

Abstract
The loss of descending serotonin (5-HT) to the spinal cord contributes to muscle spasms in chronic spinal cord injury (SCI). Hyperexcitable motoneurons receive long-lasting excitatory postsynaptic potentials (EPSPs), which activate their persistent inward currents to drive muscle spasms. Deep dorsal horn (DDH) neurons with bursting behavior could be involved in triggering the EPSPs due to loss of inhibition in the chronically 5-HT-deprived spinal cord. Previously, in an acutely transected preparation, we found that bursting DDH neurons were affected by administration of the 5-HT1B/1D receptor agonist zolmitriptan, which suppressed their bursts, and by N-methyl-d-aspartate (NMDA), which enhanced their bursting behavior. Nonbursting DDH neurons were not influenced by these agents. In the present study, we investigate the firing characteristics of bursting DDH neurons following chronic spinal transection at T10 level in adult mice and examine the effects of replacing lost endogenous 5-HT with zolmitriptan. Terminal experiments using our in vitro preparation of the sacral cord were carried out ~10 wk postransection. Compared with the acute spinal stage of our previous study, DDH neurons in the chronic stage became more responsive to dorsal root stimulation, with burst duration doubling with chronic injury. The suppressive effects of zolmitriptan were stronger overall, but the facilitative effects of NMDA were weaker. In addition, the onset of DDH neuron activity preceded ventral root output and the firing rates of DDH interneurons correlated with the integrated long-lasting ventral root output. These results support a contribution of the bursting DDH neurons to muscle spasms following SCI and inhibition by 5-HT.
NEW & NOTEWORTHY We investigate the firing characteristics of bursting deep dorsal horn (DDH) neurons following chronic spinal transection. DDH neurons in the chronic stage are different from those in the acute stage as noted by their increase in excitability overall and their differing responses serotonin (5-HT) and N-methyl-d-aspartate (NMDA) receptor agonists. Also, there is a strong relationship between DDH neuron activity and ventral root output. These results support a contribution of the bursting DDH neurons to muscle spasms following chronic spinal cord injury (SCI).
Keywords: DDH, deep dorsal horn neurons, 5-HT, N-methyl-d-aspartate, NMDA, SCI, serotonin, spinal cord injury
INTRODUCTION
A reduction of raphespinal serotonin (5-HT) fibers can drive exaggerated sensory transmission and thus muscle spasms in the chronically transected spinal cord (Baker and Chandler 1987; Bellardita et al. 2017; Bennett et al. 2004; Hadjiconstantinou et al. 1984; Li et al. 2004a; Murray et al. 2010). At this chronic stage of spinal cord injury (SCI), long-lasting excitatory postsynaptic potentials (long EPSPs) activate persistent inward currents (PICs) of hyperexcitable motoneurons to trigger muscle spasms (Bennett et al. 2004; Li et al. 2004a). Zolmitriptan, which is a selective 5-HT1B/1D receptor agonist, can suppress long EPSPs and muscle spasms in chronic SCI rats, without affecting PICs intrinsic to motoneurons (Murray et al. 2011). Our previous study in the acute spinal stage suggests that these long EPSPs may be mediated by deep dorsal horn (DDH) neurons that exhibit burst firing, as zolmitriptan can suppress the bursting behavior of these DDH neurons while not affecting nonbursting neighbors (Thaweerattanasinp et al. 2016). In contrast to zolmitriptan, NMDA can moderately facilitate the firing properties of the bursting DDH neurons in the acutely transected spinal cord (Thaweerattanasinp et al. 2016), possibly contributing to long EPSPs and spasms following SCI, especially considering that the long EPSPs are strongly NMDA dependent (Lin et al. 2019).
Previously, we examined deep dorsal horn interneuron activity in an acute spinal transection model, but immense changes occur within the spinal cord after chronic SCI. This study therefore characterizes the firing properties of the bursting DDH neurons in chronic SCI and evaluates how these properties differ from those observed in acute SCI (Thaweerattanasinp et al. 2016). Using the in vitro sacral cord preparation and extracellular electrophysiology recordings, we examine DDH neuronal responses to dorsal root stimulation during administration of zolmitriptan or NMDA following the 10-wk period of transection recovery. Then, we compare the firing properties of the bursting neurons with the bursting neurons from the acute transection study for all drug conditions. From acute to chronic stages of spinal transection, we found that the bursting neurons become slightly more excitable and more sensitive to inhibition by zolmitriptan. In contrast, the NMDA-induced facilitation seen in the acute spinal stage is reduced in the chronic stage. Also, there is a relationship between DDH activity and ventral root activity in animals with chronic SCI. The onset of DDH neuron activity precedes ventral root output, and the firing rates of the DDH interneurons correlate with the integrated ventral root output observed in animals with chronic SCI. Therefore, the current study supports the contribution of bursting DDH neurons to generation of long EPSPs and muscle spasms following chronic SCI.
MATERIALS AND METHODS
Animal models.
Adult mice (C57BL/6; aged > 10 wk; n = 55) received complete spinal transection at T10 vertebral level, which corresponds to T11–T12 spinal segments. The animals were allowed to recover from the injury for 10 wk during which they were subject to a weekly assessment of locomotor recovery based on the Basso, Beattie, Bresnahan (BBB) score for mouse and the Basso Mouse Scale (BMS; Basso et al. 2006; Joshi and Fehlings 2002). Only the animals that exhibited little or no hindlimb movement with some isolated joint movements (BBB score ≤ 7; Joshi and Fehlings 2002) (BMS score ≤2; Basso et al. 2006) after 10 wk were selected (n = 30) for the sacral cord preparation and extracellular recordings of DDH neurons. All experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Northwestern University and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Sacral cord preparation and experimental protocol.
The same procedures of sacral cord preparation and experimental protocols were used in the current study as in the acute transection study (see details in Thaweerattanasinp et al. 2016). Briefly, the whole sacral cord (S1–S3) was removed from chronically injured animals with a secondary complete transection at the junction of L6 and S1 spinal segments. Then, the isolated sacral cord was maintained in vitro in a recording chamber for extracellular recordings of DDH neurons and ventral roots. Electrode penetration was made at the dorsal surface of the sacral cord to position the electrode tips in the vicinity of DDH neurons at 100–500 µm from the dorsal surface, which corresponds to lamina 3 to 5 (Watson et al. 2008). Threshold was defined as the lowest stimulus where we see a monosynaptic reflex. At a given stimulus intensity, the corresponding sacral dorsal roots were electrically stimulated five times at intervals of 30 s for four stimulus intensities (i.e., 1×, 2×, 5×, and 10×, magnitudes of threshold). Bath administration of NMDA (15 and 20 μM) or zolmitriptan (1 μM) was used to study the drug effects on the firing properties of DDH neurons in response to dorsal root stimulation. Ventral root activity was also recorded from the isolated sacral cord during dorsal root stimulation to measure the activity of motoneuron populations. In the experiments where we compared DDH firing with ventral root output, we used a bath administration of low doses of bicuculline (1 μM) and strychnine (0.5 μM) to induce modest reduction of inhibitory circuits in the sacral cord and thus promote stronger spasms (Tysseling et al. 2017) in response to dorsal root stimulation. This addition was necessary as it seems that when the spinal cord injury is moved to a more clinically relevant area such as the thoracic cord, sacral recordings require disinhibition to see strong long latency reflexes (LLRs).
