

Abstract
Human induced pluripotent stem cells (hiPSCs) have generated a great deal of attention owing to their capacity for self‐renewal and differentiation into the three germ layers of the body. Their discovery has facilitated a new era in biomedicine for understanding human development, drug screening, disease modeling, and cell therapy while reducing ethical issues and risks of immune rejection associated with traditional embryonic stem cells. Bioreactor‐based processes have been the method of choice for the efficient expansion and differentiation of stem cells in controlled environments. Current protocols for the expansion of hiPSCs use horizontal impeller, paddle, or rocking wave mixing method bioreactors which require large static cell culture starting populations and achieve only moderate cell fold increases. This study focused on optimizing inoculation, agitation, oxygen, and nutrient availability for the culture of hiPSCs as aggregates in single‐use, low‐shear, vertical‐wheel bioreactors. Under optimized conditions, we achieved an expansion of more than 30‐fold in 6 days using a small starting population of cells and minimal media resources throughout. Importantly, we showed that that this optimized bioreactor expansion protocol could be replicated over four serial passages resulting in a cumulative cell expansion of 1.06E6‐fold in 28 days. Cells from the final day of the serial passage were of high quality, maintaining a normal karyotype, pluripotent marker staining, and the ability to form teratomas in vivo. These findings demonstrate that a vertical‐wheel bioreactor‐based bioprocess can provide optimal conditions for efficient, rapid generation of high‐quality hiPSCs to meet the demands for clinical manufacturing of therapeutic cell products.
Keywords: bioreactor, expansion, human induced pluripotent stem cells (hiPSCs), low‐shear, serial‐passage
Under optimized conditions in vertical‐wheel bioreactors, we achieved an expansion of human induced pluripotent stem cells of more than 30‐fold in 6 days using a small starting population of cells and minimal media resources throughout. Importantly, we showed that this optimized bioreactor expansion protocol could be replicated over four serial passages resulting in a cumulative cell expansion of 1.06E6‐fold in just 28 days.
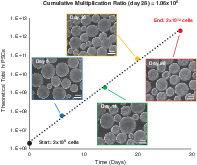
Significance statement.
This study has developed a new method to grow human induced pluripotent stem cells in large quantities through serial passaging in vertical‐wheel bioreactors. Cells were cultured from small starting numbers, in optimized conditions, resulting in economical, reproducible culture techniques for high‐quality populations. These advances will have significant economic and practical applications in stem cell therapies.
1. INTRODUCTION
Due to their proliferation capacity and ability to differentiate into any cell type in the body, pluripotent stem cells (PSCs) have generated significant interest in the fields of regenerative medicine, drug discovery, and disease modeling. 1 , 2 , 3 , 4 The ability to derive a wide range of cell types from a single‐cell source makes PSCs an ideal platform for cell‐based therapies. 5 The discovery of human induced pluripotent stem cells (hiPSCs) provides an important alternative to traditional human embryonic stem cells (hESCs)—overcoming ethical barriers associated with an embryo cell source and reducing the risk of immune rejection associated with allogenic treatments. 2 , 6 Despite the increased demand for the use of hiPSCs, challenges with large‐scale expansion to meet clinically relevant cell quantities have prevented advances within the field. To date, approximately 300 clinical trials have involved human mesenchymal stem cells to treat diseases including neurological disorders and strokes, 7 more than 10 clinical trials have been approved for the use of hESCs, but only a few clinical applications involve the use of hiPSCs. 2 , 8 Recent clinical trials include iPSC‐derived mesenchymal stem cells for treatment of steroid‐resistant acute graft vs host disease (Cynata Therapeutics), 9 iPSC‐derived dopaminergic progenitors to treat Parkinson's disease (Kyoto University), 10 and clonal master characterized iPSCs for treatment of advanced solid tumors (Fate Therapeutics). 11 The number of high‐quality cells required for treatment ranges from 109 to 1012 depending on the therapeutic target, with therapeutic efficacy directly correlating to cell dose. It is suggested that for myocardial infraction, 1 × 109 to 2 × 109 cardiomyocytes would be required to replace damaged cardiac tissue in an adult, while 1 × 1010 hepatocytes would be required for treatment of hepatic failure. 12
At a small scale, hiPSCs are most often cultured as adherent monolayer colonies on an undefined matrix coated flask. This is not a feasible approach to achieve clinically relevant numbers and possesses a high degree of variability. 13 , 14 , 15 Bioreactor‐based processes have been the method of choice for efficient expansion and differentiation of cells in controlled environments. Compared with static culture, bioreactor‐based processes reduce labor and operating costs, improve process monitoring and control, and create a well‐mixed environment promoting greater cell densities per unit volume. 16 While some studies have investigated the expansion of hiPSCs in bioreactors, many have not been optimized in a systematic manner, and the effect of variations in bioprocess parameters on product output has not been fully examined. These studies, summarized in Table 1, 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 have achieved only moderate cell fold increases with most requiring high cell seeding densities (2 × 105 to 1 × 106 cells/mL) that must be cultured through a lengthy and resource intensive static expansion phase prior to bioreactor inoculation. Early publications achieve a maximum of 6‐fold expansion in 4 to 7 days 17 while recent publications achieve a maximum of 10‐ to 13‐fold expansion in 5 days 24 and 10‐ to 16‐fold expansion in 7 days. 25 In comparison, bioreactor studies examining the expansion of mouse PSCs achieve maximums of 31‐fold expansion in 4 to 5 days, 27 , 28 54‐fold expansion in 4 days, 29 58‐fold expansion in 4 days, 30 and up to 184‐fold expansion in 11 days with controlled dissolved oxygen concentrations and continuous perfusion. 31
TABLE 1.
Literature comparison of hiPSCs cultured in bioreactor systems
| Reference | Cell lines | Media | Bioreactor size and type | Inoculation and growth platform | Inoculation density (cells/mL) | Feeding strategy | Fold expansion/days |
|---|---|---|---|---|---|---|---|
| Zweigerdt et al 17 | hCBiPSC2, hiPSOCT4eGFP | mTeSR1 | 100 mL, Horizontal‐blade spinner (Integra Biosciences) | Single‐cell inoculation, aggregate growth | 1 × 106 | Media exchange (100%) at 48 h followed by daily media exchange (100%) | 3‐ to 6‐fold/4‐7 d |
| Olmer et al 18 | hCBiPSC2 | mTeSR1 | 100 mL, Pitched blade, computer controlled reactor (DasGip) | Single‐cell inoculation, aggregate growth | 4 × 105 to 5 × 105 | Media exchange (100%) at 72 h followed by daily media exchange (100%) | 5.5‐fold/7 d |
| Abbasalizadeh et al 19 | hiPSC1, hiPSC4 |
DMEM/F12‐CM+ GlutaMAX+bFGF |
100 mL, Horizontal‐blade spinner (Integra Biosciences) | Single‐cell inoculation, aggregate growth | 2 × 105 to 1 × 106 | Media refreshment beginning at 48 h | 8‐fold/7‐10 d |
| Wang et al 20 | BC1, TNC1 | E8 | 100 mL, Glass‐ball spinner flask (CELLSPIN) | Single‐cell inoculation, aggregate growth | 4 × 105 to 5 × 105 | Media exchange (2/3) daily | 3.5‐fold/4 d |
| Elanzew et al 21 | iLB‐C‐31f‐r1 | mTeSR1 or E8 | 50 mL, Tube rotation (BioLevitator) | Single‐cell inoculation, aggregate growth | 7.5 × 104 | Media addition (10 mL) at 24, 48, and 72 h | 5‐fold/4 d |
| Haraguchi et al 22 | 201B7, 253G1 | mTeSR1 |
100 mL, Horizontal‐blade spinner (Integra Biosciences) |
Single‐cell inoculation, aggregate growth | 3 × 105 | Media exchange (100%) daily | 10‐fold/12 d |
| Badenes et al 23 | Gibco CD34+ derived | E8 | 50 mL, Horizontal‐blade spinner flask (StemSpan) | Single‐cell inoculation, polystyrene‐coated microcarrier growth | 2 × 105 to 5 × 105 | Media exchange (80%) at 72 h followed by daily media exchange (80%) | 3.5‐fold/10 d |
| Meng et al 24 | 4YF, 4YA | mTeSR1 | 100 mL, Horizontal‐blade spinner (NDS) | Preformed aggregate inoculation, aggregate growth | 2 × 104 to 8 × 104 | Media exchange (100%) on days when culture pH measured below 7.0 | 11‐ to 13‐fold/5 d |
| Kwok et al 25 | AFiPS, FSiPS |
mTeSR1 or StemMACs iPSC‐Brew |
1. 100 mL, Horizontal‐blade spinner 2. 1 L, Horizontal‐blade, single‐use bioreactor |
Single‐cell inoculation, aggregate growth | 2 × 105 |
1. Media addition (20 mL) at 48 and 72 h followed by daily media exchange (100%) 2. Media addition (250 mL) at 48 and 72 h followed by daily media exchange (100%) |
1. 16‐fold /7 d 2. 10‐fold /7 d |
| Abecasis et al 26 | ChiPSC4 | Cellartis DEF‐CS Xeno‐Free Culture Medium | 200 mL, Trapezoid paddle, computer controlled reactor (DasGip) | Single‐cell inoculation, aggregate growth | 2.5 × 105 to 5 × 105 | Perfusion system (initiated at 24 h) simulating media renewal frequency established in static monolayer culture (dilution rate = 1.3 d−1) | 19‐fold/4 d |
| This study | 4YA | mTeSR1 | 100 mL, Vertical‐wheel, single‐use spinner (PBS Biotech) | Preformed aggregate inoculation, aggregate growth | 2 × 104 | Media exchange (50 mL) at 96 h | 32‐fold/6 d |
Note: Referenced data include cell lines, media, bioreactor size and type, inoculation and growth platform, inoculation density, feeding strategy, and fold expansion/days.
Abbreviation: DMEM, Dulbecco's modified Eagle medium; hiPSCs, human induced pluripotent stem cells.
