

Abstract
Aims
Olaparib, a potent oral poly(ADP‐ribose) polymerase inhibitor, is partially hepatically cleared. We investigated the pharmacokinetics (PK) and safety of olaparib in patients with mild or moderate hepatic impairment to provide dosing recommendations.
Methods
This Phase I open‐label study assessed the PK, safety and tolerability of single doses of olaparib 300‐mg tablets in patients with advanced solid tumours. Patients had normal hepatic function (NHF), or mild (MiHI; Child–Pugh class A) or moderate (MoHI; Child–Pugh class B) hepatic impairment. Blood was collected for PK assessments for 96 hours. Patients could continue taking olaparib 300 mg twice daily for long‐term safety assessment.
Results
Thirty‐one patients received ≥1 dose of olaparib and 30 were included in the PK assessment. Patients with MiHI had an area under the curve geometric least‐squares mean (GLSmean) ratio of 1.15 (90% confidence interval 0.72, 1.83) and a GLSmean maximum plasma concentration ratio of 1.13 (0.82, 1.56) vs those with NHF. In patients with MoHI, GLSmean ratio for area under the curve was 1.08 (0.66, 1.74) and for maximum plasma concentration was 0.87 (0.63, 1.22) vs those with NHF. For patients with mild or moderate hepatic impairment, no new safety signals were detected.
Conclusion
Patients with MiHI or MoHI had no clinically significant changes in exposure to olaparib compared with patients with NHF. The safety profile of olaparib did not differ from a clinically relevant extent between cohorts. No olaparib tablet or capsule dose reductions are required for patients with MiHI or MoHI.
Keywords: advanced solid tumours, hepatic impairment, liver, olaparib, pharmacokinetics, poly(ADP‐ribose) polymerase, safety
1.
What is already known about this subject
The poly(ADP‐ribose) polymerase inhibitor olaparib is approved for treatment of patients with ovarian or breast cancer
Olaparib is partially hepatically cleared
What this study adds
Mild/moderate hepatic impairment did not significantly change exposure to olaparib vs normal hepatic function
The safety profile of olaparib did not differ to a clinically relevant extent between cohorts
Clinical significance
No olaparib dose reductions are required for patients with moderate or mild hepatic impairment
Olaparib is not recommended in patients with severe hepatic impairment
2. INTRODUCTION
Olaparib (Lynparza) is a potent oral poly (ADP‐ribose) polymerase inhibitor 1 that is approved in the USA, Europe, Japan and other countries as first‐line maintenance therapy in patients with advanced BRCA mutation (BRCAm) ovarian cancer, for women with platinum‐sensitive relapsed ovarian cancer regardless of their BRCAm status, and for patients with germline BRCAm, human epidermal growth factor receptor 2‐negative locally advanced or metastatic breast cancer. 2 , 3 , 4 , 5 , 6 Evaluation of olaparib is ongoing in ovarian, breast, prostate, pancreatic and other cancers.
The liver is involved in the clearance of many drugs through a variety of oxidative and conjugative metabolic pathways and/or through biliary excretion of unchanged drug or metabolites. Alterations of these excretory and metabolic activities by hepatic impairment can lead to drug accumulation or failure to form an active metabolite and therefore reduced efficacy. 7 Hepatic function decreases with age, but due to the high capacity of the liver, this is not considered to change drug pharmacokinetics (PK) to a clinically relevant extent. 8 Liver disease, however, is known to be a common cause of altered PK (absorption and disposition) of drugs, as well as their pharmacodynamics (efficacy and safety), through different mechanisms, with the degree of effect dependent on the severity of hepatic impairment. 7 , 8 The effects of hepatic impairment on drug PK can be difficult to predict because of consequences of shunting of blood past the liver (both porto‐systemic and intrahepatic), impaired hepatocellular function, impaired biliary excretion and decreased protein binding. 8
The elimination of olaparib and its metabolites occurs via both the hepatic and renal routes, with approximately 42% of the dose eliminated in faeces and 44% eliminated in urine. 9 Patients with cancer may have varying degrees of hepatic impairment. 10 Given that olaparib undergoes extensive hepatic metabolism (primarily by cytochrome P450 3A4/5 isozymes), 3 , 4 , 11 it is important to examine its PK profile in patients with hepatic impairment to determine the appropriate dose.
The aims of this study (D0816C00005; NCT01894243) were to evaluate the olaparib tablet formulation PK profile following a single oral dose in adult patients with advanced solid tumours and mild or moderate hepatic impairment (Part A), and to evaluate the safety and tolerability profile of single (Part A) and multiple (Part B) doses of olaparib in these patients compared with patients with normal hepatic function. This study also supported label dose recommendations for the olaparib tablet and capsule formulations in patients with hepatic impairment.
3. METHODS
3.1. Materials
Olaparib was supplied by AstraZeneca (Cambridge, UK). Covance (Harrogate, UK) performed the PK analyses of olaparib. 12
3.2. Study design
This 2‐part, nonrandomised, open‐label, parallel‐group, multicentre, Phase I study was conducted in adult patients with advanced solid tumours who had normal hepatic function, mild or moderate hepatic impairment (ClinicalTrials.gov NCT01894243). Twenty‐two sites in 6 countries (Belgium, Czech Republic, France, Korea, The Netherlands and UK) were included.
In Part A, patients with normal hepatic function or mild or moderate hepatic impairment received a single oral dose of olaparib 300 mg (2 × 150 mg tablets) for evaluation of olaparib PK, safety and tolerability. Patients had to fast from 1 hour before to 2 hours after olaparib dosing and were not permitted to consume grapefruit‐containing products between 48 hours before and 96 hours after their olaparib dose or, throughout the study, any potent or moderate CYP3A inhibitors or inducers.
Part B of the study evaluated long‐term safety of olaparib in patients with mild or moderate hepatic impairment compared with those with normal hepatic function with continued access to olaparib tablets (300 mg twice daily). Patients could take olaparib with a light meal or snack; food does not significantly affect olaparib PK. 12 No PK analyses were performed.
The protocol was approved by the independent ethics committees or institutional review boards of all investigational sites. The study was performed in accordance with the Declaration of Helsinki, Good Clinical Practice, and the AstraZeneca policy on Bioethics. 13
3.3. Patients
Eligible patients were aged ≥18 years and had a confirmed (histologically or, where appropriate, cytologically) malignant solid tumour refractory or resistant to standard therapy for which no suitable therapy exists (excluding patients with gastric, gastro‐oesophageal or oesophageal cancer or symptomatic uncontrolled brain metastases). Patients with hepatic metastases and/or hepatocellular carcinoma (HCC) were eligible for the study, provided the hepatic metastases or HCC was not the sole reason for any changes in liver function satisfying the criteria for mild or moderate hepatic impairment (classified by Child–Pugh criteria in line with European Medicines Agency recommendations 8 ; Tables S1 and S2). Patients with severe hepatic impairment were not included in this study because it would not be ethical to treat these patients with a drug that is extensively metabolised by the liver.