The magnitude of the ventral root long latency reflex (LLR) was determined from the integral of the ventral root output starting at 500 ms and continuing until its cessation. We also quantified the integral of the ventral root output from 40 to 100 ms as an index of the effect of the long lasting EPSP, which we termed the long polysynaptic reflex (LPR). The first 40 ms of the ventral root output are dominated by the EPSPs from activation of group Ia afferents), giving the short polysynaptic reflex (SPR). The average firing rate of each DDH neuron was quantified in these same time periods (SPR, LPR, and LLR). These studies were undertaken on a background of modest block of inhibitory circuits in the sacral cord to promote stronger spasms (Tysseling et al. 2017).
Data analysis.
The same data analysis protocol was used in the current study as in the acute transection study (see details in Thaweerattanasinp et al. 2016) to measure evoked spike count, field potential, burst duration (for the bursting neurons), first-spike latency, and spontaneous firing rate for the firing properties of DDH neurons. Evoked spike count was measured in each trial by counting the number of spikes within the first 1-s time window from a stimulus onset after determining the first evoked spike. Measuring first-spike latency in each trial helped identify the first evoked spike by taking the time difference between the stimulus onset and the onset of the first evoked spike (maximally capped at 30 ms). Only the nonzero values of first-spike latency were used for statistical analyses. Field potential was measured from a peak-to-peak voltage of a hyperpolarization that usually happened shortly after a stimulus artifact. Only nonzero values of field latency were used for statistical analyses. Zero field latency occurred when there were no field potentials observed at all for measuring the field latency at all stimulating intensities in some recording trials of our experiments. These recording trials tended to have no evoked spikes observed for measuring the first-spike latency as well. Moreover, we calculated spontaneous firing rate in each trial from the number of spikes within the last 1 s before a stimulus onset. Specifically for the bursting neurons (see results), burst duration was measured in the same software by taking the time difference between the first and last evoked spikes. For a recorded DDH neuron, the mean of each firing property was calculated from the measurements in the five trials (or fewer in cases with some missed trials) of each stimulus intensity. Moreover, the final mean of evoked spike count was corrected for poststimulus spontaneous spikes by subtracting the mean of spontaneous firing rate (within 1 s before stimulus) from the precorrected mean of evoked spike count (within 1 s after stimulus).
Because of the unbalanced data sets, a two-way unbalanced ANOVA via multiple regression analysis (α = 0.05) was used for all comparisons of the firing properties from DDH neuron types, drug conditions, or recovery periods from the spinal transection at four stimulus intensities. If necessary, a post hoc pairwise comparison (t test) of group means (α = 0.05) with the Bonferroni correction was further used to specify significant differences.
Spike2 (CED) software was used for rectifying and averaging (i.e., smoothing) the raw data of the ventral root activity. Then, three components of the polysynaptic ventral root reflexes were quantified as the area under the curve (i.e., definite integral) in the order of their latency as described in Murray et al. (2011): the short polysynaptic reflex (SPR; 10–40 ms poststimulus); the long polysynaptic reflex (LPR; 40–500 ms poststimulus) corresponding to the long EPSP; and the long-lasting reflex (LLR; 500–4,000 ms poststimulus) associated with motoneuronal persistent inward currents (PIC). For each reflex component, mean ventral root activity was computed for all five dorsal root stimuli at each stimulus intensity. Background ventral root activity was measured over 800 ms before the first dorsal root stimulus and was subtracted from the mean ventral root activity to obtain the final reflex response. Measured as the area under the curve (mV·s in units), the final reflex response for each component could have a positive, negative, or zero value, corresponding to increased activity after dorsal root stimulation, decreased activity, or no reflex response. Correlation between the bursting neuron firing activity and integral of the ventral root activity was analyzed using linear regression analysis (α = 0.05).
When comparing the effect of drugs on bursting neurons in chronic and acute SCI, we used the present data set and our previously obtained acute data set (Thaweerattanasinp et al. 2016). All comparisons between the two states were based on absolute changes from predrug control conditions. Because of the large number of data with zero values in control conditions, absolute changes were used instead of relative changes for the comparisons of the drug effects with the baselines between acute and chronic transection studies. However, it is important to note that relative values are not the same as absolute values, thus possibly reaching different conclusions based on different calculation methods. Another consideration with the acute versus chronic comparison is that in the acute studies, some zolmitriptan experiments included more than one DDH neuron per animal. The exclusion of these additional neurons in acute transection from the data analysis did not overturn any results of the statistical comparisons in which all the neurons were included. Accordingly, we described the statistical results with the inclusion of all the bursting neurons. 62 DDH neurons were recorded from 30 adult mice with chronic spinal transection via extracellular recordings. Fifty of these DDH neurons from 24 adult mice were investigated during the bath administration of either NMDA (n = 23) or zolmitriptan (n = 27).
RESULTS
Firing response types of DDH neurons following chronic spinal transection.
The primary focus of our studies was on the effects of chronic SCI on the firing patterns and drug-sensitivities of DDH neurons that responded with a burst of spikes to single-pulse (0.2 ms) stimulation of the dorsal root (see examples in Figs. 2A and 4A). These are the cells most likely contributing to spasms (Bellardita et al. 2017; Murray et al. 2011; Thaweerattanasinp et al. 2016). A total of 52 bursting neurons were studied in the present chronic SCI study and systematically compared with bursting neurons in acute SCI from our previous study (Thaweerattanasinp et al. 2016).
Fig. 2.

Effects of N-methyl-d-aspartate (NMDA) on the firing properties of the bursting neurons following chronic spinal transection. A: representative recording traces showing the effect of NMDA (bottom) on a bursting neuron, compared with that in control (top), on dorsal root stimulation (0.2 ms; arrow). B–G: group data showing the effects of NMDA on the firing properties (mean marked by X) of the bursting neurons in response to increasing stimulus intensity, compared with those during control and washout conditions. B: evoked spike count during control (n = 20 for all intensities), NMDA (n = 21; except n = 23 at 10×), and washout (n = 18 for all intensities) conditions. C: field potential during control (n = 20 for all intensities), NMDA (n = 21; except n = 23 at 10×), and washout (n = 18 for all intensities) conditions. D: field latency (only nonzero values) during control (n = 20 for all intensities), NMDA (n = 21; except n = 23 at 10×), and washout (n = 18 for all intensities) conditions. E: burst duration during control (n = 20 for all intensities), NMDA (n = 21; except n = 23 at 10×), and washout (n = 18 for all intensities) conditions. F: first-spike latency (only nonzero values) during control (n = 20; except n = 19 at 1×), NMDA (n = 18, 1×; n = 19, 2×; n = 21, 5×; n = 23, 10×), and washout (n = 16, 1×; n = 15, 2×; n = 18, 5× and 10×) conditions. G: spontaneous firing rate during control (n = 20 for all intensities), NMDA (n = 21; except n = 23 at 10×), and washout (n = 18 for all intensities) conditions. Significant difference from control (*P < 0.017) and washout (†P < 0.017) conditions using a post hoc pairwise comparison (t test) of group means with the Bonferroni correction is shown. The box and whisker plots show the minimum, first quartile, median, third quartile, and maximum, without outliers.
Fig. 4.