In this study, we focused on optimizing bioprocess variables including inoculation, agitation, oxygen content, and nutrient availability for hiPSCs cultured as aggregates in single‐use, vertical‐wheel bioreactors. A graphical outline highlighting the experimental design leading to an optimized process for hiPSC bioreactor serial passaging and quality testing is shown in Figure 1. The PBS Biotech vertical‐wheel bioreactor, is a novel system that combines radial and axial flow components producing more uniform distributions of hydrodynamic forces, as well as lower shear stress, compared with traditional horizontal‐blade based bioreactors, 32 , 33 shown in Figure 1. We hypothesized that this unique bioreactor system would provide a more optimal environment for the efficient, rapid generation of high‐quality hiPSCs to meet the demands for manufacturing of therapeutic cell products. Our optimized protocol in the vertical‐wheel bioreactor used preformed aggregates seeded at low inoculation densities in defined, serum‐free medium. We achieved a 32‐fold expansion in 6 days with minimal feeding required (a single, 50% media exchange on day 4). Over four serial passages, clinically relevant cell numbers could be achieved (over 2E11 hiPSCs from a single cryopreserved vial of 2E6 cells) that maintain a normal karyotype, positive expression of pluripotency markers, and the functional ability to form teratomas in vivo.
FIGURE 1.
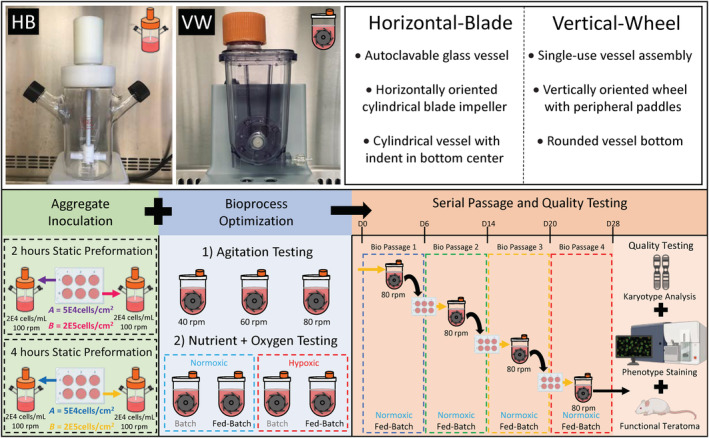
Schematic comparison of horizontal‐blade and vertical‐wheel bioreactors used in the experimental design of this study. This study focused on optimizing inoculation, agitation, oxygen content, and nutrient availability for hiPSCs cultured as aggregates in vertical‐wheel bioreactors. Best conditions from these optimization experiments were used for a serial passage and quality testing experiment. hiPSCs, human induced pluripotent stem cells
2. MATERIALS AND METHODS
The cell cultures used in our experiments were hiPSC line 4YA, derived from infant fibroblasts and reprogrammed using retrovirus four factors (OSKM) with EOS reporter. These cells were obtained from Dr James Ellis' laboratory at the University of Toronto (Toronto, Canada). The cells underwent numerous static expansion cycles to develop a cryogenically preserved cell bank. Cultures used in this study were between passage numbers 40 and 45.
2.1. Static culture
For expansion prior to inoculation in suspension culture, hiPSCs were grown in T‐75 flasks (Cat#156599, Thermo Fisher Scientific) maintained under standard culture conditions (37°C and 5% CO2). Flasks were coated with feeder‐free substrate hESC‐qualified Matrigel (Cat#354277, Corning Life Sciences) in DMEM/Hams F‐12 (Cat#10‐090‐CV, Corning Life Sciences) for 2 hours at room temperature. The cells were inoculated into T‐75 flasks at a density of 20 000 cells/cm2 with 12 mL/flask mTeSR1 medium (Cat#85851, STEMCELL Technologies) supplemented with 10 μM Y‐27632 (Cat#72304, STEMCELL Technologies). Daily medium replacements were carried out, excluding the addition of Y‐27632. When approximately 80% confluence was reached (3‐4 days), cells were passaged and reseeded at a 1:6 culture areas split ratio. Cultures were washed once with Ca2+ and Mg2+ free PBS and treated with 5 mL/flask 0.5 mM EDTA·4Na Accutase (Cat#07922, STEMCELL Technologies) supplemented with 10 μM Y‐27632 and left at 37°C in the incubator for 5 minutes. Flasks were observed under the microscope to confirm cell detachment, and medium supplemented with 10 μM Y‐27632 was added at a 1:1 ratio of Accutase to dilute the enzyme. The cell solution was transferred to a conical tube to be centrifuged at 300g for 5 minutes. The supernatant was discarded, and the cell pellet was resuspended in medium supplemented with 10 μM Y‐27632 to be inoculated into Matrigel‐coated flasks.
2.2. Suspension culture
Dynamic suspension vessels used in this study include 100 mL working volume, horizontal‐blade, glass bioreactors (Corning Style Spinner Flask, NDS Technologies, Inc.) and 100 mL working volume, single‐usevertical‐wheel bioreactors (PBS Biotech, Ltd.). Constant mixing was maintained in culture at standard conditions of 37°C and 5% CO2. For experiments studying cell growth under hypoxic conditions, N2 and CO2 was used to maintain the ambient gas environment at 5% CO2 and 3% O2 in constrained chambers within the incubator. hiPSCs were cultured at 100 mL working volume at batch conditions with mTeSR1 medium supplemented with 10 μM Y‐27632. Experiments studying the effects of nutrient availability in repeated‐batch conditions involved 50 mL medium replacements with mTeSR1 (excluding Y‐27632). To perform the medium exchange, bioreactors were brought under a laminar flow hood where cell aggregates were allowed to settle for 5 minutes. Following aggregate settlement, 50 mL was aspirated from the surface of the bioreactor and added to a conical tube to be centrifuged at 300g for 3 minutes. Medium from the conical tube was discarded and the remaining aggregate cell pellet was resuspended with fresh medium to be added back into the bioreactor.
Preformed hiPSC aggregates were used to inoculate bioreactors at a density of 20 000 cells/mL. Preformed aggregates were produced in nonadherent, six‐well suspension culture plates (Cat#657‐185, Greiner CELLSTAR) with 2.0 mL/well mTeSR1 supplemented with Y‐27632. To optimize the preforming aggregate protocol, we studied the effects of seeding density per well and incubation time prior to bioreactor inoculation. Wells were inoculated at densities of 50 000, 100 000, and 200 000 cells/cm2 and left in the incubator for 1, 2, 3, and 4 hours. Cell clumps were then mechanically broken up through gentle pipetting four to five times using a P‐1000 micropipette and inoculated into the bioreactors for further analysis.
2.3. Aggregate dissociation and aggregate sizing
To study the growth kinetics of hiPSCs in suspension culture, daily samples were taken with cell counts performed in duplicates. Samples of 1.0 to 5.0 mL were removed using a serological pipette during bioreactor agitation to minimize settling of the aggregates. The samples were centrifuged at 300g for 5 minutes. The supernatant was discarded, and the cell pellet was resuspended in 1.0 mL of Accutase and left in a 37°C water bath for 4 to 7 minutes. The cell solution was then gently pipetted three times followed by the addition of 1.0 mL of medium used dilute the enzyme. The sample was centrifuged at 500g for 5 minutes, the supernatant was discarded, and the cell pellet was resuspended in 0.5 to 1.0 mL medium. Two 200 μL aliquots were taken from each cell sample for viable cell counts using the NucleoCounter NC‐200 (ChemoMetec, Denmark), an automated cassette counter, which analyzes samples stained with fluorescent dyes Acridine Orange and 4′,6‐diamidino‐2‐phenylindole (DAPI). These counts were used to calculate apparent growth rate, doubling time, and fold expansion using Equation (1):
| (1) |
where X 1 and X 2 are the viable cell densities (cells/mL) at the beginning and end of the time interval Δt. The “overall” apparent specific growth rate, μ (h−1), is the value obtained when X 1 is taken to be the cell density at inoculation and X 2 is the cell density at the end of the culture period, assuming cells grow exponentially during the culture period. The doubling time (hours) and fold expansion can be calculated using Equations (2) and (3), respectively:
| (2) |
| (3) |
To determine average aggregate size and size distributions, 1.5 mL samples were removed using a serological pipette from the bioreactors and added into 12‐well plates for visualization. Images were taken using a Zeiss Axiovert 25 microscope (Carl Zeiss) with AxioVision software used for measurements. Aggregates were defined as multicellular spheroids with a diameter greater than 50 μm. The diameter for each aggregate was determined by taking the average of the greatest length across the aggregate and the length perpendicular to the greatest length. A minimum of 100 aggregates were sized per condition.
2.4. Suspension culture serial passage
Serial passaging from bioreactor to bioreactor was performed every 6 to 8 days of repeated‐batch culture. Aggregate cell samples were removed from each bioreactor and dissociated into single cells using the aggregate dissociation method described above. Single cells were counted and inoculated into six‐well suspension culture wells at a concentration of 200 000 cells/cm2 in 2.0 mL/well mTeSR1 supplemented with Y‐27632. Wells were left in the incubator for 4 hours to preform cell clumps which were inoculated into the next bioreactor at a concentration of 20 000 cells/mL. This serial passage procedure was repeated three times.
2.5. High‐performance liquid chromatography mass spectrometry
For metabolomic analysis, two 1.0 mL samples were taken daily from each bioreactor condition and centrifuged at 300g for 5 minutes to separate the culture media from the cell pellet. Two hundred microliters of samples of the culture medium were then diluted 1:10 in LC‐MS grade methanol (Cat# 1.06035, Sigma). Macromolecules were pelleted by centrifugation at 13000 rpm for 5 minutes (Biofuge Pico, Sorvall) to extract the supernatant. The extracted supernatant was diluted 1:2 in cell culture grade water and stored at −20°C for 8 hours prior to running high‐performance liquid chromatography mass spectrometry (HPLC‐MS). Samples were analyzed using a Thermo Fisher Vanquish UHPLC system integrated with a Thermo Fisher TSQ Quantum mass spectrometer. Data were processed using MAVEN. Statistical analysis and visual representation of the data were done in R and MATLAB.