Additional eligibility criteria and exclusion criteria are detailed in the Supplementary Materials.
Three patient groups were included: normal hepatic function (total bilirubin ≤1.5 × institutional upper limit of normal [ULN]; albumin and prothrombin time within normal limits; aspartate aminotransferase and alanine aminotransferase ≤2.5 × institutional ULN unless liver metastases were present, in which case it had to be ≤5 × ULN; negative for serum hepatitis B surface antigen and hepatitis C antibody; and no evidence of ascites [unless related to disease under study] or encephalopathy); stable mild hepatic impairment (Child–Pugh class A: 5–6 points); and stable moderate hepatic impairment (Child–Pugh class B: 7–9 points).
Patients in the hepatic impairment cohorts were required to have stable mild globally impaired hepatic function for ≥1 month before the start of the study or stable moderate globally impaired hepatic function for ≥2 weeks before the start of the study, based on the Child–Pugh classification system in accordance with the Committee for Medicinal Products for Human Use (CHMP/EWP/2339/02). 8
In each cohort, where possible, patients were matched for body mass index and age and were selected so that there was a similar proportion of males and females. Recruitment of the normal hepatic function and mild hepatic impairment cohorts occurred before that of the moderate hepatic impairment cohort; ≥3 months of safety information from ≥3 patients with mild hepatic impairment was required prior to recruiting patients with moderate hepatic impairment.
3.4. Study objectives
Assessment of the PK of olaparib tablets following a single oral dose of 300 mg to patients with advanced solid tumours and mild or moderate hepatic impairment compared with patients with normal hepatic function was the primary objective (Part A). Evaluation of the safety and tolerability of olaparib tablets in patients with advanced solid tumours and mild or moderate hepatic impairment vs normal hepatic function (Parts A and B) was the secondary objective. Investigation of changes in the protein binding of olaparib and subsequent effects on its PK profile in patients with varying degrees of hepatic function (Part A) were exploratory objectives.
3.5. Pharmacokinetic assessments
PK analysis methods are detailed in the Supplementary Materials. In each patient cohort, the PK parameters determined for olaparib included: maximum plasma concentration (Cmax), time to Cmax (tmax), area under the plasma concentration–time curve from zero to the last measurable time point (AUC0–t), area under the plasma concentration–time curve from zero extrapolated to infinity (AUC), apparent plasma clearance following oral administration (CL/F), terminal elimination half‐life (t½), and apparent volume of distribution following oral administration (Vz/F). Plasma protein binding at 1 hour after dosing was used to calculate free Cmax (Cmax of unbound olaparib), free AUC (AUC of unbound olaparib) and unbound CL/F (CL/F of unbound olaparib), which were exploratory PK parameters.
3.6. Safety assessments
Adverse events (AEs) were reported throughout the study, graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4 (NCI‐CTCAE v4). All serious AEs (SAEs) and AEs related to treatment were followed up to resolution. Clinical laboratory, vital signs and physical examination parameters were evaluated. Safety was analysed in the safety analysis set, and analyses were descriptive.
3.7. Statistical analysis
The study was sized to provide adequate information to assess the effects of hepatic impairment on the PK of olaparib, while exposing as few patients as possible to procedures and treatment. Overall, 24 evaluable patients (8 per arm) was considered adequate to provide reasonable precision around the estimate of the magnitude of the effects (see Supplementary Materials).
Based on an estimate of between‐patient standard deviation for loge‐transformed AUC of 0.531 from a study of olaparib tablet monotherapy (300 mg), 14 8 evaluable patients per arm were determined to provide approximately 80% power to reject the null hypothesis of a clinically significant increase in exposure (i.e. to rule out a 100% increase), indicated by a 1‐sided 95% confidence limit for the ratio of geometric least‐squares means (GLSmeans) of AUC or Cmax < 2.0, in patients with mild or moderate hepatic impairment compared with those with normal hepatic function. Recruitment of approximately 30 patients was planned to ensure that there were 8 evaluable patients with full PK sampling in each group.
Following loge transformation, the PK parameters AUC and Cmax were analysed separately using an analysis‐of‐variance model, fitting for hepatic function group (moderate or mild hepatic impairment, or normal hepatic function). The ratio of GLSmeans and 2‐sided 90% confidence intervals (CIs) for AUC and Cmax comparing patients with moderate or mild hepatic impairment against those with normal hepatic function were estimated from the models. A clinically relevant effect on the exposure of olaparib due to hepatic impairment was determined if the upper confidence limit for the ratios of AUC and Cmax exceeded 2 (implying a doubling in exposure).
Descriptive statistics and parameters of PK data were summarised and calculated using Phoenix WinNonlin (Certara USA, Inc., Princeton, NJ, USA).
The PK analysis set in Part A consisted of all patients who received the olaparib dose and had full PK sampling up to 96 hours postdose; the safety analysis population in both parts consisted of all patients who received at least 1 dose of olaparib and had available postdose data.
3.8. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.
4. RESULTS
4.1. Part A
4.1.1. Patients
Between March 2014 and March 2017, 52 patients were screened and 31 entered the study, received treatment and completed Part A of the study; 13 patients with normal hepatic function, 10 patients with mild hepatic impairment and 8 patients with moderate hepatic impairment. The most common reason for exclusion was that eligibility criteria were not met (n = 20).
Demographic and baseline characteristics were slightly unbalanced between groups, which would be expected with small cohorts (Table 1). Six, 7 and 4 patients with normal hepatic function, mild and moderate hepatic impairment had hepatic metastases at the time of their most recent progression, respectively. Ascites were present in 4 patients in the moderate hepatic impairment group (3 patients had slight ascites and 1 patient had moderate ascites). Global liver impairment causality for patients with mild or moderate liver impairment is detailed in Table S2.
TABLE 1.