Effects of zolmitriptan on the firing properties of the bursting neurons following chronic spinal transection. A: representative recording traces showing the effect of zolmitriptan (Zol; bottom) on a bursting neuron, compared with that in control (top), upon dorsal root stimulation (0.2 ms; arrow). B–G: group data showing the effects of zolmitriptan on the firing properties (mean marked by X) of the bursting neurons in response to increasing stimulus intensity, compared with those during control and washout conditions. B: evoked spike count during control (n = 23 for all intensities), zolmitriptan (same as control), and washout (n = 21 for all intensities) conditions. C: field potential during control (n = 23 for all intensities), zolmitriptan (same as control), and washout (n = 21 for all intensities) conditions. D: field latency (only nonzero values) during control (n = 23 for all intensities), zolmitriptan (same as control), and washout (n = 21 for all intensities) conditions. E: burst duration during control (n = 23 for all intensities), zolmitriptan (same as control), and washout (n = 21 for all intensities) conditions. F: first-spike latency (only nonzero values) during control (n = 23; except n = 21 at 1×), zolmitriptan (n = 15, 1×; n = 20, 2×; n = 21, 5×; n = 20, 10×), and washout (n = 15, 1×; n = 18, 2×; n = 19, 5× and 10×) conditions. G: spontaneous firing rate during control (n = 23 for all intensities), zolmitriptan (n = 22 for all intensities), and washout (n = 21 for all intensities) conditions. Significant difference from zolmitriptan (*P < 0.017) and washout (†P < 0.017) conditions using a post hoc pairwise comparison (t test) of group means with the Bonferroni correction is shown. The box and whisker plots show the minimum, first quartile, median, third quartile, and maximum, without outliers.
Bursting neurons become more excitable in chronic SCI.
We found that chronic SCI induced a strong increase in excitability of bursting interneurons. The response latency of the first spike in the chronic stage was shorter in chronic (3.85 ± 0.17 ms; means ± SE) than acute SCI (4.51 ± 0.20 ms) (Fig. 1A; two-way unbalanced ANOVA, P = 0.023). The field potential was significantly smaller (chronic: 1.00 ± 0.03 mV vs. acute: 1.37 ± 0.05 mV; P = 8.51 × 10−9), and the field latency was significantly longer (chronic: 1.53 ± 0.05 ms vs. acute: 1.37 ± 0.03 ms; P = 1.19 × 10−3) (Fig. 1, B and C). They also have significantly longer burst durations (Fig. 1D) than those in acute transection (almost twice as long, 151.66 ± 17.61 ms vs. 88.95 ± 9.78 ms; P = 5.41 × 10−4). The doubling of the burst duration that we find here is important because motoneuron and interneuron PICs are more effectively activated by longer EPSPs, making sensory input more likely to produce long spasms. Bursting neurons in chronic SCI, however, did not produce significantly more evoked spikes (defined as the number of spikes within the first 1-s time window, see materials and methods) than bursting neurons in acute transection (chronic: 4.71 ± 0.31 spikes vs. acute: 3.59 ± 0.46 spikes) in response to single pulse stimulation of the dorsal root at low intensity (Fig. 1E; P = 0.090) and did not now show increased spontaneous firing (chronic: 0.12 ± 0.06 Hz vs. acute: 0.10 ± 0.04 Hz) (Fig. 1F; P = 0.78). In Fig. 1, G and H, we show the distribution comparisons between bursting neurons in acute and chronic transection with respect to mean field potential (Fig. 1G) and field latency (Fig. 1H) measured across various depths from the spinal dorsal surface to suggest changes in the spinal circuitry surrounding the bursting neurons over time after SCI, with the center of neural activity approximately at 200- to 400-µm deep from the dorsal surface.
Fig. 1.

Comparisons of the firing properties of the bursting neurons following acute and chronic spinal transection. A–F: group data comparing firing properties (mean marked by X) of the bursting neurons following acute and chronic spinal transection in response to increasing stimulus intensity in control. A: first-spike latency (only nonzero values) for acute (n = 64, 1×; n = 92, 2×; n = 96, 5×; n = 94, 10×) and chronic (n = 49; except n = 46 at 1×) spinal transection. B: field potential for acute (n = 96; except n = 95 at 10×) and chronic (n = 49 for all intensities) spinal transection. C: field latency (only nonzero values) for acute (n = 65, 1×; n = 93, 2×; n = 96, 5×; n = 94, 10×) and chronic (n = 49 for all intensities) spinal transection. D: burst duration for acute (n = 96; except n = 95 at 10×) and chronic (n = 49 for all intensities) spinal transection. E: evoked spike count for acute (n = 96; except n = 95 at 10×) and chronic (n = 49 for all intensities) spinal transection. F: spontaneous firing rate for acute (n = 96; except n = 95 at 10×) and chronic (n = 49 for all intensities) spinal transection. G and H: distribution comparisons between the bursting neurons following acute (n = 96) and chronic (n = 49) spinal transection as plots of mean field potential (G) and field latency (H) against depths measured from the dorsal surface. Significant difference (*P < 0.05; †P < 0.01; ‡P < 0.001) using a two-way unbalanced ANOVA is shown. The box and whisker plots show the minimum, first quartile, median, third quartile, and maximum, without outliers.
NMDA had mixed effects on bursting neurons after chronic SCI.
Of the total of 52 bursting neurons recorded in chronic SCI, 23 neurons were studied during bath administration of NMDA (n = 21 at 15 µM; n = 2 at 20 µM). The combined effects of both concentrations were assessed. Effects were mixed, as illustrated in Fig. 2. Regardless of the drug conditions, increasing stimulus intensity did increase evoked spike count (P = 7.45 × 10−4) and field potential magnitude (P = 5.16 × 10−11), without significantly affecting field latency (P = 0.81). Note that field latency was already very short, suggesting a disynaptic connection. NMDA did significantly increase first-spike latency (Fig. 2F; P = 0.004), delaying it (4.30 ± 0.24 ms) compared with control (3.62 ± 0.18 ms; Bonferroni-corrected pairwise t test, P = 0.011) and washout (3.44 ± 0.14 ms; P = 0.002). The only change that indicates a facilitatory effect of NMDA was its action on spontaneous firing rate (Fig. 2G; P = 1.85 × 10−4), increasing it (0.69 ± 0.20 Hz) compared with control (0.05 ± 0.03 Hz; P = 4.20 × 10−4) and washout (0.01 ± 0.01 Hz; P = 3.09 × 10−4). Evoked spike count, field potential magnitude, and field latency were not significantly different in the NMDA condition as compared with controls and washout (two-way unbalanced ANOVA; P = 0.13; P = 0.38; P = 0.31, respectively) (Fig. 2).
NMDA effects on bursting neurons are weaker in chronic than acute SCI.
Figure 3 shows that NMDA had different effects on evoked spike count in the two states (Fig. 3A), with the stronger facilitatory effect in acute transection (0.70 ± 0.24 spikes) than in the chronic state (−0.94 ± 0.26 spikes; two-way unbalanced ANOVA, P = 9.07 × 10−6). The drug also enhanced the burst duration of the bursting neurons (Fig. 3D) in acute transection (107.01 ± 24.97 ms) more strongly than in chronic transection (0.67 ± 21.74 ms; P = 0.002). NMDA, however, was more effective in delaying the firing of the surrounding population of cells shown in field latency (Fig. 3C) in the chronic (0.18 ± 0.03 ms) than acute state (0.05 ± 0.01 ms; P = 3.23 × 10−4). The drug also delayed the first-spike latency of our target neurons, bursting neurons (Fig. 3E), in chronic transection (0.78 ± 0.16 ms) significantly more than that in acute transection (0.29 ± 0.13 ms; P = 0.018). There were no significant differences in the NMDA effects on the field potential (Fig. 3B; P = 0.48) in the vicinity of the bursting neurons or on the spontaneous firing rate (Fig. 3F; P = 0.63) of the bursting neurons in acute transection from those in chronic transection. These results show that NMDA’s effects on bursting neuron excitability were weaker in the chronic state and that this change was accompanied by an increase in latency of stimulation-induced changes in firing.
Fig. 3.