2.6. Immunocytochemistry
For each sample, a calculated volume of bioreactor‐generated aggregates containing 1E6 cells per stain were enzymatically dissociated into a single‐cell suspension, using the dissociation method described above, and resuspended in fixation buffer (Cat#FC001, R&D Systems) to be incubated for 15 minutes at room temperature. Dissociated cells were rinsed twice with PBS then resuspended in 200 μL of permeabilization buffer (Cat#FC005, R&D Systems) with 1 μg/106 cells antibody stain and 1 μM/106 cells nuclei stain and incubated for 1 hour at room temperature. Conjugated antibody stains for TRA‐1‐60 (Ca # FAB4770P, R&D Systems) and Nanog (Cat#MABD24A4, Millipore Sigma) were used along with the nuclei stain To‐Pro‐3 Iodide Nucleic Acid Stain (Cat#T3605, Thermo Fisher). Cells were then rinsed twice with PBS and imaged using a Carl Zeiss Laser Scanning Microscope 700 with lasers at 488 and 639 nm and corresponding filter sets.
2.7. Flow cytometry
For each sample, a calculated volume of bioreactor‐generated aggregates containing 1E6 cells per stain were enzymatically dissociated as previously described and divided into microcentrifuge tubes to be stained. The single‐cell suspensions were resuspended in PBS with 1 μL/106 cells Live/Dead Fixable Blue Dead Cell Stain (Cat#L34961, Thermo Fisher) and incubated for 30 minutes at room temperature. Samples were then centrifuged at 2000 rpm for 5 minutes (Biofuge Pico, Sorvall), washed with PBS, and resuspended in PBS with SSEA‐4 antibody stain (Cat#FAB1435F, R&D Systems) at a concentration of 10 μL/106 cells and incubated for 30 minutes at 4°C. These samples were then centrifuged, washed with PBS, and incubated with fixation buffer for 15 minutes at room temperature. Fixed samples were then centrifuged, washed with PBS, and incubated for 30 minutes at 4°C with permeabilization buffer and OCT‐4 intracellular stain (Cat#IC1759N‐100UG, R&D Systems) at a concentration of 1 μg/106 cells. Isotype controls were added for fluorescein isothiocyanate (FITC) (Cat#11‐4742‐42, Thermo Fisher) and for Alexa Fluor 700 (Cat#IC013N, R&D Systems). Immunostained hiPSCs were subjected to fluorescence‐activated cell sorting using a BD LSRII with four lasers: 405, 561, 488, and 640 (BD Bioscience) equipped with the BD Diva version 6.1.3. software. A minimum of 10 000 events were collected per sample, and analysis of the acquired cells was performed using appropriate area and width of side and forward scatter gates to avoid cellular debris.
2.8. Karyotyping
Samples of bioreactor‐generated aggregates were incubated in 10 mL bioreactors in medium supplemented with 0.1 μg/mL KaryoMax Colcemid (Cat#15212012, Thermo Fisher Scientific) for 4 hours. The aggregates were then enzymatically dissociated as previously described. Single cells were collected by centrifugation, suspended in 0.075 M KCl hypotonic solution (Cat#P217‐500, Fischer Scientific), and incubated at 37°C for 25 minutes. Cells were then fixed with 3:1 methanol:acetic acid solution (Cat#A412‐4, Fisher Scientific, Cat#AX0073, EMD) and chromosome preparations were G‐bands by Trypsin and Giemsa (GTG)‐banded using standard cytogenetic techniques. Karyograms were analyzed according to the ISCN standards at ~450 band resolution using the Ikaros karyotyping system (Metasystems).
2.9. Teratoma formation
Severe combined immune deficiency (SCID) Beige mice were obtained from Charles River (Wilmington, Massachusetts) and stored in a single‐barrier animal facility (Cumming School of Medicine, University of Calgary). All mice were fed adlibitum and maintained on a normal day/night cycle. All procedures carried out were approved by the University of Calgary animal care committee (protocol no. AC16‐0043). Mice were anesthetized using veterinary isoflurane and 1.2E6 dissociated hiPSCs in 100 μL of PBS were injected subcutaneously into the inner thigh of each leg of four mice (eight injections total). After 6weeks, the mice were sacrificed, and the tissue mounds were excised. The excised tissue was fixed in 4% preformed aggregate (PFA) for 24 hours. The tissue was then processed overnight using an automatic tissue processor and embedded in paraffin wax. The tissue was sectioned using a microtome at 10 μm thickness and stained with H&E. The tissue sections were imaged using the Zeiss Axio Scan microscope (Zeiss, Germany).
2.10. Statistics
Statistical analysis was done using GraphPad Prism (v6.0). A one‐way analysis of variance (ANOVA) followed by Tukey's multiple comparison test was used for all growth curve and aggregate size comparisons. Time‐series metabolomic data sets were compared by analyte at single time points using Mann‐Whitney (for two groups) or Kruskal‐Wallis (for more than two groups) tests to assess multiple unmatched populations under each experimental condition. Cell samples were collected from n = 4 stirred suspension bioreactors at each condition. The P values were set at .05 and all graphs are presented with a ±SEM.
3. RESULTS
3.1. Bioreactor inoculation with preformed aggregates
To optimize the protocol for the generation of preformed aggregates used in bioreactor inoculation, we first studied the effects of cell seeding density and incubation time in nonadherent suspension wells. Seeding densities of 50 000, 100 000, and 200 000 cells/cm2 were explored, with incubation times of 1, 2, 3, and 4 hours. At the end of the incubation period, the cell clumps were mechanically dissociated with gentle pipetting to break up the clusters into preformed aggregates as presented in Figure 2A. At lower incubation times, most of the cell clusters dissociated into single cells or clumps comprised less than 10 cells. Higher incubation times produced larger preformed aggregates with fewer single cells present. The largest preformed aggregates were obtained at the seeding density of 200 000 cells/cm2 with an incubation time of 4 hours.
FIGURE 2.
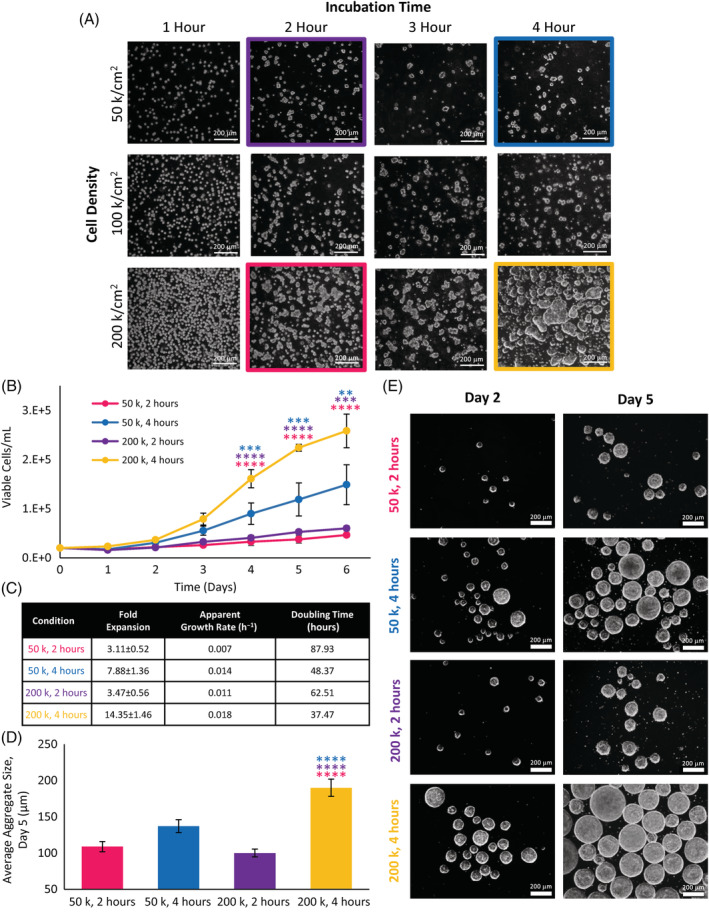
A, Preformed aggregates in six‐well suspension culture plates at various seeding densities and incubation times. B, Cell growth and, C, corresponding growth kinetic values of selected preformed aggregates seeded into horizontal‐blade bioreactors. D, Average aggregate diameters from selected preformed conditions on day 5 of bioreactor culture and, E, phase contrast microscope images of aggregates on day 2 and day 5 of culture in horizontal‐blade bioreactors. Statistical significance equivalent to **P ≤ .01, ***P ≤ .001, ****P ≤ .0001
To obtain a representative picture of what combined effects preformation density and time would have on downstream aggregate formation and growth kinetics, a subset of 4 of 12 preformation conditions (highlighted in Figure 2A) were inoculated into the bioreactors. Preformed aggregates recovered from the six‐well suspension plates seeded at the densities of 50 000 and 200 000 cells/cm2 with incubation times of 2 and 4 hours were inoculated at a density of 20 000 cells/mL into NDS horizontal‐blade bioreactors operated at 100 rpm. Cells were cultured under batch conditions for a period of 6 days. Incubation time had the greatest effect on cell growth, with bioreactors inoculated with preformed aggregates that were incubated for 4 hours resulting in significantly greater growth than bioreactors inoculated with preformed aggregates incubated for 2 hours, as shown in Figure 2B. The greatest cell growth was obtained with the inoculation of preformed aggregates seeded at 200 000 cells/cm2 for a 4 hours incubation period. This condition resulted in the highest fold expansion of 14.35 ± 1.46 with the greatest apparent specific growth rate of 0.018 h−1 and the shortest doubling time of 37.47 hours, as presented in Figure 2C. The average aggregate sizes were compared on day 5. The largest aggregates generated were 214 ± 6 μm (200 k, 4 hours) and the smallest aggregates generated were 100 ± 5 μm (200 k, 2 hours), as seen in Figure 2D,E. It is evident from the phase contrast images that morphologically healthy spheroids were formed under each bioreactor condition. The average aggregate size from the 200 k, 4 hours bioreactor condition was significantly greater than all other preformed conditions, corresponding to significantly greater cell densities on days 4 to 6 of culture.