Summary of baseline patient characteristics
| Normal hepatic function (n = 13) | Mild hepatic impairment (n = 10) | Moderate hepatic impairment (n = 8) | |
|---|---|---|---|
| Median age (range), y | 59 (43–66) | 58 (41–70) | 67 (52–78) |
| Sex, n (%) | |||
| Male | 5 (38.5) | 6 (60) | 6 (75) |
| Female | 8 (61.5) | 4 (40) | 2 (25) |
| Race, n (%) | |||
| Caucasian | 13 (100) | 10 (100) | 7 (87.5) |
| Asian | 0 | 0 | 1 (12.5) |
| ECOG performance status, n (%) a | |||
| 0 | 8 (61.5) | 5 (50) | 1 (12.5) |
| 1 | 5 (38.5) | 5 (50) | 6 (75) |
| 2 | 0 | 0 | 1 (12.5) |
| Primary tumour location, n (%) | |||
| Breast | 3 (23.1) | 0 | 0 |
| Head and neck b | 1 (7.7) | 0 | 0 |
| Liver | 0 | 2 (20) | 6 (75) |
| Prostate | 1 (7.7) | 1 (10) | 0 |
| Renal | 0 | 1 (10) | 0 |
| Pancreas | 1 (7.7) | 0 | 0 |
| Ovary | 3 (23.1) | 1 (10) | 0 |
| Colon | 1 (7.7) | 2 (20) | 1 (12.5) |
| Biliary tract | 1 (7.7) | 0 | 1 (12.5) |
| Primary peritoneal | 1 (7.7) | 0 | 0 |
| Other c | 1 (7.7) | 3 (30) | 0 |
| Overall disease classification, n (%) a | |||
| Metastatic | 10 (76.9) | 9 (90) | 3 (37.5) |
| Locally advanced | 2 (15.4) | 0 | 3 (37.5) |
| Both | 1 (7.7) | 1 (10) | 2 (25) |
| Median ALT, μkat L −1 (range) a | 0.5 (0.2–1.9) | 1.3 (0.1–4.4) | 0.7 (0.3–2.3) |
| Median AST, μkat L −1 (range) a | 0.5 (0.2–1.1) | 1.0 (0.4–2.3) | 1.0 (0.6–3.2) |
| Median GGT, μkat L −1 (range) a | 2.1 (0.2–12.6) | 5.2 (0.3–30.8) | 7.3 (1.0–19.4) |
| Median bilirubin, μmol L −1 (range) a | 6.0 (4–19) | 12.8 (3–15) | 24.5 (10–45) |
| Median albumin, g L −1 (range) a | 41.0 (34–46) | 39.0 (30–46) | 27.0 (2–37) |
At baseline for Part A.
Including nasopharynx, larynx and trachea.
Other included cholangiocarcinoma, duodenum, colorectal and unknown (all n = 1).
ALT, alanine aminotransferase; AST, aspartate aminotransferase; ECOG, Eastern Cooperative Oncology Group; GGT, γ‐glutamyl transferase.
One patient in the moderate hepatic impairment group had elevated serum creatinine (within the inclusion criteria limits) during screening and renal failure before receiving olaparib, which were considered to have affected the outcomes for this patient. At the time of screening, this patient had no history of ascites, encephalopathy, or hepatorenal syndrome, and the patient's physical examination was normal, without evidence of ascites or peripheral oedema. Their medical history did not include chronic kidney disease, and according to the investigator, the patient's renal failure was detected before enrolment and was as a cause of worsening hepatic function.
Overall in Part A, 4 patients had 5 protocol deviations, of whom only 1 patient (with deviations of unable to swallow orally administered medication and gastrointestinal disorder/resection likely to interfere with olaparib absorption, and disallowed surgery [pancreatic duodenectomy]) was excluded from PK analyses. The remaining 3 patients had protocol deviations that were not considered to have an impact on the PK analyses. All patients were included in the safety analysis set.
4.2. Pharmacokinetics
The olaparib geometric mean (Gmean) plasma concentration–time profile for each hepatic status group is shown in Figure 1. Compared with patients with normal hepatic function, in patients with mild hepatic impairment, olaparib exposure (Gmean AUC, AUC0–t and Cmax) was slightly increased, whereas in patients with moderate hepatic impairment, AUC and AUC0–t were slightly increased and Cmax was slightly decreased (Table 2); tmax was similar for all cohorts; CL/F was similar across all cohorts; and VZ/F was slightly decreased in patients with mild or moderate hepatic impairment compared with patients with normal hepatic function (Table 2).
FIGURE 1.
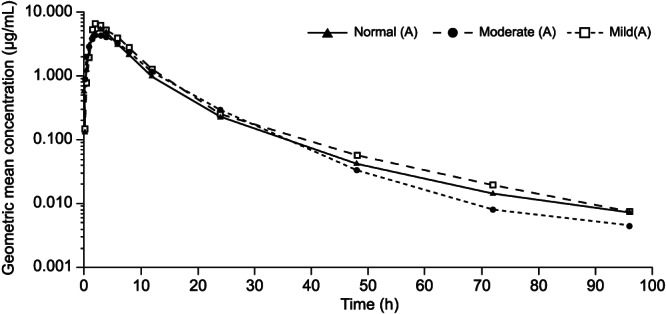
Geometric mean plasma concentration of olaparib over time (log scale) in patients with mild and moderate hepatic impairment or normal hepatic function following a single dose of olaparib 300 mg (tablet formulation)
TABLE 2.
Pharmacokinetics of a single dose of olaparib 300 mg (tablet formulation)
| Parameter a | Normal hepatic function (n = 13) | Mild hepatic impairment (n = 9 b ) | Moderate hepatic impairment (n = 8) |
|---|---|---|---|
| Cmax, μg mL−1 | 7.3 (47.6) | 8.3 (39.9) | 6.4 (49.9) |
| tmax, h | 1.5 (0.9–6) | 2.1 (1.5–6.1) | 1.5 (0.5–6) |
| AUC, μg·h mL−1 | 52.3 (65.6) | 60.3 (48.2) | 56.3 (97.8) |
| AUC0–t, μg·h mL−1 | 51.8 (64.2) | 59.6 (46.8) | 56.0 (96.3) |
| t½, h | 20.3 (12.5) | 18.4 (9.6) | 14.4 (4.0) |
| CL/F, L h−1 | 6.6 (3.2) | 5.4 (2.1) | 6.6 (3.8) |
| VZ/F, L | 166.4 (91.7) | 134.5 (67.4) | 129.0 (81.7) |
| Free Cmax, μg mL−1 | 0.7 (49.4) c | 0.7 (43.5) c | 0.6 (58.3) |
| Free AUC, μg·h mL−1 | 4.8 (46.8) c | 5.0 (52.4) c | 5.5 (91.4) |
| Unbound CL F−1, L h−1 | 68.0 (28.1) c | 68.2 (39.7) c | 69.4 (51.9) |
Cmax, AUC and AUC0–t values are geometric means (geometric coefficient of variation, %), CL/F, VZ/F and t½ values are arithmetic means (standard deviation), and tmax values are medians (range).
One patient was excluded as they underwent cephalic duodenopancreatectomy before study entry, which may have affected olaparib absorption.
For the group with normal hepatic function, n = 11; for the group with mild hepatic impairment, n = 8.
