

Abstract
Introduction:
The postmortem examination still represents the reference standard for detecting the pathological nature of chronic neurodegenerative diseases (NDD). This approach displays intrinsic conceptual limitations since NDD represent a dynamic spectrum of partially overlapping phenotypes, shared pathomechanistic alterations that often give rise to mixed pathologies.
Areas covered:
We scrutinized the international clinical diagnostic criteria of NDD and the literature to provide a roadmap toward a biomarker-based classification of the NDD spectrum. A few pathophysiological biomarkers have been established for NDD. These are time-consuming, invasive, and not suitable for preclinical detection. Candidate screening biomarkers are gaining momentum. Blood neurofilament light-chain represents a robust first-line tool to detect neurodegeneration tout court and serum progranulin helps detect genetic frontotemporal dementia. Ultrasensitive assays and retinal scans may identify Aβ pathology early, in blood and the eye, respectively. Ultrasound also represents a minimally invasive option to investigate the substantia nigra. Protein misfolding amplification assays may accurately detect α-synuclein in biofluids.
Expert opinion:
Data-driven strategies using quantitative rather than categorical variables may be more reliable for quantification of contributions from pathophysiological mechanisms and their spatial-temporal evolution. A systems biology approach is suitable to untangle the dynamics triggering loss of proteostasis, driving neurodegeneration and clinical evolution.
Keywords: Alzheimer’s disease, biomarkers, cerebral amyloid angiopathy, Parkinson disease, amyotrophic lateral sclerosis
1. Introduction
Proteostasis is a complex interplay of subcellular molecular networks deputed to ensure a dynamic equilibrium between protein folding quality control and clearance machineries.
Experimental models of aging indicate that the structural and functional integrity of each proteostasis network account for longevity and health-span whereas aging is associated with a risk of proteostasis failure and neurodegeneration.
In this regard, NDD associated with proteinopathies represent a broad spectrum of diseases characterized by loss of proteostasis and accumulation of misfolded and aggregated proteins [1]. At present, NDD are classified depending on the type of misfolded protein depositions [2,3].
The gold standard for a definite NDD diagnosis has been postmortem examination since the introduction of the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA [4]) criteria for Alzheimer’s disease (AD), in 1984. Indeed, NDD clinical diagnostic criteria are expected to provide high accuracy compared to their reference standard (essentially autopsy data). Nevertheless, this approach has several practical and conceptual limitations. First, postmortem diagnosis is not applicable to detect NDD in the early or preclinical phases when prospective disease-modifying treatments are potentially more effective. Secondly, this is a final result of in vivo dynamic pathophysiological process acting differently in terms of space and time with undetectable onset and progression. In fact, postmortem examination often reveals composite patterns of multi-proteinopathies associated with diverse premortem clinical phenotypes [5].
NDD are a spectrum, as demonstrated by their overlapping phenotypes, shared pathophysiologies, and mixed pathologies [6,7]. Mounting evidence from autopsy studies shows transactive response DNA-binding protein 43 KDa (TDP-43) and alpha-synuclein (α-syn) co-pathologies in brains of individuals meeting pathological diagnostic criteria for AD. A mixed misfolded deposition of amyloid-beta (Aβ) and α-syn proteins was also described in the central nervous system (CNS) of several amyotrophic lateral sclerosis (ALS) patients [8]. Currently, NDD involve a broad disease spectrum, with classification criteria representing at times limiting artificial boundaries [9,10]. Further, this categorization remains descriptive and mainly focused on the late-stage clinical phenotypic picture, while the underlying pathophysiological processes begin decades prior to symptomatic onset. Relevant exceptions are represented by specific research criteria for preclinical and prodromal AD [11–16] as well as prodromal Parkinson’s disease (PD) [17]. Indeed, from the failure of several disease-modifying drug trials in AD we understood that in order to have a chance to modify disease progression, interventions need to be implemented early.
Starting from a dissertation of the international consensus and diagnostic criteria, our aim is to find pathophysiological commonalities and provide a roadmap toward a comprehensive biomarker-based diagnostic representation of idiopathic chronic progressive NDD. In this regard, we will not provide an exhaustive systematic review of biomarkers developments in NDD. Rather, we seek to critically discuss syndromic and pathological overlaps among NDD, especially focusing on candidate biomarkers that may refine patient stratifications and early detection, or allow screening for targetable pathophysiological pathways, irrespective of clinical diagnosis.
1.1. Alzheimer disease: a heterogeneous clinicopathological entity
AD is the most common pathology associated with dementia and its treatment represents a major health-care challenge. AD is considered a complex, polygenic multifactorial disorder [18]. Autosomal-dominant genetic AD represents <1% of cases (Table 1) and APOE-ε4 is the major established risk factor for sporadic AD [19]. Several large-scale genome-wide association studies (GWAS) and meta-analyzes of GWAS have been executed, in the last decade [20,21]. Moreover, whole-exome sequencing, whole-genome sequencing, and targeted sequencing have led to the identification of contributions from rare genetic variants to late-onset AD [22]. Besides the three identified causal genetic mutations (amyloid precursor protein [APP], presenilin 1 and 2 [PSEN1 and PSEN2]) for autosomal dominant early-onset AD, more than 40 susceptibility genes/loci have been identified for late-onset AD [22–26]. From a pathological perspective, diverse kinds and patterns of changes have been demonstrated including α-syn and TDP-43 depositions in one-third and one-fifth of AD autopsy cases, respectively [27], in addition to varying degrees of cerebrovascular disease, including cerebral amyloid angiopathy (CAA) [28]. However, the pathologic hallmarks of AD remain: a) extracellular neuritic plaques, primarily containing Aβ42 peptide aggregates [29] and b) intraneuronal neurofibrillary tangles, mainly composed of abnormally phosphorylated protein tau (3R/4R tau) [28,30,31].
Table 1.
Diagnostic accuracy of international criteria for chronic neurodegenerative diseases.
| Clinical syndroms | Accuracy of diagnostic criteria* | Estimated prevalence (N/100,000) | Primary pathology | Main genes mutations | Pathophysiological biomarkers | Topographic/Phenotypic biomarkers (typical brain regions of atrophy or hypometabolism) | Candidate pathophysiological biomarkers |
|---|---|---|---|---|---|---|---|
| AD | Sensitivity 81% Specificity 71% [4,32] | 5000 | Amyloid Plaques, NFTs (3R/4R tau) | PSEN1 PSEN2 APP |
Reduced CSF Aβ42 (Aβ42:Aβ40) Increased CSF p-tau and t-tau Increased tracer retention on amyloid PET Increased tracer retention on tau PET |
MRI/FDG-PET: Medial temporal lobe atrophy (hippocampi, entorhinal cortex, amygdala). Hypometabolism in bilateral temporal parietal regions Note: Structural (FDG-PET, DTI) and Functional (fMRI) techniques identify atypical AD phenotypes (fvAD, IvAD, PCA) depending on cerebral areas and networks involved |
Aβ42, Aβ40, Aβ42: Aβ40 (blood) B-Amyloid retina scanning |
| CAA | Sensitivity highly variable Specificity 87.5–100% [71] |
Unknown, suspected high prevalence | Amyloid deposits (mostly Aβ40 peptide) in leptomeningeal/cortical/cerebellar arteries wall and smooth muscle cells | APP CST3 |
NA | MRI: cortical, cortico-subcortical microbleeds, cortical superficial siderosis (T2*MRI), small vessels disease (centrum semiovale), lobar (or cerebellar) hemorrhages | Aβ42, Aβ40, Aβ42: Aβ40 (blood) |
| PD | Sensitivity 91% Specificity 98% [17,76,77] |
1500 | A-synuclein in LBs (mainly midbrain/pons) | SNCA LRRK2 PARK2 PINK1 DJ1 |
NA | SPECT: Nigrostriatal dopaminergic degeneration | Transcranial sonography CSF α-syn (Protein misfolding amplification assays) |
| DLB | Sensitivity 83% Specificity 95% [105,106] |
1000 | A-synuclein in LBs (diffuse in brain) | SNCA LRRK2 |
NA | SPECT: Nigrostriatal dopaminergic degeneration FDG-PET: hypometabolism on occipital regions and sparing of PCC. MIBG myocardial scintigraphy: Low uptake |
Transcranial sonography CSF α-syn (Protein misfolding amplification assays) |
|
bvFTD ~ 60% of FTD |
Sensitivity of 85% Specificity of 95% [142] |
11 (all FTD) | TDP-43 B (50%) 3R tau (40%) FUS (5–10%) |
C90RF72 MAPT GRN Heritability (40–45%) |
NA | MRI/FDG-PET: Frontal and/or anterior temporal atrophy/hypometabolism (frontoinsular region is especially indicative of frontotemporal dementia) | Progranulin (blood) CSF Poly(GP) |
|
PPA nfvPPA ~ 25% of FTD |
NA | 11 (all FTD) 5 |
4R tau | GRN C90RF72 Heritability (5%) |
NA | MRI/FDG-PET: Left posterior frontoinsular atrophy/hypometabolism | Progranulin (blood) CSF Poly(GP) |
| svPPA ~20% of FTD | NA | 4 | TDP-43 C | GRN C90RF72 Heritability (< 1%) |
NA | MRI/FDG-PET: Anterior temporal lobe atrophy/hypometabolism | Progranulin (blood) CSF Poly(GP) |
| PSP | Sensitivity 87.9% Specificity 85.7% [110,111] |
5 | 4R tau (tufted astrocytes) | MAPT | NA | SPECT: Nigrostriatal dopaminergic degeneration MRI/FDG-PET: Midbrain atrophy/hypometabolism |
NA |
| CBD | Unreliable accuracy [123,124] |
Unknown | 4R tau (astrocytic plaques) | NA | NA | (MRI/FDG-PET not included in diagnostic criteria: Asymmetric frontoparietal atrophy) | NA |
| ALS | Sensitivity 57% Specificity 99% [224] | 3 | TDP-43 SOD1 (2%) | C9ORF72 TARDBP FUS SOD1 |
NA | Electrophysiological tests (low motor neuron degeneration) | NFL (blood): massive increase T2*MRI (cortical primary motor hypointensities) |
| MSA | Accuracy 79% [128,129] | 5 | A-synuclein (oligodendrocytes cytoplasmic inclusion) | COQ2 | NA | SPECT: Nigrostriatal dopaminergic degeneration MRI/FDG-PET: Atrophy of putamen, middle cerebellar peduncle, pons, or cerebellum. Hypometabolism in putamen, brainstem, or cerebellum Orthostatic hypotension |
NA |
with postmortem validation as reference standard
Abbreviations: Aβ40= Amyloid-Beta 40; Aβ42= Amyloid-Beta 42; AD= Alzheimer’s Disease; ALS= Amyotrophic Lateral Sclerosis; APP= Amyloid protein precursor; α-syn= alpha-synuclein; BRI-2= type II Transmembrane Protein gene; bvFTD= behavioral variant Frontotemporal Dementia; CAA= Cerebral Amyloid Angiopathy; CBD= Corticobasal Degeneration; C9ORF72= Chromosome 9 Open Reading Frame 72; DLB= Dementia with Lewy Bodies; COQ2= OH-benzoate Polyprenyltransferase gene; CST3= (Cystatin C gene); DJ1= Protein deglycase DJ-1 gene, also known as Parkinson Protein 7 (PARK7) DTI= Diffusion Tensor Imaging; fvAD= frontal variant of Alzheimer’s Disease; FTD= Frontotemporal Dementia; FUS= Fused in Sarcoma DNA-binding protein; GRN= Progranulin (Granulin Precursor); LBs= Lewy bodies; LRRK2= Leucine-Rich Repeat Kinase 2; lvAD= logopenic variant of Alzheimer’s Disease; MAPT= Microtubule Associated Protein Tau; MSA= Multiple System Atrophy; NfL= Neurofilament Light Chain; NFTs= Neurofibrillary Tangles; nfv-PPA= non-fluent variant Primary Progressive Aphasia; PARK2= Parkinson Protein 2 (Parkin) gene; PCA= Posterior Cortical Atrophy; PCC= Posterior Cingulate Cortex; PD= Parkinson’s Disease; PINK1= PTEN Induced Kinase 1 gene; PPA= Primary Progressive Aphasia; PSEN 1= presenilin-1; PSEN 2= presenilin-2; PSP= Progressive Supranuclear Palsy; SNCA= Synuclein Alpha gene; TDP-43= Transactive Response DNA-binding Protein 43 KDa; SOD1= Superoxide Dismutase 1; sv-PPA= semantic-variant Primary Progressive Aphasia.
The typical phenotype in AD is characterized by an early impairment of episodic memory [4], as defined by the NINCDS-ADRDA criteria in 1984 [4]. The clinical diagnosis of probable AD based on this phenotype has a fairly good accuracy (Table 1). Since 2007, the clinical criteria have been revised by the International Working Group (IWG) [13–15] and the National Institute on Aging–Alzheimer Association (NIA-AA) [12,16,32,33]. In particular, progressive refinements led to a simplified clinical diagnosis of AD in the IWG-2 criteria [15]. The previous diagnostic categorical distinction between prodromal AD (alternatively mild cognitive impairment [MCI] due to AD for NIA-AA framework [16]) and AD dementia was replaced by a novel unified categorical diagnostic group irrespective of the severity of cognitive impairment, combined with in vivo evidence of AD pathology [15]. Decreased cerebrospinal fluid (CSF) concentrations of Aβ42 combined with increased hyperphosphorylated tau (p-tau), or total tau (t-tau) proteins are surrogates of AD pathophysiology [12,15,16,32]. An additional pathophysiological biomarker is the increased signal of cerebral amyloid-PET uptake. By contrast, brain atrophy and glucose hypometabolism detected in some cerebral regions measured by brain MRI (e.g. hippocampal volume) and 2-deoxy-2-[fluorine-18]fluoroD-glucose ([18F]-FDG)-PET (e.g. parietotemporal and posterior cuneus areas) are currently considered as later features occurring across AD phenotypes and useful for disease progression monitoring [9,34]. Additional imaging techniques that could refine the identification of AD subtypes and monitor disease progression are Diffusion Tensor Imaging (DTI) MRI sequences and task-free functional MRI [35]. These imaging modalities track different structural and functional patterns of brain network disintegration, respectively [35].
Typical or hippocampal, and atypical AD presentations have been established. The former shows episodic memory impairment fulfilling the original NINCDS-ADRDA, the latter involves different cognitive domains (language, visual, praxis, or executive problems) occurring earlier than memory deficits [24]. The rise in diagnoses of atypical AD phenotypes and mixed AD forms (e.g. evidence of AD and coexisting parkinsonism or cerebrovascular disease) may relate to the integration of pathophysiological biomarkers. The IWG-2 criteria and the NIA-AA guidelines identified: an occipitotemporal variant characterized by a predominant, and progressive impairment of visuoperceptive functions; a biparietal variant, defined by the presence of an early, predominant, and progressive difficulty with visuospatial function; a logopenic variant with impairment of single word retrieval and repetition of sentences; and a frontal variant characterized by behavioral changes such as apathy, behavioral disinhibition, and executive dysfunction [15]. Frontal and logopenic variants are AD phenotypes that can clinically overlap with behavioral variant of frontotemporal dementia (bvFTD) and primary progressive aphasia (PPA) described below. Posterior cortical atrophy (PCA) typically starts at a younger age (before age 60) than typical AD, similarly to the other atypical AD variants [36], with patients exhibiting a progressive impairment in object localization in the visual field among other visual processing deficits; memory decline usually occurs later [36,37]. In 2017, a consensus classification of PCA clinicopathological features was published [37]. Briefly, the early occurrence of visual symptoms alterations is mandatory. Neuroimaging should show mainly occipital, parietal or occipito-parietal bilateral atrophy (MRI) and a specific bilateral parieto-occipital hypometabolism (FDG-PET). In this consensus, the concept of ‘Pure PCA’ vs ‘PCA-plus’ was introduced in order to capture the phenotype of patients that present with typical PCA symptoms while also meeting core criteria for other NDD [37]. CSF Aβ42, t-tau and p-tau levels in ‘pure PCA’ are indistinguishable from those of typical AD [38]. However, ‘pure PCA’ phenotypes may also be associated with non-AD-pathologies, such as Lewy body disease (LBD), corticobasal degeneration (CBD) or even prion disease pathological features [36].