Comparisons of N-methyl-d-aspartate (NMDA) effects on the firing properties of the bursting neurons following acute and chronic spinal transection. A–F: group data comparing absolute changes from controls of NMDA effects (i.e., 15 µM NMDA minus control; mean marked by X) on the firing properties of the bursting neurons following acute and chronic spinal transection in response to increasing stimulus intensity. A: evoked spike count for acute (n = 21 for all intensities) and chronic (n = 20 for all intensities) spinal transection. B: field potential for acute (n = 21 for all intensities) and chronic (n = 20 for all intensities) spinal transection. C: field latency for acute (n = 16, 1×; n = 19, 2×; n = 21, 5× and 10×) and chronic (n = 20 for all intensities) spinal transection. D: burst duration for acute (n = 21 for all intensities) and chronic (n = 20 for all intensities) spinal transection. E: first-spike latency for acute (n = 12, 1×; n = 19, 2×; n = 20, 5× and 10×) and chronic (n = 17, 1×; n = 18, 2×; n = 20, 5× and 10×) spinal transection. F: spontaneous firing rate for acute (n = 21 for all intensities) and chronic (n = 20 for all intensities) spinal transection. Significant difference (*P < 0.05; †P < 0.01; ‡P < 0.001) using a two-way unbalanced ANOVA is shown. The box and whisker plots show the minimum, first quartile, median, third quartile, and maximum, without outliers.
The 5HT1b/d agonist zolmitriptan strongly suppressed firing in bursting neurons in the chronic state.
Of a total number of 52 bursting neurons, 27 neurons were studied for their firing properties during the bath administration of zolmitriptan (1 µM; Fig. 4). Washout of zolmitriptan is difficult (Murray et al. 2011), and we found no significant differences in all the firing properties between zolmitriptan and washout conditions. Therefore, we performed the recording protocol with zolmitriptan administration in only one bursting neuron from each animal.
Zolmitriptan significantly decreased the evoked spike count of the bursting neurons (Fig. 4B; two-way unbalanced ANOVA, P = 3.37 × 10−10; 2.06 ± 0.22 spikes), compared with control (4.80 ± 0.43 spikes; Bonferroni-corrected pairwise t test, P = 5.50 × 10−10). Zolmitriptan also significantly decreased the burst duration of the bursting neurons (Fig. 4E; P = 5.65 × 10−8; 33.43 ± 8.77 ms) compared with control (115.17 ± 17.18 ms; P = 5.28 × 10−7). Moreover, a significant interaction between drug condition and stimulus intensity occurred (P = 0.04), revealing that zolmitriptan prevented the effect of increasing stimulus intensity in increasing burst duration seen in the control state (5×: predrug 160.63 ± 38.40 ms vs. postdrug 44.18 ± 23.26 ms; P = 3.00 × 10−4; 10×: predrug 185.77 ± 46.22 ms vs. postdrug 33.39 ± 9.52 ms; P = 2.74 × 10−6). These results clearly show zolmitriptan is potent in decreasing the excitability of bursting neurons in chronic SCI.
Zolmitriptan also significantly delayed the field latency from the vicinity of bursting neurons (Fig. 4D; P = 0.005), from a predrug value of 1.49 ± 0.06 ms to 1.81 ± 0.08 ms (P = 0.002). The drug significantly delayed the first-spike latency of the bursting neurons (Fig. 4F; P = 0.002), from a predrug value of 3.99 ± 0.30 ms to 5.58 ± 0.36 ms (P = 0.001). However, zolmitriptan had no significant effect on the field potential magnitude from the vicinity of the bursting neurons (Fig. 4C; P = 0.10) or on the spontaneous firing rate of the bursting neurons (Fig. 4G; P = 0.33). Therefore, the effects of zolmitriptan in delaying the field latency and first-spike latency further support its potential in suppressing the firing behavior of bursting neurons in chronic SCI with some effects on the timing of excitatory synaptic inputs to bursting neurons.
Zolmitriptan effects on bursting neurons are stronger in the chronic than in the acute state.
As opposed to NMDA actions, the overall suppressive effects of zolmitriptan become stronger on the bursting neurons as SCI transitions from acute to chronic stages (Fig. 5). Zolmitriptan suppressed the evoked spike count of bursting neurons (Fig. 5A) in chronic transection (−2.74 ± 0.32 spikes) significantly more than it did in acute transection (−1.19 ± 0.24 spikes; two-way unbalanced ANOVA, P = 9.96 × 10−5). The drug also suppressed the field potential from the vicinity of bursting neurons (Fig. 5B) in chronic transection (−0.09 ± 0.02 mV) significantly more than that in acute transection (0.01 ± 0.04 mV; P = 0.031). Zolmitriptan delayed the field latency from the vicinity of the bursting neurons (Fig. 5C) in chronic transection (0.32 ± 0.05 ms) significantly more than that in acute transection (0.13 ± 0.03 ms; P = 0.001). In addition, the drug delayed the first-spike latency of bursting neurons (Fig. 5E) in chronic transection (1.84 ± 0.31 ms) significantly more than that in acute transection (0.80 ± 0.15 ms; P = 0.003). However, as zolmitriptan barely affected the spontaneous firing rate of bursting neuron (Fig. 4G), its ability to suppress the spontaneous firing rate of the bursting neurons (Fig. 5F) in chronic transection (0.08 ± 0.06 Hz) became significantly weaker than that in acute transection (−0.11 ± 0.05 Hz; P = 0.014). There was no significant difference in the zolmitriptan effect on the burst duration of bursting neurons in acute transection from that in chronic transection (P = 0.36). Overall, the effect of zolmitriptan in chronic SCI compared with acute SCI is mixed, but it is clear from the increased latency and decreased evoked spike count data that there is a net increase in efficacy.
Fig. 5.

Comparisons of zolmitriptan (Zol) effects on the firing properties of the bursting neurons following acute and chronic spinal transection. A–F: group data comparing absolute changes from controls of zolmitriptan effects (i.e., zolmitriptan minus control; mean marked by X) on the firing properties of the bursting neurons following acute and chronic spinal transection in response to increasing stimulus intensity. A: evoked spike count for acute (n = 24; except n = 25 at 10×) and chronic (n = 23 for all intensities) spinal transection. B: field potential for acute (n = 24; except n = 25 at 10×) and chronic (n = 23 for all intensities) spinal transection. C: field latency for acute (n = 16, 1×; n = 22, 2×; n = 23, 5×; n = 24, 10×) and chronic (n = 23 for all intensities) spinal transection. D: burst duration for acute (n = 24; except n = 25 at 10×) and chronic (n = 23 for all intensities) spinal transection. E: first-spike latency for acute (n = 16, 1×; n = 21, 2×; n = 22, 5×; n = 21, 10×) and chronic (n = 14, 1×; n = 20, 2×; n = 21, 5×; n = 20, 10×) spinal transection. F: spontaneous firing rate for acute (n = 24; except n = 25 at 10×) and chronic (n = 22 for all intensities) spinal transection. Significant difference (*P < 0.05; †P < 0.01; ‡P < 0.001) using a two-way unbalanced ANOVA is shown. The box and whisker plots show the minimum, first quartile, median, third quartile, and maximum, without outliers.
Correlation of DDH bursting neuron firing and long latency responses in ventral roots.
If the bursting neurons in the DDH contribute to generation of spasms in chronic spinal injury, then their firing patterns should correlate with spasms. To evaluate this possibility, we combined simultaneous recordings of the firing patterns of the DDH interneurons with recordings of long-lasting reflexes (LLRs) in ventral roots (n = 30 from 5 animals). Previous studies have shown that LLRs provide a good in vitro model of spasms (Bennett et al. 2004). LLRs are initiated by a long lasting EPSP (~500 ms) in motoneurons, which then activates motoneuronal PICs to produce sustained firing, generating LLRs lasting up to several seconds. An example of an LLR from the ventral root is illustrated in Fig. 6, A and B, top traces, along with the simultaneously recorded firing pattern of a DDH interneuron Fig. 6, A and B, bottom traces. The reflex and the interneuron firing patterns show good temporal correspondence.
