MS/MS analysis. The tryptic BTK peptide, QRP, consisted of 21 amino acids (position number 467 to 487 in BTK) with Cys481 bound to branebrutinib or BMT‐250433 as the surrogate peptides for LC–MS/MS quantitation of QBBTK and DBBTK, respectively. As each peptide is measured in the assay without reference to predose samples and the result of the assay is a ratio of the peptides, each sample is independent of the other samples. Ultra‐high performance LC was performed on a Waters CORTECS UPLC C18+ column with a gradient elution. MS detection was performed with positive electrospray ionisation on a Triple Quad 5500 system in MRM mode. The DBBTK and QBBTK reference standards were prepared from recombinant BTK by chemical reaction of recombinant BTK with an excess of branebrutinib or the quencher, respectively. The blood lysate treated with an excess of quencher (= 0 nM of DBBTK) was used for the preparation of DBBTK standard curve, and the blood lysate treated with an excess of branebrutinib (= 0 nM QBBTK) was used for the preparation of QBBTK standard curve. LLOQs were 0.125 nM for QBBTK and 0.250 nM for DBBTK, with an upper limit of quantification of 12.5 nM for both analytes. The detailed procedure has been published. 22 Based on the concentrations of DBBTK and the free BTK (detected as QBBTK), the BTK occupancy was determined using the following formula:
Additional measurements were conducted in the MAD part for B‐cell subsets and in both parts for immunoglobulins.
In the SAD part of the study, samples for PD assessment were collected at predose (t = 0) and at 1, 2, 8, 12, 16, 24, 36, 48, 60, 72, 84, 96, 108, 120, 132, 144, 156 and 168 hours postdose. In the MAD part, samples for PD assessment were collected at predose (t = 0) on Day 1, 2, 3, 5, 8 and 14, at 1, 2, 4, 8 and 12 hours postdose on Day 1, at 2 and 12 hours postdose on Days 2 and 5 and at 1, 6, 12, 16, 24, 36, 48, 60, 72, 84, 96, 108, 120, 132, 144, 156, 168, 240 and 336 hours after the last dose on Day 14. PD samples for the JMAD part were collected at predose (t = 0) on Days 1, 2, 5 and 14, at 12 hours postdose on Day 1 and at 1, 6, 12, 16, 24, 36, 48, 60, 72, 84, 96, 108, 120, 132, 144, 156, 168, 240 and 336 hours after the last dose on Day 14.
2.8. Data and statistical analysis
Descriptive statistics reported continuous variables using arithmetic and/or geometric means, standard deviations (SDs), medians, minimum, and maximums. Categorical variables were summarised as counts and percentages. Statistics for PK parameters were analysed by treatment and study day. Plots were generated to show PK and PD parameters over time. SAS version 9.2 or similar version was used for statistical analyses, tabulations and graphical presentations. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology. 23
2.9. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
3. RESULTS
3.1. Analysis population
The SAD part of the study included 40 participants (active dosing: n = 30; placebo dosing: n = 10). A total of 39 (97.5%) completed the study; 1 participant (2.5%) in the 1‐mg group discontinued for personal reasons. The MAD part included a total of 32 participants (active dosing: n = 24; placebo dosing: n = 8), of whom 31 (96.9%) completed the study. One participant (3.1%) in the 10‐mg QD group discontinued due to a serious AE (psychotic disorder) related to a pre‐existing, but not disclosed, condition that was not considered related to study treatment. All 24 participants (active dosing: n = 18; placebo dosing: n = 6) in the JMAD part completed the study. A summary of participant demographics is shown in Table S1 (supporting information).
3.2. Safety
All AEs were mild to moderate in severity with 1 exception: on Day 10, 1 serious AE of psychosis (presenting as irritability and paranoid behaviour) that led to study discontinuation was reported in a male participant aged 27 years in the 10‐mg cohort of the MAD part. This participant had a history of previously treated psychosis that was not disclosed at the time of enrolment, and was considered not related to study drug. The participant improved following treatment with antipsychotic medications. There were no obvious associations between this SAE and an elevated PK. No other serious AEs or AEs leading to discontinuation were reported.
A summary of AEs is shown in Table 1. The most frequently reported AE in both the SAD and MAD parts was a headache (17.5 and 28.1% of participants, respectively). Upper respiratory tract infections were reported in 5 (15.6%) participants in the MAD study. A total of 2 AEs of abdominal pain were reported in the MAD part; in both cases the AEs were not associated with abnormalities in amylase or lipase values and resolved without treatment. Across both parts of the trial, no dose‐related trend in AEs was observed (Table S2 in the supporting information).
TABLE 1.
