

Abstract
Lymphocytes such as T‐cells can be genetically transduced to express a synthetic chimeric antigen receptor (CAR) that re‐directs their cytotoxic activity against a tumour‐expressed antigen of choice. Autologous (patient‐derived) CAR T‐cells have been licensed to treat certain relapsed and refractory B‐cell malignancies, and numerous CAR T‐cell products are in clinical development. As living gene‐modified cells, CAR T‐cells exhibit unique pharmacokinetics, typically proliferating within the recipient during the first 14 days after administration before contracting in number, and sometimes exhibiting long‐term persistence. The relationship between CAR T‐cell dose and exposure is highly variable, and may be influenced by CAR design, patient immune function at the time of T‐cell harvest, phenotype of the CAR T‐cell product, disease burden, lymphodepleting chemotherapy and subsequent immunomodulatory therapies. Recommended CAR T‐cell doses are typically established for a specific product and indication, although for some products, stratification of dose based on disease burden may mitigate toxicity while maintaining efficacy. Re‐evaluation of CAR T‐cell dosing may be necessary following changes to the lymphodepleting regimen, for different disease indications, and following significant manufacturing changes, if product comparability cannot be demonstrated. Dose escalation trials have typically employed 3 + 3 designs, although this approach has limitations, and alternative phase I trial designs may facilitate the identification of CAR T‐cell doses that strike an optimal balance of safety, efficacy and manufacturing feasibility.
Keywords: chimeric antigen receptor therapy, drug interactions, drug toxicity, immunologic dose–response relationship, pharmacokinetics, phase 1 clinical trials
1. INTRODUCTION
Chimeric antigen receptor (CAR) T‐cells are viable T‐lymphocytes that have been genetically modified to express a synthetic receptor, which redirects T‐cell cytotoxicity against a target of choice. Two CAR T‐cell products, tisagenlecleucel (Kymriah, Novartis) and axicabtagene–ciloleucel (Yescarta, Gilead), both targeting the B‐cell antigen CD19, have been licensed for treatment of relapsed and refractory (r/r) B‐cell malignancies, 1 and many other CAR T‐cell products are in clinical development. 2
CARs have a modular design, permitting countless variations of specificity and function. Considerable variation between CAR T‐cell products can also arise from the phenotype of T‐cells used for CAR T‐cell manufacture, the method of gene modification, promoters and other features of the transgene that affect CAR expression, methods of T‐cell stimulation, expansion and culture, and formulation and delivery of the final product.
Upon intravenous administration to recipients, CAR T‐cells can become activated through binding to the CAR target on tumour (or on normal) cells, and proliferate to a variable degree. Following a period of activation and expansion, CAR T‐cells become exhausted, and contract in number. A population of CAR T‐cells may persist long‐term within the recipient, with the potential to mediate both durable antitumour efficacy and toxicity.
This review provides a brief background to CAR T‐cell therapies, discusses the principal methods used to quantify CAR T‐cells, summarises current knowledge of CAR T‐cell pharmacokinetics, and lists factors known to influence CAR T‐cell pharmacokinetics and pharmacodynamics. Key considerations for the design and interpretation of CAR T‐cell dose escalation trials, and for selection of an optimal dose, will be discussed.
2. BACKGROUND
2.1. History of CAR T‐cells
The use of autologous T‐lymphocytes as a cancer therapy is not new, with reports of dramatic clinical responses among some recipients of lymphokine‐activated killer cells and expanded tumour‐infiltrating lymphocytes since the 1980s. 3 , 4 , 5 However, most recipients do not respond to these adoptive T‐cell therapies, and it is possible that many individuals with cancer lack a T‐cell population capable of recognising and eradicating their tumour.
T‐cells can be redirected to target an antigen of choice by introducing a gene encoding a synthetic receptor, overcoming reliance on the endogenous T‐cell receptor (TCR) repertoire. CARs combine elements of an antibody (the B‐cell receptor) to bind to an antigen, and a component of the TCR complex to signal to the T‐cell. 6 , 7
So‐called first‐generation CARs combined an extracellular antigen recognition domain (single chain variable fragment, scFv) derived from a monoclonal antibody linked via a hinge and transmembrane domain to the intracellular CD3ζ region of the TCR complex (Figure 1A). 8 While first‐generation CARs led to T‐cell activation upon binding to the target of the scFv, the T‐cells exhibited only limited proliferation and function. 9
FIGURE 1.
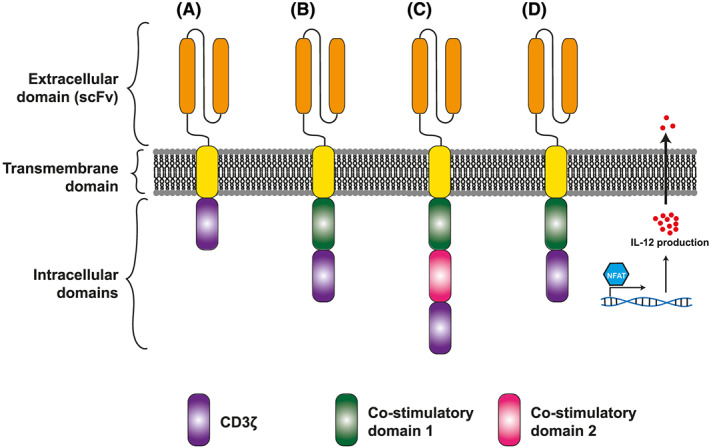
Schematic representation of chimeric antigen receptors. (A) First‐generation chimeric antigen receptors (CARs) signal via CD3ζ only, and elicit limited T‐cell effector function against target antigen. (B) Second‐generation CARs, employed in the licensed CAR T‐cell products tisagenlecleucel and axicabtagene–ciloleucel, employ a single intracellular costimulatory domain such as 4‐1BB or CD28. (C) Third‐generation CARs employ 2 costimulatory domains in sequence. (D) Bicistronic constructs allow expression of a second protein, such as the T‐cell stimulatory cytokine interleukin (IL)‐12, alongside a CAR. scFv, single chain variable fragment
Second‐generation CARs add an intracellular costimulatory domain, often derived from the T‐cell costimulatory molecules CD28 or 4‐1BB, and result in more robust T‐cell proliferation and function in response to CAR binding. 10 , 11 , 12 The licensed CAR T‐cell therapies tisagenlecleucel and axicabtagene–ciloleucel both employ second‐generation CAR constructs, incorporating costimulatory domains from 4‐1BB and CD28, respectively (Figure 1B).
A myriad of other CARs have been produced, including CARs containing alternative costimulatory domains, and third‐generation CARs combining 2 intracellular costimulatory domains (Figure 1C). 13 In addition, bicistronic transgenes can be introduced into T‐cells to drive expression of a second protein alongside the CAR, such as a T‐cell‐stimulating cytokine, or a protein that facilitates CAR T‐cell detection, enrichment or depletion (Figure 1D). 14
2.2. Manufacture of CAR T‐cells
A typical manufacturing process for CAR T‐cells is summarised in Figure 2. Patient or donor leucocytes are obtained by leukapheresis, from which T‐cells are isolated with immunomagnetic beads. T‐cells are activated and genetically transduced with a CAR‐encoding transgene in vitro. Transduction can be achieved using vectors derived from human retro‐ or lentiviruses (incorporating safety modifications to limit virulence and prevent viral replication). An alternative is genetic transduction using a transposon/transposase system, such as Sleeping Beauty or PiggyBac. 15
FIGURE 2.