Fig. 6.
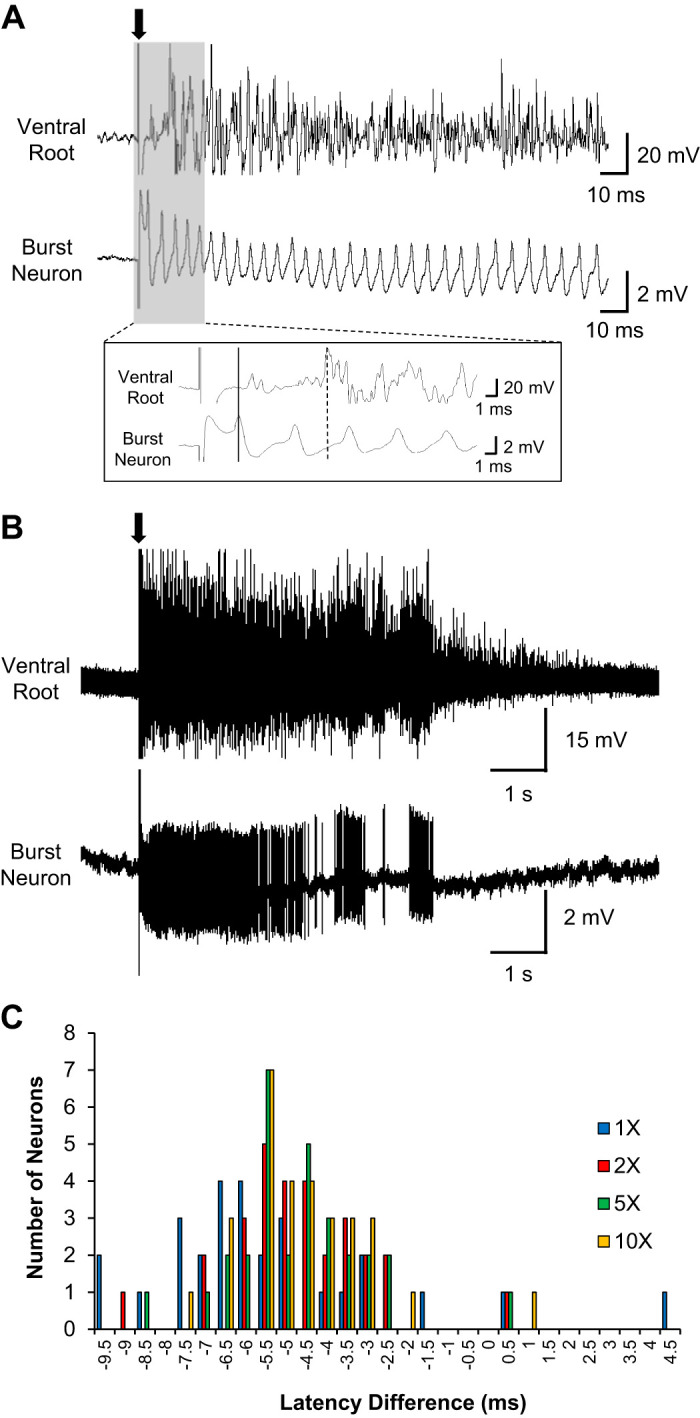
Temporal correspondence between the ventral root reflex and bursting neuron firing pattern. A: representative simultaneous recording traces of the ventral root and bursting neuron upon dorsal root stimulation (0.2 ms; arrow). Inset: expanded time scale of the gray area, illustrating the early onset of the bursting neuron firing (solid line) preceding the reflex output from the ventral root (dash line). Bath application of bicuculline (1 µM) and strychnine (0.5 µM) was used to promote stronger spasms by blocking inhibitory circuits in the sacral cord. B: compressed time scale of the same simultaneous recording traces in A. C: distribution of latency differences [as the number of deep dorsal horn (DDH) neurons, n = 30] between the first spike peak of the bursting neuron firing and the first peak of the ventral root reflex at 4 different stimulus intensities. Negative values of the latency difference mean the onset of the bursting neuron firing precedes the reflex output from the ventral roots, and vice versa for positive values.
The expanded time scale in Fig. 6A shows that the onset of firing of the DDH neuron preceded the output from the ventral roots. This relative timing was a consistent finding, with the DDH neuronal latency being [5.09 ± 0.77 ms at 1× (n = 28); 3.72 ± 0.33 ms at 2× (n = 29); 3.48 ± 0.29 ms at 5× (n = 30); 3.37 ± 0.30 ms at 10× (n = 30)] and the ventral root latency being [10.71 ± 0.43 ms at 1× (n = 28); 8.62 ± 0.25 ms at 2× (n = 29); 8.41 ± 0.20 ms at 5× (n = 30); 8.14 ± 0.16 ms at 10× (n = 30)] as shown in Fig. 6C, where 25 out of 30 DDH neurons (~83%) had their firing onset preceding the reflex output from the ventral roots. The average firing rates of the DDH interneurons during the LPR correlated with the LLR magnitude for all stimulation intensities, as illustrated in Fig. 7A. In contrast, the DDH firing during the SPR did not correlate with any phase of the ventral root output, which is consistent with the insensitivity of the motoneuron PIC to short-lasting EPSPs (<50 ms). The average firing of DDH neurons during the LLR period was similarly correlated with the integral of the LLR (Fig. 7B), as was the total duration of firing of DDH interneurons (Fig. 7C). These results support the concept that the DDH bursting interneurons may be involved in the initiation and generation of spasms.
Fig. 7.

Correlation between the bursting neuron firing activity and integral of the long-lasting reflex (iLLR). The bursting neuron firing activity is positively correlated with the integrated ventral root output during the long latency reflex (LLR) at 4 different stimulus intensities using linear regression analysis. A: positive correlation of average firing rate of the bursting neurons during the long polysynaptic reflex (LPR) with integrated ventral root output during the subsequent LLR at 4 different stimulus intensities (A1–A4). Note the significant correlations at all stimulus intensities [R2 = 0.277, P = 0.003 at 1× (A1); R2 = 0.154, P = 0.032 at 2× (A2); R2 = 0.257, P = 0.004 at 5× (A3); and R2 = 0.153, P = 0.033 at 10× (A4)]. B: positive correlation of average firing rate of the bursting neurons with integrated ventral root output during the LLR at the same stimulus intensities as in A (B1–B4). Note the significant correlations at 1× (B1; R2 = 0.146, P = 0.041), 5× (B3; R2 = 0.175, P = 0.021), and 10× (B4; R2 = 0.138, P = 0.043) stimulus intensities. C: positive correlation of total firing duration of the bursting neurons with integrated ventral root output during the LLR at the same stimulus intensities as in A and B (C1–C4). Note the significant correlations at 1× (C1; R2 = 0.171, P = 0.026) and 5× (C3; R2 = 0.206, P = 0.012) stimulus intensities.
Fig. 7.

—Continued.
When SCI is performed in the thoracic cord, the ventral root response is not as robust in the sacral cord as compared with studies that use sacral injury and record in the sacral cord. Blocking inhibition by administering GABAA receptor antagonist bicuculline and glycine receptor antagonist strychnine allows for LLRs that are similar to those when recording sacral ventral roots after a sacral SCI. Therefore, as stated in the materials and methods, we used bicuculline and strychnine where we compared DDH bursting interneuron firing with ventral root outputs (Fig. 7) and performed a separate analysis of DDH firing with and without disinhibition. Under the influence of bicuculline and strychnine, the bursting neurons produced significantly more evoked spikes (Fig. 8A; P = 7.72 × 10−15; 56.08 ± 8.00 spikes) and longer burst duration (Fig. 8D; P = 6.52 × 10−15; 834.09 ± 103.38 ms) than those without the drugs (4.71 ± 0.31 spikes; 151.66 ± 17.61 ms, respectively). The drugs also reduced field potential magnitude from the vicinity of the bursting neurons (Fig. 8B; P = 3.64 × 10−7; 0.76 ± 0.04 mV), compared with no drugs (1.00 ± 0.03 mV). However, there were no significant differences in field latency (Fig. 8C; P = 0.89), first-spike latency (Fig. 8E; P = 0.80), and spontaneous firing rate (Fig. 8F; P = 0.35).