With a wealth of research over the past decade, the definition of AD is gradually shifting from that of a syndrome to a biological construct and continuum [33,39]. The unbiased descriptive categorization proposed by the NIA-AA recognizes three general groups of biomarkers, indicative of amyloid deposition (A), pathologic tau (T), and neurodegeneration (N) (ATN). The committee agreed that only pathophysiological biomarkers should be considered for a consistent AD diagnosis across stages. Notably, the mere presence of amyloid deposition (A+ state) has been now termed as Alzheimer’s pathologic change, within the AD continuum. Cerebral tau-PET was added to the array of biomarkers compared to the previous diagnostic AD criteria [33]. However, although tau-PET ligands (AV1451 tracer) bind preferentially to paired helical filament tau [40], false-positive binding in non-tauopathy disorders such as in frontotemporal dementia (FTD) due to chromosome 9 open reading frame 72 gene (C9ORF72) expansions [41] have been described.
A series of biomarker categories and profiles, independent from clinical symptoms, were developed to chart the different pathological phases across the AD continuum (ATN system). Noteworthy, the ATN biomarker categorization is intentionally open to incorporate novel biomarkers. For instance, α-syn may represent an additional pathophysiological fluid biomarker for AD and can be added to the matrix. In particular, CSF total levels of α-syn (t-α-syn) are higher in AD and prodromal AD compared to both cognitive normal controls and synucleinopathies such as PD, Parkinson’s disease dementia (PDD) and DLB [42]. On the other hand, the oligomeric and phosphorylated forms of α-syn (o-α-syn, and p-α-syn) are significantly reduced in AD patients compared to synucleinopathies [42]. A recent meta-analysis showed that α-syn measurement using standard ELISA methods does not achieve significant diagnostic accuracy in discriminating AD from synucleinopathies [42]. Innovative techniques such as Real Time quaking-induced conversion test assay (RT-QuIC [43]) and the Protein misfolding cyclic amplification (PMCA [44]) (these techniques amplify and detect α-syn proteins in CSF and brain extracts conceptually as polymerase chain reaction fragment analysis) are promising tools for the identification of subgroups of AD individuals with α-syn co-pathology, namely AD patients [43] with Lewy body variant.
Despite an increased accuracy of up to 90% for the diagnosis of AD using core biomarkers, all disease-modifying therapies of AD dementia have failed [45]. Therefore, early recruitment into clinical trials and early treatment of preclinical AD with altered pathophysiological biomarkers may well enhance the probability of success in modifying the disease course.
As a consequence, minimally invasive and globally accessible screening tools with high negative predictive value in individuals at high risk of AD could help to exclude the majority of subjects not needing more complex evaluations [46]. Noninvasive and cost-effective examinations such as retinal imaging or peripheral blood biomarker analyses may be used for the initial broad screening phase within a multi-step diagnostic process to diagnose and differentiate NDD. If made reproducible, their being easily repeatable, make them ideal biomarkers of disease progression and response monitoring to disease-modifying treatments. Growing evidence indicates that AD is not confined to the brain but also affects the retina [47]. Inspired by the successful detection of the amyloid pathological hallmarks in the retina and given the accessibility, safety, and low cost of high-resolution noninvasive retinal imaging [47–51], an in vivo approach for visualizing Aβ deposits in living subjects have been recently developed [52–54] (Figure 1). Indeed, similar to brain plaques, retinal Aβ deposits are more frequent in AD patients as compared with controls, and their burden correlates tightly with brain-plaque burden [50]. Since 2016 advanced high-sensitive techniques [55] have significantly improved the detection of peripheral plasma/serum biomarkers for NDD. In particular, several promising results from independent study groups indicate that blood Aβ peptides, alone or in combination, and blood p-tau have diagnostic accuracy to detect AD pathophysiology almost to a comparable extent as more sophisticated and expensive amyloid-PET investigations [56–59], as early as during the preclinical stage of subjective memory complaints [60]. In addition, peripheral blood neurofilament light chain (NFL), a marker of axonal degeneration, is evolving to be a robust candidate biomarker to detect early AD-related neurodegenerative progression well discriminating AD subjects from controls (AUROC = 0.87) [61]. High baseline plasma NFL concentration could be predictive of subsequent cognitive decline in the preclinical phase of AD [62] and is also potentially useful as a prognostic biomarker since faster increase in NFL levels have been associated with worsening in cognitive score in MCI subjects [63]. In this regard, blood NFL is promising, in prospective, as monitoring biomarker in disease-modifying trials.
Figure 1.

Identification of Aβ plaques and vascular amyloidosis in postmortem retinas from Alzheimer’s disease patients. A-D. Representative micrographs of retinal flatmounts of AD patients relative to age- and sex-matched cognitively normal (CN) controls, immunostained against Aβ42 (DAB-12F4). AD patients’ retinas accumulate both abluminal (B, B’) and vascular (D) Aβ42-containing deposits. E. Quantitative immunohistochemical analysis of retinal 12F4+Aβ42 plaques in AD patients vs. CN controls. Adopted from Koronyo et al. JCI Insight 2017 with permission of ASCI via Copyright Clearance Center.
1.2. Cerebral amyloid angiopathy: when neurodegeneration starts from circulation
CAA is the main cause of primitive lobar hemorrhage, and the risk of recurrence varies, on average, between 7% and 12% per year. In autopsy studies, these are detected in 20–40% of non-demented and in 50–60% of demented elderly patients, coexisting with AD up to 85–95% [64]. Attention has been focused on amyloid perivascular drainage impairment and, therefore, an inefficient clearance (glymphatic system) related to the integrity of the brain microcirculation or astrocytes [65,66]. Ultimately, the interstitial fluid blockage can lead to the enlargement of perivascular Virchow-Robin spaces frequently associated also with deep small vessels disease [67].
The clinical history of a patient affected by CAA can include lobar cerebral (or sometimes, cerebellar) hemorrhages (ICH) and post-hemorrhagic dementia, cognitive decline or vascular dementia without ICH, in addition to transient neurological focal deficits (amyloid spells). The latter can mimic transient ischemic attacks leading physicians to administer antithrombotic drugs [68].
Most CAA cases are sporadic. Rarely, causative genes related to the amyloid precursor proteins may be recognized (Table 1) [69]. APOE polymorphisms seem to be associated with different disease subtypes: ε4 with non-ICH forms and abundance of neurofibrillary tangles, ε2 with cortical siderosis and lobar hemorrhages [68,70].
The clinical diagnosis of CAA requires meeting the Boston criteria [71]. These indicate that specific cerebrovascular disease patterns, detected with brain MRI using particular sequences (e.g., Gradient Echo (GE) T2*, Susceptibility Weighted Imaging (SWI)), distinguish CAA from the hypertensive arteriopathy (or non-amyloid microangiopathy). Actually, patients affected with CAA are typically found to harbor strictly lobar cerebral microbleeds (CMB), essentially posterior white matter hyperintensities (WMH), cortical superficial siderosis (CSS), and enlarged perivascular spaces in centrum semiovale. Importantly, cerebral lacunes in deep white matter are rare. By contrast, the typical CMB due to hypertensive arteriopathy are localized in basal ganglia and frequently in combination with multiple ischemic lacunes [72]. CSS is usually absent, and WMH are variably distributed throughout the brain.
The original Boston criteria have been recently modified to increase diagnostic accuracy in vivo [71]. Substantially, they differ from the original ones, especially when classifying probable from possible CAA, considering the presence of CSS in addition to the ICH included in the old criteria. The standard diagnosis remains essentially postmortem neuropathology or analyzing tissue samples from evacuated ICH. When validated in clinical practice in a hospital-based or population-based MRI-neuropathology studies, the original criteria have an optimal specificity but a low sensitivity that increases with the revised criteria [71] (Table 1). Most importantly, a diagnostic challenge emerges when neurologists investigate the etiology of a single lobar hemorrhage without CMB or an isolated siderosis (criterium of ‘possible’ CAA). This has relevant therapeutic implications especially whether starting (or not) an anticoagulant therapy, in the presence of atrial fibrillation. The cerebral PET with 18F-Florbetapir or C-Pittsburgh compound B (PiB) showed a sensitivity of 79% and a specificity of 78% [73] in a meta-analysis of 106 cases and 151 controls (healthy or typical hemorrhages). Another meta-analysis including CAA cases, controls, and AD patients reported a greater distribution rate of the tracer in the occipital regions in CAA compared to AD but not to controls [74]. Notably, study results indicated that amyloid-PET tracers do not significantly distinguish CAA from AD cases.
Currently, there are no reliable fluid biomarkers supporting a CAA diagnosis. The majority of studies have investigated AD related core pathophysiological pathways due to existing commonalities. In particular, a recent meta-analysis [75] showed that the Aβ40/Aβ42 ratio was significantly lower, while t-tau was higher in CAA compared to controls. Moreover, CSF Aβ40 concentrations were lower in CAA compared to AD patients whereas Aβ42, t-tau, and p-tau were comparable. In conclusion, AD and CAA have largely overlapping CSF core biomarkers profiles. We could not find significant data on specific blood diagnostic biomarkers. High-sensitivity techniques measuring blood Aβ40/Aβ42 in AD patients, however, demonstrated good diagnostic accuracy in predicting cerebral amyloid load compared to cerebral amyloid-PET and CSF biomarkers concentrations [56,58]. Eventually, given the pathological similarities between CAA and AD, we suggest the design of specific studies measuring Aβ40 and Aβ42 peptides in CAA.
1.3. Parkinson’s disease
PD is the second most prevalent neurodegenerative disease next to AD and is associated with progressive loss of dopaminergic neurons in the pars compacta of the substantia nigra (SN). A minority of cases have a genetic cause (Table 1).
The presence of a parkinsonian syndrome is the clinical hallmark of PD. According to current diagnostic criteria [76], parkinsonism is defined by the presence of bradykinesia with either rest tremor or rigidity. Early diagnosis of PD may be challenging but supportive criteria, absolute exclusion criteria, and ‘red flags’ were proposed to improve diagnostic accuracy. In particular dopaminergic treatment responsiveness and levodopa-induced dyskinesia onset support PD diagnosis. Using UK Parkinson’s Disease Society Brain Bank clinical diagnostic criteria [77], clinical diagnosis was confirmed in 76% of autopsy cases [78], whereas applying revised criteria [17,79] an optimal accuracy of 90% was obtained [80]. In the elderly, concomitant AD pathology was reported in 48.9% of cases [81].
In the last decades, non-motor symptoms and signs of PD gained momentum and were used to support diagnosis in a novel proposal of diagnostic criteria. The onset of non-motor features may precede motor symptoms and a few clinical non-motor markers were included in the research diagnostic criteria for prodromal PD [17]. In particular, prodromal REM sleep behavior disorder (RBD) proven with polysomnography, and olfactory loss are associated with a high likelihood of future PD. Prodromal criteria were recently validated in a longitudinal study on RBD subjects: 39.7% of individuals converted to PD/DLB at 4-year follow-up with 81.3% sensitivity and 67.9% specificity for conversion [82].
In addition to clinical biomarkers, neuroimaging and biochemical biomarkers have been developed. Normal functional neuroimaging of the presynaptic dopaminergic system (assessed with PET or SPECT) represents an absolute exclusion criterion for PD [76] whereas a clearly abnormal tracer uptake was included in the list of PD prodromal biomarkers. Conventional brain MRI excluded secondary parkinsonism, but advanced techniques might improve the diagnostic workup of degenerative parkinsonism. Noteworthy, the use of SWI with 3T and 7T MRI allowed visualization of SN anatomy, differentiating PD patients from controls with an accuracy of 86% and 96%, respectively [83]. In a large longitudinal cohort of healthy subjects, transcranial ultrasound identified SN hyperechogenicity at baseline in 14 out of 17 who subsequently developed PD [84]. Overall, transcranial sonography represents a potential screening tool to support clinical diagnosis (sensitivity = 84% and specificity = 85%), according to a recent meta-analysis [85].
Pathological biomarkers are lacking, but several molecules are currently under investigation. A-syn and NFL might have a potential role in differential diagnosis between PD and controls, but also between PD and atypical parkinsonism (AP). Regarding CSF AD biomarkers, lower CSF Aβ42 levels are considered a potential predictor of cognitive decline in longitudinal studies [86]; recent multicenter data from the Parkinson’s Progression Markers Initiative (PPMI) cohort would confirm these results [87]. Moreover, reduced CSF Aβ42 baseline levels could be an independent predictor for early L-dopa-resistant gait impairment and psychosis [88,89].
CSF α-syn (oligomeric or total) is a candidate biomarker for all synucleinopathies [90]. The current consensus is that CSF α-syn levels are lower in PD than controls and similar to multiple system atrophy (MSA) and DLB [91]. A meta-analysis [92] confirmed the significantly lower CSF t-α-syn levels and higher CSF concentration of both o-α-syn and p-α-syn in PD patients compared to controls. Any consistent differences in CSF t-α-syn levels were demonstrated between PD and DLB as well as PD and MSA patients [93]. According to previous findings, some authors hypothesize that CSF α-syn measured with traditional ELISA techniques is unlikely to be a diagnostic biomarker [94]. In addition, conflicting results were obtained investigating its potential role as a prognostic biomarker for both motor and cognitive progression [95–97].
A meta-analysis [98] reported that CSF NFL is higher in AP than PD and has an almost optimal accuracy in discriminating PD from AP (AUROC = 0.89). These results were confirmed in a larger review and meta-analysis [99] reporting overlapping CSF NFL concentrations among PD, PDD, DLB, and control groups, and increased levels in AP.
Mounting data indicate that blood NFL measurement may be an effective and minimally invasive biomarker to discriminate among parkinsonian syndromes: a class III study demonstrated that blood NFL levels discriminate between PD and AP with comparable accuracy to CSF levels of these proteins. Moreover, MSA, progressive supranuclear palsy (PSP), and patients with corticobasal syndrome (CBS) are found to have higher blood NFL levels compared to PD and controls [100]. Results from a Class II study also confirmed that serum NFL concentrations provide an excellent accuracy in discriminating PD from controls (sensitivity = 86% and specificity = 85%) and charted a tight correlation with the CSF NFL levels as reported in other studies on different NDD. According to overall results serum NFL levels differentiate AP from PD with high accuracy, supporting its use in the diagnostic workup of parkinsonian syndromes. Furthermore, at 3-year follow-up serum NFL levels showed a negative association with both motor and cognitive performances, measured as tandem gait test, H&Y score, and MMSE, respectively, suggesting a potential prognostic value of NFL [101].
1.4. Lewy body dementia
DLB belongs to the clinical spectrum of Lewy bodies (LB) disorders, characterized by accumulation of pathogenic α-syn in the brain. However, concomitant AD pathology can be detected in up to 40% of patients with PD, PDD, and DLB clinical diagnoses [102–104].
Dementia is the main clinical feature, with memory impairment usually not prominent in early stages, with deficits mainly affecting attention, executive functions, and visuoperceptual abilities. Core clinical features include fluctuating cognition, recurrent visual hallucinations, RBD, and features of parkinsonism. The onset of parkinsonism should be concomitant or follow the onset of dementia. The so-called 1-year rule between dementia and parkinsonism onset is currently recommended, although controversial, to discriminate DLB and PDD. Supportive clinical features include among others sensitivity to antipsychotic agents, postural instability, syncope or transient unresponsiveness, delusions [105].