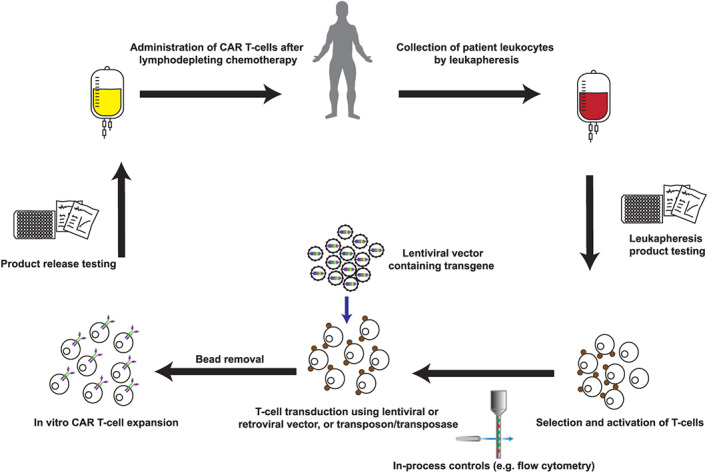
Manufacture of chimeric antigen receptor (CAR) T‐cells. Manufacture of autologous CAR T‐cells typically begins with patient blood leucocytes obtained by leukapheresis. A sample of this input product is sent for screening tests, while T‐cells are purified and activated using immunomagnetic beads or plate‐bound antibodies. In‐process controls may include purity checks by flow cytometry. A transgene is introduced using a lentiviral or retroviral vector, or a transposon/transposase system. CAR T‐cells are expanded in vitro using optimised cell culture conditions and in the presence of cytokines. Product release testing is required before CAR T‐cells are released for administration to the patient, typically following lymphodepleting chemotherapy
Following transduction, T‐cells are expanded in vitro in the presence of specific cytokines. Various in‐process controls are applied, and following manufacture, each CAR T‐cell product undergoes product release testing. Product release criteria typically include tests to ensure viability, identity, purity, microbiological sterility and stability of the CAR T‐cells. 16 In addition to product release criteria, additional tests are often conducted to assess the proportion of various T‐cell subsets within the CAR T‐cell product, and to evaluate CAR T‐cell function, or potency. 17
3. QUANTIFYING CAR T‐CELLS
CAR T‐cell pharmacokinetics are most commonly determined by quantitative polymerase chain reaction (qPCR) for the transgene, or by direct enumeration of CAR‐expressing T‐cells using flow cytometry, using peripheral blood or plasma samples.
3.1. Quantitative polymerase chain reaction
The number of CAR T‐cells can be estimated by qPCR for the CAR‐encoding transgene. A transgene‐encoding plasmid is serially diluted to generate a standard curve. After appropriate corrections for qPCR reaction efficiency and DNA quantity input, the number of transgene copies per unit DNA can be determined within CAR T‐cell recipient whole blood 18 , 19 or peripheral blood mononuclear cells. 20
Advantages of quantifying CAR T‐cells by qPCR are that it is a sensitive method, can be applied to stored DNA samples and will detect CAR DNA even if the CAR has been downregulated on the T‐cell surface. Disadvantages are that qPCR can be time consuming, limiting applicability for real‐time analysis, and that it does not distinguish between cells that express the CAR and T‐cells that harbour 1 or more CAR transgene copies but are incapable of expressing the receptor.
3.2. Flow cytometry
Flow cytometry is routinely used in clinical laboratories to quantify lymphocyte subsets in blood. A cell suspension is labelled with fluorescent reagents (often monoclonal antibodies), then analysed to determine fluorescence characteristics on a per cell basis. Several reagents can be used to detect surface expression of CARs, including monoclonal antibodies specific for the scFv domain of the CAR, 21 protein L 22 or the target antigen itself. 23 For some products, CAR T‐cells can be enumerated by detecting a second protein encoded alongside the CAR, such as a truncated EGFR polypeptide. 24
Advantages of flow cytometry over qPCR include detection only of T‐cells expressing the CAR protein, and the potential to simultaneously assess expression of other proteins on CAR T‐cells on a per‐cell basis. This allows, for example, the separate enumeration of CD4+ and CD8+ subsets of CAR T‐cells, and the determination of memory phenotype of circulating CAR T‐cells. Disadvantages of flow cytometry include a lack of assay standardisation between laboratories, difficulty distinguishing low‐level CAR expression from background fluorescence, the need for intact cells to perform the assay and, in some instances, a paucity of available reagents for CAR detection. Compared to qPCR, flow cytometry is relatively insensitive, limiting its utility for the detection of low‐level CAR T‐cell persistence. For example, Maude et al. reported the detection of persisting CAR T‐cells by qPCR, but not by flow cytometry, up to 2 years after therapy. 25
3.3. Imaging CAR T‐cells
A general limitation of both flow cytometry and qPCR is that they are typically applied only liquid samples, and do not fully reflect CAR T‐cell tissue distribution. CAR T‐cells are expected to traffic to, and potentially proliferate and persist within, tissue and tumour locations. While tissue biopsies can provide proof of principal of CAR T‐cell tissue infiltration, 26 invasive tests are unsuitable for serial monitoring in the clinic.
The direct labelling of CAR T‐cells with isotopes or superparamagnetic particles can enable their localisation using γ‐camera, positron emission tomography (PET) or magnetic resonance imaging. 20 , 27 , 28 However, the requirement for a relatively large number of labelled cells in close proximity to detect a signal, dilution of the label during CAR T‐cell proliferation, and loss of the label during CAR T‐cell death, can all limit the sensitivity and specificity of imaging, especially beyond the first few days after CAR T‐cell administration. An alternative approach is to administer a PET tracer that detects a second protein encoded by the transgene. This can enable, for example, PET imaging of a truncated PSMA expressed on CAR T‐cells, or of viral thymidine kinase activity within CAR T‐cells. 29 , 30 These techniques can inform CAR T‐cell pharmacokinetics in research settings.
4. CAR T‐CELL PHARMACOKINETICS
Unlike conventional drugs, CAR T‐cells typically proliferate within the recipient, resulting in a highly variable relationship between dose and exposure. CAR T‐cells are usually administered intravenously and, following initial localisation to the lung, may redistribute to the spleen and bone marrow within hours. 20 , 27 , 31 It is assumed that, like normal T‐cells, CAR T‐cells are capable of distributing widely into other tissues, including to tumour sites, but pharmacokinetic data are usually limited to circulating CAR T‐cell numbers.
In clinical experience of second‐generation anti‐CD19 CAR T‐cell therapies, intravenous administration of CAR T‐cells is followed by a brief decline in circulating levels, possibly reflecting distribution to tissue sites, followed by a rapid increase in numbers following activation and proliferation of the CAR T‐cells. 32 Circulating CAR T‐cell numbers frequently peak within 2 weeks of administration, after which they decline in number at a variable rate (Figure 3). 33
FIGURE 3.