Summary of AEs
| SAD part | MAD part | JMAD part | ||||
|---|---|---|---|---|---|---|
| Placebo (n = 10) | Branebrutinib (n = 30) | Placebo (n = 8) | Branebrutinib (n = 24) | Placebo (n = 6) | Branebrutinib (n = 18) | |
| Any AE * | 5 (50.0) | 18 (60.0) | 7 (87.5) | 17 (70.8) | 2 (33.3) | 7 (38.9) |
| Dizziness | 0 (0.0) | 3 (10.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Headache | 3 (30.0) | 4 (13.3) | 1 (12.5) | 8 (33.3) | 0 (0.0) | 1 (5.6) |
| Upper respiratory tract infection | 0 (0.0) | 3 (10.0) | 1 (12.5) | 4 (16.7) | 2 (33.3) | 0 (0.0) |
| Oral herpes | 0 (0.0) | 1 (3.3) | 0 (0.0) | 3 (12.5) | 1 (16.7) | 0 (0.0) |
| Nausea | 0 (0.0) | 2 (6.7) | 1 (12.5) | 2 (8.3) | 0 (0.0) | 0 (0.0) |
| Rash | 0 (0.0) | 2 (6.7) | 1 (12.5) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Myalgia | 0 (0.0) | 2 (6.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Dysmenorrhoea | 0 (0.0) | 2 (6.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (11.1) |
| Tension headache | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (8.3) | 0 (0.0) | 0 (0.0) |
| Throat irritation | 0 (0.0) | 2 (6.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
Data are presented as n (%). AEs that occurred at greater frequencies in one treatment group than in the other and occurred in 1 or more participants were included
Incidence of any AEs, SAD part: 0.3 mg (n = 4), 1 mg (n = 5), 3 mg (n = 2), 10 mg (n = 5), 30 mg (n = 2); MAD part: 0.3 mg (n = 1), 1 mg (n = 5), 3 mg (n = 6), 10 mg (n = 5); JMAD part: 0.3 mg (n = 2), 3 mg (n = 3), 10 mg (n = 2).
AE: adverse event; JMAD: MAD in first‐generation Japanese participants; MAD: multiple‐ascending dose; SAD: single‐ascending dose
3.3. PK
Branebrutinib was rapidly absorbed following oral administration, with the Cmax occurring within the first hour (Figure 1 and Table 2) following a single dose. Elimination of branebrutinib also occurred rapidly, with a t1/2 of 1.2–1.7 hours, dropping to undetectable levels within 24 hours after single or multiple doses. No accumulation was seen following administration of multiple doses; all trough observed concentrations were below the LLOQ. Systemic exposure was dose‐proportional following administration of single or multiple doses, and average plasma concentrations over the dosing interval were consistent with the given dose level (Table 2). The PK profile of branebrutinib in the Japanese cohort was consistent with that of the non‐Japanese cohort.
FIGURE 1.
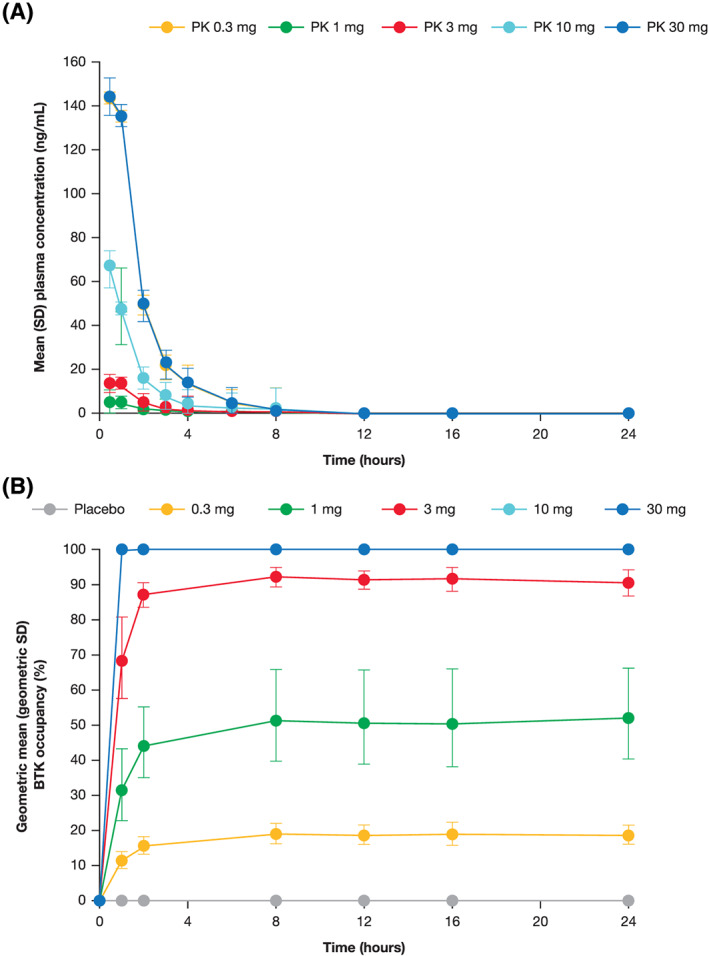
(A) Plasma concentration and (B) BTK occupancy following a single dose of branebrutinib. BTK: Bruton's tyrosine kinase; PK: pharmacokinetics; SD: standard deviation. Error bars for BTK occupancy represent geometric SDs
TABLE 2.