AUC, area under the plasma concentration–time curve from zero extrapolated to infinity; AUC0–t, area under the plasma concentration–time curve from zero to the last measurable time point; CL/F, apparent plasma clearance following oral administration; Cmax, maximum plasma concentration; tmax, time to Cmax; t½, terminal half‐life; Vz/F, apparent volume of distribution following oral administration.
Variability in exposure to olaparib was generally high for all cohorts (geometric coefficient of variation >40%), with the highest variability seen in the moderate hepatic impairment group largely because of 1 patient whose Gmean Cmax and AUC were 1.75‐fold and 5.41‐fold higher, respectively, than the Gmean for that cohort.
Compared with patients with normal hepatic function, the geometric least‐squares mean (GLSmean) Cmax for olaparib was 13% higher in patients with mild hepatic impairment (GLSmean ratio 1.13; 90% CI 0.82, 1.56) and 13% lower in patients with moderate hepatic impairment (GLSmean ratio 0.87; 90% CI 0.63, 1.22; Figure 2a) and the GLSmean AUC for olaparib was 15% higher in patients with mild hepatic impairment (GLSmean ratio 1.15; 90% CI 0.72, 1.83) and 8% higher in patients with moderate hepatic impairment (GLSmean ratio 1.08; 90% CI 0.66, 1.74; Figure 2b). As the 2‐sided 90% CIs for these GLSmean ratios included 1 and the upper limits of the 90% CIs (equivalent to the 1‐sided upper 95% CIs) were <2.0 (indicating that the exposure was less than double in the hepatic impairment groups), the changes in exposure were not considered clinically relevant.
FIGURE 2.
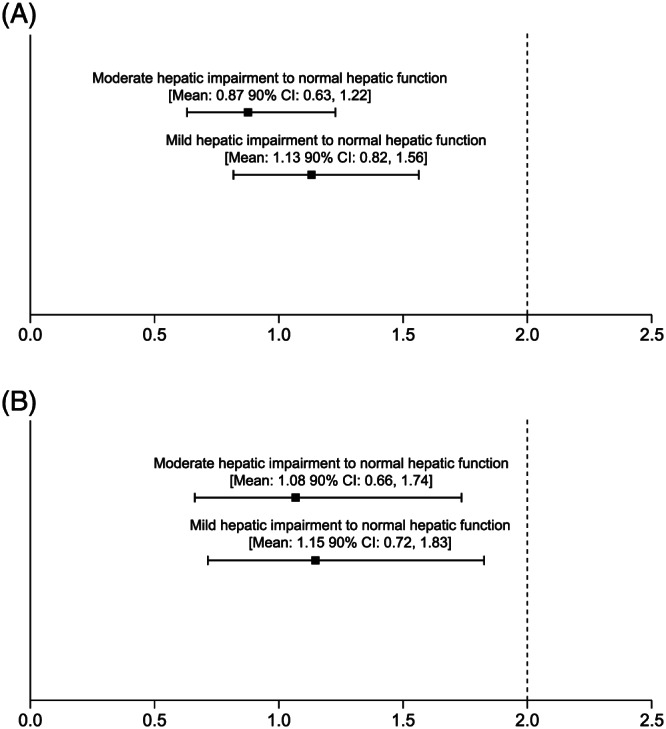
Geometric least‐squares mean ratios (90% confidence interval [CI]) for (A) maximum plasma concentration and (B) area under the plasma concentration–time curve from zero extrapolated to infinity in patients with mild or moderate hepatic impairment vs those with normal hepatic function following a single dose of olaparib 300 mg (tablet formulation)
Mild or moderate hepatic impairment had no obvious effect on the protein binding of olaparib, therefore the total plasma exposures were representative of free olaparib (free Cmax and free AUC; Table 2). Linear regression analysis suggested that there was little correlation between functional markers of liver status (prothrombin time) and olaparib Cmax (R 2 = 0.039), AUC (R 2 = 0.077) or plasma clearance (R 2 = 0.077). There was no obvious relationship for any of the other Child–Pugh parameters investigated, including bilirubin and albumin.
4.3. Safety and tolerability
In Part A of the study, all patients received 1 dose of olaparib tablet 300 mg. Twenty (64.5%) patients experienced at least 1 AE, of which 3 patients experienced a grade 3 AE (moderate hepatic impairment group, renal failure [n = 1] and lymphopenia [n = 1]; mild hepatic impairment group, hypertension [n = 1]; Table 3). Six (19.4%) patients had at least 1 AE that was considered causally related to olaparib (nausea, n = 3; anaemia, asthenia, decreased appetite and muscle spasm, n = 1 each).
TABLE 3.
Incidence of adverse events (AEs) following a single dose of olaparib 300 mg (tablet formulation; part A) a
| AE, n (%) | Normal hepatic function (n = 13) | Mild hepatic impairment (n = 10) | Moderate hepatic impairment (n = 8) | Total (n = 31) |
|---|---|---|---|---|
| Any AE | 10 (76.9) | 5 (50) | 5 (62.5) | 20 (64.5) |
| Nausea | 4 (30.8) | 0 | 0 | 4 (12.9) |
| Diarrhoea | 3 (23.1) | 0 | 0 | 3 (9.7) |
| Anaemia | 1 (7.7) | 1 (10) | 2 (25) | 3 (9.7) |
| Constipation | 1 (7.7) | 1 (10) | 0 | 2 (6.5) |
| Abdominal pain | 1 (7.7) | 0 | 1 (12.5) | 2 (6.5) |
| Asthenia | 0 | 1 (10) | 1 (12.5) | 2 (6.5) |
| Pain in extremity | 1 (7.7) | 0 | 1 (12.5) | 2 (6.5) |
| Mouth ulceration | 1 (7.7) | 0 | 0 | 1 (3.2) |
| Fatigue | 1 (7.7) | 0 | 0 | 1 (3.2) |
| Headache | 1 (7.7) | 0 | 0 | 1 (3.2) |
| Rash pruritic | 1 (7.7) | 0 | 0 | 1 (3.2) |
| Hepatic pain | 0 | 1 (10) | 0 | 1 (3.2) |
| Urinary tract infection | 0 | 1 (10) | 0 | 1 (3.2) |
| ALT increased | 0 | 1 (10) | 0 | 1 (3.2) |
| Hyperglycaemia | 0 | 1 (10) | 0 | 1 (3.2) |
| Erythema | 0 | 1 (10) | 0 | 1 (3.2) |
| Pruritus | 0 | 1 (10) | 0 | 1 (3.2) |
| Hypertension | 0 | 1 (10) | 0 | 1 (3.2) |
| Lymphopenia | 0 | 0 | 1 (12.5) | 1 (3.2) |
| Bilirubin increased | 0 | 0 | 1 (12.5) | 1 (3.2) |
| Decreased appetite | 0 | 0 | 1 (12.5) | 1 (3.2) |
| Muscle spasms | 0 | 0 | 1 (12.5) | 1 (3.2) |
| Insomnia | 0 | 0 | 1 (12.5) | 1 (3.2) |
| Renal failure | 0 | 0 | 1 (12.5) | 1 (3.2) |
| Any serious AE | 1 (7.7) | 0 | 1 (12.5) | 2 (6.5) |
| Abdominal pain | 1 (7.7) | 0 | 0 | 1 (3.2) |
| Renal failure | 0 | 0 | 1 (12.5) | 1 (3.2) |
| Any AE of grade ≥ 3 | 0 | 1 (10) | 2 (25) | 3 (9.7) |
| Hypertension | 0 | 1 (10) | 0 | 1 (3.2) |
| Lymphopenia | 0 | 0 | 1 (12.5) | 1 (3.2) |
| Renal failure | 0 | 0 | 1 (12.5) | 1 (3.2) |
AEs were coded according to Medical Dictionary for Regulatory Activities‐preferred terms.