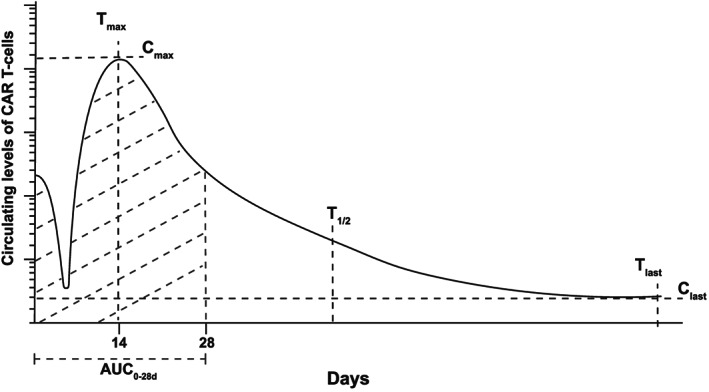
Chimeric antigen receptor (CAR) T‐cell pharmacokinetics. For second‐generation anti‐CD19 CAR T‐cells for B‐cell malignancies, an initial drop in CAR T‐cell levels after infusion is followed by a period of CAR T‐cell expansion. Peak levels (Cmax), typically occur within 14 days (tmax); t1/2 represents the half‐life, tlast represents the time at which the last measurable level (Clast) was recorded. AUC0‐28d represents the area under the concentration‐time curve during the first 28 days after CAR T‐cell administration. The y‐axis represents a log‐scale in terms of differences in the CAR T‐cell concentrations
The maximum CAR T‐cell level (Cmax) and area under the concentration–time curve between the time of CAR T‐cell administration and day 28 (AUC0‐28d) are frequently used as measures of early CAR T‐cell exposure. 32 Both reflect early CAR T‐cell expansion, and are associated with clinical response to therapy, with higher AUC0‐28d values among those who respond to second‐generation anti‐CD19 CAR T‐cell products, than among nonresponders. 32 , 34
In some recipients, CAR T‐cells persist long‐term, as determined by qPCR or, for anti‐CD19 CAR T‐cell products, as inferred by persistent depletion of normal B‐cells (B‐cell aplasia; an on‐target, off‐tumour toxicity). CAR T‐cells can persist longer than 4 years after therapy, 35 although persistence rates are variable, and may depend upon the CAR target, costimulatory domains within the CAR, CAR T‐cell phenotype, and patient and disease characteristics. 33 , 36
4.1. Factors influencing pharmacokinetics
4.1.1. Lymphodepleting chemotherapy
Treatments that deplete circulating lymphocytes increase systemic levels of T‐cell‐simulating cytokines such as IL‐7 and IL‐15, 37 , 38 , 39 and have long been employed to augment expansion of subsequently‐administered T‐cells. 40 Lymphodepletion before administration of CAR T‐cells results in improved CAR T‐cell expansion, persistence and antitumour activity. 41 , 42 Lymphodepleting chemotherapies are widely employed before CAR T‐cell administration within clinical trials and in routine practice, 43 , 44 reported regimens including cyclophosphamide with fludarabine, cyclophosphamide alone and bendamustine. 34 , 35 , 45 , 46 , 47
While the dramatic effect of lymphodepleting chemotherapy on early CAR T‐cell pharmacokinetics is principally attributed to increased levels of cytokines, 48 , 49 lymphodepletion may also facilitate long‐term CAR T‐cell persistence: lymphodepleting chemotherapy can prevent immunological rejection of CAR T‐cells due to immune responses against murine‐derived or human epitopes of the CAR. 49 , 50
As CAR T‐cell pharmacokinetics are strongly influenced by the selection and dose of lymphodepleting chemotherapy administered beforehand, package inserts for both tisagenlecleucel and axicabtagene–ciloleucel recommend specific cyclophosphamide and fludarabine dosing schedules. 51 , 52
4.1.2. Tumour burden
Although supported by serum cytokines, early expansion of CAR T‐cells in vivo is driven by CAR binding to tumour (and normal) cells that express the CAR target. Among patients with B‐cell acute lymphoblastic leukaemia (B‐ALL), the presence of >5% leukaemic blasts in the bone marrow was associated with higher Cmax and higher CAR T‐cell levels 28 days after CAR T‐cell infusion. 45 Similarly, in B‐cell non‐Hodgkin lymphoma (B‐NHL), expansion of both second‐ and third‐generation CAR T‐cells was greater in patients treated at the time of active lymphoma, than among those treated following autologous stem cell transplantation, when tumour burden was minimal. 53
4.1.3. CAR target and scFv
While the ideal CAR target might be exclusively and uniformly expressed at a high level on tumour cells, yet not expressed on normal cells, 54 the targets most successfully employed for CAR T‐cell therapy of B‐cell cancers do not meet these criteria: CD19, CD22 and B‐cell maturation antigen (BCMA) are all expressed on normal as well as malignant cells. Indeed, exposure to target antigen on normal cells may contribute to CAR T‐cell expansion and persistence or, conversely, CAR T‐cell exhaustion. 55 Fortunately, the on‐target off‐tumour toxicities of B‐cell aplasia and hypogammaglobulinaemia (reduction of normal immunoglobulin due to depletion of plasma cells) are both manageable.
For a given antigenic target, the scFv domain, which determines CAR binding affinity and specificity, can be expected to influence CAR T‐cell pharmacokinetics and pharmacodynamics. 56 A higher rate of severe neurotoxicity was observed in recipients of an anti‐CD19 CAR T‐cell therapy incorporating an scFv from the SJ25C1 antibody clone than those who received a product using the scFv from the FMC63 clone, although the relative contribution of the scFv compared to other factors remains uncertain. 34 , 57
A particular challenge is identification of suitable targets for CAR T‐cell therapy of solid cancers, for which complete response rates are very low. 58 Identification of uniformly expressed solid tumour antigens capable of supporting robust CAR T‐cell activation, expansion and on‐target cytotoxicity remains an issue, alongside measures to overcome tumour‐related immunosuppression and to assure CAR T‐cell infiltration into the tumour. 59
4.1.4. Costimulatory domains
The most frequently employed costimulatory domains within CARs are CD28 and 4‐1BB. 13 The CD28 domain is associated with rapid CAR T‐cell expansion and an effector memory phenotype, 13 , 60 while the 4‐1BB costimulatory domain is associated with slower initial CAR T‐cell expansion, enhanced persistence and a central memory phenotype. 13 , 60 Many other costimulatory domains have been reported, and their selection may influence CAR T‐cell pharmacokinetics and pharmacodynamics. 13
Third‐generation CAR T‐cells employ 2 costimulatory domains, and are associated with greater CAR T‐cell expansion and persistence than second‐generation counterparts both preclinically, and in a small clinical trial. 53 Whether this will translate to improved clinical outcomes is not yet clear.
4.1.5. CAR T‐cell phenotype
Based on the expression of specific surface markers, normal T‐cells can be divided into various subsets. These include CD4+ and CD8+ T‐cells, and various memory and more highly differentiated effector T‐cell subsets.
While most CAR T‐cell products can be expected to contain a variable mixture of CD4+ and CD8+ T‐cells, some advocate for CAR T‐cell products of defined CD4:CD8 ratio, citing improved efficacy and toxicity, 61 albeit at the expense of a more involved manufacturing process. 62
In retrospective analyses, CAR T‐cell products bearing a greater fraction of cells with memory phenotypes, or expressing higher levels of genes associated with T‐cell memory, are associated with improved expansion and persistence. 63 , 64 Some manufacturers employ manufacturing steps to select for specific memory T‐cell subsets, or modify culture conditions during in vitro CAR T‐cell expansion, to raise the fraction of CAR T‐cells expressing memory markers. 65 , 66 , 67 , 68 , 69
4.2. Drug interactions
As a cellular product, CAR T‐cells are not expected to exhibit typical pharmacokinetic interactions. However, the significant impact of prior lymphodepleting chemotherapy on CAR T‐cell pharmacokinetics has already been discussed, and subsequent immunomodulatory treatments, including those used to manage CAR T‐cell toxicities, could affect CAR T‐cell expansion, persistence or function.
4.2.1. Anti‐IL‐6 therapy
Cytokine release syndrome (CRS) and immune effector cell‐associated neurotoxicity syndrome (ICANS) are among the most significant CAR T‐cell‐related toxicities. 70 The pathogenesis of CRS in particular appears to involve the activation of myeloid cells by CAR T‐cell‐derived IL‐6, 71 and anti‐IL‐6 therapy with the monoclonal antibodies tocilizumab or siltuximab can lead to rapid clinical improvement. 70 , 72
While preclinical studies suggest that IL‐6 is not directly involved in CAR T‐cell lysis of tumour cells, 73 an adverse impact of IL‐6 blockade on CAR T‐cell pharmacokinetics or pharmacodynamics is a theoretical concern. Reassuringly, clinical experience suggests that clinical response rates are not adversely affected by tocilizumab administration. 34 , 46 For example, in a retrospective analysis of children receiving anti‐CD19 CAR T‐cells for B‐ALL, early tocilizumab use did not appear to affect Cmax, AUC0‐28d, CAR T‐cell persistence, or complete response rate. 74 Similarly, a retrospective analysis of a cohort of adults with B‐NHL reported that tocilizumab had no effect on CAR T‐cell Cmax. 75 Pending the results of prospective trials, these observations can be considered encouraging, and concerns about CAR T‐cell pharmacokinetics or function should not deter the use of anti‐IL‐6 therapies to manage toxicities.