Fig. 8.

Comparisons of the firing properties of the bursting neurons following chronic spinal transection in control and bicuculline/strychnine cocktail. A–F: group data comparing firing properties (mean marked by X) of the bursting neurons following chronic spinal transection in response to increasing stimulus intensity in control and bicuculline/strychnine cocktail. A: evoked spike count in control (n = 30; except n = 29 at 1×) and drug cocktail (n = 49 for all intensities). B: field potential in control (n = 30; except n = 29 at 1×) and drug cocktail (n = 49 for all intensities). C: field latency (only nonzero values) in control (n = 30; except n = 29 at 1×) and drug cocktail (n = 49 for all intensities). D: burst duration in control (n = 30; except n = 29 at 1×) and drug cocktail (n = 49 for all intensities). E: first-spike latency (only nonzero values) in control (n = 28, 1×; n = 29, 2×; n = 30, 5×; n = 30, 10×) and drug cocktail (n = 49; except n = 46 at 1×). F: spontaneous firing rate in control (n = 30; except n = 29 at 1×) and drug cocktail (n = 49 for all intensities). Significant difference (‡P < 0.001) using a two-way unbalanced ANOVA is shown. The box and whisker plots show the minimum, first quartile, median, third quartile, and maximum, without outliers.
DISCUSSION
Our goal was to assess the effects of 5-HT1B/1D and NMDA receptor activation on the firing characteristics of bursting DDH neurons following a complete-transection chronic SCI and to compare these effects to those observed after acute SCI (Thaweerattanasinp et al. 2016). In the following discussion, we consider the possibility that bursting DDH neurons may contribute to generation of long EPSPs and muscle spasms as the spinal injury transitions from acute to chronic stages.
This study had three main findings. First, bursting neurons become slightly more excitable following chronic SCI. Second, bursting neurons still experience significant changes in their firing activity during zolmitriptan and NMDA administration following chronic SCI. However, zolmitriptan more strongly suppresses the bursting neurons after chronic SCI by decreasing evoked spike count and field potential magnitude and by delaying field latency and first-spike latency to a greater degree than observed after acute SCI. In contrast, NMDA very slightly facilitates the bursting neurons, but this facilitation is weaker following chronic SCI, seen only in a significant increase in spontaneous firing. Finally, bursting DDH neurons may contribute to spasms by initiating the intrinsic motoneuron activity as well as helping to generate the spasm itself.
Bursting DDH neurons become more excitable following SCI.
The more excitable bursting neurons could be a major contributing factor in generation of long EPSPs in motoneurons and muscle spasms following chronic SCI. Not only do bursting neurons continue to fire multiple spikes in response to sensory inputs (Fig. 1E), the bursting neurons fire for longer durations (Fig. 1D) and show faster responses (Fig. 1A) to a single-pulse dorsal root stimulation over time following SCI. In addition, the bursting neurons receive increased excitatory synaptic drive because the amplitude of spontaneous excitatory postsynaptic currents recorded from DDH neurons is increased over time following SCI (Rank et al. 2015). The study of Rank et al. (2015) found no differences in both passive and active membrane properties of DDH neurons between acute and chronic SCI. Thus increased DDH firing in response to sensory stimulation after chronic SCI may be in part due to increased sensory transmission to the DDH neurons, similar to the increased sensory transmission to motoneurons after chronic SCI (Li et al. 2004a; Lucas-Osma et al. 2019). Other studies, however, did find DDH membrane property differences. Therefore, another possibility is that that altered expression of specific ion channels could trigger the increased excitability of the bursting neurons in chronic SCI. The expression of Nav1.3 voltage-gated sodium channels has been found to be upregulated in hyperexcitable dorsal horn neurons (laminae I–VI) following chronic contusive SCI (Hains et al. 2003). Nav1.3 channels can generate PICs and respond with large ramp currents to slow depolarizing stimuli below action potential threshold (Cummins et al. 2001). Moreover, the channels can rapidly recover from inactivation, thus making them suitable for sustaining high-frequency firing in neurons with increased excitability (Cummins et al. 2001). Reducing the expression of Nav1.3 protein via antisense oligodeoxynucleotides attenuates the hyperexcitability of dorsal horn neurons following SCI (Hains et al. 2003), and the effects are reversible after ceasing the antisense administration (Hains et al. 2003). Therefore, these studies suggest a functional relationship between Nav1.3 expression and hyperexcitability of dorsal horn neurons following SCI (Cummins et al. 2001; Hains et al. 2003) that might be responsible for the increased excitability of bursting neurons described in this study. Clearly, more studies are required.
Besides the bursting neurons, the dorsal horn networks in the vicinity of the bursting neurons may experience synaptic or cellular plasticity over time following SCI, since the field potential becomes smaller in magnitude (Fig. 1B) and slower in latency (Fig. 1C) from acute to chronic spinal transection, showing a change in the way the dorsal horn networks are responding to sensory input. These changes in the magnitude and latency of the field potential recorded from the vicinity of the bursting neurons may arise from an increased synaptic activity of inhibitory interneurons that mediate presynaptic inhibition of sensory afferents or excitatory interneurons to the bursting neurons, or postsynaptic inhibition of the bursting neurons. For example, a subpopulation of inhibitory GABAergic interneurons in lamina I has been found to exhibit an increase in their excitability following chronic spinal transection (Dougherty and Hochman 2008). The increased excitability of inhibitory GABAergic interneurons may function as a compensatory homeostatic response (Dougherty and Hochman 2008) to exaggerated sensory transmission following SCI-induced loss of descending 5-HT inhibitory control (Baker and Chandler 1987; Bellardita et al. 2017; Bennett et al. 2004; Hadjiconstantinou et al. 1984; Li et al. 2004b; Millan 2002; Murray et al. 2010; Yoshimura and Furue 2006) or indirectly increase sensory transmission (Lucas-Osma et al. 2019).
Another possibility is that there could be a complete shift in the bursting neuron population in chronic SCI. In this study, we are studying the firing patterns of bursting interneurons after chronic SCI and comparing them to those that burst in response to sensory input after acute SCI. We cannot necessarily identify them as the exact same neuronal population, but they all have the same bursting response to sensory input as a potential trigger for motoneurons. Future studies will include a full analysis of the interneuronal subpopulations based on firing properties and tell us if there is a shift in the subpopulations in chronic SCI.
Bursting neurons react differently to pharmacology postchronic SCI.