Pharmacokinetic parameters following a single or multiple doses of branebrutinib
| SAD part (day 1) | MAD part (day 14) | JMAD part (day 14) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose (mg) | 0.3 (n = 6) | 1 (n = 6) | 3 (n = 6) | 10 (n = 6) | 30 (n = 6) | 0.3 | 1 | 3 | 10 | 0.3 | 3 | 10 |
| GM C max (ng mL −1 ) | 1.7 | 4.6 | 14.2 | 66.6 | 179 | 2.2 (n = 6) | 5.9 (n = 6) | 17.8 (n = 6) | 60.5 (n = 5) | 2.0 (n = 6) | 16.3 (n = 6) | 97.2 (n = 6) |
| Median T max (hr) | 0.5 | 0.5 | 0.8 | 0.5 | 0.5 | 1 (n = 6) | 0.5 (n = 6) | 0.5 (n = 6) | 0.5 (n = 5) | 0.5 (n = 6) | 0.5 (n = 6) | 0.5 (n = 6) |
| GM AUC inf (ng hr mL −1 ) | 2.4 | 8.1 | 24.5 | 100 | 282 | 3.7 (n = 5) | 10.5 (n = 6) | 30.6 (n = 6) | 101.0 (n = 5) | 3.2 (n = 5) | 27.4 (n = 6) | 139.0 (n = 6) |
| Mean t 1/2 (hr) | 1.22 | 1.59 | 1.73 | 1.56 | 1.63 | 1.30 (n = 5) | 1.66 (n = 6) | 1.64 (n = 6) | 1.74 (n = 5) | 0.91 (n = 5) | 1.51 (n = 6) | 1.70 (n = 6) |
| GM CLT/F (L/hr) | 126 | 124 | 122 | 99.6 | 106 | 81.3 (n = 5) | 95.5 (n = 6) | 98.1 (n = 6) | 99.5 (n = 5) | 94.0 (n = 5) | 109 (n = 6) | 72.2 (n = 6) |
| GM Vz/F (L) | 216 | 255 | 304 | 221 | 245 | NA | NA | NA | NA | NA | NA | NA |
AUC: area under the curve; AUCinf: AUC to time infinity; AUCtau: AUC to the end of the dosing period; CLT/F: apparent total body clearance; Cmax: maximum serum concentration; GM: geometric mean; JMAD: MAD in first‐generation Japanese participants; MAD: multiple‐ascending dose; NA: not available; SAD: single‐ascending dose; Tmax: time to reach maximum concentration; Vz/F: apparent volume of distribution at terminal phase
3.4. PD
As a covalent inhibitor, the key PD endpoint is the proportion of the target occupied by branebrutinib. In the SAD part, maximal BTK occupancy increased with a single dose of up to 10 mg. Dosing with 10 mg of branebrutinib produced approximately 100% occupancy throughout 24 hours (Figure 1). Occupancy was maintained after branebrutinib concentrations fell below the LLOQ (Figures 1 and 2), returning towards baseline over 7 days of observation (Figure 2). BTK occupancy remained roughly constant 24 hours following dosing and continued to increase after PK measurements dropped below the LLOQ in some cases, indicating that branebrutinib could continue to have an active PD effect at concentrations around or below the LLOQ in some participants. Interparticipant variability (as shown by the SDs) in BTK occupancy was low, and decreased as BTK occupancy approached 100%.
FIGURE 2.
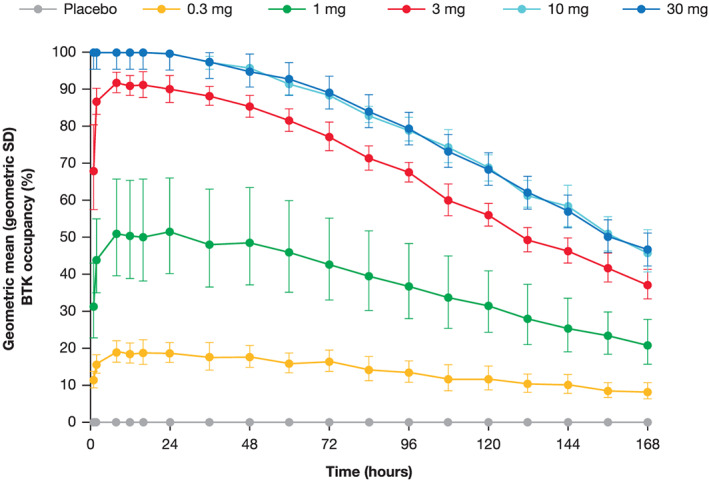
BTK occupancy over time following a single dose of branebrutinib. BTK: Bruton's tyrosine kinase; SD: standard deviation. Error bars represent geometric SDs
With additional doses of branebrutinib, BTK occupancy accumulated until reaching a dose‐dependent steady state between Day 2 and Day 14 (Figure 3). Steady‐state PD was achieved in all doses. At steady state, minimal peak to trough fluctuations in BTK occupancy were observed. Interparticipant variation was low and decreased as BTK occupancy approached 100% (Figures 3 and 4). After the end of dosing, BTK occupancy decayed at a consistent rate, irrespective of dose or maximum level of BTK occupancy, consistent with synthesis of new BTK and degradation of occupied BTK. BTK occupancy in the MAD part was consistent between the Japanese cohort and the non‐Japanese cohort (Figure 5).
