

Abstract
Aims
To identify, characterize and compare all Food and Drug Administration (FDA) and European Medicines Agency (EMA) approvals that included real‐world data on efficacy from expanded access (EA) programmes.
Methods
Cross‐sectional study of FDA (1955–2018) and EMA (1995–2018) regulatory approval documentation. We automated searching for terms related to EA in 22,506 documents using machine learning techniques. We included all approvals where EA terms appeared in the regulatory documentation. Our main outcome was the inclusion of EA data as evidence of clinical efficacy. Characterization was based on approval date, disease area, orphan designation and whether the evidence was supportive or pivotal.
Results
EA terms appeared in 693 out of 22,506 (3.1%) documents, which referenced 187 approvals. For 39 approvals, data from EA programmes were used to inform on clinical efficacy. The yearly number of approvals with EA data increased from 1.25 for 1993–2013 to 4.6 from 2014–2018. In 13 cases, these programmes formed the main evidence for approval. Of these, patients in EA programmes formed over half (median 71%, interquartile range: 34–100) of the total patient population available for efficacy evaluation. Almost all (12/13) approvals were granted orphan designation. In 8/13, there were differences between regulators in approval status and valuation of evidence. Strikingly, 4 treatments were granted approval based solely on efficacy from EA.
Conclusion
Sponsors and regulators increasingly include real‐world data from EA programmes in the efficacy profile of a treatment. The indications of the approved treatments are characterized by orphan designation and high unmet medical need.
Keywords: drug regulation, effectiveness, evidence‐based medicine, health policy
What is already known about this subject
Patients are demanding speedier access to investigational treatments.
Industry and regulators are increasingly incorporating data collected outside of clinical trials in decision making.
Little is known about how, why and whether real‐world data from expanded access may lead to regulatory approvals.
What this study adds
Techniques from artificial intelligence can be applied to process approval documentation for health policy analyses.
Real‐world data from expanded access programmes are increasingly accepted as evidence by regulators when collecting data in controlled settings is infeasible.
Thirty‐nine Food and Drug Administration/European Medicines Agency approvals rely on real‐world data from expanded access programmes to constitute the efficacy profile of a treatment. In 4 cases, these data were the sole source of evidence.
1. INTRODUCTION
Patients suffering from seriously debilitating or life‐threatening conditions who are not eligible for further treatments or any clinical trials, may resort to expanded access (EA): preapproval access to investigational treatments. EA, also known as early access, preapproval access or compassionate use, 1 is the formal regulation adopted by the Food and Drug Administration (FDA) in 1987, 2 propelled by the human immunodeficiency virus (HIV)/acquired immunodeficiency syndrome crisis. In the USA, the FDA regulates this process of formalized non‐clinical trial access while in the European Union (EU) the responsibility lies with individual member states. 3 The exact conditions, types (single patient, group, protocolized, emergency) and definitions of EA vary between member states. 4 The numbers of requests for EA are growing 5 and state and federal legislation, such as Right‐to‐Try laws in the US, stress the need and interest of patients in having earlier access to medicines that are still under clinical investigation.
Also of interest, and closely related to EA, is the field of real‐world data (RWD). RWD are “information on health care that is derived from multiple sources outside typical clinical research settings”. 6 Recent publications and regulatory frameworks have boosted the promise of RWD. 7 , 8 , 9 It can come in many forms and shapes, such as electronic health records, social media or claims databases.
EA programmes are generally considered to be a source of RWD. 10 Historically, however, EA programmes were only deemed fit for treatment and not for research. Although the primary purpose of EA is treatment, scholars have argued that there is a moral obligation to collect outcome data in all cases where patients are treated with investigational medicine. 11 , 12 , 13 The debate on combining data collection and EA has substantially increased, 14 , 15 , 16 with FDA officials confirming, beginning 2018, their willingness to review data from EA programmes to support drug applications. 11
Considering the increasing interest in both EA and RWD, the question arises of whether alternative routes of access to novel treatments can provide clinical information and impact regulatory decision making. In this research, we systematically assess the role of RWD from EA programmes in the regulatory approval process of the FDA and the European Medicines Agency (EMA), comparing and characterizing all approvals that utilize RWD on efficacy from EA programmes.
2. METHODS
In the USA, the FDA oversees both EA programmes and marketing authorizations. In the EU, EA is supervised by individual member states, whereas marketing authorizations are granted by the EMA via the centralized procedure. Therefore, to obtain an overview of whether data from EA programmes were used for submissions, we downloaded the Drugs@FDA database 17 and the EMA medicines overview 18 on 1 May 2019. For the FDA database, we downloaded the application documents (labels and reviews) associated with all approvals available in the database. Next, we retrieved documents from the drug approval packages sites. For the EMA, for each approved drug, we saved the scientific discussion, label and/or public assessment report that are listed in the database. Figure 1 gives a schematic overview of our method.
FIGURE 1.