A limited number of ‘indicative biomarkers’ are reported in the recently revised criteria [105,106]: the reduced dopamine transporter uptake in basal ganglia (demonstrated with SPECT or PET), the reduced uptake on meta-iodo-benzyl-guanidine (MIBG) myocardial scintigraphy, and a polysomnographic diagnosis of RBD. Medial temporal lobe structural preservation on CT/MRI scan, reduced FDG-PET hypometabolism in occipital region and/or cingulate gyrus (island sign), and EEG abnormalities characterized by prominent posterior slowwave activity with periodic fluctuations in the pre-alpha/theta range are defined as only ‘supportive biomarkers’ (Table 1).
Results from CSF studies have confirmed the relevant role of AD pathology in DLB patients. Levels of t-tau, p-tau, and Aβ42/Aβ38 ratio can be helpful in the differential diagnosis between DLB and AD, whereas, similarly to PD, low CSF Aβ42 levels predict the development of cognitive impairment. In a large study on DLB patients, low baseline levels of CSF Aβ42 were associated with a more rapid cognitive decline, measured with the MMSE (2-year follow-up). CSF t-α-syn levels [107] are overlapping in PD and MSA patients, but lower than in controls [104]. Most interestingly, CSF t-α-syn levels are higher in AD [42] compared to PD, PDD, and DLB individuals [91,108]. Recently, RT-QuIC and PMCA showed an average accuracy of 80% in distinguishing PD and DLB patients from controls. CSF NFL levels are similar to those measured in PD, PDD, and controls but lower compared to MSA, PSP, and CBS [99].
1.5. Atypical parkinsonism
Early differential diagnosis between PD and AP can be challenging. A retrospective postmortem study evaluated accuracy of in vivo clinical diagnosis performed by general neurologists: in the 75.3% of cases diagnosis of PD was confirmed (sensitivity = 89.2% and specificity = 57.8%) whereas lower accuracy was reported for PSP and MSA (sensitivity 52.9% and 64.3%, and specificity 100% and 99.0%, respectively). The overall results showed an overdiagnosis of PD compared to movement disorder specialists [109].
The most common PD mimics are MSA and PSP while the lowest diagnostic accuracy accounts for CBS.
1.5.1. Progressive supranuclear palsy
PSP is a rare sporadic 4-repeats (4R) tauopathy with onset >40 years and chronic progression. Diagnostic criteria were recently revised [110,111] since the previous National Institute of Neurological Disorders and Stroke and the Society for Progressive Supranuclear Palsy (NINDS/SPSP) criteria have been published in 1996 [112] which had excellent specificity for the most classical phenotype of PSP but low sensitivity for the atypical ones in light of the broad spectrum of phenotypes in PSP. Four functional domains (ocular motor dysfunction, postural instability, akinesia, and cognitive dysfunction) have been identified as clinical predictors. The presence of predominant clinical features suggestive for a different type of dementia (e.g. impairment of episodic memory) or parkinsonism (e.g. autonomic failure or visual hallucinations) are considered mandatory exclusion criteria. Additional clinical cues include levodopa-resistance, hypokinetic/spastic dysarthria, dysphagia, and photophobia.
Supportive features are predominant midbrain atrophy (MRI) or hypometabolism (FDG-PET) and post-synaptic striatal dopaminergic degeneration (SPECT/PET) (Table 1). The use of combined MRI indexes like the magnetic resonance parkinsonism index (MRPI) [113] improved differential diagnosis between PD and PSP, but with variable accuracy among studies [114–116]. Advanced MRI techniques such as the use of SWI for qualitative evaluation of SN failed to distinguish between PD and AP [117]. More encouraging results were obtained with quantitative techniques (QSM) [118,119].
In the current Movement Disorder Society (MDS) criteria fluid biomarkers help in excluding alternative NDD. Hence, the observation of a typical AD CSF pattern can help to distinguish patients with primary AD neuropathology that can mimic PSP.
Similarly to other AP, PSP patients have higher serum NFL levels compared to controls [120], thus strengthening its role as diagnostic biomarker in differentiating PD from AP. Interestingly, a positive association with motor/cognitive symptoms progression has been also reported. Indeed, high baseline NFL concentrations were associated with a subsequent faster motor and cognitive decline [121], as well as a shorter survival [120]. These evidences support an intriguing prognostic role of serum NFL for PSP patients.
1.5.2. Corticobasal syndrome
CBS is included in the group of 4R-tauopathies (Table 1). The term CBS is used to denote a syndrome, whereas CBD refers to pathologically confirmed cases. Misdiagnosis rate with previous criteria was extremely high [122]. The current diagnostic criteria [123], based on pathologically proven cases, identified four main phenotypes, according to the new concept of a spectrum of taurelated NDD with overlapping phenotypes: CBS, frontal behavioral-spatial syndrome, the non-fluent/agrammatic variant of PPA (nfvPPA) and the PSP syndrome (PSPS).
Clinical features of CBS include the asymmetric presentation of limb rigidity or akinesia, limb dystonia, limb myoclonus, possibly associated with orobuccal or limb apraxia, cortical sensory deficit, alien limb phenomena. These criteria were subsequently applied to clinical cases with postmortem examination and failed to demonstrate an improved in vivo diagnostic accuracy compared to the previous one, with only 47% of cases fulfilling criteria for CBS at presentation, and 68% at the final follow-up [124]. Differential diagnosis can be challenging mainly regarding PSP and also AD where CBS phenotype has been reported in up to 3.5% of pathologically proven cases [125]. Structural/functional imaging biomarkers may support clinical diagnosis but with limited improvement of diagnostic accuracy [126].
Among biofluid biomarkers CSF NFL levels are higher in CBS than in PD [99]. Unfortunately, CSF and blood NFL remain unspecific surrogates of AP since they can distinguish CBS from PD, but do not allow to discriminate one AP subtype from one other [127].
1.5.3. Multiple system atrophy
Current diagnostic criteria define MSA [128] as a rare NDD included in the group of synucleinopathies (Table 1).
Diagnosis of probable MSA requires the presence of autonomic failure involving urinary incontinence, with erectile dysfunction in males, or orthostatic decrease of blood pressure within 3 min of standing (at least 30 mmHg systolic or 15 mmHg diastolic). Symptoms must be associated with a poorly levodopa-responsive parkinsonism or with a cerebellar syndrome. Cerebellar (c-MSA) and parkinsonian (p-MSA) MSA phenotypes share few clinical features, especially Babinski sign with hyperreflexia, and stridor. P-MSA is essentially a rapidly progressive parkinsonism with poor response to levodopa, and c-MSA a cerebellar syndrome characterized by gait/limbs ataxia, dysarthria, and oculomotor dysfunction. The main features supporting MSA diagnosis include orofacial dystonia, postural deformities, inspiratory sighs, severe dysphonia/dysarthria, snoring [129].
Cerebral MRI and FDG-PET imaging features suggestive for MSA are reported in Table 1. Overall, conventional MRI improved accuracy compared to clinical diagnosis but suboptimal sensitivity values were obtained in pathologically confirmed cases (76.9% radiological vs 61.5% clinical diagnosis) [130]. No specific morphometric indices are currently available. The use of advanced sequences like SWI-MRI was proposed to improve differential diagnosis but qualitative evaluation of SN failed to discriminate MSA patients [131]. A neuropathological study confirmed a frequent misdiagnosis with DLB (37%), PSP (29%), and PD (15%) cases [132].
As mentioned for other AP, CSF NFL levels are significantly higher than in PD and controls [99]. Noteworthy c-MSA reported higher blood NFL concentrations compared to sporadic adult-onset ataxia, an MSA mimic (AUROC = 0.74).
1.6. The frontotemporal dementia spectrum
The term FTD indicates a group of clinically, genetically and pathologically heterogeneous NDD which invariably progress to dementia and typically are associated with misfolded protein aggregates in neural cells and atrophy of frontal and/or anterior temporal lobes. It is a relatively common cause of dementia, being the second most common cause of young-onset dementia after AD [133] (Table 1). A family history of dementia occurs up to 40% of all cases of FTD, although a clear autosomal-dominant transmission is present in only 10% of patients [134]. The most common genetic causes of FTD currently known are represented by mutations, in order of frequency, of C9ORF72 gene, microtubule-associated protein tau (MAPT) gene, and of granulin precursor (GRN) gene [135].
Three broad clinical presentations of FTD are recognized: bvFTD, PPA(s), and the motor-FTD syndromes detailed above (CBD syndromes and PSP). The PPAs are subdivided on the basis of the prevalent language deficit involved in nfvPPA, and semantic-variant PPA (svPPA). As noted above, the logopenic variant (lvPPA) is an atypical AD subtype, rather than FTD [136]. The most common clinical type of FTD is bvFTD, covering over half of the cases [137]. Differently from PPA where a quite fair relation between phenotype and underlying pathology (nfvPPA with FTD-tau and svPPA with FTD-TDP) has been described, bvFTD has a poor phenotype/pathological association [138].
Three main pathological subtypes are identified [35]. FTD with TDP-43 aggregates (FTD-TDP) is the most frequent, genetically associated with the C9ORF72 and GRN mutations. There is a clinical, genetic, and pathological heterogeneity in TDP-43 proteinopathies. On this basis, four TDP-43 aggregates (A, B, C, and D) have been distinguished [139]. Type A cases are associated with bvFTD or nfvPPA, along with mutations in GRN. Type B cases with bvFTD including motor neuron disease (MND) and mutations of C9ORF72. Type C cases with bvFTD or svPPA are generally sporadic. Finally, the type D cases are associated with the mutation of the valosine-containing protein (VCP) gene [140]. FTD with tau depositions (FTD-Tau), also referred to as Pick’s disease (3R) or PSP and CBD (4R) cover about 40% of all cases of FTD [141]. Tau is abnormally hyperphosphorylated, dissociated from microtubules, and prone to form aggregates within neurons and glia. Genetic forms are linked to MAPT mutations. FTD with FUS aggregates represents a remaining 5–10% of cases [35].
1.6.1. The behavioral variant of FTD
According to the new diagnostic criteria, the main clinical core features of bvFTD include the early appearance of behavioral disinhibition, apathy or inertia, loss of sympathy or empathy, perseverative/stereotyped or compulsive/ritualistic behavior, hyperorality and dietary changes, executive deficits with relative sparing of memory and visuospatial functions. For the diagnosis of probable bvFTD, at least three of these symptom groups are required [142].
Typically, FTD patients have a low insight into their symptoms [136] and early functional impairment [143], related to prominence in behavioral deficits. BvFTD usually progresses faster than AD [144], and decreased survival is associated often with MND [145].
Genetic testing and neuroimaging are currently available tools to confirm the clinical diagnosis [136]. Conventional MRI and cerebral FDG-PET support a bvFTD diagnosis though they do not identify a specific underlying pathology (Table 1). Functional imaging is more sensitive than structural MRI in detecting early pathological changes in bvFTD and FDG-PET should be used in the early diagnosis of bvFTD for its high negative predictive value (0.78) [146,147]. Conversely, amyloid-PET tracers can help to distinguish FTD from atypical variants of AD (frontal and logopenic variants) [148].
Despite the recent revision of the diagnostic criteria improved accuracy (Table 1) [142,149], the diagnosis of bvFTD remains challenging in the early phases of the late-onset bvFTD where specificity remains low [150]. BvFTD shows significant overlap with other NDD and with several psychiatric disorders. Notably, around 12.5% of bvFTD patients [151] develop signs of MND [152,153], and around 20% parkinsonian syndromes [154], especially CBS and PSP [155], suggesting common underlying pathophysiological pathways. Moreover, early bvFTD is often misdiagnosed as a psychiatric disorder, especially depression, bipolar disorder, and schizophrenia [156], or with personality disorders (borderline, antisocial, and schizoid personality [136]) when these have late onset [157]. Psychiatric disorders fulfilling the core clinical diagnostic criteria of bvFTD are classified as frontotemporal dementia phenocopies. These cases are not progressive and with normal brain MRI and PET findings [158]. In other cases, bvFTD phenocopies may also show a very slow-progressive cognitive impairment that makes challenging the FTD diagnostic workup [159]. Eventually, they may represent a slow sporadic or genetic form of bvFTD [159].
Recent imaging studies reported encouraging results for an earlier and more accurate diagnosis although not at the level of the individual subject. DTI sequences measuring the white matter integrity are an interesting biomarker of structural brain networks and potentially more sensitive than traditional MRI sequences in detecting early and subtle FTD changes. Actually, white matter alterations precede gray matter atrophy involving different brain regions depending on the clinical FTD subtype [160]. The white matter fibers most involved in bvFTD are the uncinate fasciculus, cingulum bundle, and genu of the corpus callosum [161,162]. Interestingly, a greater loss of integrity of the white matter has been found in the presence of tau rather than TDP-43 pathology [163]. F-MRI investigates functional network connectivity and has revealed very early changes in presymptomatic mutation carriers, even up to 10 years before the expected onset [164,165]. The patterns of functional network connectivity identified differences among bvFTD, AD patients and controls [166]. More precisely, reduced connectivity in frontoinsular and/or anterior cingulate cortex, part of the so-called ‘salience network,’ is the most common finding in patients with bvFTD [167].
Currently, reliable fluid biomarkers are not yet clinically available to significantly improve bvFTD diagnosis. The combination of CSF p-tau, t-tau proteins, Aβ peptides, and their ratios only rules out the frontal variant of AD [168,169]. Nevertheless, several fluid biomarkers are under investigation for bvFTD [170,171]. NFL is the most promising candidate biomarker for FTD, especially for disease monitoring and prognosis [160,171]. Recent evidence proved that blood and CSF levels of NFL are tightly related and significantly higher in FTD subjects than in controls [172]. This difference is more pronounced for bvFTD subtypes [173,174], and NFL concentrations are associated with disease severity, progression, and survival [175,176]. Moreover, some evidence show that NFL levels are similar in presymptomatic FTD individuals and healthy controls while they increase during the symptomatic stage [177]. In the symptomatic phase, NFL increases in parallel with brain atrophy progression and the development of bvFTD symptoms [178]. CSF NFL concentrations have been shown to be higher in patients harboring TDP-43 compared to tau [177]. CSF NFL concentrations were also higher in bvFTD compared to both AD [179,180] and psychiatric subjects [181]. Noteworthy, the best discrimination may be with non-NDD. Indeed, compared to controls, blood NFL was found to be significantly higher in concentration in patients with bvFTD but not in patients with psychiatric disease [181]. The strong correlation between blood and CSF NFL levels makes blood NFL a suitable screening biomarker tool for FTD and a potential disease monitoring proxy [182].
Regarding other fluid biomarkers, gene-specific biomarkers are the most robust. Progranulin and poly(GP) are the most studied, potentially useful in the differential diagnosis between tau and TDP-43 subtypes [183]. Poly(GP) is an abnormal dipeptide repeat protein detectable only in the CSF of C9ORF72 mutation carriers, that develop the TDP-43 pathologic subtype of FTD (TDP type B). Most importantly, given that poly(GP) is negative in patients with tau pathology, it may play a role in stratifying FTD into distinct pathophysiological pathways. However, poly(GP) is present only in the symptomatic phase of FTD with very modest correlation with neurodegeneration [184].
CSF and plasma progranulin concentrations are lower (around 25–40%) in GRN mutation carriers compared to healthy controls and FTD non-mutation carriers, and allow to discriminate mutation carriers from non-carriers with high sensitivity (96–100%) and specificity (93–100%) [185–189]. This biomarker is associated with TDP-43 pathology (TDP type A). Hence, progranulin may be a reliable gene-specific biomarker for GRN mutation carriers and [171] could help in screening of familiar cases, in treatment monitoring of GRN mutation carriers and finally in the discrimination of FTD pathologic subtypes [160].
