

Abstract
The development of immune checkpoint inhibitors (ICI) represents a major milestone in immune‐oncology. Over the years these agents have demonstrated efficacy in an increasing array of malignancies. Despite this success however, significant challenges remain. Novel approaches to both drug development and trial design are required to incorporate the unique pharmacokinetic and pharmacodynamic properties of ICIs. Further, it has also been established that the benefit of ICIs is limited to only a subset of patients. The molecular interactions between native immune cells and tumorigenesis and progression represent an active area of biomarker research, and elucidating the mechanisms of response and resistance is crucial to develop rational trial designs for the next wave of immune‐oncology (IO) clinical trials, particularly in patients with primary and/or acquired resistance. Efforts are now being made to integrate both biological and clinical information using novel multi‐omic approaches which are now being developed to further elucidate the molecular signatures associated with IO treatment response and resistance and enable rational drug development and trial design processes. As such, precision IO and the ability to deliver patient‐specific choices for ICI monotherapies or combination therapies has become an increasingly tangible goal. We herein describe the current landscape in ICI drug development and discuss the challenges and future directions in this exciting and evolving era in immune‐oncology.
Keywords: biomarkers, clinical trials, drug development, immune checkpoint inhibitors, immuno‐oncology
1. INTRODUCTION
Over the last decade, advances in our understanding of the intricacies of our immune system has resulted in unprecedented and practice‐changing innovation in the field of immune‐oncology (IO), with the development of novel approaches that has resulted in a paradigm shift, where we now aim to modulate our own immune response to target cancer cells. Immune checkpoints are the regulators of our immune system, and encompass a group of regulatory surface proteins on immune cells that are crucial for self‐tolerance, responsible for preventing excessive stimulation and subsequent autoimmune responses. Immune checkpoint inhibitors (ICIs) target these molecules and exploit the exquisite specificity and potency of the hosts' immune response to “release the brakes” on this normally stringent regulatory process, unleashing T‐cell‐mediated cancer cytotoxicity. 1 ICIs such as programmed cell death‐1 (PD‐1)/programmed cell death ligand‐1 (PD‐L1) inhibitors and cytotoxic T‐lymphocyte associated protein‐4 (CTLA‐4) inhibitors have revolutionized cancer therapeutics and are now the new standard of care in multiple solid and haematologic malignancies in both curative and palliative settings. To date seven of these immunomodulatory monoclonal antibodies (mAbs) have received regulatory approval; ipilimumab, pembrolizumab, nivolumab, atezolizumab, avelumab, durvalumab, and, most recently, cemiplimab 2 (Figure 1).
FIGURE 1.

Time of FDA approval of ICIs in solid tumours (https://www.fda.gov/drugs, accessed November 14, 2019). Abbreviations: 1 = first line; 2 = second line; 3 = third line; A = any line; RCC = renal cell carcinoma; NSCLC = non‐small cell lung cancer; non‐squam = non‐squamous; PDL1 = programmed death‐ligand 1; HNSCC = head and neck squamous cell carcinoma; MSI‐H = microsatellite instability‐high; dMMR = deficient mismatch repair; Met = metastatic; HCC = hepatocellular carcinoma; GEJ = gastroesophageal; SCC = squamous cell carcinoma; HR/IR = high risk/intermediate risk; ES SCLC = extensive stage small cell lung cancer; TNBC = triple negative breast cancer
In addition to ICIs, other IO agents that have had success targeting different immune modulatory pathways directly include cytokines (Interleukin‐2, interferon‐α) and cancer vaccines (Sipuleucel‐T), both capable of inducing an immune response (active immunity) (Figure 2). Passive immunotherapies, or those that stimulate a patient's intrinsic immune response, include oncolytic viruses (Talimogene laherparepvec [T‐VEC]) and cell‐based therapies (Adoptive T‐cell therapy (e.g., tumour‐infiltrating lymphocytes [TIL], T‐cell receptors [TCR], chimeric antigen receptor T‐cells [CAR‐T]), the discussion of which is beyond the scope of this review. The field of IO continues to evolve at a breathtaking pace, and the increase in the number of these agents under development has resulted in a reform of the drug development enterprise. 3
FIGURE 2.

Summary of immunotherapeutic agents
Despite this exciting breakthrough, clinical efficacy of ICIs as monotherapy has been limited to only a small subset of patients. Overall, the response rates for the majority of solid tumour types treated with ICI monotherapy fall below 50%. 4 Combination strategies that target various phases of the cancer–immunity cycle are now being explored in an effort to induce a synergistic effect as well as enhancing the anti‐tumour activity of these agents. This approach has already demonstrated improved overall survival (OS) when compared to monotherapy in advanced melanoma and advanced renal cell carcinoma (RCC), albeit with increased toxicity. 5 , 6
It is, however, necessary to remain vigilant and proceed with caution when administering combination ICIs, as it has been well published that ICIs are associated with a unique toxicity profile. These immune‐related adverse events (irAEs), which arise from immunologic enhancement, may affect any organ system including skin, gastrointestinal, endocrine organs, liver, and lungs. This toxicity has been associated with significant morbidity and, indeed, mortality. 7
As such, biomarkers that can predict the efficacy of ICI therapy and the development of irAEs could guide patient selection and inform decision making in the drug development and clinical trial design process by distinguishing between responders and non‐responders. Numerous studies on predictive biomarkers have been described and include immune cell infiltration, peripheral blood analyses, copy number alterations, neoantigen clonality, mutational landscape and mismatch repair (MMR) deficiency. 8 To date, however, PD‐L1 expression as a predictive biomarker has been the best described. Its role in the accelerated approval of ICIs has been critical, along with novel surrogate endpoints such as response rate. However, it has also been demonstrated in melanoma that negative expressors can also benefit and obtain durable responses, suggesting that PD‐L1 expression alone may not be able to fully capture the complexities of the tumour microenvironment (TME) and its changes under the treatment selection pressure of immunotherapy. 9 Novel approaches that combine other potential biomarkers at serial timepoints are now being explored in an effort to generate a more complete molecular signature that may predict response and resistance. This may be coupled with more innovative and integrated computational omics approaches to better characterize the TME and its changes over the course of treatment. Finally, it is also important to recognize the financial burden associated with these agents, which has significant pharmacoeconomic implications. Thus methods of improving patient selection and enabling patient‐specific choices for ICI monotherapy or combination therapy is paramount.
Novel and innovative clinical trial design strategies are required to address these issues and adapt to the IO paradigm shift. Recognition of the variant biological properties of IO agents, which have distinct pharmacokinetic (PK) and pharmacodynamic (PD) characteristics compared to chemotherapy, and the understanding that the conventional correlative relationship between toxicity and efficacy does not apply, is crucial to early phase trial design. PK/PD modelling now plays an increasingly important role in the decision‐making process during drug development. Traditional clinical trial endpoints, including OS and progression‐free survival (PFS), may be limited in their ability to predict long‐term survival of patients treated with ICIs. As such, there is a growing need to adapt and implement novel approaches to clinical trial design that address the unique clinical and statistical considerations arising from these new IO treatment models. 10 In this review we describe the current landscape and challenges in ICI drug development and discuss novel approaches to trial design that are now being applied in in the field of immune‐oncology.
2. CURRENT BASIS FOR DRUG DEVELOPMENT
The evolving landscape of oncological drug development has resulted in a demand for more innovative approaches to clinical trial design, particularly in early drug development. The objectives for preclinical evaluation remain similar: (1) identification of the pharmacological characteristics of the development molecule, (2) determination of an initial safe and starting dose for human studies, and (3) understanding the toxicological profile of the development molecule. 11 However, the development of cancer immunotherapies present unique challenges in translating preclinical data to the clinic, namely cell lines or animal models that do not adequately mimic the tumour or its microenvironment, human immune response, or the predisposition to develop resistance. 11
Beyond the preclinical setting, drug development has historically followed an assiduously designed pathway of sequential trials with an increasing number of patients, systematically generating data pertaining to both the safety profile and clinical activity of a novel therapeutic agent. 12 However traditional approaches to early phase trial design were developed during the era of chemotherapy, and thought to be unsuitable for modern ICI development due to varying biological properties which may influence the ability to correctly define the recommended phase II dose (RP2D) or identify adverse events (AEs). Historically, increasing doses of a drug were sequentially administered until a pre‐specified rate of dose‐limiting toxicity (DLT) was reached, and the dose immediately below that was considered the maximum tolerated dose (MTD). 10 , 13 This approach, however, is unsuitable for ICIs. DLTs associated with cytotoxic chemotherapy agents are usually observed early in the course of treatment, and the MTD may be defined at this point. ICIs, however, are administered over longer durations, which can result in irAEs occurring outside of this short‐term evaluation window. This makes it difficult to fully elucidate the chronic toxicity profile, resulting in an incomplete portrayal of tolerability. In a pooled analysis of 576 patients with advanced melanoma who received nivolumab, the median onset time of various treatment‐related adverse events of any grade ranged from 5.0 weeks for skin toxicities to 15.1 weeks for renal toxicities. 14
The assumption that the MTD will provide the greatest therapeutic benefit was driven by the notion that cytotoxic chemotherapy treatments will directly inhibit tumour cell growth. The long‐standing belief that “more is better” for efficacy and “more is worse” for toxicity has now been rendered obsolete with the introduction of IO agents. Early development of ICIs have transitioned from identifying the MTD to instead identifying the minimum effective, or optimal biologic dose (OBD). Novel, adaptive phase 1 trial designs that incorporate the establishment of an OBD range have been implemented successfully, as in the case of pembrolizumab, where the OBD of 2 mg/kg and maximum administered dose of 10 mg/kg were explored, with the former demonstrating a similar anti‐tumour response in this range. 15 , 16 , 17 , 18 However, OBD range will be challenging to define with novel IO agents unless a robust, biologically‐relevant, PD biomarker exists. 19 This provides an opportunity for PK/PD‐based dose recommendations to come to the fore.