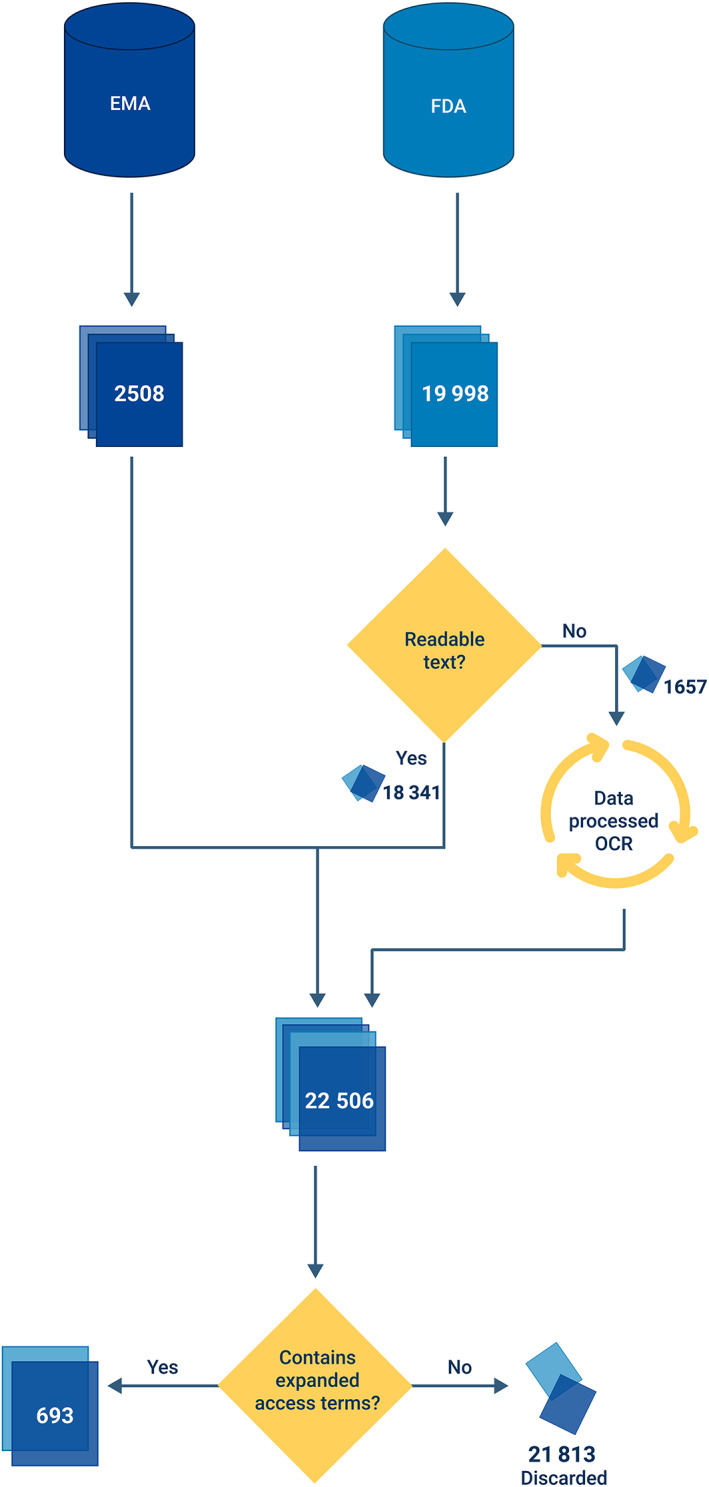
Flowchart of automated candidate search. We searched through all FDA and EMA documentation for expanded‐access related terms (compassionate use, expanded access, early access, preapproval access, named patient and managed access). When these terms appear, the document is considered a candidate. For scanned files, optical character recognition was used
As some of the FDA documents are scanned files, we first performed optical character recognition using Google's Tesseract engine, 19 to extract text from the scans and subsequently process the extracted text.
To find candidate documents, i.e. documents that mention EA, we searched for the related terms: compassionate use, expanded access, early access, preapproval access, named patient and managed access. We expected these terms to appear if the data from an associated EA programme were used in the submission package. When at least 1 of these terms appeared in the document, the associated submission package possibly included EA data for the approval. Therefore, we assessed these candidates manually to determine whether data from EA were used in a supportive/pivotal manner, or whether the mention of EA‐related terms was not in support of efficacy. The manual assessment was performed by T.B.P. and in case of doubt discussed with C.A.U.‐d.G.
As all (preapproval) data concerning patient safety are reported for purposes of pharmacovigilance, we focused on data from efficacy. Patients in EA programmes are never randomized, and the absence of a direct control group makes it challenging to draw sound conclusions on efficacy. Nonetheless, this makes it even more attractive to understand the reasons that led to acceptance of EA programmes as source of evidence.
Duplicates are removed from our data set. Duplication in this sense occurs when an approved treatment consists of multiple (recurring) compounds and the underlying data is duplicated. If no new data from EA were used, we removed such duplicates.
To determine whether EA data were included in the clinical efficacy profile, we followed 2 criteria. First, the data from the EA programme must have been mentioned under the section clinical efficacy in the medical/summary review (FDA) or scientific discussion/public assessment report (EMA). In addition, we studied the impact of the evidence. If the EA data were mentioned under the pivotal/main studies, we considered the data to have a pivotal (P) level of evidence. If not, we labelled the evidence as supportive (S). For all candidate documents, we considered related approved treatment. Figure 2 illustrates our review procedure.