1.6.2. Primary progressive aphasia
PPAs are rare [190] clinical syndromes resulting from the selective neurodegeneration in the cerebral networks responsible for language control localized in the language-dominant cerebral hemisphere (the left hemisphere up to 90% of cases).
International consensus guidelines on PPA assume a 2-steps clinical diagnosis supported by imaging or associated with definite pathology at postmortem examination or definite pathogenic mutation [191,192]. First, core criteria for PPA require: (a) a subtle and progressive language impairment, which should be (b) the main source of interference in performing the activities of daily living (for at least the first 1–2 years), (c) isolated in the initial phase and prominent throughout the whole course of the disease.
Three main clinical variants can be identified according to variant-specific patterns of language impairment: nfvPPA, svPPA, and lvPPA. A small fraction of cases are labeled as ‘unclassifiable PPA’ or ‘mixed PPA’ [191]. These include the PPA phenotypes arising from genetic mutations, such as GRN mutation, that often do not fit in the diagnostic boxes made initially for sporadic disease.
In brief, language assessment requires evaluation of speech production features (grammar, motor speech, sound errors, and word-finding pauses), repetition, single-word and syntax comprehension, confrontation naming, semantic knowledge, and reading/spelling.
Essential features of nfvPPA are at least one of the following: (a) agrammatism, namely shortening and simplification of phrases, misuse of grammatical morphemes like articles and conjunctions, and (b) effortful speech, which results slow and labored, with inconsistent speech sound errors and distortions (apraxia of speech). Individuals may eventually develop parkinsonism, either in the form of PSP or CBS [193]. Nevertheless, generalized motor symptoms should be excluded from the nfvPPA diagnosis. Atrophy at MRI or FDG-PET hypometabolism at the level of left posterior fronto-insular region (inferior frontal gyrus particularly) is mandatory (Table 1). Regarding the underlying pathology, nfvPPA is the most heterogeneous of the three PPA variants, being FTD-tau pathology the most commonly associated (from 50% to 88% of the cases), followed by FTD-TDP (8–31%, type A more frequently), and AD pathology (4–19%) [194–197]. Pathogenetic mutations in FTDassociated genes (C9ORF72, GRN, MAPT) have been found in approximately 5% of nfvPPA cases [198], although more frequently subjects with these mutations show a bvFTD phenotype [134].
A diagnosis of svPPA requires the presence of both anomia and impaired single-word comprehension. Impairment in naming is usually severe, particularly if compared to relative sparing of other language features such as phrase repetition and speech production. Single-word comprehension deficit typically involves unfamiliar or atypical items, at least at the beginning. For imaging supported diagnosis see Table 1 [191,199]. This variant has been associated with TDP-43 pathology (67–100%) usually of type C [194,196,197,200]. Genetic forms of svPPA (GRN, MAPT, C9ORF) are more rarely reported than in nfvPPA (1.6%) [198].
LvPPA, related to AD neuropathology rather than FTD pathological entities, is the most recently defined of the three variants [201], resulting from phonological loop dysfunction. Core clinical features need both (a) impaired single-word retrieval in spontaneous speech and naming and (b) impaired repetition of sentences and phrases. For imaging supported diagnosis, brain MRI and FDG-PET should demonstrate atrophy and/or hypometabolism, respectively, in left posterior perisylvian or parietal areas [191,194,202]. Very importantly, patients with lvPPA show an AD pathology in most of the postmortem cases reported (54–100%) [194–197]. Hence, IWG-2 criteria recently included lvPPA due to AD pathology among the atypical presentations of AD [15]. Genetic forms (GRN, TARDBP) have been recognized very rarely in lvPPA (1.1%) [198].
Patterns of language impairment essentially reflect areas of cerebral dysfunction (and the relative control on specific language aspects) rather than specific underlying neuropathology. Nevertheless, the preferential association of each PPA phenotype with recurrent pathologic patterns indicates a specific vulnerability of certain brain areas and networks to different misfolded protein accumulations [203].
Specific diagnostic biomarkers are necessary. Currently, only core biomarkers of AD pathology [197,204–206] can be considered reliable pathologic biomarkers. DTI can help to discriminate PPA subtypes according to different spatial patterns of white matter damage [160,207], and specific network alterations are also found in svPPA at fMRI [208]. Higher CSF and serum NFL levels were reported in nfvPPA and svPPA compared to controls, but also compared to lvPPA [175,209–211]. Beyond specific diagnostic relevance, CSF and serum NFL levels in nfvPPA and svPPA were proven to correlate with disease severity, cognitive worsening, and cerebral atrophy [209,210]. Notably, low CSF p/t-tau ratio may contribute to the discrimination of nfvPPA and svPPA from controls, and it shows correlation with survival [209]. Since neither high CSF NFL levels nor low p/t-tau ratio is exclusive of svPPA and nfvPPA but have been found also in other FTD forms and NDD like AD and DLB [99,172,175,212], identification of variant-specific biomarkers is mandatory.
1.7. Amyotrophic lateral sclerosis
ALS is a rare, aggressive, and phenotypically heterogeneous [213] NDD affecting mainly cortical, brainstem, and spinal motor neurons [214]. ALS is a complex disease sharing many of its biological and clinical features with FTD [215].
An abnormal deposition of TDP-43 is the pathological hallmark in about 95% of ALS subjects [216]. The ALS pathogenesis is multifaceted and multiple neurodegenerative pathways often concur [214,217]. Indeed, ALS may be familiar and sporadic with the former representing up to 20% of cases, with more than 30 genes described so far [218]. The increasing causal genes detection shed light on the ALS underlying pathophysiological mechanisms [217]. Notably, the ALS genetic network is tangled and challenging since it includes oligogenic inheritance, non-concordant penetrance, different expressivity, and genetic pleiotropy [219]. Nevertheless, the mutation of four genes is responsible for ~70% of familial ALS cases (Table 1).
In 1994, the El Escorial criteria [220] stated the need of both upper and lower motoneuron clinical signs of degeneration in three out of four parts (bulbar, cervical, thoracic, and lumbosacral regions) of the body for a definite diagnosis of ALS; simplifying, probable and possible diagnosis should be posed when two or one regions are involved, respectively. The presence of isolated lower motoneuron signs allows only a suspected ALS diagnosis. These criteria have an overall excellent specificity but a low sensitivity especially for early ALS diagnosis. In this regard, these criteria were revised (Airlie House Criteria) in 2000 [221] introducing electrophysiological evidence of upper and lower motoneuron degeneration as supportive of ALS diagnosis. However, only with the Awaji-Shima criteria in 2008 the electrophysiological alterations became surrogates of clinical signs, and this significantly increased the diagnostic sensitivity (sensitivity = 57%, significantly higher compared to the revised El Escorial criteria (45%)) especially in the earlier ALS stages [222,223], and in bulbar as well as limb-onset subtypes, still maintaining very high specificity (~ 99%) [224].
The clinical presentation of ALS is complex and difficult to categorize. ALS phenotypes include a classic form (70% of cases including prevalent bulbar (35%) and spinal subtypes (65%)), an isolated bulbar localization (5%), and slow-progressive ALS with only lower motor neuron signs (including isolated flail legs and flail arms syndrome) or primary ALS which need an exclusive involvement of upper motor neurons for at least 4 years (10%). Rare clinical pictures (3% of ALS) are also described and are characterized by generalized cachessia and respiratory failure due to an early dramatic involvement of the diaphragm [214]. In about half of ALS individuals neuropsychological deficits were described, leading to frank dementia in the FTD spectrum (ALS-FTD) in 10–15% of cases [225]. Recently, specific diagnostic criteria for ALS cognitive impairment were proposed [226,227] and subsequently revised [215]. In brief, ALS subjects with cognitive impairment who do not fulfill criteria for FTD (in the majority of the cases the bvFTD phenotype, but one-third out of ALS-FTD may show a predominant language profile [228]) are classified as ALS with behavioral modifications (ALSbi) or with cognitive impairment (ALSci). In the latter, evidence of executive dysfunction and alterations in verbal fluency or both is predominant. The revised criteria in 2017 identify a further subtype with both behavioral and cognitive impairment (ALScbi) [215] maybe representing a transitional stage to FTD as MCI in AD [229]. Noteworthy, isolated memory impairment should be considered with caution as it is rare (4%) and not yet sufficiently characterized to be causally associated with ALS [225]. Notably, in ALS with cognitive deficits, the mutation of C9ORF72 is more probable than in ALS patients without cognitive impairment [230].
The clinical picture of ALS is diverse encompassing, apart from the mandatory motor involvement, also sensory, cerebellar and/or extrapyramidal systems up to frank parkinsonian signs [231,232]. Moreover, ALS phenotypes differ in rate of progression, survival, age of onset, sight of onset, pattern of symptoms distribution, prevalence of upper or lower motor signs [213]. Finally, a disease description by discrete and qualitative categories (e.g. bulbar or spinal ALS) is misleading, though practical for a clinical classification. Instead, a systematic and possibly multicenter collection of quantitative variables including cognitive and motor scores using standardized clinical scale (e.g. the Revised Amyotrophic Lateral Sclerosis Functional Rating Scale, namely ALSFRS-R), imaging and molecular biomarkers, electrophysiological data, genetics information, could implement ALS stratification. In particular, as recently proposed for AD clinical syndromes [233], clustering analysis [213] with non-a priori determination of the number of categories, could provide an innovative, data-driven stratification of ALS patients potentially useful for a correct patients recruitment in disease-modifying trials.
Since the revised El Escorial diagnostic criteria have been published in 1994 [220], considerable advances were made in ALS biomarker identification. Several promising neurophysiological, neuropsychological, imaging, and biofluid biomarkers have been assessed. Neurophysiological markers of lower motoneuron are currently incorporated in the body of the diagnostic criteria for ALS and have been shown to increase sensitivity [223] especially in early disease phase. Extensive psychometric evaluation in the diagnostic workup of ALS-FTD spectrum [215,227] detects frequent subtle cognitive alterations in course of the disease.
Although not yet included among ALS diagnostic criteria, advanced neuroimaging techniques may help to identifying preclinical or prodromal ALS upper motoneuron signs [234]. Cortical hypointensities on T2-weighted MRI sequences measuring ferritin microglia deposition are sensitive early proxies of the motor cortex involvement in ALS subjects [235,236]. Also, a reduction of the neuronal metabolite N-acetylaspartate concentration relative to choline and creatine levels by Proton spectroscopy of the brain [237–240] is considered nowadays as an accurate indicator of upper motor neuron disruption and has been used in a clinical trial [236].
Up to now, the most interesting candidate biomarker for ALS is NFL [7,241,242]. Initially measured in CSF, this biomarker has been extensively investigated also in blood dating back to 2016, when ultrasensitive techniques offered for a sensitivity 25 and 125 folds higher than electrochemiluminescence and traditional ELISA methods, respectively [243,244].
CSF NFL is high in ALS subjects showing excellent diagnostic accuracy (AUROC > 0.90) in differentiating ALS from normal controls and individuals with other neurological diseases including ALS mimics (primary lateral sclerosis, Kennedy’s disease, cervical spondylosis, and axonal polyneuropathies such as multifocal motor neuropathy with conduction block) [245–247]. Noteworthy, NFL is not specific (pathognomonic) since it indicates axonal degeneration but its massive release is an ALS hallmark compared to other chronic neurological diseases. Moreover, CSF NFL has prognostic value distinguishing ALS fast from slow progressors and correlating with motor symptoms. Indeed, ALS patients with CSF NFL concentration in the third and upper quartile group revealed a shorter survival [176]. Finally, bulbar and spinal phenotype reported similar NFL concentrations and higher than flail arm or leg syndrome and progressive muscular atrophy [248]. Noteworthy, in another study the CSF phosphorylated neurofilament heavy chain (p-NFH) showed even better diagnostic accuracy in differentiating ALS from ALS-mimics than CSF NFL [245]. CSF p-NFH positively correlated with CSF YKL-40, a marker of glia activation and neuroinflammation [249]. Increased CSF YKL-40 levels distinguish ALS from ALS mimics with optimal accuracy [250], and is associated with symptoms progression and shorter survival [251]. Serum NFL concentrations are significantly increased in ALS showing an optimal diagnostic accuracy in discriminating ALS individuals from controls (AUROC > 0.90) [252]. Serum NFL also correlated with disease progression, and higher concentration is associated with shorter survival [252–254]. Being a reliable peripheral proxy of ALS neurodegeneration with low-invasiveness and feasible repeatability it represents an excellent candidate biomarker as first-step diagnostic tool, prognostic surrogate, and treatment monitoring.
2. Conclusions
Clinical syndromes of NDD are highly heterogeneous and overlapping as a given proteinopathy can be found to be associated with multiple phenotypes and one phenotype may result from the coexistence of several proteinopathies. Pure or single pathologic hallmarks can be found in NDD, but mixed, or co-pathologies, including diverse kinds of concurrent cerebrovascular disease, especially in elderly, are more common than previously thought. In general, any given phenotype of chronic NDD is not specific for single pathology and this should be taken into account especially in clinical practice. In prospective, classical phenotypes of NDD currently considered distinct might be merged to constitute larger, and conceptually more flexible, clinical groups definable ‘macrophenotypes’ (e.g. merging AD and DLB in a single clinical entity). Indeed, the concept of ‘macro-phenotype’ might more properly reflect common underlying pathophysiological mechanisms of neurodegeneration than traditional clinical definitions (e.g. AD and DLB phenotypes share ß-amyloid and α-synuclein pathologies) (Figure 2). Moreover, use of autopsy examinations as effective and robust diagnostic gold standards pose problems since they are in effect static and final representations of complex, dynamic pathophysiological processes, evolving over years to decades.
Figure 2.
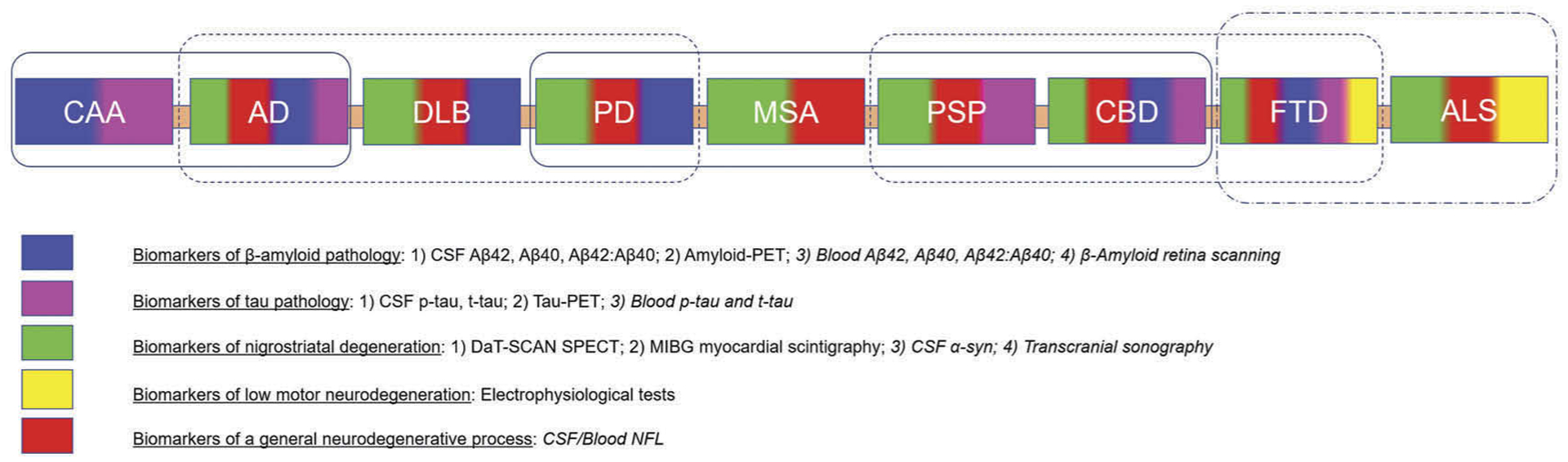
Phenotypes of major neurodegenerative diseases, and their established and candidate pathophysiological biomarkers. The clinical phenotypes of primary neurodegenerative diseases are represented as colored box. Each color corresponds to a set of biomarkers (see legend) that can capture pathological components of these neurodegenerative diseases according to the diagnostic criteria. The biomarkers shown in cursive are potential and emerging candidate biomarkers that are being validated and are not yet included in the existing diagnostic criteria. Smoothed boxes group clinical phenotypes with shared pathophysiological components (macro-phenotypes).