3. CLINICAL PHARMACOLOGY: PHARMACOKINETICS AND PHARMACODYNAMICS
PK and PD can inform on the relationship between drug concentration and its biologic effects, specifically drug absorption, distribution, metabolism, and excretion. The PK of approved monoclonal antibodies (mAbs) such as ICIs substantially differ in complexity compared to small molecule drugs. 20 The PK of mAbs are similar to that of endogenous immunoglobulin G (IgG), and after administration, distribution is limited by their size and hydrophilicity, thus body composition becomes less of an issue. 3 , 20 , 21 , 22 , 23 Clearance of mAbs is governed by various physiological mechanisms, and again differs from clearance of small molecule drugs. MAbs are too large to be cleared via renal or hepatic routes, thus elimination occurs via two alternative pathways. 20 , 24 The first, which appears to predominate, is a nonspecific (Fc‐mediated) route of degradation within plasma and tissues via proteolytic catabolism, which accounts for a linear elimination PK. The second, hastier method is more specific and target mediated, involving receptor‐mediated endocytosis and degradation, accounting for nonlinear elimination PK. 20 , 25 Clearance may also be expedited via the development and formation of antidrug antibodies (ADAs), which facilitate the uptake and endocytic degradation of ICIs. 4 The PD characteristics also are different. While the toxicity of small molecules may be caused by an on‐target or off‐target mechanism, the toxicity associated with immunotherapies is largely due to indirect activation of immunogenicity, often associated with a delayed onset of most of the reported irAEs.
In recent times PK/PD modelling has become a particularly useful tool in enabling a more efficient IO drug development process. Only a fraction (approximately 5%) of the novel agents demonstrating anticancer activity in preclinical development are ultimately approved for use in the clinical arena. 26 Drug modelling has considerable economic value with its potential ability to save on costs by determining which compounds should progress forward to later phase trials and which compounds should not. PK/PD modelling refers to an exploratory analysis, integrating preclinical information and employing mathematical and statistical models to describe disease progression, PK, and PD. These models augment our understanding of the relationship between exposure and response, as well as the dynamic changes between these relationships as a result of drug intake. These models can optimize the design of clinical trials, guide the dose and regimen that should be further evaluated, improve patient selection, and facilitate a better understanding about the potential clinical relevance of preclinical efficacy data. 27 , 28 , 29 , 30
The clinical development and approval of pembrolizumab highlights the importance of modelling in ICI drug development, exemplifying a modern, seamless drug development strategy that facilitated the rapid testing and approval of a novel, highly promising ICI under the umbrella of a single clinical trial. 12 , 31 , 32 KEYNOTE‐001 (NCT01295827) was a multicentre, open‐label phase I clinical trial enrolling patients with progressive, locally advanced, or metastatic carcinoma, melanoma, or non‐small cell lung carcinoma (NSCLC). 16 , 33 A total of 135 patients with metastatic melanoma received either the MTD, pembrolizumab every 2 weeks at a dose of 10 mg/kg, or pembrolizumab every 3 weeks at a dose of 2 mg/kg or 10 mg/kg. The high level of clinical activity observed in these patients led to an increase in the sample size, and additional cohorts of patients with other types of cancer were added. Ultimately over 1200 patients were treated on this trial in order to further evaluate dose characteristics, overall response rates (ORR), and disease control rates (DCR). 12 , 33 , 34 The ORR and safety profiles were similar for both doses tested, though the peak drug exposure was higher for the 10 mg/kg dose cohort. 35 , 36 , 37 Despite the safety of the higher dosage studied (10 mg/kg every 2 weeks), model‐based characterization of the PK of pembrolizumab indicated an absence of covariate effects and suggested that robust clinical activity would be observed from a dosage of 2 mg/kg every 3 weeks. 17 , 18 This dosage, therefore, was recommended to be tested for clinical efficacy in additional clinical trials. The unprecedented clinical efficacy observed in this cohort was sufficient to support accelerated approval of pembrolizumab at 2 mg/kg every 3 weeks for unresectable or metastatic melanoma in September 2014, only 3 years after the trial began and 3 months after the publication of the phase 1 data. 33 , 34 Subsequently data from the NSCLC cohort led to accelerated approval of pembrolizumab for metastatic PD‐L1+ NSCLC. Modelling and simulation were crucial components in expediting approval by characterizing dosing and clinical pharmacology of pembrolizumab, sparing the need for extensive dose‐finding and dedicated clinical pharmacology studies. 38
4. DOSING STRATEGY
Determination of first‐in‐human (FIH) starting dose for anticancer drugs is traditionally based on preclinical toxicology testing in the appropriate, most sensitive species and then is converted to the human‐equivalent dose and finally the recommended safe dose. 3 , 39 , 40 This approach is challenging for biologics due to the differences in target binding, distribution, and catabolism capacities between humans and animals. 41 In general, mAbs have shown to be well tolerated; however, they can be highly toxic when dose and schedule are not diligently scrutinized, as evidenced by the events observed with an immunomodulatory anti‐CD28 superagonist mAb (TGN1412), which resulted in six patients requiring admission to intensive care for life‐threatening cytokine‐release syndrome (CRS). 42 One approach accepted and recommended by both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) is to consider the minimum anticipated biological effect level (MABEL) for the FIH starting dose to ensure a balance of both safety and pharmacological activity. 43
Traditionally most mAbs are administered in a body–size‐based dosing schedule instead of a fixed dose for all patients, based on a perceived contribution of body size in PK variability. 23 , 44 All the currently approved checkpoint inhibitors were initially developed and approved by the FDA as body weight‐based dosing regimens, apart from atezolizumab, which was approved as a flat dose. However, because of the specific properties of mAbs (selective mode of action, with substantial/broad therapeutic window) and the advantages of fixed/flat dosing (increased convenience/easier dose preparation and improved safety resulting from a reduced chance for dosing errors), the weight‐based dosing paradigm has recently been re‐evaluated. 44
Following the initial body weight‐based approvals, the need for this type of dosing was reassessed for both pembrolizumab and nivolumab based on population PK modelling and therapeutic window information derived from exposure–response analyses, as well as demonstrated clinical safety at doses up to 10 mg/kg. 3 , 15 These assessments showed that 200 mg and 2 mg/kg doses provided similar exposure distributions for pembrolizumab and that the benefit–risk profile of nivolumab 240 mg every 2 weeks was comparable to 3 mg/kg every 2 weeks. An analysis comprising samples from 2993 patients on KEYNOTE studies 001, 002, 006, 010, and 024 found that the PK of pembrolizumab was consistent across weight‐based and fixed‐dose regimens and demonstrated a flat exposure–response relationship, and these findings were confirmed in randomized clinical trials demonstrating no statistically significant differences in outcomes (ORR, PFS, and OS) between the two dose options (2 vs 10 mg/kg) in melanoma and mNSCLC. 16 , 34 , 36 , 45 , 46
Pembrolizumab was approved as a flat dose for all indications subsequent to melanoma, and most recently six supplemental Biologics License Applications (sBLAs) have been approved to update the dosing schedule to 400 mg given in 30‐minute infusions every 6 weeks. These indications include unresectable or metastatic melanoma, adjuvant treatment of patients with melanoma with involvement of lymph node(s) following complete resection, adult and paediatric patients with recurrent locally advanced or metastatic Merkel cell carcinoma, adult and paediatric patients with refractory classical Hodgkin lymphoma, or those who have relapsed after ≥3 prior lines of therapy, adult and paediatric patients with refractory primary mediastinal large B‐cell lymphoma, or who have relapsed following ≥2 lines of prior therapy, patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction adenocarcinoma whose tumours express PD‐L1 (CPS ≥1) and patients with hepatocellular carcinoma who have been previously treated with sorafenib.
The change to flat dosing regimens has significant economic implications, as mAbs are expensive drugs and have a significant impact on the healthcare budget. Fixed dosing results in easier administration and less wastage. 19 Goldstein et al. evaluated the economic impact of pembrolizumab using personalized dosing (2 mg/kg) vs fixed dosing (200 mg) in the first line setting of metastatic NSCLC. 47 It was postulated that the FDA‐approved dose of 200 mg for all patients may be unnecessarily high, given that the average weight in the United States is 82 kg. Thus, if dosed at 2 mg/kg, an appropriate dose for the average American adult would be 164 mg. In this study, it was suggested that, by employing personalized dosing, it may be possible to save $0.825 billion annually and that this shift is unlikely to impact clinical outcomes.
5. NOVEL ICI APPROACHES TO PRECISION ONCOLOGY
The success of immunotherapy has generated new clinical questions, including the appropriate use of immunotherapy in earlier stage disease, duration of therapy, novel biomarkers for patient selection, rational combination regimens and the development of novel surrogate endpoints that reflect early clinical benefit of ICI therapy. The translation of preclinical data into the clinic is hampered by the major limitation that cell lines or animal models cannot adequately represent the TME or human immune response, consequently most of our insight into the mechanisms of ICIs is generated from early phase clinical trials. Overcoming these challenges and defining which subgroups will derive the most benefit is one of the major challenges facing cancer drug development in oncology in general and in IO specifically (Figure 3).
FIGURE 3.
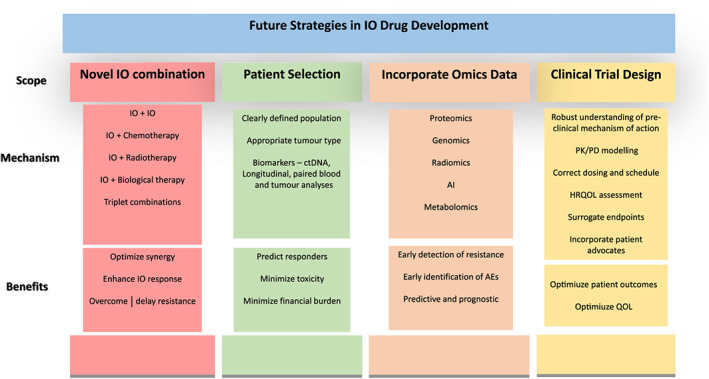
Innovative strategies in IO drug development
5.1. Biomarkers
While the introduction of ICIs has been a most welcome addition to the treatment armamentarium in various advanced malignancies, there remains limitations to their clinical application due to a lack of effective, predictive biomarkers. Despite impressive clinical outcomes associated with immunotherapy, there remains a substantial percentage of patients that do not receive any clinical benefit. Coupled with the risk of therapeutic toxicity and escalating costs, the current challenge is to identify these non‐responders as early as possible, and then select the best combinatorial approach from the numerous IO drugs available. The successful development of clinically significant biomarkers depends upon three features: their biological role with respect to malignant transformation and tumour progression; the ability to detect them with robust, reliable, and clinically applicable analytical genomic tests; and their prognostic or predictive value, as validated in clinical trials. 48 The value of biomarkers in the therapeutic process has already demonstrated clinical utility, for example in the case of anti‐HER2 therapies for HER2 amplified breast cancer and EGFR inhibitors for EGFR mutated NSCLC. 49 , 50
To date PD‐L1 expression in the pre‐treatment TME represents the most well‐researched potential biomarker of response to ICI therapy. As discussed previously, early clinical trials investigating PD‐1 blockade showed that immunohistochemistry (IHC) detection of tumour cell PD‐L1 expression was associated with therapeutic response, and its expression played a critical role in the accelerated approval of ICIs. 51 The emergence and approval of pembrolizumab in PD‐L1 positive (>50% positive tumour cells by IHC) second‐line previously treated NSCLC was predominantly based on responses observed in an expanded cohort of the phase 1 Keynote 001 study, and this has now been extended to the first‐line setting. 14 , 15 A recent meta‐analysis involving 41 clinical trials and 6664 patients with advanced solid tumours investigated the predictive value of tumour and tumour‐infiltrating immune cell PD‐L1 expression by IHC assays (Dako 28‐8, Dako 22C3, Ventana SP142, Ventana SP263, Dako clone 73‐10) and demonstrated that PD‐L1 expression was predictive of tumour response across all tumour types (odds ratio [OR] 2.26, 95% confidence interval [CI] 1.85–2.75, P < 0.001). 52
However, there remain controversies about its predictive value, as other clinical studies have also found that a subset of patients, for example in melanoma, with low or negative PD‐L1 expression, can also benefit from PD‐1/PD‐L1 inhibitors. 9 It has been postulated that some of the limitations surrounding PD‐L1 testing in current clinical practice are likely related to variability of the aforementioned assays and antibodies used to detect PD‐L1, the thresholds used to define positivity, and the heterogeneous and dynamic expression of PD‐L1 on TME cell types. 53 , 54 Ultimately while PD‐L1 expression may give an early indication as to the potential of treatment response, it alone cannot adequately capture the complexity of the TME and its interactions and consistently predict clinical benefit from ICIs.