FIGURE 3.
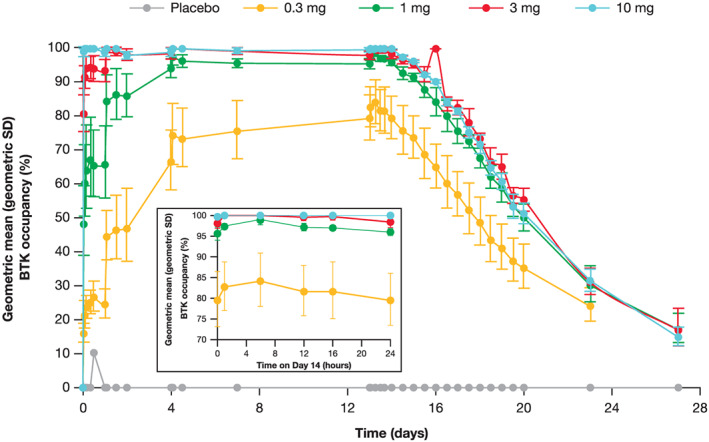
BTK occupancy following multiple doses of branebrutinib (inset shows occupancy on day 14). BTK: Bruton's tyrosine kinase; SD: standard deviation. Error bars represent geometric SDs
FIGURE 4.
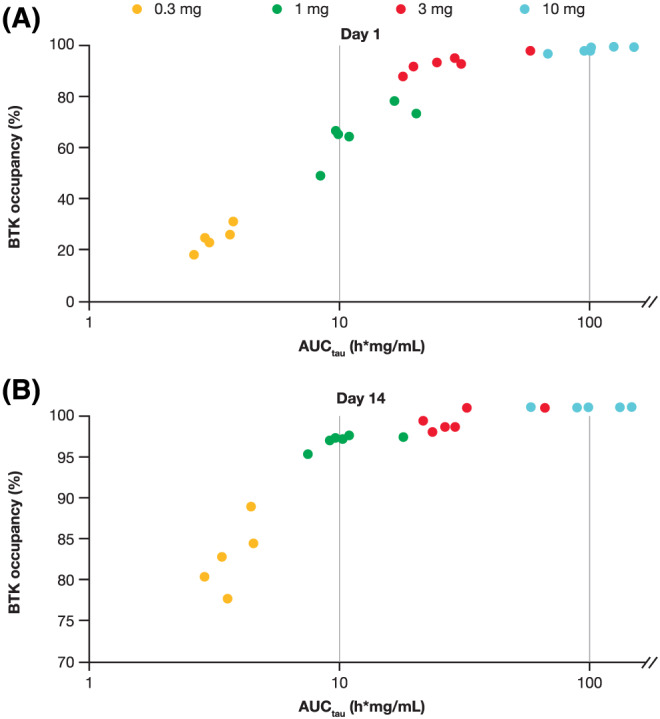
Branebrutinib exposure and BTK occupancy on (A) day 1 and (B) day 14 (MAD arm). AUCtau: area under the curve to the end of the dosing period; MAD: multiple‐ascending dose; BTK: Bruton's tyrosine kinase
FIGURE 5.
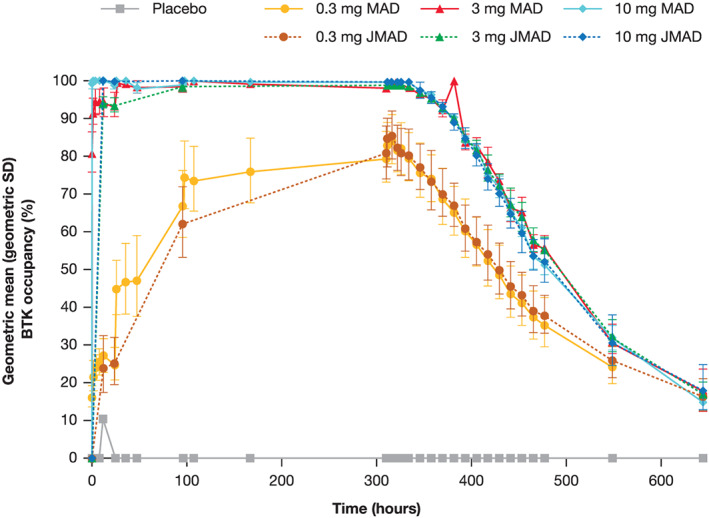
BTK occupancy over time (MAD versus JMAD parts). BTK: Bruton's tyrosine kinase; JMAD: MAD in first‐generation Japanese participants; MAD: multiple‐ascending dose; QD: once daily; SD; standard deviation. Error bars for BTK occupancy represent geometric SDs
PD were correlated to exposure (AUC) following a single dose of branebrutinib. In the MAD part, a clear relationship was observed between AUCtau and BTK occupancy on Days 1 and 14; however, the relationship on Day 1 was different from that on Day 14, with greater BTK occupancy observed with 0.3–3‐mg doses of branebrutinib on Day 14 due to the indirect PK/PD relationship and the temporal accumulation of occupied BTK (Figure 4).