4.2.2. Corticosteroids
High‐dose corticosteroids induce T‐cell apoptosis, 76 and are a recommended treatment for refractory CRS and severe ICANS. 72
Retrospective studies have not associated corticosteroid administration with a significant reduction in CAR T‐cell Cmax, AUC0‐28d or clinical response rates. 34 , 74 However, corticosteroid use was restricted to patients with severe tocilizumab‐refractory CRS or ICANS, among whom brisk CAR T‐cell expansion may have already been well‐established, 72 , 77 and an impact of corticosteroids on early CAR T‐cell expansion, or on persistence or durability of antitumour responses, is not excluded. 77 Accordingly, many clinical protocols restrict corticosteroid use after CAR T‐cell therapies to specific settings, in which the benefits are likely to outweigh the risks.
4.2.3. Checkpoint blockade
Like normal T‐cells, CAR T‐cells are thought to become exhausted following prolonged exposure to antigen, characteristically upregulating expression of the inhibitor programme cell death‐1 (PD‐1) receptor. 78
The function of normal antitumour T‐cells can be enhanced by therapies that prevent PD‐1 binding to its receptor, providing rationale for combining antibodies that block PD‐1, such as pembrolizumab or nivolumab, with CAR T‐cell therapies. 59 In preclinical studies, PD‐1 blockade enhances CAR T‐cell expansion and function, 78 , 79 and anecdotally, delayed rises in CAR T‐cell levels and clinical responses have been observed among patients receiving anti‐PD1 therapies for progressive B‐NHL and B‐ALL following disease progression after anti‐CD19 CAR T therapy. 80 , 81 , 82
5. OPTIMISING CAR T‐CELL DOSE
5.1. Interindividual variability
The licensed, and the majority of investigational, CAR T‐cell therapies are manufactured from autologous (patient‐derived) T‐cells. As such, a CAR T‐cell product will be influenced by a patient's immunological status before leukapheresis. 83 Patient factors, such as age, underlying disease and the number of circulating T‐lymphocytes influence the probability of successful CAR T‐cell manufacture. 84 Moreover, even when CAR T‐cell products are successfully manufactured and meet product release criteria, characteristics of the starting T‐cells may influence subsequent CAR T‐cell pharmacokinetics and pharmacodynamics. For example, a higher frequency of a memory subset of CD8+ T‐cells within leukapheresis products was associated with clinical response to subsequent anti‐CD19 CAR T‐cell therapy for chronic lymphocytic leukaemia. 64 Additionally, preclinical data suggest that interindividual differences in the immunological milieu at the time of CAR T‐cell administration can influence CAR T‐cell pharmacokinetics. 85 Therefore, when seeking to optimise CAR T‐cell dosing, it must be acknowledged that interindividual variation in CAR T‐cell pharmacokinetics is likely to be considerable, and that the factors governing this variation are still being explored.
5.2. Dose−exposure and dose–response relationships
CAR T‐cell doses are frequently provided as the number of viable CAR‐expressing T‐cells per kg recipient body weight, although some studies report CAR T‐cell doses per m2 recipient body surface area, or as the total dose administered. Because CAR T‐cells proliferate within the recipient, a linear relationship between CAR T‐cell dose and exposure cannot be assumed. Moreover, because CAR T‐cells are typically delivered as a single infusion, there is little opportunity to adjust exposure once the therapy has been administered.
Among patients with r/r B‐ALL receiving second‐generation anti‐CD19 CAR T‐cells after fludarabine and cyclophosphamide lymphodepletion, a dose of 2 × 105 CAR T‐cells kg−1 was associated with later attainment of a lower Cmax than a 10‐fold higher dose. 50 Similarly, among individuals with r/r myeloma receiving second‐generation anti‐BCMA CAR T‐cells, minimal or no CAR T‐cell expansion was observed at the 2 lowest dose levels, while robust expansion was observed after higher doses. 86 A dose–exposure relationship has also been reported for third‐generation anti‐CD19 CAR T‐cells: Cmax was lower among recipients of 2 × 107 CAR T‐cells m−2 than those receiving ≥1 × 108 cells m−2. 87
Conversely, within a dose range of 5 × 105 to 1 × 107 CAR T‐cells kg−1 Gardner et al. did not observe a relationship between dose and CAR T‐cell expansion in children with r/r B‐ALL, 88 while among patients receiving tisagenleuceucel for r/r B‐NHL, no relationship between dose and exposure (AUC0‐28d and Cmax) was seen within a total dose range of 6 × 107 to 6 × 108 cells. 89
In B‐cell malignancies including B‐ALL, B‐NHL, chronic lymphocytic leukaemia and myeloma, CAR T‐cell exposure has been associated with clinical response. 32 , 34 For example, both among children receiving tisagenlecleucel for B‐ALL and adults receiving axicabtagene–ciloleucel for B‐NHL, Cmax and AUC0‐28d were significantly greater in responders than in nonresponders. 32 , 34 An association between Cmax and clinical response rate was also reported among recipients of anti‐BCMA CAR T‐cells for myeloma. 47 , 90 In contrast, the JULIET trial of tisagenlecleucel in adults with B‐NHL did not report an association between AUC0‐28d and clinical response. 91
Both Cmax and AUC0‐28d reflect short‐term CAR T‐cell exposure but, for some malignancies, long‐term CAR T‐cell persistence may be required for durable remission. 33 CAR T‐cell persistence is thought particularly important in B‐ALL, as persistence correlates with improved disease‐free survival, 25 , 32 , 92 and CD19+ leukaemic relapse is a frequent occurrence among individuals who lose anti‐CD19 CAR T‐cell persistence. 93 In B‐NHL, the relationship between CAR T‐cell persistence and long‐term outcomes is less clear: the majority of axicabtagene–ciloleucel recipients for B‐NHL remaining free of disease at 24 months show recovery of normal B‐cell populations, 81 , 94 suggesting that CAR T‐cell persistence is not a prerequisite for durable remission in this disease.
6. DOSE−TOXICITY RELATIONSHIP
The onset of CRS is typically within 1–2 weeks of CAR T‐cell administration, coinciding with brisk CAR T‐cell expansion. 95 , 96 Several studies report that CAR T‐cell recipients who developed severe CRS had higher Cmax and/or AUC0‐28d, compared to those who did not, 45 , 47 , 50 , 89 although not all trials have observed this. 34 , 97
Neurotoxicity (ICANS), can occur concurrently with CRS, or in isolation. Anti‐CD19 CAR T‐cell trials in both r/r B‐ALL and B‐NHL have reported that severe ICANS is associated with higher AUC0‐28d and/or Cmax. 34 , 50 , 70 , 71 , 72 , 96 , 97 In contrast, among recipients of tisagenlecleucel for r/r B‐ALL, no significant association between CAR T‐cell expansion and severe neurological adverse events was reported. 32 The discrepancy between trials may be accounted for by differences in toxicity grading and management protocols, different product or disease characteristics, or because blood CAR T‐cell levels may not reflect expansion of, and cytokine production by, CAR T‐cells within tissues (including the central nervous system).
6.1. Can a therapeutic window be identified?
In B‐cell malignancies, in vivo CAR T‐cell expansion is a major determinant of exposure, and lack of expansion is associated with lack of clinical response. There appears to be a threshold dose of CAR T‐cells, beneath which robust expansion, and a clinical response, is unlikely. This threshold is likely to vary between products and treatment indications, and with patient factors, such as tumour burden and immune function. Above this threshold, escalating the CAR T‐cell dose may have little additional benefit in terms of response rate, but could potentially add to the risk of severe CRS or ICANS.