Tumour mutation burden (TMB) has recently emerged as another promising biomarker, and represents the total number of somatic mutations found in the DNA of cancer cells. This has been facilitated by the increasing development and implementation of next generation sequencing (NGS) technologies. The translational significance of TMB assessment is derived from its link to tumour immunogenicity and its subsequent prognostic and predictive values. 55 Mutational load, and in particular, nonsynonymous mutations, in cancer cells may generate neoantigens, novel protein epitopes specific to tumours that may be presented on the tumour cell surface by major histocompatibility complex (MHC) molecules. 56 These neoantigens are not subject to immune tolerance and allow for an adaptive immune response by the host, and increased production is thought to render a tumour more immunogenic, thus increasing its likelihood of IO response. Consistent with this hypothesis, a high TMB has been shown to predict response to ICI in a diverse range of cancer types. 56 , 57 Furthermore, a high TMB was shown to be associated with an enhanced benefit from ICIs as well as combined anti‐PD‐1–anti‐CTLA4 therapy, compared with patients with low TMB. 56 , 57 , 58 , 59 However as with PD‐L1 concentrations, high TMB alone does not guarantee response to ICI, as patients with low TMB have been shown to respond to ICI and vice versa. 60 To date, TMB assessment is not standardized, and its use has been limited by significant costs, a long turnaround time and expertise required for NGS data interpretation. 61
Microsatellite instability (MSI) is another clinically relevant biomarker used to predict IO response. Microsatellites (MSs) are short stretches of DNA (usually 1–6 nucleotides long) tandemly repeated throughout the genome. 62 MSI occurs when the genome gains or loses one or more repeats. The DNA MMR system is responsible for correcting these errors, and its key proteins include MLH1, MSH2, PMS2, MSH6, or epithelial cellular adhesion molecule. 63 , 64 Germline or somatic mutations in any of these genes, or hypermethylation in the promoter of the MLH1 gene, result in defective MMR (dMMR) and subsequent inability to repair errors that occur during DNA replication. These errors tend to occur predominantly in MS regions, and tumours with these errors are regarded as MSI‐high (H).
As a result of dMMR, both the number of mutations and neoantigens are higher in tumours with this defect than in those with intact, or proficient, MMR. As in the case of TMB, the increased number of neoantigens may be expected to render tumours more immunogenic. This hypothesis was tested by Le et al., who evaluated response to pembrolizumab in 41 patients with advanced metastatic carcinoma with or without dMMR. 65 Objective response was found in 4 of 10 (40%) patients with dMMR CRC, in 7 of 9 (78%) patients with dMMR non‐CRC, but in none of 18 patients with intact MMR. Subsequent studies across a broad range of different cancer types confirmed these findings. 66 , 67 Another study using combined data from MSI‐H/dMMR tumours across 15 different cancer types enrolled in single‐arm studies showed that pembrolizumab induced a complete or partial response in 40% of 149 patients. 67 Based on these findings, MSI became the first biomarker used to select patients for ICI in a tissue/site‐agnostic manner, with FDA approval of pembrolizumab for the second or later lines of therapy for metastatic, MSI‐high tumours. However, only approximately 5% of all solid tumours have been found to have dMMR, and the low prevalence thus limits its use as a broad‐based predictive biomarker for immunotherapy.
Peripheral blood biomarkers have also been studied but are less established. Lymphocytes are perhaps the most frequently studied peripheral blood component as predictors of response to immunotherapy. 68 , 69 A strong positive association between clinical response and the degree of lymphocytosis immediately after therapy has been reported. In a study of ipilimumab treatment, increasing absolute lymphocyte counts (ALCs) during treatment were significantly associated with both disease control and survival. 70
Elevated serum lactate dehydrogenase (LDH) levels have demonstrated a negative predictive value, with limited long‐term benefit from ipilimumab treatment found among patients with baseline serum LDH greater than twice the upper limit of normal in a cohort of 166 patients from the Netherlands. 71 Simeone et al. also found that decreasing levels of serum LDH and CRP between baseline and the end of ipilimumab treatment at week 12 were significantly associated with both disease control and survival for 95 patients treated with ipilimumab. 70
Additional proposed markers of response include neutrophil‐to‐lymphocyte ratio (NLR), which has been reported to increase over time in non‐responders and decrease in responders, and inflammatory cytokines, which have also been correlated with response rates and toxicity 72 , 73 , 74 , 75 , 76 , 77 , 78 (Figure 3). Myeloid‐derived suppressor cell (MDSC) levels have been used to predict therapy response or resistance to ipilimumab treatment in metastatic melanoma patients, where increased frequency was associated with decreased efficacy, and vice versa. 79 , 80 Furthermore, the efficacy of ICI treatment has been reported to be negatively correlated with increased MDSC frequency and function in other malignancies, including lung and colorectal cancer. 81 , 82 , 83
Measurements of single biomarkers at static timepoints, however, may not accurately reflect the complex, dynamic network of interactions of the TME. Martens et al. recently reported a composite analysis suggesting that a signature of low LDH, absolute monocyte counts, high relative lymphocyte counts, eosinophil counts, and regulatory T cells (Tregs) are associated with favourable outcomes among 209 patients, suggesting that evaluating multiple dynamic cell populations may be necessary to achieve predictive power (Figure 4). 84 Additional insight into tumour dynamics may be gained by pairing longitudinal, peripheral blood and tumour monitoring over the course of treatment. Previous clinical studies of ICIs have highlighted the importance of assessment of early on‐treatment immune signatures in predicting responses to therapy, as seen with CD8 T cells and ICOS positive CD4 T cells after ipilimumab treatment and CD3 and CD8 T cells after anti‐PD1 treatment. 85 , 86
FIGURE 4.
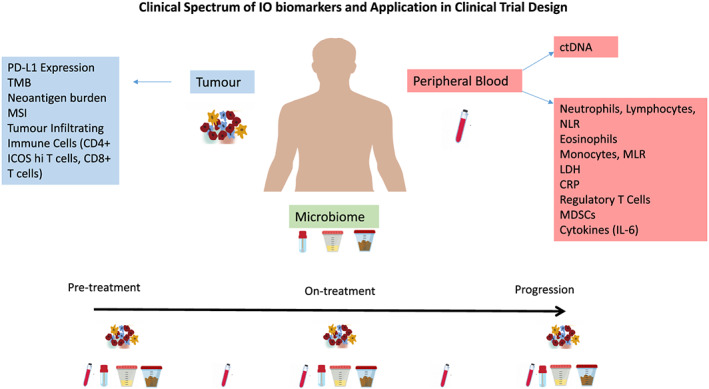
Current biomarkers under investigation for IO agents. Longitudinal analyses of serial blood, tumour and microbiome samples may provide further insight into tumour dynamics under the treatment selection pressure of immunotherapy
The acquisition of multiple, on treatment tissue biopsies, however, is a significant challenge in routine clinical practice because it may increase morbidity, and may not be feasible in all cases. Liquid biopsies, which involve the genomic analysis of circulating tumour DNA (ctDNA), could minimize these issues. 48 This approach has been shown to be associated with tumour burden, and high levels of ctDNA are an indicator of poor prognoses in patients with various types of cancer. 69 , 87 Lee et al. demonstrated that melanoma patients with a persistently elevated ctDNA during treatment with anti‐PD‐1 antibodies were associated with a worse response and shorter PFS and OS. 88 Novel clinical trial strategies are now emerging that incorporate ctDNA to further evaluate its role as a predictive biomarker. The INSPIRE trial (NCT02644369) added incentive and momentum for future biomarker studies by facilitating a dynamic assessment of the antitumor immune response with the aim of providing further insight into the mechanisms of ICI response and resistance in multiple cohorts of patients treated with pembrolizumab. 89 This phase II, multi‐cohort basket trial involved 106 patients treated across five cohorts: (a) squamous cell carcinoma of the head and neck (SCCHN) (n = 19), (b) triple negative breast cancer (TBNC) (n = 22), (c) epithelial ovarian cancer type II (EOC) (n = 21), (d) malignant melanoma (MM) (n = 12), and (e) mixed advanced solid tumours (n = 32). This unique study demonstrated that serial biopsies are informative and feasible in many cases, and establishing the quantity and character of immune cells from both blood and tumour samples at serial timepoints can further inform on the spatial relationship of inflammatory mediators and potentially establish a role for peripheral blood sampling as a potential surrogate for tumour biopsies.