3.2. Effect of vertical‐wheel bioreactor agitation on growth kinetics and aggregate sizes
The following studies optimizing expansion conditions in the PBS vertical‐wheel bioreactor were inoculated with preformed aggregates generated at 200 000 cells/cm2 for a period of 4 hours. We first compared the effect of agitation rate on cell growth and aggregate growth, inoculating vertical‐wheel bioreactors at 40, 60, and 80 rpm. All conditions resulted in cell growth with an initial lag phase, and cell growth reaching a plateau by day 6, as shown in Figure 3B. The maximum multiplication ratios obtained were between 9.9 and 12.7, with no significant difference between the agitation rates.
FIGURE 3.
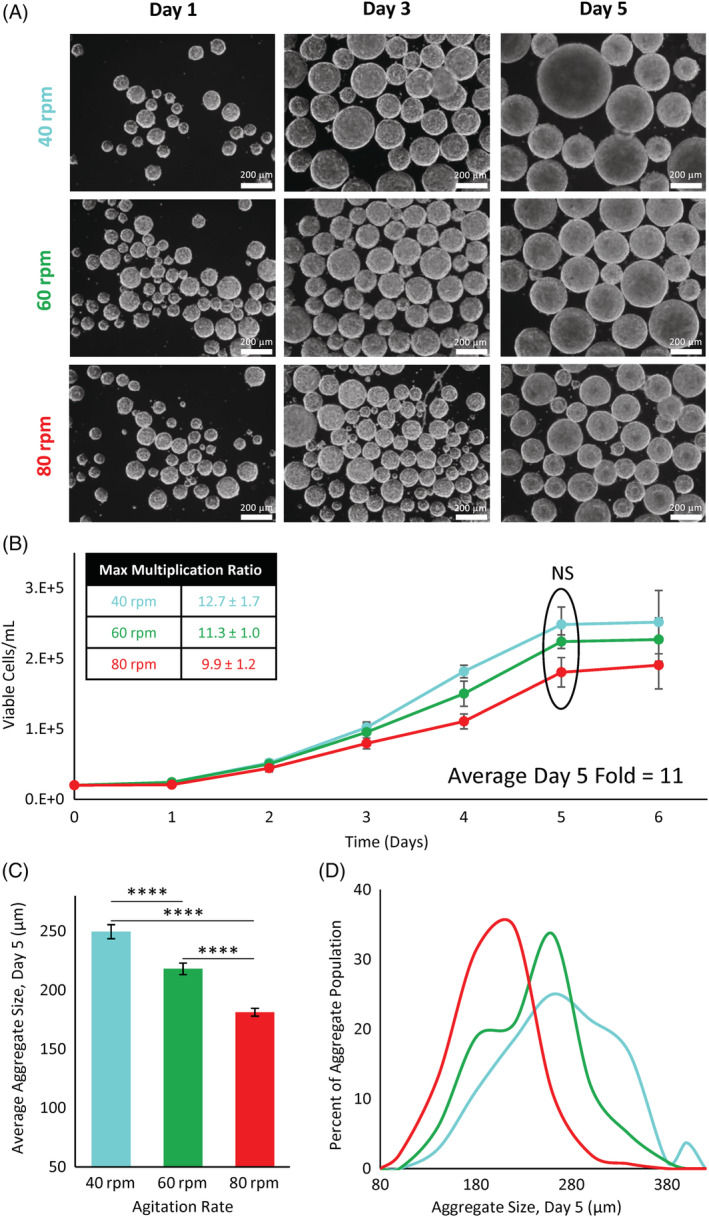
A, Phase contrast microscope images of hiPSCs cultured in vertical‐wheel reactors at agitation rates of 40, 60, and 80 rpm. B, Cell growth and corresponding maximum multiplication ratios of hiPSCs cultured in vertical‐wheel reactors for 6 days at 40, 60, and 80 rpm. C, Average day 5 aggregate size and, D, aggregate size distributions of hiPSCs cultured in vertical‐wheel reactors at 40, 60, and 80 rpm. Statistical significance equivalent to ****P ≤ .0001. hiPSCs, human induced pluripotent stem cells
We also studied the effect of agitation rate on aggregate size over the culture period. As expected, the aggregate size increased from day 1 to day 5, reflected in the phase contrast images in Figure 3A. We compared the average aggregate sizes at the end of the growth phase (day 5). The average aggregate diameters were 250 ± 5.9 μm at 40 rpm followed by 218 ± 4.9 μm at 60 rpm, and 181 ± 3.3 μm at 80 rpm, as shown in Figure 3C. When comparing the aggregate size distributions, the 40 rpm condition resulted in a wide distribution of aggregate sizes, whereas the 80 rpm condition produced a more narrow distribution of aggregate sizes, as presented in Figure 3D. For this reason, together with no significant difference in cell growth between the agitation rates, 80 rpm was used in subsequent experiments, providing more flexibility for additional aggregate growth with greater homogeneity in aggregate size.
3.3. Effect of oxygen and nutrient availability on growth kinetics and metabolism
To further improve hiPSC growth kinetics, we studied the effects of dissolved oxygen content and nutrient availability through a factorial design experiment. Cells were cultured in normoxic conditions at 21% O2, and in hypoxic conditions at 3% O2. To manipulate nutrient availability, cells were cultured in batch conditions, and in fed‐batch conditions where a 50% medium change was performed on day 4. Preformed aggregates were inoculated into PBS vertical‐wheel bioreactors at 80 rpm. The growth curves, shown in Figure 4A, were similar with initial lag phase and overlapping growth up to day 4 when batch‐feeding was performed. Static controls (matrigel‐coated T‐flasks seeded at 20 000 cells/mL) were included for an initial growth platform comparison, shown in Figure S2A. Under 21% O2, batch conditions, hiPSC cultured in static reached a maximum fold expansion of 9.51 ± 0.38 on day 5. This is comparable to the fold expansion reached on day 5 in the vertical‐wheel bioreactor cultured under 21% O2, batch conditions (13.33 ± 1.22). While the exponential growth rate between the two culture platforms is similar, there is a reduced lag phase present in bioreactor culture. After day 5, hiPSCs cultured in static become surface area limited and die off, whereas hiPSCs cultured in the vertical‐wheel bioreactor continue to grow. In the vertical‐wheel bioreactors, the 21% O2 conditions achieved the greatest growth, resulting in maximum fold expansions of 31.63 ± 1.92 (fed‐batch) and 16.01 ± 0.41 (batch), compared with 3% O2 conditions where the growth plateaued around day 4 giving maximum fold expansions of 12.48 ± 0.96 (fed‐batch) and 9.23 ± 1.22 (batch). This is shown along with the corresponding apparent growth rates and doubling times in Figure 4C. The aggregate growth and morphological health depicted in the phase contrast images in Figure 4D are reflective of the growth kinetic patterns. On day 3, there are no noticeable differences in aggregate morphology between normoxic and hypoxic culture conditions. On day 6, the normoxic aggregates continued to grow, with the fed‐batch condition producing the largest aggregates. In comparison, the aggregates in hypoxic conditions appeared to be disintegrating. The hypoxic batch condition was primarily composed of single cells, and the hypoxic fed‐batch aggregates were smaller and possessed rough edges. With the promising results of the normoxic fed‐batch condition, we examined additional samples from day 6 by chromosome analysis and immunocytochemistry. Metaphases obtained from the expanded hiPSCs demonstrated a normal karyotype at the level of resolution achieved, as shown in Figure 4B. Our dissociated aggregates were successfully stained with the pluripotency surface marker TRA‐1‐60, presented in Figure 4E.
FIGURE 4.