Identifying a CAR T‐cell dose that is sufficient to allow robust CAR T‐cell expansion and efficacy in the majority of recipients, without undue elevation of severe toxicity risk, may be challenging. Figure 4 illustrates 1 model, in which the therapeutic window can be conceptualised as the range between a minimum effective dose (MED) and a maximum tolerated dose (MTD). This window may be difficult to determine, may be narrow, and is likely to depend upon disease indication, and product and patient characteristics. 33
FIGURE 4.
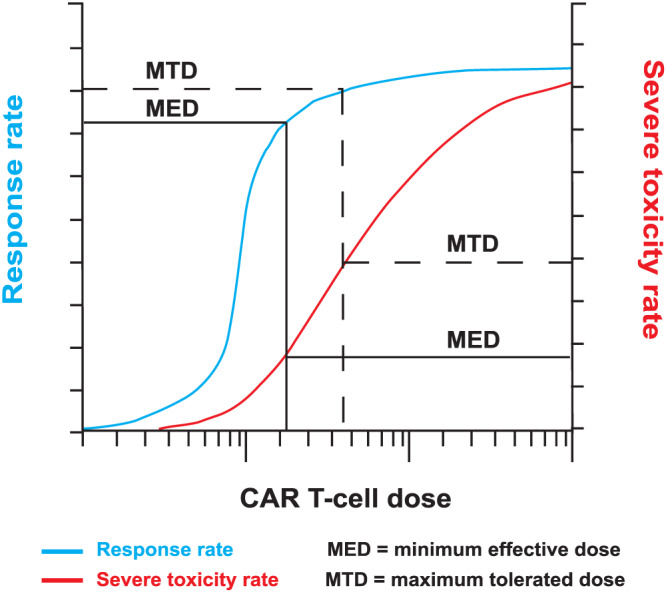
Selecting an optimal chimeric antigen receptor (CAR) T‐cell dose. Beneath a certain dose threshold, CAR T‐cells may fail to expand within the recipient, and both response and severe toxicity are unlikely. Above this threshold, clinical efficacy may increase rapidly until a minimum effective dose is reached, beyond which there may be little further improvement in response rate with increasing dose. In contrast, the rate of severe toxicities may continue to rise as the dose is increased. Phase I trial designs that seek to determine the minimum effective dose as well as the maximum tolerated dose may facilitate selection of a dose that maximises response rate without undue toxicity risk
The relationship between initial CAR T‐cell dose and long‐term persistence of the infused CAR T‐cell, and between persistence and relapse‐free survival is poorly defined, and is also likely to vary by CAR T‐cell construct and by clinical indication.
6.2. Risk‐stratified dosing
One way to overcome a narrow therapeutic window for CAR T‐cell therapies is to stratify the dose according to toxicity risk. 96 Reasoning that a heavy disease burden was associated with greater CAR T‐cell expansion and toxicity risk, Turtle et al. stratified CAR T‐cell doses in adults with B‐ALL according to pretreatment bone marrow leukaemia burden, reporting retention of antitumour efficacy without excessive toxicity. 45 Similarly, doses of CAR T‐cells directed against the plasma cell antigen BCMA have been stratified based on bone marrow myeloma burden. 47 , 86 While lymphoma disease bulk has been associated with elevated CRS and ICANS risk, 98 it is not yet clear whether or not modifying CAR T‐cell dose can retain efficacy while reducing toxicity risk in B‐NHL. 50 , 96
Various pretreatment biomarkers have been associated with elevated CRS and ICANS risk, including serum lactate dehydrogenase, C‐reactive protein, ferritin and markers of endothelial activation. 33 , 38 , 57 , 77 , 96 Whether these can be used to stratify CAR T‐cell dosing is not yet known.
6.3. Repeated dosing
In the face of marked interindividual variability in CAR T‐cell pharmacokinetics and toxicity rates, the notion of within‐patient dose escalation is appealing, allowing each recipient to first receive a low CAR T‐cell dose, to be observed for toxicities, and to receive higher doses if no toxicity is observed. Limitations of this approach include the potential need to repeatedly administer lymphodepleting chemotherapy, with its inherent toxicities.
Omitting lymphodepletion may be particularly problematic for repeated CAR T‐cell dosing, as repeated CAR T‐cell exposure can provoke immunological responses against the CAR, limiting subsequent CAR T‐cell expansion and persistence. 45 , 92 , 99 This may be mitigated by employing humanised rather than murine scFv within CAT T‐cell constructs 100 , 101 within CAR T‐cell constructs, but even fully humanised constructs can elicit anti‐CAR immune responses. 102
6.4. Dose escalation trials
Many CAR T‐cell dose escalation trials use a traditional 3 + 3 design, whereby up to 3 subjects are treated at a given dose, and the dose escalated only if fewer than a predefined number of dose limiting toxicities occur. 52 , 86 , 92 , 103 A review of CAR T‐cell trials registered at ClinicalTrials.gov up to the end of 2016, found that the typical dose escalation trial covered a 2‐log (100‐fold) dose range, typically within the range 106–109 total CAR T‐cells per subject. 2 Doses are frequently escalated in half‐log (approximately 3‐fold) to log (10‐fold) steps. 47 , 52 , 53 , 87 , 92 Dose expansion cohorts may be employed to further characterise toxicity profiles and to aid design of subsequent efficacy trials. 47 , 87 , 90
A number of factors impact dose escalation approaches, including lymphodepleting regimen, disease indication and manufacturing process. 2 , 34 , 50 , 97 For example, Turtle et al. conducted a separate anti‐CD19 CAR T‐cell dose escalation following incorporation of fludarabine into the lymphodepletion regimen, observing greater CAR T‐cell expansion and higher clinical response rates in the fludarabine‐treated cohorts. 50 Many dose escalation trials now incorporate uniform fludarabine and cyclophosphamide lymphodepletion at each dose step. 37 , 41 , 47 , 53
Manufacturing changes also have potential to impact the safety and efficacy of a CAR T‐cell product, 102 , 104 and may necessitate additional dose finding studies if product comparability cannot be assured. 105
6.4.1. Beyond MTD
Dose escalation trials classically aim to identify a MTD by assessing for dose limiting toxicities. 47 , 53 , 87 However, if there is a threshold dose above which CAR T‐cells are likely expand within the recipient, the MTD may be higher than the dose needed for maximal clinical efficacy (Figure 4). 106 Selecting an unnecessarily high CAR T‐cell dose could adversely affect both manufacturing feasibility and toxicity risk. Instead, the determination of a MED, taking account of pharmacokinetic and/or response rate as well as toxicity rates, may be a preferable goal for dose‐finding CAR T‐cell trials. 107
Phase I trial designs that incorporate both safety and efficacy data have been proposed for adoptive cellular therapies. These include Toxicity Equivalence Range and Modified Toxicity Probability Interval designs. 108 , 109 Chiuzan et al. propose a 2 stage design, in which an initial dose escalation stage incorporating a surrogate outcome (such as pharmacokinetics) is followed by second adaptive randomisation stage, which assigns subjects to all dose levels showing signals of efficacy, within safety constraints defined during stage 1. 110 A general limitation is that interindividual variability in CAR T‐cell products and their pharmacokinetics, and a potentially narrow therapeutic window, may limit the ability to determine a MED within small phase I CAR T‐cell clinical trials. 111
6.5. Cost effectiveness and off‐the‐shelf products
List prices for the licensed autologous CAR T‐cell products axicabtagene–ciloleucel and tisagenlecleucel are high, at USD $373 000 and $475 000, respectively. 112 In addition to this, funders must consider the costs of T‐cell harvest procedures, lymphodepleting chemotherapy, CAR T‐cell administration, monitoring, toxicity management and, for some recipients, immunoglobulin replacement for B‐cell aplasia. 113 These costs are likely to limit the availability of CAR T‐cell therapies in the short term, at least pending the outcomes of phase III trials.