While the BTK occupancy assay was optimised to determine the proportion of BTK occupied, the total amount of BTK can also be calculated. In both the SAD and MAD parts, total BTK over time was similar regardless of dose and occupancy achieved (Figure 6, Figure S1 in the supporting information). The t1/2 of BTK occupancy was calculated to be between 115 and 154 hours in the MAD panels (Table S3). There was no obvious effect of branebrutinib on peripheral blood B‐cell populations in the MAD part (Table S4 in the supporting information) or serum immunoglobulin levels in both parts (Table S5 in the supporting information).
FIGURE 6.
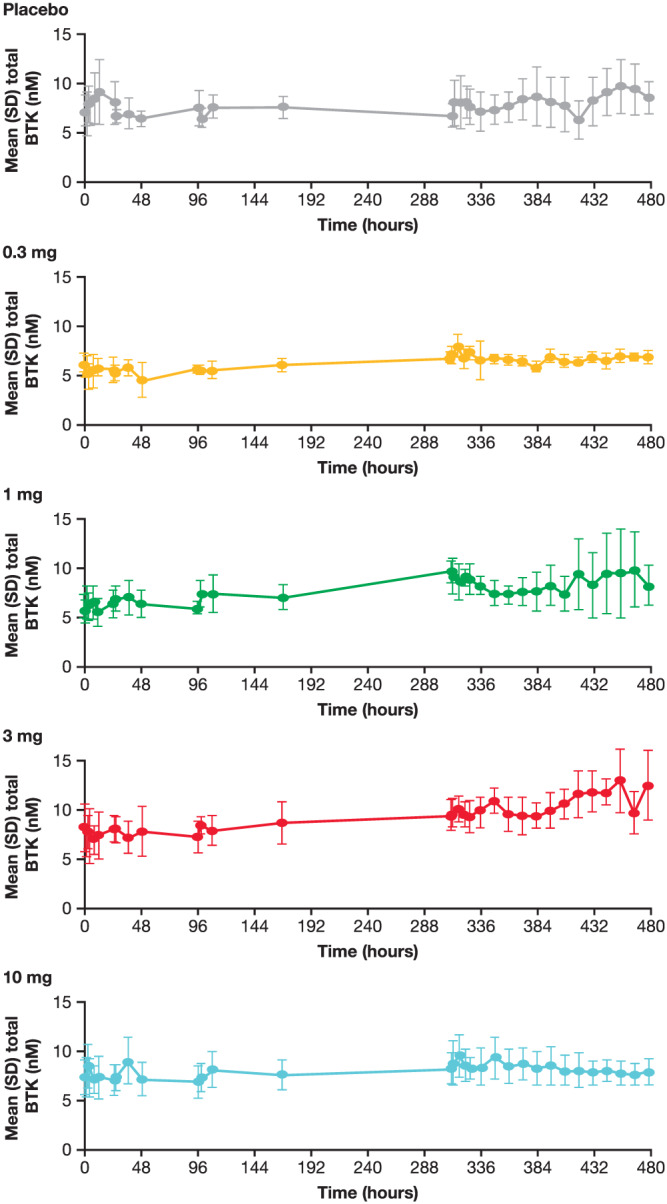
Total BTK levels over time following multiple doses of branebrutinib. BTK: Bruton's tyrosine kinase; SD: standard deviation
4. DISCUSSION
In this study of healthy participants, the PK and PD of branebrutinib were dose‐proportional and predictable. Exposure to branebrutinib was short and was low relative to the administered dose due to the drug's short t1/2 and apparent first pass metabolism (indicated by its rapid elimination and low exposure compared with expected values). PD effects persisted significantly longer than the exposure of branebrutinib due to the comparatively slow turnover of BTK.
No changes in laboratory or physical examinations or ECGs were identified. No dose‐dependent AEs, drug‐related serious AEs or drug‐related AEs that led to discontinuation were observed. Events were all mild to moderate. One serious AE of psychosis that led to study discontinuation was reported in the 10‐mg cohort of the MAD part; this was deemed unrelated to branebrutinib exposure. Preclinical studies have demonstrated that branebrutinib has very low brain penetration (<5% of plasma concentration). 21 An issue with covalent kinase inhibitors has been binding to and inhibition of epidermal growth factor receptors (EGFRs). Compared with agents such as ibrutinib, a BTK inhibitor that is also a potent irreversible inhibitor of the EGFR family of kinases, 24 branebrutinib lacks activity against EGFR kinases. 21 In this study, rash, an AE often associated with EGFR inhibitors, 25 occurred rarely and without notable differences between the placebo (one event in the MAD part) and active dosing groups (two events in the SAD part). Thus, consistent with the in vitro assessment, no sign of EGFR inhibition was observed in vivo. As sample sizes are small, it is important to note that the overall safety of a compound cannot be inferred by the absence of events occurring in a first‐in‐human trial. Further studies are needed to fully establish the risk–benefit profile in patient populations.