Graded using CTCAE v4.0. ALT, alanine aminotransferase; CTCAE v4.0, Common Terminology Criteria for Adverse Events version 4.
Two patients experienced an SAE, neither of which were considered related to olaparib, 1 moderate hepatic impairment patient reported grade 3 renal failure and 1 normal hepatic function patient had grade 2 abdominal pain. No deaths or discontinuations because of AEs were reported.
4.4. Part B
4.4.1. Patients
All patients completed Part A of the study, entered Part B and discontinued treatment during Part B because of: disease progression (n = 16, 52%), lack of response (n = 7, 23%), an AE (n = 3), death (n = 3), subject decision (n = 1) and other reasons (n = 1).
The mean (total and actual) duration of olaparib exposure was similar in patients with normal hepatic function (155.8 and 150.7 days, respectively) and mild hepatic impairment (132.3 and 130.2 days); however, it was evidently lower in those with moderate hepatic impairment (42.0 and 34.4 days).
4.4.2. Safety and tolerability
In Part B of the study, 30 (96.8%) patients experienced at least 1 AE, and grade ≥ 3 AEs were reported in 13 (41.9%) patients (Table 4). Twenty‐one (67.7%) patients had ≥1 AE that was considered to be causally related to olaparib (n = 9 [69.2%] in the normal hepatic function group, n = 7 [70%] in the mild hepatic impairment group and n = 5 [62.5%] in the moderate hepatic impairment group) and 6 (19.4%) patients had a grade ≥ 3 AE considered causally related to olaparib (n = 1 [7.7%], n = 3 [30%] and n = 2 [25%], respectively). Table S3 details grade ≥ 3 AEs by Child–Pugh classification.
TABLE 4.
Incidence of all‐grade adverse events (AEs) occurring in 3 or more patients overall and grade ≥ 3 AEs following multiple dosing of olaparib 300 mg twice daily (tablet formulation; Part B) a
| AE, n (%) | Normal hepatic function (n = 13) | Mild hepatic impairment (n = 10) | Moderate hepatic impairment (n = 8) | Total (n = 31) | ||||
|---|---|---|---|---|---|---|---|---|
| All grades | Grade ≥ 3 | All grades | Grade ≥ 3 | All grades | Grade ≥ 3 | All grades | Grade ≥ 3 | |
| Any AE | 13 (100) | 4 (30.8) | 10 (100) | 4 (40) | 7 (87.5) | 5 (62.5) | 30 (96.8) | 13 (41.9) |
| Nausea | 7 (53.8) | 0 | 5 (50) | 0 | 2 (25) | 0 | 14 (45.2) | 0 |
| Anaemia | 2 (15.4) | 0 | 5 (50) | 3 (30) | 3 (37.5) | 2 (25) | 10 (32.3) | 5 (16.1) |
| Fatigue | 6 (46.2) | 0 | 1 (10) | 0 | 1 (12.5) | 1 (12.5) | 8 (25.8) | 1 (3.2) |
| Decreased appetite | 2 (15.4) | 1 (7.7) | 2 (20) | 1 (10) | 3 (37.5) | 1 (12.5) | 7 (22.6) | 3 (9.7) |
| Vomiting | 5 (38.5) | 0 | 1 (10) | 0 | 1 (12.5) | 0 | 7 (22.6) | 0 |
| Asthenia | 2 (15.4) | 1 (7.7) | 3 (30) | 1 (10) | 1 (12.5) | 1 (12.5) | 6 (19.4) | 3 (9.7) |
| Lymphopenia | 2 (15.4) | 0 | 2 (20) | 0 | 1 (12.5) | 0 | 5 (16.1) | 0 |
| Thrombocytopenia | 1 (7.7) | 0 | 3 (30) | 1 (10) | 1 (12.5) | 1 (12.5) | 5 (16.1) | 2 (6.5) |
| Back pain | 3 (23.1) | 0 | 0 | 0 | 1 (12.5) | 0 | 4 (12.9) | 0 |
| Ascites | 0 | 0 | 0 | 0 | 3 (37.5) | 0 | 3 (9.7) | 0 |
| ALP increased | 2 (15.4) | 0 | 1 (10) | 0 | 0 | 0 | 3 (9.7) | 0 |
| Constipation | 2 (15.4) | 0 | 0 | 0 | 1 (12.5) | 0 | 3 (9.7) | 0 |
| Dyspnoea | 1 (7.7) | 0 | 2 (20) | 1 (10) | 0 | 0 | 3 (9.7) | 1 (3.2) |
| Musculoskeletal pain | 3 (23.1) | 0 | 0 | 0 | 0 | 0 | 3 (9.7) | 0 |
| Upper abdominal pain | 3 (23.1) | 0 | 0 | 0 | 0 | 0 | 3 (9.7) | 0 |
AEs were coded according to Medical Dictionary for Regulatory Activities‐preferred terms.
Graded using CTCAE v4.0. ALP, alkaline phosphatase; CTCAE v4.0, Common Terminology Criteria for Adverse Events version 4.
The most common all‐grade AEs in all cohorts of Part B were nausea (n = 14 [45.2%]), anaemia (n = 10 [32.3%]) and fatigue (n = 8 [25.8%]) and the most common grade ≥ 3 AEs were anaemia (n = 5 [16.1%]), decreased appetite and asthenia (n = 3 each [9.7%]). In the normal hepatic function group, the most common AEs were nausea (n = 7 [53.8%]) and fatigue (n = 6 [46.2%]); in the mild hepatic impairment cohort anaemia and nausea (n = 5 [50%] each); and in the moderate hepatic impairment group they were anaemia, ascites and decreased appetite (n = 3 [37.5%] each).