Optimising CAR T‐cell dosing can maximise the feasibility of product manufacture, and possibly reduce toxicity risk. However, the need to produce, test, store, release and transport a personalised cellular product limits economy of scale. One solution is to develop off the shelf CAR‐transduced cellular products, in which CAR‐expressing cells are manufactured from a pool of healthy donors, from umbilical cord blood or from a cell line. The risk of donor‐vs‐recipient graft vs host disease due to allogeneic T‐cell administration can be addressed through a variety of strategies, such as by selecting tissue type‐matched donors, by expressing CARs in cells that lack a conventional T‐cell receptor (such as natural killer cells or innate‐like T‐cells), or by deleting the T‐cell receptor locus. 114 , 115 , 116 , 117 If successful, off‐the‐shelf CAR‐transduced cellular therapies may overcome a many of the logistical, feasibility, cost and product variability issues of autologous CAR T‐cell therapies. Each off‐the‐shelf CAR‐transduced cellular product will, of course, require its own dose optimisation studies.
7. CONCLUSION
As cellular gene‐transduced therapies, CAR T‐cells exhibit unusual pharmacokinetic properties, including variable degrees of expansion within the recipient, and variable long‐term persistence. The factors associated with interindividual variation in pharmacokinetics are still under investigation, but optimising CAR T‐cell dose may require consideration of underlying disease, tumour burden, lymphodepleting chemotherapy, recipient immune function, and characteristics of the manufactured CAR T‐cell product itself.
Clinical experience of autologous CAR T‐cell therapies other than those directed against CD19 is limited. CAR‐transduced cells directed against alternative target antigens, manufactured using alternative methods, transduced to express additional proteins, manufactured from cell lines or from allogeneic donors, or administered directly into tumour sites rather than intravenously may exhibit very different pharmacokinetic characteristics to those outlined in this review.
In the future, stratification of CAR T‐cell doses based on product characteristics or disease burden, the adoption of phase I trial designs that incorporate pharmacokinetic or efficacy data, and the development of CAR T‐cells with intrinsically reduced potential for toxicity, could all help clinicians and researchers optimise CAR T‐cell dosing, widen the therapeutic window and improve the availability of this emerging cancer immunotherapy.
Nomenclature of Targets and Ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
COMPETING INTERESTS
N.D., P.G. and R.W. are employees of the Malaghan Institute of Medical Research, which is the Sponsor of an investigator‐initiated phase I CAR T‐cell trial(ClinicalTrials.gov reference NCT04049513), of which R.W. is the Principal Investigator. The authors have no proprietary or financial interest in the trial or the investigational product. R.W. is in receipt of a ClinicalPractitioner Research Fellowship from the Health Research Council of NewZealand (HRC), is Principal Investigator of an HRC grant to investigate the mechanism of CAR T‐cell co‐stimulatory domains, and has received honoraria from Janssen and AbbVie.
ACKNOWLEDGEMENTS
The authors wish to acknowledge funding support from the Freemasons CAR T‐cell Research Programme and the Health Research Council of New Zealand (grants 19/139 and19/816).
Dasyam N, George P, Weinkove R. Chimeric antigen receptor T‐cell therapies: Optimising the dose. Br J Clin Pharmacol. 2020;86:1678–1689. 10.1111/bcp.14281
REFERENCES
- 1. Boyiadzis MM, Dhodapkar MV, Brentjens RJ, et al. Chimeric antigen receptor (CAR) T therapies for the treatment of hematologic malignancies: clinical perspective and significance. J Immunother Cancer. 2018;6(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hartmann J, Schussler‐Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells‐challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. 2017;9(9):1183‐1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosenberg SA, Lotze MT, Muul LM, et al. Observations on the systemic administration of autologous lymphokine‐activated killer cells and recombinant interleukin‐2 to patients with metastatic cancer. N Engl J Med. 1985;313(23):1485‐1492. [DOI] [PubMed] [Google Scholar]
- 4. Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor‐infiltrating lymphocytes. Science. 1986;233(4770):1318‐1321. [DOI] [PubMed] [Google Scholar]
- 5. Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor‐infiltrating lymphocytes and Interleukin‐2 in the immunotherapy of patients with metastatic melanoma ‐ a preliminary‐report. N Engl J Med. 1988;319(25):1676‐1680. [DOI] [PubMed] [Google Scholar]
- 6. Kuwana Y, Asakura Y, Utsunomiya N, et al. Expression of chimeric receptor composed of immunoglobulin‐derived V regions and T‐cell receptor‐derived C regions. Biochem Biophys Res Commun. 1987;149(3):960‐968. [DOI] [PubMed] [Google Scholar]
- 7. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin‐T‐cell receptor chimeric molecules as functional receptors with antibody‐type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024‐10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody‐binding domains and the gamma or zeta subunits of the immunoglobulin and T‐cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kowolik CM, Topp MS, Gonzalez S, et al. CD28 costimulation provided through a CD19‐specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66(22):10995‐11004. [DOI] [PubMed] [Google Scholar]
- 10. Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T‐lymphocyte cytotoxicity and proliferation directed by a single chimeric TCR zeta/CD28 receptor. Nat Biotechnol. 2002;20(1):70‐75. [DOI] [PubMed] [Google Scholar]
- 11. Finney HM, Lawson AD, Bebbington CR, Weir AN. Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol. 1998;161(6):2791‐2797. [PubMed] [Google Scholar]
- 12. Finney HM, Akbar AN, Lawson AD. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. 2004;172(1):104‐113. [DOI] [PubMed] [Google Scholar]
- 13. Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunology. 2019;8(5):e1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tokarew N, Ogonek J, Endres S, von Bergwelt‐Baildon M, Kobold S. Teaching an old dog new tricks: next‐generation CAR T cells. Br J Cancer. 2019;120(1):26‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vormittag P, Gunn R, Ghorashian S, Veraitch FS. A guide to manufacturing CAR T cell therapies. Curr Opin Biotechnol. 2018;53:164‐181. [DOI] [PubMed] [Google Scholar]
- 16. Roddie C, O'Reilly M. Dias Alves pinto J, Vispute K, Lowdell M. manufacturing chimeric antigen receptor T cells: issues and challenges. Cytotherapy. 2019;21(3):327‐340. [DOI] [PubMed] [Google Scholar]
- 17. Wang X, Riviere I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. 2016;3:16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl med. 2011;3(95). 95ra73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kochenderfer JN, Somerville RPT, Lu TY, et al. Long‐duration complete remissions of diffuse large B cell lymphoma after anti‐CD19 chimeric antigen receptor T cell therapy. Mol Ther. 2017;25(10):2245‐2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ritchie DS, Neeson PJ, Khot A, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21(11):2122‐2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jena B, Maiti S, Huls H, et al. Chimeric antigen receptor (CAR)‐specific monoclonal antibody to detect CD19‐specific T cells in clinical trials. PLoS ONE. 2013;8(3):e57838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zheng Z, Chinnasamy N, Morgan RA. Protein L: a novel reagent for the detection of chimeric antigen receptor (CAR) expression by flow cytometry. J Transl med. 2012;10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Oliveira SN, Wang J, Ryan C, Morrison SL, Kohn DB, Hollis RP. A CD19/fc fusion protein for detection of anti‐CD19 chimeric antigen receptors. J Transl med. 2013;11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X, Chang WC, Wong CW, et al. A transgene‐encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J med. 2014;371(16):1507‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. O'Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII‐directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl med. 2017;9(399):eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Weist MR, Starr R, Aguilar B, et al. PET of adoptively transferred chimeric antigen receptor T cells with (89)Zr‐Oxine. J Nucl med. 2018;59(10):1531‐1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meier R, Golovko D, Tavri S, et al. Depicting adoptive immunotherapy for prostate cancer in an animal model with magnetic resonance imaging. Magn Reson med. 2011;65(3):756‐763. [DOI] [PubMed] [Google Scholar]