A key issue in developing covalent binding compounds is understanding the accumulation of compound‐bound target and also the steady‐state target occupancy in the multiple dose setting. In the MAD part of this study, 100% BTK occupancy was observed throughout the dosing interval in the 10‐mg dose group irrespective of the number of doses. In all other dose groups, BTK occupancy increased with each additional dose until steady state was achieved. This profile was expected based upon the decay in BTK occupancy observed in the SAD study.
Preclinical models 16 , 26 , 27 and clinical experience 28 with covalent BTK inhibitors suggest that high levels of BTK occupancy are required for therapeutic effect. Efficacy is expected to occur at BTK occupancy of >90%; thus, at steady state, doses ≥1 mg are projected to be efficacious. Similarly, loss of pharmacological activity would be expected 2–3 days after the last dose, and resolution of target inhibition–mediated AEs would begin. BTK occupancy reached steady state at different times depending upon dose group; from Day 1 in the 10‐mg dose group to Day 14 in the 0.3‐mg dose group. At steady state, a flat profile of BTK occupancy was observed with small differences between the peak and trough BTK occupancy. The PD observations in participants of Japanese descent were nearly identical; thus, both the behaviour of the assay and the compound were robust. Steady state is reached quickly in the potentially efficacious dose range, making a loading dose unnecessary.
The relationship between PK and PD of branebrutinib was explored in both parts of the study. In the SAD part, maximal and 24‐hour (trough) BTK occupancies were directly related to the exposure over time. As expected, unlike a reversible compound, there was no direct relationship between PK concentration and PD. In the MAD part, although plasma concentrations rapidly dropped to below the LLOQ, the slow turnover of existing BTK and synthesis of new enzyme resulted in the accumulation of BTK occupancy. Thus, although AUC on Day 14 was similar to that seen on Day 1, maximal and 24‐hour BTK occupancies increased to a higher equilibrium level at steady state. On Day 14 there was a clear relationship between exposure and PD, albeit a different relationship than on Day 1; a loss of the difference between the occupancy at the lower doses and the higher doses was seen. The variation in observed BTK occupancy was small compared with PD typically seen in human studies, probably due to compound‐specific qualities, including rapid binding and high potency. In addition, the assay reduces the level of analytical variation typically seen, with greater resolution and consistency than previously reported. 16 , 29 Further, the asymptotic approach to 100% occupancy limits the possibility of variation. When comparing the MAD and JMAD groups, both PK and PD results matched well, despite several months of separation between measurements.
The numerically lower t1/2 noted with the 0.3‐mg branebrutinib dose, compared with higher doses, may be a result of target‐mediated drug disposition and the LLOQ of the assay. Nonlinearity is seen in target‐mediated drug disposition models as elimination by binding to a target is saturable (due to the finite number of targets on the cell surface). 30 As such, elimination of the 0.3‐mg dose may occur largely via target‐mediated elimination whereas, at higher doses, BTK is partially saturated, leading to mixed elimination.
Branebrutinib acts as a pulse label of BTK and, thus, the t1/2 of BTK can be estimated based upon the washout of the label. After multiple doses achieving steady state, we estimated the t1/2 of BTK to be between 115 and 154 hours, considerably longer than previous estimates, 16 which were based upon purified B cells, as compared with whole blood used in this analysis. Further, previous assays only measured free BTK with resulting occupancy expressed relative to a predose sample; any error in the predose measurement of total BTK would cause systemic inaccuracy. The assay used here measured occupied and free BTK simultaneously from the same sample with minimal manipulation in the sample collection steps. As BTK is expressed in a wide variety of blood cells, the results of these experiments represent the aggregated outcome of all the cells in whole blood. If the rate of BTK synthesis or degradation in any major cell type was significantly different from the others, the decay curve would be uneven. Whether the occupancy observed in peripheral blood accurately represents the occupancy in cells of secondary lymphoid organs is not precisely known. However, over a broad range of BTK occupancies the decay is consistent, suggesting there are no significant effects of recirculation from secondary lymphoid organs to peripheral blood.
While the assay used here was not optimised to provide a precise measurement of the concentration of BTK for each sample, it does provide a good estimate of BTK concentration within a population. No changes in the total BTK concentrations were observed in either part of the study (Figure 6 and Figure S1 in the supporting information). The lack of dose, occupancy or time‐related change in total BTK suggest no compensatory mechanisms exist in response to BTK inhibition. Based on these data, no hysteresis in PD effect would be expected.