5.2. IO combination therapy
Administering a second IO agent, with the aim of inducing synergistic activity, may further optimize response to IO drugs. There is ample evidence that immune checkpoint blockade is most effective primarily in “hot” tumours, or those that are already recognized by the immune system (due to a pre‐existing CD8+ T cell infiltrate), and less efficacious in “cold” tumours. The lack of a spontaneous tumour‐directed immune response in the latter may be due to tumour antigens that are almost indistinct from self‐antigens, resulting in more inconspicuous tumour cell features. 90 Mechanisms to convert a non‐T‐cell‐inflamed, or “cold” tumour into a T‐cell‐inflamed, “hot” tumour are an active area of research and include novel vaccines, oncolytic virus approaches, stimulation of co‐stimulatory molecules, targeted therapy, radiation/chemotherapy, and adoptive cell therapy (T cells, CARs). 90 This approach could be particularly promising for patients with either primary or acquired resistance to single agent anti‐PD‐1/PD‐L1 therapy. The complex toxicity and efficacy profiles associated with combination therapy and potential interactions, however, makes initial dose finding challenging, but patterns of side effects and proposals for safe starting doses have begun to be delineated for combination strategies. 91 , 92 , 93 , 94
The first FDA approval of combination therapy involved two ICIs, nivolumab plus ipilimumab, indicated for unresectable or metastatic melanoma. 95 There was a strong mechanistic basis for testing this combination; while both are ICIs targeting negative regulators of the immune response, they act on nonredundant regulatory pathways. 96 CTLA‐4 acts in the lymphoid system, during the priming phase, while PD‐1 and PD‐L1 act downstream during the effector phase in the TME. 97 Mechanisms of resistance to PD‐1 and CTLA‐4 inhibitors could therefore potentially be overcome with a combination approach. As monotherapies, these agents demonstrated significant clinical benefit with marked improvements in PFS and OS. In combination, these agents augmented that anti‐tumour response with even more impressive results. Recent updates from the CHECKMATE 067 trial reported a median OS of more than 60 months (median not reached) in the nivolumab‐plus‐ipilimumab group compared to 36.9 and 19.9 months in the nivolumab monotherapy and ipilimumab monotherapy arms respectively. Five‐year OS was reported at a rate of 52% in the combination arm compared with 44% in the nivolumab group and 26% in the ipilimumab group. 5
With increasing use of IO combination therapy, it is reasonable to anticipate that concessions from standard dosing and scheduling are likely to be required to achieve therapeutic potential of combination regimens with an acceptable risk–benefit. 90 The combination of standard doses of the aforementioned ICIs (ipilimumab [3 mg/kg] and nivolumab [3 mg/kg]) was poorly tolerated, resulting in substantial incremental toxicity without improvement in clinical benefit. 95 The initial phase I study of ipilimumab/nivolumab reported grade 3/4 drug‐related AEs in 53% of patients across the range of doses tested, while rates of grade 3/4 AEs in the subsequent randomized phase III trial were 55% in patients treated with the combination versus 27.3% or 16.3% among patients treated with either ipilimumab or nivolumab alone, respectively. 95 , 98 However, standard doses of ipilimumab (3 mg/kg) could be combined safely with doses of nivolumab up to 1 mg/kg, and vice versa (nivolumab [3 mg/kg] combined with ipilimumab up to 1 mg/kg). More recently the CheckMate 511 phase IIIb/IV trial raised the nivolumab dose to 3 mg/kg and lowered the ipilimumab dose to 1 mg/kg, and resulted in an improved safety profile without compromising efficacy. 99
There is an increasing number of new clinical trials in development combining IO agents with other IO agents, and combinations with chemotherapy, radiation therapy, and targeted therapies are also being explored. As of April 19, 2020, there are currently 181 early phase clinical trials investigating IO agents in combination with other IO agents and other treatment modalities such as chemotherapy, radiation therapy, and biologics (Table 1) (www.clinicaltrials.gov). Chemotherapy has the potential to activate the immune system through multiple pathways. 100 Cytotoxic cell death and subsequent antigen release may provide immune stimulation, and cyclophosphamide has been shown to reduce the number of circulating Tregs, a key mediator in immunosuppression. 101 Gemcitabine has also been shown to reduce MDSCs which also have immunosuppressive properties. 102 Radiotherapy has been reported to stimulate the immune response; indeed, the well‐recognized abscopal response appears to be immune‐mediated. 103 Direct cytotoxicity by ionizing radiation leads to increased neoantigen expression and the induction of inflammatory cytokines that attract T cells and dendritic cells (DCs). 104
TABLE 1.
Summary of active phase 1 clinical trials investigating IO agents in combination with other IO agents, chemotherapy, radiation therapy and biological therapeutics (www.clinicaltrials.gov)
| Combination regimens | Number of phase 1 trials | Treatment setting (number of active trials) | Tumour types (number of active phase 1 trials) |
|---|---|---|---|
| IO + IO | 70 | Early (5) Advanced (65) | Advanced Solid Tumours (42), Lung (5), Breast (4), GI (5), CNS (1), Gyn (2), HN (2), Sarcoma (1), Melanoma/Skin (4), GU (4) |
| IO + chemotherapy | 37 | Early (8) Advanced (29) | Advanced Solid Tumours (5), Lung (5), Breast (8), GI (13), CNS (1), Gynae (1), HN (1), Sarcoma (1), GU (1), Melanoma/Skin (1) |
| IO + radiation therapy | 35 | Early (7) Advanced (28) | Advanced Solid Tumours (6), Lung (6), Breast (1), GI (4), CNS (6), Gynae (3), HN (3), Melanoma/Skin (1), GU (5) |
| IO + biological therapy | 39 | Early (0) Advanced (39) | Advanced Solid Tumours (8), Lung (6), Breast (1), GI (7), Gynae (3), HN (2), Melanoma/Skin (2), GU (8), CNS (2) |
Abbreviations: IO = immuno‐oncology, GI = gastrointestinal; CNS = central nervous system; Gynae = gynaecological; HN = head and neck; GU = genitourinary.
Oncolytic viruses are treatments that have been developed to replicate in tumour cells and release antigenic proteins. The most advanced oncolytic virus is talimogene laherparepvec (T‐VEC), a novel intralesional therapy that gained FDA approval in October 2015 based on early results from the OPTiM study. 105 A phase 1 trial in patients with advanced melanoma combining T‐VEC with ipilimumab led to response rates of 50% and 18 month OS 67%. 106 Adverse events were manageable and resembled those of mostly ipilimumab, with 26% grade 3–4 AEs reported. Phase II trials combining T‐VEC and pembrolizumab are underway (NCT02965716, NCT04068181).
5.3. Novel immune checkpoint targets
Despite the clinical success obtained by “first generation” ICIs, many patients have unsatisfactory responses or relapse after initial benefit. Advances in our understanding of the complex network of regulatory interactions in the TME have given us further insight into the underlying mechanisms of resistance and relapse. It is increasingly evident that multiple modulatory checkpoints, either inhibitory or stimulatory, play a role and provide attractive new targets for innovative therapeutic strategies. 107 , 108 , 109
Targeting metabolites in the TME and their associated immunosuppressive pathways has become a particularly promising area of research. To facilitate immune escape, tumours may produce anti‐inflammatory cytokines, recruit regulatory immune subsets, and produce immunosuppressive metabolites. 110 CD73 is an ectoenzyme that catalyses the generation of extracellular adenosine, a potent immunosuppressive molecule that has emerged as one such therapeutic target. Adenosine is a regulatory autocrine and paracrine factor that accumulates in the TME, influencing immune activity, angiogenesis, and metastasis, and is recognized as a major immuno‐metabolomic checkpoint in tumours. 111 Under physiological conditions, it serves as an immunoregulatory molecule to protect normal tissues from uncontrolled inflammation, can impair antitumour immunity, both through the attenuation of protective immune cells including T cells, NK cells, and DCs, and by enhancing the suppressive capacity of Tregs, and MDSCs. 112 Upon apoptotic or necrotic cell death, tumour cells release adenosine triphosphate (ATP) into the extracellular space, resulting in a pro‐inflammatory response. To prevent an immune reaction stimulated by cell death, tissues express a cluster of differentiation CD39 and CD73 to enzymatically convert ATP to adenosine monophosphate (AMP), and AMP to adenosine, respectively. Adenosine then induces a localized immunosuppressive response through pleotropic effects upon multiple immune cell types. One mechanism by which tumours may have evolved to evade the immune system is via overexpression of CD73, resulting in an immunosuppressive microenvironment and this has been associated with poor prognosis in multiple cancer types. 113 , 114 , 115 , 116 , 117 , 118 It has been postulated that targeted immunotherapy to inhibit CD73 activity can potentially reduce adenosine production, thus augmenting the host response. Several trials combining anti‐CD73 with ICIs are underway (NCT03334617, NCT02740985).
Indoleamine 2,3‐dioxygenases (IDO1 and IDO2) and tryptophan 2,3‐dioxygenase (TDO) are tryptophan catabolic enzymes that catalyse the conversion of tryptophan into kynurenine. The depletion of tryptophan and the increase in kynurenine exert immunosuppressive effects by activating Tregs and MDSCs, suppressing the functions of effector T and NK cells, and promoting immune tolerance. 119 , 120 , 121 IDO is expressed by most human cancers and in clinical studies its expression levels correlate with lower OS, PFS, and response to chemotherapy, radiotherapy, and immunotherapy. 107 , 122 , 123 Despite promising results in early‐phase clinical trials as part of combination therapy with PD‐1 checkpoint inhibitors in a range of tumour types, a phase III study of the IDO1‐selective inhibitor epacadostat in combination with between the epacadostat‐treated group versus placebo in patients with metastatic melanoma. 124 The interest in IDO1 inhibitors subsequently waned; however, novel approaches to inhibit this pathway continue to be explored. 125
CD40, a tumour necrosis factor (TNF)‐receptor superfamily member, is primarily expressed on antigen‐presenting cells (APCs), including DCs, B cells, macrophages, and monocytes, as well as non‐hematopoietic cells and subsets of cancer cells. 126 , 127 Ligation of CD40 with CD40L (CD154) results in direct activation of APCs, which involves upregulation of co‐stimulatory and MHC molecules and production of pro‐inflammatory cytokines. This is a key step in generating an antitumour immune response, and there is evidence that agonistic CD40 antibodies facilitate rejection of established tumours in different mouse models of cancer. 126 CD40‐targeting antibodies with varying binding affinity have been evaluated in clinical trials, including selicrelumab (RG7876), dacetuzumab, APX 005M, ChiLob 7/4, and lucatumumab. 128 , 129 While toxicity appears manageable and durable anticancer responses were observed, clinical activity was modest, with response rates of <20% observed. 128 Nevertheless, clinical efforts are now exploring combination regimens of anti‐CD40 with ICIs (NCT03502330, NCT03123783, and NCT02706353).