- 29. Minn I, Huss DJ, Ahn HH, et al. Imaging CAR T cell therapy with PSMA‐targeted positron emission tomography. Sci Adv. 2019;5(7):eaaw5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keu KV, Witney TH, Yaghoubi S, et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci Transl med. 2017;9(373):eaag2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wallace PK, Palmer LD, Perry‐Lalley D, et al. Mechanisms of adoptive immunotherapy: improved methods for in vivo tracking of tumor‐infiltrating lymphocytes and lymphokine‐activated killer cells. Cancer Res. 1993;53(10 Suppl):2358‐2367. [PubMed] [Google Scholar]
- 32. Mueller KT, Maude SL, Porter DL, et al. Cellular kinetics of CTL019 in relapsed/refractory B‐cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. 2017;130(21):2317‐2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Milone MC, Bhoj VG. The pharmacology of T cell therapies. Mol Ther Methods Clin Dev. 2018;8:210‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T‐cell therapy in refractory large B‐cell lymphoma. N Engl J med. 2017;377(26):2531‐2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl med. 2015;7(303). 303ra139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Park JH, Geyer MB, Brentjens RJ. CD19‐targeted CAR T‐cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127(26):3312‐3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kochenderfer JN, Somerville RPT, Lu T, et al. Lymphoma remissions caused by anti‐CD19 chimeric antigen receptor T cells are associated with high serum Interleukin‐15 levels. J Clin Oncol. 2017;35(16):1803‐1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hirayama AV, Gauthier J, Hay KA, et al. The response to lymphodepletion impacts PFS in patients with aggressive non‐Hodgkin lymphoma treated with CD19 CAR T cells. Blood. 2019;133(17):1876‐1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor‐specific CD8+ T cells. J Exp med. 2005;202(7):907‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8(4):299‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brentjens RJ, Riviere I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19‐targeted T cells in patients with relapsed or chemotherapy refractory B‐cell leukemias. Blood. 2011;118(18):4817‐4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116(19):3875‐3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brudno JN, Kochenderfer JN. Chimeric antigen receptor T‐cell therapies for lymphoma. Nat Rev Clin Oncol. 2018;15(1):31‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Susanibar Adaniya SP, Cohen AD, Garfall AL. Chimeric antigen receptor T cell immunotherapy for multiple myeloma: a review of current data and potential clinical applications. Am J Hematol. 2019;94(S1):S28‐s33. [DOI] [PubMed] [Google Scholar]
- 45. Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR‐T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123‐2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B‐cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brudno JN, Maric I, Hartman SD, et al. T cells genetically modified to express an anti‐B‐cell maturation antigen chimeric antigen receptor cause remissions of poor‐prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267‐2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mishra A, Sullivan L, Caligiuri MA. Molecular pathways: interleukin‐15 signaling in health and in cancer. Clin Cancer Res. 2014;20(8):2044‐2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Turtle CJ, Riddell SR, Maloney DG. CD19‐targeted chimeric antigen receptor‐modified T‐cell immunotherapy for B‐cell malignancies. Clin Pharmacol Ther. 2016;100(3):252‐258. [DOI] [PubMed] [Google Scholar]
- 50. Turtle CJ, Hanafi LA, Berger C, et al. Immunotherapy of non‐Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19‐specific chimeric antigen receptor‐modified T cells. Sci Transl Med. 2016;8(355). 355ra116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. USFDA . YESCARTA (axicabtagene ciloleucel). Package Insert ‐ YESCARTA 2018; https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/yescarta-axicabtagene-ciloleucel.
- 52. Schubert ML, Schmitt A, Sellner L, et al. Treatment of patients with relapsed or refractory CD19+ lymphoid disease with T lymphocytes transduced by RV‐SFG.CD19.CD28.4‐1BBzeta retroviral vector: a unicentre phase I/II clinical trial protocol. BMJ Open. 2019;9(5):e026644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ramos CA, Rouce R, Robertson CS, et al. In vivo fate and activity of second‐ versus third‐generation CD19‐specific CAR‐T cells in B cell non‐Hodgkin's lymphomas. Mol Ther. 2018;26(12):2727‐2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu B, Yan L, Zhou M. Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am J Cancer Res. 2019;9(2):228‐241. [PMC free article] [PubMed] [Google Scholar]
- 55. Cheadle EJ, Hawkins RE, Batha H, O'Neill AL, Dovedi SJ, Gilham DE. Natural expression of the CD19 antigen impacts the long‐term engraftment but not antitumor activity of CD19‐specific engineered T cells. J Immunol. 2010;184(4):1885‐1896. [DOI] [PubMed] [Google Scholar]
- 56. Guedan S, Calderon H, Posey AD Jr, Maus MV. Engineering and Design of Chimeric Antigen Receptors. Mol Ther Methods Clin Dev. 2019;12:145‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gust J, Hay KA, Hanafi LA, et al. Endothelial activation and blood‐brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR‐T cells. Cancer Discov. 2017;7(12):1404‐1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Grigor EJM, Fergusson D, Kekre N, et al. Risks and benefits of chimeric antigen receptor T‐cell (CAR‐T) therapy in cancer: a systematic review and meta‐analysis. Transfus med Rev. 2019;33(2):98‐110. [DOI] [PubMed] [Google Scholar]
- 59. Mardiana S, Solomon BJ, Darcy PK, Beavis PA. Supercharging adoptive T cell therapy to overcome solid tumor‐induced immunosuppression. Sci Transl Med. 2019;11(495):eaaw2293. [DOI] [PubMed] [Google Scholar]
- 60. Kawalekar OU, O'Connor RS, Fraietta JA, et al. Distinct signaling of Coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380‐390. [DOI] [PubMed] [Google Scholar]
- 61. Sommermeyer D, Hudecek M, Kosasih PL, et al. Chimeric antigen receptor‐modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30(2):492‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rouce RH, Heslop HE. Equal opportunity CAR T cells. Blood. 2017;129(25):3275‐3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. McLellan AD, Ali Hosseini Rad SM. Chimeric antigen receptor T cell persistence and memory cell formation. Immunol Cell Biol. 2019;97(7):664‐674. [DOI] [PubMed] [Google Scholar]
- 64. Fraietta JA, Lacey SF, Orlando EJ, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24(5):563‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Klebanoff CA, Finkelstein SE, Surman DR, et al. IL‐15 enhances the in vivo antitumor activity of tumor‐reactive CD8+ T cells. Proc Natl Acad Sci U S A. 2004;101(7):1969‐1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xu Y, Zhang M, Ramos CA, et al. Closely related T‐memory stem cells correlate with in vivo expansion of CAR.CD19‐T cells and are preserved by IL‐7 and IL‐15. Blood. 2014;123(24):3750‐3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl med. 2016;8(320). 320ra323 [DOI] [PubMed] [Google Scholar]
- 68. Hinrichs CS, Spolski R, Paulos CM, et al. IL‐2 and IL‐21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111(11):5326‐5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Singh H, Figliola MJ, Dawson MJ, et al. Reprogramming CD19‐specific T cells with IL‐21 signaling can improve adoptive immunotherapy of B‐lineage malignancies. Cancer Res. 2011;71(10):3516‐3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T‐cell therapy ‐ assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Norelli M, Camisa B, Barbiera G, et al. Monocyte‐derived IL‐1 and IL‐6 are differentially required for cytokine‐release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739‐748. [DOI] [PubMed] [Google Scholar]
- 72. Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Singh N, Hofmann TJ, Gershenson Z, et al. Monocyte lineage‐derived IL‐6 does not affect chimeric antigen receptor T‐cell function. Cytotherapy. 2017;19(7):867‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gardner RL, Leger KJ, Annesley CE, et al. Decreased rates of severe CRS seen with early intervention strategies for CD19 CAR‐T cell toxicity management. ASH; 2016.
- 75. Locke FL, Neelapu SS, Bartlett NL, et al. Preliminary results of prophylactic Tocilizumab after Axicabtageneciloleucel (axi‐cel; KTE‐C19) treatment for patients with Refractory,Aggressive non‐Hodgkin lymphoma (NHL). ASH; 2017.