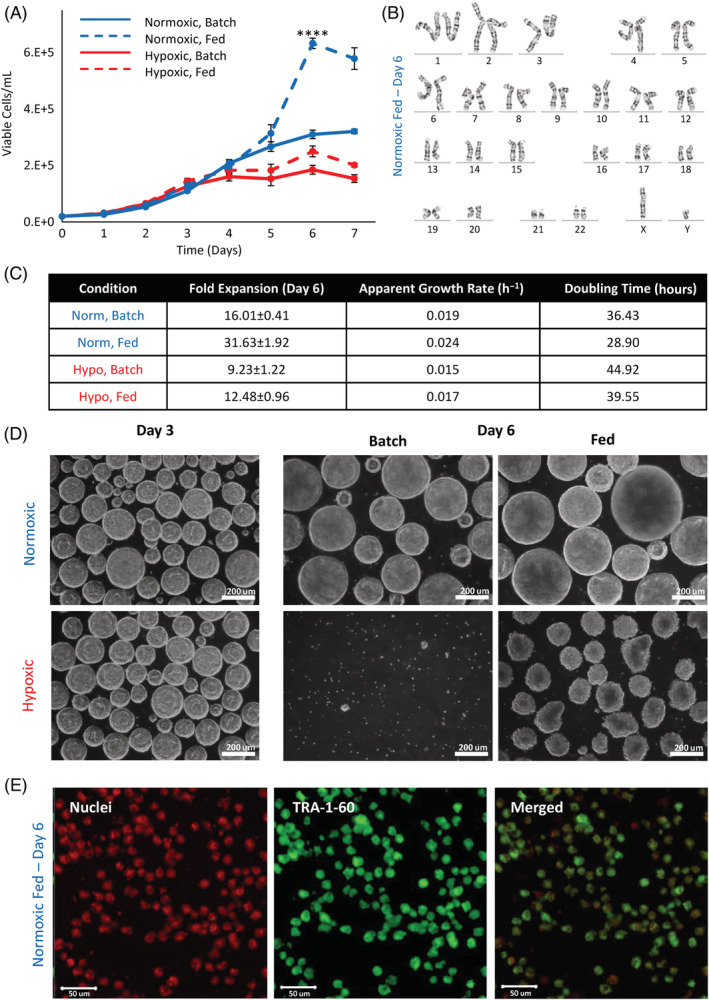
A, Cell growth of hiPSCs cultured in vertical‐wheel reactors under normoxic (21% O2) or hypoxic (3% O2) ambient oxygen conditions with no media exchange (batch) or a 50% media exchange on day 4 of culture (fed). B, Karyogram analysis of hiPSCs cultured for 6 days in the vertical‐wheel reactor under normoxic, fed conditions. C, Corresponding growth kinetic values and, D, phase contrast microscope images of hiPSCs cultured in vertical‐wheel reactors under the various oxygen (normoxic or hypoxic) and nutrient (batch or fed) conditions. E, TRA‐1‐60 confocal staining of hiPSCs cultured for 6 days in the vertical‐wheel reactor under normoxic, fed conditions. Statistical significance equivalent to ****P ≤ .0001. hiPSCs, human induced pluripotent stem cells
To assess the effects of available oxygen and nutrients on metabolic pathway activity, untargeted HPLC‐MS was performed for all samples. Metabolite peaks were selected from the raw dataset by measurable amplitude above the baseline. For the dilution used, 49 metabolites were identified, shown in Figure S1. Of these 49 metabolites, six provided evidence to suggest distinct utilization of metabolic pathways dependent on oxygen availability. Glucose and glutamine levels remained relatively consistent between conditions, shown in Figure 5A,B. On day 4, levels of glucose and glutamine for the fed‐batch conditions were significantly greater than batch conditions then decreased such that there was no difference between conditions on days 5 to 7. This indicates that the nutrients were rapidly consumed by the cells in the exponential growth phase, contributing to the significant increase in expansion for fed‐batch conditions. Detectable pyruvate levels were also similar, indicating that glycolysis occurred at similar rates for all conditions, shown in Figure 5C. Lactic acid production was markedly increased in the hypoxic conditions, with significantly reduced levels observed in normoxia batch and fed‐batch conditions, shown in Figure 5D. The reduced lactic acid observed in 21% O2 conditions, coupled with increased cell growth suggest that the cells used an aerobic pathway alternative to anaerobic fermentation to meet their energetic demands. Alpha‐ketoglutarate and succinate, which are key metabolites in aerobic respiration, were also evaluated as shown in Figure 5E,F. Significantly higher levels of both metabolites were observed in normoxic (21% O2) cultures, again suggesting utilization of aerobic respiration to a much greater extent than in hypoxia (3% O2). The pH of all bioreactor conditions remained above 7.3 for the first 3 days of culture, as shown in Figure S2B. While the pH of the normoxic (21% O2) fed‐batch bioreactor culture remained above 7.0 throughout the 6 days of culture, the pH of the hypoxic (3% O2) batch bioreactor culture fell to 6.3 by day 6 of culture. As a pH between 7.0 and 7.4 is considered within healthy range for hiPSC culture, a single 50% media exchange on day 4 of normoxic (21% O2) culture was deemed sufficient. 34
FIGURE 5.
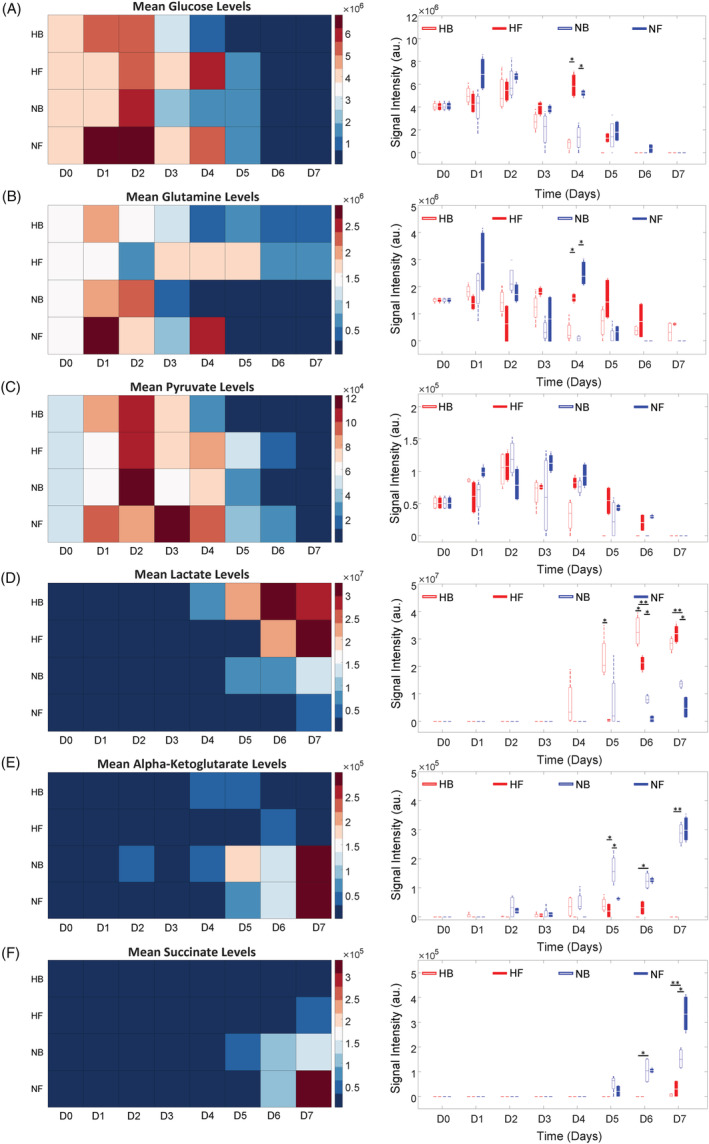
Heat map and signal intensity levels of, A, glucose, B, glutamine, C, pyruvate, D, lactate, E, alpha‐ketoglutarate, and, F, succinate levels taken from hiPSCs cultured in vertical‐wheel bioreactors for 7 days under normoxic (N) or hypoxic (H) ambient oxygen conditions with no media exchange, B, or a 50% media exchange on day 4 of culture, F. Statistical significance equivalent to *P ≤ .05, **P ≤ .01. hiPSCs, human induced pluripotent stem cells
3.4. Vertical‐wheel bioreactor serial passaging growth kinetics and quality testing
To explore the long‐term maintenance of hiPSCs expanded as aggregates, we studied the growth kinetics throughout several serial passages in PBS vertical‐wheel bioreactors. We implemented the successes from previous experiments using normoxic fed‐batch conditions at 80 rpm to culture hiPSCs from passages 42 to 45 over the course of 28 days, where the hiPSCs first occurrence in suspension culture was designated as P1 (passage 42). Comparing the growth between different passages, P1 and P3 demonstrated similar growth rates involving feeding after 4 days of culture and were passaged after 6 days of culture when cell growth was beginning to plateau, as shown in Figure 6A. An increased lag phase was observed for P2 and P4 cultures, where an additional feeding was implemented on day 6 and the culture period was extended to 8 days. Each passage achieved a maximum cell density over 6E5 cells/mL while maintaining viability over 85%, as shown by the dashed lines in Figure 6A. At the end of each passage, morphologically healthy aggregates were produced, as shown in Figure 6D. Maximum fold expansions between 30 and 34 were achieved each passage, as presented in Figure 6B. To account for the fold expansion over the total culture period, the cumulative multiplication ratio and theoretical total hiPSCs produced were calculated, shown in Figure 6C. At the end of the 28‐day culture period, we obtained a cumulative multiplication ratio of 1.06E6, resulting in over 2E12 hiPSCs when a vial of 2E6 cells were used as a starting cell population (and if all cells were passaged each culture period).
FIGURE 6.
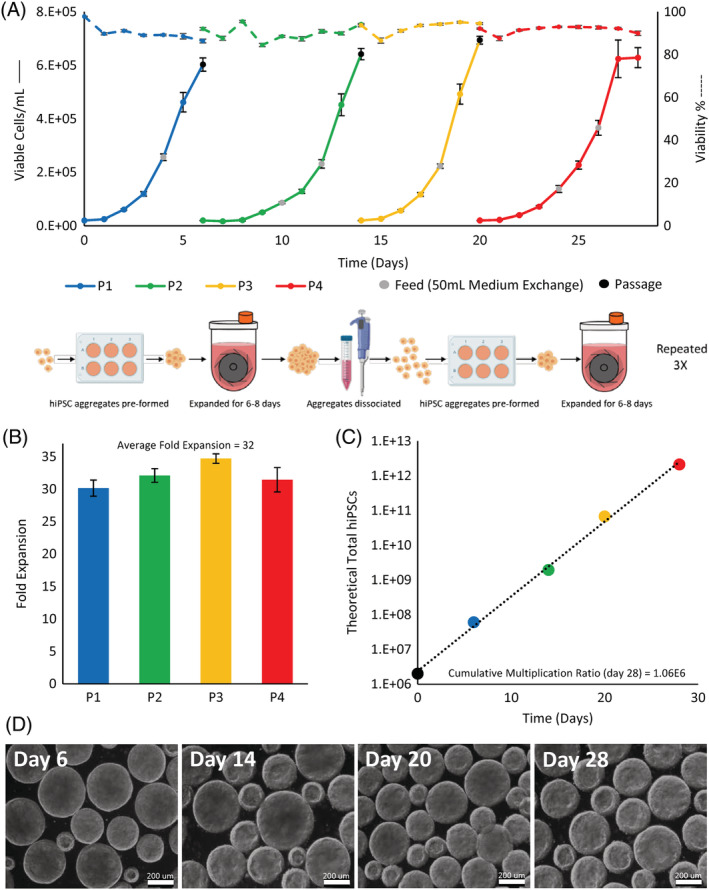
A, Cell growth and viability of hiPSCs cultured over four serial passages in vertical‐wheel reactors (total of 28 days). B, Fold expansion calculated using the maximum cell density from each passage and, C, theoretical total hiPSCs generated over the serial passage period. D, Aggregate phase contrast microscope images taken at the end of each bioreactor passage. hiPSCs, human induced pluripotent stem cells
Additional samples were collected at the end of the 28‐day culture period to assess genomic stability and to determine phenotypic and functional pluripotency quality. Following the multiple serial passages in the vertical‐wheel bioreactors, the hiPSCs presented with a normal chromosome complement, as shown in Figure 7C. Through immunocytochemistry, we demonstrated that the cells maintained expression for the important pluripotency markers TRA‐1‐60 and Nanog, shown in Figure 7A, and flow cytometry results demonstrated positive expression of SSEA‐4 (98.1 ± 0.93) and Oct‐4 (98.1 ± 1.07), shown in Figure 7B. Teratoma analysis, shown in Figure 8A to D, demonstrated that the hiPSCs retained full pluripotency after bioreactor culture. Tissues from all three germ layers—ectoderm, endoderm, and mesoderm—were found to be present.