Finally, V‐domain Ig suppressor of T cell activation (VISTA) is a negative immune‐checkpoint structurally analogue to PD‐L1, mainly expressed by APC, myeloid cells but also T lymphocytes and Tregs. 107 , 130 Its expression on both the myeloid and T lymphocyte lineages, which is consistent across tumour types, coupled to its important role in regulating innate and adaptive immune responses, makes VISTA a unique and attractive target for immunotherapeutics. In preclinical models it was confirmed that, even if structurally analogue to PD‐L1, the role of VISTA is functionally distinct from that the PD‐1/PD‐L1 axis in controlling T cell activation. 131 Inhibition of VISTA results in enhanced T cell infiltration, concomitantly with a reduction of MDSCs, and beneficial antitumour responses enhanced by combination with anti‐PD‐1/PD‐L1. 131 , 132 , 133
5.4. Incorporation of omics data into IO clinical trial design
Over the last several years, increased understanding of the molecular pathogenesis of various malignancies has been coupled with the development and integration of high throughput bioinformatic “omics” technologies, allowing for large‐scale analyses that provide further insight into the molecular signatures underlying a diverse range of malignancies, and also offering the potential for identification of novel therapeutic targets. There already exist multiple comprehensive genome‐wide profiling platforms such as the Catalogue of Somatic Mutations in Cancer (COSMIC), the Cancer Genome Atlas (TCGA) and the American Association for Cancer Research (AACR) Project Genomics Evidence Neoplasia Information Exchange (GENIE), that have aided in the detection of novel molecular targets. Beyond genomics, additional omics approaches such as epigenomics and transcriptomics have been increasingly investigated to further elucidate the complexity of biological systems at different dimensions. While these single‐level omics approaches may shed further light on epigenetic alterations and molecular subtyping of tumours based on protein expression, they are limited in their ability to fully portray the relationship between molecular signatures and the phenotypic manifestation of the hallmarks of cancer. 134 However, by integrating multi‐omics approaches, there exists an opportunity to further expose the intricate molecular mechanisms underlying these phenotypic manifestations. The Immune Resistance Interrogation Study (IRIS) is one such initiative that aims to incorporate multi‐omics to elucidate the mechanisms behind ICI primary and acquired resistance (NCT04243720).
Transcriptomics is gaining increasing recognition, with techniques such as RNA sequencing (RNA‐seq) enabling scrutiny of expression profiles and assessment of the impact of their alterations, which may aid in disease classification and progression. In contrast to the static genome, the transcriptome may be more informative as it exhibits dynamic changes depending on cellular, environment, extracellular, and developmental stimuli. Recent efforts to include transcriptomics, together with genomic profiling, in clinical trial design strategies have been met with success. 135
In addition, the field of metabolomics and its role in IO has garnered increasing attention in recent years, and there has been renewed interest in its role as a potential modulator of cancer metabolism, which may further inform on phenotype. 136 Several studies have reported the role of tumour metabolism in cancer development and therapeutic response and resistance, and recently the role of glycolysis has come to the forefront. 137 , 138 , 139 Jiang et al. recently reported glycolytic activity was likely correlated with active immune signatures in various cancers and highly glycolytic tumours presented an immune‐stimulatory TME. 140 They found that glycolytic activity enhances PD‐L1 expression on tumour cells and promotes anti‐PD‐1/PD‐L1 immunotherapy response, suggesting a role as a potential predictive biomarker. Further, Cascone et al. identified tumour glycolysis as a pathway associated with immune resistance in melanoma. 137 Recent research efforts have focused on identifying tumour‐specific metabolite profiles using different biological sample types and a variety of novel metabolomic platforms and technologies, some of which have been shaped by increasing recognition of oral host–microbiome interactions. 141
The human body, particularly the oral cavity and gut, is host to rich and taxonomically diverse multi‐species microbial communities. The microbiota plays an important role in regulating immune function and providing protection from pathogens. Disturbances in the symbiotic relationship between host and microbiota, referred to as dysbiosis, may alter the community composition and result in altered metabolism that has subsequently been implicated in the pathogenesis of various malignancies. 142 , 143 Recently, chemoradiotherapy (CRT) has been implicated in dysbiosis, where increases of potentially pathogenic species were found in patients with locally advanced oropharyngeal squamous cell carcinoma (SCC). 144 Differences in species population has been reported in both IO responders and non‐responders. For example in melanoma patients whose baseline microbiota was enriched with Faecalibacterium genus and other Firmicutes showed a longer PFS and OS than those whose baseline microbiota was enriched with Bacteroides upon ipilimumab treatment. 145 One substudy from the Checkmate‐141 trial, however, explored the oral microbiota as a predictive biomarker in patients with advanced SCC of the head and neck treated with nivolumab and reported no significant correlation with treatment efficacy or survival. 146 Recent studies have also suggested that the immune microbiome plays a role in the development of toxicity. 147 , 148 , 149 As such attempts to manipulate the gut microbiota to further elucidate the mechanisms of IO response and toxicity are ongoing (NCT03686202, NCT03838601).
Finally, there has been increasing development and integration of novel computational omics methods into the IO clinical arena, particularly pertaining to artificial intelligence (AI). Machine learning (ML) is an AI tool that can process enormous amounts of imported data, enabling classification with predictive capabilities, as it has been shown to predict genotypes associated with poor prognosis. 150 AI may be applied to cellular phenotype detection and classification to determine the presence of a particular disease or its outcome. Image‐based phenotype detection has been developed to overcome the limitations of the classical methods of microscopic visual inspection of tissues, which are time‐consuming, prone to subjectivity and unable to keep up with the data collected from high‐throughput studies. 151 These novel techniques can determine immune responses such as macrophage activation and lymphocyte infiltration, which has particular relevance in IO, where different lesions can have different TMEs resulting in heterogeneous response patterns. These radiomic signatures have shown promise in multiple tumour types, demonstrating both prognostic and predictive features. 152 , 153 , 154 , 155 Leiserson et al. developed a multifactorial model for response to ICIs, utilizing an ML method that automatically selects informative features from imported data, and incorporates clinical, tumour, and immunologic features into its model to simultaneously predict clinical benefit. 156 More recently Trebeschi et al. analysed 1055 primary and metastatic lesions from 203 patients with advanced melanoma and NSCLC undergoing anti‐PD1 therapy, and performed an AI‐based characterization of each lesion on CT scans. The study reported associations between radiomic characteristics and immunotherapy response. 157
5.5. Health related quality of life (HRQOL) and patient‐reported outcomes
Quality of life (QOL) is a complex, abstract, and multidimensional idea that describes an individual's satisfaction with life in the domains considered most vital. HRQOL restricts the complex concept of QOL to aspects of life specifically related to an individual's health, and that can potentially be altered by healthcare. 158 , 159 Determinants of HRQOL include general health (physical, social, and psychosocial) as well as disease‐ and treatment‐related effects, such as disease symptoms and treatment‐related side effects. 160 A comprehensive and complete evaluation of the benefit of IO agents would also include an assessment of their impact on HRQOL in addition to traditional overall and progressive free survival endpoints. Baseline HRQOL measures have been correlated with prognosis, and inclusion of HRQOL as either secondary or exploratory endpoints in clinical trials has increased over the last several years. 161 However, clinical trials do not typically have HRQOL as a primary endpoint and thus studies are not adequately powered or sized to demonstrate differences in HRQOL. To date, general HRQOL tools such as EORTC QLQC30 and the FACT‐General (FACT‐G) are used in clinical trials of IO agents to measure HRQOL. The first tool developed specifically to measure HRQOL in patients treated with IO agents is the FACT‐ICM. 162 This instrument was developed in accordance with the FDA guidelines and is undergoing validity testing in longitudinal clinical trials.
The justification for a specific IO tool has been the unique side effect profile that IO therapies have compared with chemotherapy and radiation. The wide range of immune‐related adverse events (irAEs) is well recognized and highlights the importance of accurate toxicity collection on clinical trials. Currently, toxicities occurring in clinical trials are captured and reported using the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), and while it has been a valuable tool in assessing toxicity, recent studies have shown that patient symptoms may be under‐reported by clinicians. 13 To address this issue, the NCI has developed the patient‐reported outcome version of the CTCAE, known as the PRO‐CTCAE. This instrument has been incorporated into clinical trials to complement physician‐captured adverse events, including studies testing IO agents. PROs can provide comprehensive information about the patients' experiences on IO therapies, which would aid in the assessment of benefit from immune treatments.
6. CONCLUSION
Recent advances in cancer immunotherapy have demonstrated unprecedented long‐term clinical benefit for cancer patients, and these agents have rapidly become the new standard of care in the treatment and management of many advanced tumour types. This success has challenged the existing paradigm for clinical research and drug development. Combination immunotherapy represents the future of cancer treatment, with the aim of expanding the spectrum of IO responders, improving the quality of response and survival without compromising quality of life.
The success of this combination approach is dependent on our ability to manage new challenges in drug development and trial design. The design of the right pre‐clinical experiments and their subsequent translation into the clinic, optimizing dosing strategy and regimen, improving patient selection and early detection, and management of toxicities are some of the barriers that must be overcome. The need for innovative, validated biomarkers is crucial to identify and select the most suitable patients for IO therapy, and blood, tumour, and other biomarker samples should be longitudinally explored and prospectively evaluated during IO trials. As biomarker‐based techniques mature, we can gain further insight into the mechanisms of the dynamic TME under the treatment selection pressure of immunotherapy, identify predictors of response and resistance, and further refine patient selection. Pre‐emptive strategies may then be developed which may guide decision making with regard to escalation of therapy in the face of primary or acquired resistance. The incorporation of omics data can further delineate the complexity of the anti‐tumour immune response, and may have profound implications in the area of precision IO.
6.1. Nomenclature of Targets and Ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, 163 and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16. 164
COMPETING INTERESTS
G.W. and J.D. state that they have no conflicts of interest to declare. A.H. has had consulting and advisory roles at Genentech Inc., Hoffmann La Roche Inc., Merck Serono S.A., GlaxoSmithKline Inc., Bristol‐Myers Squibb Company, Novartis Pharmaceuticals Canada Inc., Boston Biomedical Inc., Boehringer Ingelheim International GmbH, AstraZeneca Pharmaceuticals LP, MedImmune LL, and Pfizer Inc. He has received research funding from Karyopharm and Novartis. A.S. has held consultant and advisory roles at Merck, Bristol‐Myers Squibb, Novartis, Oncorus, and Janssen. She has received grant and research support for clinical trials from Novartis, Bristol‐Myers Squibb, Symphogen AstraZeneca/Medimmune, Merck, Bayer, Surface Oncology, Northern Biologics, Janssen Oncology/ Johnson & Johnson, Roche, Regeneron, Alkermes, and Array Biopharma.