- 76. Herold MJ, McPherson KG, Reichardt HM. Glucocorticoids in T cell apoptosis and function. Cell Mol Life Sci. 2006;63(1):60‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Brudno JN, Kochenderfer JN. Recent advances in CAR T‐cell toxicity: mechanisms, manifestations and management. Blood Rev. 2019;34:45‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yoon DH, Osborn MJ, Tolar J, Kim CJ. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR‐Ts): combination or built‐in CAR‐T. Int J Mol Sci. 2018;19(2):340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. John LB, Kershaw MH, Darcy PK. Blockade of PD‐1 immunosuppression boosts CAR T‐cell therapy. Oncoimmunology. 2013;2(10). e26286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hill BT, Roberts ZJ, Xue A, Rossi JM, Smith MR. Rapid tumor regression from PD‐1 inhibition after anti‐CD19 chimeric antigen receptor T‐cell therapy in refractory diffuse large B‐cell lymphoma. Bone Marrow Transplant. 2019. 10.1038/s41409-019-0657-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schuster SJ, Svoboda J, Chong EA, et al. Chimeric antigen receptor T cells in refractory B‐cell lymphomas. N Engl J Med. 2017;377(26):2545‐2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Maude SL, Hucks GE, Seif AE. The effect of pembrolizumab in combination with CD19‐targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL). Paper presented at: American Society of Clinical Oncology 2017.
- 83. Fesnak AD. The challenge of variability in chimeric antigen receptor T cell manufacturing. Regener Eng Transl Med. 2019. 10.1007/s40883-019-00124-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tuazon SA, Li A, Gooley T, et al. Factors affecting lymphocyte collection efficiency for the manufacture of chimeric antigen receptor T cells in adults with B‐cell malignancies. Transfusion. 2019;59(5):1773‐1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Scarfo I, Maus MV. Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment. J Immunother Cancer. 2017;5:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ali SA, Shi V, Maric I, et al. T cells expressing an anti‐B‐cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128(13):1688‐1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Enblad G, Karlsson H, Gammelgard G, et al. A phase I/IIa trial using CD19‐targeted third‐generation CAR T cells for lymphoma and leukemia. Clin Cancer Res. 2018;24(24):6185‐6194. [DOI] [PubMed] [Google Scholar]
- 88. Gardner RA, Finney O, Annesley C, et al. Intent‐to‐treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322‐3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Awasthi RTSCea . Clinical pharmacology of tisagenlecleucel (CTL019) in patients with relapsed/refractory (r/r) diffuse large B‐cell lymphoma (DLBCL). American Association of Cancer Research Annual Meeting; 2018.
- 90. Raje N, Berdeja J, Lin Y, et al. Anti‐BCMA CAR T‐cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380(18):1726‐1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B‐cell lymphoma. N Engl J Med. 2018;380(1):45‐56. [DOI] [PubMed] [Google Scholar]
- 92. Lee DW, Kochenderfer JN, Stetler‐Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose‐escalation trial. Lancet. 2015;385(9967):517‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Frey NV. Chimeric antigen receptor T cells for acute lymphoblastic leukemia. Am J Hematol. 2019;94(S1):S24‐s27. [DOI] [PubMed] [Google Scholar]
- 94. Locke FL, Ghobadi A, Jacobson CA, et al. Long‐term safety and activity of axicabtagene ciloleucel in refractory large B‐cell lymphoma (ZUMA‐1): a single‐arm, multicentre, phase 1‐2 trial. Lancet Oncol. 2019;20(1):31‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Teachey DT, Lacey SF, Shaw PA, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T‐cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6(6):664‐679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hay KA, Hanafi LA, Li D, et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor‐modified T‐cell therapy. Blood. 2017;130(21):2295‐2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Park JH, Riviere I, Gonen M, et al. Long‐term follow‐up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Borchmann P, Tam CS, Jäger U, et al. An updated analysis of JULIET, a global pivotal phase 2 trial of tisagenlecleucel in adult patients with relapsed or refractory (r/r) diffuse large b‐cell lymphoma (DLBCL). Paper presented at: The 23rd Congress of EHA2018; Stockholm.
- 99. Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19‐specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16(9):1245‐1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zola H, MacArdle PJ, Bradford T, Weedon H, Yasui H, Kurosawa Y. Preparation and characterization of a chimeric CD19 monoclonal antibody. Immunol Cell Biol. 1991;69(Pt 6):411‐422. [DOI] [PubMed] [Google Scholar]
- 101. Qian L, Li D, Ma L, et al. The novel anti‐CD19 chimeric antigen receptors with humanized scFv (single‐chain variable fragment) trigger leukemia cell killing. Cell Immunol. 2016;304‐305:49‐54. [DOI] [PubMed] [Google Scholar]
- 102. Alabanza L, Pegues M, Geldres C, et al. Function of novel anti‐CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. 2017;25(11):2452‐2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Storer BE. Design and analysis of phase I clinical trials. Biometrics. 1989;45(3):925‐937. [PubMed] [Google Scholar]
- 104. Salmikangas P, Kinsella N, Chamberlain P. Chimeric antigen receptor T‐cells (CAR T‐cells) for cancer immunotherapy ‐ moving target for industry? Pharm Res. 2018;35(8):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Seimetz D, Heller K, Richter J. Approval of first CAR‐Ts: have we solved all hurdles for ATMPs? Cell Med. 2019;11:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Li DH, Whitmore JB, Guo W, Ji Y. Toxicity and efficacy probability interval Design for Phase I Adoptive Cell Therapy Dose‐Finding Clinical Trials. Clin Cancer Res. 2017;23(1):13‐20. [DOI] [PubMed] [Google Scholar]
- 107. Wages NA, Chiuzan C, Panageas KS. Design considerations for early‐phase clinical trials of immune‐oncology agents. J Immunother Cancer. 2018;6(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ananthakrishnan R, Green S, Li D, LaValley M. Extensions of the mTPI and TEQR designs to include non‐monotone efficacy in addition to toxicity for optimal dose determination for early phase immunotherapy oncology trials. Contemp Clin Trials Commun. 2018;10:62‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ananthakrishnan R, Green S, Li D, LaValley M. 2D (2 dimensional) TEQR design for determining the optimal dose for safety and efficacy. Contemp Clin Trials Commun. 2019;16:100461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Chiuzan C, Garrett‐Mayer E, Nishimura M. An adaptive dose‐finding design based on both safety and immunologic responses in cancer clinical trials. Stat Biopharm Res. 2018;10(3):185‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Watanabe K, Kuramitsu S, Posey AD Jr, June CH. Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front Immunol. 2018;9:2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Hay AE, Cheung MC. CAR T‐cells: costs, comparisons, and commentary. J Med Econ. 2019;22(7):613‐615. [DOI] [PubMed] [Google Scholar]
- 113. Lin JK, Muffly LS, Spinner MA, Barnes JI, Owens DK, Goldhaber‐Fiebert JD. Cost effectiveness of chimeric antigen receptor T‐cell therapy in multiply relapsed or refractory adult large B‐cell lymphoma. J Clin Oncol. 2019;37(24):2105‐2119. [DOI] [PubMed] [Google Scholar]
- 114. Qasim W, Zhan H, Samarasinghe S, et al. Molecular remission of infant B‐ALL after infusion of universal TALEN gene‐edited CAR T cells. Sci Transl Med. 2017;9(374):eaaj2013. [DOI] [PubMed] [Google Scholar]
- 115. Liu E, Marin D, Banerjee P, et al. Use of CAR‐transduced natural killer cells in CD19‐positive lymphoid tumors. N Engl J Med. 2020;382(6):545‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Xu X, Huang W, Heczey A, et al. NKT cells Coexpressing a GD2‐specific chimeric antigen receptor and IL15 show enhanced in vivo persistence and antitumor activity against Neuroblastoma. Clin Cancer Res. 2019;25(23):7126‐7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC‐derived natural killer cells engineered with chimeric antigen receptors enhance anti‐tumor activity. Cell Stem Cell. 2018;23(2):181‐192. e185 [DOI] [PMC free article] [PubMed] [Google Scholar]