Ten (32.3%) patients experienced an SAE (3 patients each in the groups with normal hepatic function and mild hepatic impairment, and 4 patients in the moderate hepatic impairment group), 5 of which were considered by the investigator to be related to olaparib (Table 5).
TABLE 5.
Serious adverse events reported in Part B of the study
| Serious adverse event details | Considered by the investigator as related to olaparib | Olaparib dose modification | |
|---|---|---|---|
| Normal hepatic impairment | |||
| Patient 1 |
Grade 2 seizure lasting 1 day; the patient recovered Grade 3 hyponatraemia; the patient was described as recovering |
No | No olaparib dose changes were required for these events |
| Patient 2 |
Grade 3 respiratory tract infection on day 87 of olaparib dosing and lasted 7 days; the patient recovered after an olaparib dose interruption Grade 3 sepsis on day 129 of olaparib dosing and lasted for 8 days; the patient recovered |
No | Olaparib dose reduction for grade 3 respiratory tract infection |
| Patient 3 | Grade 3 ileus on day 53 of olaparib dosing, lasted for 10 days and resolved following an olaparib dose interruption | No | Olaparib dose interruption |
| Mild hepatic impairment | |||
| Patient 1 |
Grade 4 thrombocytopenia after 13 days of olaparib dosing Grade 2 unstable angina which lasted for 3 days; the patient recovered |
The event of thrombocytopenia was considered related to olaparib | Olaparib was discontinued following the event of grade 4 thrombocytopenia |
| Patient 2 | Grade 3 asthenia and grade 3 decreased appetite, both on day 14 of olaparib dosing and lasted for 4 days; the patient recovered following an olaparib dose reduction | Yes | Olaparib dose reduction |
| Patient 3 |
Grade 3 anaemia on day 37 of olaparib dosing, lasted for 82 days and resolved following an olaparib dose reduction Grade 4 anaemia on day 120 of olaparib dosing and lasted for 14 days; the patient recovered following an olaparib dose reduction |
Yes | Olaparib dose reduction |
| Moderate hepatic impairment | |||
| Patient 1 | Grade 5 hepatic failure after 23 days of olaparib dosing and was terminal | No | NA |
| Patient 2 | Grade 4 thrombocytopenia on day 53 of olaparib dosing and lasted for 3 days; the patient recovered after olaparib was discontinued | Yes | Olaparib was discontinued |
| Patient 3 | Grade 4 pneumonia after 8 days of olaparib dosing | No | Olaparib was discontinued |
| Patient 4 |
Grade 3 normochromic normocytic anaemia on day 61 of olaparib dosing, lasted for 12 days and resolved following olaparib dose interruption Grade 2 pyelonephritis on day 109 of olaparib dosing and lasted for 13 days; the patient recovered |
The event of normochromic normocytic anaemia was considered related to olaparib | Olaparib was interrupted following the event of grade 3 normochromic normocytic anaemia |
Four patients discontinued olaparib because of an AE (all of which were SAEs). One patient in the group with mild hepatic impairment because of thrombocytopenia and 3 patients in the group with moderate hepatic impairment (n = 1 each for thrombocytopenia, pneumonia and terminal hepatic failure). The 2 discontinuations because of thrombocytopenia were considered by the investigator as related to olaparib treatment.
In total, 6 patients died during Part B of the study, 2 because of an AE (1 each in normal hepatic function and moderate hepatic impairment group), with neither death being considered related to olaparib. Four deaths were related to the disease under investigation.
There were no additional clinically significant changes relating to vital signs, electrocardiogram recordings and physical examination during Part B.
5. DISCUSSION
Liver disease is known to be a common cause of altered PK of drugs, with the degree of effect being dependent on the severity of hepatic impairment. 7 , 8 Olaparib is eliminated through hepatic metabolism, with approximately 42 and 44% of the dose eliminated in faeces and urine, respectively. 9 In patients with mild renal impairment, there was a small increase in exposure to olaparib which was not considered clinically relevant, and in those with moderate renal impairment, olaparib exposure was increased by 44%; these patients should be carefully monitored and, if required, the tablet dose should be adjusted to 200 mg twice daily. 15 Given that olaparib undergoes extensive hepatic metabolism, 3 , 4 , 11 is eliminated through the liver 9 and patients with cancer may have different degrees of hepatic impairment, 10 we evaluated the PK profile of olaparib in patients with normal hepatic function compared with those with mild or moderate hepatic impairment, to determine their appropriate olaparib dose.
To be eligible for this study, patients had to have globally impaired mild or moderate hepatic function. Hepatic impairment resulting in changes in drug disposition is largely related to liver cirrhosis and fibrosis (i.e. factors affecting the ability of the drug to enter the hepatocyte, rather than drug metabolism). As the liver has a very large capacity for drug metabolism, a reduction in the amount of healthy functioning liver (because of the presence of a tumour) would not necessarily have an impact on drug clearance; regulatory authorities recommend hepatic impairment PK studies be conducted in appropriate patients such as those with globally impaired hepatic function based on the Child–Pugh classification. 7 , 8 Since the initiation of this study, the Food and Drug Administration has released draft guidance for the use of the Child–Pugh classification system for hepatic impairment studies and now suggests that pre‐existing liver disease, as well as epidemiological, genetic, environmental and historical variables should also be ascertained to gauge hepatic drug transformation capacity. 16 Patients with hepatic metastases and/or HCC could participate in the current study, providing that these were not the sole reason for changes in liver function satisfying the mild or moderate hepatic impairment criteria. The use of the Child–Pugh classification system in this study may have biased patient selection towards recruiting more patients with HCC in the groups with hepatic impairment. However, patients with hepatic metastases and/or HCC were only eligible if this was not the sole reason for liver function changes and they had globally impaired hepatic function. This was specified because the type of hepatic impairment that affects drug disposition is related to liver cirrhosis and fibrosis, i.e. to factors affecting the ability of the drug to enter the hepatocyte, rather than drug metabolism per se. 17 , 18 HCC and hepatic metastases do not have the same effect on general hepatocyte permeability that is seen in global hepatic impairment (e.g. related to cirrhosis or fibrosis). Since the liver has a very large capacity for drug metabolism, a reduction in the amount of healthy functioning liver (due to the presence of carcinoma/metastases) would not necessarily have any impact on drug clearance. 17 , 18 The aim of this study was to determine the exposure of olaparib in patients with globally impaired hepatic function, and therefore Child–Pugh criteria were determined to be the most appropriate classification to use to determine the degree of impairment for an individual patient.