The bursting neurons continue to show significant changes in their firing activity during zolmitriptan and NMDA administration following chronic spinal transection. Via activation of 5-HT1B/1D receptors, zolmitriptan can suppress long EPSPs in motoneurons and muscle spasms without affecting the intrinsic membrane properties of motoneurons following chronic SCI (Murray et al. 2011). Our study demonstrates that zolmitriptan suppresses the bursting neurons (Fig. 4A) by reducing their evoked spike count (Fig. 4B) and burst duration (Fig. 4E) and by delaying their first-spike latency (Fig. 4G). The drug also suppresses the dorsal horn networks in the vicinity of the bursting neurons by delaying their field latency (Fig. 4D). The shorter burst duration and longer first-spike latency of the bursting neurons during zolmitriptan administration could also reflect changes in the EPSPs underlying the excitation of the bursting neurons. Perhaps zolmitriptan affects the sensory afferents that provide excitatory synaptic inputs to the bursting neurons, as recently shown for sensory transmission to motoneurons (Lucas-Osma et al. 2019). Interestingly, the main receptor that zolmitriptan activates (5-HT1D receptor) is confined nearly entirely to C fibers, and thus the action of zolmitriptan on DDH neurons is likely indirect by changes in spontaneous C-fiber activity, which in turn alters low threshold 1a afferent transmission (Lucas-Osma et al. 2019). When compared with its effects in acute SCI (Thaweerattanasinp et al. 2016), zolmitriptan suppresses the bursting neurons and the dorsal horn networks in their vicinity with even stronger effects over time following SCI (Fig. 5) by reducing their evoked spike count (Fig. 5A) and field potential (Fig. 5B) and by delaying their field latency (Fig. 5C) and first-spike latency (Fig. 5E) to a greater degree. The increase in strength of the zolmitriptan effects could result from an increase in sensitivity of 5-HT1B receptors (Honda et al. 2006; Husch et al. 2012; Sawynok and Reid, 1994) although the underlying mechanism remains unclear as 5-HT1B/1D receptors are unlikely develop supersensitivity after SCI (D’Amico et al. 2013). Although the mechanism underlying the anti-spastic effect of zolmitriptan in the dorsal horn is not well understood, there is plasticity in the spinal cord that allows zolmitriptan efficacy to increase in chronic SCI.
On the other hand, activation of NMDA receptors slightly facilitates the bursting neurons following chronic spinal transection (Fig. 2A). NMDA enhances the spontaneous firing rate (Fig. 2G) despite slightly delaying the first-spike latency (Fig. 2F) of the bursting neurons. When compared with the acute transection study (Thaweerattanasinp et al. 2016), NMDA facilitates the bursting neurons and the dorsal horn networks in their vicinity by enhancing their evoked spike count (Fig. 3A) and burst duration (Fig. 3D) and by delaying their field latency (Fig. 3C) and first-spike latency (Fig. 3E); however, the effects are weaker in chronic SCI (Fig. 3). The decrease in strength of the NMDA effects could result from an increase in inhibitory synaptic drive to the bursting neurons from hyperexcitable inhibitory interneurons, such as GABAergic interneurons in lamina I. These neurons are involved in and regulate, albeit insufficiently, NMDA receptor-mediated central sensitization and chronic pain following SCI (Hao et al. 2004; Wang et al. 2005; Woolf and Costigan 1999; Woolf and Salter 2000). Nevertheless, the reduced facilitative effects of NMDA on the bursting neurons may contribute to the generation of long EPSPs and muscle spasms following SCI.
Understanding the neuronal circuits involved in spasms after chronic SCI allows us to better understand the mechanism and, therefore, find targets for therapy. The current standard treatment for spasms, baclofen, works through presynaptic inhibition of the sensory axons. Our strategy would prevent interneurons from firing, which is an alternative approach. Increased spinal sensitivity to zolmitriptan may be a good thing therapeutically as new versions of the drug may be pursued for reduced side effects. The effects, or lack thereof, of the NMDA system may indicate that circuitry sensitive to NMDA, including those in locomotor circuits, may not be involved in spasms.
Bursting DDH interneurons are involved in spasms.
This study further supports the roles of bursting DDH interneurons in contributing to long EPSPs and to muscle spasms following SCI. Both the bursting firing pattern and the timing of the firing just before ventral root activity (Fig. 6) are both consistent with the initiation of a long EPSP. In addition, there is a significant correlation between DDH firing and overall ventral root activity (Fig. 7). This study shows that bursting DDH interneurons are sensitive to zolmitriptan, which has been seen to decrease spasms in rodents (Murray et al. 2011) and humans (D’Amico et al. 2013). All of this is consistent with the bursting DDH interneurons driving spasms. Although our correlation results are positive overall, our results also suggest that there is some heterogeneity within the deep dorsal horn population with some bursting neurons having very strong correlation with ventral root activity and others with little correlation. Our studies will continue to identify subpopulations based on activity and pharmacology while others are using genetic approaches such as the many subclasses of dorsal interneurons that have recently been identified by RNA sequencing methods (Häring et al. 2018). It is possible the bursting neurons in our study overlap with some of the deeper classes of glutamatergic interneurons identified in that work (e.g., Glu13–15), but further studies are required. We already know that the excitatory V3 propriospinal interneuron (identified by the developmental transcription factor Sim1) is essential for triggering muscle spasms, as optogenetically activating or inhibiting these neurons turns on or off LLRs and associate spasms (Lin et al. 2019). It is likely that the DDH burst neurons include these V3 neurons, as they are located in the DDH.
As noted in results, the use of bicuculline and strychnine is required to consistently show strong LLRs in the ventral root when stimulating the dorsal root. Interestingly, the bursting DDH interneurons that we propose to be involved in spasms have greater excitability when bicuculline and strychnine are present. Since this cocktail is needed for LLRs and also increases the excitability of the bursting DDH neurons, this lends additional support to the excitability of the bursting DDH interneurons being involved in spasms.
Conclusions.
Our study shows the potential contribution of bursting DDH neurons to generation of long EPSPs and muscle spasms following chronic SCI. By investigating the firing activity of the bursting neurons following chronic spinal transection and comparing it to the activity of these neurons following acute spinal transection, we show that the bursting neurons become slightly more excitable following chronic SCI. They also experience stronger suppressive effects of zolmitriptan but weaker facilitative effects of NMDA on their firing activity over time following SCI, which gives us information regarding mechanism and for future therapeutics. Nevertheless, future studies are necessary for investigating the functional connectivity of the bursting neurons within the spinal networks and ultimately for identifying their actual roles in the generation of long EPSPs and muscle spasms.
GRANTS
This work was supported by National Institute of Neurological Disorders and Stroke (NINDS) Grants NS-047567 and NS-089313 and Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) Grant HD-084672.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.T., M.C.T., D.J.B., C.H., and V.M.T. conceived and designed research; T.T. and D.B. performed experiments; T.T. analyzed data; T.T., D.B., M.C.J., M.C.T., D.J.B., C.H., and V.M.T. interpreted results of experiments; T.T. and V.M.T. prepared figures; T.T., C.H., and V.M.T. drafted manuscript; T.T., D.B., M.C.J., M.C.T., D.J.B., C.H., and V.M.T. edited and revised manuscript; T.T., D.B., M.C.J., M.C.T., D.J.B., C.H., and V.M.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Alyssa Puritz for help with animal care and Jack Miller for assistance with laboratory and equipment management.