Watson GA, Doi J, Hansen AR, Spreafico A. Novel strategies in immune checkpoint inhibitor drug development: How far are we from the paradigm shift? Br J Clin Pharmacol. 2020;86:1753–1768. 10.1111/bcp.14355
REFERENCES
- 1. Littman DR. Releasing the brakes on cancer immunotherapy. Cell. 2015;162(6):1186‐1190. [DOI] [PubMed] [Google Scholar]
- 2. Fares CM, Allen EMV, Drake CG, Allison JP, Hu‐Lieskovan S. Mechanisms of resistance to immune checkpoint blockade: why does checkpoint inhibitor immunotherapy not work for all patients? Am Soc Clin Oncol Educ Book. 2019;39:147‐164. [DOI] [PubMed] [Google Scholar]
- 3. Sheng J, Srivastava S, Sanghavi K, et al. Clinical pharmacology considerations for the development of immune checkpoint inhibitors. J Clin Pharmacol. 2017;57(Suppl 10):S26‐S42. [DOI] [PubMed] [Google Scholar]
- 4. Centanni M, Moes D, Troconiz IF, Ciccolini J, van Hasselt JGC. Clinical pharmacokinetics and pharmacodynamics of immune checkpoint inhibitors. Clin Pharmacokinet. 2019;58(7):835‐857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Larkin J, Chiarion‐Sileni V, Gonzalez R, et al. Five‐year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381(16):1535‐1546. [DOI] [PubMed] [Google Scholar]
- 6. Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal‐cell carcinoma. N Engl J Med. 2018;378(14):1277‐1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang DY, Salem JE, Cohen JV, et al. Fatal toxic effects associated with immune checkpoint inhibitors: a systematic review and meta‐analysis. JAMA Oncol. 2018;4(12):1721‐1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50(12):1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Teixido C, Gonzalez‐Cao M, Karachaliou N, Rosell R. Predictive factors for immunotherapy in melanoma. Ann Transl Med. 2015;3(15):208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wages NA, Chiuzan C, Panageas KS. Design considerations for early‐phase clinical trials of immune‐oncology agents. J Immunother Cancer. 2018;6(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morrissey KM, Yuraszeck TM, Li CC, Zhang Y, Kasichayanula S. Immunotherapy and novel combinations in oncology: current landscape, challenges, and opportunities. Clin Transl Sci. 2016;9(2):89‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Emens LA, Butterfield LH, Hodi FS Jr, Marincola FM, Kaufman HL. Cancer immunotherapy trials: leading a paradigm shift in drug development. J Immunother Cancer. 2016;4(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Minasian L, Rosen O, Auclair D, Rahman A, Pazdur R, Schilsky RL. Optimizing dosing of oncology drugs. Clin Pharmacol Ther. 2014;96(5):572‐579. [DOI] [PubMed] [Google Scholar]
- 14. Weber JS, Hodi FS, Wolchok JD, et al. Safety profile of nivolumab monotherapy: a pooled analysis of patients with advanced melanoma. J Clin Oncol. 2017;35(7):785‐792. [DOI] [PubMed] [Google Scholar]
- 15. Sachs JR, Mayawala K, Gadamsetty S, Kang SP, de Alwis DP. Optimal dosing for targeted therapies in oncology: drug development cases leading by example. Clin Cancer Res. 2016;22(6):1318‐1324. [DOI] [PubMed] [Google Scholar]
- 16. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non‐small‐cell lung cancer. N Engl J Med. 2015;372(21):2018‐2028. [DOI] [PubMed] [Google Scholar]
- 17. Elassaiss‐Schaap J, Rossenu S, Lindauer A, et al. Using model‐based "learn and confirm" to reveal the pharmacokinetics‐pharmacodynamics relationship of pembrolizumab in the KEYNOTE‐001 trial. CPT Pharmacometrics Syst Pharmacol. 2017;6(1):21‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahamadi M, Freshwater T, Prohn M, et al. Model‐based characterization of the pharmacokinetics of pembrolizumab: a humanized anti‐PD‐1 monoclonal antibody in advanced solid tumors. CPT Pharmacometrics Syst Pharmacol. 2017;6(1):49‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wages NA, Slingluff CL Jr, Petroni GR. A phase I/II adaptive design to determine the optimal treatment regimen from a set of combination immunotherapies in high‐risk melanoma. Contemp Clin Trials. 2015;41:172‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Keizer RJ, Huitema AD, Schellens JH, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493‐507. [DOI] [PubMed] [Google Scholar]
- 21. Scott AM, Allison JP, Wolchok JD. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012;12:14. [PMC free article] [PubMed] [Google Scholar]
- 22. Hanley MJ, Abernethy DR, Greenblatt DJ. Effect of obesity on the pharmacokinetics of drugs in humans. Clin Pharmacokinet. 2010;49(2):71‐87. [DOI] [PubMed] [Google Scholar]
- 23. Hendrikx J, Haanen J, Voest EE, Schellens JHM, Huitema ADR, Beijnen JH. Fixed dosing of monoclonal antibodies in oncology. Oncologist. 2017;22(10):1212‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mould DR, Green B. Pharmacokinetics and pharmacodynamics of monoclonal antibodies: concepts and lessons for drug development. BioDrugs. 2010;24(1):23‐39. [DOI] [PubMed] [Google Scholar]
- 25. Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548‐558. [DOI] [PubMed] [Google Scholar]
- 26. Hutchinson L, Kirk R. High drug attrition rates—where are we going wrong? Nat Rev Clin Oncol. 2011;8(4):189‐190. [DOI] [PubMed] [Google Scholar]
- 27. Garralda E, Dienstmann R, Tabernero J. Pharmacokinetic/pharmacodynamic modeling for drug development in oncology. Am Soc Clin Oncol Educ Book. 2017;37:210‐215. [DOI] [PubMed] [Google Scholar]
- 28. Thiel C, Schneckener S, Krauss M, et al. A systematic evaluation of the use of physiologically based pharmacokinetic modeling for cross‐species extrapolation. J Pharm Sci. 2015;104(1):191‐206. [DOI] [PubMed] [Google Scholar]
- 29. Aarons L, Karlsson MO, Mentre F, et al. Role of modelling and simulation in phase I drug development. Eur J Pharm Sci. 2001;13(2):115‐122. [DOI] [PubMed] [Google Scholar]
- 30. Rajman I. PK/PD modelling and simulations: utility in drug development. Drug Discov Today. 2008;13(7‐8):341‐346. [DOI] [PubMed] [Google Scholar]
- 31. Theoret MR, Pai‐Scherf LH, Chuk MK, et al. Expansion cohorts in first‐in‐human solid tumor oncology trials. Clin Cancer Res. 2015;21(20):4545‐4551. [DOI] [PubMed] [Google Scholar]
- 32. Bates SE, Berry DA, Balasubramaniam S, Bailey S, LoRusso PM, Rubin EH. Advancing clinical trials to streamline drug development. Clin Cancer Res. 2015;21(20):4527‐4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti‐PD‐1) in melanoma. N Engl J Med. 2013;369(2):134‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Robert C, Ribas A, Wolchok JD, et al. Anti‐programmed‐death‐receptor‐1 treatment with pembrolizumab in ipilimumab‐refractory advanced melanoma: a randomised dose‐comparison cohort of a phase 1 trial. Lancet. 2014;384(9948):1109‐1117. [DOI] [PubMed] [Google Scholar]
- 35. Ribas A, Hamid O, Daud A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315(15):1600‐1609. [DOI] [PubMed] [Google Scholar]
- 36. Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator‐choice chemotherapy for ipilimumab‐refractory melanoma (KEYNOTE‐002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16(8):908‐918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robert C, Schachter J, Long GV, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521‐2532. [DOI] [PubMed] [Google Scholar]
- 38. de Greef R, Elassaiss‐Schaap J, Chatterjee M, et al. Pembrolizumab: role of modeling and simulation in bringing a novel immunotherapy to patients with melanoma. CPT Pharmacometrics Syst Pharmacol. 2017;6(1):5‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bai S, Jorga K, Xin Y, et al. A guide to rational dosing of monoclonal antibodies. Clin Pharmacokinet. 2012;51(2):119‐135. [DOI] [PubMed] [Google Scholar]
- 40. Cook N, Hansen AR, Siu LL, Abdul Razak AR. Early phase clinical trials to identify optimal dosing and safety. Mol Oncol. 2015;9(5):997‐1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saber H, Gudi R, Manning M, Wearne E, Leighton JK. An FDA oncology analysis of immune activating products and first‐in‐human dose selection. Regul Toxicol Pharmacol. 2016;81:448‐456. [DOI] [PubMed] [Google Scholar]
- 42. Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti‐CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018‐1028. [DOI] [PubMed] [Google Scholar]
- 43. Zhao L, Ren TH, Wang DD. Clinical pharmacology considerations in biologics development. Acta Pharmacol Sin. 2012;33(11):1339‐1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Freshwater T, Kondic A, Ahamadi M, et al. Evaluation of dosing strategy for pembrolizumab for oncology indications. J Immunother Cancer. 2017;5(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD‐L1‐positive, advanced non‐small‐cell lung cancer (KEYNOTE‐010): a randomised controlled trial. Lancet. 2016;387(10027):1540‐1550. [DOI] [PubMed] [Google Scholar]
- 46. Reck M, Rodriguez‐Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med. 2016;375(19):1823‐1833. [DOI] [PubMed] [Google Scholar]
- 47. Goldstein DA, Gordon N, Davidescu M, et al. A phamacoeconomic analysis of personalized dosing vs fixed dosing of pembrolizumab in firstline PD‐L1‐positive non‐small cell lung cancer. J Natl Cancer Inst. 2017;109(11). 10.1093/jnci/djx063 [DOI] [PubMed] [Google Scholar]
- 48. Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina‐Pinelo S, Paz‐Ares L. Current challenges in cancer treatment. Clin Ther. 2016;38(7):1551‐1566. [DOI] [PubMed] [Google Scholar]
- 49. Slamon DJ, Leyland‐Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783‐792. [DOI] [PubMed] [Google Scholar]
- 50. Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin‐paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947‐957. [DOI] [PubMed] [Google Scholar]
- 51. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med. 2012;366(26):2443‐2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Khunger M, Hernandez AV, Pasupuleti V, et al. Programmed cell death 1 (PD‐1) ligand (PD‐L1) expression in solid tumors as a predictive biomarker of benefit from PD‐1/PD‐L1 axis inhibitors: a systematic review and meta‐analysis. JCO Precis Oncol. 2017;1:1‐15. [DOI] [PubMed] [Google Scholar]