In this study, we found a 13% increase and 13% decrease in olaparib Cmax and a 15% and 8% increase in olaparib AUC seen in patients with mild or moderate hepatic impairment, respectively, vs those with normal hepatic function; these variations in olaparib exposure were not considered clinically relevant. 19 , 20 Variation in olaparib exposure was notable in the moderate hepatic impairment group; however, this was largely because of 1 patient who had increased exposure to olaparib (indicated by AUC0–t and Cmax values; see the Supplementary Materials for further information on this patient).
Plasma protein binding can be altered in patients with hepatic impairment. However, the protein binding of olaparib appeared unaffected by mild or moderate hepatic impairment, meaning that total plasma exposures were representative of free drug. In addition, we found no correlation between olaparib exposure and clearance and prothrombin time (a functional measure of liver status).
Besides affecting CYP3A expression and activity, hepatic impairment has been reported to be associated with various pathogenic factors, including alterations in drug absorption, plasma protein binding, liver blood flow, portal–systemic shunting, biliary excretion, enterohepatic circulation, and renal clearance, many of which may affect the PK of olaparib. 21 , 22 In addition, in patients with moderate hepatic impairment, cirrhosis may decrease gastrointestinal absorption because of portal hypertension (congestion) and decreased blood flow in intestinal mucosa. 23 , 24 In the moderate hepatic impairment cohort, 1 patient had moderate ascites and 3 patients had slight ascites (a complication of portal hypertension). The patient with moderate ascites had the lowest Cmax in this cohort, and the combined Gmean Cmax value for patients with ascites (n = 4) was lower than in patients without ascites (n = 4) in the same cohort (5.32 vs 7.71 μg mL−1). Although this finding was based on a small number of patients, the potential for reduced absorption of olaparib in patients with ascites cannot be ruled out. The decrease in olaparib absorption could counteract the theoretical effect of decreased metabolism, resulting in little change in the net exposure to olaparib in patients with moderate hepatic impairment compared with those with normal hepatic function. In addition, it has also been reported that there is a lack of effect of moderate hepatic impairment on the PK of substrates that are largely cleared by CYP3A. 25 , 26 Taken together, the results of the current study and those previously reported suggest that mild or moderate hepatic impairment will have no clinically significant effect on exposure to olaparib.
The AE profile following single and multiple olaparib tablet dosing was consistent with the known olaparib safety profile, 27 , 28 , 29 the most common all‐grade AEs following continuous olaparib tablet dosing in Part B were nausea, anaemia and fatigue. There was a slight apparent increase in the incidence of AEs during Part B in the cohort of patients with normal hepatic function; this is likely to be attributable to the extended duration of study participation in this group compared with the hepatic impairment cohorts. A similar observation of slightly increased AEs occurring in patients with extended olaparib exposure was observed in the renal impairment study of olaparib. 15 Overall, in this Phase I trial in patients with advanced solid tumours, there were no clinically relevant differences in the safety profile of olaparib in patients with normal hepatic impairment and those with mild or moderate hepatic impairment.
In conclusion, patients with mild or moderate hepatic impairment had no clinically significant changes in their exposure to olaparib when compared with patients with normal hepatic function. No new safety signals were observed in patients with hepatic impairment, and the safety profile of multiple olaparib dosing did not differ to a clinically relevant extent between patients with normal hepatic function and those with mild or moderate hepatic impairment. As such, no olaparib tablet or capsule dose reductions are required for patients with mild or moderate hepatic impairment. 3 , 6 Olaparib is not recommended in patients with severe hepatic impairment because safety and PK have not been studied in these patients.
COMPETING INTERESTS
Christian Rolfo has received research grants from Pfizer (Lung Cancer Research Foundation), Guardant Health and Biomarker and consulting fees/honorarium from Mylan and Oncompass, has attended advisory boards for ARCHER, Inivata and MD Serono, and has participated in speaker bureaus for MSD, and AstraZeneca. Jan H.M. Schellens is employed by, and holds stock in, Modra Pharmaceuticals, has a patent with oral taxanes, and has a consultancy with Debiopharm. Judith de Vos‐Geelen has received nonfinancial support from BTG and Servier, has served as a consultant for Shire and has received institutional research funding from Servier. Thomas Decaens has consulted for Bristol Myers Squibb, Ipsen, AstraZeneca and Sirtex, has attended advisory boards for Bristol Myers Squibb, Ipsen, AstraZeneca, Sirtex, Bayer, Eisia and Roche, and has received research grants from ArQule and Genoscience. Jean‐Yves Blay has received institutional research support from AstraZeneca. Antoine Italiano has received a research grant from AstraZeneca. Maria Learoyd and Gershon Locker are employees of, and hold stock in, AstraZeneca. Wendy Bannister is a contractor for AstraZeneca. Nicolas Isambert, L. Rhoda Molife, Alain Ravaud, Oliver Rosmorduc, Rebecca Kristeleit, Myung‐Ah Lee, Regina Demlova and Katerina Kopeckova report no conflicts of interest.
CONTRIBUTORS
C.R. was involved in the collection and interpretation of the data and developed the first draft of the manuscript. N.I., A.I., L.R.M., J.H.M.S., J.‐Y.B., T.D., R.K., O.R., R.D., M.‐A.L., A.R., K.K. and J.d.V.‐G. were involved in the collection and interpretation of the data. C.R. and M.L. were involved in the study design and interpretation of the data. G.L. was involved in the interpretation of the data and development of the manuscript. W.B. performed statistical analyses. All authors critically reviewed previous drafts of the manuscript and approved the final version.
Supporting information
TABLE S1 Child–Pugh classification system
TABLE S2 Global liver impairment causality for patients with mild or moderate hepatic impairment included in the study
TABLE S3 Grade ≥ 3 adverse events causally related to olaparib, by Child–Pugh classification
ACKNOWLEDGEMENTS
The authors would like to thank the patients, their families, and all investigators and study personnel involved. Medical writing assistance was provided by Claire Routley PhD from Mudskipper Business Ltd, funded by AstraZeneca and Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (MSD).
This study was sponsored by AstraZeneca and is part of an alliance between AstraZeneca and MSD. AstraZeneca was involved in the study design, data collection, data analysis and data interpretation. MSD also provided input into data interpretation. Medical writing support was funded by AstraZeneca and MSD.
Rolfo C, Isambert N, Italiano A, et al. Pharmacokinetics and safety of olaparib in patients with advanced solid tumours and mild or moderate hepatic impairment. Br J Clin Pharmacol. 2020;86:1807–1818. 10.1111/bcp.14283
Present Address L. Rhoda Molife, MSD, London, UK.
The authors confirm that the PI for this paper is Professor Christian Rolfo and that he had direct clinical responsibility for patients.