FIGURE 2.
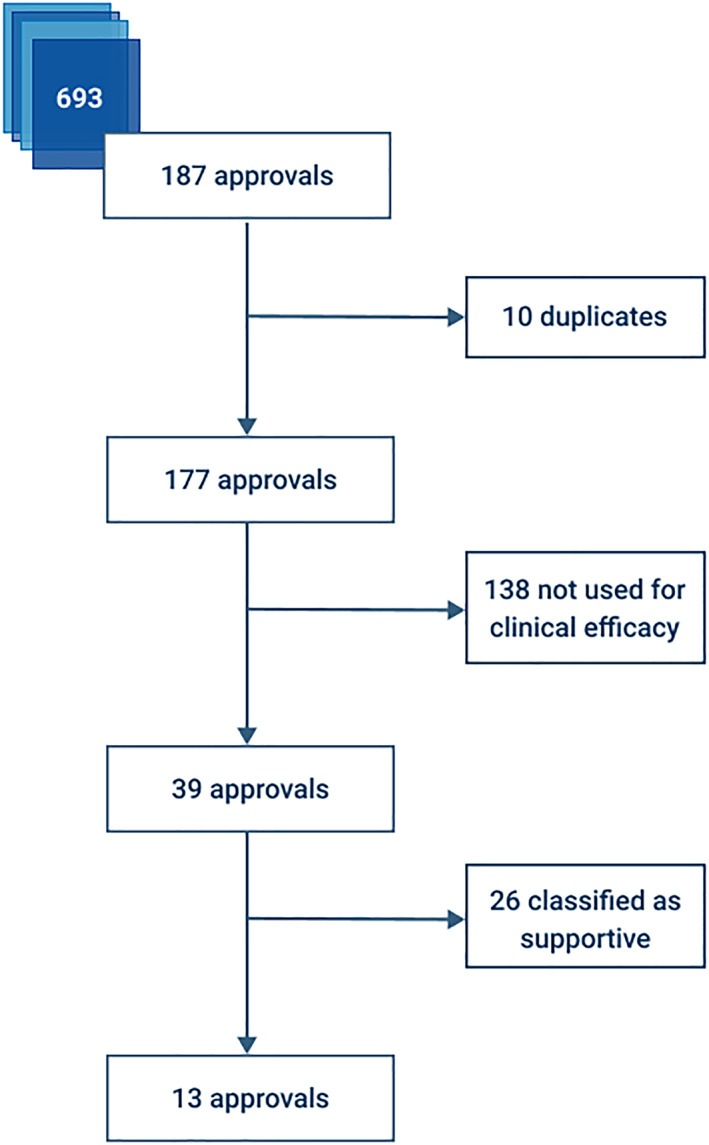
Flowchart of review process. We manually reviewed and deduplicated 187 approvals related to the candidate documents. All approvals that used expanded access (n = 39) to support clinical efficacy were analysed. For 13 approvals, the evidence from expanded access was the pivotal source of evidence
Finally, we further inspected the group of approvals that included RWD from EA programmes in terms of disease areas, orphan designation, timing of marketing approval and the number of patients in the EA programmes relative (n EA) to the total number of patients (N) in the trials. We used descriptive statistics to describe our data set. To compare our subset of approvals to regular approvals with respect to the number of orphan designations, we used χ2 tests with a 2‐sided significance level of .05. To detect a trend over time in the yearly numbers of approvals, we used a Spearman rank correlation test with a 2‐sided significance level of .05.
3. RESULTS
In total, 693 out of 22,506 scanned documents contained terms related to EA. The number of documents is skewed between agencies (2,508 EMA, 19,998 FDA), but this is mainly due to the nature of documentation. The FDA database distinguishes between medical reviews, chemistry reviews, pharmacology reviews, microbiology reviews, statistical review summary reviews and even all‐version updates thereof. The EMA merges the content of reviews in public assessment reports and scientific discussions. These 693 documents referenced 187 unique drug approvals, 126 from the FDA and 93 from the EMA (32 overlap). The FDA database contains documentation dating back to 1955. The first EMA documentation is available since 1995.
As a first step, we removed 10 duplicates, leaving 177 approvals. For example, the safety profile of tenofovir disoproxil mentions a “compassionate use program”. 20 This is repeated in all documentation regarding highly active antiretroviral therapy in the treatment of HIV, of which tenofovir disoproxil is part.
Second, we determined whether data from EA programmes was used to back the profile of clinical efficacy of the treatment. This was the case in 22% (39/177) of all approvals. The FDA considered efficacy data in 25 cases, the EMA in 24 (10 overlapped). Interestingly, nearly 3/4 (29/39) of these drugs were granted orphan designation. We encountered the first use of RWD from EA in 1993. From the beginning of 1993 to the end of 2013, the average number of approvals that included EA‐efficacy data per year was 1.24 (standard deviation 1.09) vs 4.6 (standard deviation 1.14) from 2014–2018. We observe a clear increase over the years with a Spearman correlation of 0.40 (P = .042). Figure 3 shows the distribution of these approvals by the EMA and the FDA, alongside their level of evidence, in a Venn diagram. Figure 4 displays the date of marketing authorization for these approvals. The complete list of approvals can be found in the Supplementary Files.