Reliable biomarkers included in core diagnostic criteria of various NDD that have the ability to trace specific pathophysiological mechanisms are few (Figure 2), and mainly pertaining to AD. Concerning parkinsonism, SPECT with DAT scan can reveal relatively early dopaminergic nigrostriatal fibers degeneration. Cardiac [123I]-MIBG scintigraphy may be supportive to refine the diagnosis distinguishing parkinsonian phenotypes due to synucleinopathy from those due to non-synucleinopathies. Concerning cognitive impairment, Aβ peptides and tau levels in CSF together with brain binding of amyloid and tau-PET ligands are consistent surrogates of AD pathology and confirmative or exclusionary in the diagnostic workup. Advanced structural and fMRI techniques as well as cerebral FDG-PET should be considered useful markers of intracerebral networks disintegration and/or neuronal loss reflecting phenotypes but not specific pathophysiological process. On the other hand, conventional MRI is needed to rule out mimics and T2* sequences specifically support the diagnosis of CAA with relevant clinically therapeutic implications. Similarly, to the above mentioned topographic and progression markers, electrophysiological tests though useful to exclude ALS mimics are not specific of ALS pathophysiological mechanisms.
Currently, effective validated screening biomarkers in preclinical or prodromal phases of NDD are lacking. For screening, a biomarker needs a high negative predictive value. Ideally, the first step should help select high-risk individuals that would undergo additional, more expensive or invasive diagnostic studies. In this regard, an optimal screening tool should be minimally invasive, fast, easily repeatable, and costeffective. Several biomarkers can serve this purpose (Figure 2). First, although it is not specific, blood NFL levels, that tightly correlate with CSF NFL expression is the most promising for a multistep diagnostic approach [255], especially in selected populations at risk for neurodegeneration such as in elderly, familial cases, and subjects with diabetes and some psychiatric diseases. Noteworthy, a massive NFL increase is highly suggestive of acutely aggressive and rapidly progressive NDD, such as ALS. Also, serum progranulin levels in suspected FTD patients could precisely identify individuals carrying GRN mutations. In select cases, Poly(GP) depositions in CSF may be effective to identify and monitor FTD patients with C9ORF72 mutation.
Further studies should confirm if the novel ultrasensitive techniques may be applied to blood to recognize the amyloid-driven pathophysiological processes in the CNS. For instance, in the near future a blood screening test evaluating a peripheral amyloid increase suggestive of CAA might be proposed for older patients before starting any anticoagulants to assess the risk of potential cerebral bleeding. In this regard, a noninvasive retinal scan allowing a serial detection at any time of specific patterns of amyloid deposition – suggestive of AD and CAA – as well as signs of cerebrovascular disease, might help screen the typically mixed neurodegenerative and cerebrovascular pathological profiles in elderly. Transcranial sonography as potential low-cost screening tool could early detect SN alteration as an indirect sign of synucleinopathy in selected population such as people with RBD. Importantly, blood biomarkers, noninvasive retinal scanning, and ultrasounds, being easily repeatable, may be considered as progression and monitoring treatment biomarkers as well. Finally, although α-syn strains measured in CSF with traditional ELISA methods do not represent robust diagnostic biomarkers, the very recent advance of in vitro protein misfolding amplification assays (RT-QuIC and PMCA) to measure CSF α-syn prion-like seeds is promising to identify synucleinopathies, mixed DLB-AD pathologies, and the Lewy body variant of AD [256]. In prospective, the last decade has seen a surge in studies of novel molecular pathways in aging and NDD. Amongst these, new evidence suggesting cell to cell transmission of NDD pathologies via extracellular vesicles, such as exosomes (30–−50 nm) is beginning to emerge [257]. Methods for isolation of exosomes from human biospecimen have been developed and quantification of AD neuropathologic markers such as tau, Aß, and p-tau from exosomes suggests utility of exosomal cargo as noninvasive biomarkers of disease [258–261]. In addition to proteins, exosomes carry non-coding, microRNA (miRNA) from one cell to another. These miRNAs have potent regulatory functions, thereby representing an important cellcell communication in health and disease. Their quantification across disease spectrum can serve as unique noninvasive biomarkers of molecular pathway dysregulations across NDD [262,263]. In addition to exosomal cargo, cell-free miRNAs and circular RNA (circRNA) are under investigation as novel biomarkers across NDD and in AD [264]. However, more work remains to be done before these novel molecular markers can be connected to clinical phenotypes and mapped onto disease states and stages.
3. Expert opinion
NDD are biologically and genetically heterogeneous entities characterized by complex pathomechanistic alterations with non-linear spatial and temporal evolution [265].
Traditional neuropathological investigations as well as breakthrough studies conducted using advanced high-throughput technologies, and a systems-level integration of multiple biological signatures, indicate a wide spectrum of pathophysiological processes giving rise to a given clinical phenotype or even just brain aging [8,34,266]. In advanced age [8], it is well established that pathophysiological commonalities – due to (epi)genetic and biological aberrant pathway(s) – are shared across divergent brain diseases and phenotypes [265,267].
Biomarker-based and systems biology approaches [9,268,269] are expected to guide the dynamic detection and quantification of target druggability, the in-vivo demonstration of proof-of-mechanism, as well as the prediction of drug resistance mechanisms [270–272]. To follow, biomarker panels will offer a flexible tool for the accurate selection of individuals likely to benefit from pathway (mechanism)-based pharmacological therapies regardless of the downstream clinical manifestations.
Going beyond the traditional ‘one marker, one disease, one diagnosis, one drug’ construct, the use of biomarker-guided individualized health-care algorithms represents a crucial step that can advance neurology toward benchmarks set for precision medicine. Based on the continuum from aging to neurodegenerative disease, through intermediate stages of compensation and resilience to incipient pathomechanistic alterations [266,273], we argue that next-generation therapeutic strategies should aim at enhancing resilience pathways that ensure normal cognitive functions during aging, in combination with treatments targeting pathophysiological changes associated with diverse dementia syndromes [265].
A conceptual shift toward a novel unifying framework characterizing and mapping all NDD is still at its infancy. Traditional and unconnected clinical classifications can hardly depict the complex multifaceted NDD spectrum with an important disconnect with underlying pathophysiology. Similarly, the systematic strategy of stratifying patients with dichotomic biomarker classifications (e.g. positive versus negative for a certain biomarker) may be confounding, with the risk to add further arbitrary separations along the NDD spectrum.
Artificial intelligence-based techniques, including unsupervised clustering strategies, have the potential [233] to enable the identification of multiple nonlinear associations among quantitative variables systematically collected in subjects. These variables can encompass: i) clinical scores, exploring cognitive, behavioral, and motor functions, ii) topographic and pathophysiological biomarkers [213,233], as well as genetic variants. In theory, NDD may be classified based on the combination of clinical and genetic/biological data to segregate large-scale heterogeneous populations into consistent ‘clusters,’ i.e. from bigdata to ‘smartdata’ [274,275].
Article highlights.
The clinical diagnostic guidelines of neurodegenerative diseases (NDD) based on post-mortem evaluations as reference standard do not reflect the dynamic continuum of overlapping pathophysiological abnormalities, mixed pathologies, and the shared anatomic, phenotypic constituents.
Few pathophysiological biomarkers are reported in the NDD diagnostic criteria
CSF Aβ42, t-tau, p-tau concentrations or, alternatively, cerebral amyloid-PET and tau-PET retention, essentially confirm or exclude an Alzheimer’s disease pathology
SPECT with datscan detects specific dopaminergic denervation; electrophysiological tests identify lower motor neuron degeneration
Altered patterns of cerebral atrophy or hypometabolism on conventional MRI and FDG-PET, respectively, reflect phenotypes but not pathophysiologies
Protein misfolding amplification assays may accurately detect αsynuclein prion-like seeds in synucleinopathies and serum progranulin some genetic forms of frontotemporal dementia.
Ultrasensitive techniques measuring blood Aβ42 and NFL concentrations might represent screening tools in select populations
Other potential screening tools are retina scanning and ultrasound to non-invasively investigate β-amyloid and substantia nigra
Strategies combining clinical and biomarker information to gather individuals from large heterogeneous cohorts into consistent clusters might improve classifications of NDD across the wide spectrum of presentations, and aid the uncovering of pathophysiological causes and identification of impactful therapies
Funding
NIH/NIA R01 AG055865 and R01 AG056478 awards (M.K.H.).
HH is an employee of Eisai Inc. During his previous work (until April 2019) he was supported by the AXA Research Fund, the ‘Fondation partenariale Sorbonne Université’ and the ‘Fondation pour la Recherche sur Alzheimer,’ Paris, France.
AV is an employee of Eisai Inc.
Footnotes
Declaration of interest
AV is an employee of Eisai Inc. and serves as Associate Editor for the Journal of Alzheimer’s disease; he received lecture honoraria from Roche, MagQu LLC, and Servier. HH is an employee of Eisai Inc. and serves as Senior Associate Editor for the Journal Alzheimer’s & Dementia; he received lecture fees from Biogen, Roche, and Servier, research grants from Pfizer, Avid, and MSD Avenir (paid to the institution), travel funding from Functional Neuromodulation, Axovant, Eli Lilly and company, Takeda and Zinfandel, GE-Healthcare and Oryzon Genomics, consultancy fees from Qynapse, Jung Diagnostics, Cytox Ltd., Axovant, Anavex, Takeda and Zinfandel, GE Healthcare, Oryzon Genomics, and Functional Neuromodulation, and participated in scientific advisory boards of Functional Neuromodulation, Axovant, Eisai, Eli Lilly and company, Cytox Ltd., GE Healthcare, Takeda and Zinfandel, Oryzon Genomics and Roche Diagnostics.
- In Vitro Multiparameter Determination Method for The
- Diagnosis and Early Diagnosis of Neurodegenerative Disorders Patent Number: 8916388
- In Vitro Procedure for Diagnosis and Early Diagnosis of Neurodegenerative Diseases Patent Number: 8298784
- Neurodegenerative Markers for Psychiatric Conditions Publication Number: 20120196300
- In Vitro Multiparameter Determination Method for The
- Diagnosis and Early Diagnosis of Neurodegenerative Disorders Publication Number: 20100062463
- In Vitro Method for The Diagnosis and Early Diagnosis of Neurodegenerative Disorders Publication Number: 20100035286
- In Vitro Procedure for Diagnosis and Early Diagnosis of Neurodegenerative Diseases Publication Number: 20090263822
- In Vitro Method for The Diagnosis of Neurodegenerative Diseases Patent Number: 7547553
- CSF Diagnostic in Vitro Method for Diagnosis of Dementias and Neuroinflammatory Diseases Publication Number: 20080206797
- In Vitro Method for The Diagnosis of Neurodegenerative Diseases Publication Number: 20080199966
- Neurodegenerative Markers for Psychiatric Conditions Publication Number: 20080131921
KHM is a co-founding member and consultant for NeuroVision Imaging (NVI), also a co-inventor on the following patent as a scientist and has received no royalties: Optical method for the detection of Alzheimer’s disease. Patent Number: 9839699.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewers Disclosure
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Jellinger KA. Basic mechanisms of neurodegeneration: a critical update. J Cell Mol Med. 2010;14:457–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kovacs GG. Molecular pathological classification of neurodegenerative diseases: turning towards precision medicine. Int J Mol Sci. 2016;17:pii:E189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldacci F, Lista S, Garaci F, et al. Biomarker-guided classification scheme of neurodegenerative diseases. J Sport Health Sci. 2016;5:383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology. 1984;34:939–944. [DOI] [PubMed] [Google Scholar]; The first ever published criteria on a neurodegenerative disease.
- 5.Baldacci F, Lista S, O’Bryant SE, et al. Blood-based biomarker screening with agnostic biological definitions for an accurate diagnosis within the dimensional spectrum of neurodegenerative diseases. Methods Mol Biol. 2018;1750:139–155. [DOI] [PubMed] [Google Scholar]; This review discuss the need for a novel classification of neurodegenerative diseases.
- 6.Kovacs GG, Milenkovic I, Wohrer A, et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol. 2013;126:365–384. [DOI] [PubMed] [Google Scholar]
- 7.Baldacci F, Lista S, Vergallo A, et al. A frontline defense against neurodegenerative diseases: the development of early disease detection methods. Expert Rev Mol Diagn. 2019;19:559–563. [DOI] [PubMed] [Google Scholar]
- 8.Robinson JL, Lee EB, Xie SX, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain. 2018;141:2181–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hampel H, Toschi N, Babiloni C, et al. Revolution of Alzheimer precision neurology. passageway of systems biology and neurophysiology. J Alzheimers Dis. 2018;64:S47–s105. [DOI] [PMC free article] [PubMed] [Google Scholar]; This is a fundamental article on the relationship between neurodegeneration, system biology and precision medicine.
- 10.Hampel H, O’Bryant SE, Durrleman S, et al. A precision medicine initiative for Alzheimer’s disease: the road ahead to biomarker-guided integrative disease modeling. Climacteric J Int Menopause Soc. 2017;20:107–118. [DOI] [PubMed] [Google Scholar]
- 11.Jack CR Jr., Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimer’s Dementia. 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the national institute on aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia. 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]; The first Alhzheimer’s disease guidelines for a preclinical diagnosis.
- 13.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6:734–746. [DOI] [PubMed] [Google Scholar]; The first Alzheimer’s disease criteria reporting molecular biomarkers.
- 14.Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9:1118–1127. [DOI] [PubMed] [Google Scholar]
- 15.Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 2014;13:614–629. [DOI] [PubMed] [Google Scholar]; The most recent Alzheimer’s disease clinical diagnostic criteria including pathophysiological biomarkers.
- 16.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the national institute on aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dementia. 2011;7:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]; The concept and the importance of mild cognitive impairment due to Alzheimer’s disease is stated out.
- 17.Berg D, Postuma RB, Adler CH, et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord. 2015;30:1600–1611. [DOI] [PubMed] [Google Scholar]; The concept of Prodromal Parkinson’s disease is stated out.
- 18.Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses. 2004;63:8–20. [DOI] [PubMed] [Google Scholar]
- 19.Belloy ME, Napolioni V, Greicius MD. A quarter century of APOE and Alzheimer’s disease: progress to date and the path forward. Neuron. 2019;101:820–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pimenova AA, Raj T, Goate AM. Untangling genetic risk for Alzheimer’s disease. Biol Psychiatry. 2018;83:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freudenberg-Hua Y, Li W, Davies P. The role of genetics in advancing precision medicine for Alzheimer’s disease-a narrative review. Front Med (Lausanne). 2018;5:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamboh MI. A brief synopsis on the genetics of Alzheimer’s disease. Current Genet Med Rep. 2018;6:133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. [DOI] [PubMed] [Google Scholar]
- 24.Scheltens P, Blennow K, Breteler MM, et al. Alzheimer’s disease. Lancet. 2016;388:505–517. [DOI] [PubMed] [Google Scholar]
- 25.Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang B, Gaiteri C, Bodea LG, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rabinovici GD, Carrillo MC, Forman M, et al. Multiple comorbid neuropathologies in the setting of Alzheimer’s disease neuropathology and implications for drug development. Alzheimer’s Dementia. 2017;3:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serrano-Pozo A, Frosch MP, Masliah E, et al. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1: a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Armstrong RA. The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Folia Neuropathol. 2009;47:289–299. [PubMed] [Google Scholar]
- 30.Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. [DOI] [PubMed] [Google Scholar]
- 31.Sanabria-Castro A, Alvarado-Echeverria I, Monge-Bonilla C. Molecular pathogenesis of Alzheimer’s disease: an update. Ann Neurosci. 2017;24:46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the national institute on aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Demen. 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jack CR Jr., Bennett DA, Blennow K, et al. Contributors, NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hampel H, Vergallo A, Aguilar LF, et al. Precision pharmacology for Alzheimer’s disease. Pharmacol Res. 2018;130:331–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elahi FM, Miller BL. A clinicopathological approach to the diagnosis of dementia. Nat Rev Neurol. 2017;13:457–476. [DOI] [PMC free article] [PubMed] [Google Scholar]; A milestone review for an integrate approach to neurodegenerative dementia.