REFERENCES
- Baker LL, Chandler SH. Characterization of postsynaptic potentials evoked by sural nerve stimulation in hindlimb motoneurons from acute and chronic spinal cats. Brain Res 420: 340–350, 1987. doi: 10.1016/0006-8993(87)91255-8. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. A Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma 23: 635–659, 2006. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Bennett DJ, Sanelli L, Cooke CL, Harvey PJ, Gorassini MA. Spastic long-lasting reflexes in the awake rat after sacral spinal cord injury. J Neurophysiol 91: 2247–2258, 2004. doi: 10.1152/jn.00946.2003. [DOI] [PubMed] [Google Scholar]
- Bellardita C, Caggiano V, Leiras R, Caldeira V, Fuchs A, Bouvier J, Low P, Kiehn O. Spatiotemporal correlation of spinal network dynamics underlying spasms in chronic spinalized mice. eLife 6: e23011, 2017. doi: 10.7554/elife.23011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Aglieco F, Renganathan M, Herzog RI, Dib-Hajj SD, Waxman SG. Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci 21: 5952–5961, 2001. doi: 10.1523/JNEUROSCI.21-16-05952.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amico JM, Li Y, Bennett DJ, Gorassini MA. Reduction of spinal sensory transmission by facilitation of 5-HT1B/D receptors in noninjured and spinal cord-injured humans. J Neurophysiol 109: 1485–1493, 2013. doi: 10.1152/jn.00822.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty KJ, Hochman S. Spinal cord injury causes plasticity in a subpopulation of lamina I GABAergic interneurons. J Neurophysiol 100: 212–223, 2008. doi: 10.1152/jn.01104.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjiconstantinou M, Panula P, Lackovic Z, Neff NH. Spinal cord serotonin: a biochemical and immunohistochemical study following transection. Brain Res 322: 245–254, 1984. doi: 10.1016/0006-8993(84)90114-8. [DOI] [PubMed] [Google Scholar]
- Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci 23: 8881–8892, 2003. doi: 10.1523/JNEUROSCI.23-26-08881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao JX, Kupers RC, Xu XJ. Response characteristics of spinal cord dorsal horn neurons in chronic allodynic rats after spinal cord injury. J Neurophysiol 92: 1391–1399, 2004. doi: 10.1152/jn.00121.2004. [DOI] [PubMed] [Google Scholar]
- Häring M, Zeisel A, Hochgerner H, Rinwa P, Jakobsson JE, Lönnerberg P, La Manno G, Sharma N, Borgius L, Kiehn O, Lagerström MC, Linnarsson S, Ernfors P. Neuronal atlas of the dorsal horn defines its architecture and links sensory input to transcriptional cell types. Nat Neurosci 21: 869–880, 2018. doi: 10.1038/s41593-018-0141-1. [DOI] [PubMed] [Google Scholar]
- Honda M, Tanabe M, Ono H. Spinal cord injury-specific depression of monosynaptic spinal reflex transmission by l-5-hydroxytryptophan results from loss of the 5-HT uptake system and not 5-HT receptor supersensitivity. Exp Neurol 202: 258–261, 2006. doi: 10.1016/j.expneurol.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Husch A, Van Patten GN, Hong DN, Scaperotti MM, Cramer N, Harris-Warrick RM. Spinal cord injury induces serotonin supersensitivity without increasing intrinsic excitability of mouse V2a interneurons. J Neurosci 32: 13145–13154, 2012. doi: 10.1523/JNEUROSCI.2995-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi M, Fehlings MG. Development and characterization of a novel, graded model of clip compressive spinal cord injury in the nouse: Part 1. Clip design, behavioral outcomes, and histophathology. J Neurotrauma 19: 175–190, 2002. doi: 10.1089/08977150252806947. [DOI] [PubMed] [Google Scholar]
- Li Y, Gorassini MA, Bennett DJ. Role of persistent sodium and calcium currents in motoneuron firing and spasticity in chronic spinal rats. J Neurophysiol 91: 767–783, 2004a. doi: 10.1152/jn.00788.2003. [DOI] [PubMed] [Google Scholar]
- Li Y, Lucas-Osma AM, Black S, Stephens MJ, Fenrich KK, Fouad K, Bennett DJ. Sensory transmission to motoneurons is facilitated by cold-sensing C fibres expressing TRPM8 and 5-HT1D receptors: links between management of spasticity and migraines. Proceedings from the 46th Annual Meeting of the Society for Neuroscience, November 12–16, 2016. San Diego, CA, 2016, abstract 612.29. [Google Scholar]
- Li Y, Li X, Harvey PJ, Bennett DJ. Effects of baclofen on spinal reflexes and persistent inward currents in motoneurons of chronic spinal rats with spasticity. J Neurophysiol 92: 2694–2703, 2004b. doi: 10.1152/jn.00164.2004. [DOI] [PubMed] [Google Scholar]
- Lin S, Li Y, Lucas-Osma AM, Hari K, Stephens MJ, Singla R, Heckman CJ, Zhang Y, Fouad K, Fenrich KK, Bennett DJ. Locomotor-related V3 interneurons initiate and coordinate muscles spasms after spinal cord injury. J Neurophysiol 121: 1352–1367, 2019. doi: 10.1152/jn.00776.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas-Osma AM, Li Y, Murray K, Lin S, Black S, Stephens MJ, Ahn AH, Heckman CJ, Fenrich KK, Fouad K, Bennett DJ. 5-HT1D receptors inhibit the monosynaptic stretch reflex by modulating C-fiber activity. J Neurophysiol 121: 1591–1608, 2019. doi: 10.1152/jn.00805.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol 66: 355–474, 2002. doi: 10.1016/S0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Murray KC, Nakae A, Stephens MJ, Rank M, D’Amico J, Harvey PJ, Li X, Harris RL, Ballou EW, Anelli R, Heckman CJ, Mashimo T, Vavrek R, Sanelli L, Gorassini MA, Bennett DJ, Fouad K. Recovery of motoneuron and locomotor function after spinal cord injury depends on constitutive activity in 5-HT2C receptors. Nat Med 16: 694–700, 2010. doi: 10.1038/nm.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KC, Stephens MJ, Rank M, D’Amico J, Gorassini MA, Bennett DJ. Polysynaptic excitatory postsynaptic potentials that trigger spasms after spinal cord injury in rats are inhibited by 5-HT1B and 5-HT1F receptors. J Neurophysiol 106: 925–943, 2011. doi: 10.1152/jn.01011.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank MM, Flynn JR, Battistuzzo CR, Galea MP, Callister R, Callister RJ. Functional changes in deep dorsal horn interneurons following spinal cord injury are enhanced with different durations of exercise training. J Physiol 593: 331–345, 2015. doi: 10.1113/jphysiol.2014.282640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawynok J, Reid A. Spinal supersensitivity to 5-HT1, 5-HT2 and 5-HT3 receptor agonists following 5,7-dihydroxytryptamine. Eur J Pharmacol 264: 249–257, 1994. doi: 10.1016/0014-2999(94)00465-X. [DOI] [PubMed] [Google Scholar]
- Thaweerattanasinp T, Heckman CJ, Tysseling VM. Firing characteristics of deep dorsal horn neurons after acute spinal transection during administration of agonists for 5-HT1B/1D and NMDA receptors. J Neurophysiol 116: 1644–1653, 2016. doi: 10.1152/jn.00198.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tysseling VM, Klein DA, Imhoff-Manuel R, Manuel M, Heckman CJ, Tresch MC. Constitutive activity of 5-HT2C receptors is present after incomplete spinal cord injury but is not modified after chronic SSRI or baclofen treatment. J Neurophysiol 118: 2944–2952, 2017. doi: 10.1152/jn.00190.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Kawamata M, Namiki A. Changes in properties of spinal dorsal horn neurons and their sensitivity to morphine after spinal cord injury in the rat. Anesthesiology 102: 152–164, 2005. doi: 10.1097/00000542-200501000-00024. [DOI] [PubMed] [Google Scholar]
- Watson C, Paxinos G, Kayalioglu G. The Spinal Cord. New York: Academic Press, 2008. [Google Scholar]
- Woolf CJ, Costigan M. Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci USA 96: 7723–7730, 1999. doi: 10.1073/pnas.96.14.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science 288: 1765–1769, 2000. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Furue H. Mechanisms for the anti-nociceptive actions of the descending noradrenergic and serotonergic systems in the spinal cord. J Pharmacol Sci 101: 107–117, 2006. doi: 10.1254/jphs.CRJ06008X. [DOI] [PubMed] [Google Scholar]