- 53. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science. 2015;348(6230):124‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Balar AV, Weber JS. PD‐1 and PD‐L1 antibodies in cancer: current status and future directions. Cancer Immunol Immunother. 2017;66(5):551‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Galuppini F, Dal Pozzo CA, Deckert J, Loupakis F, Fassan M, Baffa R. Tumor mutation burden: from comprehensive mutational screening to the clinic. Cancer Cell Int. 2019;19(1):209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Buttner R, Longshore JW, Lopez‐Rios F, et al. Implementing TMB measurement in clinical practice: considerations on assay requirements. ESMO Open. 2019;4(1):e000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Samstein RM, Lee C‐H, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wolchok JD, Chiarion‐Sileni V, Gonzalez R, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377(14):1345‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hellmann MD, Callahan MK, Awad MM, et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small‐cell lung cancer. Cancer Cell. 2018;33(5):853‐61.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mouw KW, Goldberg MS, Konstantinopoulos PA, D'Andrea AD. DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017;7(7):675‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shindo Y, Hazama S, Tsunedomi R, Suzuki N, Nagano H. Novel biomarkers for personalized cancer immunotherapy. Cancers (Basel). 2019;11(9):1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Duffy MJ, Crown J. Biomarkers for predicting response to immunotherapy with immune checkpoint inhibitors in cancer patients. Clin Chem. 2019;65(10):1228‐1238. [DOI] [PubMed] [Google Scholar]
- 63. Baretti M, Le DT. DNA mismatch repair in cancer. Pharmacol Ther. 2018;189:45‐62. [DOI] [PubMed] [Google Scholar]
- 64. Ryan E, Sheahan K, Creavin B, Mohan H, Winter D. The current value of determining the mismatch repair status of colorectal cancer: a rationale for routine testing. Crit Rev Oncol Hematol. 2017;116:38‐57. [DOI] [PubMed] [Google Scholar]
- 65. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372(26):2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ruiz‐Bañobre J, Goel A. DNA mismatch repair deficiency and immune checkpoint inhibitors in gastrointestinal cancers. Gastroenterology. 2019;156(4):890‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability‐high solid tumors. Clin Cancer Res. 2019;25(13):3753‐3758. [DOI] [PubMed] [Google Scholar]
- 68. Spencer KR, Wang J, Silk AW, Ganesan S, Kaufman HL, Mehnert JM. Biomarkers for immunotherapy: current developments and challenges. Am Soc Clin Oncol Educ Book. 2016;35:e493‐e503. [DOI] [PubMed] [Google Scholar]
- 69. Nakamura Y. Biomarkers for immune checkpoint inhibitor‐mediated tumor response and adverse events. Front Med (Lausanne). 2019;6:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Simeone E, Gentilcore G, Giannarelli D, et al. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol Immunother. 2014;63(7):675‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kelderman S, Heemskerk B, van Tinteren H, et al. Lactate dehydrogenase as a selection criterion for ipilimumab treatment in metastatic melanoma. Cancer Immunol Immunother. 2014;63(5):449‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bilen MA, Martini DJ, Liu Y, et al. The prognostic and predictive impact of inflammatory biomarkers in patients who have advanced‐stage cancer treated with immunotherapy. Cancer. 2019;125(1):127‐134. [DOI] [PubMed] [Google Scholar]
- 73. Bridge JA, Lee JC, Daud A, Wells JW, Bluestone JA. Cytokines, chemokines, and other biomarkers of response for checkpoint inhibitor therapy in skin cancer. Front Med (Lausanne). 2018;5:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Daud AI, Loo K, Pauli ML, et al. Tumor immune profiling predicts response to anti‐PD‐1 therapy in human melanoma. J Clin Invest. 2016;126(9):3447‐3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jacquelot N, Roberti MP, Enot DP, et al. Predictors of responses to immune checkpoint blockade in advanced melanoma. Nat Commun. 2017;8(1):592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Soda H, Ogawara D, Fukuda Y, et al. Dynamics of blood neutrophil‐related indices during nivolumab treatment may be associated with response to salvage chemotherapy for non‐small cell lung cancer: a hypothesis‐generating study. Thorac Cancer. 2019;10(2):341‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tarhini AA, Zahoor H, Lin Y, et al. Baseline circulating IL‐17 predicts toxicity while TGF‐beta1 and IL‐10 are prognostic of relapse in ipilimumab neoadjuvant therapy of melanoma. J Immunother Cancer. 2015;3(1):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tavakkoli M, Wilkins CR, Mones JV, Mauro MJ. A novel paradigm between leukocytosis, G‐CSF secretion, neutrophil‐to‐lymphocyte ratio, myeloid‐derived suppressor cells, and prognosis in non‐small cell lung cancer. Front Oncol. 2019;9:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fleming V, Hu X, Weber R, et al. Targeting myeloid‐derived suppressor cells to bypass tumor‐induced immunosuppression. Front Immunol. 2018;9:398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Meyer C, Cagnon L, Costa‐Nunes CM, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63(3):247‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hansen GL, Gaudernack G, Brunsvig PF, Cvancarova M, Kyte JA. Immunological factors influencing clinical outcome in lung cancer patients after telomerase peptide vaccination. Cancer Immunol Immunother. 2015;64(12):1609‐1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Limagne E, Euvrard R, Thibaudin M, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX‐bevacizumab drug treatment regimen. Cancer Res. 2016;76(18):5241‐5252. [DOI] [PubMed] [Google Scholar]
- 83. Chesney JA, Mitchell RA, Yaddanapudi K. Myeloid‐derived suppressor cells—a new therapeutic target to overcome resistance to cancer immunotherapy. J Leukoc Biol. 2017;102(3):727‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Martens A, Wistuba‐Hamprecht K, Yuan J, et al. Increases in absolute lymphocytes and circulating CD4+ and CD8+ T cells are associated with positive clinical outcome of melanoma patients treated with ipilimumab. Clin Cancer Res. 2016;22(19):4848‐4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ng Tang D, Shen Y, Sun J, et al. Increased frequency of ICOS+ CD4 T cells as a pharmacodynamic biomarker for anti‐CTLA‐4 therapy. Cancer Immunol Res. 2013;1(4):229‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wargo JA, Reddy SM, Reuben A, Sharma P. Monitoring immune responses in the tumor microenvironment. Curr Opin Immunol. 2016;41:23‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Heitzer E, Ulz P, Geigl JB. Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem. 2015;61(1):112‐123. [DOI] [PubMed] [Google Scholar]
- 88. Lee JH, Long GV, Boyd S, et al. Circulating tumour DNA predicts response to anti‐PD1 antibodies in metastatic melanoma. Ann Oncol. 2017;28(5):1130‐1136. [DOI] [PubMed] [Google Scholar]
- 89. Clouthier DL, Lien SC, Yang SYC, et al. An interim report on the investigator‐initiated phase 2 study of pembrolizumab immunological response evaluation (INSPIRE). J Immunother Cancer. 2019;7(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J Immunother Cancer. 2017;5(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Liu S, Nikanjam M, Kurzrock R. Dosing de novo combinations of two targeted drugs: towards a customized precision medicine approach to advanced cancers. Oncotarget. 2016;7(10):11310‐11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Nikanjam M, Liu S, Kurzrock R. Dosing targeted and cytotoxic two‐drug combinations: lessons learned from analysis of 24,326 patients reported 2010 through 2013. Int J Cancer. 2016;139(9):2135‐2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Nikanjam M, Liu S, Yang J, Kurzrock R. Dosing three‐drug combinations that include targeted anti‐cancer agents: analysis of 37,763 patients. Oncologist. 2017;22(5):576‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Nikanjam M, Patel H, Kurzrock R. Dosing immunotherapy combinations: analysis of 3,526 patients for toxicity and response patterns. Oncoimmunology. 2017;6(8):e1338997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Larkin J, Chiarion‐Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Spranger S, Gajewski T. Rational combinations of immunotherapeutics that target discrete pathways. J Immunother Cancer. 2013;1(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Naidoo J, Page DB, Li BT, et al. Toxicities of the anti‐PD‐1 and anti‐PD‐L1 immune checkpoint antibodies. Ann Oncol. 2015;26(12):2375‐2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lebbe C, Meyer N, Mortier L, et al. Evaluation of two dosing regimens for nivolumab in combination with ipilimumab in patients with advanced melanoma: results from the phase IIIb/IV CheckMate 511 trial. J Clin Oncol. 2019;37(11):867‐875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol. 2011;8(3):151‐160. [DOI] [PubMed] [Google Scholar]
- 101. van der Most RG, Currie AJ, Mahendran S, et al. Tumor eradication after cyclophosphamide depends on concurrent depletion of regulatory T cells: a role for cycling TNFR2‐expressing effector‐suppressor T cells in limiting effective chemotherapy. Cancer Immunol Immunother. 2009;58(8):1219‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013;39(1):74‐88. [DOI] [PubMed] [Google Scholar]
- 103. Demaria S, Ng B, Devitt ML, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys. 2004;58(3):862‐870. [DOI] [PubMed] [Google Scholar]
- 104. Reits EA, Hodge JW, Herberts CA, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259‐1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780‐2788. [DOI] [PubMed] [Google Scholar]
- 106. Puzanov I, Milhem MM, Minor D, et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB‐IV melanoma. J Clin Oncol. 2016;34(22):2619‐2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Donini C, D'Ambrosio L, Grignani G, Aglietta M, Sangiolo D. Next generation immune‐checkpoints for cancer therapy. J Thorac Dis. 2018;10(Suppl 13):S1581‐S1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Burugu S, Dancsok AR, Nielsen TO. Emerging targets in cancer immunotherapy. Semin Cancer Biol. 2018;52(Pt 2):39‐52. [DOI] [PubMed] [Google Scholar]
- 109. Dempke WCM, Fenchel K, Uciechowski P, Dale SP. Second‐ and third‐generation drugs for immuno‐oncology treatment—the more the better? Eur J Cancer. 2017;74:55‐72. [DOI] [PubMed] [Google Scholar]
- 110. Allard B, Turcotte M, Stagg J. CD73‐generated adenosine: orchestrating the tumor‐stroma interplay to promote cancer growth. J Biomed Biotechnol. 2012;2012:485156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. 2017;17(12):709‐724. [DOI] [PubMed] [Google Scholar]