FIGURE 7.
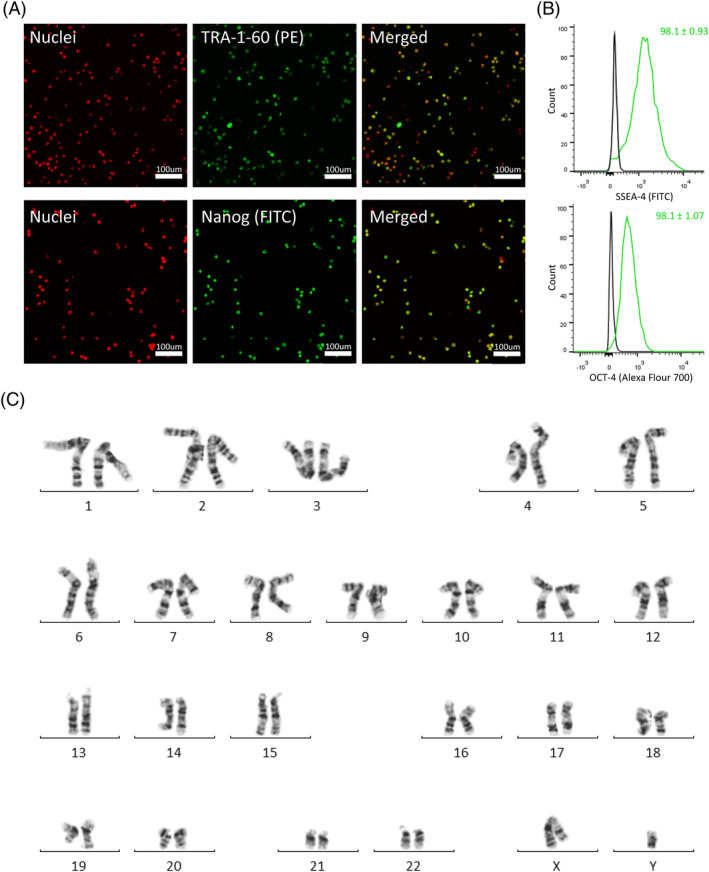
A, Confocal images of hiPSCs stained with pluripotency markers TRA‐1‐60 and Nanog, B, representative flow cytometry histograms of hiPSCs stained for SSEA‐4 and Oct‐4, and, C, karyogram analysis of hiPSCs taken from the final day of the vertical‐wheel bioreactor serial passage (day 28). hiPSCs, human induced pluripotent stem cells
FIGURE 8.
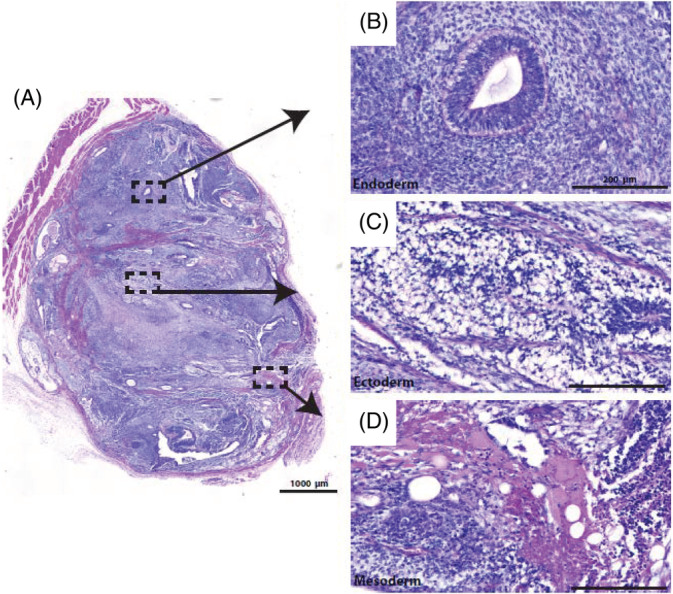
A, Teratomas show tissue types from all three germ layers: B, endoderm; C, ectoderm; D, mesoderm
4. DISCUSSION
PSCs are capable of unlimited proliferative capacity invitro and the ability to differentiate into any cell type of the body makes them an ideal cell source for applications including disease modeling, drug discovery, and cell therapy. Recently, hiPSCs have provided an important alternative to hESCs, overcoming ethical barriers associated with cell source, and the potential to reduce the risk of immune rejection by providing an autologous treatment option. 6 , 35 The potential of hiPSC‐based therapies, however, depends on the successful translation of laboratory culture protocols into clinically compliant manufacturing strategies. Approximately 1010 to 1012 cells per treatment are necessary for most applications, requiring large‐scale stem cell production and the development of robust manufacturing practices and quality testing. 12 Bioreactors provide an attractive solution to address the bioprocess bottleneck of large‐scale stem cell production. In addition to the advantage of scalability, bioreactors allow for process monitoring, control, and reproducibility which contribute to the quality, safety, and efficacy of clinical applications of stem cells. 7 , 12 , 36 While the suspension culture of human pluripotent stem cells (hPSCs) has been studied extensively using rocking platforms 37 , 38 and traditional horizontal‐blade, stirred suspension bioreactors, 17 , 19 , 22 , 23 , 24 , 25 the use of vertical‐wheel bioreactors for culture of hPSCs has not been studied in great detail. 33 , 39 The geometry of the scalable, single‐use bioreactor consists of a large vertical‐wheel with pitched inner blades that rotate along the vertical plane about a horizontal axis. 40 The system combines radial and axial flows that provide agitation and particle suspension with a more uniform distribution of hydrodynamic forces. This results in efficient mixing, reduced shear rates and power input compared to traditional horizontal‐blade, stirred suspension bioreactors. 33 , 40 It was, therefore, our hypothesis that hiPSC expansion as uniform aggregates from low inoculation densities could be greatly enhanced using the novel vertical‐wheel bioreactor.
hiPSCs can be cultured as suspended aggregates, where cell‐cell adhesion and secretion of endogenous extracellular matrix (ECM) stimulates attachment and growth. 8 Many groups that have successfully cultured PSCs as aggregates emphasize that aggregate size is an important parameter to control and can affect cell health, phenotype, and differentiation potential. 3 , 8 , 17 , 26 , 41 , 42 We aimed to develop a protocol to preform aggregates prior to inoculation into bioreactors, allowing for low inoculation density seeding. The initial generation and size control of aggregates is important in maintaining long‐term culture, greatly affecting the quality of the final cell product. 8 Our method for the generation of preformed aggregates was developed by seeding single cells into nonadherent suspension wells in the incubator to allow cells to cluster and form clumps. To optimize this method, we explored the effects of seeding density per well and incubation time. Our results indicated that longer incubation time had the greatest effect on the success of aggregate formation and resulted in enhanced proliferation. We achieved a 14‐fold expansion in 6 days batch culture when aggregates were preformed at a density of 200 000 cells/cm2 for 4 hours and inoculated into horizontal‐blade, stirred suspension bioreactors. This is comparable to 11‐ to 13‐fold expansion in 5 days achieved by Meng et al 24 where the group optimized a variety of culture conditions in the same bioreactors. It should be noted that aggregate preformation in suspension well plates complicates the overall protocol scalability. A scalable protocol to harvest a seed train bioreactor culture and then passage the harvested cells into a larger bioreactor should be further developed and optimized. The aggregates grown in the seed train bioreactor could be partially dissociated into small clumps, which are then passaged into a larger bioreactor without the aggregate preformation process outside the bioreactor. Alternatively, it may be practical to dissociate the aggregates grown in the seed train bioreactor into single cells for harvest, which are then passaged into a larger bioreactor in which they are allowed to form aggregates either under no agitation or at a low seeding agitation rate during an initial time period. Then, once small sizes of aggregates are formed, a normal culture agitation rate is implemented to support cell growth while controlling the aggregate sizes at a desired range. Developing and optimizing a scalable harvest/passage protocol should require a systematic study to investigate several process variables to assess their effect on cell harvest and subsequent cell growth and aggregation formation in the passaged culture.