FIGURE 3.
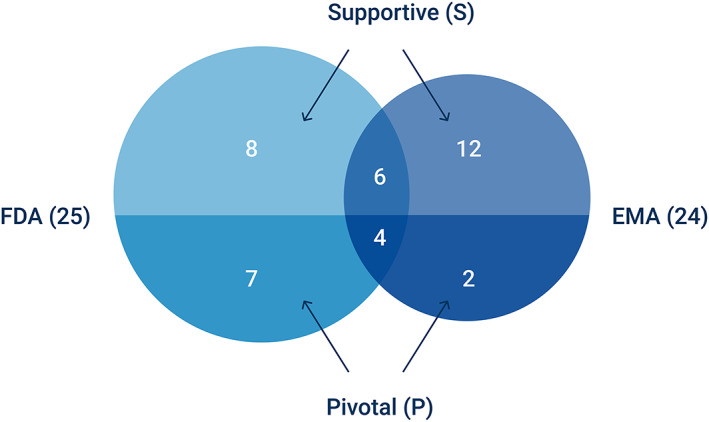
Venn‐diagram of approvals where the Food and Drug Administration (FDA) and/or European Medicines Agency (EMA) relied on data from expanded access programmes to form the clinical efficacy profile. The level of evidence associated to these data by either regulator could be pivotal or supportive
FIGURE 4.
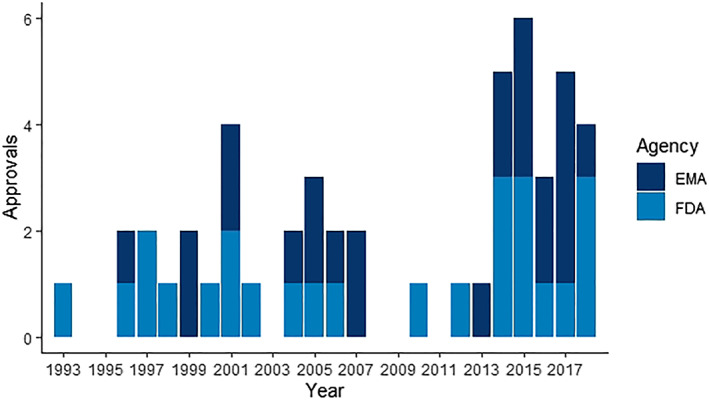
Bar chart of dates of marketing authorization of 25 Food and Drug Administration (FDA) and 24 European Medicines Agency (EMA) approvals that relied on real‐world data from expanded access for the clinical efficacy profile
3.1. Expanded access as pivotal evidence
We further investigate the approvals where RWD from EA programmes played a pivotal role. This was the case in 1 third (13/39) of the group mentioned in the previous paragraph. Table 1 gives an overview of these approvals. Twelve out of 13 received orphan designation. This is significantly higher if we compare it to regular approvals. For example, EMA assigned orphan designation to 134 out of 1,111 all‐time approved drugs vs 12 out of 13 drugs in the pivotal group (P < .0001). If we characterize by indication, just under half of the approvals (6/13) concerned treatments for metabolic disorders. The remainder are divided between haemato‐oncology (3 indications), infectious diseases (2 indications) and overdosing (2 indications), all covering areas of high unmet medical need.
TABLE 1.
Overview of all Food and Drug Administration (FDA) and European Medicines Agency (EMA) approvals that rely in a pivotal way on real‐world data from expanded access (EA) programmes
| Generic name | Indication | FDA a | EMA | Studies b | n EA/N c |
|---|---|---|---|---|---|
| Amphotericin B | Fungal infections | 1997 | N | 10 CT (n = 2038), 1 EA (n = 133) | 0.06 (133/2171) |
| Anagrelide | Essential thrombocythaemia | 1997 | 2004 | 2 SACT (n = 35 + 254), 1EA (n = 245) | 0.45 (245/538) |
| Cholic acid | Inborn errors of bile acid metabolism | 2015 | 2015 | 2 EA (n = 63 + 22) | 1.00 (85/85) |
| Clarithromycin | Mycobacterium avium complex | 1993 | N d | 1 RCT (n = 154), 1 SACT (n = 25), 1 EA (n = 469) | 0.72 (469/648) |
| Dinutuximab β | Neuroblastoma | N e | 2017 | 1 RCT (n = 370), 1 SACT (n = 44), 1 EA (n = 54) | 0.12 (54/468) |
| Fish oil triglycerides | Parenteral nutrition‐associated cholestasis | 2018 | N | 1 SACT (n = 144), 1 EA (n = 37) | 0.20 (37/181) |
| Glucarpidase | Elevated metrotrexate levels | 2012 | W | 1 SACT (n = 147), 1 EA (n = 22) | 0.13 (22/169) |
| Lutetium oxodotreotide | Neuroendocrine tumours | 2018 | 2017 | 1 RCT (n = 229), 1 EA (n = 558) | 0.71 (558/787) |
| Nitisinone | Tyrosinaemias | 2002 | 2005 | 1 EA (n = 207) | 1.00 (207/207) |
| Sodium phenylacetate/benzoate | Acute hyperammonaemia in urea cycle disorders | 2005 | N | 1 EA (n = 316) | 1.00 (316/316) |
| Uridine triacetate | Fluoruacil or capeticabine overdose | 2015 | N | 2 EA (n = 75 + 60) | 1.00 (135/135) |
| Velmanase α | Alpha‐mannosidosis | N | 2018 | 1 RCT (n = 25), 1 EA (n = 35) | 0.58 (35/60) |
| Vestronidase α | Mucopolysaccharidosis VII | 2017 | 2018 | 2 SACT (n = 3 + 12), 1 EA (n = 2) f | 0.12 (2/17) |
Year of EMA/FDA approval (if applicable). W: withdrawn; N: not approved.