- 112. Vigano S, Alatzoglou D, Irving M, et al. Targeting adenosine in cancer immunotherapy to enhance T‐cell function. Front Immunol. 2019;10:925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Inoue Y, Yoshimura K, Kurabe N, et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non‐small‐cell lung cancer. Oncotarget. 2017;8(5):8738‐8751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Lu XX, Chen YT, Feng B, Mao XB, Yu B, Chu XY. Expression and clinical significance of CD73 and hypoxia‐inducible factor‐1alpha in gastric carcinoma. World J Gastroenterol. 2013;19(12):1912‐1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Turcotte M, Spring K, Pommey S, et al. CD73 is associated with poor prognosis in high‐grade serous ovarian cancer. Cancer Res. 2015;75(21):4494‐4503. [DOI] [PubMed] [Google Scholar]
- 116. Wang M, Zhao J, Zhang LM, et al. Combined erlotinib and cetuximab overcome the acquired resistance to epidermal growth factor receptors tyrosine kinase inhibitor in non‐small‐cell lung cancer. J Cancer Res Clin Oncol. 2012;138(12):2069‐2077. [DOI] [PubMed] [Google Scholar]
- 117. Yang Q, Du J, Zu L. Overexpression of CD73 in prostate cancer is associated with lymph node metastasis. Pathol Oncol Res. 2013;19(4):811‐814. [DOI] [PubMed] [Google Scholar]
- 118. Yu YI, Wang W, Song L, et al. Ecto‐5′‐nucleotidase expression is associated with the progression of renal cell carcinoma. Oncol Lett. 2015;9(6):2485‐2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Liu M, Wang X, Wang L, et al. Targeting the IDO1 pathway in cancer: from bench to bedside. J Hematol Oncol. 2018;11(1):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Mellor AL, Keskin DB, Johnson T, Chandler P, Munn DH. Cells expressing indoleamine 2,3‐dioxygenase inhibit T cell responses. J Immunol. 2002;168(8):3771‐3776. [DOI] [PubMed] [Google Scholar]
- 121. Yan Y, Zhang G‐X, Gran B, et al. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. Journal of Immunology (Baltimore, M: 1950). 2010;185(10):5953‐5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Theate I, van Baren N, Pilotte L, et al. Extensive profiling of the expression of the indoleamine 2,3‐dioxygenase 1 protein in normal and tumoral human tissues. Cancer Immunol Res. 2015;3(2):161‐172. [DOI] [PubMed] [Google Scholar]
- 123. Uyttenhove C, Pilotte L, Theate I, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3‐dioxygenase. Nat Med. 2003;9(10):1269‐1274. [DOI] [PubMed] [Google Scholar]
- 124. Long GV, Dummer R, Hamid O, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO‐301/KEYNOTE‐252): a phase 3, randomised, double‐blind study. Lancet Oncol. 2019;20(8):1083‐1097. [DOI] [PubMed] [Google Scholar]
- 125. Labadie BW, Bao R, Luke JJ. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan‐kynurenine‐aryl hydrocarbon axis. Clin Cancer Res. 2019;25(5):1462–1471. 10.1158/1078-0432.CCR-18-2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Vonderheide RH. Prospect of targeting the CD40 pathway for cancer therapy. Clin Cancer Res. 2007;13(4):1083‐1088. [DOI] [PubMed] [Google Scholar]
- 127. Kashyap AS, Schmittnaegel M, Rigamonti N, et al. Optimized antiangiogenic reprogramming of the tumor microenvironment potentiates CD40 immunotherapy. Proc Natl Acad Sci. 2020;117(1):541‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Hassan SB, Sørensen JF, Olsen BN, Pedersen AE. Anti‐CD40‐mediated cancer immunotherapy: an update of recent and ongoing clinical trials. Immunopharmacol Immunotoxicol. 2014;36(2):96‐104. [DOI] [PubMed] [Google Scholar]
- 129. Vonderheide RH. The immune revolution: a case for priming, not checkpoint. Cancer Cell. 2018;33(4):563‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. ElTanbouly MA, Croteau W, Noelle RJ, Lines JL. VISTA: a novel immunotherapy target for normalizing innate and adaptive immunity. Semin Immunol. 2019;42:101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Liu J, Yuan Y, Chen W, et al. Immune‐checkpoint proteins VISTA and PD‐1 nonredundantly regulate murine T‐cell responses. Proc Natl Acad Sci. 2015;112(21):6682‐6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Le Mercier I, Chen W, Lines JL, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res. 2014;74(7):1933‐1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lines JL, Sempere LF, Broughton T, Wang L, Noelle R. VISTA is a novel broad‐spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res. 2014;2(6):510‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Chakraborty S, Hosen MI, Ahmed M, Shekhar HU. Onco‐multi‐OMICS approach: a new frontier in cancer research. Biomed Res Int. 2018;2018:9836256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Rodon J, Soria JC, Berger R, et al. Genomic and transcriptomic profiling expands precision cancer medicine: the WINTHER trial. Nat Med. 2019;25(5):751‐758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Johnson CH, Spilker ME, Goetz L, Peterson SN, Siuzdak G. Metabolite and microbiome interplay in cancer immunotherapy. Cancer Res. 2016;76(21):6146‐6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Cascone T, McKenzie JA, Mbofung RM, et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 2018;27(5):977‐987. e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Nakazawa MS, Keith B, Simon MC. Oxygen availability and metabolic adaptations. Nat Rev Cancer. 2016;16(10):663‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Jiang Z, Liu Z, Li M, Chen C, Wang X. Increased glycolysis correlates with elevated immune activity in tumor immune microenvironment. EBioMedicine. 2019;42:431‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Shin JM, Kamarajan P, Fenno JC, Rickard AH, Kapila YL. Metabolomics of head and neck cancer: a mini‐review. Front Physiol. 2016;7:526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Rea D, Coppola G, Palma G, et al. Microbiota effects on cancer: from risks to therapies. Oncotarget. 2018;9(25):17915‐17927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Oliva M, Spreafico A, Taberna M, et al. Immune biomarkers of response to immune‐checkpoint inhibitors in head and neck squamous cell carcinoma. Ann Oncol. 2019;30(1):57‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Bernal MO, Schneeberger PHH, Taylor R, et al. Role of the oral and gut microbiota as a biomarker in locoregionally advanced oropharyngeal squamous cell carcinoma (ROMA LA‐OPSCC). J Clin Oncol. 2019;37(15 Suppl):6045. [Google Scholar]
- 145. Chaput N, Lepage P, Coutzac C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017;28(6):1368‐1379. 10.1093/annonc/mdx108 [DOI] [PubMed] [Google Scholar]
- 146. Ferris RL, Blumenschein G, Harrington K, et al. Abstract CT022: evaluation of oral microbiome profiling as a response biomarker in squamous cell carcinoma of the head and neck: analyses from CheckMate 141. Cancer Res. 2017;77(13 Suppl):CT022‐CT. [Google Scholar]
- 147. Pitt JM, Vetizou M, Waldschmitt N, et al. Fine‐tuning cancer immunotherapy: optimizing the gut microbiome. Cancer Res. 2016;76(16):4602‐4607. [DOI] [PubMed] [Google Scholar]
- 148. Dubin K, Callahan MK, Ren B, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint‐blockade‐induced colitis. Nat Commun. 2016;7(1):10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Sivan A, Corrales L, Hubert N, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti‐PD‐L1 efficacy. Science. 2015;350(6264):1084‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Yu J, Hu Y, Xu Y, et al. LUADpp: an effective prediction model on prognosis of lung adenocarcinomas based on somatic mutational features. BMC Cancer. 2019;19(1):263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Jabbari P, Rezaei N. Artificial intelligence and immunotherapy. Expert Rev Clin Immunol. 2019;15(7):689‐691. [DOI] [PubMed] [Google Scholar]
- 152. Pavillon N, Hobro AJ, Akira S, Smith NI. Noninvasive detection of macrophage activation with single‐cell resolution through machine learning. Proc Natl Acad Sci U S A. 2018;115(12):E2676‐E2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Sun R, Limkin EJ, Vakalopoulou M, et al. A radiomics approach to assess tumour‐infiltrating CD8 cells and response to anti‐PD‐1 or anti‐PD‐L1 immunotherapy: an imaging biomarker, retrospective multicohort study. Lancet Oncol. 2018;19(9):1180‐1191. [DOI] [PubMed] [Google Scholar]
- 154. Coroller TP, Agrawal V, Huynh E, et al. Radiomic‐based pathological response prediction from primary tumors and lymph nodes in NSCLC. J Thorac Oncol. 2017;12(3):467‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Braman N, Prasanna P, Whitney J, et al. Association of peritumoral radiomics with tumor biology and pathologic response to preoperative targeted therapy for HER2 (ERBB2)‐positive breast cancer. JAMA Netw Open. 2019;2(4):e192561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Leiserson MDM, Syrgkanis V, Gilson A, et al. A multifactorial model of T cell expansion and durable clinical benefit in response to a PD‐L1 inhibitor. PLoS One. 2018;13(12):e0208422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Trebeschi S, Drago SG, Birkbak NJ, et al. Predicting response to cancer immunotherapy using noninvasive radiomic biomarkers. Ann Oncol. 2019;30(6):998‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Apolone G, De Carli G, Brunetti M, Garattini S. Health‐related quality of life (HR‐QOL) and regulatory issues. An assessment of the European Agency for the Evaluation of Medicinal Products (EMEA) recommendations on the use of HR‐QOL measures in drug approval. Pharmacoeconomics. 2001;19(2):187‐195. [DOI] [PubMed] [Google Scholar]
- 159. Arpinelli F, Bamfi F. The FDA guidance for industry on PROs: the point of view of a pharmaceutical company. Health Qual Life Outcomes. 2006;4(1):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Michael M, Tannock IF. Measuring health‐related quality of life in clinical trials that evaluate the role of chemotherapy in cancer treatment. CMAJ. 1998;158(13):1727‐1734. [PMC free article] [PubMed] [Google Scholar]
- 161. Anagnostou V, Yarchoan M, Hansen AR, et al. Immuno‐oncology trial endpoints: capturing clinically meaningful activity. Clin Cancer Res. 2017;23(17):4959‐4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Hansen AR, Ala‐leppilampi K, McKillop C, et al. 1276PDevelopment of the functional assessment of cancer therapy‐immune checkpoint modulator (FACT‐ICM): a scale to measure quality of life in cancer patients treated with ICMs. Ann Oncol. 2019;30(Supplement_5). [Google Scholar]
- 163. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 2018;46(D1):D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Alexander SPH, Kelly E, Marrion N, et al. The Concise Guide to PHARMACOLOGY 2017/18: Overview. Br J Pharmacol. 2017;174(Supp 1):S1‐S16. [DOI] [PMC free article] [PubMed] [Google Scholar]