- 36.Crutch SJ, Lehmann M, Schott JM, et al. Posterior cortical atrophy. Lancet Neurol. 2012;11:170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crutch SJ, Schott JM, Rabinovici GD, et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 2017;13:870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ossenkoppele R, Schonhaut DR, Baker SL, et al. Tau, amyloid, and hypometabolism in a patient with posterior cortical atrophy. Ann Neurol. 2015;77:338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jack CR Jr., Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;87:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266:2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bevan-Jones RW, Cope TE, Jones SP, et al. [(18)F]AV-1451 binding is increased in frontotemporal dementia due to C9orf72 expansion. Ann Clin Transl Neurol. 2018;5:1292–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Twohig D, Nielsen HM. alpha-synuclein in the pathophysiology of Alzheimer’s disease. Mol Neurodegener. 2019;14:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fairfoul G, McGuire LI, Pal S, et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol. 2016;3:812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shahnawaz M, Tokuda T, Waragai M, et al. Development of a biochemical diagnosis of Parkinson disease by detection of alpha-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017;74:163–172. [DOI] [PubMed] [Google Scholar]
- 45.Van Bulck M, Sierra-Magro A, Alarcon-Gil J, et al. Novel approaches for the treatment of Alzheimer’s and Parkinson’s disease. Int J Mol Sci. 2019;20:pii:E719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Bryant SE. Blood biomarkers for use in Alzheimer disease-moving from “if” to “how?”. JAMA Neurol. DOI: 10.1001/jamaneurol.2019.0845(2019) [DOI] [PubMed] [Google Scholar]
- 47.Hart NJ, Koronyo Y, Black KL, et al. Ocular indicators of Alzheimer’s: exploring disease in the retina. Acta Neuropathol. 2016;132:767–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, et al. Identification of amyloid plaques in retinas from Alzheimer’s patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. NeuroImage. 2011;54(Suppl 1):S204–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koronyo Y, Salumbides BC, Black KL, et al. Alzheimer’s disease in the retina: imaging retinal abeta plaques for early diagnosis and therapy assessment. Neurodegener Dis. 2012;10:285–293. [DOI] [PubMed] [Google Scholar]
- 50.Koronyo Y, Biggs D, Barron E, et al. Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer’s disease. JCI Insight. 2017;2: pii:93621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.La Morgia C, Ross-Cisneros FN, Koronyo Y, et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann Neurol. 2016;79:90–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Javaid FZ, Brenton J, Guo L, et al. Visual and ocular manifestations of Alzheimer’s disease and their use as biomarkers for diagnosis and progression. Front Neurol. 2016;7:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Katz B, Rimmer S, Iragui V, et al. Abnormal pattern electroretinogram in Alzheimer’s disease: evidence for retinal ganglion cell degeneration? Ann Neurol. 1989;26:221–225. [DOI] [PubMed] [Google Scholar]
- 54.Yanagisawa D, Shirai N, Amatsubo T, et al. Relationship between the tautomeric structures of curcumin derivatives and their Abetabinding activities in the context of therapies for Alzheimer’s disease. Biomaterials. 2010;31:4179–4185. [DOI] [PubMed] [Google Scholar]
- 55.Andreasson U, Blennow K, Zetterberg H. Update on ultrasensitive technologies to facilitate research on blood biomarkers for central nervous system disorders. Alzheimers Dement (Amst). 2016;3:98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature. 2018;554:249–254. [DOI] [PubMed] [Google Scholar]
- 57.Nabers A, Perna L, Lange J, et al. Amyloid blood biomarker detects Alzheimer’s disease. EMBO Mol Med. 2018;10:pii:E8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Palmqvist S, Janelidze S, Stomrud E, et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease-related beta-amyloid status. JAMA Neurol. DOI: 10.1001/jamaneurol.2019.1632(2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mielke MM, Hagen CE, Xu J, et al. Plasma phospho-tau181 increases with Alzheimer’s disease clinical severity and is associated with tau- and amyloid-positron emission tomography. Alzheimer’s Dementia. 2018;14:989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vergallo A, Megret L, Lista S, et al. Plasma amyloid beta 40/42 ratio predicts cerebral amyloidosis in cognitively normal individuals at risk for Alzheimer’s disease. Alzheimer’s Dementia. 2019;15:764–775. [DOI] [PubMed] [Google Scholar]
- 61.Mattsson N, Andreasson U, Zetterberg H, et al. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2017;74:557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hu H, Chen KL, Ou YN, et al. Neurofilament light chain plasma concentration predicts neurodegeneration and clinical progression in nondemented elderly adults. Aging (Albany NY). 2019;11:6904–6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mattsson N, Cullen NC, Andreasson U, et al. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2019;76:791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keage HA, Carare RO, Friedland RP, et al. Population studies of sporadic cerebral amyloid angiopathy and dementia: a systematic review. BMC Neurol. 2009;9:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Carare RO, Hawkes CA, Jeffrey M, et al. Review: cerebral amyloid angiopathy, prion angiopathy, CADASIL and the spectrum of protein elimination failure angiopathies (PEFA) in neurodegenerative disease with a focus on therapy. Neuropathol Appl Neurobiol. 2013;39:593–611. [DOI] [PubMed] [Google Scholar]
- 66.Carare RO, Kalaria R. Cerebrovascular pathology: the dark side of neurodegeneration. Acta Neuropathol. 2016;131:641–643. [DOI] [PubMed] [Google Scholar]
- 67.van Veluw SJ, Biessels GJ, Bouvy WH, et al. Cerebral amyloid angiopathy severity is linked to dilation of juxtacortical perivascular spaces. J Cereb Blood Flow and Metab. 2016;36:576–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Charidimou A, Martinez-Ramirez S, Shoamanesh A, et al. Cerebral amyloid angiopathy with and without hemorrhage: evidence for different disease phenotypes. Neurology. 2015;84:1206–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Biffi A, Greenberg SM. Cerebral amyloid angiopathy: a systematic review. J Clin Neurol. 2011;7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Greenberg SM, Rebeck GW, Vonsattel JP, et al. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol. 1995;38:254–259. [DOI] [PubMed] [Google Scholar]
- 71.Greenberg SM, Charidimou A. Diagnosis of cerebral amyloid angiopathy: evolution of the boston criteria. Stroke. 2018;49:491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]; A fundamental paper toward an improving of the diagnostic criteria of Cerebral Amiloid Angiopathy.
- 72.Charidimou A, Boulouis G, Gurol ME, et al. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain. 2017;140:1829–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Charidimou A, Farid K, Baron JC. Amyloid-PET in sporadic cerebral amyloid angiopathy: a diagnostic accuracy meta-analysis. Neurology. 2017;89:1490–1498. [DOI] [PubMed] [Google Scholar]
- 74.Charidimou A, Farid K, Tsai HH, et al. Amyloid-PET burden and regional distribution in cerebral amyloid angiopathy: a systematic review and meta-analysis of biomarker performance. J Neurol Neurosurg Psychiatry. 2018;89:410–417. [DOI] [PubMed] [Google Scholar]
- 75.Charidimou A, Friedrich JO, Greenberg SM, et al. Core cerebrospinal fluid biomarker profile in cerebral amyloid angiopathy: a meta-analysis. Neurology. 2018;90:e754–e762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Postuma RB, Gagnon JF, Bertrand JA, et al. Parkinson risk in idiopathic REM sleep behavior disorder: preparing for neuroprotective trials. Neurology. 2015;84:1104–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1988;51:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hughes AJ, Daniel SE, Kilford L, et al. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]; An evergreen milestone for the Parkinson’s disease diagnosis.
- 79.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Arch Neurol. 1999;56:33–39. [DOI] [PubMed] [Google Scholar]
- 80.Hughes AJ, Daniel SE, Lees AJ. Improved accuracy of clinical diagnosis of Lewy body Parkinson’s disease. Neurology. 2001;57:1497–1499. [DOI] [PubMed] [Google Scholar]
- 81.Jellinger KA. Very old onset parkinsonism: a clinical-pathological study. Parkinsonism Relat Disord. 2018;57:39–43. [DOI] [PubMed] [Google Scholar]
- 82.Fereshtehnejad SM, Montplaisir JY, Pelletier A, et al. Validation of the MDS research criteria for prodromal Parkinson’s disease: longitudinal assessment in a REM sleep behavior disorder (RBD) cohort. Mov Disord. 2017;32:865–873. [DOI] [PubMed] [Google Scholar]
- 83.Cosottini M, Frosini D, Pesaresi I, et al. Comparison of 3T and 7T susceptibility-weighted angiography of the substantia nigra in diagnosing Parkinson disease. AJNR Am J Neuroradiol. 2015;36:461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Berg D, Behnke S, Seppi K, et al. Enlarged hyperechogenic substantia nigra as a risk marker for Parkinson’s disease. Mov Disord. 2013;28:216–219. [DOI] [PubMed] [Google Scholar]
- 85.Tao A, Chen G, Deng Y, et al. Accuracy of transcranial sonography of the substantia nigra for detection of Parkinson’s disease: a systematic review and meta-analysis. Ultrasound Med Biol. 2019;45:628–641. [DOI] [PubMed] [Google Scholar]
- 86.Siderowf A, Xie SX, Hurtig H, et al. CSF amyloid {beta} 1–42 predicts cognitive decline in Parkinson disease. Neurology. 2010;75:1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Terrelonge M Jr., Marder KS, Weintraub D, et al. CSF beta-amyloid 1–42 predicts progression to cognitive impairment in newly diagnosed Parkinson disease. J Mol Neurosci. 2016;58:88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rochester L, Galna B, Lord S, et al. Decrease in Abeta42 predicts dopa-resistant gait progression in early Parkinson disease. Neurology. 2017;88:1501–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ffytche DH, Pereira JB, Ballard C, et al. Risk factors for early psychosis in PD: insights from the Parkinson’s progression markers initiative. J Neurol Neurosurg Psychiatry. 2017;88:325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Constantinides VC, Paraskevas GP, Emmanouilidou E, et al. CSF biomarkers beta-amyloid, tau proteins and a-synuclein in the differential diagnosis of Parkinson-plus syndromes. J Neurol Sci. 2017;382:91–95. [DOI] [PubMed] [Google Scholar]
- 91.Gao L, Tang H, Nie K, et al. Cerebrospinal fluid alpha-synuclein as a biomarker for Parkinson’s disease diagnosis: a systematic review and meta-analysis. Int J Neurosci. 2015;125:645–654. [DOI] [PubMed] [Google Scholar]
- 92.Eusebi P, Giannandrea D, Biscetti L, et al. Diagnostic utility of cerebrospinal fluid alpha-synuclein in Parkinson’s disease: a systematic review and meta-analysis. Mov Disord. 2017;32:1389–1400. [DOI] [PubMed] [Google Scholar]
- 93.Zhou B, Wen M, Yu WF, et al. The diagnostic and differential diagnosis utility of cerebrospinal fluid alpha -synuclein levels in Parkinson’s disease: a meta-analysis. Parkinson’s Dis. 2015;2015:567386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kalia LV. Diagnostic biomarkers for Parkinson’s disease: focus on alpha-synuclein in cerebrospinal fluid. Parkinsonism Relat Disord. 2019;59:21–25. [DOI] [PubMed] [Google Scholar]
- 95.Hall S, Surova Y, Ohrfelt A, et al. Longitudinal measurements of cerebrospinal fluid biomarkers in Parkinson’s disease. Mov Disord. 2016;31:898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pagano G, De Micco R, Yousaf T, et al. REM behavior disorder predicts motor progression and cognitive decline in Parkinson disease. Neurology. 2018;91:e894–e905. [DOI] [PubMed] [Google Scholar]
- 97.Backstrom DC, Eriksson Domellof M, Linder J, et al. Cerebrospinal fluid patterns and the risk of future dementia in early, incident Parkinson disease. JAMA Neurol. 2015;72:1175–1182. [DOI] [PubMed] [Google Scholar]
- 98.Ge F, Ding J, Liu Y, et al. Cerebrospinal fluid NFL in the differential diagnosis of parkinsonian disorders: A meta-analysis. Neurosci Lett. 2018;685:35–41. [DOI] [PubMed] [Google Scholar]
- 99.Bridel C, van Wieringen WN, Zetterberg H, et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta-analysis. JAMA Neurol. DOI: 10.1001/jamaneurol.2019.1534(2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hansson O, Janelidze S, Hall S, et al. Blood-based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology. 2017;88:930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marques TM, van Rumund A, Oeckl P, et al. Serum NFL discriminates Parkinson disease from atypical parkinsonisms. Neurology. 2019;92:e1479–e1486. [DOI] [PubMed] [Google Scholar]
- 102.Colom-Cadena M, Grau-Rivera O, Planellas L, et al. Regional overlap of pathologies in Lewy body disorders. J Neuropathol Exp Neurol. 2017;76:216–224. [DOI] [PubMed] [Google Scholar]
- 103.Irwin DJ, Xie SX, Coughlin D, et al. CSF tau and beta-amyloid predict cerebral synucleinopathy in autopsied Lewy body disorders. Neurology. 2018;90:e1038–e1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Parnetti L, Paciotti S, Farotti L, et al. Parkinson’s and Lewy body dementia CSF biomarkers. Clin Chim Acta. 2019;495:318–325. [DOI] [PubMed] [Google Scholar]
- 105.McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with lewy bodies: fourth consensus report of the DLB consortium. Neurology. 2017;89:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]; The last international consensus for the diagnosis of Dementia with Lewy body.
- 106.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with lewy bodies: third report of the DLB consortium. Neurology. 2005;65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 107.Abdelnour C, van Steenoven I, Londos E, et al. Alzheimer’s disease cerebrospinal fluid biomarkers predict cognitive decline in Lewy body dementia. Mov Disord. 2016;31:1203–1208. [DOI] [PubMed] [Google Scholar]
- 108.Sako W, Murakami N, Izumi Y, et al. Reduced alpha-synuclein in cerebrospinal fluid in synucleinopathies: evidence from a meta-analysis. Mov Disord. 2014;29:1599–1605. [DOI] [PubMed] [Google Scholar]
- 109.Joutsa J, Gardberg M, Roytta M, et al. Diagnostic accuracy of parkinsonism syndromes by general neurologists. Parkinsonism Relat Disord. 2014;20:840–844. [DOI] [PubMed] [Google Scholar]
- 110.Hoglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. 2017;32:853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]; A consensus for the diagnosis of Progressive Supranuclear Palsy (typical and atypical phenotypes).
- 111.Ali F, Martin PR, Botha H, et al. Sensitivity and specificity of diagnostic criteria for progressive supranuclear palsy. Mov Disord. 2019;34:1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47:1–9. [DOI] [PubMed] [Google Scholar]; The international consensus for the diagnosis of Progressive Supranuclear Palsy (typical phenotype).