Main studies that provided information on efficacy. SACT = single arm clinical trial; EA = expanded access; (R)CT = (randomized) controlled trial.
Ratio of patients in expanded access (n EA) to total number of patients in main studies (N)
Clarithromycin is approved in individual member states, before the introduction of the centralized procedure.
Dinutixumab α is marketed in the USA (Unituxin) but not anymore in the EU (replaced by β). As α and β are not exactly equal, we opted not to compare α and β.
The EMA did not consider the 2 patients in EA for clinical efficacy.
The median ratio of patients from EA programmes to the total patient population (n EA/N) that pivotally reinforced the efficacy profile was 71% (interquartile range: 34–100). In absolute terms, this varies from only 2 (vestronidase alfa) up to 558 patients (lutetium oxodotreotride). Even though this a small sample, the former 2 patients formed 12% (2/17) of the total patient population in pivotal studies. By contrast, 558 patients comprised 71% (558/787) of the total patient population, meaning that almost ¾ of the patient population was treated under EA programmes. Although 558 is the largest number of patients we observed in the pivotal group, we have encountered EA programmes containing >13 000 patients (stavudine) that provided information on efficacy with a supportive level of evidence.
3.2. Expanded access as sole evidence
Strikingly, the evidence from EA programmes was the only evidence in 4 cases: (i) sodium phenylacetate and sodium benzoate (FDA); (ii) uridine triacetate (FDA); (iii) cholic acid (FDA/EMA); and (iv) nitisinone (FDA/EMA). We describe these approvals here in more detail.
The combination of sodium phenylacetate and sodium benzoate (i) is indicated for the acute treatment of hyperammonaemia in patients with urea cycle disorders, a rare disorder causing dangerously elevated ammonia levels. The observed treatment effect was considerable; historical control data showed a 48% survival rate, whereas 80% of the patients treated under EA with sodium phenylacetate and sodium benzoate survived. 21
Uridine triacetate (ii) treats patients following 5‐fluoruoacil or capecitabine overdose. Overdosing can lead to life threatening toxicities; uridine triacetate was therefore administered under emergency EA. Historical control data indicated that 16% of patients receiving only supportive care survived. In the EA programme of uridine triacetate, survival rate was 97%. 22
Cholic acid (iii) is approved for the life‐long treatment of bile acid synthesis disorders. It replaces the abnormal bile acids produced by patients with inborn errors in primary bile acid synthesis. Effectiveness was established by comparing changes in bile acid levels before and after treatment. The submission package only included RWD from EA programmes, because “the indications for which the product in question is intended are encountered so rarely that the applicant cannot reasonably be expected to provide comprehensive evidence, and that it would be contrary to generally accepted principles of medical ethics to collect such information.” 23
Nitisinone (iv) is a treatment for hereditary tyrosinaemia type 1 and it prevents the flawed conversion of tyrosine. The EA programme was coordinated from Sweden and patients were treated in 87 different hospitals in 25 countries. Marketing approval was granted “in view of the rare occurrence and seriousness of the disease, the lack of therapeutic alternatives and the obvious clinical efficacy.” 24
4. DISCUSSION
Having analysed all available approval documentation, we observe that expanded access programmes can provide information on clinical efficacy that impacts regulatory decision making. Furthermore, we find that sponsors and regulators increasingly include RWD from EA programmes in the efficacy profile of an approval package. The indications of these approvals are characterized by their orphan designation and high unmet medical need, a specific group of conditions. Although the use of RWD may seem a novel application, our study shows that, in the case of EA, RWD was already used before the year 2000. The EA programmes propelled by acquired immunodeficiency syndrome activism already led to the use of RWD: in 1996, for abacavir (FDA) or 1997 for stavudine (EMA): both compounds are used in the management of HIV.
The specific circumstances in which RWD can complement or even substitute RCTs 25 , 26 , 27 are in line with the motivations behind the use of RWD from EA programmes. One of these conditions arises when randomization is unethical, for example due to a large unmet medical need. 25 By design, this prerequisite is also a criterion of EA: EA programmes are only for patients in dire need of unapproved treatments. A second situation occurs when further randomized controlled research is infeasible, e.g. when patient populations are small due to low disease incidence. 26 Our study shows that indeed, orphan designations characterize RWD use from EA. As the rising interest in personalized medicine results in sample sizes becoming even smaller, data from EA may become increasingly important. Although in most cases data collected from EA programmes complement data from conventional clinical trials, in 4 approvals in our research, EA data were the sole source of evidence. The absence of other approved therapies, the rarity of the condition and the large observed treatment effect formed the extraordinary circumstances that led to the approval without evidence from (randomized) clinical trials. In particular conditions it may be challenging to collect data in conventional highly controlled settings and therefore collection of RWD from EA programmes may be crucial.