Branebrutinib was well tolerated and no safety concerns were identified following exposure to single or multiple doses in healthy participants. Further studies are required as the tolerability profile among target patients may differ from that of the healthy participants in this study. PK, PD and safety profiles of branebrutinib were similar in Japanese and non‐Japanese participants. High levels of BTK occupancy were achieved with low exposures of branebrutinib. Overall, the PK, PD, safety and tolerability findings of this study support further clinical development of branebrutinib in immune‐mediated diseases. Branebrutinib appears to have better safety and tolerability than less selective BTK inhibitors that have already been approved, as the rash typical of EGFR inhibition was not observed even with 14 days of 100% BTK occupancy.
COMPETING INTERESTS
All authors are employees and stockholders of Bristol Myers Squibb and receive salary and stock grants commensurate with employment.
CONTRIBUTORS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published.
Study conception and design: I.M.C., M.N., B.H., I.G., D.M.G.
Acquisition of data: I.M.C., M.N., A.L., N.Z., B.H., I.G.
Analysis and interpretation of data: I.M.C., M.N., S.K., A.L., B.H., I.G., D.M.G.
Supporting information
TABLE S1 Participant demographics
TABLE S2 Overall adverse events by dose
TABLE S3 Half‐life (hours) in the pharmacodynamics population
TABLE S4 Percent change of total peripheral blood B‐cell population from baseline
TABLE S5 Serum immunoglobulin change from baseline
FIGURE S1 Total Bruton's tyrosine kinase levels over time following single doses of branebrutinib
ACKNOWLEDGEMENTS
This study was sponsored by Bristol Myers Squibb. Professional medical writing and editorial assistance was provided by Andy Shepherd, PhD, Bu Reinen, PhD and Michele Springer, and was funded by Bristol Myers Squibb.
Catlett IM, Nowak M, Kundu S, et al. Safety, pharmacokinetics and pharmacodynamics of branebrutinib (BMS‐986195), a covalent, irreversible inhibitor of Bruton's tyrosine kinase: Randomised phase I, placebo‐controlled trial in healthy participants. Br J Clin Pharmacol. 2020;86:1849–1859. 10.1111/bcp.14290
Ian M. Catlett and Miroslawa Nowak should be considered joint first author.
As this study was conducted by a pharmaceutical company (Bristol Myers Squibb), the principal investigator, Jason Lickliter, was an employee of the clinical research organisation contracted to conduct the research. He was not involved in the design of the study or in the analysis or interpretation of data; as such, he is not included as an author on this manuscript. Rather, the sponsor's Responsible Medical Officer, Miroslawa Nowak, is the co‐lead author of the manuscript.
DATA AVAILABILITY STATEMENT
Bristol Myers Squibb policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html
REFERENCES
- 1. Mano H. Tec family of protein‐tyrosine kinases: an overview of their structure and function. Cytokine Growth Factor Rev. 1999;10(3‐4):267‐280. [DOI] [PubMed] [Google Scholar]
- 2. Smith CI, Baskin B, Humire‐Greiff P, et al. Expression of Bruton's agammaglobulinemia tyrosine kinase gene, BTK, is selectively down‐regulated in T lymphocytes and plasma cells. J Immunol. 1994;152:557‐565. [PubMed] [Google Scholar]
- 3. Satterthwaite AB, Witte ON. The role of Bruton's tyrosine kinase in B‐cell development and function: a genetic perspective. Immunol Rev. 2000;175:120‐127. [PubMed] [Google Scholar]
- 4. Di Paolo JA, Huang T, Balazs M, et al. Specific Btk inhibition suppresses B cell‐ and myeloid cell‐mediated arthritis. Nat Chem Biol. 2011;7(1):41‐50. [DOI] [PubMed] [Google Scholar]
- 5. Corneth OBJ, Verstappen GMP, Paulissen SMJ, et al. Enhanced Bruton's tyrosine kinase activity in peripheral blood B lymphocytes from patients with autoimmune disease. Arthritis Rheumatol. 2017;69:1313‐1324. [DOI] [PubMed] [Google Scholar]
- 6. Kong W, Deng W, Sun Y, et al. Increased expression of Bruton's tyrosine kinase in peripheral blood is associated with lupus nephritis. Clin Rheumatol. 2018;37(1):43‐49. [DOI] [PubMed] [Google Scholar]
- 7. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205‐2219. [DOI] [PubMed] [Google Scholar]
- 8. Diaz de Stahl T, Andren M, Martinsson P, Verbeek JS, Kleinau S. Expression of FcgammaRIII is required for development of collagen‐induced arthritis. Eur J Immunol. 2002;32(10):2915‐2922. [DOI] [PubMed] [Google Scholar]
- 9. Jongstra‐Bilen J, Puig Cano A, Hasija M, Xiao H, Smith CI, Cybulsky MI. Dual functions of Bruton's tyrosine kinase and Tec kinase during Fcgamma receptor‐induced signaling and phagocytosis. J Immunol. 2008;181:288‐298. [DOI] [PubMed] [Google Scholar]