- 113.Quattrone A, Nicoletti G, Messina D, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology. 2008;246:214–221. [DOI] [PubMed] [Google Scholar]
- 114.Hussl A, Mahlknecht P, Scherfler C, et al. Diagnostic accuracy of the magnetic resonance Parkinsonism index and the midbrain-to-pontine area ratio to differentiate progressive supranuclear palsy from Parkinson’s disease and the Parkinson variant of multiple system atrophy. Mov Disord. 2010;25:2444–2449. [DOI] [PubMed] [Google Scholar]
- 115.Longoni G, Agosta F, Kostic VS, et al. MRI measurements of brainstem structures in patients with Richardson’s syndrome, progressive supranuclear palsy-parkinsonism, and Parkinson’s disease. Mov Disord. 2011;26:247–255. [DOI] [PubMed] [Google Scholar]
- 116.Moller L, Kassubek J, Sudmeyer M, et al. Manual MRI morphometry in Parkinsonian syndromes. Mov Disord. 2017;32:778–782. [DOI] [PubMed] [Google Scholar]
- 117.Frosini D, Ceravolo R, Tosetti M, et al. Nigral involvement in atypical parkinsonisms: evidence from a pilot study with ultra-high field MRI. J Neural Transm. 2016;123:509–513. [DOI] [PubMed] [Google Scholar]
- 118.Sjostrom H, Granberg T, Westman E, et al. Quantitative susceptibility mapping differentiates between parkinsonian disorders. Parkinsonism Relat Disord. 2017;44:51–57. [DOI] [PubMed] [Google Scholar]
- 119.Mazzucchi S, Frosini D, Costagli M, et al. Quantitative susceptibility mapping in atypical Parkinsonisms. Neuroimage Clin. 2019;24:101999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Donker Kaat L, Meeter LH, Chiu WZ, et al. Serum neurofilament light chain in progressive supranuclear palsy. Parkinsonism Relat Disord. 2018;56:98–101. [DOI] [PubMed] [Google Scholar]
- 121.Rojas JC, Karydas A, Bang J, et al. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Clin Transl Neurol. 2016;3:216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ling H, O’Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain. 2010;133:2045–2057. [DOI] [PubMed] [Google Scholar]
- 123.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]; The international guidelines for the diagnosis of Corticobasal Degeneration.
- 124.Alexander SK, Rittman T, Xuereb JH, et al. Validation of the new consensus criteria for the diagnosis of corticobasal degeneration. J Neurol Neurosurg Psychiatry. 2014;85:925–929. [DOI] [PMC free article] [PubMed] [Google Scholar]; Poor accuracy of the recent guidelines for Corticobasal Degeneration diagnosis.
- 125.Sakae N, Josephs KA, Litvan I, et al. Clinicopathologic subtype of Alzheimer’s disease presenting as corticobasal syndrome. Alzheimer’s Dementia. 2019;15:1218–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ceravolo R, Rossi C, Cilia R, et al. Evidence of delayed nigrostriatal dysfunction in corticobasal syndrome: a SPECT follow-up study. Parkinsonism Relat Disord. 2013;19:557–559. [DOI] [PubMed] [Google Scholar]
- 127.Svenningsson P Corticobasal degeneration: advances in clinicopathology and biomarkers. Curr Opin Neurol. 2019;32:597–603. [DOI] [PubMed] [Google Scholar]
- 128.Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]; The international consensus for the diagnosis of Multiple System Atrophy.
- 129.Miki Y, Foti SC, Asi YT, et al. Improving diagnostic accuracy of multiple system atrophy: a clinicopathological study. Brain. 2019;142:2813–2827. [DOI] [PubMed] [Google Scholar]
- 130.Massey LA, Micallef C, Paviour DC, et al. Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord. 2012;27:1754–1762. [DOI] [PubMed] [Google Scholar]
- 131.Kim JM, Jeong HJ, Bae YJ, et al. Loss of substantia nigra hyperintensity on 7 tesla MRI of Parkinson’s disease, multiple system atrophy, and progressive supranuclear palsy. Parkinsonism Relat Disord. 2016;26:47–54. [DOI] [PubMed] [Google Scholar]
- 132.Koga S, Aoki N, Uitti RJ, et al. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology. 2015;85:404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ratnavalli E, Brayne C, Dawson K, et al. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. [DOI] [PubMed] [Google Scholar]
- 134.Rohrer JD, Guerreiro R, Vandrovcova J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73:1451–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Le Ber I Genetics of frontotemporal lobar degeneration: an up-date and diagnosis algorithm. Rev Neurol (Paris). 2013;169:811–819. [DOI] [PubMed] [Google Scholar]
- 136.Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386:1672–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Johnson JK, Diehl J, Mendez MF, et al. Frontotemporal lobar degeneration: demographic characteristics of 353 patients. Arch Neurol. 2005;62:925–930. [DOI] [PubMed] [Google Scholar]
- 138.Snowden J, Neary D, Mann D. Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol. 2007;114:31–38. [DOI] [PubMed] [Google Scholar]
- 139.Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lee EB, Porta S, Michael Baer G, et al. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 2017;134:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]; Guidelines for the diagnosis of Frontotemporal Dementia (behavioral variant).
- 143.Pasquier F, Richard F, Lebert F. Natural history of frontotemporal dementia: comparison with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2004;17:253–257. [DOI] [PubMed] [Google Scholar]
- 144.Rascovsky K, Salmon DP, Lipton AM, et al. Rate of progression differs in frontotemporal dementia and Alzheimer disease. Neurology. 2005;65:397–403. [DOI] [PubMed] [Google Scholar]
- 145.Garcin B, Lillo P, Hornberger M, et al. Determinants of survival in behavioral variant frontotemporal dementia. Neurology. 2009;73:1656–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Grimmer T, Wutz C, Alexopoulos P, et al. Visual versus fully automated analyses of 18F-FDG and amyloid pet for prediction of dementia due to Alzheimer disease in mild cognitive impairment. J Nucl Med. 2016;57:204–207. [DOI] [PubMed] [Google Scholar]
- 147.Arbizu J, Festari C, Altomare D, et al. Clinical utility of FDG-PET for the clinical diagnosis in MCI. Eur J Nucl Med Mol Imaging. 2018;45:1497–1508. [DOI] [PubMed] [Google Scholar]
- 148.Engler H, Santillo AF, Wang SX, et al. In vivo amyloid imaging with PET in frontotemporal dementia. Eur J Nucl Med Mol Imaging. 2008;35:100–106. [DOI] [PubMed] [Google Scholar]
- 149.Harris JM, Gall C, Thompson JC, et al. Sensitivity and specificity of FTDC criteria for behavioral variant frontotemporal dementia. Neurology. 2013;80:1881–1887. [DOI] [PubMed] [Google Scholar]
- 150.Vijverberg EG, Dols A, Krudop WA, et al. Diagnostic accuracy of the frontotemporal dementia consensus criteria in the late-onset frontal lobe syndrome. Dement Geriatr Cogn Disord. 2016;41:210–219. [DOI] [PubMed] [Google Scholar]
- 151.Burrell JR, Kiernan MC, Vucic S, et al. Motor neuron dysfunction in frontotemporal dementia. Brain. 2011;134:2582–2594. [DOI] [PubMed] [Google Scholar]
- 152.Lillo P, Garcin B, Hornberger M, et al. Neurobehavioral features in frontotemporal dementia with amyotrophic lateral sclerosis. Arch Neurol. 2010;67:826–830. [DOI] [PubMed] [Google Scholar]
- 153.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–1079. [DOI] [PubMed] [Google Scholar]
- 154.Le Ber I, Guedj E, Gabelle A, et al. Demographic, neurological and behavioural characteristics and brain perfusion SPECT in frontal variant of frontotemporal dementia. Brain. 2006;129:3051–3065. [DOI] [PubMed] [Google Scholar]
- 155.Kertesz A, McMonagle P, Blair M, et al. The evolution and pathology of frontotemporal dementia. Brain. 2005;128:1996–2005. [DOI] [PubMed] [Google Scholar]
- 156.Woolley JD, Khan BK, Murthy NK, et al. The diagnostic challenge of psychiatric symptoms in neurodegenerative disease: rates of and risk factors for prior psychiatric diagnosis in patients with early neurodegenerative disease. J Clin Psychiatry. 2011;72:126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nascimento C, Nunes VP, Diehl Rodriguez R, et al. A review on shared clinical and molecular mechanisms between bipolar disorder and frontotemporal dementia. Prog Neuropsychopharmacol Biol Psychiatry. 2019;93:269–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kipps CM, Hodges JR, Hornberger M. Nonprogressive behavioural frontotemporal dementia: recent developments and clinical implications of the ‘bvFTD phenocopy syndrome’. Curr Opin Neurol. 2010;23:628–632. [DOI] [PubMed] [Google Scholar]
- 159.Baker M, Mackenzie IR, Pickering-Brown SM. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. [DOI] [PubMed] [Google Scholar]
- 160.Meeter LH, Kaat LD, Rohrer JD, et al. Imaging and fluid biomarkers in frontotemporal dementia. Nat Rev Neurol. 2017;13:406–419. [DOI] [PubMed] [Google Scholar]
- 161.Mahoney CJ, Ridgway GR, Malone IB, et al. Profiles of white matter tract pathology in frontotemporal dementia. Hum Brain Mapp. 2014;35:4163–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Mahoney CJ, Simpson IJ, Nicholas JM. Longitudinal diffusion tensor imaging in frontotemporal dementia. Ann Neurol. 2015;77:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.McMillan CT, Irwin DJ, Avants BB, et al. White matter imaging helps dissociate tau from TDP-43 in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. 2013;84:949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rohrer JD, Nicholas JM, Cash DM, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the genetic frontotemporal dementia initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. 2015;14:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lee SE, Sias AC, Mandelli ML, et al. Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. NeuroImage Clin. 2017;14:286–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhou J, Greicius MD, Gennatas ED, et al. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer’s disease. Brain. 2010;133:1352–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Filippi M, Agosta F, Scola E, et al. Functional network connectivity in the behavioral variant of frontotemporal dementia. Cortex. 2013;49:2389–2401. [DOI] [PubMed] [Google Scholar]
- 168.Bian H, Van Swieten JC, Leight S, et al. CSF biomarkers in frontotemporal lobar degeneration with known pathology. Neurology. 2008;70:1827–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Paterson RW, Slattery CF, Poole T, et al. Cerebrospinal fluid in the differential diagnosis of Alzheimer’s disease: clinical utility of an extended panel of biomarkers in a specialist cognitive clinic. Alzheimer’s Res Ther. 2018;10:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Katisko K, Cajanus A, Korhonen T, et al. Prodromal and early bvFTD: evaluating clinical features and current biomarkers. Front Neurosci. 2019;13:658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zetterberg H, van Swieten JC, Boxer AL, et al. Review: fluid biomarkers for frontotemporal dementias. Neuropathol Appl Neurobiol. 2019;45:81–87. [DOI] [PubMed] [Google Scholar]
- 172.Pijnenburg YA, Verwey NA, van der Flier WM, et al. Discriminative and prognostic potential of cerebrospinal fluid phosphoTau/tau ratio and neurofilaments for frontotemporal dementia subtypes. Alzheimers Dement (Amst). 2015;1:505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Niikado M, Chrem-Mendez P, Itzcovich T, et al. Evaluation of cerebrospinal fluid neurofilament light chain as a routine biomarker in a memory clinic. J Gerontol A Biol Sci Med Sci. 2019;74:442–445. [DOI] [PubMed] [Google Scholar]
- 174.Rohrer JD, Woollacott IO, Dick KM, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016;87:1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Skillback T, Farahmand B, Bartlett JW, et al. CSF neurofilament light differs in neurodegenerative diseases and predicts severity and survival. Neurology. 2014;83:1945–1953. [DOI] [PubMed] [Google Scholar]
- 176.Skillback T, Mattsson N, Blennow K, et al. Cerebrospinal fluid neurofilament light concentration in motor neuron disease and frontotemporal dementia predicts survival. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:397–403. [DOI] [PubMed] [Google Scholar]
- 177.Meeter LH, Dopper EG, Jiskoot LC, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol. 2016;3:623–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Steinacker P, Anderl-Straub S, Diehl-Schmid J, et al. Serum neurofilament light chain in behavioral variant frontotemporal dementia. Neurology. 2018;91:e1390–e1401. [DOI] [PubMed] [Google Scholar]
- 179.Sjogren M, Rosengren L, Minthon L, et al. Cytoskeleton proteins in CSF distinguish frontotemporal dementia from AD. Neurology. 2000;54:1960–1964. [DOI] [PubMed] [Google Scholar]
- 180.de Jong D, Jansen RW, Pijnenburg YA, et al. CSF neurofilament proteins in the differential diagnosis of dementia. J Neurol Neurosurg Psychiatry. 2007;78:936–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Al Shweiki MR, Steinacker P, Oeckl P, et al. Neurofilament light chain as a blood biomarker to differentiate psychiatric disorders from behavioural variant frontotemporal dementia. J Psychiatr Res. 2019;113:137–140. [DOI] [PubMed] [Google Scholar]
- 182.Wilke C, Preische O, Deuschle C, et al. Neurofilament light chain in FTD is elevated not only in cerebrospinal fluid, but also in serum. J Neurol Neurosurg Psychiatry. 2016;87:1270–1272. [DOI] [PubMed] [Google Scholar]
- 183.Perry DC, Brown JA, Possin KL, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. 2017;140:3329–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Meeter LHH, Gendron TF, Sias AC, et al. Poly(GP), neurofilament and grey matter deficits in C9orf72 expansion carriers. Ann Clin Transl Neurol. 2018;5:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ghidoni R, Benussi L, Glionna M, et al. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology. 2008;71:1235–1239. [DOI] [PubMed] [Google Scholar]
- 186.Morenas-Rodriguez E, Cervera-Carles L, Vilaplana E, et al. Progranulin protein levels in cerebrospinal fluid in primary neurodegenerative dementias. J Alzheimers Dis. 2016;50:539–546. [DOI] [PubMed] [Google Scholar]
- 187.Meeter LH, Patzke H, Loewen G, et al. Progranulin levels in plasma and cerebrospinal fluid in granulin mutation carriers. Dement Geriatr Cogn Dis Extra. 2016;6:330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Ghidoni R, Stoppani E, Rossi G, et al. Optimal plasma progranulin cutoff value for predicting null progranulin mutations in neurodegenerative diseases: a multicenter Italian study. Neurodegener Dis. 2012;9:121–127. [DOI] [PubMed] [Google Scholar]
- 189.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. [DOI] [PubMed] [Google Scholar]
- 190.Magnin E, Demonet JF, Wallon D, et al. Primary progressive aphasia in the network of french alzheimer plan memory centers. J Alzheimers Dis. 2016;54:1459–1471. [DOI] [PubMed] [Google Scholar]
- 191.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]; Guidelines for the diagnosis of Primary Progressive Aphasias.