Interestingly, there is no guidance on the collection and analysis of data within EA programmes. This seems rather odd, as our results show that RWD have been used for some time, there is an increasing demand of patients and physicians for preapproval access to investigational treatments, and, recently, pharmaceutical industry demanded such guidance. 28 Some European countries prohibit the collection of data in an EA setting, stating that “no other data except pharmacovigilance data can be gathered which will only be used for the evaluation of the UMN (red: Unmet Medical Need/expanded access) program.” 29 Well‐designed data collection in clinical trials should be considered a prerequisite for scientifically sound conclusions. EA programmes harbour inherent flaws—such as data quality issues—and, first and foremost, the lack of randomization. Although this should always be kept in mind, it does not mean that the information from patients treated under EA should simply be ignored. On the contrary, various approvals rest on data from single‐arm clinical trials. 30 Although patients in EA programmes are not eligible for clinical trials, this does not imply that their treatment data do not qualify for analysis. Data from every patient could provide useful insights and moreover, our study shows that the EMA has considered data from these patients critical in specific approvals. A paradoxical situation arises when individual member states do not allow RWD collection during EA programmes, yet the EMA uses these RWD in decision making. Harmonization across regulators and individual member states should solve these paradoxes.
We encountered differences in regulatory decisions. In 7 cases, the FDA considered data pivotal to the approval whereas the EMA had not approved the product (6 cases) or the data were merely considered supportive (1 case). Conversely, the FDA has not (yet) approved 2 products whereas the EMA has. Despite international drug regulation harmonization efforts 31 , 32 there is still room for regulatory cooperation across the Atlantic.
Considering our observations in a greater context it appears conventional lines between treatment and research are becoming blurred. This is true for both the field of RWD as a whole and for RWD from EA in particular. An example of the former are administrative data used for analyses: data that were not collected with the purpose of research are now found at the heart of an analysis. Similarly for the latter, where EA programmes were traditionally also meant exclusively for treatment, data collection and thereby research has become a reality. The changing position of EA patients from treatment subjects to (partly) research subjects, provides a challenge for bioethicists. 11 , 12 , 33
When it comes to comparisons between RCTs and RWD, both have their merits. On the one hand, the control of variability and assurance of data quality in RCTs leads to valid results. On the other hand, these trials target specific homogenous patient populations, e.g. younger and with fewer comorbidities, which limits the generalizability of findings. 26 , 27 RWD represents a more heterogenous or real‐life population, conclusions drawn on RWD are arguably more applicable in day‐to‐day clinical settings. 6 , 9 Finding the right balance between RWD and RCTs can become an interesting topic for (bio)statisticians, (pharmaco)epidemiologists, regulators and industry.
Awareness of the potential value of RWD from EA should facilitate that these data are used appropriately. This helps pharmaceutical industry and regulators determine whether EA—and associated RWD—is useful. For patients, this would hopefully result in speedier access to more diverse treatments.
4.1. Limitations and future research
Previous literature focused on the legal and ethical implications of EA 3 , 34 , 35 , 36 , 37 , 38 or attempted to characterize (US) EA programmes in terms of associated clinical holds, impact on product labelling, acceptance rates or dates of initiation. 5 , 39 , 40 , 41 , 42 This study is the first to systematically identify, compare and categorize all EMA and FDA approvals that rely on RWD from EA. This differs from previous literature comparing approvals, focusing on the absence of randomized controlled trials. 30 Additionally, to analyse the entire history of RWD use from EA, no time limit was used. We are limited by the fact that the Drugs@FDA database includes only drugs and therapeutics (no other biologics) and consistently included reviews only after 1997.
Using recent advances in artificial intelligence to facilitate the processing of documents, we were able to analyse many approvals. As only approvals that appeared EA‐related were assessed manually, the possibility remains that cases where EA data were in fact used were missed because these terms did not appear in the documentation. Although it is unlikely that such terms would not appear in relevant documents, our numbers therefore form a lower bound of the real number of cases where EA data were used for approvals.
Future research could focus on statistical implications of combining data from EA programmes and controlled trials. Additionally, we have only investigated the use of EA efficacy data for regulators. Its influence on other stakeholders such as payors or drug developers is a subject that could be pursued through further research.
5. CONCLUSION
EA programmes can generate real‐world evidence prior to drug approval. EMA and FDA increasingly utilize RWD from EA in regulatory decision making. The treatments in these approval decisions involved orphan designations and high unmet medical need.
COMPETING INTERESTS
Tobias Polak received an unrestricted grant from myTomorrows for a part‐time doctorate trajectory. Tobias Polak works part‐time as an RWD lead for myTomorrows. Joost van Rosmalen declares no conflict of interest. Carin Uyl‐de Groot has received unrestricted grants from Boehringer Ingelheim, Astellas, Celgene, Sanofi, Janssen‐Cilag, Bayer, Amgen, Genzyme, Merck, Glycostem Therapeutics, Astra Zeneca, Roche and Merck.