DATA AVAILABILITY STATEMENT
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca's data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
REFERENCES
- 1. Gunderson CC, Moore KN. Olaparib: an oral PARP‐1 and PARP‐2 inhibitor with promising activity in ovarian cancer. Future Oncol. 2015;11(5):747‐757. [DOI] [PubMed] [Google Scholar]
- 2. European Medicines Agency . Lynparza (olaparib): An overview of Lynparza and why it is authorised in the EU. 2019. Available at: https://www.ema.europa.eu/en/documents/overview/lynparza-epar-medicine-overview_en.pdf.
- 3. European Medicines Agency . Lynparza summary of product characteristics. 2014. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003726/WC500180151.pdf
- 4. FDA . Lynparza prescribing information. 2014. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/206162lbl.pdf.
- 5. European Medicines Agency . CHMP assessment report on extension of marketing authorisation grouped with a variation: Lynparza. 2018. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/003726/WC500249582.pdf
- 6. FDA . Lynparza prescribing information (revised September 2018). 2018. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208558s006lbl.pdf.
- 7. FDA . Guidance for industry. Pharmacokinetics in patients with impaired hepatic function: study design, data analysis, and impact on dosing and labeling. 2003. Available at: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072123.pdf
- 8. European Medicines Agency . Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with impaired hepatic function. 2005. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-impaired-hepatic-function_en.pdf
- 9. Ang JE, Clarkson‐Jones JA, Swaisland H, et al. A mass balance study to investigate the metabolism, excretion and pharmacokinetics of [14C]‐olaparib (AZD2281) in patients with advanced solid tumours refractory to standard treatments. Eur J Cancer Suppl. 2010;8(7):128‐129. [Google Scholar]
- 10. Superfin D, Iannucci AA, Davies AM. Commentary: oncologic drugs in patients with organ dysfunction: a summary. Oncologist. 2007;12(9):1070‐1083. [DOI] [PubMed] [Google Scholar]
- 11. Dirix L, Swaisland H, Verheul HMW, et al. Effect of itraconazole and rifampin on the pharmacokinetics of olaparib in patients with advanced solid tumors: results of two phase I open‐label studies. Clin Ther. 2016;38(10):2286‐2299. [DOI] [PubMed] [Google Scholar]
- 12. Rolfo C, Swaisland H, Leunen K, et al. Effect of food on the pharmacokinetics of olaparib after oral dosing of the capsule formulation in patients with advanced solid tumors. Adv Ther. 2015;32(6):510‐522. [DOI] [PubMed] [Google Scholar]
- 13. AstraZeneca . Global policy: bioethics. 2016. Available at: https://www.astrazeneca.com/content/dam/az/PDF/2016/Bioethics_policy.pdf
- 14. Mateo J, Moreno V, Gupta A, et al. An adaptive study to determine the optimal dose of the tablet formulation of the PARP inhibitor olaparib. Target Oncol. 2016;11(3):401‐415. [DOI] [PubMed] [Google Scholar]
- 15. Rolfo C, de Vos‐Geelen J, Isambert N, et al. Pharmacokinetics and safety of olaparib in patients with advanced solid tumours and renal impairment. Clin Pharmacokinet. 2019;58(9):1165‐1174. [DOI] [PubMed] [Google Scholar]
- 16. FDA . Cancer clinical trial eligibility criteria: patients with organ dysfunction or prior or concurrent malignancies guidance for industry. 2019. Available at: https://www.fda.gov/media/123745/download.
- 17. Talal AH, Venuto CS, Younis I. Assessment of hepatic impairment and implications for pharmacokinetics of substance use treatment. Clin Pharmacol Drug Dev. 2017;6(2):206‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Palatini P, De Martin S. Pharmacokinetic drug interactions in liver disease: an update. World J Gastroenterol. 2016;22(3):1260‐1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pilla Reddy V, Bui K, Scarfe G, Zhou D, Learoyd M. Physiologically based pharmacokinetic modeling for olaparib dosing recommendations: bridging formulations, drug interactions, and patient populations. Clin Pharmacol Ther. 2019;105(1):229‐241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou D, Li J, Learoyd M, et al. Efficacy and safety exposure‐response analysis of olaparib capsule and tablet formulations in oncology patients. Clin Pharmacol Ther. 2019;105(6):1492‐1500. [DOI] [PubMed] [Google Scholar]
- 21. Johnson TN, Boussery K, Rowland‐Yeo K, Tucker GT, Rostami‐Hodjegan A. A semi‐mechanistic model to predict the effects of liver cirrhosis on drug clearance. Clin Pharmacokinet. 2010;49(3):189‐206. [DOI] [PubMed] [Google Scholar]
- 22. Bupsilondingen FV, Gonzalez D, Tucker AN, Derendorf H. Relevance of liver failure for anti‐infective agents: from pharmacokinetic alterations to dosage adjustments. Ther Adv Infect Dis. 2014;2(1):17‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ono C, Hsyu PH, Abbas R, Loi CM, Yamazaki S. Application of physiologically based pharmacokinetic modeling to the understanding of bosutinib pharmacokinetics: prediction of drug‐drug and drug‐disease interactions. Drug Metab Dispos. 2017;45(4):390‐398. [DOI] [PubMed] [Google Scholar]
- 24. Sarfeh IJ, Aaronson S, Lombino D, et al. Selective impairment of nutrient absorption from intestines with chronic venous hypertension. Surgery. 1986;99(2):166‐169. [PubMed] [Google Scholar]
- 25. Bui K, She F, Sostek M. The effects of mild or moderate hepatic impairment on the pharmacokinetics, safety, and tolerability of naloxegol. J Clin Pharmacol. 2014;54(12):1368‐1374. [DOI] [PubMed] [Google Scholar]
- 26. Khalilieh S, Yee KL, Liu R, et al. Moderate hepatic impairment does not affect doravirine pharmacokinetics. J Clin Pharmacol. 2017;57(6):777‐783. [DOI] [PubMed] [Google Scholar]
- 27. Friedlander M, Matulonis U, Gourley C, et al. Long‐term efficacy, tolerability and overall survival in patients with platinum‐sensitive, recurrent high‐grade serous ovarian cancer treated with maintenance olaparib capsules following response to chemotherapy. Br J Cancer. 2018;119(9):1075‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pujade‐Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum‐sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT‐Ov21): a double‐blind, randomised, placebo‐controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274‐1284. [DOI] [PubMed] [Google Scholar]
- 29. Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523‐533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Child–Pugh classification system
TABLE S2 Global liver impairment causality for patients with mild or moderate hepatic impairment included in the study
TABLE S3 Grade ≥ 3 adverse events causally related to olaparib, by Child–Pugh classification
Data Availability Statement
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca's data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.