- 10. Kleinau S, Martinsson P, Heyman B. Induction and suppression of collagen‐induced arthritis is dependent on distinct fcgamma receptors. J Exp Med. 2000;191(9):1611‐1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hata D, Kawakami Y, Inagaki N, et al. Involvement of Bruton's tyrosine kinase in FcepsilonRI‐dependent mast cell degranulation and cytokine production. J Exp Med. 1998;187(8):1235‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuehn HS, Swindle EJ, Kim MS, Beaven MA, Metcalfe DD, Gilfillan AM. The phosphoinositide 3‐kinase‐dependent activation of Btk is required for optimal eicosanoid production and generation of reactive oxygen species in antigen‐stimulated mast cells. J Immunol. 2008;181:7706‐7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Whang JA, Chang BY. Bruton's tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Drug Discov Today. 2014;19(8):1200‐1204. [DOI] [PubMed] [Google Scholar]
- 14. Lee SH, Kim T, Jeong D, Kim N, Choi Y. The tec family tyrosine kinase Btk regulates RANKL‐induced osteoclast maturation. J Biol Chem. 2008;283(17):11,526‐11,534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shinohara M, Koga T, Okamoto K, et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell. 2008;132(5):794‐806. [DOI] [PubMed] [Google Scholar]
- 16. Evans EK, Tester R, Aslanian S, et al. Inhibition of Btk with CC‐292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346(2):219‐228. [DOI] [PubMed] [Google Scholar]
- 17. Vargas L, Hamasy A, Nore BF, Smith CI. Inhibitors of BTK and ITK: state of the new drugs for cancer, autoimmunity and inflammatory diseases. Scand J Immunol. 2013;78(2):130‐139. [DOI] [PubMed] [Google Scholar]
- 18. Burger JA. Bruton's tyrosine kinase (BTK) inhibitors in clinical trials. Curr Hematol Malig Rep. 2014;9(1):44‐49. [DOI] [PubMed] [Google Scholar]
- 19. Kim YY, Park KT, Jang SY, et al. HM71224, a selective Bruton's tyrosine kinase inhibitor, attenuates the development of murine lupus. Arthritis Res Ther. 2017;19:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pan Z, Scheerens H, Li SJ, et al. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem. 2007;2(1):58‐61. [DOI] [PubMed] [Google Scholar]
- 21. Watterson SH, Liu Q, Beaudoin Bertrand M, et al. Discovery of branebrutinib (BMS‐986195): a strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo inactivation of Bruton's tyrosine kinase (BTK). J Med Chem. 2019;62(7):3228‐3250. [DOI] [PubMed] [Google Scholar]
- 22. Zheng N, Catlett IM, Taylor K, et al. Determination of real time in vivo drug receptor occupancy for a covalent binding drug as a clinical pharmacodynamic (PD) biomarker by immunocapture‐LC‐MS/MS. Anal Chem. 2019;91(13):8443‐8452. [DOI] [PubMed] [Google Scholar]
- 23. Curtis MJ, Alexander S, Cirino G, et al. Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol. 2018;175(7):987‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao W, Wang M, Wang L, et al. Selective antitumor activity of ibrutinib in EGFR‐mutant non‐small cell lung cancer cells. J Natl Cancer Inst. 2014;106:dju204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perez‐Soler R, Delord JP, Halpern A, et al. HER1/EGFR inhibitor‐associated rash: future directions for management and investigation outcomes from the HER1/EGFR inhibitor rash management forum. Oncologist. 2005;10(5):345‐356. [DOI] [PubMed] [Google Scholar]
- 26. De Lucca GV, Shi Q, Liu Q, et al. Small molecule reversible inhibitors of Bruton's tyrosine kinase (BTK): structure–activity relationships leading to the identification of 7‐(2‐Hydroxypropan‐2‐yl)‐4‐[2‐methyl‐3‐(4‐oxo‐3,4‐dihydroquinazolin‐3‐yl)phenyl]‐9H‐carbazole‐1‐carboxamide (BMS‐935177). J Med Chem. 2016;59(17):7915‐7935. [DOI] [PubMed] [Google Scholar]
- 27. Haselmayer P, Camps M, Liu‐Bujalski L, et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease models. J Immunol. 2019;202(10):2888‐2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Montalban X, Arnold DL, Weber MS, et al. Placebo‐controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med. 2019;380(25):2406‐2417. [DOI] [PubMed] [Google Scholar]
- 29. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI‐32765) has significant activity in patients with relapsed/refractory B‐cell malignancies. J Clin Oncol. 2013;31(1):88‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dua P, Hawkins E, van der Graaf PH. A tutorial on target‐mediated drug disposition (TMDD) models. CPT Pharmacometrics Syst Pharmacol. 2015;4:324‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Participant demographics
TABLE S2 Overall adverse events by dose
TABLE S3 Half‐life (hours) in the pharmacodynamics population
TABLE S4 Percent change of total peripheral blood B‐cell population from baseline
TABLE S5 Serum immunoglobulin change from baseline
FIGURE S1 Total Bruton's tyrosine kinase levels over time following single doses of branebrutinib
Data Availability Statement
Bristol Myers Squibb policy on data sharing may be found at https://www.bms.com/researchers-and-partners/independent-research/data-sharing-request-process.html