CONTRIBUTORS
T.B.P., J.v.R. and C.A.U.‐d.G. contributed to the concept and design, T.B.P. acquired and analysed the data and drafted the manuscript, J.v.R. and C.A.U.‐d.G revised the manuscript.
Supporting information
DATA S1 Supporting Information
ACKNOWLEDGEMENTS
The code and analysed datasets are available on request and can be found at research.mytomorrows.com and on the GitHub of the corresponding author. The authors thank David Cucchi, Noor Gieles and Simon de Wijs for proof reading. This study was supported by an unrestricted grant from myTomorrows.
Polak TB, van Rosmalen J, Uyl – de Groot CA. Expanded Access as a source of real‐world data: An overview of FDA and EMA approvals. Br J Clin Pharmacol. 2020;86:1819–1826. 10.1111/bcp.14284
There was no principal investigator for this study as it did not concern medical research with human subjects.
REFERENCES
- 1. Kimberly LL, Beuttler MM, Shen M, Caplan AL, Bateman‐House A. Pre‐approval access terminology. Ther Innov Regul Sci. 2017;51(4):494‐500. 10.1177/2168479017696267 [DOI] [PubMed] [Google Scholar]
- 2. Young FE, Norris JA, Levitt JA, Nightingale SL. The FDA's new procedures for the use of investigational drugs in treatment. JAMA J am Med Assoc. 1988;259:2267‐2270. 10.1001/jama.1988.03720150043034 [DOI] [PubMed] [Google Scholar]
- 3. Darrow JJ, Sarpatwari A, Avorn J, Kesselheim AS. Practical, legal, and ethical issues in expanded access to investigational drugs. N Engl J Med. 2015;372(3):279‐286. 10.1056/NEJMhle1409465 [DOI] [PubMed] [Google Scholar]
- 4. Balasubramanian G, Morampudi S, Chhabra P, Gowda A, Zomorodi B. An overview of compassionate use programs in the european union member states. Intractable Rare Dis Res. 2016;5(4):244‐254. 10.5582/irdr.2016.01054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jarow JP, Lemery S, Bugin K, Khozin S, Moscicki R. Expanded access of investigational drugs: the experience of the Center of Drug Evaluation and Research over a 10‐year period. Ther Innov Regul Sci. 2016;50(6):705‐709. 10.1177/2168479016656030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sherman RE, Anderson SA, Dal Pan GJ, et al. Real‐world evidence — what is it and what can it tell us? N Engl J Med. 2016;375:2293‐2297. 10.1056/NEJMsb1609216 [DOI] [PubMed] [Google Scholar]
- 7. Stower H. The promise of real‐world data. Nat Med. 2019. 10.1038/d41591-019-00010-z [DOI] [PubMed] [Google Scholar]
- 8. Food and Drug Administration . Submitting Documents Using Real‐World Data and Real‐World Evidence to FDA for Drugs and Biologics Guidance for Industry DRAFT GUIDANCE 2019.
- 9. Blommestein H. The added value of real‐world evidence. Erasmus University Rotterdam, 2016. [Google Scholar]
- 10. Leveraging Real‐World Treatment Experience from Expanded Access Protocols. 2018.
- 11. Chapman CR, Moch KI, McFadyen A, et al. What compassionate use means for gene therapies. Nat Biotechnol. 2019;37(4):352‐355. 10.1038/s41587-019-0081-7 [DOI] [PubMed] [Google Scholar]
- 12. Bunnik EM, Aarts N, van de Vathorst S. Little to lose and no other options: ethical issues in efforts to facilitate expanded access to investigational drugs. Health Policy. 2018;122(9):977‐983. 10.1016/j.healthpol.2018.06.005 [DOI] [PubMed] [Google Scholar]
- 13. Fountzilas E, Said R, Tsimberidou AM. Expanded access to investigational drugs: balancing patient safety with potential therapeutic benefits. Expert Opin Investig Drugs. 2018;27(2):155‐162. 10.1080/13543784.2018.1430137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rawson K. Expanded Access Data Can Support Approval Decisions, US FDA Says n.d. https://pink.pharmaintelligence.informa.com/PS124296/Expanded-Access-Data-Can-Support-Approval-Decisions-US-FDA-Says (accessed June 19, 2019).
- 15. Sutter S. Expanded Access Programs Eyed For Data‐Gathering Purposes n.d. https://pink.pharmaintelligence.informa.com/PS122926/Expanded-Access-Programs-Eyed-For-DataGathering-Purposes%0D.
- 16. Usdin S. Beyond compassionate use: the case for using expanded access protocols to generate real world data. n.d. https://www.biocentury.com/biocentury/regulation/2017-09-29/case-using-expanded-access-protocols-generate-real-world-data (accessed June 9, 2019).
- 17. FDA . Drugs@FDA n.d. https://www.fda.gov/drugs/drug-approvals-and-databases/drugsfda-data-files (accessed May 1, 2019).
- 18. EMA . EMA: Download medicine data. n.d. https://www.ema.europa.eu/en/medicines/download-medicine-data (accessed May 1, 2019).