We then translated the optimized preforming aggregate protocol into vertical‐wheel bioreactors. Although growth kinetics between the different agitation rates (40, 60, and 80 rpm) were not significantly different, different aggregate size distributions were observed. We chose to continue further experiments using the agitation rate of 80 rpm due to the narrow size distribution obtained, and to select for a smaller average aggregate size. Aggregate size homogeneity is an important parameter in establishing a reproducible cell product, influencing cell phenotype, differentiation potential, and overall cell quality. 3 , 43 Smaller aggregates are also desired to limit physiochemical gradients throughout the aggregates thereby producing uniform cell populations. Above 200 to 350 μm in diameter nutrient and oxygen diffusion into the aggregates can be limited, resulting in cell necrosis. 3 , 8 Additionally, for practical reasons, smaller aggregates are preferred for ease of frequent dissociation in controlling aggregate sizes over long‐term culture. 3 , 8 , 26
Once we established a baseline in culturing hiPSCs as aggregates in the vertical‐wheel bioreactors, we explored the effects of dissolved oxygen and nutrient availability to improve the overall growth kinetics. Low oxygen culture conditions have been used to successfully improve growth of hPSCs, 26 , 41 suggesting that reduced oxygen concentrations are more representative of physiological levels. 12 At 3% O2, our hiPSCs grew similar to 21% O2 conditions up to day 4 of culture, when cell growth reached a plateau under reduced oxygen conditions. Following 6 days of culture in hypoxic conditions, the batch culture aggregates disintegrated into single cells and the fed‐batch aggregates appeared morphologically unhealthy with reduced size and rugged edges. It is possible that as the aggregates became larger (past day 4 of culture), further oxygen diffusion was required to penetrate to the center. Likely, the oxygen level experienced by the cells at the center was reduced below 3% resulting in an environment unable to sustain cell proliferation and function. In contrast, cells cultured at 21% O2 resulted in superior growth with the fed‐batch condition achieving a 32‐fold expansion in 6 days. This was almost triple the 11‐ to 13‐fold expansion achieved by Meng et al in the optimized horizontal‐blade, stirred suspension bioreactor study. 24 This success was achieved with a single 50% medium replacement on day 4. Imposing minimal intervention and resource requirements has significant implications on minimizing the cost of the process when translating it to a larger scale. Designing an economical manufacturing process is a significant hurdle in the successful translation of cell therapies to clinics and markets. In comparison, static hiPSC culture requires a daily medium exchange, and other published hiPSC bioreactor studies use 80% to 100% daily media exchanges or perfusion systems following 48 hours of culture, 17 , 19 , 22 , 25 , 26 proving much more resource intensive and costly. It should be noted that in our study, a medium replacement was implemented on day 4 of culture; however, we observed a decrease in growth rate between days 3 and 4. We could potentially offset this decreased growth rate by performing the medium replacement a day earlier to achieve our target cell density in a shorter timeframe. Our metabolic analysis suggests that the significantly higher cell growth observed in 21% O2 conditions is largely due to the improved energy production capabilities of the cell population in the presence of sufficient oxygen to undergo aerobic respiration. This environmental conditioning is counter to the physiological niche that early embryo cells experience in vivo, and their behavior in vitro catalyzes greater questions about the overall phenotype of the cell populations. The literature suggests that PSCs stringently manage metabolic pathway activity to regulate histone modification, reactive oxygen species generation, and precursor production for biosynthetic demands within their niche. 22 Cytogenetic analysis demonstrated that the cells cultured in 21% O2 maintain a normal karyotype. Hypoxic (3% O2) conditions within the reactor give rise to high levels of lactic acid, which remain high even after a 50% medium exchange. Coupled with a significant decrease in pH, the acidity of the environment contributes to changes in cellular activity. Clearly, maintaining the cell population in a state of ideal metabolic pathway utilization would provide the greatest cell number while maintaining the desired phenotype of the cells. Further work should be done to characterize how metabolic activity influences pluripotent gene expression and drives interactions within the regulatory networks of the cell population.
To fully explore the potential of translating hiPSCs toward clinical applications and applying bioprocessing scale‐up techniques, serial passages will be necessary to maintain long‐term cultures and to manufacture clinically relevant numbers of cells. We endeavored to apply our optimized expansion strategies to successfully culture hiPSCs over four serial passages spanning over 28 days. While final cell concentration was consistent between passages and cell viability remained high, an increased lag phase was observed in passage 2 and passage 4 of bioreactor culture. As cells are harvested at the end of exponential growth phase, glucose levels are reduced, and precursor metabolites become undetectable. This information suggests that the epigenetic activity of the cells is at a major transition point. It is possible that passaging several hours earlier or later could alter the lag characteristics in the following passage, as the behavior of the cells is transitioning to compensate for nutrient depletion at the end of the exponential phase. This is something that should be considered and carefully observed to further increase passage to passage reproducibility. Each culture period obtained an average 32‐fold expansion achieving a cumulative multiplication ratio of 1.06E6 and theoretically producing a clinically relevant total of 2E12 hiPSCs. This was achieved with the small starting population of 2 million cells (20 000 cells/mL). Nearly all published hiPSC bioreactor studies, summarized in Table 1, require large cell seeding densities (2 × 105 to 1 × 106 cells/mL) that must be cultured through a lengthy and resource intensive static expansion phase prior to bioreactor inoculation. With hiPSC reprogramming efficiencies still extremely low (between 0.2% and 1.0%), the smaller the cell quantity required for the manufacturing and expansion phase the better. 44 The ability to (a) achieve clinically relevant cell numbers in a short period of time (b) starting from a small population and (c) with minimal resource requirements offers significant implications for the potential of translating manufacturing of hiPSCs to clinic and market. Additionally, the quality of our hiPSCs was not sacrificed during serial passaging and prolonged culture. It should be noted that this study was completed with a single hiPSC line, therefore differences in population dynamics are possible for other hiPSC lines. After 28 days in suspension culture, the hiPSCs maintained in vivo differentiation potential and normal karyotypes despite the high cell passage number (passage 42‐45). This has been an industrial challenge, especially for higher passage number hiPSCs. A major concern in the application of hiPSC‐derived products is the risk of tumorigenicity, as PSCs can accumulate karyotypic abnormalities with lengthened culture periods. 2 , 35 To translate their potential toward clinical applications, hiPSC products will need continuous, rigorous screening for genetic alterations and instability that is innate from their development. By selecting for an optimal hydrodynamic environment and applying frequent dissociation, we were able to manage the growth and size of our hiPSC aggregates in vertical‐wheel bioreactors to prevent uncontrolled differentiation associated with larger heterogeneous aggregate populations.
5. CONCLUSIONS
To fully explore the potential of translating human iPSCs toward clinical applications, it is likely that serial passages will be necessary to maintain long‐term cultures and manufacture clinically relevant numbers of cells. We have developed bioprocessing strategies to successfully expand high‐quality hiPSCs that maintain genomic stability at the chromosomal level, phenotypic and functional pluripotency characteristics in single‐use, vertical‐wheel bioreactors. This study focused on optimizing inoculation, agitation, oxygen, and nutrient availability for the culture of hiPSCs as aggregates. Our optimized protocol uses preformed aggregates seeded at low inoculation densities in defined, serum‐free medium. Under optimized conditions, we achieved an expansion of more than 30‐fold in 6 days using a small starting population of cells with minimal media resources throughout. Importantly, we demonstrated that this optimized bioreactor expansion protocol could be replicated over four serial passages resulting in a cumulative cell expansion of 1.06E6‐fold in 28 days. This development of a robust bioprocess for the efficient expansion of high‐quality hiPSCs will pave the way to translating production to clinical and manufacturing settings.
6. CONFLICT OF INTEREST
Y.H. and S.J. are employees of PBS Biotech, Inc. B.L. is CEO and cofounder of PBS Biotech, Inc. These collaborating authors participated in the development of the bioreactors used in this article as well as the experimental concept design and data review. PBS Biotech, Inc. provided financial support for the researchers to complete the study. This does not alter the authors' adherence to all the policies of the journal. All other authors declared no conflicts of interest.
7. AUTHOR CONTRIBUTIONS
B.S.B., T.S.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; J.C.: collection and/or assembly of data, data analysis and interpretation, manuscript writing; T.D.: collection and/or assembly or data, manuscript writing; E.L.R., T.R.: collection and/or assembly of data, data analysis and interpretation; L.L.: collection and/or assembly of data; R.K.: conception and design, data analysis and interpretation, financial support, final approval of manuscript; I.L., B.A., D.E.R., S.J., Y.H., B.L., M.S.K.: conception and design, financial support, final approval of manuscript.
8.
Supporting information
Supplementary Figure 1 Organization of the metabolites studied in a global heatmap. Columns represent signal intensity for all metabolites in a single sample. Rows are grouped by culture replicate
Supplementary Figure 2 A) Fold expansion over 7 days of batch culture in static and bioreactor conditions B) Change in culture pH over 6 days of culture in the vertical‐wheel bioreactor system
ACKNOWLEDGMENTS
We thank Laurie Kennedy and Yiping Liu at the Flow Cytometry Core Facility for their assistance. This project was funded by the NSERC Discovery Grant Program and Collaborative Health Research Projects (NSERC Partnered). B. S. B. was funded by the Vanier Canada Graduate Scholarship Program. T. R. acknowledges the support of the Alberta Children's Hospital Research Institute Clinical Research Fellowship. We thank the Stem Cell Network for their generous and continued support in trainee development. Some graphics used in the Figures 1 and 6 were created with biorender.com. Metabolomics data were acquired by Ryan Groves at the Calgary Metabolomics Research Facility (CMRF), which is supported by the International Microbiome Centre and the Canada Foundation for Innovation. I. L. is supported by an Alberta Innovates Translational Health Chair.