- 192.Mesulam MM. Primary progressive aphasia. Ann Neurol. 2001;49:425–432. [PubMed] [Google Scholar]
- 193.Matias-Guiu JA, Cabrera-Martin MN, Moreno-Ramos T, et al. Clinical course of primary progressive aphasia: clinical and FDG-PET patterns. J Neurol. 2015;262:570–577. [DOI] [PubMed] [Google Scholar]
- 194.Spinelli EG, Mandelli ML, Miller ZA, et al. Typical and atypical pathology in primary progressive aphasia variants. Ann Neurol. 2017;81:430–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chare L, Hodges JR, Leyton CE, et al. New criteria for frontotemporal dementia syndromes: clinical and pathological diagnostic implications. J Neurol Neurosurg Psychiatry. 2014;85:865–870. [DOI] [PubMed] [Google Scholar]
- 196.Harris JM, Gall C, Thompson JC, et al. Classification and pathology of primary progressive aphasia. Neurology. 2013;81:1832–1839. [DOI] [PubMed] [Google Scholar]
- 197.Bergeron D, Gorno-Tempini ML, Rabinovici GD, et al. Prevalence of amyloid-beta pathology in distinct variants of primary progressive aphasia. Ann Neurol. 2018;84:729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Ramos EM, Dokuru DR, Van Berlo V, et al. Genetic screen in a large series of patients with primary progressive aphasia. Alzheimer’s Dementia. 2019;15:553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chan D, Fox NC, Scahill RI, et al. Patterns of temporal lobe atrophy in semantic dementia and Alzheimer’s disease. Ann Neurol. 2001;49:433–442. [PubMed] [Google Scholar]
- 200.Mesulam MM, Weintraub S, Rogalski EJ, et al. Asymmetry and heterogeneity of Alzheimer’s and frontotemporal pathology in primary progressive aphasia. Brain. 2014;137:1176–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gorno-Tempini ML, Brambati SM, Ginex V, et al. The logopenic/phonological variant of primary progressive aphasia. Neurology. 2008;71:1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Rohrer JD, Ridgway GR, Crutch SJ, et al. Progressive logopenic/phonological aphasia: erosion of the language network. NeuroImage. 2010;49:984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Seeley WW, Crawford RK, Zhou J, et al. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Santangelo R, Coppi E, Ferrari L, et al. Cerebrospinal fluid biomarkers can play a pivotal role in the diagnostic work up of primary progressive aphasia. J Alzheimers Dis. 2015;43:1429–1440. [DOI] [PubMed] [Google Scholar]
- 205.Paraskevas GP, Kasselimis D, Kourtidou E, et al. Cerebrospinal fluid biomarkers as a diagnostic tool of the underlying pathology of primary progressive aphasia. J Alzheimers Dis. 2017;55:1453–1461. [DOI] [PubMed] [Google Scholar]
- 206.Norise C, Ungrady M, Halpin A, et al. Clinical correlates of Alzheimer’s disease cerebrospinal fluid analytes in primary progressive aphasia. Front Neurol. 2019;10:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Agosta F, Scola E, Canu E, et al. White matter damage in frontotemporal lobar degeneration spectrum. Cereb Cortex. 2012;22:2705–2714. [DOI] [PubMed] [Google Scholar]
- 208.Agosta F, Galantucci S, Valsasina P, et al. Disrupted brain connectome in semantic variant of primary progressive aphasia. Neurobiol Aging. 2014;35:2646–2655. [DOI] [PubMed] [Google Scholar]
- 209.Meeter LHH, Vijverberg EG, Del Campo M, et al. Clinical value of neurofilament and phospho-tau/tau ratio in the frontotemporal dementia spectrum. Neurology. 2018;90:e1231–e1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Scherling CS, Hall T, Berisha F, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol. 2014;75:116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Steinacker P, Semler E, Anderl-Straub S, et al. Neurofilament as a blood marker for diagnosis and monitoring of primary progressive aphasias. Neurology. 2017;88:961–969. [DOI] [PubMed] [Google Scholar]
- 212.Hannibal L Nitric oxide homeostasis in neurodegenerative diseases. Curr Alzheimer Res. 2016;13:135–149. [DOI] [PubMed] [Google Scholar]
- 213.Al-Chalabi A, Hardiman O, Kiernan MC, et al. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 2016;15:1182–1194. [DOI] [PubMed] [Google Scholar]
- 214.van Es MA, Hardiman O, Chio A, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390:2084–2098. [DOI] [PubMed] [Google Scholar]; This is a very interesting review on the need of a data-driven classification for Amyotrophic Lateral Sclerosis.
- 215.Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:153–174. [DOI] [PMC free article] [PubMed] [Google Scholar]; The recent criteria for a diagnosis of dementia and cognitive imparment in Amyotrophic Lateral Sclerosis is stated out.
- 216.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- 217.Hardiman O, Al-Chalabi A, Chio A, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071. [DOI] [PubMed] [Google Scholar]
- 218.Chia R, Chio A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 2018;17:94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Chio A, Traynor BJ. Motor neuron disease in 2014Biomarkers for ALS–in search of the Promised Land. Nat Rev Neurol. 2015;11:72–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on motor neuron diseases/amyotrophic lateral sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis”workshop contributors. J Neurol Sci. 1994;124 (Suppl):96–107. [DOI] [PubMed] [Google Scholar]
- 221.Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotrophic lateral sclerosis and other motor neuron disorders. 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 222.de Carvalho M, Dengler R, Eisen A, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119:497–503. [DOI] [PubMed] [Google Scholar]
- 223.Carvalho MD, Swash M. Awaji diagnostic algorithm increases sensitivity of El Escorial criteria for ALS diagnosis. Amyotroph Lateral Scler. 2009;10:53–57. [DOI] [PubMed] [Google Scholar]; Electrophysiological alterations become a diagnostic clinical surrogate of Amyotrophic Lateral Sclerosis.
- 224.Geevasinga N, Menon P, Scherman DB, et al. Diagnostic criteria in amyotrophic lateral sclerosis: A multicenter prospective study. Neurology. 2016;87:684–690. [DOI] [PubMed] [Google Scholar]
- 225.Phukan J, Elamin M, Bede P, et al. The syndrome of cognitive impairment in amyotrophic lateral sclerosis: a population-based study. J Neurol Neurosurg Psychiatry. 2012;83:102–108. [DOI] [PubMed] [Google Scholar]
- 226.Strong MJ. The syndromes of frontotemporal dysfunction in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2008;9:323–338. [DOI] [PubMed] [Google Scholar]
- 227.Strong MJ, Grace GM, Freedman M, et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10:131–146. [DOI] [PubMed] [Google Scholar]
- 228.Tan RH, Guennewig B, Dobson-Stone C, et al. The underacknowledged PPA-ALS: A unique clinicopathologic subtype with strong heritability. Neurology. 2019;92:e1354–e1366. [DOI] [PubMed] [Google Scholar]
- 229.Iazzolino B, Pain D, Peotta L, et al. Validation of the revised classification of cognitive and behavioural impairment in ALS. J Neurol Neurosurg Psychiatry. 2019;90:734–739. [DOI] [PubMed] [Google Scholar]
- 230.Sabatelli M, Marangi G, Conte A, et al. New ALS-related genes expand the spectrum paradigm of amyotrophic lateral sclerosis. Brain Pathol. 2016;26:266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.McCluskey L, Vandriel S, Elman L, et al. ALS-Plus syndrome: non-pyramidal features in a large ALS cohort. J Neurol Sci. 2014;345:118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Brooks BR. ALS-Plus - where does it begin, where does it end? J Neurol Sci. 2014;345:1–2. [DOI] [PubMed] [Google Scholar]
- 233.Toschi N, Lista S, Baldacci F, et al. Biomarker-guided clustering of Alzheimer’s disease clinical syndromes. Neurobiol Aging. 2019;83:42–53. [DOI] [PubMed] [Google Scholar]; An example of an a posteriori data-driven stratfication of ALzheimer’s disease patients.
- 234.Blasco H, Vourc’h P, Pradat PF, et al. Further development of biomarkers in amyotrophic lateral sclerosis. Expert Rev Mol Diagn. 2016;16:853–868. [DOI] [PubMed] [Google Scholar]
- 235.Endo H, Sekiguchi K, Shimada H, et al. Low signal intensity in motor cortex on susceptibility-weighted MR imaging is correlated with clinical signs of amyotrophic lateral sclerosis: a pilot study. J Neurol. 2018;265:552–561. [DOI] [PubMed] [Google Scholar]
- 236.Verber NS, Shepheard SR, Sassani M, et al. Biomarkers in motor neuron disease: a state of the art review. Front Neurol. 2019;10:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Ellis CM, Simmons A, Andrews C, et al. A proton magnetic resonance spectroscopic study in ALS: correlation with clinical findings. Neurology. 1998;51:1104–1109. [DOI] [PubMed] [Google Scholar]
- 238.Schuff N, Rooney WD, Miller R, et al. Reanalysis of multislice (1)H MRSI in amyotrophic lateral sclerosis. Magn Reson Med. 2001;45:513–516. [DOI] [PubMed] [Google Scholar]
- 239.Mitsumoto H, Ulug AM, Pullman SL, et al. Quantitative objective markers for upper and lower motor neuron dysfunction in ALS. Neurology. 2007;68:1402–1410. [DOI] [PubMed] [Google Scholar]
- 240.Agosta F, Spinelli EG, Filippi M. Neuroimaging in amyotrophic lateral sclerosis: current and emerging uses. Expert Rev Neurother. 2018;18:395–406. [DOI] [PubMed] [Google Scholar]
- 241.Gaetani L, Blennow K, Calabresi P, et al. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. 2019;90:870–881. [DOI] [PubMed] [Google Scholar]
- 242.Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14:577–589. [DOI] [PubMed] [Google Scholar]
- 243.Blennow K A review of fluid biomarkers for Alzheimer’s disease: moving from CSF to blood. Neurol Ther. 2017;6:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Kuhle J, Barro C, Andreasson U, et al. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clin Chem Lab Med. 2016;54:1655–1661. [DOI] [PubMed] [Google Scholar]
- 245.Poesen K, De Schaepdryver M, Stubendorff B, et al. Neurofilament markers for ALS correlate with extent of upper and lower motor neuron disease. Neurology. 2017;88:2302–2309. [DOI] [PubMed] [Google Scholar]
- 246.Feneberg E, Oeckl P, Steinacker P, et al. Multicenter evaluation of neurofilaments in early symptom onset amyotrophic lateral sclerosis. Neurology. 2018;90:e22–e30. [DOI] [PubMed] [Google Scholar]
- 247.Lu CH, Macdonald-Wallis C, Gray E, et al. Neurofilament light chain: A prognostic biomarker in amyotrophic lateral sclerosis. Neurology. 2015;84:2247–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Gaiani A, Martinelli I, Bello L, et al. Diagnostic and prognostic biomarkers in amyotrophic lateral sclerosis: neurofilament light chain levels in definite subtypes of disease. JAMA Neurol. 2017;74:525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Baldacci F, Lista S, Palermo G, et al. The neuroinflammatory biomarker YKL-40 for neurodegenerative diseases: advances in development. Expert Rev Proteomics. 2019;16:593–600. [DOI] [PubMed] [Google Scholar]
- 250.Thompson AG, Gray E, Thezenas ML, et al. Cerebrospinal fluid macrophage biomarkers in amyotrophic lateral sclerosis. Ann Neurol. 2018;83:258–268. [DOI] [PubMed] [Google Scholar]
- 251.Illan-Gala I, Alcolea D, Montal V, et al. CSF sAPPbeta, YKL-40, and NfL along the ALS-FTD spectrum. Neurology. 2018;91:e1619–e1628. [DOI] [PubMed] [Google Scholar]
- 252.Verde F, Steinacker P, Weishaupt JH, et al. Neurofilament light chain in serum for the diagnosis of amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2019;90:157–164. [DOI] [PubMed] [Google Scholar]
- 253.Thouvenot E, Demattei C, Lehmann S, et al. Serum neurofilament light chain at time of diagnosis is an independent prognostic factor of survival in amyotrophic lateral sclerosis. Eur J Neurol. 2020;27:251–257. [DOI] [PubMed] [Google Scholar]
- 254.Gille B, De Schaepdryver M, Goossens J, et al. Serum neurofilament light chain levels as a marker of upper motor neuron degeneration in patients with Amyotrophic Lateral Sclerosis. Neuropathol Appl Neurobiol. 2019;45:291–304. [DOI] [PubMed] [Google Scholar]
- 255.Hampel H, O’Bryant SE, Molinuevo JL, et al. Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat Rev Neurol. 2018;14:639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Candelise N, Schmitz M, Da Silva Correia SM, et al. Applications of the real-time quaking-induced conversion assay in diagnosis, prion strain-typing, drug pre-screening and other amyloidopathies. Expert Rev Mol Diagn. 2017;17:897–904. [DOI] [PubMed] [Google Scholar]
- 257.Asai H, Ikezu S, Tsunoda S, et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci. 2015;18:1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Fiandaca MS, Kapogiannis D, Mapstone M, et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case-control study. Alzheimer’s Dementia. 2015;11:600–607.e601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Goetzl EJ, Boxer A, Schwartz JB, et al. Low neural exosomal levels of cellular survival factors in Alzheimer’s disease. Ann Clin Transl Neurol. 2015;2:769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Hamlett ED, Goetzl EJ, Ledreux A, et al. Neuronal exosomes reveal Alzheimer’s disease biomarkers in Down syndrome. Alzheimer’s Dementia. 2017;13:541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Coleman BM, Hill AF. Extracellular vesicles–Their role in the packaging and spread of misfolded proteins associated with neurodegenerative diseases. Semin Cell Dev Biol. 2015;40:89–96. [DOI] [PubMed] [Google Scholar]
- 262.Kumar S, Reddy PH. Are circulating microRNAs peripheral biomarkers for Alzheimer’s disease? Biochim Biophys Acta. 2016;1862:1617–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.D’Anca M, Fenoglio C, Serpente M, et al. Exosome determinants of physiological aging and age-related neurodegenerative diseases. Front Aging Neurosci. 2019;11:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Jain G, Stuendl A, Rao P, et al. A combined miRNA-piRNA signature to detect Alzheimer’s disease. Transl Psychiatry. 2019;9:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Hampel H, Lista S, Neri C, et al. Time for the systems-level integration of aging: resilience enhancing strategies to prevent Alzheimer’s disease. Prog Neurobiol. 2019;181:101662. [DOI] [PubMed] [Google Scholar]
- 266.Perez-Nievas BG, Stein TD, Tai HC, et al. Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain. 2013;136:2510–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Dourlen P, Kilinc D, Malmanche N, et al. The new genetic landscape of Alzheimer’s disease: from amyloid cascade to genetically driven synaptic failure hypothesis? Acta Neuropathol. 2019;138:221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Castrillo JI, Lista S, Hampel H, et al. systems biology methods for Alzheimer’s disease research toward molecular signatures, subtypes, and stages and precision medicine: application in cohort studies and trials. Methods Mol Biol. 2018;1750:31–66. [DOI] [PubMed] [Google Scholar]
- 269.Castrillo JI, Oliver SG. Alzheimer’s as a systems-level disease involving the interplay of multiple cellular networks. Methods Mol Biol. 2016;1303:3–48. [DOI] [PubMed] [Google Scholar]
- 270.Cummings J, Fox N, Vellas B, et al. Biomarker and clinical trial design support for disease-modifying therapies: report of a survey of the EU/US: Alzheimer’s disease task force. J Prev Alzheimer’s Dis. 2018;5:103–109. [DOI] [PubMed] [Google Scholar]
- 271.Kozakov D, Hall DR, Napoleon RL, et al. New frontiers in druggability. J Med Chem. 2015;58:9063–9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Wu F, Ma C, Tan C. Network motifs modulate druggability of cellular targets. Sci Rep. 2016;6:36626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Elman JA, Oh H, Madison CM, et al. Neural compensation in older people with brain amyloid-beta deposition. Nat Neurosci. 2014;17:1316–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Geerts H, Dacks PA, Devanarayan V, et al. Big data to smart data in Alzheimer’s disease: the brain health modeling initiative to foster actionable knowledge. Alzheimers Dement. 2016;12:1014–1021. [DOI] [PubMed] [Google Scholar]; A very innovative paper on the potential exploitation of Bigdata in Medicine.
- 275.Haas M, Stephenson D, Romero K, et al. Big data to smart data in Alzheimer’s disease: real-world examples of advanced modeling and simulation. Alzheimers Dement. 2016;12:1022–1030. [DOI] [PubMed] [Google Scholar]; A very innovative paper on the potential exploitation of Bigdata in Medicine.