- 19. Smith R. An overview of the tesseract OCR engine. Proc. Int. Conf. Doc. Anal. Recognition, ICDAR, 2007. 10.1109/ICDAR.2007.4376991. [DOI]
- 20. EMA . Viread: European Public Assessment Report. n.d.
- 21. FDA . NDA 20–645: Ammonul ‐ Medical Review(s). 2005.
- 22. Ison G, Beaver JA, McGuinn WD, et al. FDA approval: uridine triacetate for the treatment of patients following fluorouracil or Capecitabine overdose or exhibiting early‐onset severe toxicities following Administration of these Drugs. Clin Cancer Res. 2016;22(18):4545‐4549. 10.1158/1078-0432.CCR-16-0638 [DOI] [PubMed] [Google Scholar]
- 23. EMA . Public Assessment Report: Kolbam. 2015.
- 24. EMA . Scientific Discussion: Orfadin. 2005.
- 25. Eichler HG, Bloechl‐Daum B, Bauer P, et al. “Threshold‐crossing”: a useful way to establish the counterfactual in clinical trials? Clin Pharmacol Ther. 2016;100(6):699‐712. 10.1002/cpt.515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berger ML, Sox H, Willke RJ, et al. Good practices for real‐world data studies of treatment and/or comparative effectiveness: recommendations from the joint ISPOR‐ISPE special task force on real‐world evidence in health care decision making. Pharmacoepidemiol Drug Saf. 2017;26(9):1033‐1039. 10.1002/pds.4297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dreyer NA. Advancing a framework for regulatory use of real‐world evidence: when real is reliable. Ther Innov Regul Sci. 2018;52(3):362‐368. 10.1177/2168479018763591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brennan Z. Gilead and Novartis Seek to Expand What FDA Considers as Real‐World Data and Real‐World Evidence. n.d. https://www.raps.org/news-and-articles/news-articles/2019/7/gilead-and-novartis-seek-to-expand-what-fda-consid (accessed August 12, 2019).
- 29. FAQ's ‐ Unmet Medical Need. n.d. https://www.fagg-afmps.be/sites/default/files/content/faq_1.5_20190726.pdf (accessed August 12, 2019).
- 30. Hatswell AJ, Baio G, Berlin JA, Irs A, Freemantle N. Regulatory approval of pharmaceuticals without a randomised controlled study: analysis of EMA and FDA approvals 1999–2014. BMJ Open. 2016;6(6) e011666. 10.1136/bmjopen-2016-011666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mutual Recognition Agreement (MRA) – FDA. n.d. https://www.fda.gov/international-programs/international-arrangements/mutual-recognition-agreement-mra (accessed August 12, 2019).
- 32. Mutual recognition agreements (MRA) – EMA. n.d. https://www.ema.europa.eu/en/human-regulatory/research-development/compliance/good-manufacturing-practice/mutual-recognition-agreements-mra#united-states-section (accessed August 12, 2019).
- 33. Bunnik EM, Aarts N, van de Vathorst S. The changing landscape of expanded access to investigational drugs for patients with unmet medical needs: ethical implications. J Pharm Policy Pract. 2017;10:10 10.1186/s40545-017-0100-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hyry HI, Manuel J, Cox TM, Roos JCP. Compassionate use of orphan drugs. Orphanet J Rare Dis. 2015;10:100 10.1186/s13023-015-0306-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Joffe S, Lynch HF. Federal Right‐to‐try Legislation — threatening the FDA's public health Mission. N Engl J Med. 2018;378(8):695‐697. 10.1056/NEJMp1714054 [DOI] [PubMed] [Google Scholar]
- 36. Bateman‐House A, Robertson CT. The Federal Right to try act of 2017—a wrong turn for access to investigational drugs and the path forward. JAMA Intern Med. 2018;178:321 10.1001/jamainternmed.2017.8167 [DOI] [PubMed] [Google Scholar]
- 37. Jacob JA. Questions of safety and fairness raised as right‐to‐try movement gains steam. JAMA. 2015;314(8):758‐760. 10.1001/jama.2015.7691 [DOI] [PubMed] [Google Scholar]
- 38. Folkers KM, Bateman‐House A. Improving expanded access in the United States. Ther Innov Regul Sci. 2018;52(3):285‐293. 10.1177/2168479018759661 [DOI] [PubMed] [Google Scholar]
- 39. Jarow JP, Moscicki R. Impact of expanded access on FDA regulatory action and product labeling. Ther Innov Regul Sci. 2017;51(6):787‐789. 10.1177/2168479017707800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Puthumana J, Miller JE, Kim J, Ross JS. Availability of investigational medicines through the US Food and Drug Administration's expanded access and compassionate use programs. JAMA Netw Open. 2018;1(2) e180283. 10.1001/jamanetworkopen.2018.0283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miller JE, Ross JS, Moch KI, Caplan AL. Characterizing expanded access and compassionate use programs for experimental drugs. BMC Res Notes. 2017;10(1):350 10.1186/s13104-017-2687-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McKee AE, Markon AO, Chan‐Tack KM, Lurie P. How often are drugs made available under the Food and Drug Administration's expanded access process approved? J Clin Pharmacol. 2017;57:S136‐S142. 10.1002/jcph.960 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DATA S1 Supporting Information