Borys BS, So T, Colter J, et al. Optimized serial expansion of human induced pluripotent stem cells using low‐density inoculation to generate clinically relevant quantities in vertical‐wheel bioreactors. STEM CELLS Transl Med. 2020;9:1036–1052. 10.1002/sctm.19-0406
Funding information Alberta Children's Hospital Research Institute Clinical Research Fellowship; Vanier Canada Graduate Scholarship Program; Collaborative Health Research Projects (NSERC Partnered); NSERC Discovery Grant Program
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Takahashi K, Yamanaka S. Induced pluripotent stem cells in medicine and biology. Development. 2013;140:2457‐2461. [DOI] [PubMed] [Google Scholar]
- 2. Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16:115‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kropp C, Massai D, Zweigerdt R. Progress and challenges in large‐scale expansion of human pluripotent stem cells. Process Biochem. 2017;59:244‐254. [Google Scholar]
- 4. Scudellari M. How iPS cells changed the world. Nature. 2016;534:310‐312. [DOI] [PubMed] [Google Scholar]
- 5. Steiner D, Khaner H, Cohen M, et al. Derivation, propagation and controlled differentiation of human embryonic stem cells in suspension. Nat Biotechnol. 2010;28:361‐364. [DOI] [PubMed] [Google Scholar]
- 6. Revilla A, González C, Iriondo A, et al. Current advances in the generation of human iPS cells: implications in cell‐based regenerative medicine. J Tissue Eng Regen Med. 2016;10:893‐907. [DOI] [PubMed] [Google Scholar]
- 7. Liu N, Zang R, Yang S‐T, Li Y. Stem cell engineering in bioreactors for large‐scale bioprocessing. Eng Life Sci. 2014;14:4‐15. [Google Scholar]
- 8. Sart S, Bejoy J, Li Y. Characterization of 3D pluripotent stem cell aggregates and the impact of their properties on bioprocessing. Process Biochem. 2017;59:276‐288. [Google Scholar]
- 9. Guhr A, Kobold S, Seltmann S, Seiler Wulczyn AEM, Kurtz A, Löser P. Recent trends in research with human pluripotent stem cells: impact of research and use of cell lines in experimental research and clinical trials. Stem Cell Reports. 2018;11:485‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stoddard‐Bennett T, Pera RR. Treatment of Parkinson's disease through personalized medicine and induced pluripotent stem cells. Cells. 2019;8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hu Y, Tian Z, Zhang C. Natural killer cell‐based immunotherapy for cancer: advances and prospects. Engineering. 2019;5:106‐114. [Google Scholar]
- 12. Serra M, Brito C, Correia C, Alves PM. Process engineering of human pluripotent stem cells for clinical application. Trends Biotechnol. 2012;30:350‐359. [DOI] [PubMed] [Google Scholar]
- 13. Ludwig TE, Levenstein ME, Jones JM, et al. Derivation of human embryonic stem cells in defined conditions. Nat Biotechnol. 2006;24:185‐187. [DOI] [PubMed] [Google Scholar]
- 14. Reubinoff BE, Pera MF, Fong C‐Y, Trounson A, Bongso A. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat Biotechnol. 2000;18:399‐404. [DOI] [PubMed] [Google Scholar]
- 15. Richards M, Fong C‐Y, Chan W‐K, Wong PC, Bongso A. Human feeders support prolonged undifferentiated growth of human inner cell masses and embryonic stem cells. Nat Biotechnol. 2002;20:933‐936. [DOI] [PubMed] [Google Scholar]
- 16. Rodrigues CAV, Fernandes TG, Diogo MM, da Silva CL, Cabral JMS. Stem cell cultivation in bioreactors. Biotechnol Adv. 2011;29:815‐829. [DOI] [PubMed] [Google Scholar]
- 17. Zweigerdt R, Olmer R, Singh H, Haverich A, Martin U. Scalable expansion of human pluripotent stem cells in suspension culture. Nat Protoc. 2011;6:689‐700. [DOI] [PubMed] [Google Scholar]
- 18. Olmer R, Lange A, Selzer S, et al. Suspension culture of human pluripotent stem cells in controlled, stirred bioreactors. Tissue Eng Part C Methods. 2012;18:772‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abbasalizadeh S, Larijani MR, Samadian A, Baharvand H. Bioprocess development for mass production of size‐controlled human pluripotent stem cell aggregates in stirred suspension bioreactor. Tissue Eng Part C Methods. 2012;18:831‐851. [DOI] [PubMed] [Google Scholar]
- 20. Wang Y, Chou B‐K, Dowey S, He C, Gerecht S, Cheng L. Scalable expansion of human induced pluripotent stem cells in the defined xeno‐free E8 medium under adherent and suspension culture conditions. Stem Cell Res. 2013;11:1103‐1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Elanzew A, Sommer A, Pusch‐Klein A, Brüstle O, Haupt S. A reproducible and versatile system for the dynamic expansion of human pluripotent stem cells in suspension. Biotechnol J. 2015;10:1589‐1599. [DOI] [PubMed] [Google Scholar]
- 22. Haraguchi Y, Matsuura K, Shimizu T, Yamato M, Okano T. Simple suspension culture system of human iPS cells maintaining their pluripotency for cardiac cell sheet engineering. J Tissue Eng Regen Med. 2015;9:1363‐1375. [DOI] [PubMed] [Google Scholar]
- 23. Badenes SM, Fernandes TG, Rodrigues CAV, Diogo MM, Cabral JMS. Microcarrier‐based platforms for in vitro expansion and differentiation of human pluripotent stem cells in bioreactor culture systems. J Biotechnol. 2016;234:71‐82. [DOI] [PubMed] [Google Scholar]
- 24. Meng G, Liu S, Poon A, Rancourt DE. Optimizing human induced pluripotent stem cell expansion in stirred‐suspension culture. Stem Cells Dev. 2017;26:1804‐1817. [DOI] [PubMed] [Google Scholar]
- 25. Kwok CK, Ueda Y, Kadari A, et al. Scalable stirred suspension culture for the generation of billions of human induced pluripotent stem cells using single‐use bioreactors. J Tissue Eng Regen Med. 2018;12:e1076‐e1087. [DOI] [PubMed] [Google Scholar]
- 26. Abecasis B, Aguiar T, Arnault É, et al. Expansion of 3D human induced pluripotent stem cell aggregates in bioreactors: bioprocess intensification and scaling‐up approaches. J Biotechnol. 2017;246:81‐93. [DOI] [PubMed] [Google Scholar]
- 27. Cormier JT, zur Nieden NI, Rancourt DE, et al. Expansion of undifferentiated murine embryonic stem cells as aggregates in suspension culture bioreactors. Tissue Eng. 2006;12:3233‐3245. [DOI] [PubMed] [Google Scholar]
- 28. Zur Nieden NI, Cormier JT, Rancourt DE, et al. Embryonic stem cells remain highly pluripotent following long term expansion as aggregates in suspension bioreactors. J Biotechnol. 2007;129:421‐432. [DOI] [PubMed] [Google Scholar]
- 29. Borys BS, Le A, Roberts EL, et al. Using computational fluid dynamics (CFD) modeling to understand murine embryonic stem cell aggregate size and pluripotency distributions in stirred suspension bioreactors. J Biotechnol. 2019;304:16‐27. [DOI] [PubMed] [Google Scholar]
- 30. Shafa M, Sjonnesen K, Yamashita A, et al. Expansion and long‐term maintenance of induced pluripotent stem cells in stirred suspension bioreactors. J Tissue Eng Regen Med. 2012;6:462‐472. [DOI] [PubMed] [Google Scholar]
- 31. Fernandes‐Platzgummer A, Diogo MM, Lobato da Silva C, Cabral JMS. Maximizing mouse embryonic stem cell production in a stirred tank reactor by controlling dissolved oxygen concentration and continuous perfusion operation. Biochem Eng J. 2014;82:81‐90. [Google Scholar]
- 32. Croughan MS, Giroux D, Fang D, et al. Novel single‐use bioreactors for scale‐up of anchorage‐dependent cell manufacturing for cell therapies. Stem Cell Manuf. 2016;105‐139. [Google Scholar]
- 33. Rodrigues CA, Silva TP, Nogueira DE, et al. Scalable culture of human induced pluripotent cells on microcarriers under xeno‐free conditions using single‐usevertical‐wheel™ bioreactors. J Chem Technol Biotechnol. 2018;93:3597‐3606. [Google Scholar]
- 34. Dakhore S, Nayer B, Hasegawa K. Human pluripotent stem cell culture: current status, challenges, and advancement. Stem Cells Int. 2018;2018:7396905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neofytou E, O'Brien CG, Couture LA, Wu JC. Hurdles to clinical translation of human induced pluripotent stem cells. J Clin Invest. 2015;125:2551‐2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat Rev Mol Cell Biol. 2016;17:194‐200. [DOI] [PubMed] [Google Scholar]
- 37. Ting S, Chen A, Reuveny S, Oh S. An intermittent rocking platform for integrated expansion and differentiation of human pluripotent stem cells to cardiomyocytes in suspended microcarrier cultures. Stem Cell Res. 2014;13:202‐213. [DOI] [PubMed] [Google Scholar]
- 38. Davis BM, Loghin ER, Conway KR, Zhang X. Automated closed‐system expansion of pluripotent stem cell aggregates in a rocking‐motion bioreactor. SLAS Technol. 2018;23:364‐373. [DOI] [PubMed] [Google Scholar]
- 39. Nogueira DES, Rodrigues CAV, Carvalho MS, et al. Strategies for the expansion of human induced pluripotent stem cells as aggregates in single‐useVertical‐Wheel™ bioreactors. J Biol Eng. 2019;13:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sousa MFQ, Silva MM, Giroux D, et al. Production of oncolytic adenovirus and human mesenchymal stem cells in a single‐use, Vertical‐Wheel bioreactor system: impact of bioreactor design on performance of microcarrier‐based cell culture processes. Biotechnol Prog. 2015;31:1600‐1612. [DOI] [PubMed] [Google Scholar]
- 41. Jenkins MJ, Farid SS. Human pluripotent stem cell‐derived products: advances towards robust, scalable and cost‐effective manufacturing strategies. Biotechnol J. 2015;10:83‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Otsuji TG, Bin J, Yoshimura A, et al. A 3D sphere culture system containing functional polymers for large‐scale human pluripotent stem cell production. Stem Cell Reports. 2014;2:734‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Borys BS, Roberts EL, Le A, et al. Scale‐up of embryonic stem cell aggregate stirred suspension bioreactor culture enabled by computational fluid dynamics modeling. Biochem Eng J. 2018;133:157‐167. [Google Scholar]
- 44. Anokye‐Danso F, Trivedi CM, Juhr D, et al. Highly efficient miRNA‐mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 2011;8:376‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Organization of the metabolites studied in a global heatmap. Columns represent signal intensity for all metabolites in a single sample. Rows are grouped by culture replicate
Supplementary Figure 2 A) Fold expansion over 7 days of batch culture in static and bioreactor conditions B) Change in culture pH over 6 days of culture in the vertical‐wheel bioreactor system
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.